

Abstract
Genome-wide association studies (GWAS) have identified hundreds of primarily non-coding disease-susceptibility variants that further need functional interpretation to prioritize and discriminate the disease-relevant variants. We present a comprehensive genome-wide non-coding variants prioritization scheme followed by validation using Pyrosequencing and TaqMan assays in asthma. We implemented a composite Functional Annotation Score (cFAS) to investigate over 32,000 variants consisting of 1,525 GWAS-lead asthma-susceptibility variants and their LD proxies (r2≥0.80). Functional annotation pipeline in cFAS revealed 274 variants with significant score at 1% false discovery rate. This study implicates a novel locus 4p16 (SLC26A1) with eQTL variant (rs11936407) and known loci in 17q12–21 and 5q22 which encode ORM1-lie protein 3 (ORMDL3, rs406527 and rs12936231) and Thymic stromal lymphopoietin (TSLP, rs3806932 and rs10073816) epithelial gene, respectively. Follow-up validation analysis through pyrosequencing of CpG sites in and nearby rs4065275 and rs11936407 showed genotype dependent hypomethylation on asthma cases compared with healthy controls. Prioritized variants are enriched for asthma-specific histone modification associated with active chromatin (H3K4me1 and H3K27ac) in T cells, B cells, lung, and immune related interferon gamma signaling pathways. Our findings, together with those from prior studies, suggest that SNPs can affect asthma by regulating enhancer activity, and our comprehensive bioinformatics and functional analysis could lead to biological insights into asthma pathogenesis.
Keywords: Variant Prioritization, Functional Annotation, GTEx, Roadmap Epigenomics, Pyrosequencing, TaqMan assays
GRAPHICAL ABSTRACT
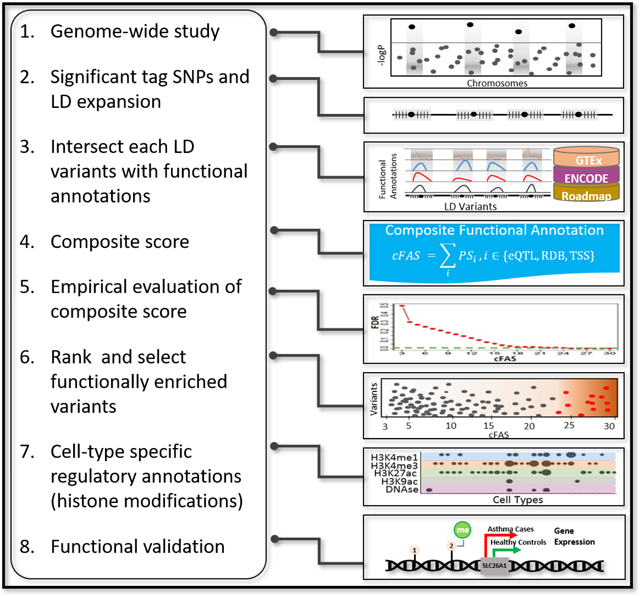
INTRODUCTION
Asthma is a complex respiratory disease characterized by multiple clinical symptoms such as wheeze, breathlessness, chest tightness, and cough. It is one of the most common non-communicable chronic disease that affects both children and adults worldwide. It affects over 6 million children in the U.S., leading to more than 3.5 million exacerbations, 135,000 hospitalizations, and around 14 million missed school days each year (Zahran et al. 2011). Over the decade, large scale genome-wide studies have identified hundreds of genetic variants that potentially contribute to asthma (Buniello et al. 2019) and provided a rich genome-wide atlas of disease-susceptibility variants. However, the functional mechanism of these variants and hence, their role in asthma is not well understood. The main strength of genome-wide analyses is their ability to systematically explore novel variants associated with a disease. However, the genome-wide SNP arrays do not necessarily represent the causal variants that underlie the molecular mechanism of the association (Hindorff et al. 2009). Other SNPs in high linkage disequilibrium (LD) block with the index SNPs can be causal for the disease (Schaub et al. 2012). The majority of genome-wide variants and their LD surrogates localize to non-coding intergenic and intronic regulatory regions rather than to protein-coding regions. Thus, their major role is rather to regulate changes in the gene expression of a critical gene through multiple mechanisms involving RNA splicing, transcription factor binding, chromatin openness measured by DNase I hypersensitivity, DNA methylation, miRNA recruitment, and histone modifications (Freedman et al. 2011; Maurano et al. 2012). Many of these regulatory variants operate in a tissue- and cell-type-specific manner (Dimas et al. 2009; Freedman et al. 2011; Hindorff et al. 2009; Maurano et al. 2012; Schaub et al. 2012). Hence, the functional characterization of the variants is critical for understanding the functional mechanism of these variants on asthma. Therefore, functional annotations and interpretation are important to translate the statistical findings into actionable functional mechanism and biology on the prognosis of the diseases.
Recently, several large-scale consortia such as the ENCODE, Genotype-Tissue Expression (GTEx), and NIH Roadmap Epigenomic have provided a rich atlas of functional annotations of non-coding variants (Consortium 2012; Lonsdale et al. 2013; Roadmap Epigenomics et al. 2015). Multiple annotations from these resources can be combined for functional characterization of the non-coding variants which in turn can be used for prioritization of non-coding variants (RegulomeDB (Boyle et al. 2012), GWAVA (Ritchie et al. 2014)). As the GWAS-discovered susceptibility variants for asthma spans over hundreds of loci, potentially mapped to several thousand SNPs consisting of both index and LD SNPs, primarily non-coding, functional prioritization is warranted to discriminate the most plausible functional variants from the benign variants. Concurrently, several tools were built to leverage the functional annotation resources and prioritize non-coding variants, however these tools are primarily utilizing the regulatory annotations. Addition to the regulatory annotations, eQTL annotations can be informative to characterize the variation in gene expression levels, and thus the incorporation of comprehensive eQTL analysis may strengthen prioritization of non-coding variants in asthma.
In this article, we sought to identify, prioritize and validate the functionally enriched asthma-susceptibility GWAS variants using a heuristic algorithm that composite the functional annotation scores followed by pyrosequencing. To obtained a comprehensive list of disease-susceptibility variants for prioritization, we searched and extracted asthma-associated variants from three public repository databases including NHGRI-EBI GWAS Catalog (MacArthur et al. 2017), dbGaP (Mailman et al. 2007), and GRASP (Leslie et al. 2014). We further performed three GWASs (CAMP-CARE, STAMPEED, and GABRIEL) in a total of 29,371 samples from European and African ancestry. Altogether, we obtained 1,525 asthma-associated risk-variants with significance level p-value < 1×10−5. In addition, we investigated the 1000 Genomes Project (Genomes Project et al. 2015) for SNPs in high LD (r2 ≥ 0.8) with the lead SNPs, resulting in a total of 32,161 SNPs. Using the cFAS scheme for functional annotation and prioritization, we identified 274 variants associated with asthma. We further prioritize these variants for tissue-specific eQTL, regulatory elements and epigenomic marks. Finally, we conducted functional validation analysis using pyrosequencing and demonstrated that selected SNP variants (rs4065275 and rs11936407) located in the known and novel locus prioritized by cFAS were hypomethylated in a genotype dependent manner when comparing asthma cases with healthy controls. Also, variants nearby the prioritized SNPs were hypomethylated in cases when compared to controls. Our results showed that the prioritized SNPs are highly co-localized in the DNase I hypersensitive sites, the enhancer and promoter regions marked by histone methylation and acetylation, and in protein binding regions.
Materials and methods
Comprehensive discovery of asthma-associated risk variants
Asthma-associated variants from three different genome-wide association studies and from three public databases were included and investigated as described below and outlined in Fig 1A.
Fig 1. Workflow of asthma associated variants discovery and prioritization scheme.
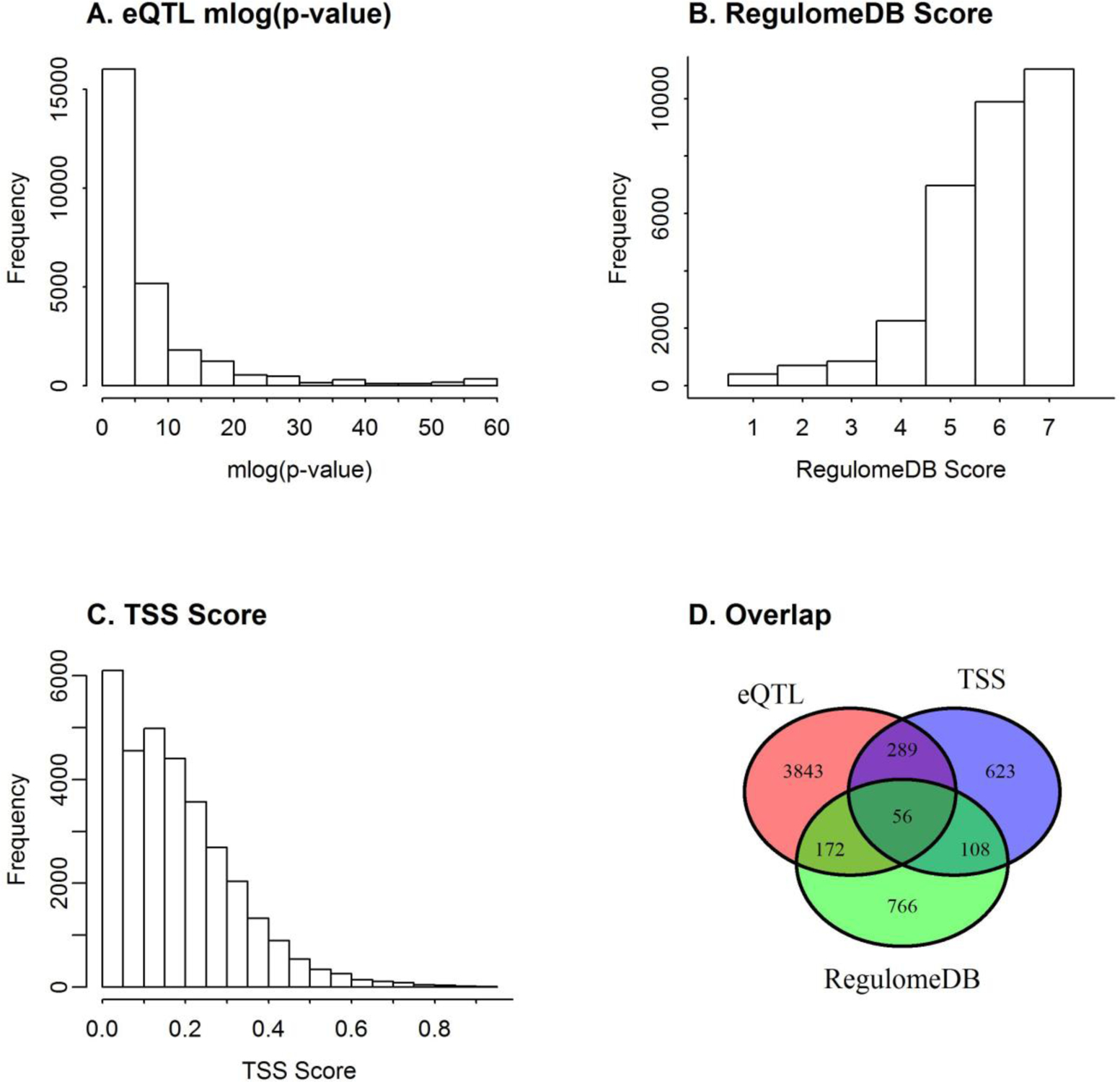
A) Flow chart shows the different sources of asthma-associated SNPs and selection process discovering asthma-associated variants. AA = African American, EA = European American. B) Manhattan plots for GWAS-lead asthma variants. Asthma associated SNPs with significance cutoff p-value < 1E-5 from multiple GWAS studies and public catalogs were used. C) Variant prioritization step. Schematic approach of variant annotation and assigning composite functional annotation score (cFAS). D) Manhattan plot of the composite cFAS. Horizontal dashed line shows the cutoff score cFAS = 22 used for the variant prioritization
GWAS public repository databases.
We searched three different GWAS repositories for asthma studies - NHGRI-EBI catalog published GWAS (MacArthur et al. 2017), the NHLBI GRASP catalog (Leslie et al. 2014), and Phenotype-Genotype Integrator (PheGenI (Ramos et al. 2014)) from dbGap. Using the GWAS catalog reported cutoff (p-values < 1×10−5), a total of 1,472 SNPs were extracted (accessed date: 7/20/2019).
CAMP-CARE genome-wide study.
Genotype and phenotype data from the Children Asthma Management Program (CAMP) and Childhood Asthma Research and Education (CARE) networks were accessed from dbGaP under accession number phs000166.v2.p1 with proper approval (Mailman et al. 2007). The dataset consists of genotype data for 429 parent-offspring trios of European ancestry (334 CAMP and 95 CARE) and 52 trios of African American ancestry (42 CAMP and 10 CARE). All the participants were genotyped on Affymatrix 6.0 chip, and the SNPs were filtered using the quality control (QC) criteria (missingness ≥ 15% and the Hardy-Weinberg Disequilibrium p < 1×10−5). Upon QC completion, the family-based transmission disequilibrium test (TDT) for affected offspring trio design was run using PLINK 1.90 (Purcell et al. 2007). Quantile-Quantile (QQ) plots were produced and checked for each ancestry. From the final association result, we obtained 56 SNPs.
STAMPEED genome-wide study.
With proper authorization under dbGaP accession phs000355.v1.p1, we accessed STAMPEED datasets, which comprised summary results of the case-control GWAS for African American (AA) and European American (EA) ancestry.(Mailman et al. 2007) Participants in STAMPEED were one of the three studies (Chicago Asthma Genetic (CAG) study, the NHLBI Collaborative Studies on the Genetics of Asthma (CSGA), and the Severe Asthma Research Center (SARP), which jointly enrolled 541 case and 451 control subjects from African American ancestry and 843 case and 580 control subjects from European ancestry (Mailman et al. 2007). All subjects were genotyped on Illumina 1Mv1 Chip. SNPs were filtered following standard QC criteria – (i) call rate < 95%, (ii) Hardy-Weinberg equilibrium p-value >1×10−5. Following the QC filter, a case-control GWAS for AA and EA data was carried out using the logistic regression model adjusting for age, sex, and population structure (the first two principal components for EA, and local ancestry estimates for AA study) using PLINK 1.90 (Purcell et al. 2007). In total, 4 SNPs from AA ancestry and 3 SNPs from EA were selected.
GABRIEL genome-wide study.
The GABRIEL asthma GWAS dataset consisted of 23 different studies with a total of 10,365 cases and 16,110 control samples from European ancestry (Moffatt et al. 2010). Genotyping was carried out using Illumina Human610 quad array except in the GABRIEL phase I study. The GABRIEL Phase I subjects were genotyped on Illumina Hap300K array, and the untyped SNPs were imputed to be included in the meta-analysis. SNPs were filtered and removed for further analysis using the following QC criteria: (i) genotype missing rate ≥ 3% in cases and controls, (ii) minor allele frequency < 5% in controls, and (iii) Hardy-Weinberg P-value < 1×10−4. Genome wide association study was carried out on study-by-study basis using logistic regression model. The first two principal components for European population structure were included as covariates in the association analysis (Moffatt et al. 2010). A total of 124 SNPs were included.
SNPs in linkage disequilibrium.
In total, we obtained 1,525 asthma-associated variants from the three asthma GWAS and publicly deposited sources (Fig 1B, Supplementary Table S1). We further expanded the asthma-susceptibility list with LD variants from the 1000 Genome Project Phase III retrieved using LDlink tool(Machiela and Chanock 2015). Since the risk SNPs in the public databases stem from GWASs with different ethnic backgrounds, we obtained all LD SNPs (r2 ≥ 0.80) of five major continental populations from The 1000 Genomes Project Phase III (Genomes Project et al. 2015). This LD expansion process resulted in ‘asthma-LD set’ of 32,162 SNPs (consisting of both GWAS-lead variants and their high LD surrogates).
Functional annotations, correlation, and concordance analysis
To prioritize the asthma-associated variants for their potential functional relevance in disease pathogenies, we developed the composite functional annotation score (cFAS) based on the three functional annotation databases: (i) Genotype-Tissue Expression (GTEx) project v7 (Lonsdale et al. 2013), (ii) RegulomeDB (Boyle et al. 2012), and (iii) Genome-wide Annotations of Variants (GWAVA) (Ritchie et al. 2014). The selection of these resources was based on public availability and complementary information provided by each and their uniqueness in functional information for scoring scheme. Also, these bases provide a rich annotations of the non-coding variants with the most comprehensive coverage of regulatory variants. GTEx is uniquely suited for comprehensive evaluation of eQTL variants. RegulomeDB uses some combinatorial approach to categorize SNPs into discrete levels of Category 1 to Category 7 with Category 1, 2, 3 are further subdivide into sub-categories. According to RegulomeDB scoring mechanism, SNPs stronger evidence of potentially functional will be assigned to a lower category. SNPs in Category 1 and 2 are considered to be functionally enriched (Boyle et al. 2012). GWAVA employs a random forest approach to assign a unique functional predictive score for all markers from the 1000 Genomes Project. The GWAVA TSS score ranges from 0 to 1 with score ≥ 0.5 suggested to be potentially functional (Ritchie et al. 2014). Detail description of the annotation databases is provided in the Supplementary Text.
Correlation:
The Spearman rank correlation coefficient was used to investigate the similarity among the tools and is defined as:
where xi and yi are the rankings of i-th variant from the two methods; and are the respective average rankings of xi and yi. The correlation coefficient (r) ranges from −1 to 1; with r closer to 1 or −1 indicating a monotonically increasing or decreasing relationship and r closer to 0 signifying weak or no relationship.
Concordance rate:
The concordance rate measures the proportion of top ranked and shared variants among the eQTL, GWAVA TSS score, and RegulomeDB category. Variants were ranked from most to least significant and compared using overlap analysis for different numbers of ranked variants (n =100, 200, 300, 500, 750, and 1000). If m is the number of overlapped variants within the top t percentile (t = 1, 2, 3… 100) in eQTL, GWAVA TSS score, and RegulomeDB, and N is the total number of variants analyzed, then the concordance rate (ζ) is defined as:
Composite functional annotation and variant prioritization
The asthma-LD set was annotated using the composite functional annotation score (cFAS), a heuristic approach of integrating the GTEx, RegulomeDB, and GWAVA TSS annotations. Briefly, we first transformed the eQTL, RegulomeDB, and GWAVA TSS annotation scores into comparable ranking system that portray into a similar rating as represented by the respective scores. To this end, we defined three priority scores, PSeQTL, PSRDB, and PSTSS; each ranging from 1 to 10 as described below.
The cFAS was computed as the sum of the three priority scores as defined below:
The asthma-LD set is prioritized using the cFAS and variants were ranked from highest to lowest ranking. For an empirical evaluation of cFAS, we constructed a positive set, consisting of disease-susceptibility variants from GWAS catalog with the significant p-value <1×10−5 (accessed July 20,2019), and a negative set, consisted of common variants from the 1000 Genome Project excluding variants in the positive set and/or in high LD (r2 ≥ 0.8) with the positive set. We compared the distribution of cFAS among the positive set and negative set to compute the empirical FDR. For a given annotation score θ (regardless of the method), let of SNPs with scores equal or extreme to θ in the negative set (# of false positives) and of SNPs with scores equal or extreme to θ in the positive set (# of true positives), then the empirical FDR (FDRθ) is computed as: . The cFAS cutoff corresponding to FDR ≤ 0.01 was used for selection of the prioritize asthma variants.
Functional characterization of the prioritized SNPs
We explored different functional annotation resources including ENCODE and Roadmap Epigenome on chromatin states and protein binding annotations, histone modification, gene expression, sequence conservation, regulatory and protein binding affinities for further characterization of variants prioritized using cFAS. We used HaploReg v4.1 tool to extract the eQTL, regulatory, and epigenomic annotation results (Ward and Kellis 2016).
Tissue-specific eQTLs associated to prioritized SNPs.
We extracted tissue-specific gene-expression results from 14 different eQTL studies including the GTEx project. The list of databases interrogated from the tool was detailed in the work of Ward and Kellis (Ward and Kellis 2016). We extracted all available association results with p-value < 0.05 from the HaploReg (See Supplementary Text).
Enrichment analysis.
To understand the functional role of the asthma-prioritized variants, we further investigated whether the prioritized variants are enriched for cell type specific histone peaks such as H3K4me1/H3K4me3 and H3K27ac/H3K9ac marks and DNase I-hypersensitive sites. For the background variants, we downloaded variants form the GWAS catalog and randomly selected 3000 variants. Next, we extracted the cell-type specific annotations on four histone peaks (H3K4me1, H3K4me3, H3K27ac, and H3K9ac), DNase-I mark, and binding proteins for the prioritized and background variants. The enrichment of the asthma-prioritized variants relative to the background variants were evaluated using the binomial test as described below. Let n0 and m0 be the overlap of an annotation with the background and the prioritized variants on respectively and let n and m be the total number of variants in the background and in the prioritized set, respectively. To assess the statistical significance, the p-value is computed as:
To correct for the multiple testing, we computed the FDR using the Benjamini-Hochberg method (Benjamini and Hochberg 1995).
The enrichment analysis of histone peaks and DNase-I mark were performed on individual cell-types which consisted of 127 reference epigemones from the Roadmap Epigenome and ENCODE projects extracted using HaploReg v4.1 (see Supplementary Text).
Pathway analyses.
To further explore biological pathways and functional clustering of the variants identified by cFAS, we applied ConsensusPathDB (Herwig et al. 2016). The candidate variants were further mined by Literature Lab™ from Acumenta Biotech for functional clustering analysis (http://acumenta.com/). Literature Lab is an interface between experimentally derived gene lists and scientific literature in a curated vocabulary of 24,000 biological and biochemical terms. It employs statistical and clustering analysis on over 15.73 million PubMed abstracts (since 01/01/90 until present) to identify pathways and diseases. The analysis engine compares statistically the submitted gene set to 1,000 randomly generated gene sets to identify terms that are associated with the gene set more than by chance alone.
Bisulfite Pyrosequencing to validate cFAS based functional variants.
DNA samples from 60 children (30 asthmatics and 30 controls) from the Cincinnati metro-area were selected for analysis. Cohort description, inclusion and exclusion criteria were described elsewhere (Baye et al. 2011; Butsch Kovacic et al. 2012). Demographic information about the samples specific to this study is found in Supplementary Table S2. Genomic DNA was bisulfite treated and purified using the EZ DNA methylation-Gold Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s specifications. Targeted regions were amplify using the Pyromark PCR Kit (Qiagen, Valencia, CA, USA) following manufacturer’s protocol. Pyrosequencing was carried out using Pyro Gold reagents with a PyroMark vacuum prep workstation and a PyroMark Q96 MD instrument (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Generated pyrograms were automatically analyzed by the Pyro Q-CpG methylation analysis software (Qiagen, Valencia, CA, USA). The Human Methylated & Non-methylated DNA Set (Zymo Research, Irvine, CA, USA) was used to validate all assays. Pyrosequencing assay design primers and analysis sequence can be found in Supplementary Table S3. SNP genotyping was carried out using TaqMan genotyping assays (Life Technologies, NY, USA) for SNPs rs4065275 and rs11936407. Fisher’s exact test was used to compare the difference in DNA methylation between asthmatics and healthy controls. A P-value of less than 0.05 was considered significant.
Results
Discovery of asthma-associated risk variants
GWAS-associated asthma-risk variants were obtained from three public databases –(GWAS catalog (MacArthur et al. 2017), GRASP catalog (Leslie et al. 2014), and dbGap (Mailman et al. 2007), accessed on July 20, 2019), and three genome-wide association analyses (CAMP-CARE, STAMPEED, and GABRIEL) (see Q-Q plot in Supplementary Fig S1A–S1E). Using the GWAS catalog reported cutoff p-value (p < 1×10−5), we obtained a total 1,472 SNPs from public repositories and 187 SNPs from the three GWAS (Fig 1A), which combined resulted in 1,525 unique set of asthma-associated variants (Supplementary Table S1). These variants were found to be distributed across the genome with chromosomes 1, 6 and 17 highly represented (Fig 1B, Fig S1F).The functional annotation of each SNP with RefSeq gene information was conducted using ANNOVAR (Wang et al. 2010). Of the 1,525 SNPs, we found 50 SNPs (3.2 %) in the exonic region in consistent to previous report (Maurano et al. 2012). Further analysis showed that about 665 (44%) and 658 (43%) SNPs are intronic and intergenic, respectively.
GWAS-lead and high-LD variants
We searched the 1000 Genome Project Phase III data (Genomes Project et al. 2015) using the LDlink tool (Machiela and Chanock 2015) for SNPs with high LD (r2 ≥ 0.8) with the GWAS-lead set of asthma-associated SNPs, and a total of 32,161 SNPs were identified (Fig 1C).
Variant annotation, correlation and overlap analysis
The distributions of the different annotation scores among the 32,161 SNPs variants are shown in Fig 2. Approximately one third of the variants were found with eQTL signal with nominal p-value < 1E-5 in one or more tissues from GTEx (Fig 2A). When evaluated based on the default criteria of RegulomeDB or TSS, vast majority of the SNPs were identified with weak or no functional evidence (Fig 2B–2C). Among the 32,161 SNPs, there were 1,102 SNPs (~ 3.4%) with RegulomeDB category 1 or 2 and similar number of SNPs (1,076, ~3.35%) with TSS score ≥ 0.5; only 15% of the functional variants based on the TSS were also categorized as the functional variants based on the RegulomeDB. To compare the top signals among the three annotation approaches, we used stringent criteria of p-value < 1E-12 to be considered as strong eQTL signals. There were 4,360 SNPs (~13%) with eQTL p-value < 1E-12. We found 56 SNPs overlapped among these three annotation resources, (Fig 2D).
Fig 2. Distribution of functional annotation scores.
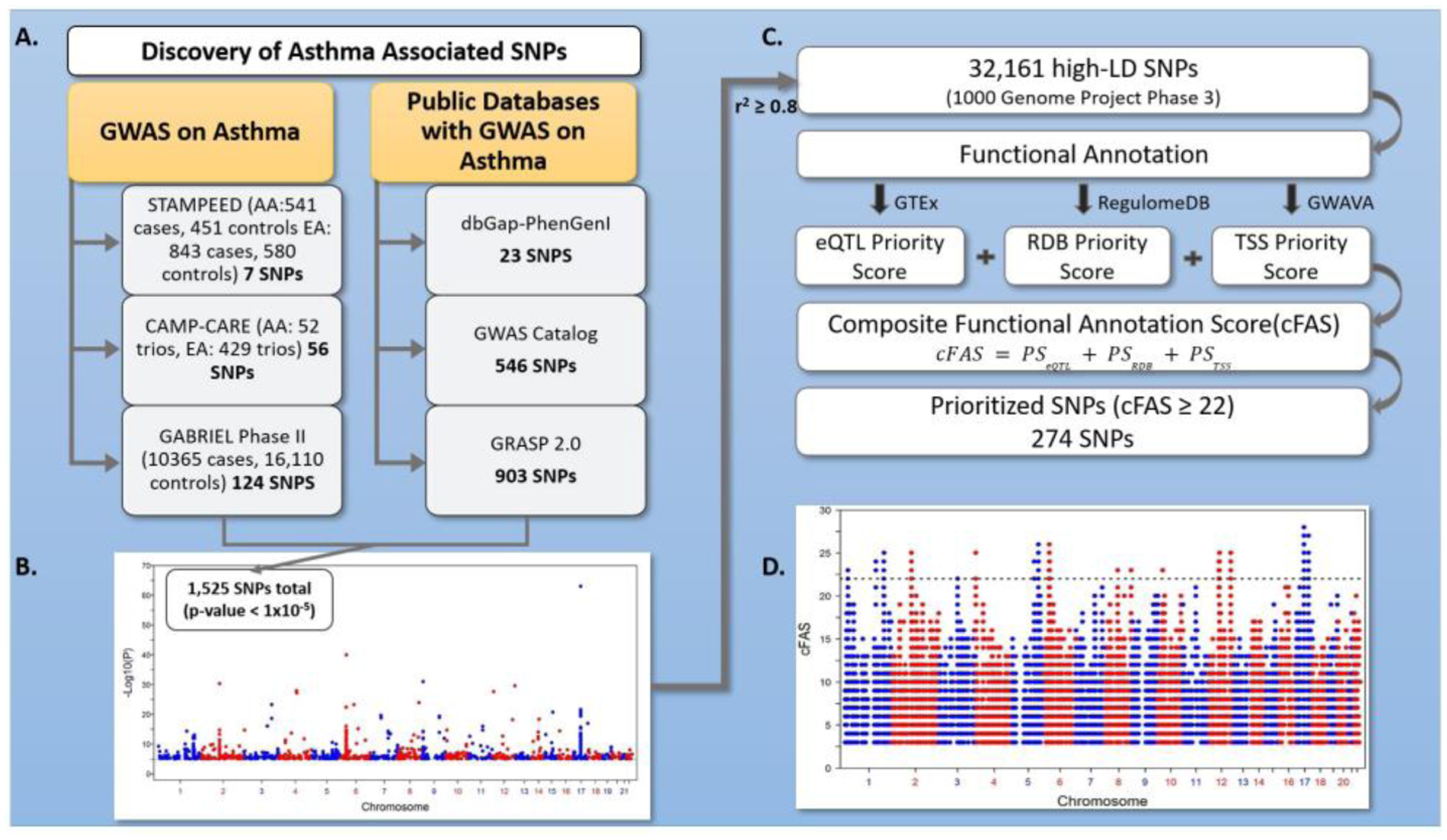
A) Histogram of eQTL p-value from the GTEx project. B) Bar plot of the RegulomeDB scores. C) Histogram of the GWAVA TSS score. D) Venn diagram showing the overlap of variants with eQTL p-value < 1E-12, RegulomeDB score ≤ 2, and TSS ≥ 0.5.
Next, we computed the pairwise Spearman rank correlation coefficients between the annotation scores to study the level of similarity. The pairwise distribution of the annotation scores showed high discrepancies in the ranking of variants among the three annotation scores (Supplementary Fig S2). There was a mild correlation between the RegulomeDB and TSS scores (r = 0.356), however, eQTL was weakly correlated to both RegulomeDB (r = 0.1354) and TSS (r = 0.0775). These discrepancies are pointing towards the significance of a composite score that mines the complementary evidence and combines the annotations from these tools to rank and prioritize risk-variants.
To gain insights into the variants shared among the eQTL, RegulomeDB and TSS scores, we performed an overlap analysis of the top-ranked variants in each method (n = 100, 200, 300, 500, 750, and 1000). For the top 100 SNPs, no SNP was overlapped among the three annotations. Among the top 1000 SNPs from each annotation, only 12 SNPs were overlapped (Supplementary Fig S3). The low overlap among the various functional annotation resources implies the need to develop a composite score using multiple annotation resources.
Composite functional annotation score (cFAS) and variant prioritization
Given the low number of overlap among the three functional annotation resources, we integrated all of them into a single composite. The composite cFAS scheme allowed us not only to comprehensively mine the annotation information into one score, but also provide a better control of the FDR to prioritize variants with strong, unified and complementary functional evidence. We first mapped the annotation scores into respective priority scores: eQTL → PSeQTL, RegulomeDB → PSRDB, and TSS → PSTSS. Each priority score ranges from 1 to 10 with higher score reflecting higher degree of functional evidence. Then, cFAS is defined as the sum of the three priority scores, i.e., cFAS = PSeQTL + PSRDB + PSTSS, and ranges from 3 to 30 [See the Materials and Methods section for details]. With this approach, cFAS integrates the annotation evidence from GTEx, RegulomeDB, and GWAVA resources into a single score and prioritizes the SNPs for their functional importance.
cFAS value was assigned to 32,161 SNPs, and the variants were ranked based on this score. The cFAS score varied from 3 to 28 (with no SNP receiving the maximum possible value of 30)(Supplementary Table S4).We evaluated the cFAS approach using empirical FDR by comparing cFASs between the association set and control set, each consisting of more than 29,000 variants as described above (See the Materials and Methods section for details). Supplementary Table S5 summarized the FDR for all possible cFAS values. Results showed that cFAS ≥ 16 resulted in FDR < 0.05 and cFAS ≥ 22 achieved more stringent FDR < 0.01.
To select the top prioritized asthma-variants for further functional characterization, we chose the cutoff cFAS ≥ 22 corresponding to FDR ≤ 0.01. Using the cutoff cFAS ≥ 22, we obtained 274 SNPs across the genome (Supplementary Table S6). The cFAS prioritized asthma variants were distributed across multiple genomic loci (Fig 1D). Chromosomes 6 and 17 were the two most enriched chromosomes with 138 and 60 prioritized variants, respectively. Among the prioritized set, only 38 SNPs were among the GWAS-lead asthma-associated SNPs, and the rest were in high LD with one or more of the GWAS-lead SNPs. This clearly pointed out the benefit of incorporating LD in the search of functional SNPs. All 134 prioritized SNPs in chromosome 6 were located in 6p21 locus that contained the major histone complexity (MHC) region. Nine of the 134 SNPs in the locus were GWAS-lead SNPs and rest of the SNPs were LD SNPs. Out of 60 SNPs in chromosome 17, 49 (22 GWAS-lead) variants were located in known asthma linked region 17q12-q21 containing several highly replicated asthma genes such as PGAP3, ORMDL3, GSDMA, ZPBP2, and GSDMB. Among the highly represented loci with cFAS ≥ 22 were 2q12.1 (10 SNPs), 5q31.1 (12 SNPs), 12q24.13 (15 SNPs), 12q13.2 (15 SNPs), and 17q23.3 (11 SNPs). Other genomic loci identified with at least one SNP with cFAS ≥ 22 were 1p36.13 (4 SNPs), 1q23.3 (2 SNPs), 1q32.1 (4 SNPs), 3q12.1 (2 SNPs), 4p16.3 (2 SNPs), 5q22.1 (2 SNPs), 8q13.1 (2 SNPs), 8q22.22 (1 SNP), 10p12.1 (1 SNP), and 12q13.13 (4 SNPs) (Fig 1D, Supplementary Table S6).
Comparison of cFAS with existing genome-wide functional annotation tools
To investigate the performance of cFAS and other three individual annotation approaches on the GWAS-based variants, we compared the false discovery rate (FDR) among the annotation scores computed using a GWAS catalog-based association set and a random control set [see the Materials and method section for details]. The FDR is defined as the ratio FP/(TP +FP), TP = True Positive, FP = False Positive. TP and FP are defined as the number of variants selected under a given criteria in the association set and in the control set, respectively. The FDR was found to be less than 0.05 for eQTL cutoff of p-value < 1E-12, but the FDRs were 0.14 for RegulomeDB ≤ 2 and 0.198 for TSS ≥ 0.5, which were several-fold larger than nominally used significance level of FDR ≤ 0.05 (Fig S4A – S4C). The FDR for cFAS ≥ 22 was 0.01 (Fig S4D). We further compared the performance of cFAS with other tools using the receiver operating curve (ROC) (Fig 3). Using the association and control sets, the area under curve (AUC) value for cFAS was 0.8289 which was higher than that for TSS (AUC = 0.7747) and RegulomeDB (AUC = 0.7146). We further investigated the performance of the combined priority scores of TSS and RDB without the eQTL priority score. The AUC for the composite score based on TSS priority score (PRTSS) and RDB priority score (PRRDB) was 0.7934. This showed that incorporating eQTL for functional annotation improved the performance of composite score.
Fig 3. Comparison of several functional prioritization tools performance.
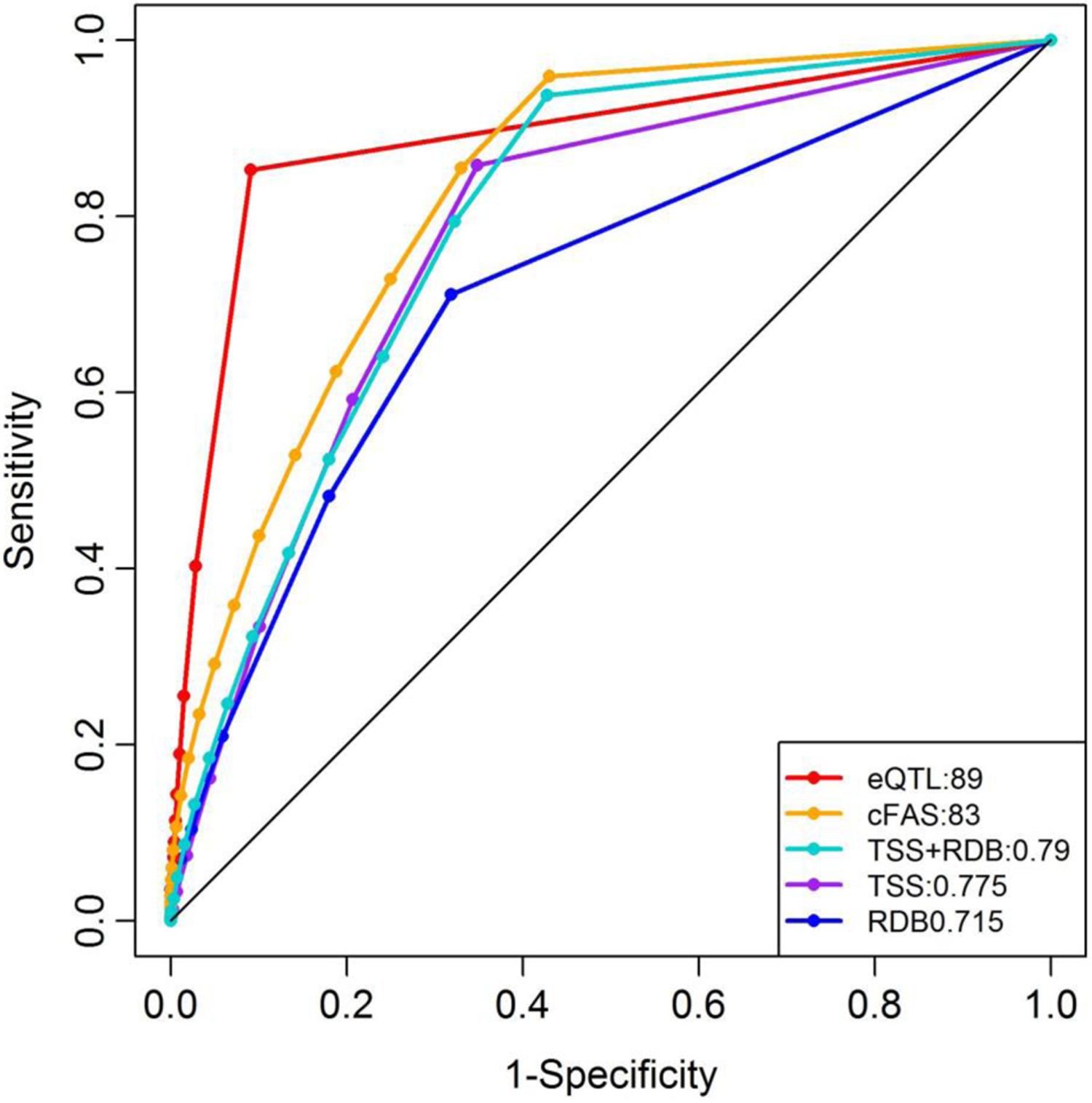
Receiver Operating Curves (ROC) for eQTL, TSS, RDB, TSS+RDB, and cFAS based on the GWAS association and random control sets. The area under the curve (AUC) is shown in the legend next to the name of each approach. eQTL = Expression quantitative trait loci, TSS = Transcription start site score from GWAVA tool, RDB = RegulomeDB, TSS+RDB = Combination of TSS and RDB prioritization scores, cFAS = Composite functional annotation score.
Tissue-specific eQTLs associated to prioritized variants
We further interrogated the 274 prioritized variants with tissue-specific eQTL databases. Several immune- and lung-related tissues were enriched in eQTLs (Supplementary Fig S5). Whole blood was the top represented tissue with 248 eQTL associated to gene expression level of 82 genes. Whole blood is one of the most extensively studied tissue types in asthma since many derivatives of this tissue such as T cells, B cells, eosinophils, neutrophils, and monocytes are known to play important roles in this disease (Fahy 2009). Other tissues with high frequency of eQTL include lung, thyroid, adipose, muscles, and skin (Supplementary Fig S5). The list of candidate loci along with the eQTLs are provided in Supplementary Table S7. Several HLA-class genes from the major histocompatibility complex (MHC) region were highly represented in the tissue specific eQTL database. For instance, there were 108 eQTLs mapped to HLA-DQA2 gene across 45 different tissues (Supplementary Table S8). Notably, several studies have linked the HLA-DQ region with asthma (Lasky-Su et al. 2012; Movahedi et al. 2008). Besides the HLA-class genes, the top 5 represented genes based on the number of significant eQTLs were in the 17q12–21 locus (ORMDL3, GSDMB, GSDMA, MED24, ZPBP2), with 46 eQTLs identified for GSDMA (Supplementary Table S8). The 17q12–21 regions are previously known to be associated with asthma, and our results further highlight the particular significance of these regions (Vicente et al. 2017). A recent study of the region 17q21 using chromatin conformation (4C-seq) assays has identified the variant rs12936231 as a functional variants leading to increased expression of ORDLM3 in the asthmatics (Schmiedel et al. 2016). Note that the intronic variant rs12936231 is the top cFAS variant with score 28. Other highly replicated asthma loci that were identified in our study include loci in 1q32 (CHI3L1), 2q12.1 (IL18R1), 5q22 (TSLP–WDR36), and 5p31 (IL5–RAD50). Our results also identified several novel loci such as 4p16 region (DGQK–SLC26A1–IUDA) and 12q13 (RAB5B–SOUX–ERBB3) with potential functional association with asthma.
Epigenetic and regulatory annotation in asthma-associated variants
Histone modifications (H3K9me1, H3K9me3, H3K27ac, H3K9ac) and DNase I hypersensitivity marks extracted from the Roadmap Epigenomic projects on 127 cell types were investigated (Supplementary Table S9). In particular, 270 and 243 SNPs were co-localized at the H3K4me1 and H3K4me3 histone modification marks, respectively. Similarly, 215 SNPs were located in the DNase I hypersensitive sites. Additionally, 166 SNPs were predicted to be located on the protein binding sites, and 233 SNPs were predicted to affect at least one sequence motif. There were 31 SNPs in the conserved region across mammals based on either GERP (Davydov et al. 2010) or Siphy score (Garber et al. 2009). These results suggest that the vast majority of the non-coding regulatory variants may not be conserved but they reside in the enhancer regions and the open chromatin regions marked by DNase 1 hypersensitive sites (Coetzee et al. 2010; Freedman et al. 2011). Next, we conducted an enrichment analysis of the variants for protein binding regions, histone modification marks, and DNase I mark.
Enrichment of the prioritized variants in protein biniding sites
About 100 proteins with binding affinity mapped to the prioritized variants were identified. Compared with the enrichment of these proteins with a random set of ~ 3,000 disease-associated SNPs from GWAS Catalog, 62 of these proteins were found be significantly enriched at adjusted p-value (adj. p) ≤ 0.05. POL2–4H8 (target gene POLR2A; adj. p < 1.73E-99), POLII complex (RNA Polymerase II; adj. p = 5.98E-76), and RFX5 (adj. p = 2.53E-39) were the top most enriched proteins (Supplementary Table S10). Nuclear factor-kappaB (NF-KB; adj. p = 6.29E-16), Jun Proto-Oncogene(cJUN; adj. p = 8E-5), , and members of signal transduction-activated transcription factors (STAT1, adj. p = 0.0016; STAT2, adj. p = 0.0003; STAT3, adj. p = 3.38E-7) have been reported to involve in asthmatic inflammation (Barnes and Adcock 1998). The CCCTC-binding factor (CTCF, adj. p = 3.76E-18) was previously revealed to interact with the IZKF3-ORMDLA3 region leading to increased expression of ORMDL3 in asthmatics (Schmiedel et al. 2016). Other TFs with enriched binding affinities such as ERG1 (adj. p = 7.29E-10), ELF1 (adj. p = 1.75E-18), EBF1 (adj. p = 0.0009), and GATA family were also reported to be involved in the differentiation of the T and B cells and in the regulation of expression of genes that are important in inflammation (Chan et al. 2009; Garrett-Sinha 2013; Gashler and Sukhatme 1995; Griffin et al. 2013; Karwot et al. 2008; Prasad et al. 2015; Russell and Garrett-Sinha 2010; Silverman et al. 2001). We identified 99 protein-coding genes with cis- eQTL variants in the binding regions of the enriched proteins (Supplementary Fig S6). Our analysis linked protein-coding genes with multiple proteins bound to cFAS-prioritized cis-eQTL variants. Genes from 6p21 (HLA-DQA1, HLA-DQA2, HLA-DRB1, HLA-DQB1) and 17q12–21 (GSDMA, GSDMB, ORMDL3, ZPBP2) were among the most enriched regions for cis-eQTL variants and protein binding sites. Additionally, multiple genes located in 4p16.3 (DGKQ, SLC26A1, and IUDA) and 12q13.2 (RAB5B, RPS26, SOUX) were also among the highly represented genes with proteins bounds to the prioritized eQTL variants.
Enrichment of the prioritized variants in cell-type specific epigenetic marks
To further investigate the enrichment of the prioritized variants on the cell-type specific regulatory regions, we tested the enrichment of the cFAS variants on five different regulatory regions including four histone modification marks (H3K4me1, HeK4me3, H3K27ac, and H3K9ac) and DNase I hypersensitive sites on the 127 reference epigenomes from the Roadmap and ENCODE projects using two different background variants. First, the enrichment of cFAS variants were compared against the background set consisting of ~3,000 disease-implicated variants randomly selected from GWAS catalog. Fig 4A summarizes the results of the enrichment analysis on the 127 reference epigenomes. The results showed the cFAS variants were significantly enriched on the selected regulatory regions across different cell types with adjusted p-value < 0.05. A strong enrichment signal of H3K4me1 mark was observed in immune related cell types such as T cells, lung, B cells, epithellial, digestive tissues and muscles, but brain, fetal, and other embryonic and cultured cells exhibited lesser extend of enrichments. The enrichment of H3K4me3 marked regions were extensively observed across all cell types than H3K4me1 marked regions. These results highlight potential enhancer regulated mechanisms on immune-related cells and tissues in asthma symptom (Heintzman et al. 2009). Second, we constructed a background set of variants with high cFAS score. For this purpose, we selected the GWAS catalog variants with cFAS ≥ 16, which in our empirical evaluation, represented cFAS score with significant FDR < 0.05. When compared with functionally similar set of variants, we can identify specific cell types that the prioritized variants are likely to regulate the functional mechanism through regulatory activities leading to asthma. Results showed T cells, B cells and lung were selectively enriched for the enhancers, promoters and DNase marks, with 31 distinct epigenomes were significantly enriched for at least one of the five chromatic marks tested at multiple testing adjusted p-value < 0.05 (Fig 4B). GM12878 lymphoblastiod is found to be enriched for all four histone marks and DNAse mark. Lung is significantly enriched for H3K4me1 (adj. p = 5.55E-9) and H3K27ac (adj. p = 0.01) marks. Multiple derivatives of T-cells and B-cells were also enriched for the enhancer and promoter marks (Fig 4B).
Fig 4. Functional annotation enrichment analysis.
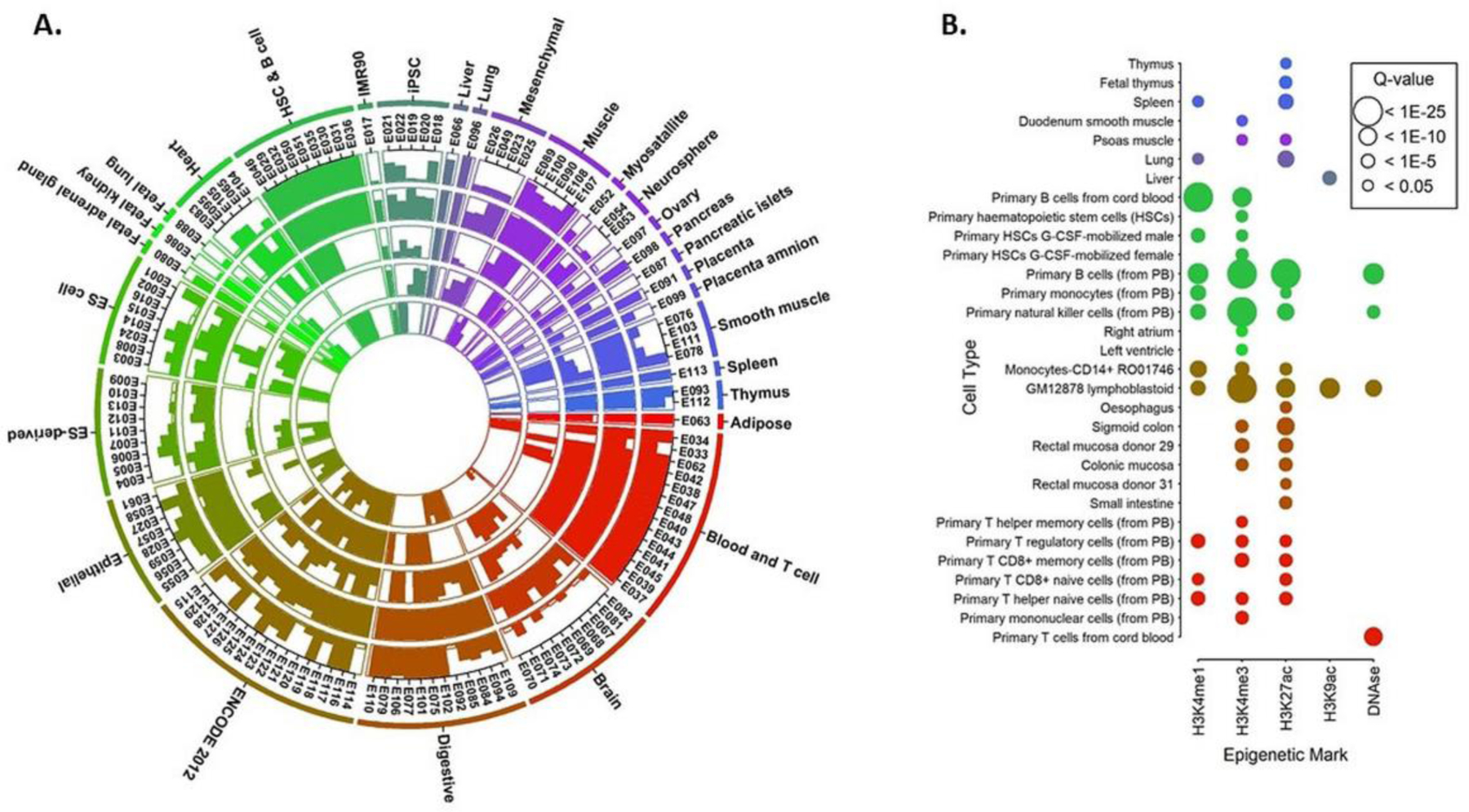
A) Circular plot shows the enrichment of the prioritized variants on 127 reference epigenomes from the Roadmap and ENCODE project against a random set of GWAS variants. The outermost circle represents the tissue/cell line types on different colors. Moving inwards, the circles represent the enrichment results for the H3K4me1, H3K4me3, H3K27ac, H3K9ac, and DNase I hypersensitive marks. Solid bars indicate enrichment of the prioritized variants on the corresponding reference epigenome is significant at FDR < 0.05. Height of the bar represents the –log10(p) of the p-value from the enrichment analysis. B) Cell type specific enrichment of the histone marks and DNase I hypersensitive marks against a prioritized set of GWAS variants with cFAS ≥ 16 as the background set. The size of circle is proportional to the adjusted p-value. Q-value = adjusted p-value using Benjamini-Hochberg procedure.
Hypomethylation based on genotype is specific to asthma patients and not healthy controls
To validate the prioritized cFAS variants found the 4p16 locus and the known 17q12–21 locus, we use DNA from a cohort of 30 asthma patients and 30 healthy controls for pyrosequencing to assess methylation of two variants in the respective regions. Hypomethylation based on genotype was detected in both variants in asthma cases when compared to controls (rs4065274 GG p-value= 0.041, AG p-value= 4.43 × 10−5; rs11936407 CC p-value= 0.041). Fig 5A shows the percent methylation of asthma cases versus healthy controls based on the different genotypes for rs4065275. On average the GG genotype in rs4065275 SNP was 48.6 and 58.7% methylated in asthma cases and healthy controls, respectively. Also, the AG genotype in this SNP variant had an average methylation of 22.1 and 29.1% in asthma cases and healthy controls, respectively. Also, an additional variant located upstream from rs4065275 was hypomethylated in asthma cases when compared to healthy controls (CpG site 1 P-value= 4.94 × 10−3, Supplementary Fig S7A). For the prioritized cFAS variant located in the novel locus 4p16 (rs11936407), Fig 5B shows a comparison of the percent methylation in asthma cases versus healthy controls based on the CC genotype. Average methylation for the CC genotype in asthma cases was 93.1% compared with 94.7% in healthy controls. Two additional variants upstream of this locus also showed hypomethylation in asthma cases when compared to healthy controls (CpG site 1 p-value= 0.002, CpG Site 2 p-value= 1.11 × 10−4, Supplementary Fig S7B). Interestingly, allele frequencies for both cFAS variants were similar for asthma cases and healthy controls leading us to believe that changes in methylation potentially play an important role in altering gene expression when comparing asthma cases with healthy controls.
Fig 5. Methylation of select cFAS variants rs4065275 and rs11936407.
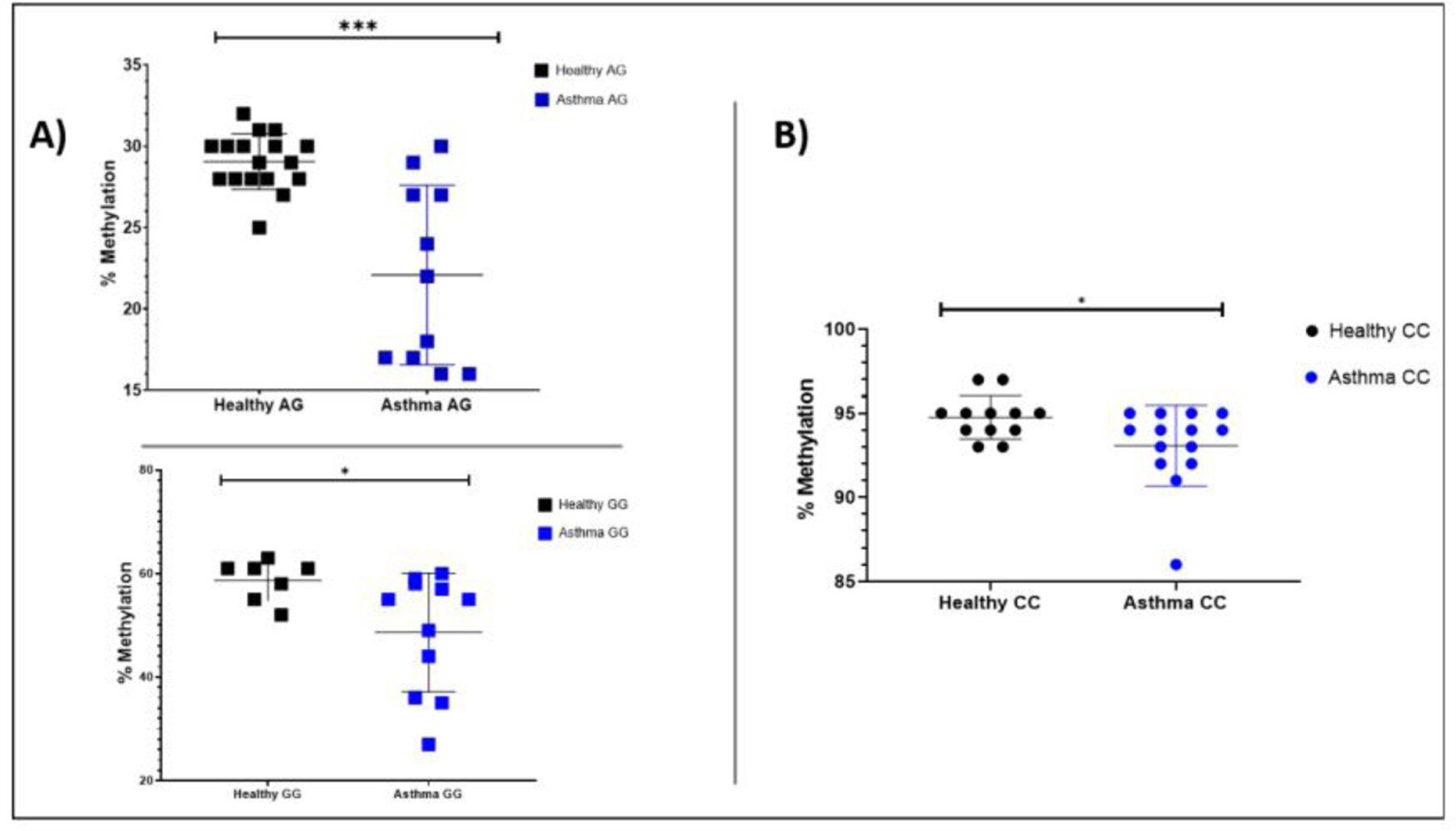
Plots show genotype specific hypomethylation at the selected sites among asthma patients compared to healthy controls. A) Scatter plots show the differences in percent methylation between asthma cases and healthy controls for the AG (top panel) and GG (bottom panel) genotypes at SNP rs4065275., respectively. B) Scatter plot shows the differences in percent methylation between asthma cases and healthy controls for the CC genotype at SNP rs11936407. * p-value < 0.50, *** p-value < 0.001.
Shared etiology of asthma associated variants with other diseases/traits
To determine the shared genetic etiology of asthma with other diseases/traits, we obtained association results from the GWAS catalog using an expanded set of asthma variants comprising the prioritized set and their LD-surrogate lead variants. The expanded set consisted of 399 SNPs, the combination of the 274 prioritized SNPs and the high LD tagging variants from the 1,525 GWAS-lead variants. Following Wang et al. (Wang et al. 2015), autoimmune diseases constituted the largest pleiotropic class with 16 different phenotypes sharing the asthma variants. Ulcerative colitis shared the most variants with asthma with seven variants. Other diseases with 3 or more overlapping SNPs included type 2 diabetes, allergy, and systemic sclerosis. In total, we found 57 SNPs overlapped with one or more of the 49 different disease/traits ontology classes (Fig 6).
Fig 6. Shared etiology analysis.
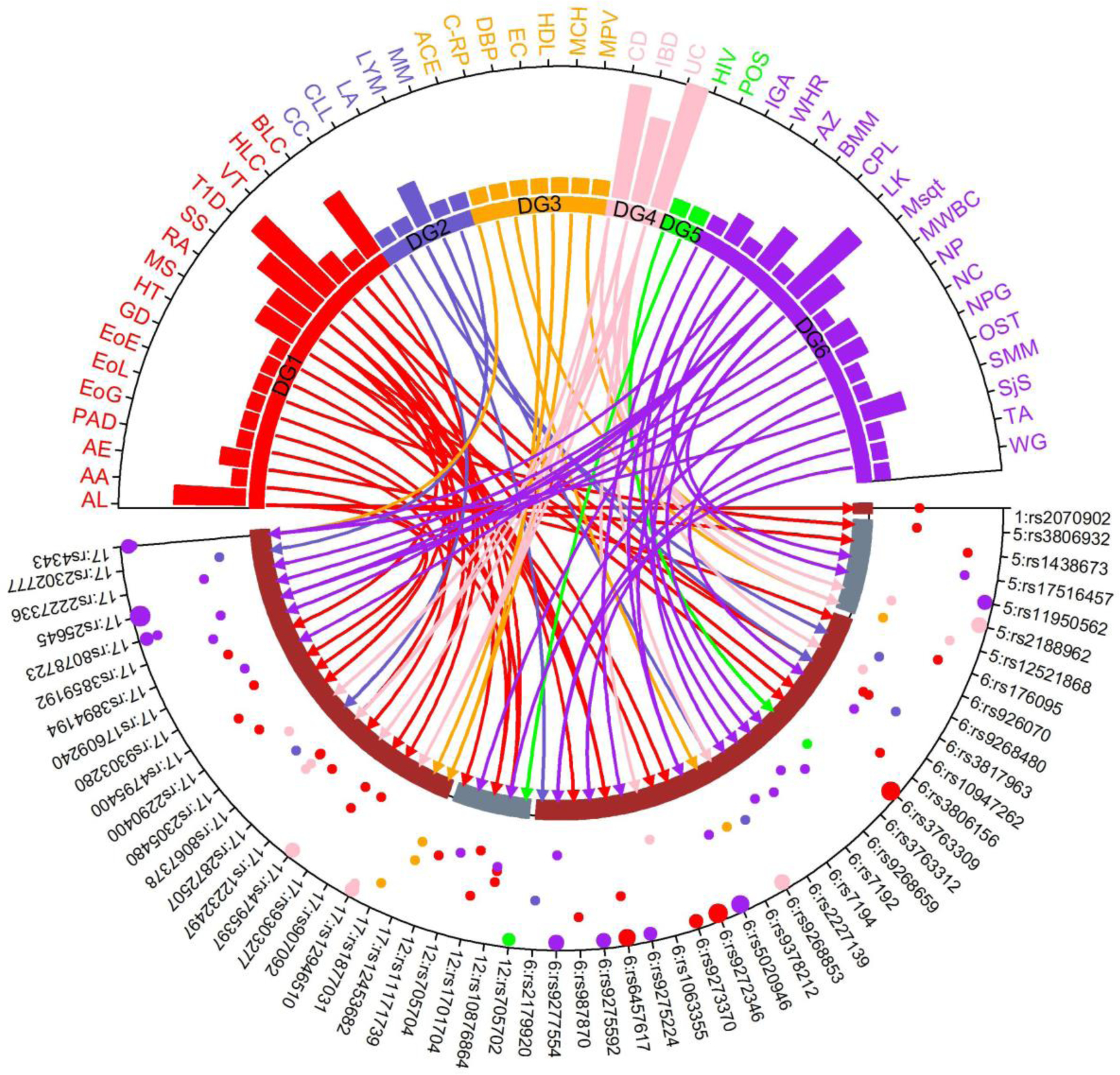
Circular plot shows mapping of asthma risk variants and diseases sharing the asthma risk-variants. The upper half of the plot shows the list of diseases and the bottom half shows the overlapped SNPs. The bars in the plot show the number of SNPs overlapped between the disease and asthma. Diseases are classified into 8 different groups following Wang et al. except the CD, UC, and IBD are grouped into digestive group (Wang et al. 2015). The y-axis of the lower part of plot shows the –log(p) of the significance p-value of the association between the disease and the SNPs. If the –log(p) > 25, a larger circular dot is used with the radius proportion to the ratio – log(p)/25. The connector lines from the diseases to SNPs show the disease-SNP map with color code reflecting the disease group.
Discussion
In this study, we presented a systematic and comprehensive approach to analyze and prioritize genome-wide variants associated with asthma by developing a novel composite scoring scheme called Composite Functional Annotation Score (cFAS). Several methods have been recently developed to utilize various functional annotation resources in predicting the functional role of genome-wide variant (Li et al. 2017; Li et al. 2016). However, such approaches fail to incorporate the rich complementary information contained in the various annotation resources (Lu et al. 2017). Therefore, testing one annotation at a time might be suboptimal, and it would be ideal to incorporate multiple annotations together in order to identify disease-associated variants. The advantage of cFAS is that it combines the eQTL annotation from a large eQTL database of GTEx consortium with the regulatory annotations from RegulomeDB and GWAVA into a single composite score. Thus, cFAS prioritize variants with stronger functional evidence that might have been missed by other methods. We annotated 32,161 asthma-associated variants (both genome-wide lead and their LD surrogate variants) and identified a prioritized set of 274 variants by using the cFAS score. Only 38 SNPs of the prioritized set are genome-wide lead SNPs, and the rest are in high LD with genome-wide lead SNPs. Although GWAS-lead SNPs are the primary source of association signals, our findings echoed prior observations that lead SNPs or SNPs in high LD (r2 ≥ 0.8) could be equally functional (Schaub et al. 2012). As the LD surrogates may control the regulatory mechanism independent to the lead variants, our results further highlighted the importance of incorporating LD in the search of functional variants (Schaub et al. 2012). Literature-driven functional cluster analysis of the genes mapped to the prioritized variants yielded asthma, and asthma and respiratory-related disease clusters (Fig 7). We performed gene set overrepresentation analysis using ConsensusPathDB and found antigen processing and presentation, T cell co-stimulation, regulation of immune system process, defense response and innate immune response were among the most differentially enriched functional category (Supplementary Fig S8). This functional analysis further validates the relevance of our approach to prioritize variants associated with asthma.
Fig 7. Functional ontology based clustering of prioritized variants.
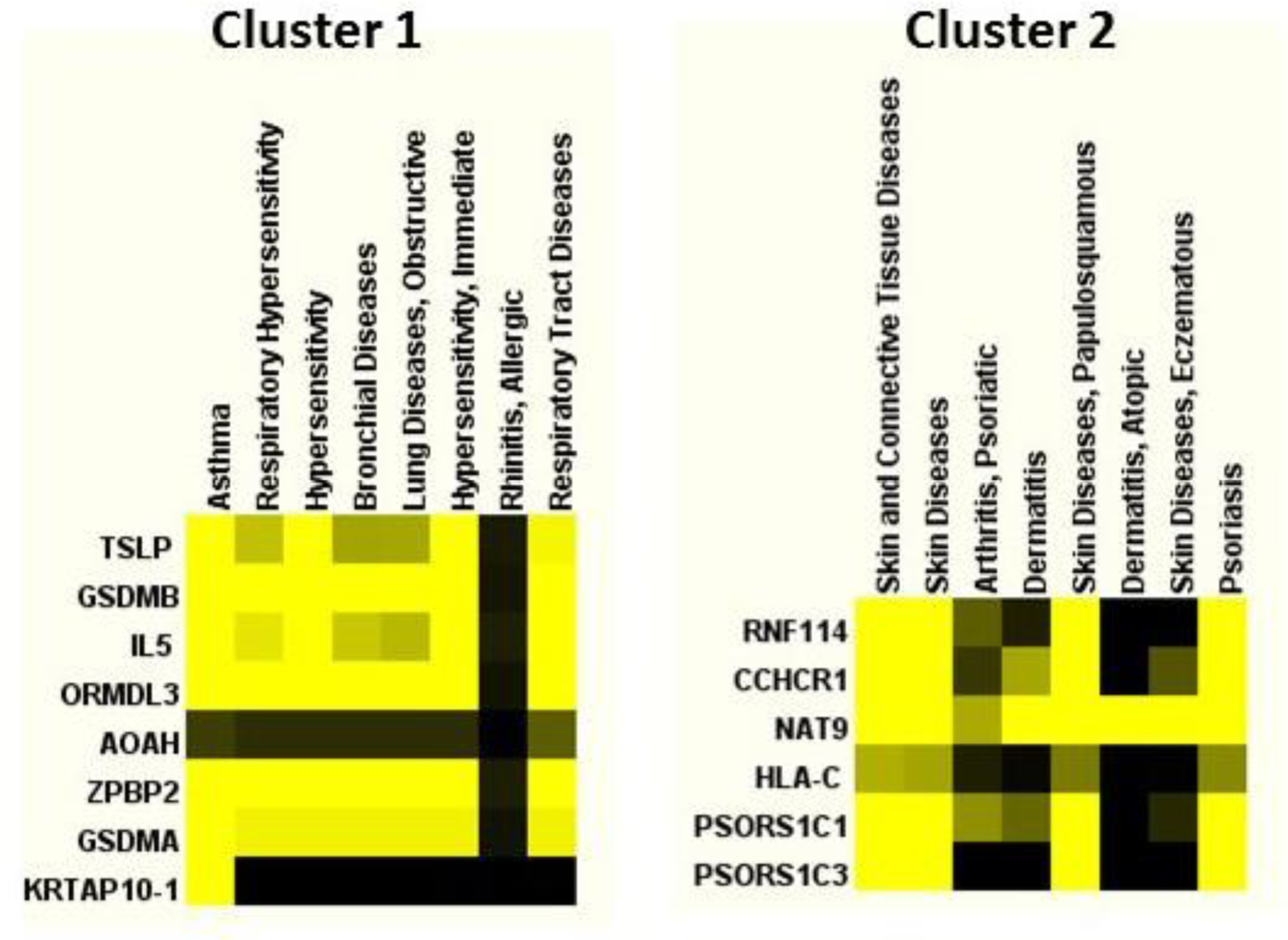
Plot shows the functional clustering analysis of asthma variants based on prioritized variants and using the Literature Lab™ from Acumenta Biotech (http://acumenta.com/).
The cFAS approach not only replicated the previously known candidate genes, but also unravel novel genes. This investigation identified two eQTL variants, rs3806932 and rs10073816 for TSLP-WDR36 region, both of which have not previously been revealed to be associated with asthma but are in high LD with a GWAS-lead variant rs1438673. The 5’ UTR variant rs3806932 is a potential enhancer, and the electrophoresis mobility shift assays (EMSAs) have paraded its ability to bind to nuclear proteins (Chen et al. 2015). Harda et al. have determined that another 5’-URT variant rs3806933 in the putative promoter region of long-form TSLP creates a binding site for the transcription factor activating protein AP-1 (Harada et al. 2009). Epigenetic annotation obtained from HaploReg have demonstrated that rs3806932 is co-located in the enhancer regions marked with H3K4me1 in multiple epigenomes including lung, primary T regulatory and helper cells, CD8+ memory and CD8+ naïve cells from peripheral bloods, epithelial cells, and smooth muscle cells. These lines of evidence point towards the potential enhancer role of rs3806932 in the transcription of TSLP. The 3’-UTR variant rs10073816 is associated to TSLP in skin, brains and other tissues, but the functional role on the transcription of TSLP genes is not known yet. Given that rs3806932 and rs10073816 mark the 5’UTR–3’URT ends of the TSLP transcript, further functional experimentations may reveals their role in regulating the transcription of TSLP in asthma. TSLP is an epithelial-cell-derived cytokine important in initiating allergic inflammation (Torgerson et al. 2011). Genome-wide investigation has led to an important clinical application for the susceptibility role of TSLP with the development of a novel therapeutic agent, a human anti-TSLP monoclonal immunoglobulin G2-lambda antibody (AMG 157 or tezepelumab), which binds human TSLP and prevents receptor interaction. In clinical studies, tezepelumab reduces allergen-induced bronchoconstriction and airway inflammation (ClinicalTrials.gov number, NCT01405963) (Gauvreau et al. 2014).
We also identified novel candidate regions such as the 4p16 locus. The lead SNP rs3796622 at this locus is associated with asthma (S1 Table), but it is not a cFAS-prioritized SNP. However, we identified two cFAS-prioritized SNPs (rs11936407, and rs3806756) in high LD with rs3796622. Using eQTL analysis in lung and whole blood, we found that all the LD SNPs showed significant eQTL (p-value < 1 ×10−8). Supplementary Fig S9 illustrates the eQTL results for the gene DGKQ, SLC26A1, and IDUA on lung from GTEx (Lonsdale et al. 2013). The SNPs in the DGKQ, SLC26A1, and IDUA genes were associated with lung and whole blood and were also co-located in the enhancers, promoters, and DNase marks in multiple epigenomes and predicted to exhibit protein binding affinities and altering motif effects.
The prioritized variants exhibited tissue-specific enrichment of enhancers marked with H3K4me1 peaks in T and B cell types, thymus, lung, and digestive tissues. Previous studies have also demonstrated that histone modification marks such as H3K4me1 and H3K27ac harbor disease-associated variants in cell-type specific manner (Gerasimova et al. 2013; Jiang et al. 2015). The epigenetic modification marks such as H3K4me1, H3K4me3, H3K27ac, and H3K9ac are highly informative about the TF binding affinities (Hnisz et al. 2013; Liu et al. 2015; Trynka et al. 2013). Hence, the interrogation of cis-regulatory regions such as those marked with histone modification peaks and overlapped to disease risk variants may lead to identifying the TF binding sites for disease-associated gene transcription and pinpointing the causal variants and disease pathogenesis. The experimental analysis of the 17q12–12 locus by Schmiedel et al using chromatic conformation assays (4C-seq) supports this interrogation (Schmiedel et al. 2016). The latter investigation has manifested that the intronic variant rs4065275 of ORMDL3 potentially introduces CTCF-binding site while the other asthma risk SNP rs12936231 in the intronic region of ZPBP2 disrupts the CTCF-binding site in CD4+ T cells in the region enriched for H3K27ac and H3K4me1 marks. Such combination of CTCF binding sites is associated to increased interaction between enhancers of the transcription factor IKZF3 (several kb upstream of ORMDL3) and ORMDL3 promoters, causing increased expression of ORMDL3 among the carrier of risk alleles from two markers primarily in the immune-related cells (Schmiedel et al. 2016; Stein et al. 2018). ORMDL3 is known to regulate endoplasmic reticulum-mediated calcium signaling and encode for ORM1-lie protein 3 and gasdermin-like protein. It results in the unfolded protein response (UPR), which is thought to trigger an inflammatory response in addition to being involved in immune cell migration, pro-inflammatory cytokine production, and allergen-induced asthma pathologies (Cantero-Recasens et al. 2010). Further validation of the rs4065275 variant using TaqMan assays and pyrosequencing showed that genotype based hypomethylation is prevalent in our cohort of asthma cases when compared to healthy controls, and that this is an indicative of possible changes in gene expression that should be further studied in the future.
Our finding and other showed that a large number of asthma loci overlap with loci associated with other autoimmune diseases such as allergy, atopic dermatitis, body mass index, inflammatory bowel diseases, as well as Type I diabetes (Cotsapas et al. 2011). A recent study has reported that nearly half of loci discovered in GWAS influence risk to at least two diseases, indicating the shared genetic architecture of immune-mediated inflammatory and autoimmune diseases (Barnes 2011; Cotsapas et al. 2011). The observed clustering of multiple risk factors could be due to an overlap in the causal pathways and could suggest a shared role of the candidate variants including autoimmune and inflammatory diseases with asthma (Demenais et al. 2018; Pickrell et al. 2016; Shi et al. 2016). Grouping variants by shared etiology should give insight into the specific biological processes underlying comorbidity and disease risk. This finding provides further evidence for the growing understanding of the importance of pleiotropy in multifactorial diseases.
In critically evaluating our cFAS method, it is important to note that our approach and hence interpretations have some limitations. First, our sequential computational scheme of assigning priority scores (PSeQTL, PSRDB, and PSTSS) is based on a heuristic principle and requires further validation using independent datasets. In addition, weighing functional annotation scores based on validated functional data could further improve the scoring scheme and functional classification of the GWAS-variants. However, in the absence of truly validated functional data, we believe that it is not realistic to assign a weight to each source of functional annotation data. As we generate denser and more reliable validated functional resources including in the non-coding region, further testing and validation of cFAS algorithm using independent dataset is needed. Second, we did not weight any variants for being replicated in the GWAS as we have limited information on the biological relevance of GWAS replications. We believe that at the moment there is no strong biological evidence to weight variants based on GWAS finding and adding such weight may results in lower score for variants that might otherwise be rightfully ranked among top list and biologically relevant. But, in future, when more replications followed by functional studies on diverse tissue/cell types become available, we may be able to revise the scoring mechanism with some prior weight based on the evidence. Third, we leverage three widely available annotation resources to compute cFAS. Our choice of these functional databases was primarily driven by public availability, coverage and recent update at the time of developing this tool. As more and more datasets are becoming freely available, it is important to revisit and leverage additional functional genomic information and incorporate in cFAS scoring scheme. Last, we have used broad definition of asthma in order to be comprehensive and maximize the reported associations.
Functional annotations are independent of the phenotype, so the cFAS scoring and ranking of variants would not be affected. However, biological mechanisms of asthma may vary across the different asthma subtypes which may have effect on the functional investigation of the prioritized variants and interpretation of the results thereafter. Nevertheless, the strength of this study lies in the context-specific regulatory activity and linkage disequilibrium annotations of complementary functional evidence (cell-type specific functional and regulatory annotations for histone modifications, enhancer enrichment analysis, expression quantitative trait loci, eQTL) in to one score to prioritize the candidate variants from asthma genome-wide studies. Existing tools attempt to prioritize variants based on only regulatory annotation data and lack comprehensive eQTL resources. Our understanding of the biological mechanism of genome-wide variants has been substantially improved by characterizing the epigenomic annotations and tissue-specific eQTLs. Finally, prioritized variants were validated using TaqMan assays and pyrosequencing.
In summary, to better understand the functional role of identified loci from the gene-wide studies, we first conducted LD expansion at lead loci to increase genetic resolution, and then we intersect each variant with three functional annotation resources (ENCODE, GTEx, and NIH Roadmap Epigenomics) and develop a composite scoring method prioritize the most plausible asthma risk variants. cFAS identified both novel and previously known asthma loci. Prioritized variants were further validated using pyrosequencing and TaqMan assays to replicate their function. To our knowledge, this article is the first to report large-scale genome-wide study followed by in silico variant prioritization and validation in asthma.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grant R01HL132344. The CAMP/CARE and STAMPEED datasets were obtained from dbGap through accession number phs000166.v2.p1 and phs000355.v1.p1, respectively.
This work was supported by the National Institutes of Health (NIH) grant R01HL132344
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Availability of data and materials
dbGaP asthma data are available to download by submitting a data access request through dbGaP (accession numbers for CAMP/CARE and STAMPEED are phs000355.v1.p1 and phs000166.v2.p1, respectively).
Disclosure Statement
The authors declare no conflict of interest.
Supplemental Data
Supplemental Data include 9 figures and 12 tables
References
- Barnes KC (2011) Genetic studies of the etiology of asthma Proc Am Thorac Soc 8:143–148 doi: 10.1513/pats.201103-030MS [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM (1998) Transcription factors and asthma Eur Respir J 12:221–234 [DOI] [PubMed] [Google Scholar]
- Baye TM et al. (2011) Differences in candidate gene association between European ancestry and African American asthmatic children PLoS One 6:e16522 doi: 10.1371/journal.pone.0016522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of Royal Statistical Society Series B (Methodological) 57:298–300 [Google Scholar]
- Boyle AP et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB Genome Res 22:1790–1797 doi: 10.1101/gr.137323.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniello A et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019 Nucleic Acids Res 47:D1005–D1012 doi: 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butsch Kovacic M et al. (2012) The Greater Cincinnati Pediatric Clinic Repository: A Novel Framework for Childhood Asthma and Allergy Research Pediatr Allergy Immunol Pulmonol 25:104–113 doi: 10.1089/ped.2011.0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R (2010) The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress Hum Mol Genet 19:111–121 doi: 10.1093/hmg/ddp471 [DOI] [PubMed] [Google Scholar]
- Chan IH et al. (2009) Association of early growth response-1 gene polymorphisms with total IgE and atopy in asthmatic children Pediatr Allergy Immunol 20:142–150 doi: 10.1111/j.1399-3038.2008.00757.x [DOI] [PubMed] [Google Scholar]
- Chen G et al. (2015) Re-annotation of presumed noncoding disease/trait-associated genetic variants by integrative analyses Sci Rep 5:9453 doi: 10.1038/srep09453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee GA, Jia L, Frenkel B, Henderson BE, Tanay A, Haiman CA, Freedman ML (2010) A systematic approach to understand the functional consequences of non-protein coding risk regions Cell Cycle 9:256–259 doi: 10.4161/cc.9.2.10419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome Nature 489:57–74 doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsapas C et al. (2011) Pervasive sharing of genetic effects in autoimmune disease PLoS Genet 7:e1002254 doi: 10.1371/journal.pgen.1002254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol 6:e1001025 doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demenais F et al. (2018) Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks Nat Genet 50:42–53 doi: 10.1038/s41588-017-0014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS et al. (2009) Common regulatory variation impacts gene expression in a cell type-dependent manner Science 325:1246–1250 doi: 10.1126/science.1174148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy JV (2009) Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies Proc Am Thorac Soc 6:256–259 doi: 10.1513/pats.200808-087RM [DOI] [PubMed] [Google Scholar]
- Freedman ML et al. (2011) Principles for the post-GWAS functional characterization of cancer risk loci Nat Genet 43:513–518 doi: 10.1038/ng.840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X (2009) Identifying novel constrained elements by exploiting biased substitution patterns Bioinformatics 25:i54–i62 doi: 10.1093/bioinformatics/btp190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Sinha LA (2013) Review of Ets1 structure, function, and roles in immunity Cell Mol Life Sci 70:3375–3390 doi: 10.1007/s00018-012-1243-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gashler A, Sukhatme VP (1995) Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors Prog Nucleic Acid Res Mol Biol 50:191–224 [DOI] [PubMed] [Google Scholar]
- Gauvreau GM et al. (2014) Effects of an anti-TSLP antibody on allergen-induced asthmatic responses N Engl J Med 370:2102–2110 doi: 10.1056/NEJMoa1402895 [DOI] [PubMed] [Google Scholar]
- Genomes Project C et al. (2015) A global reference for human genetic variation Nature 526:68–74 doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova A et al. (2013) Predicting cell types and genetic variations contributing to disease by combining GWAS and epigenetic data PLoS One 8:e54359 doi: 10.1371/journal.pone.0054359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MJ, Zhou Y, Kang S, Zhang X, Mikkelsen TS, Rosen ED (2013) Early B-cell factor-1 (EBF1) is a key regulator of metabolic and inflammatory signaling pathways in mature adipocytes J Biol Chem 288:35925–35939 doi: 10.1074/jbc.M113.491936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M et al. (2009) Functional analysis of the thymic stromal lymphopoietin variants in human bronchial epithelial cells Am J Respir Cell Mol Biol 40:368–374 doi: 10.1165/rcmb.2008-0041OC [DOI] [PubMed] [Google Scholar]
- Heintzman ND et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression Nature 459:108–112 doi: 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwig R, Hardt C, Lienhard M, Kamburov A (2016) Analyzing and interpreting genome data at the network level with ConsensusPathDB Nat Protoc 11:1889–1907 doi: 10.1038/nprot.2016.117 [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits Proc Natl Acad Sci U S A 106:9362–9367 doi: 10.1073/pnas.0903103106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D et al. (2013) Super-enhancers in the control of cell identity and disease Cell 155:934–947 doi: 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Zhu L, Buck MJ, Chen Y, Carrier B, Liu T, Jarvis JN (2015) Disease-Associated Single-Nucleotide Polymorphisms From Noncoding Regions in Juvenile Idiopathic Arthritis Are Located Within or Adjacent to Functional Genomic Elements of Human Neutrophils and CD4+ T Cells Arthritis Rheumatol 67:1966–1977 doi: 10.1002/art.39135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwot R et al. (2008) Protective role of nuclear factor of activated T cells 2 in CD8+ long-lived memory T cells in an allergy model J Allergy Clin Immunol 121:992–999 e996 doi: 10.1016/j.jaci.2007.12.1172 [DOI] [PubMed] [Google Scholar]
- Lasky-Su J et al. (2012) HLA-DQ strikes again: genome-wide association study further confirms HLA-DQ in the diagnosis of asthma among adults Clin Exp Allergy 42:1724–1733 doi: 10.1111/cea.12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie R, O’Donnell CJ, Johnson AD (2014) GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database Bioinformatics 30:i185–194 doi: 10.1093/bioinformatics/btu273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Lu Q, Zhao H (2017) An evaluation of noncoding genome annotation tools through enrichment analysis of 15 genome-wide association studies Brief Bioinform doi: 10.1093/bib/bbx131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MJ et al. (2016) Predicting regulatory variants with composite statistic Bioinformatics 32:2729–2736 doi: 10.1093/bioinformatics/btw288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Jin G, Zhou X (2015) Modeling the relationship of epigenetic modifications to transcription factor binding Nucleic Acids Res 43:3873–3885 doi: 10.1093/nar/gkv255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J et al. (2013) The Genotype-Tissue Expression (GTEx) project Nat Genet 45:580–585 doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q et al. (2017) Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease PLoS Genet 13:e1006933 doi: 10.1371/journal.pgen.1006933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res 45:D896–D901 doi: 10.1093/nar/gkw1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants Bioinformatics 31:3555–3557 doi: 10.1093/bioinformatics/btv402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD et al. (2007) The NCBI dbGaP database of genotypes and phenotypes Nat Genet 39:1181–1186 doi: 10.1038/ng1007-1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA Science 337:1190–1195 doi: 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt MF et al. (2010) A Large-Scale, Consortium-Based Genomewide Association Study of Asthma New England Journal of Medicine 363:1211–1221 doi: 10.1056/NEJMoa0906312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahedi M et al. (2008) Association of HLA class II alleles with childhood asthma and Total IgE levels Iran J Allergy Asthma Immunol 7:215–220 doi:07.04/ijaai.215220 [PubMed] [Google Scholar]
- Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA (2016) Detection and interpretation of shared genetic influences on 42 human traits Nat Genet 48:709–717 doi: 10.1038/ng.3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MA et al. (2015) Ebf1 heterozygosity results in increased DNA damage in pro-B cells and their synergistic transformation by Pax5 haploinsufficiency Blood 125:4052–4059 doi: 10.1182/blood-2014-12-617282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses Am J Hum Genet 81:559–575 doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos EM et al. (2014) Phenotype-Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources Eur J Hum Genet 22:144–147 doi: 10.1038/ejhg.2013.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, Zeggini E, Flicek P (2014) Functional annotation of noncoding sequence variants Nat Methods 11:294–296 doi: 10.1038/nmeth.2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C et al. (2015) Integrative analysis of 111 reference human epigenomes Nature 518:317–330 doi: 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell L, Garrett-Sinha LA (2010) Transcription factor Ets-1 in cytokine and chemokine gene regulation Cytokine 51:217–226 doi: 10.1016/j.cyto.2010.03.006 [DOI] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M (2012) Linking disease associations with regulatory information in the human genome Genome Res 22:1748–1759 doi: 10.1101/gr.136127.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel BJ et al. (2016) 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells Nat Commun 7:13426 doi: 10.1038/ncomms13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kichaev G, Pasaniuc B (2016) Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data Am J Hum Genet 99:139–153 doi: 10.1016/j.ajhg.2016.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman ES et al. (2001) The transcription factor early growth-response factor 1 modulates tumor necrosis factor-alpha, immunoglobulin E, and airway responsiveness in mice Am J Respir Crit Care Med 163:778–785 doi: 10.1164/ajrccm.163.3.2003123 [DOI] [PubMed] [Google Scholar]
- Stein MM et al. (2018) A decade of research on the 17q12–21 asthma locus: Piecing together the puzzle J Allergy Clin Immunol doi: 10.1016/j.jaci.2017.12.974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG et al. (2011) Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations Nat Genet 43:887–892 doi: 10.1038/ng.888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants Nat Genet 45:124–130 doi: 10.1038/ng.2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente CT, Revez JA, Ferreira MAR (2017) Lessons from ten years of genome-wide association studies of asthma Clin Transl Immunology 6:e165 doi: 10.1038/cti.2017.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data Nucleic Acids Res 38:e164 doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Oehlers SH, Espenschied ST, Rawls JF, Tobin DM, Ko DC (2015) CPAG: software for leveraging pleiotropy in GWAS to reveal similarity between human traits links plasma fatty acids and intestinal inflammation Genome Biol 16:190 doi: 10.1186/s13059-015-0722-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M (2016) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease Nucleic Acids Res 44:D877–881 doi: 10.1093/nar/gkv1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahran HS, Bailey C, Garbe P (2011) Vital signs: Asthma prevalence, disease characteristics, and self-management education --- United States, 2001−−2009 vol 60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.