

Abstract
Sphingolipid metabolism is increasingly recognized as a therapeutic target in cancer due to its regulation of cell proliferation and apoptosis. The sphingolipid rheostat is proposed to control cell fate through maintaining balance between pro-apoptotic and pro-survival sphingolipids. This balance is regulated by metabolizing enzymes involved in sphingolipid production. One such enzyme, sphingosine kinase-2 (SPHK2), produces pro-survival sphingosine 1-phosphate (S1P) by phosphorylation of pro-apoptotic sphingosine. Elevated SPHK2 has been found in multiple cancer types and contributes to cell survival, chemotherapeutic resistance and apoptosis resistance. We have previously shown elevation of S1P in large granular lymphocyte (LGL) leukemia serum and cells isolated from patients. Here, we examined SPHK2 expression in LGL leukemia and found SPHK2 mRNA and protein upregulation in a majority of LGL leukemia patient samples. Knockdown of SPHK2 with siRNA in LGL leukemia cell lines decreased proliferation. Additionally, the use of ABC294640 or K145, both SPHK2-specific inhibitors, decreased viability of LGL leukemia cell lines. ABC294640 selectively-induced apoptosis in LGL cell lines and freshly isolated LGL leukemia patient cells compared to normal controls. Mechanistically, SPHK2 inhibition downregulated pro-survival myeloid cell leukemia-1 (Mcl-1) protein through proteasomal degradation. Targeting of SPHK2 therefore provides a novel therapeutic approach for the treatment of LGL leukemia.
Introduction
Large granular lymphocyte leukemia encompasses a spectrum of rare clonal lymphoproliferative disorders, all of which involve abnormal expansion of large granular lymphocytes (LGL), either cytotoxic T-lymphocytes (CTL) or natural killer (NK) cells (Lamy and Loughran, 2011). In normal adults, LGLs represent 10–15% of peripheral blood mononuclear cells (PBMCs) and can be classified into two distinct lineages as either CD3+ CTLs or CD3− NK cells. The activation of survival pathways and the evasion of apoptosis are major dysregulations seen in LGL leukemia(Leblanc et al., 2012, Steinway et al., 2014).
Sphingolipid metabolism is increasingly recognized as a therapeutic target in cancer due to its role in the regulation of cancer cell proliferation and resistance to apoptosis (Hannun and Obeid, 2018, Newton et al., 2015, Morad and Cabot, 2013, Gouaze-Andersson and Cabot, 2011, Shaw et al., 2018, Ogretmen, 2018). Sphingosine-1-phosphate (S1P) is a pro-survival sphingolipid generated from the phosphorylation of sphingosine by sphingosine kinase (SPHK) type 1 and type 2. Sphingosine kinase-2 (SPHK2) is a less-characterized isoform compared to sphingosine kinase-1 (SPHK1)(Neubauer and Pitson, 2013). In LGL leukemia, multiple survival pathways in LGLs are dysregulated and thought to be involved in leukemogenesis (Leblanc et al., 2012, Steinway et al., 2014). Interestingly, LGL leukemia cells are thought to hijack survival pathways used by their normal counterparts during activation and expansion (Lamy and Loughran, 2011, Shah et al., 2008). We have previously shown that altered sphingolipid metabolism, and more specifically SPHK1, plays a prominent role in the pathogenesis and survival of leukemic LGLs (LeBlanc et al., 2015, Shah et al., 2008, Liao et al., 2011). Similar to SPHK1, SPHK2 exhibits a pro-survival role in cancer development and progression but has not been investigated in LGL leukemia (Sun et al., 2015, Xun et al., 2015). Although both isoforms of SPHK have pro-survival roles, previous studies suggest that they exercise this function differently. SPHK1, which predominantly resides in the cytosol, is phosphorylated by ERK1/2, translocates to the cell membrane and uses membrane-bound sphingosine to produce S1P that is exported and initiates autocrine/paracrine signaling pathways through a series of S1P receptors (Pitson et al., 2003, Takabe et al., 2008). In contrast, SPHK2 is found in the ER, nucleus and mitochondria where locally produced S1P acts intracellularly through non-S1P receptor related roles such as binding to HDACs (Hait et al., 2009). S1P produced by SPHK1 has also been shown to have an intracellular role including its binding to TRAF2 (Alvarez et al., 2010).
SPHK2 has been shown to promote various solid (colorectal (Xun et al., 2015), lung (Zhang et al., 2015), bladder (Sun et al., 2015), breast (Antoon et al., 2011)) and hematologic cancers (Venkata et al., 2014) through increased tumor cell proliferation, invasion and resistance to chemotherapeutics. Ablation of SPHK2 with siRNA or ABC294640, a SPHK2-specific inhibitor, prevents tumor cell proliferation and migration in a variety of tumor cell types (Sun et al., 2015, Xun et al., 2015, Xie et al., 2018). ABC294640 is a first-in-class orally available SPHK2 selective inhibitor that has a wide range of anti-cancer, anti-inflammatory and radioprotective properties (French et al., 2010). It has successfully undergone pre-clinical development and is currently being evaluated in Phase I/II clinical studies in patients with refractory/relapsed diffuse large B-cell lymphoma (NCT NCT02229981), advanced cholangiocarcinoma (NCT03377179), and advanced solid tumors (NCT01488513), and will soon enter into a Phase II clinical study for the treatment of multiple myeloma (NCT02757326).
We previously identified that constitutive activation of STAT3 is a unifying feature in LGL leukemia and acts to upregulate anti-apoptotic proteins (Epling-Burnette et al., 2001). It has recently been shown by our lab and others that this constitutive activation is the result of somatic activating mutations in the SH2 dimerization and activation domain of the STAT3 gene in 30–40% of both NK- and T-LGL patients (Jerez et al., 2012, Koskela et al., 2012). STAT3 inhibition also significantly reduces expression of pro-survival Bcl-2 family member Mcl-1 (Epling-Burnette et al., 2001). Evidence also links S1P and SPHK1 to Mcl-1 expression (Li et al., 2007, Li et al., 2008). In AML, SPHK1 inhibition was associated with decreased survival of leukemic cells through inhibiting S1PR2 and downstream selective downregulation of the pro-survival protein Mcl-1 (Powell et al., 2017). The use of SPHK1 specific inhibitors in AML samples also led to downregulation of Mcl-1 but not Bcl-2 or Bcl-Xl (Dick et al., 2015). Additionally, inhibition of acid ceramidase, an enzyme upstream of S1P production, led to decreased expression of Mcl-1 in AML cells (Tan et al., 2016). Furthermore, targeting SPHK2 in multiple myeloma and T-cell acute lymphoblastic leukemia (T-ALL) led to downregulation of Mcl-1(Venkata et al., 2014). Therefore, a putative molecular mediator of both SPHK1- and SPHK2-targeting strategies is well established.
The present study explores the hypothesis that elevated SPHK2 promotes tumor survival in LGL leukemia. We demonstrate that inhibition of SPHK2 with siRNA or ABC294640 induced apoptosis and decreased cell viability in LGL leukemia cell lines. We further show that ABC294640 selectively induced apoptosis in PBMCs from LGL leukemia patients but had minimal effects on PBMCs from normal donors. Mechanistically, we determined that Mcl-1 is downstream of SPHK2 and that biological or pharmacological inhibition of SPHK2 enhanced Mcl-1 degradation through the proteasome. We conclude that SPHK2 is a potential therapeutic target in LGL leukemia.
Materials and methods
Reagents and cell culture
ABC294640 was purchased from Active Biochem (Maplewood, NJ). Antibodies for β-actin, Bcl-2, Bcl-xL, Mcl-1 and SPHK1 were purchased from Cell Signaling Technology Inc (Danvers, MA). Antibody for SPHK2 was purchased from Abcam (Cambridge, MA). Human NKL cells (kindly provided by Dr. Howard Young at National Cancer Institute)(Robertson et al., 1996) were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) plus 100 IU/ml IL-2 generously provided from NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann – La Roche Inc(Lahm et al., 1985) or purchased from Miltenyi. Human TL-1 cells(Ren et al., 2013) were cultured in RPMI-1640 supplemented with 10% FBS plus 200 IU/ml IL-2. All cells were maintained in a 37°C humidified 5% CO2 atmosphere incubator.
Patient characteristics and preparation of peripheral blood mononuclear cells (PBMCs)
All patients met the criteria of chronic NK-LGL (CD3−) or T-LGL (CD3+) leukemia with increased numbers of LGLs in the peripheral blood. Peripheral blood samples were obtained and informed consents signed for all LGL leukemia patients according to protocols approved by the Institutional Review Boards of Penn State Hershey Medical Center and the University of Virginia School Of Medicine. PBMCs were isolated by Ficoll-Paque gradient separation as described previously (Shah et al., 2008). Briefly, clinical blood samples were processed on the date of the clinical blood draw for local patients and 24 hours post-draw for remote patients. Whole blood was layered over Ficoll-Paque PLUS according to manufacturer’s protocols (GE Healthcare Life Sciences). Peripheral blood mononuclear cells were isolated from resulting buffy coats and rinsed with 1x PBS. Residual red blood cell contaminates were removed using RBC Lysis buffer (Qiagen). PBMCs were quantified using trypan blue and a hemocytometer. All cells appearing as non-blue under a microscope were counted as viable cells. Samples with viability > 90% were then used for further downstream experiments. Normal CD8+ and NK cells from age- and gender-matched healthy donors were obtained from the Blood Bank of the Milton S. Hershey Medical Center or Virginia Blood Services and were isolated by selection using Stemcell Technologies EasySep Human NK or CD8+ cell Enrichment Kit. For flow cytometry apoptosis assays, five of nine patients had LGL populations that ranged from 88–99% purity. Of the other four patients, one did not have a clinical flow cytometry sample available. The other three had LGL populations that ranged from 15–40% purity.
Immunoblot analysis
Protein expression levels were determined as previously described (LeBlanc et al., 2015). Briefly, whole cell lysates were harvested using 1X RIPA buffer. Lysates were centrifuged for 15 minutes at 14,400 × g at 4°C to pellet cell debris. Protein concentrations were determined using a BCA assay (Pierce, Rockford, IL). Equal amounts of protein were loaded onto Bolt 4–12% Bis-Tris gels or 3–8% Tris-Acetate gels (Life Technologies, Carlsbad, CA), separated by electrophoresis and transferred to PVDF membranes. Membranes were blocked in 5% milk or BSA in 1X TBS-T and incubated overnight at 4°C with primary antibody (1:500–1:1000). Immunodetection was performed with IgG HRP-linked secondary antibodies (1:3000) and ECL reagent on a ChemiDOC XP or ChemiDOC MP (BioRad).
Apoptosis and cell viability assays
Apoptosis induced in NKL and TL-1 cells as well as PBMCs from normal donor controls and LGL patients was detected by flow cytometry with Annexin V–PE or Annexin V-APC (BD Pharmingen) and 7-amino-actinomycin D (7-AAD) staining using 5 × 105 cells per sample in triplicate. The percentage specific apoptosis was calculated using the follow formula: Apoptosis (%) = [(% Annexin V-PE positive cells) in treatment wells] - [(% Annexin V-PE positive cells) in control wells] × 100/[(% Annexin V-PE positive cells) in control wells]. Cell viability was determined using CellTiter 96 Aqueous One Solution assay kit (Promega). The relative viable cell number was determined using a Cytation 3 Microplate Reader (Bio-TEK).
Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. RNA was quantified using a Cytation 3 Microplate Reader and complementary DNA was constructed using iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA). qPCR reactions were conducted using a CFX384 Touch Real-time PCR Detection instrument (Bio-Rad, Hercules, CA). PrimePCR primer sets (SYBR) were purchased from Bio-rad.
siRNA transfections
Pooled scramble, SPHK1 and SPHK2 siRNA were purchased from GE Dharmacon (Lafayette, CO). NKL or TL-1 cells were transfected with an Invitrogen NEON Transfection system as described by the manufacturer (Invitrogen, Carlsbad, CA). Briefly, 5 × 105-1 × 106 cells were transfected with 50 nM of siRNA using optimized electroporation parameters (NKL: 1200V 40ms 1 pulse; TL-1 1400V 30ms 1 pulse). Knockdown efficiency was assessed by real-time PCR or immunoblot for target of interest.
Microarray data
Array results were obtained by analyzing a publicly microarray data sets completed previously by our lab (Gene Expression Omnibus accession numbers GSE10631, GSE42664) (Shah et al., 2008, Loughran et al., 2015). Briefly, for GSE10631, Leukemic PBMC RNA from 30 patients was extracted for target preparation and hybridization onto Affymetrix microarrays. We also isolated PBMCs without and with enrichment for CD8+ cells from control patients. RNA from these cells (naïve or activated with phytohemagglutinin) was extracted for target preparation and hybridization onto Affymetrix microarrays. Briefly, GSE42664 analyzed patient PBMCs after Ficoll enrichment, normal PBMCs enriched for CD3+/CD8+ or normal PBMCs enriched for CD3+/CD8+/CD45RA+/CCR7- that were then extracted in Trizol.
Statistical analysis
GraphPad Prism was utilized for statistical analyses (GraphPad Software). All data are expressed as mean ± SEM. Student’s t-test, Welch’s t-test or one-way ANOVA tests were used to determine the statistical significance, and p value of 0.05 or less was considered statistically significant.
Results
SPHK2 expression is elevated in LGL leukemia
Our microarray analysis (Shah et al., 2008) shows (Figure 1A) that SPHK2 mRNA expression is significantly elevated in LGL patient samples (n = 30) compared to purified CD8+ (n = 3) samples from normal donors (p < 0.05). Patient clinical characteristics from this microarray cohort are found in Table 1. Our data revealed that 22/30 (73%) of samples had higher expression of SPHK2 than the mean SPHK2 expression from normal controls. Furthermore, activation of CD8+ cells with IL-2/PHA resulted in increased SPHK2 mRNA expression (Figure 1A). We also noted that there was a statistically significant inverse correlation between platelet count and SPHK2 expression level (p = 0.018), and a statistically significant correlation between LGL cell number and SPHK2 expression level (p = 0.034). SPHK2 expression was not correlated with either hemoglobin or absolute neutrophil count (ANC) (Figure S1). Further, we obtained similar results using microarray data from an Eastern Cooperative Oncology Group (ECOG) clinical trial in LGL leukemia (GSE42664) (Figure S2A) (Loughran et al., 2015). Interestingly, there was no significant difference in SPHK2 expression between responders and non-responders to immunosuppressive treatment or in wild-type versus STAT3 mutant patients (Figure S2B and S2C respectively). Using immunoblot analysis, we verified that SPHK2 is elevated in both NK-LGL and T-LGL leukemia patient samples (LGL leukemia cell purity > 80%) compared to normal NK or CD8+ cells isolated from PBMCs of normal donors (Figure 1B and 1C). SPHK2 expression was not observed in PBMCs from normal donors (Figure 1C). Collectively, the above data demonstrate that LGL leukemia cells express elevated SPHK2. We next sought to investigate whether SPHK2 plays a role in cell survival in LGL leukemia cells.
Figure 1: SPHK2 is overexpressed in LGL leukemia.
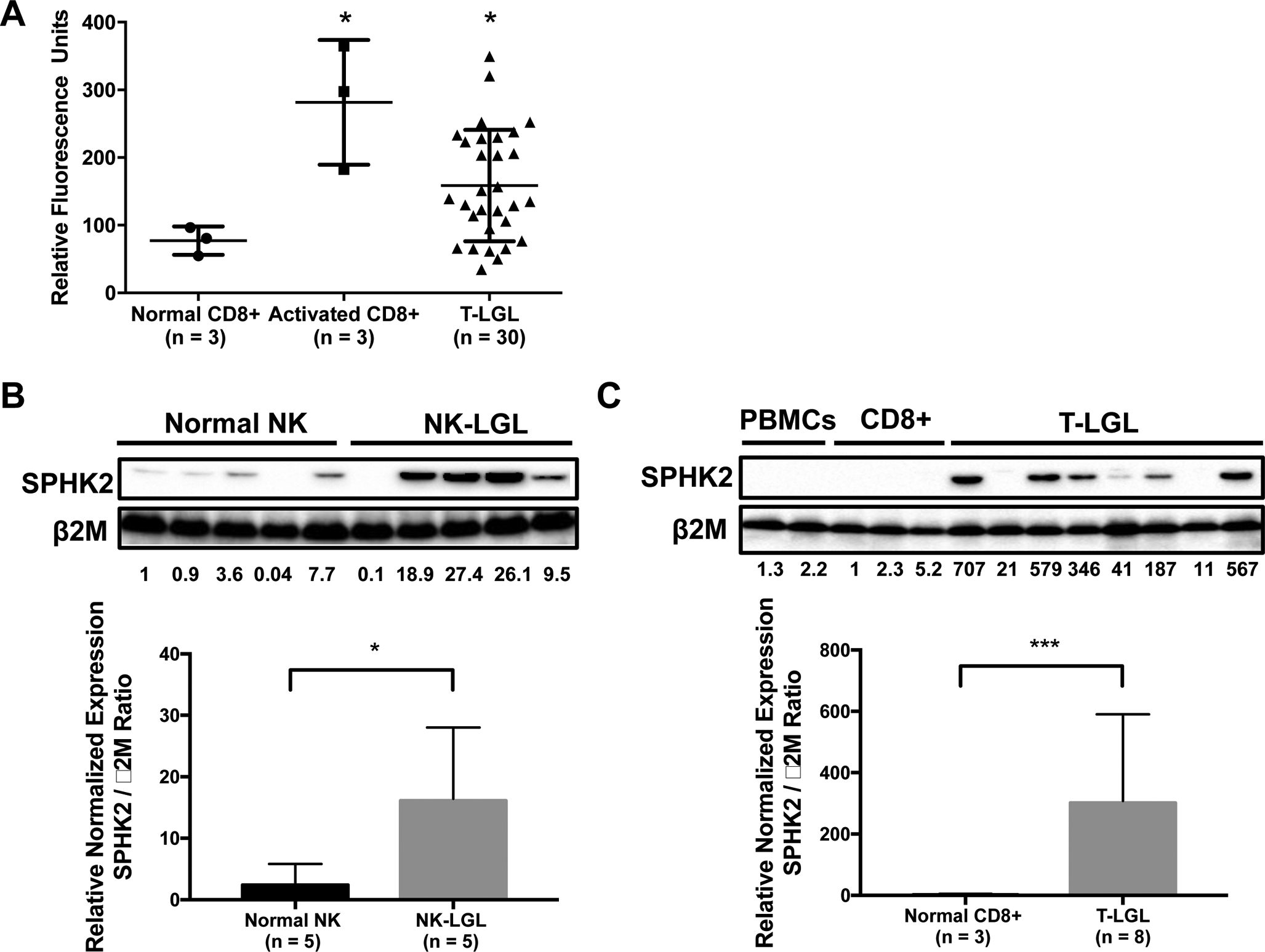
A) SPHK2 microarray mRNA expression levels from CD8+ cells from normal subjects (circles; n = 3), activated CD8+ cells from normal subjects (squares, n = 3) and T-LGL patients (triangles; n = 30) were compared. Mean + SD; *, p < 0.05 Welch’s t-test B) Immunoblot analysis of SPHK2 protein in NK cells from normal donors (n = 5) versus NK-LGL patients (n = 5) C) Immunoblot analysis of SPHK2 protein in PBMCs (n = 2) or CD8+ (n = 3) from normal donors versus T-LGL patients (LGL purity > 80%) (n = 8). Loading of protein was confirmed by probing for β2M. Quantification of SPHK2 expression relative to β2M shown in graphs. *, p < 0.05 and ***, p < 0.001; student’s t-test
Table 1:
Patient Characteristics
| Patient | Sex | Age at Dx | Hgb (g / dL) | Plt (K / uL) | WBC (K / uL) | ANC (K / uL) | ALC (K / uL) | LGL (K / uL) |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 50 | 9.6 | 556 | 7.4 | 1230 | 5350 | 1070 |
| 2 | M | 77 | 8.1 | 74 | 10.2 | 1940 | 7490 | 3150 |
| 3 | M | 71 | 9.0 | 360 | 4.4 | 2380 | 1590 | 614 |
| 4 | M | 63 | 10.7 | 126 | 9.1 | 800 | 7800 | |
| 5 | F | 63 | 15.5 | 208 | 8.2 | 920 | 6610 | 2842 |
| 6 | F | 79 | 11.6 | 253 | 13.8 | 1800 | 11700 | 10647 |
| 7 | F | 60 | 11.4 | 228 | 3.4 | 200 | 3080 | 1303 |
| 8 | F | 40 | 12.7 | 102 | 8.7 | 1800 | 6000 | |
| 9 | M | 57 | 14.5 | 119 | 22.4 | 448 | 20832 | 19998 |
| 10 | F | 57 | 14.1 | 185 | 11.4 | 1170 | 9960 | 8685 |
| 11 | M | 43 | 13.4 | 284 | 21.5 | 3270 | 17110 | 8555 |
| 12 | F | 38 | 9.0 | 213 | 4.3 | 1730 | 2250 | 536 |
| 13 | F | 72 | 12.9 | 233 | 3.7 | 370 | 2920 | 855 |
| 14 | M | 74 | 11.2 | 93 | 24.8 | 700 | 22900 | 22671 |
| 15 | F | 53 | 6.7 | 134 | 6.5 | 650 | 5470 | |
| 16 | M | 70 | 12.5 | 97 | 2.6 | 26 | 2158 | 1196 |
| 17 | M | 58 | 12 | 370 | 11.2 | 560 | 9900 | 8500 |
| 18 | F | 73 | 9.6 | 478 | 8.2 | 1886 | 5904 | 3603 |
| 19 | M | 81 | 10.5 | 49 | 9.1 | 300 | 8500 | 5100 |
| 20 | F | 53 | 9.4 | 192 | 3.8 | 114 | 3572 | 2744 |
| 21 | M | 69 | 7.0 | 59 | 3.8 | 1862 | 1558 | 756 |
| 22 | M | 66 | 7.5 | 414 | 13.8 | 800 | 11000 | |
| 23 | M | 84 | 10.0 | 346 | 3.9 | 1200 | 2100 | 1400 |
| 24 | F | 40 | 13.7 | 157 | 5.2 | 300 | 4200 | 1600 |
| 25 | F | 54 | 13.7 | 238 | 4.8 | 100 | 4000 | 2117 |
| 26 | M | 83 | 8.6 | 131 | 4.3 | 1600 | 2100 | 600 |
Genetic knockdown of SPHK2 decreases viability in LGL leukemia cell lines
We have previously shown that SPHK1 is overexpressed in LGL leukemia and pharmacologic inhibition led to decreased cell viability and increased apoptosis (LeBlanc et al., 2015). We have since been able to demonstrate that biologic inhibition of SPHK1 with siRNA in both NKL and TL-1 cells (models of NK-LGL and T-LGL leukemia, respectively) leads to decreased proliferation (Figure S3). To establish whether SPHK2 has a role in the survival of leukemic LGLs, we transfected NKL and TL-1 cells with siRNA targeting SPHK2. Cells were transfected with SPHK2-specific siRNA or control scramble siRNA and assessed for knockdown at 48 hours (mRNA) and 72 hours (protein). In NKL and TL-1 cells, there was effective knockdown of SPHK2 mRNA (Figure 2A and 2B) and protein levels (Figure 2C and 2D). Knockdown of SPHK2 in NKL and TL-1 cells significantly inhibited cell proliferation at 72h post-transfection (NKL: 35% reduction, TL-1: 30% reduction) (Figure 2E and 2F). These results suggest that SPHK2 plays an important role in LGL leukemia cell survival.
Figure 2: Knockdown of SPHK2 decreases viability of leukemic LGLs.
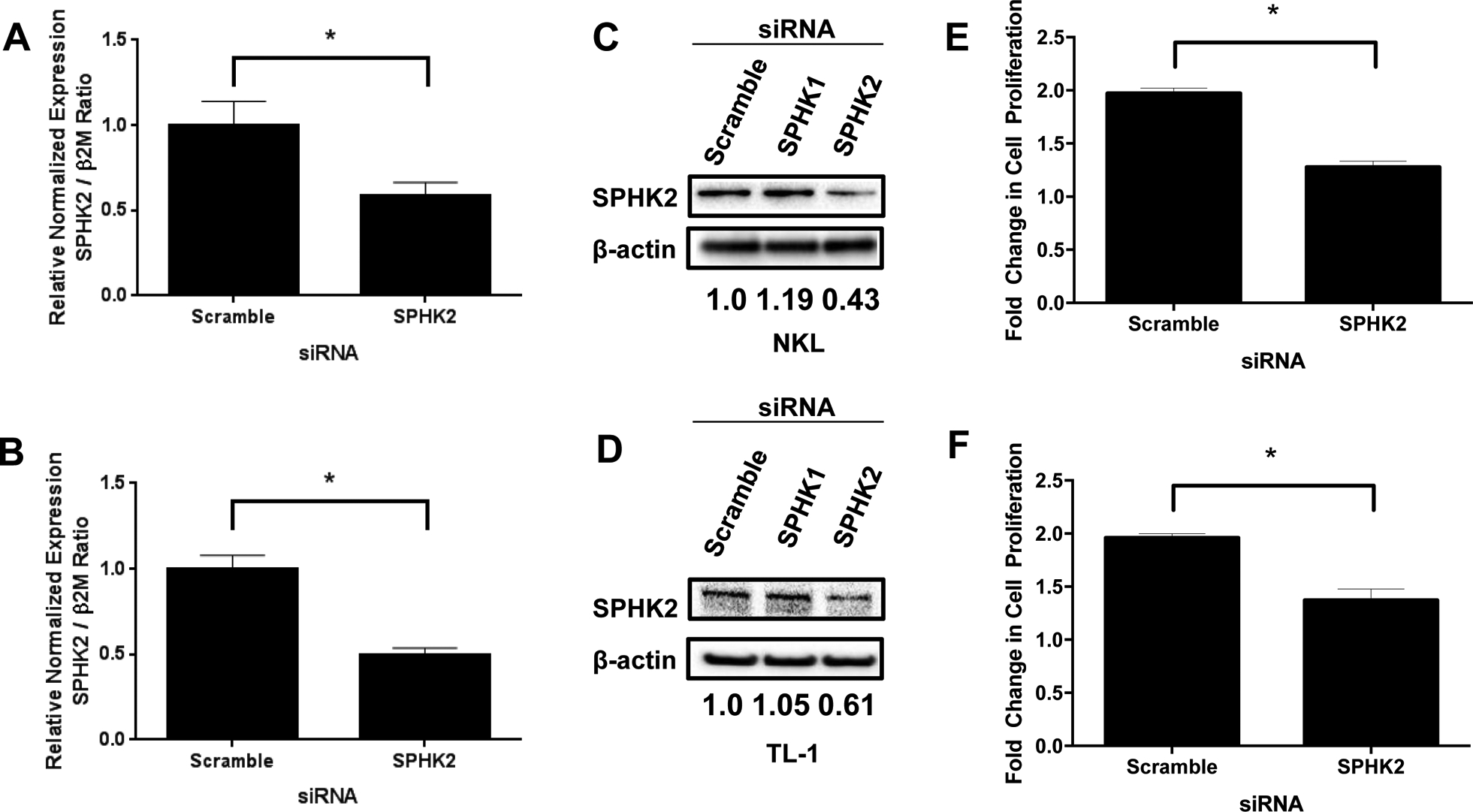
NKL or TL-1 cells were transfected with siRNA targeting SPHK2 or control (scramble) siRNA. A and B) Quantitative real-time PCR of SPHK2 knockdown in NKL (A) or TL-1 (B) cells. mRNA levels were normalized to β2M. Scramble siRNA used as control. C and D) Immunoblot analysis of SPHK2 levels in siRNA-transfected NKL (C) or TL-1 (D) cells. Loading of protein was confirmed by probing for β-actin. Normalization was performed by taking the ratio of SPHK2 to β-actin and normalizing to scramble control (Set to 1). E and F) Cell viability was assessed at 72 hours post-transfection using an MTS assay. *, p < 0.05 indicate significant difference between scramble and SPHK2 siRNA transfected cells (Student’s t-test)
SPHK2-specific inhibitors decrease viability and induces apoptosis of leukemic LGL
We next tested the effectiveness of ABC294640 in killing leukemic LGLs. ABC294640 is a nonlipid-based oral SPHK2-specific inhibitor that does not inhibit SPHK1 or a panel of other protein kinases (French et al., 2010). LGL leukemia cell lines (NKL, TL-1) treated with ABC294640 exhibited decreased viability (IC50 at 48 hours: NKL; 32.4 μM, TL-1; 17.4 μM) (Figure 3A and 3B). These concentrations are in line with established therapeutic and tolerated doses (French et al., 2010, Xun et al., 2015). We sought to confirm these results using a second SPHK2-specific inhibitor, K145 (Figure S4A and S4B) (Liu et al., 2013). K145 also decreased viability of NKL and TL-1 cells (IC50 at 48 hours: NKL; 4.24 μM, TL-1; 2.73 μM). To determine if ABC294640 induces cytotoxic effects on LGL cells, we assessed apoptosis in both NKL and TL-1 cells with flow cytometry and Annexin V/7-AAD staining. ABC294640 induced dose-dependent apoptotic cell death in both cell lines (Figure 3C and 3D). Additionally, immunoblot analysis demonstrated decreased total caspase-3 levels (35 kDa band) and increased levels of cleaved caspase-3 (15–19 kDa band) upon treatment of NKL and TL-1 cells with ABC294640, suggesting activation of caspase-dependent apoptosis (Figure 3E and 3F). Treatment of NKL or TL-1 cells with K145 also led to caspase-3 cleavage (Figure S5A and S5B) further supporting the role of SPHK2 in LGL leukemia survival.
Figure 3: ABC294640 inhibits cell proliferation and induces apoptosis in LGL leukemia cells.
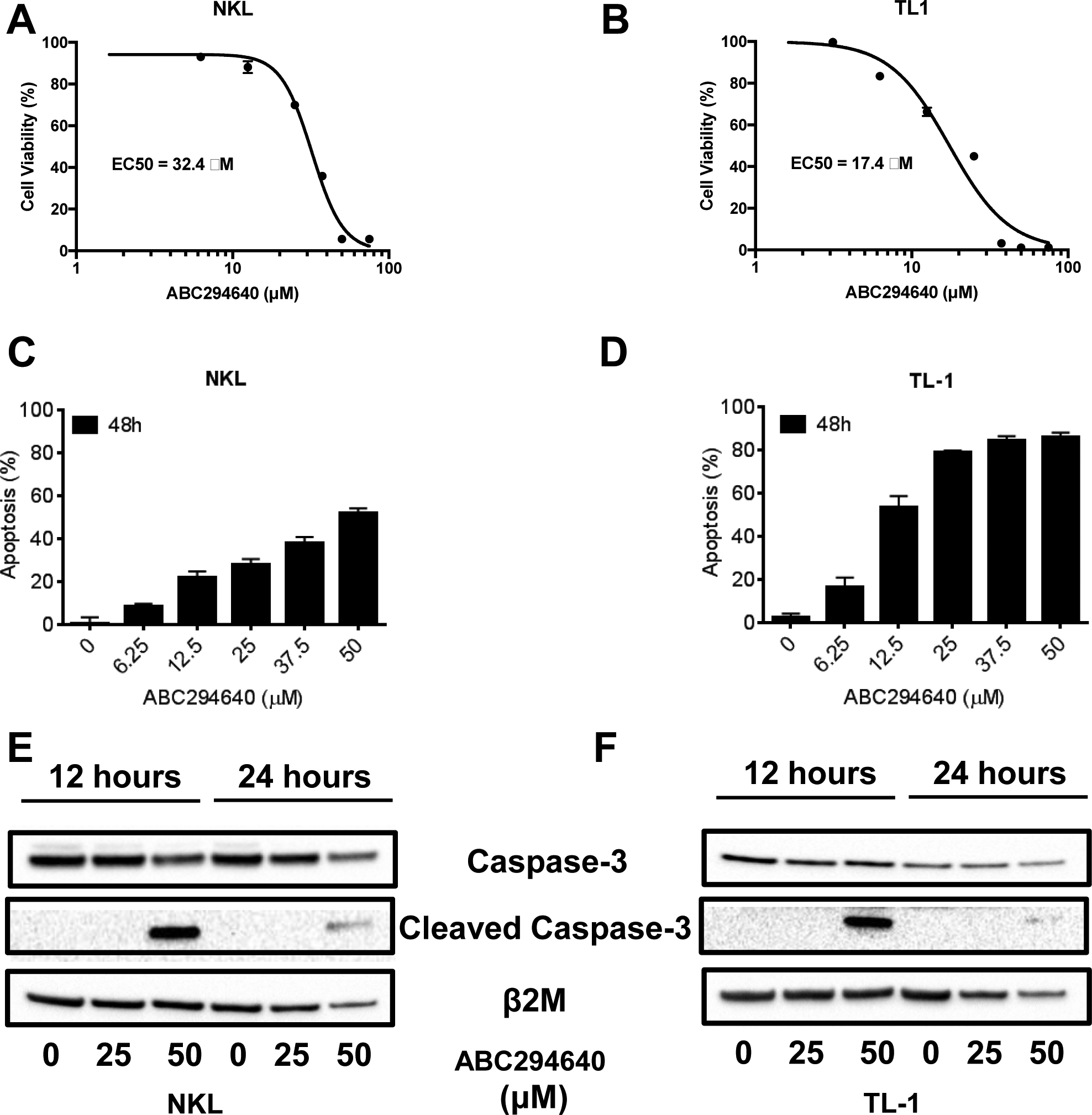
NKL and TL-1 cells were treated with increasing doses of ABC294640 and assessed for cell viability using an MTS assay or apoptosis using Annexin V/7-AAD staining by flow cytometry. A and B) MTS Assay in NKL (A) and TL-1 (B) cells. C and D) Apoptosis in NKL (C) and TL-1 (D) cells. E and F) Immunoblot analysis for Caspase-3 (35 kDa) and cleaved Caspase-3 (cCaspase-3) (15–19 kDa) of NKL (E) and TL-1 (F) cells after treatment with ABC294640. β2M used as a loading control. MTS data is shown as mean + SEM. Apoptosis data is show as mean + SEM.
We also tested the anti-tumor effects of ABC294640 on primary human leukemic LGL cells. These cells were freshly isolated from patient PBMCs (n = 9) or normal donors (n = 4). ABC294640 potently induced apoptotic cell death in leukemic LGLs with similar efficacy as in leukemic LGL cell lines. Importantly, ABC294640 had minimal apoptotic effects on freshly isolated PBMCs from normal donors (Figure 4A). ABC294640 was effective at doses as low as 12.5 μM and was dose-dependent in representative T-LGL (Figure 4B) and NK-LGL patient samples (Figure 4C). There was no difference in sensitivity of patient cells to ABC294640 when looking at STAT3 mutational status, although this was not sufficiently powered for a formal statistical evaluation (data not shown). ABC294640 effectively and selectively inhibited cell proliferation and induced apoptosis in leukemic LGL cells, demonstrating the therapeutic potential of ABC294640 in the treatment of LGL leukemia.
Figure 4: ABC294640 induces selective apoptosis in PBMCs from LGL leukemia patients.
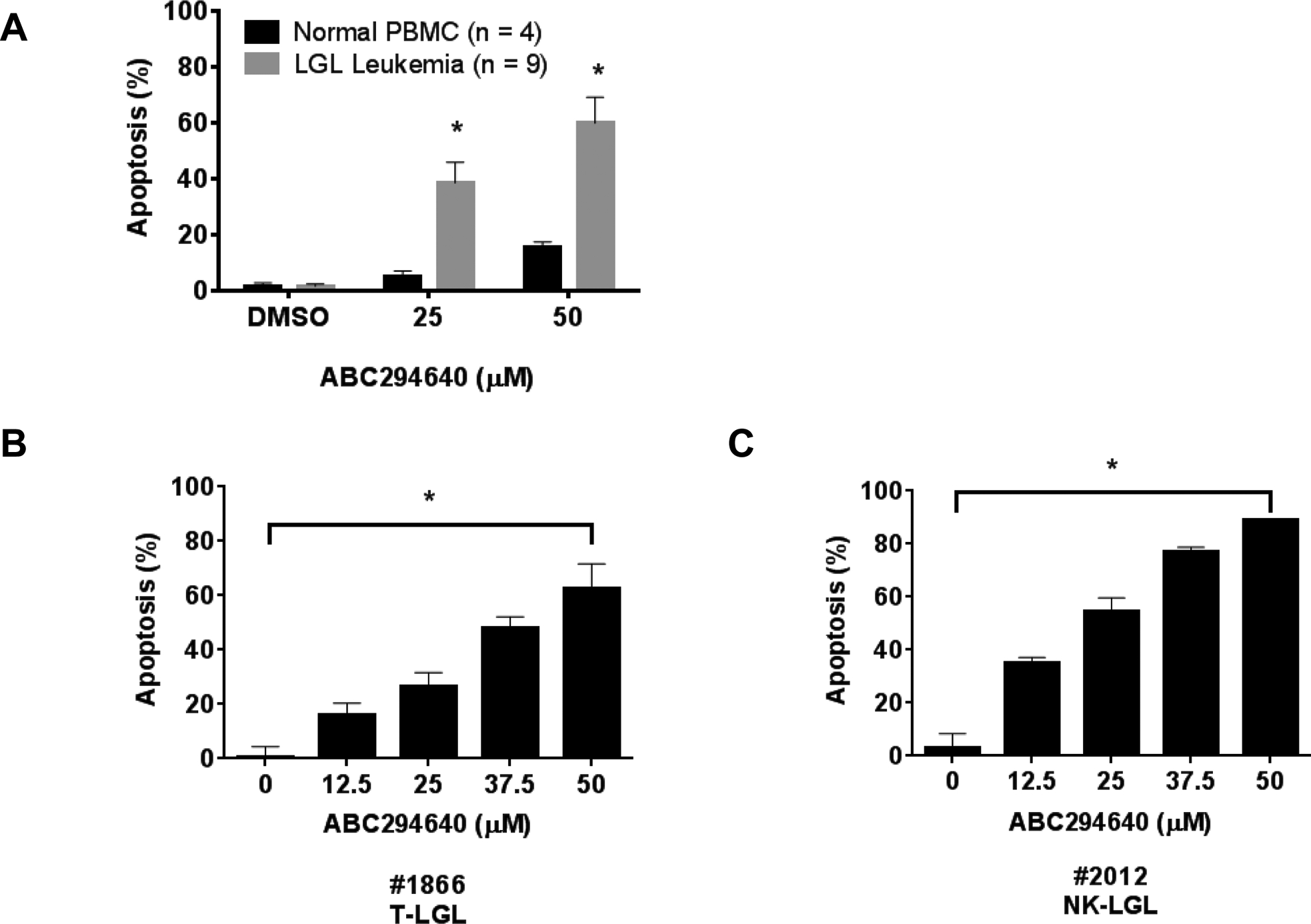
A) PBMC samples from LGL leukemia patients or PBMC samples from normal donors were treated with DMSO or ABC294640 at the indicated doses for 48 hours and assayed for apoptosis B) and C) ABC294640 induces dose-dependent apoptosis in representative T-LGL (#1866) or NK-LGL (#2012) patients. *, p < 0.05, indicate significant differences of ABC294640 of T-LGL versus normal PBMCs (Student’s t-test) or ABC294640-treated versus vehicle control (DMSO) treated cells for all doses (one-way ANOVA).
Inhibition of SPHK2 decreased Mcl-1 expression through proteasomal degradation
We sought to determine the downstream role of Bcl-2 family members while targeting SPHK2 for inhibition. We observed dose-dependent downregulation of Mcl-1, but not Bcl-2 or Bcl-XL, protein expression in both NKL (Figure 5A) and TL-1 (Figure 5B) cells treated with ABC294640. This effect was also seen in PBMCs isolated from representative NK-LGL or T-LGL patients (Figure 5C). K145 also strongly downregulated Mcl-1 and modestly reduced Bcl-2 and Bcl-XL in NKL and TL-1 cells. (Figure S5C and S5D). Transfection of both NKL and TL-1 cells with siRNA targeting SPHK2, but not SPHK1, also led to downregulation of Mcl-1 (Figure 5D). Together, these data demonstrate that SPHK2 inhibition decreases Mcl-1 in LGL leukemia.
Figure 5: SPHK2 inhibition downregulates Mcl-1 expression in LGL leukemia cells.
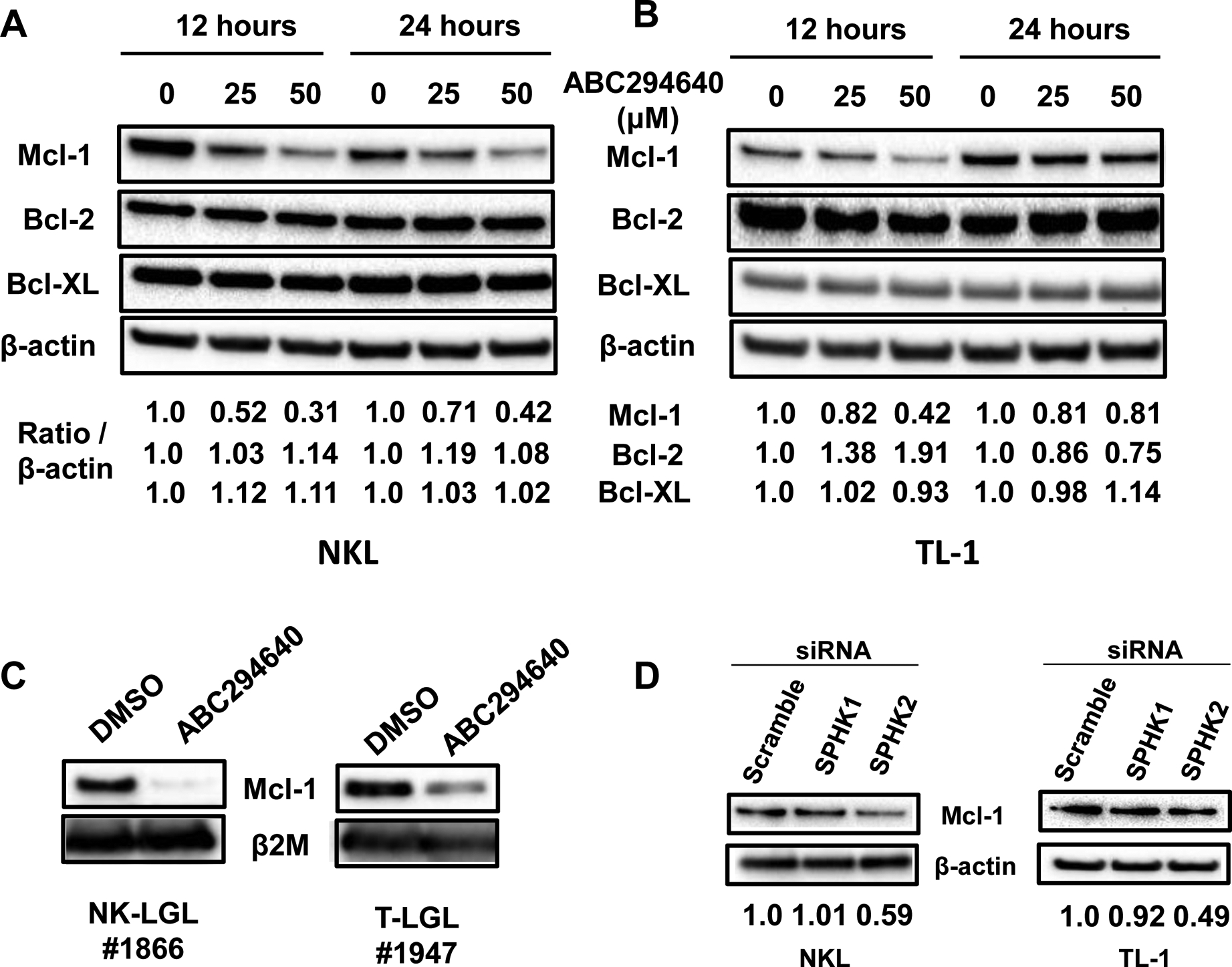
A) Immunoblot analysis of pro-survival Bcl-2 family members in NKL cells after treatment with ABC294640. β-actin was used as a loading control. Bcl-2 family member expression was normalized to β-actin B) Immunoblot analysis of pro-survival Bcl-2 family members in TL-1 cells after treatment with ABC294640. β-actin was used as a loading control. Prosurvival Bcl-2 family member expression was quantified and normalized to β-actin C) Immunoblot analysis for Mcl-1 of PBMCs were isolated from representative NK-LGL (#1866) or T-LGL (#1947) patients and treated with ABC294640 for 12 hours. β2M was used as a loading control. D) Genetic knockdown of SPHK2, but not SPHK1, downregulates Mcl-1 protein expression in NKL and TL-1 cells. Scramble siRNA used as a control.
We next investigated the mechanism by which SPHK2 inhibition induced downregulation of Mcl-1. We first set out to determine if ABC294640 treatment or siRNA targeting of SPHK2 affected MCL1 gene transcription. NKL cells were treated with ABC294640 for 16 hours or transfected with siRNA targeting SPHK2. Neither ABC294640 treatment nor siRNA inhibition of SPHK2 affected MCL1 mRNA levels (Figure 6A and 6B). This suggests that Mcl-1 downregulation by ABC294640 is not at the level of transcriptional or mRNA destabilization.
Figure 6: Inhibition of SPHK2 downregulates Mcl-1 expression via proteasomal degradation.
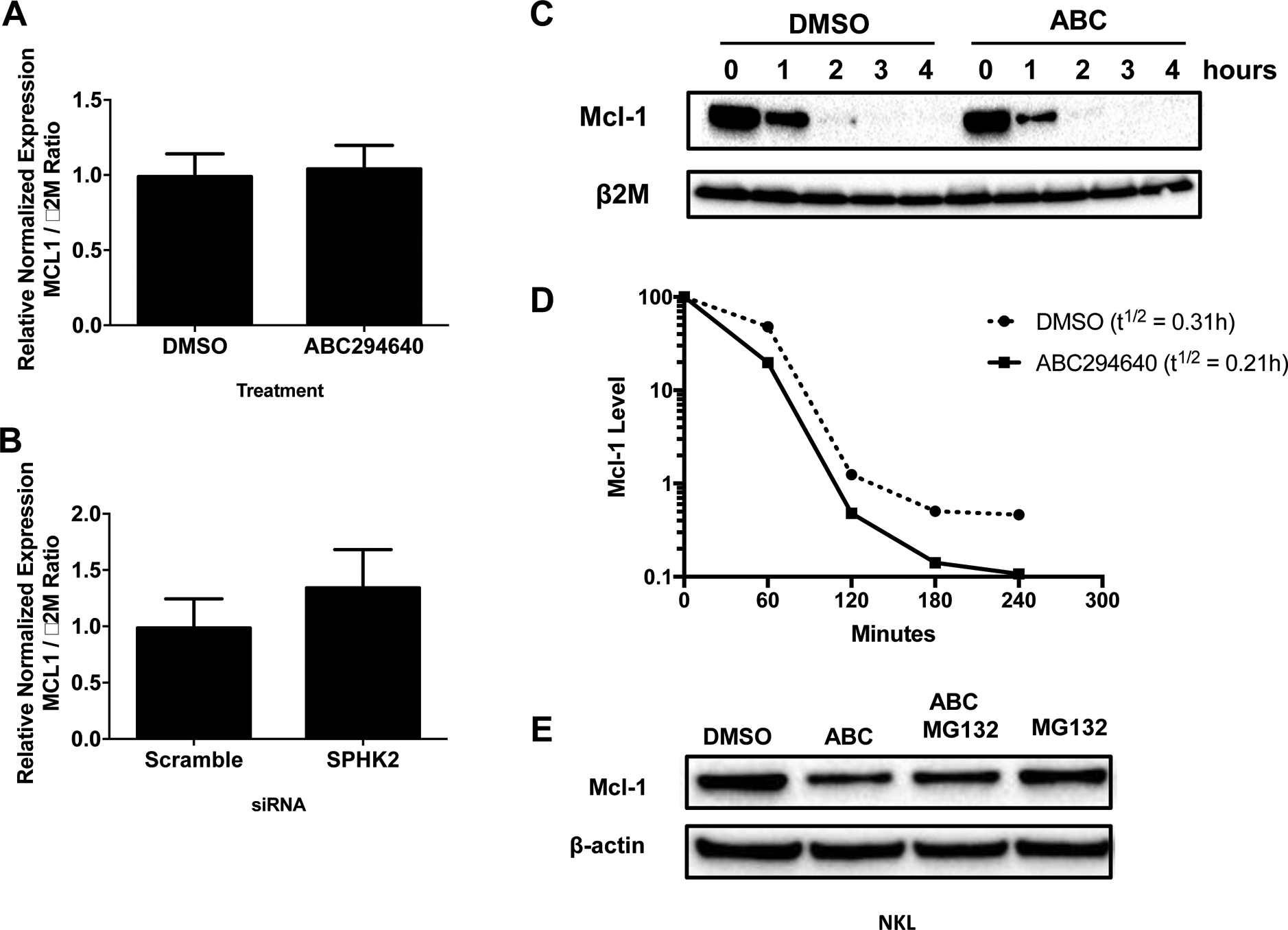
A) NKL cells were treated with 25μM of ABC294640 for 16 hours or B) transfected with siRNA targeting SPHK2 for 72 hours and mRNA extracted. Quantitative real-time PCR was performed to measure levels of Mcl-1 mRNA. Data was normalized to β2M expression. Data shown is mean + sd. C) NKL cells were treated with DMSO or 25 μM of ABC294640 for 3 hours and then cycloheximide (50 μg / mL) was added. Protein was harvested every hour for 4 hours. Mcl-1 expression was assessed by immunoblot analysis. The blots were quantified using Bio-Rad Image Lab 5.0. Mcl-1 levels were normalized to β2M which was used as a loading control. D) The graph illustrates the quantification of the immunoblots in C. E) NKL cells were pre-treated with MG132 (1 μM) for 1 hour and then 25 μM of ABC294640 or DMSO was added for 16 hours. Mcl-1 protein expression was analyzed by immunoblot. Β-actin was used as a loading control
Next, increased protein degradation was explored as a potential mechanism-of-action for ABC294640-induced Mcl-1 downregulation. We treated NKL cells for 3 hours with ABC294640 or DMSO and then added cycloheximide (CHX) to inhibit new protein synthesis. We measured Mcl-1 proteins levels by immunoblot and observed that ABC294640 treatment increased the Mcl-1 protein degradation rate (t1/2 DMSO: 0.31 hours, ABC294640: 0.21 hours) (Figure 6C and 6D). Other research groups have shown that ABC294640 can target Mcl-1 through proteasomal degradation (Venkata et al., 2014, Liao et al., 2011). We co-treated NKL cells ABC294640 and MG132, a proteasome inhibitor, and observed that MG132 partially rescued ABC294640-induced Mcl-1 degradation (Figure 6E). These data suggest that proteasome-dependent degradation contributes to the Mcl-1 loss that accompanies SPHK2 inhibition in LGL leukemia cells.
Targeting SPHK2 synergizes with BH3 mimetics to decrease cell viability
There have been multiple BH3 mimetics developed, including ABT-737 and ABT-199, which target pro-survival BCL2 family members. These agents are highly effective in tumors that have minimal dependence on Mcl-1 for cell survival (Chen et al., 2007, van Delft et al., 2006). Unfortunately, some leukemic samples, including AML and LGL leukemia, may be resistant to BH3 mimetics because of elevated expression of MCL1 (Konopleva et al., 2006). We performed preliminary studies investigating whether the enhanced Mcl-1 degradation that is mediated by SPHK2 inhibition could sensitize LGL cells to BH3 mimetics. NKL cells were resistant to ABT-199 (Figure 7A). However, in combination with ABC294640, we observed a synergistic reduction in cell viability (Figure 7B, CI < 1). This highlights the potential for combining BH3 mimetics with sphingolipid-targeted therapeutics to treat LGL leukemia.
Figure 7: Targeting SPHK2 with ABC294640 synergizes with ABT-199 to decrease proliferation in LGL leukemia cells.
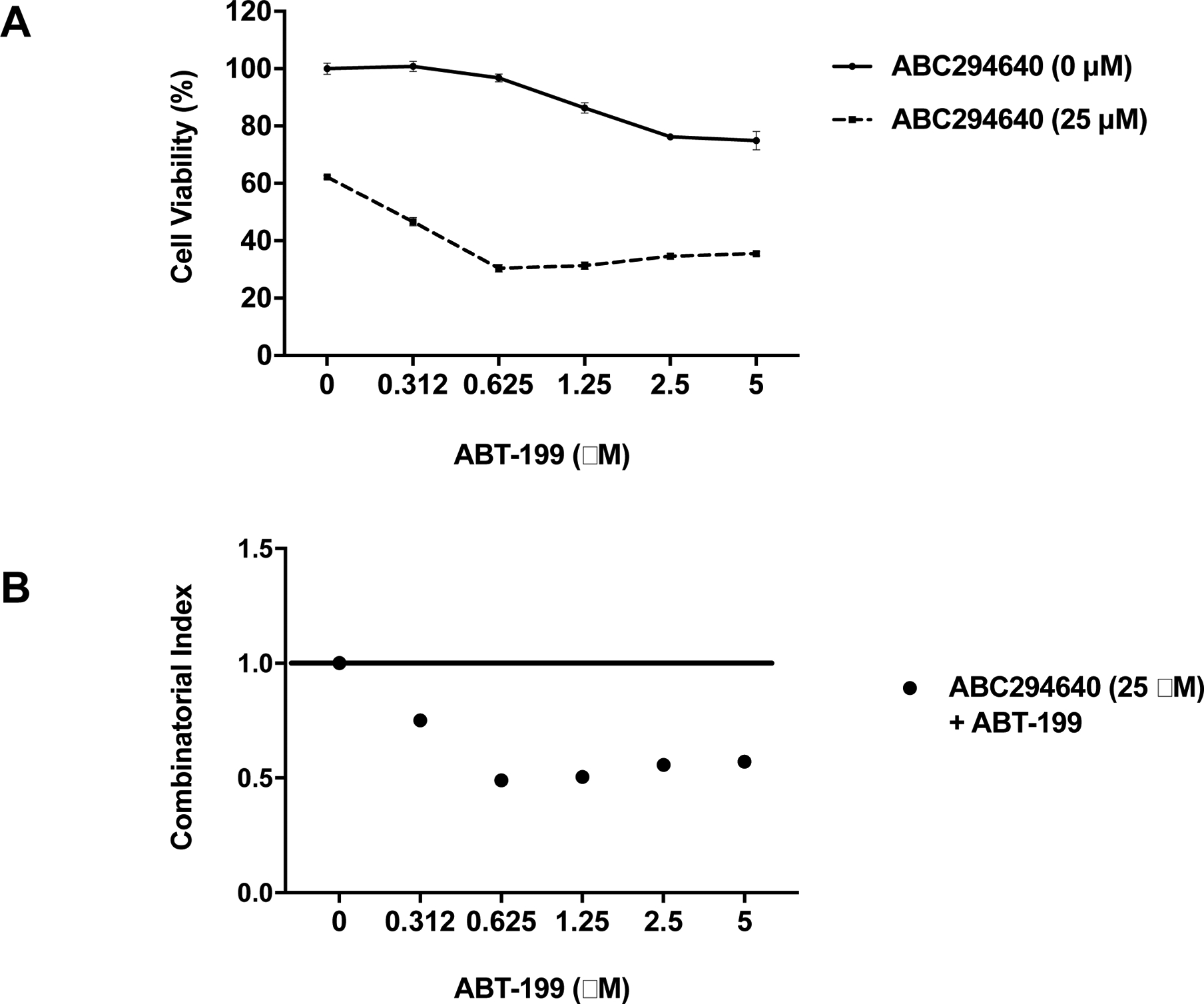
A) NKL cells were treated with increasing doses of ABT-199 in the presence or absence of 25 μM ABC294640 and cell viability was assessed using MTS assay. B) Combinatorial index (CI) was calculated for ABT-199 with ABC294640. CI values less than 1 indicate synergy between the compounds.
Discussion
Our results have identified SPHK2 as a novel therapeutic target in LGL leukemia. Using microarray analysis, we found that SPHK2 was upregulated in 22/30 (~70%) patient samples compared to CD8+ cells from normal donors. We verified this result in a second microarray dataset of an independent patient cohort from an ECOG clinical trial in LGL leukemia (Loughran et al., 2015). Furthermore, knockdown of SPHK2 with siRNA inhibited both NK- and T-LGL leukemia cell proliferation. These data demonstrate that SPHK2 has an important role in LGL leukemia. We used SPHK2-specific inhibitors, ABC294640 and K145, and for the first time showed that SPHK2 is a potential therapeutic target in LGL leukemia. We identified that pro-survival Mcl-1 levels are reduced upon inhibition of SPHK2. The identification and validation of SPHK2 and Mcl-1 as potential therapeutic targets in LGL leukemia is significant because current therapeutic regimens use non-specific immunosuppressive agents and are not curative (Lamy and Loughran, 2011, Loughran et al., 2015).
The expression of SPHK2 in our microarray analysis was found to inversely correlate with platelet count and directly correlate with LGL cell number (Shah et al., 2008). The patient characteristics of this cohort were representative of typical LGL leukemia patients with similar median age at diagnosis, gender distribution, and Hgb, ANC, platelet, WBC, ALC and LGL cell number (Lamy and Loughran, 2011, Poullot et al., 2014, Steinway et al., 2014). We did not find any correlation between SPHK2 expression and hemoglobin or absolute neutrophil count, which are the two clinical features that are most closely linked with a need for treatment. Additionally, using the microarray dataset from an ECOG prospective clinical trial using immunosuppressive therapy in LGL leukemia, we were able to compare SPHK2 expression levels between responders versus non-responders to treatment and STAT3 wild-type (WT) vs STAT3 mutant patients (Loughran et al., 2015). This comparison indicated that there is no statistically significant difference in SPHK2 levels within these groups. The key difference between the two independent patient cohort microarray datasets is that the ECOG trial patients are a subset of LGL leukemia patients that required treatment. Therefore, they may represent a different subpopulation of patients compared to the overall cohort of LGL leukemia patients. The relationship between SPHK2 expression and both platelet count and LGL leukemia cell number should be further investigated.
We demonstrated that ABC294640 effectively inhibited proliferation and induced apoptosis preferentially in LGL leukemia patient samples with minimal effects on PBMCs from normal donors. The LGL leukemia patient samples were harvested as PBMCs, however, a majority of patients had LGL cell populations representing more than 85% of cells. This supports the conclusion that the effects are specific to the LGL leukemia cells. Importantly, we did not observe a difference in effectiveness when accounting for STAT3 mutational status, although this study was not sufficiently powered to perform a statistical analysis. ABC294640 is a first-in-class SPHK2-selective inhibitor that is administered orally. ABC294640 has successfully completed a Phase I clinical study in patients with advanced solid tumors (NCT01488513). It is currently being investigated in Phase I/II clinical trials for the treatment of refractory/relapsed diffuse large-B cell lymphoma (NCT0222998), refractory / relapsed multiple myeloma (NCT02757326) and hepatocellular carcinoma (NCT02939807). ABC294640 has also shown anticancer effects in a multitude of solid tumors (Sun et al., 2015, Zhang et al., 2015). It demonstrates good pharmacokinetics, oral bioavailability and can reach plasma concentrations > 200 μM without significant toxicity (Venkata et al., 2014). This concentration is significantly higher than values that exhibited strong potency in LGL leukemia cells (12.5–50 μM), therefore establishing a foundation for the feasibility of clinical trials in LGL leukemia. Due to ABC294640 also recently being identified as a dihydroceramide desaturase (Des1) inhibitor (McNaughton et al., 2016), we verified our results using K145, a second SPHK2 inhibitor. We were able to demonstrate that K145 also decreased proliferation in LGL leukemia cells. We showed that targeting SPHK2 with ABC294640 induced apoptosis and decreased proliferation in both LGL leukemia cell lines and freshly isolated primary LGLs from patients. Importantly, ABC294640 selectively induced apoptosis in LGL leukemia patient cells but not in cells from normal donors. This highlights SPHK2 targeting with ABC294640 or K145 as a promising and selective therapeutic approach in LGL leukemia.
Mechanistically, we found that both siRNA inhibition of SPHK2 and ABC294640 treatment downregulated Mcl-1 expression through proteasomal degradation. This is similar to results reported in multiple myeloma cells treated with ABC294640 (Venkata et al., 2014). The treatment of LGL leukemia cells with proteasome inhibitors abrogated the effects of ABC294640 on Mcl-1 downregulation, suggesting that degradation is at least partly via the proteasome degradation pathway. Interestingly, we also found that K145 downregulated Mcl-1 expression, further supporting Mcl-1 as being downstream of SPHK2. We observed a modest decrease in Bcl-2 and Bcl-XL with K145 treatment that we did not see with ABC294640. The most pronounced effect was on Mcl-1 and it is possible that the downregulation of Bcl-2 and Bcl-XL is related to non-specific protein loss during cell death rather than specifically a result of SPHK2 inhibition, but further investigations are warranted. Mcl-1 is an important anti-apoptotic Bcl-2 family member that has been identified as a STAT3 transcriptional target in LGL leukemia (Epling-Burnette et al., 2001). We demonstrate that inhibition of SPHK2 targets Mcl-1, which further highlights it as an attractive therapeutic approach to impair key survival pathways (Doi et al., 2014).
Both SPHK1 and SPHK2 have been demonstrated to have a role in the pathogenesis of cancer (Takabe et al., 2008, Maceyka et al., 2012, Stevenson et al., 2011, Shida et al., 2008, Santos and Lynch, 2015, Lynch et al., 2016, Childress et al., 2017). Most studies have focused on SPHK1, however, recently there have been multiple publications suggesting that SPHK2 also primarily exhibits a pro-survival role in cancer (Takabe et al., 2008, Maceyka et al., 2012, Stevenson et al., 2011). Additionally, the role of extracellular versus intracellular S1P and the involvement of S1PRs are actively being studied (Blaho and Hla, 2014). SPHK2 has not been investigated in LGL leukemia but prior studies have shown that it can stimulate cancer cell growth (Sun et al., 2015). SPHK2 expression stimulates c-MYC upregulation in ALL cells promoting their survival and proliferation (Wallington-Beddoe et al., 2014) and SPHK2 overexpression was necessary for the survival of multiple myeloma cells (Venkata et al., 2014). We have previously published on SPHK1 in LGL leukemia (which produces S1P to be used both extracellularly and intracellularly) using the pharmacologic inhibitor SKI-178 (LeBlanc et al., 2015). At the time of publication, SKI-178 was thought to be highly specific for SPHK1 but has since been found to additionally target both SPHK2 as well as microtubule formation (Hengst et al., 2017, Dick et al., 2015, Hengst et al., 2010). We provide data in this study demonstrating that the SPHK1 also plays a role in survival of LGL leukemia cells. We have previously shown that SPHK1 is important for the survival of LGL leukemia cells through its interaction with both Bcl-2 survival pathways and STAT3 signaling. Interestingly, we observed that SKI-178 treatment did not affect Mcl-1 expression. However, in AML, pharmacologic and biologic inhibition of SPHK1 led to cell death through the downregulation of Mcl-1 but the inhibition of SPHK2 did not (Powell et al., 2017). The reasons for the differential regulation of Mcl-1 by SPHK1 versus SPHK2 are unclear. It has been suggested that the subcellular localization of SPHK2 may ultimately be responsible for the different functions of this enzyme. Regardless, it is clear in our studies that SPHK2 plays an important role in survival of leukemic LGLs.
We show preliminary results that inhibition of SPHK2 with ABC294640 can act synergistically with ABT-199 in LGL leukemia cells. The inhibition of Bcl-2 with BH3 mimetics, like ABT-737 and ABT-199, has been successful in treatment of chronic lymphocytic leukemia but less effective as a monotherapy in Mcl-1 dependent cancers such as AML (Eradat, 2019, Bose et al., 2017). The inhibition of SPHK1 in AML was shown to synergize with ABT-737 (Powell et al., 2017). In LGL leukemia, Mcl-1 is overexpressed and contributes to survival. Inhibition leads to decreased proliferation and increased apoptosis (manuscript in preparation). Not surprisingly then, we observed that LGL leukemia cells were resistant to ABT-199. The targeting of SPHK2, and likely SPHK1, to sensitize cells to BH3 mimetics is therefore a promising avenue to pursue for future studies for the treatment of LGL leukemia.
Overall, we have identified that SPHK2 is important for the survival of LGL leukemia cells. We demonstrated that SPHK2 inhibition led to decreased survival and increased apoptosis and mechanistically this occurred in part through Mcl-1 downregulation. This work highlights the potential role of targeting SPHK2 and/or Mcl-1, in the treatment of LGL leukemia.
Supplementary Material
Figure S1: Platelet count and LGL number are correlated with SPHK2 mRNA expression levels. SPHK2 microarray mRNA expression levels versus A) Hemoglobin (Hgb) B) Platelet count C) Absolute neutrophil count (ANC) and D) LGL Number.
Figure S2: SPHK2 is overexpressed in LGL leukemia but is not correlated with treatment outcomes or STAT3 mutational status. A) SPHK2 microarray mRNA expression levels from CD8+ cells from normal subjects (circles; n = 5), Temra cells from normal subjects (squares, n = 3) and T-LGL patients (triangles; n = 37) were compared. Mean + SD; **, p < 0.01 Welch’s t-test B) SPHK2 microarray mRNA expression levels comparison between Responder (n = 19) and Non-Responder (n = 18) to treatment C) SPHK2 microarray mRNA expression levels comparison between STAT3 WT (n = 21) versus STAT3 mutant (n = 16) patients.
Figure S3: Knockdown of SPHK1 decreases viability of leukemic LGLs. NKL or TL-1 cells were transfected with siRNA targeting SPHK1 or control (scramble) siRNA. A and B) Quantitative real-time PCR of SPHK1 knockdown in NKL (A) or TL-1 (B) cells. mRNA levels were normalized to β2M. Scramble siRNA used as control. C and D) Immunoblot analysis of SPHK1 levels in siRNA-transfected NKL (C) or TL-1 (D) cells. Loading of protein was confirmed by probing for β-actin. Normalization was performed by taking the ratio of SPHK1 to β-actin and normalizing to scramble control (Set to 1). E and F) Cell viability was assessed at 72 hours post-transfection using an MTS assay. *, p < 0.05 indicate significant difference between scramble and SPHK1 siRNA transfected cells (Student’s t-test).
Figure S4: K145 inhibited cell proliferation in LGL leukemia cells. NKL and TL-1 cells were treated with increasing doses of K145 and assessed for cell viability using an MTS assay. A and B) MTS Assay in NKL (A) and TL-1 (B) cells.
Figure S5: SPHK2 inhibition with K145 downregulates Mcl-1, Bcl-2 and Bcl-XL expression and leads to cleavage of caspase-3 in LGL leukemia cells. A) Immunoblot analysis of total and cleaved caspase-3 in NKL cells after treatment with K145. β-actin was used as a loading control. B) Immunoblot analysis of total and cleaved caspase-3 in TL-1 cells. C) Immunoblot analysis of pro-survival Bcl-2 family in NKL cells after treatment with K145. D) Immunoblot analysis of pro-survival Bcl-2 family in TL-1 cells after treatment with K145. β-actin was used as a loading control. Normalization was performed by taking the ratio of Mcl-1, Bcl-2 or Bcl-XL to β-actin (0 μM, Set to 1).
Acknowledgments
LGL leukemia patient samples and clinical information were obtained from the LGL Leukemia Registry at the University of Virginia with the assistance of Kendall Baab, Holly Davis, Bryna Shemo and Andrea Hines. Dr. Tom Olson and Cait Hamele profiled SH2 domain STAT3 somatic mutations in LGL leukemia patient samples. Alexander Wendling provided excellent technical support while processing patient samples. The University of Virginia School of Medicine flow cytometry core assisted with flow cytometry and data analysis. Recombinant human interleukin 2 (rh-IL-2) was kindly provided by NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann - La Roche Inc. NKL cells, a leukemic LGL NK-cell line, were kindly provided by Dr. Howard Young at the National Cancer Institute.
Funding
This research was funded by the National Cancer Institute of the National Institutes of Health under award number R01CA098472, R01CA178393, R01CA171983 and P30CA044579 (TPL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional funding was provided by the Bess Family Charitable Fund, the LGL Leukemia Foundation and a generous anonymous donor.
Footnotes
Conflict of interest disclosure
Thomas P. Loughran, Jr. is on the Scientific Advisory Board and has stock options for Keystone Nano and Bioniz Therapeutics. David J. Feith served on an advisory board for Kymera Therapeutics. There are no conflicts of interest with the work presented in this manuscript.
REFERENCES
- ALVAREZ SE, HARIKUMAR KB, HAIT NC, ALLEGOOD J, STRUB GM, KIM EY, MACEYKA M, JIANG H, LUO C, KORDULA T, MILSTIEN S & SPIEGEL S 2010. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature, 465, 1084–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANTOON JW, WHITE MD, SLAUGHTER EM, DRIVER JL, KHALILI HS, ELLIOTT S, SMITH CD, BUROW ME & BECKMAN BS 2011. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther, 11, 678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLAHO VA & HLA T 2014. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res, 55, 1596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOSE P, GANDHI V & KONOPLEVA M 2017. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma, 58, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN S, DAI Y, HARADA H, DENT P & GRANT S 2007. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res, 67, 782–91. [DOI] [PubMed] [Google Scholar]
- CHILDRESS ES, KHAREL Y, BROWN AM, BEVAN DR, LYNCH KR & SANTOS WL 2017. Transforming Sphingosine Kinase 1 Inhibitors into Dual and Sphingosine Kinase 2 Selective Inhibitors: Design, Synthesis, and in Vivo Activity. J Med Chem, 60, 3933–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICK TE, HENGST JA, FOX TE, COLLEDGE AL, KALE VP, SUNG SS, SHARMA A, AMIN S, LOUGHRAN TP JR., KESTER M, WANG HG & YUN JK 2015. The apoptotic mechanism of action of the sphingosine kinase 1 selective inhibitor SKI-178 in human acute myeloid leukemia cell lines. J Pharmacol Exp Ther, 352, 494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOI K, GOWDA K, LIU Q, LIN JM, SUNG SS, DOWER C, CLAXTON D, LOUGHRAN TP JR., AMIN S & WANG HG 2014. Pyoluteorin derivatives induce Mcl-1 degradation and apoptosis in hematological cancer cells. Cancer Biol Ther, 15, 1688–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPLING-BURNETTE PK, LIU JH, CATLETT-FALCONE R, TURKSON J, OSHIRO M, KOTHAPALLI R, LI Y, WANG JM, YANG-YEN HF, KARRAS J, JOVE R & LOUGHRAN TP JR. 2001. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest, 107, 351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERADAT H 2019. Venetoclax for the Treatment of Chronic Lymphocytic Leukemia. Curr Hematol Malig Rep. [DOI] [PubMed] [Google Scholar]
- FRENCH KJ, ZHUANG Y, MAINES LW, GAO P, WANG W, BELJANSKI V, UPSON JJ, GREEN CL, KELLER SN & SMITH CD 2010. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther, 333, 129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOUAZE-ANDERSSON V & CABOT MC 2011. Sphingolipid metabolism and drug resistance in hematological malignancies. Anticancer Agents Med Chem, 11, 891–903. [DOI] [PubMed] [Google Scholar]
- HAIT NC, ALLEGOOD J, MACEYKA M, STRUB GM, HARIKUMAR KB, SINGH SK, LUO C, MARMORSTEIN R, KORDULA T, MILSTIEN S & SPIEGEL S 2009. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science, 325, 1254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANNUN YA & OBEID LM 2018. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol, 19, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENGST JA, DICK TE, SHARMA A, DOI K, HEGDE S, TAN SF, GEFFERT LM, FOX TE, SHARMA AK, DESAI D, AMIN S, KESTER M, LOUGHRAN TP, PAULSON RF, CLAXTON DF, WANG HG & YUN JK 2017. SKI-178: A Multitargeted Inhibitor of Sphingosine Kinase and Microtubule Dynamics Demonstrating Therapeutic Efficacy in Acute Myeloid Leukemia Models. Cancer Transl Med, 3, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENGST JA, WANG X, SK UH, SHARMA AK, AMIN S & YUN JK 2010. Development of a sphingosine kinase 1 specific small-molecule inhibitor. Bioorg Med Chem Lett, 20, 7498–502. [DOI] [PubMed] [Google Scholar]
- JEREZ A, CLEMENTE MJ, MAKISHIMA H, KOSKELA H, LEBLANC F, PENG NG K, OLSON T, PRZYCHODZEN B, AFABLE M, GOMEZ-SEGUI I, GUINTA K, DURKIN L, HSI ED, MCGRAW K, ZHANG D, WLODARSKI MW, PORKKA K, SEKERES MA, LIST A, MUSTJOKI S, LOUGHRAN TP & MACIEJEWSKI JP 2012. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood, 120, 3048–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONOPLEVA M, CONTRACTOR R, TSAO T, SAMUDIO I, RUVOLO PP, KITADA S, DENG X, ZHAI D, SHI YX, SNEED T, VERHAEGEN M, SOENGAS M, RUVOLO VR, MCQUEEN T, SCHOBER WD, WATT JC, JIFFAR T, LING X, MARINI FC, HARRIS D, DIETRICH M, ESTROV Z, MCCUBREY J, MAY WS, REED JC & ANDREEFF M 2006. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell, 10, 375–88. [DOI] [PubMed] [Google Scholar]
- KOSKELA HL, ELDFORS S, ELLONEN P, VAN ADRICHEM AJ, KUUSANMAKI H, ANDERSSON EI, LAGSTROM S, CLEMENTE MJ, OLSON T, JALKANEN SE, MAJUMDER MM, ALMUSA H, EDGREN H, LEPISTO M, MATTILA P, GUINTA K, KOISTINEN P, KUITTINEN T, PENTTINEN K, PARSONS A, KNOWLES J, SAARELA J, WENNERBERG K, KALLIONIEMI O, PORKKA K, LOUGHRAN TP JR., HECKMAN CA, MACIEJEWSKI JP & MUSTJOKI S 2012. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med, 366, 1905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAHM D, LEE LK & BETTELHEIM FA 1985. Age dependence of freezable and nonfreezable water content of normal human lenses. Invest Ophthalmol Vis Sci, 26, 1162–5. [PubMed] [Google Scholar]
- LAMY T & LOUGHRAN TP JR. 2011. How I treat LGL leukemia. Blood, 117, 2764–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEBLANC F, ZHANG D, LIU X & LOUGHRAN TP 2012. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol, 8, 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEBLANC FR, LIU X, HENGST J, FOX T, CALVERT V, PETRICOIN EF 3RD, YUN J, FEITH DJ & LOUGHRAN TP JR. 2015. Sphingosine kinase inhibitors decrease viability and induce cell death in natural killer-large granular lymphocyte leukemia. Cancer Biol Ther, 16, 1830–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI QF, HUANG WR, DUAN HF, WANG H, WU CT & WANG LS 2007. Sphingosine kinase-1 mediates BCR/ABL-induced upregulation of Mcl-1 in chronic myeloid leukemia cells. Oncogene, 26, 7904–8. [DOI] [PubMed] [Google Scholar]
- LI QF, WU CT, GUO Q, WANG H & WANG LS 2008. Sphingosine 1-phosphate induces Mcl-1 upregulation and protects multiple myeloma cells against apoptosis. Biochem Biophys Res Commun, 371, 159–62. [DOI] [PubMed] [Google Scholar]
- LIAO A, BROEG K, FOX T, TAN SF, WATTERS R, SHAH MV, ZHANG LQ, LI Y, RYLAND L, YANG J, ALIAGA C, DEWEY A, ROGERS A, LOUGHRAN K, HIRSCH L, JARBADAN NR, BAAB KT, LIAO J, WANG HG, KESTER M, DESAI D, AMIN S, LOUGHRAN TP JR. & LIU X 2011. Therapeutic efficacy of FTY720 in a rat model of NK-cell leukemia. Blood, 118, 2793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU K, GUO TL, HAIT NC, ALLEGOOD J, PARIKH HI, XU W, KELLOGG GE, GRANT S, SPIEGEL S & ZHANG S 2013. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One, 8, e56471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOUGHRAN TP JR., ZICKL L, OLSON TL, WANG V, ZHANG D, RAJALA HL, HASANALI Z, BENNETT JM, LAZARUS HM, LITZOW MR, EVENS AM, MUSTJOKI S & TALLMAN MS 2014. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the eastern cooperative oncology group (E5998). Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYNCH KR, THORPE SB & SANTOS WL 2016. Sphingosine kinase inhibitors: a review of patent literature (2006–2015). Expert Opin Ther Pat, 26, 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACEYKA M, HARIKUMAR KB, MILSTIEN S & SPIEGEL S 2012. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol, 22, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNAUGHTON M, PITMAN M, PITSON SM, PYNE NJ & PYNE S 2016. Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen-independent LNCaP-AI prostate cancer cells. Oncotarget, 7, 16663–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORAD SA & CABOT MC 2013. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer, 13, 51–65. [DOI] [PubMed] [Google Scholar]
- NEUBAUER HA & PITSON SM 2013. Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J, 280, 5317–36. [DOI] [PubMed] [Google Scholar]
- NEWTON J, LIMA S, MACEYKA M & SPIEGEL S 2015. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp Cell Res, 333, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGRETMEN B 2018. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer, 18, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PITSON SM, MORETTI PA, ZEBOL JR, LYNN HE, XIA P, VADAS MA & WATTENBERG BW 2003. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J, 22, 5491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POULLOT E, ZAMBELLO R, LEBLANC F, BAREAU B, DE MARCH E, ROUSSEL M, BOULLAND ML, HOUOT R, RENAULT A, FEST T, SEMENZATO G, LOUGHRAN T & LAMY T 2014. Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann Oncol, 25, 2030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POWELL JA, LEWIS AC, ZHU W, TOUBIA J, PITMAN MR, WALLINGTON-BEDDOE CT, MORETTI PA, IAROSSI D, SAMARAWEERA SE, CUMMINGS N, RAMSHAW HS, THOMAS D, WEI AH, LOPEZ AF, D’ANDREA RJ, LEWIS ID & PITSON SM 2017. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood, 129, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REN T, YANG J, BROEG K, LIU X, LOUGHRAN TP JR. & CHENG H 2013. Developing an in vitro model of T cell type of large granular lymphocyte leukemia. Leuk Res, 37, 1737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTSON MJ, COCHRAN KJ, CAMERON C, LE JM, TANTRAVAHI R & RITZ J 1996. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol, 24, 406–15. [PubMed] [Google Scholar]
- SANTOS WL & LYNCH KR 2015. Drugging sphingosine kinases. ACS Chem Biol, 10, 225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAH MV, ZHANG R, IRBY R, KOTHAPALLI R, LIU X, ARRINGTON T, FRANK B, LEE NH & LOUGHRAN TP JR. 2008. Molecular profiling of LGL leukemia reveals role of sphingolipid signaling in survival of cytotoxic lymphocytes. Blood, 112, 770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAW J, COSTA-PINHEIRO P, PATTERSON L, DREWS K, SPIEGEL S & KESTER M 2018. Novel Sphingolipid-Based Cancer Therapeutics in the Personalized Medicine Era. Adv Cancer Res, 140, 327–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIDA D, TAKABE K, KAPITONOV D, MILSTIEN S & SPIEGEL S 2008. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets, 9, 662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINWAY SN, LEBLANC F & LOUGHRAN TP JR. 2014. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev, 28, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEVENSON CE, TAKABE K, NAGAHASHI M, MILSTIEN S & SPIEGEL S 2011. Targeting sphingosine-1-phosphate in hematologic malignancies. Anticancer Agents Med Chem, 11, 794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN E, ZHANG W, WANG L, WANG A, MA C, LEI M, ZHOU X, SUN Y, LU B, LIU L & HAN R 2015. Down-regulation of Sphk2 suppresses bladder cancer progression. Tumour Biol. [DOI] [PubMed] [Google Scholar]
- TAKABE K, PAUGH SW, MILSTIEN S & SPIEGEL S 2008. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev, 60, 181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN SF, LIU X, FOX TE, BARTH BM, SHARMA A, TURNER SD, AWWAD A, DEWEY A, DOI K, SPITZER B, SHAH MV, MORAD SA, DESAI D, AMIN S, ZHU J, LIAO J, YUN J, KESTER M, CLAXTON DF, WANG HG, CABOT MC, SCHUCHMAN EH, LEVINE RL, FEITH DJ & LOUGHRAN TP JR. 2016. Acid ceramidase is upregulated in AML and represents a novel therapeutic target. Oncotarget, 7, 83208–83222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DELFT MF, WEI AH, MASON KD, VANDENBERG CJ, CHEN L, CZABOTAR PE, WILLIS SN, SCOTT CL, DAY CL, CORY S, ADAMS JM, ROBERTS AW & HUANG DC 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell, 10, 389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKATA JK, AN N, STUART R, COSTA LJ, CAI H, COKER W, SONG JH, GIBBS K, MATSON T, GARRETT-MAYER E, WAN Z, OGRETMEN B, SMITH C & KANG Y 2014. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood, 124, 1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLINGTON-BEDDOE CT, POWELL JA, TONG D, PITSON SM, BRADSTOCK KF & BENDALL LJ 2014. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res, 74, 2803–15. [DOI] [PubMed] [Google Scholar]
- XIE V, TONG D, WALLINGTON-BEDDOE CT, BRADSTOCK KF & BENDALL LJ 2018. Sphingosine kinase 2 supports the development of BCR/ABL-independent acute lymphoblastic leukemia in mice. Biomark Res, 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XUN C, CHEN MB, QI L, TIE-NING Z, PENG X, NING L, ZHI-XIAO C & LI-WEI W 2015. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res, 34, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG C, ZHANG W, LU Y, YAN X, YAN X, ZHU X, LIU W, YANG Y & ZHOU T 2015. NudC regulates actin dynamics and ciliogenesis by stabilizing cofilin 1. Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Platelet count and LGL number are correlated with SPHK2 mRNA expression levels. SPHK2 microarray mRNA expression levels versus A) Hemoglobin (Hgb) B) Platelet count C) Absolute neutrophil count (ANC) and D) LGL Number.
Figure S2: SPHK2 is overexpressed in LGL leukemia but is not correlated with treatment outcomes or STAT3 mutational status. A) SPHK2 microarray mRNA expression levels from CD8+ cells from normal subjects (circles; n = 5), Temra cells from normal subjects (squares, n = 3) and T-LGL patients (triangles; n = 37) were compared. Mean + SD; **, p < 0.01 Welch’s t-test B) SPHK2 microarray mRNA expression levels comparison between Responder (n = 19) and Non-Responder (n = 18) to treatment C) SPHK2 microarray mRNA expression levels comparison between STAT3 WT (n = 21) versus STAT3 mutant (n = 16) patients.
Figure S3: Knockdown of SPHK1 decreases viability of leukemic LGLs. NKL or TL-1 cells were transfected with siRNA targeting SPHK1 or control (scramble) siRNA. A and B) Quantitative real-time PCR of SPHK1 knockdown in NKL (A) or TL-1 (B) cells. mRNA levels were normalized to β2M. Scramble siRNA used as control. C and D) Immunoblot analysis of SPHK1 levels in siRNA-transfected NKL (C) or TL-1 (D) cells. Loading of protein was confirmed by probing for β-actin. Normalization was performed by taking the ratio of SPHK1 to β-actin and normalizing to scramble control (Set to 1). E and F) Cell viability was assessed at 72 hours post-transfection using an MTS assay. *, p < 0.05 indicate significant difference between scramble and SPHK1 siRNA transfected cells (Student’s t-test).
Figure S4: K145 inhibited cell proliferation in LGL leukemia cells. NKL and TL-1 cells were treated with increasing doses of K145 and assessed for cell viability using an MTS assay. A and B) MTS Assay in NKL (A) and TL-1 (B) cells.
Figure S5: SPHK2 inhibition with K145 downregulates Mcl-1, Bcl-2 and Bcl-XL expression and leads to cleavage of caspase-3 in LGL leukemia cells. A) Immunoblot analysis of total and cleaved caspase-3 in NKL cells after treatment with K145. β-actin was used as a loading control. B) Immunoblot analysis of total and cleaved caspase-3 in TL-1 cells. C) Immunoblot analysis of pro-survival Bcl-2 family in NKL cells after treatment with K145. D) Immunoblot analysis of pro-survival Bcl-2 family in TL-1 cells after treatment with K145. β-actin was used as a loading control. Normalization was performed by taking the ratio of Mcl-1, Bcl-2 or Bcl-XL to β-actin (0 μM, Set to 1).