

Abstract
Introduction:
In recent years, computational polypharmacology has gained significant attention to study the promiscuous nature of drugs. Despite tremendous challenges, community wide efforts have led to a variety of novel approaches for predicting drug polypharmacology. In particular, some rapid advances using machine learning and artificial intelligence have been reported with great successes.
Areas Covered:
In this article, the authors provide a comprehensive update on the current state-of-the-art polypharmacology approaches and their applications, focusing on those reports published after our 2017 review article. The authors particularly discuss some novel, groundbreaking concepts and methods that have been developed recently and applied to drug polypharmacology studies.
Expert opinion:
Polypharmacology is evolving and novel concepts are being introduced to counter the current challenges in the field. However, major hurdles remain including incompleteness of high-quality experimental data, lack of in vitro and in vivo assays to characterize multi-targeting agents, shortage of robust computational methods, and challenges to identify the best target combinations and design effective multi-targeting agents. Fortunately, numerous national/international efforts including multi-omics and artificial intelligence initiatives as well as most recent collaborations on addressing the COVID-19 pandemic have shown significant promise to propelling the field of polypharmacology forward.
Keywords: Drug Polypharmacology, Multi-targeting Design, Drug Repurposing, Artificial Intelligence, Deep Learning, Multi-omics, Network Pharmacology, Molecular Promiscuity, Off-targets
1. Introduction
Traditionally, drug discovery efforts aim to develop candidates that are highly potent and selective toward a specific biological target. However, it has been realized that most therapeutics exert their desired effects by modulating multiple targets and pathways[1, 2, 3]. In addition, it was reported that the excessive selectivity for a single target sometimes may have fatal safety concerns[4]. The promiscuous nature of ligand binding to multiple targets is termed as polypharmacology. According to the National Library of Medicine, the definition of polypharmacology is “the design or use of pharmaceutical agents that act on multiple targets or disease pathways”. Some broad definitions of polypharmacology may also include different phenotypic or pharmacological effects. But in this review, we focus on multi-target binding, also exchangeable with terms such as drug off-targeting and molecular promiscuity. Unlike the “single drug single target” approach, polypharmacology allows to study broader impact of the interested molecules in the early stages of drug discovery and development.
Complex disorders such as cancer and Alzheimer’s disease are usually caused by complicated mechanisms with various factors including environmental and genetic elements. Polypharmacology has been suggested and emerged as a powerful and promising alternative paradigm for development of versatile therapeutic agents to meet urgent medical needs. Such agents capable of modulating multiple biological targets simultaneously display great advantages of higher efficacy, less resistance, and improved safety profile, achievable through polypharmacological monotherapies or combination therapies[5]. It is worth noting that these two approaches should not be considered as competitive or exclusive to each other. As a matter of fact, combination therapies have been playing a critical role in the current treatment regimens, in particular for complex diseases such as cancer[6]. In this review as an update of our previous work, we focus more on single polypharmacological agents since they do have their own advantages. Especially, a polypharmacological agent is expected to have a more predictable pharmacokinetic (PK) profile as compared to multiple drug-based combination therapies, and thus more superior PK and safety properties along with avoidance of potential drug-drug interactions or negative synergistic effects, as described in more details previously by Anighoro et al.[5]. In addition, polypharmacology is beneficial particularly for drug repurposing or overcome drug resistance. A well-known example is sildenafil (Viagra), originally developed to treat ischemic heart disease (IHD) and hypertension, but later becoming a blockbuster to treat erectile dysfunctions. Most recently, poziotinib and ceritinib were repurposed to treat a subset of non-small cell lung cancer (NSCLC) patients with EGFR exon-20 insertions[7] and ALK-negative lung cancer[8], respectively. More excitingly, the polypharmacology concept has also been widely employed in a variety of multi-targeting therapeutics development as game changers, including proteolysis targeting chimera (PROTAC)[9, 10, 11, 12], bispecific monoclonal antibody (BsMAb)[13], and Chimeric antigen receptor T cell (CAR-T)[14, 15]. We will have more detailed discussions on these in the present manuscript. On the other side, it is expected that interactions with unintentional targets, previous thought as dirty drugs, would most probably cause side effects and toxicities. For instance, some compounds (e.g., terfenadine and cisapride) containing basic nitrogen centers flanked by aromatic or hydrophobic groups may have promising therapeutic effects, but at the same time, they are also known as human ether-à-go-go related gene (hERG) blockers[16, 17] This would cause life-threatening ventricular tachyarrhythmia, a common reason of failure in drug safety assessment. Therefore, by considering the multifactorial nature of drugs, polypharmacology studies certainly open novel avenues to rationally design the next generation of therapeutic agents, with higher potency but less resistance and toxicity[3, 18, 19, 20, 21, 22].
While the identification of multi-targeting agents has largely been serendipitous and fortuitous in the past, recent technological advances in chemical biology and computational sciences enable rational design of drug polypharmacology. In particular, modern in vitro high-throughput/high-content screening and in vivo animal modeling techniques accelerate systematic identification of combinations of drug targets, while in silico methods, structural crystallography and medicinal chemistry allow for efficient design of multi-targeting agents. In the last decade, numerous computational methods have been developed to study molecular promiscuity[2, 3, 18, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32]. In several of our previous articles, we comprehensively reviewed such methodological development and their applications[33, 34]. Since our publications, many others also reviewed various aspects of polypharmacology[25, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44]. For instance, Amelio et al. discussed polypharmacology with focus on anticancer drugs and their targets[35]. In particularly the authors systematically presented data on approved drugs targeting kinases, histone deacetylase and DNA topoisomerases. Another article reported antitumor agents with controlled polypharmacology. Therein the authors used data-driven Fragments in Structure Environments (FRASE) approach to design multi-targeting ligands in protein pockets based on information from structural and chemogenomic databases[25]. Not only did the authors show that the designed multi-targeting ligands demonstrated activities against the targets, but their structural rationale was also confirmed using x-ray crystallography. Karuppasamy et al. put their focus on polypharmacology studies associated with NSCLC[39]. The authors provided in-depth analyses of various drug repurposing and polypharmacological approaches for developing new NSCLC treatments. Recently, Proschak et. al. published a comprehensive review on polypharmacology for rational design of multi-targeting compounds from a medicinal chemist’s perspective[43]. In this article, the authors described methods to identify suitable target combinations, optimize multi-targeting ligands, and develop different assay systems to test polypharmacological compounds. In another article, Ravikumar et al. discussed efficacy-safety balance of polypharmacology in multi-targeting drug discovery[44], with specific focus on multi-targeting monotherapies for cancer treatment.
Herein, we will focus on some novel concepts used to polypharmacology modeling, especially those reports published or updated after our last review[30], and provide some insights on their advantages and limitations. We apologize that, although we attempt to cover as many publications as possible in the present manuscript, by no means will it be all-inclusive. Table 1 provides an updated list of methods that have been used to conduct polypharmacology modeling and predictions, and many of them were either previously discussed or will be described in detail later in this review.
Table 1.
Methods and algorithms used for polypharmacology studies.
| Methods | Algorithms used | Web links |
|---|---|---|
| Docking-based methods | ||
| DOCK[45] | Geometric shape matching algorithm anchor and grow algorithm | http://dock.compbio.ucsf.edu/ |
| INVDOCK[46]* | Geometric algorithm | http://bidd.nus.edu.sg/group/softwares/invdock.htm |
| idTarget[47]* | Modified DOCK algorithm | http://idtarget.rcas.sinica.edu.tw |
| DRAR-CPI[48] | Connectivity maps with DOCK6 algorithm | http://cpi.bio-x.cn/drar/ |
| TarFisDock[49]* | Modified DOCK algorithm | http://www.dddc.ac.cn/tarfisdock/ |
| GLIDE[50]π | Stochastic search algorithm | http://www.schrodinger.com/Glide |
| FRED[51] | Stochastic search algorithm | https://docs.eyesopen.com/oedocking/fred.html |
| DeepVS[52] | Use docking experiments to identify the features relevant for the binding of a particular ligand to a given protein | https://github.com/JanainaCruz/DeepVS |
| Pocket-based Methods | ||
| SiteEngines[53] | Geometric hashing method | http://bioinfo3d.cs.tau.ac.il/SiteEngine/ |
| SuMo[54]* | Geometric matching of triplets | http://sumo-pbil.ibcp.fr/cgi-bin/sumo-welcome |
| IsoMIF Finder[55] | FLAP algorithm, Tanimoto coefficient | http://bcb.med.usherbrooke.ca/imfi.php |
| BioGPS[56] | FLAP algorithm | NA |
| PocketMatch[57] | Greedy alignment algorithm, PM scoring method | http://proline.physics.iisc.ernet.in/pocketmatch/ |
| ProBis[58] | Solvent accessible surface algorithm | http://probis.cmm.ki.si |
| APoc[59] | Pocket similarity score | http://cssb.biology.gatech.edu/APoc |
| CavBase[60] | Clique detection algorithm | http://www.ccdc.cam.ac.uk/ |
| PARIS[61] | Convolution kernel-based method | http://cbio.ensmp.fr/paris/paris.html |
| BSAlign[62] | Subgraph algorithm | http://www.aungz.com/BSAlign/index.htm |
| PharmMapper[63] | Kabsch algorithm | http://www.lilab-ecust.cn/pharmmapper/ |
| TRAPP[64] | MD simulation, ARDR, and PCA | http://trapp.h-its.org/trapp/ |
| KinomeX[65] | An online platform for the structure-based prediction of kinome-wide polypharmacology | https://kinome.dddc.ac.cn/en/ |
| Molecular Grow Methods | ||
| GANDI[66] | Genetic algorithm with tabu search | http://www.biochem-caflisch.uzh.ch/download/ |
| AutoT&T[67] | Automatic tailoring and transplanting algorithm | http://www.sioc-ccbg.ac.cn/software/att2/ |
| ReCore[68] | Geometric rank searching algorithm | https://www.biosolveit.de/ReCore/ |
| AutoGrow[69] | Click chemistry assisted evolutionary algorithm | http://autogrow.ucsd.edu |
| Chemical Similarity-based Methods | ||
| SEA[70]* | Chemical similarity, Kruskal’s algorithm | http://sea.bkslab.org |
| TarPred[71]* | Extended-connectivity fingerprint 4 (ECFP4), Tanimoto Coefficient | http://www.dddc.ac.cn/tarpred |
| SuperPred[72]* | Extended-connectivity fingerprint 4 (ECFP4), Tanimoto Coefficient | http://prediction.charite.de |
| SwissTarget[73]* | Chemical and structure similarity | http://www.swisstargetprediction.ch |
| TargetHunter[74]* | TAMOSIC algorithm | http://cbligand.org/TargetHunter |
| Omics-based Methods | ||
| CMAP[75] | Pattern matching | https://www.broadinstitute.org/cmap/ |
| STITCH[76] | Text mining | http://stitch.embl.de/ |
| LINCS Canvas Browser[77] | LINCS database browser | http://www.maayanlab.net/LINCS/LCB/ |
| Ingenuity Pathway Analysis® π | Pathway analysis | http://www.ingenuity.com/ |
| cBIOPortal[78] | A web portal to visualize genomics and proteomic data | http://www.cbioportal.org/ |
| Machine Learning-based Methods | ||
| Hit Dexter 2.0[26] | Provides machine learning models and similarity-based methods for prediction of aggregators, dark chemical matter and frequent hitters. | http://hitdexter2.zbh.uni-hamburg.de/ |
| RF-QSAR[79]* | Random Forest Algorithm | http://rfqsar.kaist.ac.kr/ |
Methods specifically designed for polypharmacological studies.
Commercial Methods
2. Update on Databases for Polypharmacology
As computational polypharmacology is an emerging field, one of the problems is data curation and lack of experimental data for model development. Recently, Leon et al. assembled a multi-targeting dataset to perform regression and classification to evaluate the effect of missing data on compound bioactivity prediction[80]. They also made some datasets progressively sparser by removing activity records. The predictive performance of their models derived from the sparse data sets were compared with models learnt from the initial dataset. It was found that the performance was decreased slowly in the beginning but decremented very fast when 80% of the data was removed.
Allaway et al. developed Drug Target Explorer, a database that allows users to query for compounds associated with a given target, targets associated with a given compound, and targets associated with molecules that are structurally similar to a given compound[81]. Similarly, Awale et al. implemented an online target prediction server, termed Polypharmacology Browser (PPB), to identify possible targets of a given compound based on the compound’s similarity to other compounds with known targets in the ChEMBL database. This makes the database useful to identify possible off-targets of drug-like compounds[82].
Also, Chen and colleagues implemented a free web-accessible database called Multiple Target Ligand Database (MTLD)[83]. It contains 2,444 Multiple Target Ligands (MTLs) that bind to 21,424 binding sites from 18,231 crystal structures. In MTLD 304 entries are approved drugs and 1,911 entries are drug-like compounds. Also, they have added new functions such as multiple conditional search and linkage visualization. Sun and co-workers recently developed ExCAPE-DB by curating and standardizing large scale chemogenomic data from PubChem and ChEMBL databases[84]. A large-scale chemical structure and bioactivity data standardization was carried out using open source cheminformatics tools. The resulting data is labeled with target activities, Entrez IDs, gene symbols, chemical descriptors and InChIKey. It is accessible for multi-target QSAR model generation and can be easily used for polypharmacology method development.
Recently, MolTarPred was developed by Peon and colleagues as a web tool for molecular target prediction[71]. It takes as input the chemical structure of the query molecule, identifies structurally similar molecules, and returns a list of predicted targets for the query molecule based on the known targets of the structurally similar molecules. Features of MolTarPred include the use of precalculated Morgan fingerprints of compounds to identify similar structures, a reliability score for identifying predictions, and the ability to compare the query molecule with the target-annotated molecules supporting a given prediction. The authors demonstrated the accuracy of MolTarPred’s prediction with numerous examples. They also analyzed four recently FDA-approved drugs, lofexidine, avatrombopag, cannabidiol and tafenoquine, and MolTarPred correctly identified known primary targets of the drugs and predicted novel targets with high reliability scores.
In our previous review, we combined the methods and databases in one table[33]. Herein we present the databases separately to emphasize the importance and challenges of data curation for polypharmacology studies. We updated the list of published databases that can be employed to develop polypharmacology prediction methods in Table 2.
Table 2.
Databases for developing polypharmacology methods.
| Database | Description | URL |
|---|---|---|
| PubChem[85] | Small molecules and their biological targets | http://pubchem.ncbi.nlm.nih.gov/ |
| ChEMBL[86] | Open large-scale bioactivity database containing information manually extracted from medicinal chemistry literature | http://www.ebi.ac.uk/chembl |
| BindingDB[87] | Database of measured binding affinities of proteins-ligand. | http://www.bindingdb.org/bind/ |
| DrugBank[88] | Repository of drug, drug–target and drug action information | http://www.drugbank.ca/ |
| Therapeutic Target Database[89] | Database of therapeutic protein and nucleic acid targets, pathway information and corresponding drugs and ligands directed at each of these targets | http://bidd.nus.edu.sg/group/ttd/ttd.asp |
| SuperTarget and Matador[90] | Database collecting information about drug–target relations, pathways and ontologies. A subset of these drugs has been annotated with additional binding information and indirect interactions as a separate resource (Matador) |
http://bioinf-apache.charite.de/supertarget_v2/ http://matador.embl.de/ |
| STITCH[76] | Resource to explore known and predicted interactions of chemicals and proteins by evidence derived from experiments, databases and literature | http://stitch.embl.de |
| Polypharma[91]* | Curated drug-target interaction and structural information from PDB database | https://imdlab.mdanderson.org/polyp/ |
| visANT[92] | Integrative analysis of biological networks | http://visant.bu.edu |
| hemProt[93] | Compilation of multiple chemical-protein annotation resources integrated with diseases and clinical outcomes | http://www.cbs.dtu.dk/services/ChemProt-2.0 |
| VNP[94] | Disease–target–drug network pharmacology relationships | http://hulab.lifemodules.org/ditad/vnpsearch/ |
| PhIN[95] | Pharmacology interaction network database | http://cadd.pharmacy.nankai.edu.cn/phin/ |
| PROMISCUOUS[96]* | Resource of drug–protein, protein–protein and drug–side effect relations | http://bioinformatics.charite.de/promiscuous |
| ToxCast and Tox21[97] | EPA’s most updated, publicly available high throughput toxicity data on thousands of chemicals. ToxCast is part of the Toxicology in the 21st Century (Tox21) federal collaboration | http://www2.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data |
| Gene Expression Atlas[98] | Gene expression patterns under different conditions | https://www.ebi.ac.uk/gxa/home |
| Genecards[99] | Gene-centric databases integrated data from more than 100 web sources | http://www.genecards.org/ |
| Gene Expression Omnibus[100] | Public database include microarray, NGS, and other high throughput functional genomics data | http://www.ncbi.nlm.nih.gov/geo/ |
| KEGG[101] | Integrated database containing both genomic and molecular levels | http://www.kegg.jp/kegg/ |
| Cancer Genome Anatomy Project (CGAP)[102] | Determination of gene expression levels of normal and cancer cells | http://cgap.nci.nih.gov/ |
| The Cancer Genome Atlas (TCGA) Data Portal[103] | Large genome sequencing data of multiple cancer types | http://cancergenome.nih.gov/ |
| 1000 Human Genome[104] | Sequencing the genomes of a large number of people | http://www.1000genomes.org/ |
| DrugCombDB[105] | Curation of 448 555 drug combinations derived from HTS assays, covering 2887 unique drugs and 124 human cancer cell lines | http://drugcombdb.denglab.org/ |
| canSAR[106] | It integrates genomic, protein, pharmacological, drug and chemical data with structural biology, protein networks and unique, comprehensive and orthogonal ‘druggability’ assessments. | http://cansar.icr.ac.uk |
| Drug Target Explorer[81]* | Allows to query for compounds associated with targets, or targets associated with compounds. | https://shiny.synapse.org/users/rallaway/polypharmacology-db/ |
| Polypharmacology Browser (PPB)[82] | Identify possible targets of compounds based on their similarity to other compounds with known targets in the ChEMBL database | http://gdbtools.unibe.ch:8080/PPB/ |
| Multiple Target Ligand Database (MTLD)[83]* | Contains 2,444 Multiple Target Ligands (MTLs) that bind to 21,424 binding sites from 18,231 crystal structures. | http://www.mtdcadd.com |
| MolTarPred[71]* | Use precalculated Morgan fingerprints to identify similar compounds and a reliability score with ability to compare the query molecule with the target-annotated molecules | http://moltarpred.marseille.inserm.fr/ |
| ExCAPE-DB[84] | Large curated drug target interaction databased generated from PubChem and ChEMBL with labels for targets and compounds. | https://solr.ideaconsult.net/search/excape/ |
Database specifically developed for polypharmacology studies.
3. Ligand-based Polypharmacology
Prediction of drug polypharmacology using ligand-based methods rely primarily on chemical structures and their bioactivities. Small molecules can be quantitatively represented with molecular descriptors or fingerprints encoding molecular properties, bond connectivity and atomic properties/types, which are employed to compare molecular similarity or build quantitative structure-activity regression/classification models.
Traditionally QSAR models are used to predict activity of small molecules against individual targets. If the QSAR models are available for all targets, then polypharmacology profiles can be predicted for any given molecule. Recently, Lee et al. used 1,121 targets to develop ligand-based target prediction models with the Random Forest method. They showed that 67.6% and 73.9% recall rates were achieved for their top 1% and top 3% targets, respectively[79]. Accuracy of such models could improve as more data becomes available.
In 2017, Schneider et al. performed a comprehensive analysis of 1.4 million bioactive compounds to identify specific molecular frameworks overrepresented in this chemical space and quantify the promiscuity of the identified frameworks[107]. They revealed the hydrogen-bond-acceptor count of a scaffold as a possible indicator of a scaffold’s potential promiscuity and suggested that easily accessible chemical scaffolds serve as templates for generation of multi-targeting compound libraries, with heterocyclic, sp3-rich frameworks particularly suited for this purpose. In 2018, Meyers et al. used a privileged structure library to show that understanding of inter-family polypharmacology is important to reduce the toxicity risks and design screening libraries[40]. Their results were based on two compounds: one was the CDK9 inhibitor CCT250006 and the other was the pirin ligand CCT245232. The findings suggest that relation between ligand similarity or dissimilarity and off-target profiles is not straightforward and privileged structures can be used to better understand these relations.
A study by Monteleone and colleagues demonstrated the application of 3D chemical descriptors to polypharmacology modeling[108]. In this study, they investigated the relationship between chemical properties relating to 3D connectivity (e.g. number of carbon atoms with a given hybridization or number of chiral centers) and the number of targets of a given molecule. Based on the number of known targets for each molecule in question, they grouped molecules into four classes: black (active on one target), gray (active on two to four targets), white (active on more than four targets), and inactive (not having any target measured by the considered assays). Additionally, they found that black and inactive compounds have higher solubility and more chiral centers, sp3 carbon atoms, and aliphatic rings, while white compounds have more double bonds and fused aromatic rings. Interestingly, some common descriptors such as MW, HBD and HBA did not perform well in distinguishing the four classes of compounds (grouped based on the number of targets). These results suggest that consideration of the target class and molecular features describing the 3D connectivity could be useful in development of drugs with polypharmacology.
4. Target-based Polypharmacology
Most of the previous literature was focused on drug promiscuity, which is a drug’s ability to bind multiple targets. However, recent work has been carried out regarding target binding-site promiscuity, as their properties are now recognized among the key factors that impact drug polypharmacology. In a recent article, Cerisier et al. characterized the promiscuity of druggable binding sites from protein-ligand complexes in the Mother of All Databases (MOAD) [37]. As expected, the sites that interact with only one ligand have pockets with features less conducive to forming protein-ligand contacts, and their ligands are highly adaptable, small, and hydrophilic. Interestingly, while the sites analyzed in this dataset were often promiscuous (80% of the sites), their ligands were not, being dedicated to a single site. In a pairwise comparison of 90,000 putative binding pockets detected in 3,700 proteins, Duran-Frigola found that 23,000 pairs of proteins have at least one similar pocket that might accommodate similar ligands, raising the possibility of using the detection of similar binding sites to identify opportunities for designing polypharmacology [109]. They further showed how to leverage these opportunities in protein-protein interaction networks related to several therapeutic classes and tumor types.
Similarly, Ehrt et al. employed automated analysis of protein binding sites to study promiscuous binding on a novel dataset of sites in complex with ligands sharing common shape and physicochemical properties [110]. They presented suitability of this dataset to benchmark novel binding site comparison methods. Moreover, they revealed promising directions for further in-depth analyses of promiscuity and druggability in a pocket-centered manner. Naderi with co-workers recently surveyed 12 tools used to quantify similarities among pockets (pocket matching) in terms of shape and chemical properties[111]. They divided the methods into five categories and discussed applications of computational pocket matching in drug repurposing and polypharmacology studies.
It is known that fragments are relatively smaller chemical entities compared to drug-like molecules, and thus they tend to be more promiscuous. Da et al. developed a computational strategy to achieve restricted polypharmacology in the design of agents selectively targeting tyrosine kinases including TYRO3, AXL, and MERTK (TAM)[25]. The approach, based on the concept of fragments in structural environments, assembles a 3D structure of the inhibitor directly in the protein pocket using relevant structural and chemogenomic information from chemical databases. As a proof of concept, the authors corroborated the structural basis for their methodology using X-ray crystallography, and they also showed that inhibitors designed using their method displayed promising antitumor activity in biochemical, cellular, and animal studies.
When the 3D structures are concerned, selection of protein conformations is critical in the design of multi-targeting drugs. In a recent study, Pinzi et al. observed that the binding site conformations of different targets might best be selected by keeping two considerations: 1) unusual protein conformations should be avoided, as they may represent low populated protein states and therefore bias compound selection; and 2) the ideal protein conformations would accommodate compounds with similar scaffolds[42]. Selecting appropriate protein conformations for rational design of polypharmacology is therefore challenging. There are even more factors that complicate the process, including the dissimilarities between two targets due to water molecules in the binding sites. For this issue, the authors recommended a priori assessment to avoid inclusion of thermodynamically unfavorable waters in the binding site while retaining waters involved in highly conserved H-bond networks. To better understand the binding of promiscuous small molecules to their targets, Gilberg et al. analyzed 192 ligands (including endogenous ligands) that formed crystallographic complexes with multiple unrelated or distantly related proteins[112]. These compounds were often more polar than other bound, non-promiscuous compounds. Interestingly, while some of these “multifamily” ligands conformationally adjusted to the binding sites of their different targets, many displayed conserved or similar binding conformations across the active sites of their targets; these ligands, however, formed different interaction hotspots in different binding sites.
5. Network Polypharmacology
Network pharmacology integrates multi-omics technologies and systems biology for drug discovery and development[113]. With a network-based insight, this field intends to systematically understand the biological foundation of complex diseases and drug effects, thus it is a promising strategy for the next generation of pharmaceutical research. A recent study by Moya-Garcia et al. integrated structural and functional information in a polypharmacological analysis associating multi-targeting drugs with CATH functional families of proteins[114]. The authors identified 81 CATH functional families that are enriched in druggable targets. Their results showed that proteins within druggable CATH functional families exhibited structural similarities including: highly conserved drug-binding sites; tending to be the central nodes in human protein functional networks; clustered together to form network neighborhoods; and less likely to be associated with drug side effects.
Recently, Wu et al. predicted drug-target interactions using network-based inference methods based on the recommendation algorithms[115]. Additionally, the authors showed that their network-based method can be utilized for elucidations of molecular and safety mechanisms. In another study, Cheng et al. employed human protein-protein interactome and studied the link between disease related proteins and drug targets to develop rational combinatorial therapy strategies[116]. The authors curated 243,603 protein-protein interactions covering 16,677 proteins and 1978 FDA approved or investigational drugs and then identified the nearest distance between drug and diseases with drug targets and disease protein information. In addition, the authors quantified the relationship between drug-target networks and drug-drug-disease networks to derive combination feasibility. Based on the analysis of complementary exposure patterns of the networks, the authors deduced that the effectiveness of drug combination is dependent on specific network topological relationship to the localized disease protein neighborhood.
Frequently the target profiles of many drugs are established early in their development and are not systematically revisited at the time of FDA approval. Thus, it is often unclear whether therapeutics with the same nominal targets, but different chemical structures are functionally equivalent. Hafner et al. [117] reported an analysis of several clinically approved CDK4/6 inhibitors using multi-omics approaches including mRNA sequencing (mRNA-seq) of drug-perturbed cell and animal models, (2) mass spectrometry for phosphoproteomics studies, (3) growth rate inhibition (GR)-based dose-response measurement, and (4) kinome-wide profiling using the commercial KINOMEscan platform from DiscoverX[118] along with kinase enrichment proteomics studies. They found that these experiments provided complementary views of target coverage with insights into drug mechanisms of action. It was revealed that palbociclib, ribociclib, and abemaciclib have substantial differences in their secondary targets and biological activities in breast cancer cell lines with varying genotypes. Multiple lines of evidence, including an in vivo xenograft model and preliminary data on patients and patient-derived cell lines treated with abemaciclib, suggest that the unique activities of abemaciclib arise from inhibition of kinases in addition to CDK4/6, notably CDK1 and CDK2, and may be therapeutically advantageous.
6. Novel Concepts for Polypharmacology.
To tackle the challenges in drug polypharmacology and rational multi-targeting molecular design, numerous efforts are being made to develop novel concepts in the field, in addition to those described above. Figure 1 illustrates several of latest transformative concepts in rational polypharmacology design with multi-targeting agents, both small molecules and biologics, some already FDA approved or in clinical trials.
Figure. 1. Current and novel concepts of polypharmacology and multi-targeting agents.
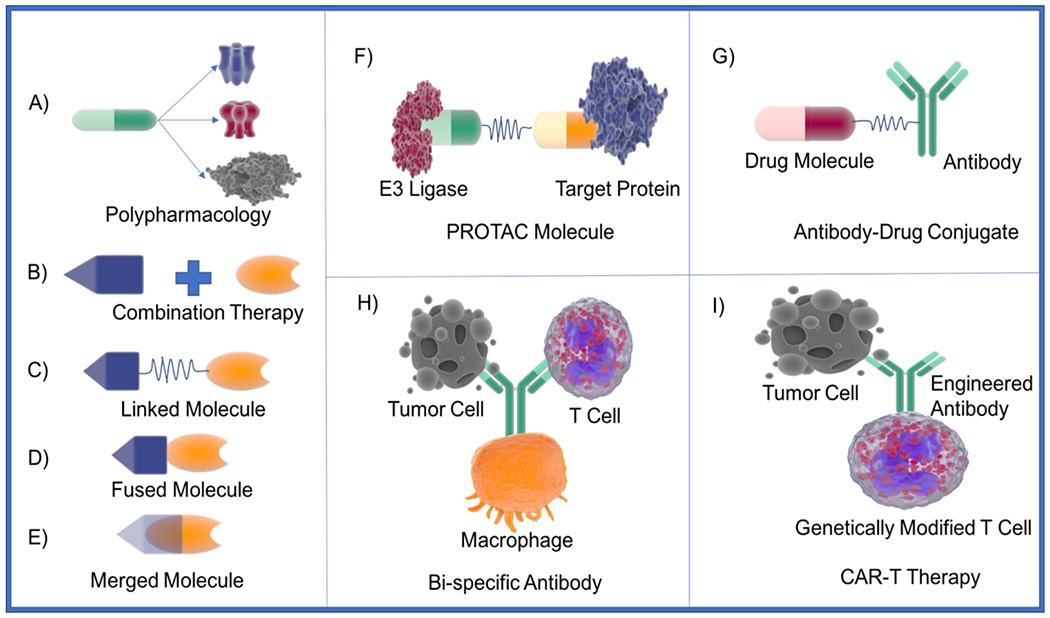
Note: more details for each concept are described in the main text.
6.1. Proteolysis Targeting Chimeras (PROTACs)
Recently the PROTAC technology has gained significant attention. PROTACs are dual targeting small molecules consisting of two covalently linked protein-binding molecules (Fig. 1F): one capable of engaging an E3 ubiquitin ligase and the other binding to the target protein meant for degradation. Recruitment of the E3 ligase to the target protein leads to ubiquitination and ultimately degradation of the target protein by the proteasome. In 2001, PROTAC molecules were developed to hijack cell’s ubiquitin-proteosome system (UPS) to degrade protein of interest. Sakamoto et al. in their study showed that their protein of interest methionine aminopeptidase-2 (MetAP-2) can be artificially recruited to Skp1-Cullin F box complex for ubiquitination and ultimately result in degradation of MetAP-2[119]. They developed the first chimeric molecule by linking with ovalicin, an angiogenesis inhibitor, with IκBα phosphopeptide recognized by F-box protein β-TRCP that was able to ubiquitinate and degrade MetAP-2 in PROTAC manner. Recent review by Konstantinidou et al. focuses on advances in the PROTAC field [120]. Especially they covered the topics based on available experimental data from crystallography, kinetics, and aspects of selectivity. In addition, the authors described various PROTAC examples including homoPROTACs, PROTACS in clinical trials, and PROTACS targeting Tau protein. Recently, the promiscuous nature of the PROTAC molecules were studied[121, 122]. Bondeson et al. studied the effects of VHL recruiting PROTAC containing a promiscuous molecule that targets >50 kinases[121]. They showed that not all kinases resulted in degradation. The authors speculated that the stability of the protein-protein interaction complex might be one of the main factors in selectivity. They also suggested that weak target protein binders could be employed in PROTACs by maximizing target-protac-ligase complex stability. In a similar study, Huang et al. developed CRBN-based multi-kinase degrader and identified 28 kinase targets that were degraded using an unbiased, multiplexed quantitative proteomic approach[122]. Additionally, the authors showed that once the PROTAC kinase profile is identified, they were able to develop specificity for proteins of interest (from PROTAC kinase profile) with ease. These studies clearly indicate the potential of, especially targeting difficult targets, with the PROTAC technologies.
6.2. Antibody-Drug Conjugates (ADCs)
Similarly, ADCs are also chimeric multi-targeting molecules, where antibodies are linked to small molecules with cell killing activities via a spacer for cancer treatments (Figure. 1G). ADCs allow to take advantage of specificity of antibodies for antigens on cancer cells and deliver the cytotoxic molecules to cancer cells. This approach helps overcome the side effects of regular chemotherapy by sparing the healthy cells. Several ADCs have been approved for cancer treatments and many are in clinical trials[123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133]. For instance, inotuzumab ozogamicin is approved for treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL)[134]. In this ADC, the cytotoxic agent ozogamicin is linked thorugh the AcBut linker to a humanized monoclonal antibody that targets CD22 receptors. Upon interaction with CD22, the conjugate gets internalized resulting in cleavage of the linker and the antibody, ultimately killing cancer cells due to the ozogamicin activity. Another approved ADC is trastuzumab emtansine developed by Genentech for treatment of HER2 positive metastatic breast cancer[135]. The humanized trastuzumab targets HER2 receptors and inhibit its homodimerization or heterodimerization, leading to cancer growth inhibition. When this antibody is conjugated with emtansine, the ADC gets internalized and kills the cancer cells as well[136].
6.3. Bispecific Monoclonal Antibody (BsMAb) and Chimeric Antigen Receptor T Cells (CAR-T)
Similar ideas have been utilized in multi-targeting biologics development, including BsMAb and CAR-T technologies. As this is beyond the scope of our review essentially focusing on polypharmacological small molecules, we only give a brief introduction to emphasize the groundbreaking biology and clinical success of the drug polypharmacology concept. Unlike ADC, BsMAb is an antibody artificially designed to simultaneously bind two different types of antigens (Figure 1H)[137]. BsMAbs can be classified into three major types: trifunctional antibody, chemically linked fragment-antigen binders, and bi-specific T cell engagers. Currently, at least two BsMAb are approved and over 85 are in clinical trials[138]. Detailed review on bispecific antibodies can be found in articles published by Konterman et al. and Labrijn et al. [13, 138]. On the other hand, CAR-T cells are genetically engineered T cells that can produce artificially designed T-cell receptors for immunotherapy[139]. The T-cell receptors in this approach are designed to bind to specific antigens on the tumor cells causing immunogenic response against the tumor cells[140]. The T-cell receptor consists of an extracellular antigen binding domain, a spacer domain between transmembrane region and antigen binding domain, a transmembrane domain, and an intracellular T-cell signaling domain (Figure 1I). The extracellular antigen binding domain targets specific antigens present on tumor cells; the spacer domain provides flexibility for antigen domain to interact with tumor cell receptors; the transmembrane domain is important for level of expression and stability; and the intracellular domain activates T-cell in response to antigen binding. The first CAR-T therapy Kymriah (tisagenlecleucel) was approved in 2017 for B-cell precursor acute lymphoblastic leukemia[141]. Currently over 364 clinical trials are ongoing for various CAR-T therapies worldwide[142].
6.4. Enhancing Molecular Promiscuity Evaluation through Assay Profiles (APs)
In an interesting study Avram et al. developed an encoding feature called assay profiles (APs)[143]. They used unique combinations of five binary assay-related features: 1. absence or presence of protein specification; 2. whether the assay is primary or confirmatory; 3. whether the assay is cellular or biochemical; 4. whether the results are derived from the scientific literature or from a high-throughput screen; and 5. whether the assay is detergent-based or not. Bioactivity data from PubChem was fit into 32 theoretical APs, and eight dominant ones were selected for analysis. For each compound, the promiscuity was measured in terms of Frequency of Hits (FoH) or the ratio between the number of times the compound was found active and the number of times it was tested. The authors showed that the source of activity data and the type of activity measurement are the primary factors defining the promiscuity of compounds. In particularly, they observed that the confirmatory assays and scientific literature reported more promiscuity (95% of the drugs with FoH scores >0.93) compared to the primary and HTS assays (95% of the compounds with FoH scores <0.11). Their analysis suggested that drug molecules are more promiscuous than non-drug molecules and the compound APs provide a novel way to study promiscuity but need further exploration.
6.5. Promiscuity Cliff Pathways
Promiscuity cliff (PC), known as compounds with significant diverse promiscuity profiles but similar chemical structures (single substitution), has been utilized in polypharmacology analyses[144, 145]. Minor structural modifications which contribute to selectivity or promiscuity may be revealed using PC analysis. Clusters with different scope and complexity can be formed by PCs using network representations. A new computational study published by Miljkovic et al. used systematic approaches to find PC pathways and extracted structure-promiscuity information from PC clusters[28]. Through identification of 600 PC clusters and related pathways generated from kinase inhibitors, this method was proved to be able to explore novel promiscuity patterns.
6.6. Artificial Intelligence for Polypharmacology Modeling
Deep learning (DL) and artificial intelligence (AI) approaches have been shown to significantly improve polypharmacology predictions. In particular, AI can take advantage of large complex data generated from HTS and multi-omics technologies and learn the patterns that are difficult to find using any other approaches[29, 146, 147, 148, 149, 150]. A general workflow of using AI techniques for polypharmacology predictions is summarized in Figure. 2. For instance, Pereira et al. recently reported an approach based on Convolutional Neural Networks (CNN) called DeepVS[52]. It uses the results from docking experiments to identify the features relevant for the binding of a particular ligand to a given protein. DeepVS generates DL-based scores for ligand-target poses based on structural data describing the complex. To validate DeepVS and compare its performance to that of other algorithms, the authors screened forty different datasets reported in the DUD-E database [151] and found that DeepVS was able to perform better than standard scoring functions. Interestingly, their study was the first to report on the application of deep learning to the rescoring of docking poses without requiring manually set parameters, making it appealing for unsupervised, large-scale virtual screening.
Figure. 2.
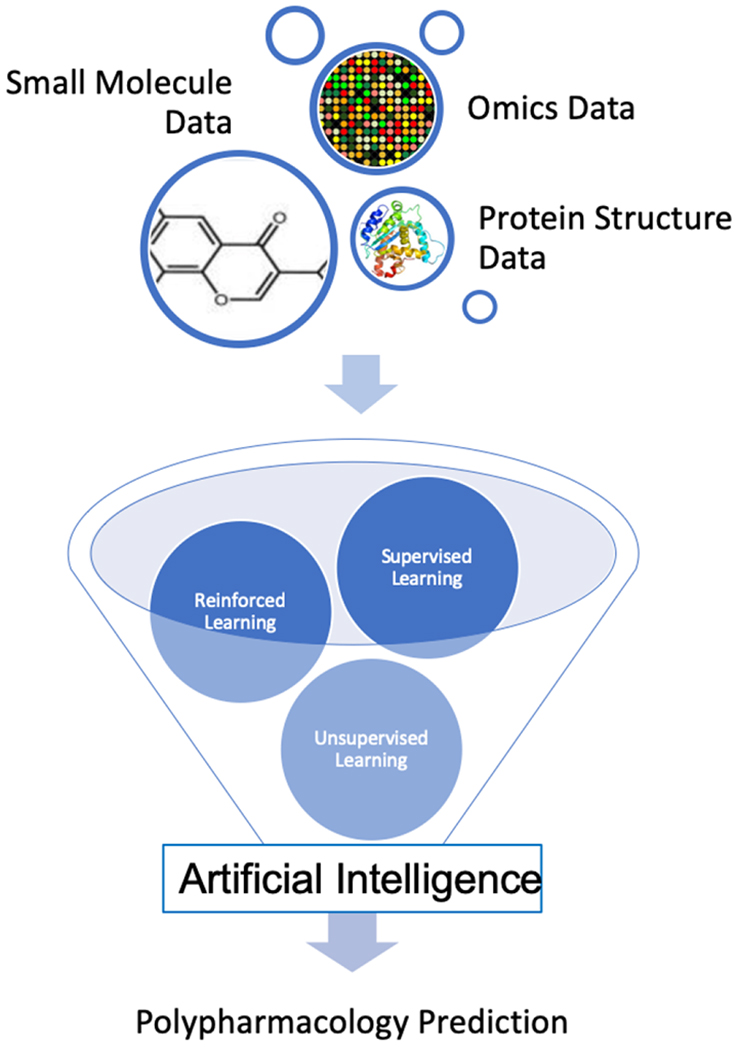
Generalized workflow of AI in polypharmacology predictions.
When considering polypharmacology, kinase inhibitors would be the best example to demonstrate the concept. Kinases are a group of very important proteins in human that play significant role in regulation of signaling pathways and physiological processes. Deregulation of kinases have been reported continuously in different cancers. AI methods have shown great promise in expediting kinome-wide profiling of small molecule inhibitors for polypharmacology studies. For instance, KinomeX, developed by Li et al., is an online platform for the structure-based prediction of kinome-wide polypharmacology [65]. The platform uses a multi-task deep neural network model trained with over 140,000 bioactivity data points for 391 kinases and has been validated both computationally and experimentally. Jones et al. also creates a neural network model which was used to investigate the human kinome[148]. They assessed numerous feature combinations to improve their models in order to outperform molecular docking and other methods, and obtained 60% increase in the performance when compared to general docking scoring functions.
Although DL and AI methods have shown great promises in predicting ligand-receptor binding affinities, they are still far away from maturation given the challenges such as availability and quality of training and validation datasets. Some recent review articles by Li et al. and Shen et al. sheds light on accuracy comparison between classical scoring functions and machine learning methods[152, 153]. Their analyses showed that AI techniques have advanced significantly, they are not always more predictive than those based on more established methods. The major outlook for the future still lies on the direct exploitation of state-of-the-art machine learning strategies. Beyond predictive accuracies, many non‐AI methods possess the advantages of much shorter training times along with easier interpretation of the models. This trend has also been observed in other disciplines. In clinical settings, even today’s best AI struggles to make sense of complex medical information. It is very tricky to encode a human doctor’s expertise in software, as demonstrated by IBM’s Watson healthcare story illustrating a cautionary tale of hubris and hype in AI doctor revolution[154]. Nevertheless, we believe in future tremendous efforts will be put forward in developing big reliable data and interpretable models that facilitate progress of basic scientific research and clinical disease treatment.
7. Application of Polypharmacology Approaches
7.1. Predict Off-targets and Side Effect/Toxicities
As mentioned, polypharmacological therapeutics may increase the risk of side effects or toxicities due to the increased number of drug-protein interactions. Large collaborative efforts such as the Library of Integrated Network-based Cellular Signatures (LINCS) project have shown that drugs affect multiple molecular pathways which may be associated with adverse drug. Several studies have, therefore, analyzed whether relationships between drugs and their effects on biological networks can be used to predict drug adverse reactions[48, 155, 156, 157]. Zitnik et al. employed graph convolutional approach based on drug-drug, protein-protein, and drug-protein interactions to predict polypharmacy side effects[158]. In this study, the authors associated every drug pair with their side effect types and used this data to construct the test and training sets. The authors then used convolutional graph neural network to calculate the probability for association between drug pair and a given side effect. Recently Zhou et al. employed drugs and proteome data to predict drug-target interactions and then using disease data they further extended their prediction of side effects[159]. Another study by the Uner group described prediction of side-effects of drugs using combination of chemical structure data and LINCS 1000 data[160]. In addition, Anda-Jáuregui et al. implemented network-based parameters to investigate the relationship of given adverse reactions with single multi-targeting drugs or drug combinations[157]. Along the same line, Kim et al. exploited off-target tissue effects to predict side effects[161]. The authors used ATC codes and drug target information to identify off-target tissues and developed a tissue protein – symptom matrix, with which the authors derived relation between tissue effects and symptoms.
7.2. Drug Repurposing
Given the extremely high cost and time-consuming nature of new drug development, repurposing of marketed drugs to treat new diseases have attracted more and more attentions. One of most promising application of polypharmacology studies is drug repurposing, and a large number of approved drugs have been studied in clinical trials for repurposed indications with great successes[8, 162]. Some of the examples initiated after 2016 are listed in Table 3.
Table 3:
Recent examples of drug repurposing
| Drug Name | Primary Indication | Repurposed Indication | Study Completion Year | Trial Number/ Article DOI |
|---|---|---|---|---|
| Paracetamol | Analgesic, Anti-pyretic | AKI in Plasmodium knowlesi Malaria | 2018 | NCT03056391 |
| Rosiglitazone | Anti-diabetic | Malaria as an adjunct therapy | 2020 | NCT02694874 |
| Niclosamide | Anti-helminthic drug | Cancer-Melanoma | 2019 | 10.1016/j.bcp.2019.08.012 |
| Niclosamide | Anti-helminthic drug | Pulmonary fibrosis | 2019 | 10.1016/j.lfs.2018 |
| Fenofibrate | Anti-Hyperlipidemia | Huntington’s Disease | 2019 | NCT03515213 |
| Nelfinavir | Anti-viral | Kaposi Sarcoma | 2020 | NCT03077451 |
| Azithromycin | Antibiotic | neonatal neuroprotection | 2019 | 10.1038/s41390-019-0408-6 |
| Tigecycline | Antibiotic | Alcohol dependency | 2018 | 10.1111/acer.13312 |
| Linezolid | Antibiotic for Vancomycin resistant strain | Multi Drug Resistant Tuberculosis | 2018 | NCT02778828 |
| Pranlukast | Asthma | Arboviral disease | 2019 | 10.1016/j.antiviral.2019.104668 |
| Azithromycin | Broad spectrum antibiotic | Pulmonary Tuberculosis | 2020 | NCT03160638 |
| Olaparib | Cancer | Septic shock | 2019 | 10.1016/j.phrs.2019 |
| Salirasib | Cancer | Malaria | 2019 | 10.1039/c9md00298g |
| Nilotinib | Chronic Myeloid Leukemia | Parkinson’s Disease | 2020 | NCT03205488 |
| Propronalol | CVS diseases | Age related osteoporosis | 2018 | NCT02467400 |
| Agomelatine | Depression | Brain injury induced by craniotomy | 2019 | 10.5582/ddt.2019.01056 |
| Metformin | Diabetes | Cancer-Melanoma | 2019 | 10.3390/cancers11020209 |
| Digoxin | Heart failure | medullablastoma | 2018 | 10.1126/scitranslmed.aat0150 |
| Manidipine dihydrochloride | Hypertension | Cytomegalovirus infection | 2018 | 10.1016/j.antiviral.2017.12.014 |
| Isradipine | Hypertension | Parkinson’s Disease | 2018 | NCT02168842 |
| Ibuprofen | Juvenile rheumatoid arthritis, osteoarthritis | Extensive Drug Resistant Tuberculosis | 2019 | NCT02781909 |
| Imatinib | Leukemia | Uncomplicated malaria | 2018 | NCT02614404 |
| Quinacrine Hydrochloride | Malaria | Ebola virus (EBOV) infection | 2019 | 10.1128/AAC.01142-19 |
| Pyronaridine tetraphosphate | Malaria | Ebola virus (EBOV) infection | 2019 | 10.1371/journal.pntd.0007890 |
| Dantrolene | Malignant hyperthermia | Alzheimer’s disease | 2019 | 10.3390/molecules24234298 |
| Disulfiram with copper | Management of alcoholism | Metastatic Breast Cancer | 2020 | NCT03323346 |
| Carvedilol | Mild to moderate heart failure | Alzheimer’s Disease | 2017 | NCT01354444 |
| Dexmedetomidine | Analgesic | Sleep|Insomnia | 2016 | NCT02818569 |
| Prazosin|Placebo | Hypertension | Alcoholism | 2016 | NCT02966340 |
| Intragastric botulinum toxin type A | Cervical Dystonia | Obesity | 2017 | NCT03079557 |
| Metformin | Diabetes | Prostate Cancer | 2017 | NCT03137186 |
| Adalimumab | Rheumatoid arthritis | Dupuytren’s Disease | 2016 | NCT03180957 |
| ATRA|Gemcitabine|Nab-paclitaxel | Non-small cell lung cancer | Pancreatic Adenocarcinoma | 2016 | NCT03307148 |
| Metformin|Acetylsalicylic acid|Olaparib|Letrozole | Diabetes | Ovarian Cancer | 2018 | NCT03378297 |
| Famotidine | Antacid | Pulmonary Arterial Hypertension | 2019 | NCT03554291 |
| Simvastatin | Dislipidemia | Dilated Cardiomyopathy | 2018 | NCT03775070 |
| Alpha-lipoic acid | Dietary imbalance | Schizophrenia|Oxidative Stress | 2019 | NCT03788759 |
| Atomoxetine | ADHD | Orthostatic Intolerance | 2018 | NCT03808740 |
| Metformin Hydrochloride | Diabetes | Chronic Kidney Diseases | 2019 | NCT03831464 |
| Pramipexole | Parkinson’s disease | Chronic Pain | 2019 | NCT03842709 |
| Magnesium Sulfate|Nimodipine | subarachnoid hemorrhage | Anticholinesterase Insecticide Poisoning | 2019 | NCT03925025 |
| Azithromycin | Antibacterial | Sarcoidosis, Pulmonary | 2019 | NCT04020380 |
| Zoledronic Acid | Hypercalcemia | Seropositive Muskuloskeletal Complaints | 2020 | NCT04115397 |
| Nicorandil | Angina Pectoris | Dementia|Mild Cognitive Impairment | 2019 | NCT04120766 |
| CEP-1347 | Parkinson’s disease | Anti-cancer stem cell drug | 2017 | 10.18632/oncotarget.22033 |
| Enzalutamide | Prostate cancer | Advanced Breast Cancer | 2021 | NCT02953860 |
| Omeprazole | Proton pump inhibitor (GERD) | Remyelination in multiple sclerosis | 2019 | 10.1016/j.brainres.2018.12.037 |
| Penfluridol | psychotic condition | Lung cancer | 2020 | 10.1016/j.biopha.2019.109598 |
| Isotretinoin | Recalcitrant Nodular acne | Relapsed/ Refractory neuroblastoma | 2019 | NCT01334515 |
| Moxifloxacin + Bedaquiline and Pyrazinamide | Respiratory infections and conjunctivitis | MDR-TB | 2018 | NCT02193776 |
| Doxycycline | Respiratory tract infection | Pulmonary tuberculosis | 2017 | NCT02774993 |
| Auranofin | Rheumatoid arthritis | Parasites | 2016 | 10.1128/AAC.01947-16 |
| Penfluridol | schizophrenia | Cancer-glioblastoma | 2019 | 10.3390/cancers11091310 |
| Eltrombopag | thrombocytopenia | Crytococcosis | 2019 | 10.1093/mmy/myz077 |
| Masitinib | Mast cell tumor | Alzheimers | 2020 | NCT01872598 |
| Remdesvir | Ebola Virus Disease | COVID19 | 2020 | NCT04257656 |
| Chloroquine | Malaria | COVID19 | 2021 | NCT04349371 |
| Hydroxychloroquine | Malaria | COVID19 | 2021 | NCT04329611 |
| Fluoxamine | Obsessive-compulsive disorder | COVID19 | 2020 | NCT04342663 |
Among others, our laboratory has significant experience of successful drug repurposing for cancer therapy. One of such examples is our recent repositioning of poziotinib[7]. EGFR Exon 20 insertion is a novel mutation observed in 10% EGFR mutant NSCLC patients[7, 163, 164]. Tumors harboring this mutation are resistant to known EGFR tyrosine kinase inhibitors (TKIs). Using computational modeling, we found that the binding pocket of EGFR Exon 20 insertions are more restricted compared to the wild type protein (Figure. 3), and thus smaller and more flexible compounds such as poziotinib, have better binding affinity to the EGFR Exon 20 insertion mutants[7]. Following pre-clinical experiments confirming our hypothesis, poziotinib showed excellent activity and selectivity in patients with EGFR exon 20 insertions in a Phase II clinical trial. Since our discovery was published, at least 4 new clinical trials have opened worldwide (NCT03318939, NCT04044170, NCT02544997, and NCT03744715) to evaluate poziotinib treatment of NSCLC and breast cancer patients with EGFR/HER2 exon 20 insertions.
Figure. 3. Poziotinib repurposing to treat EGFR exon 20 insertion patients.
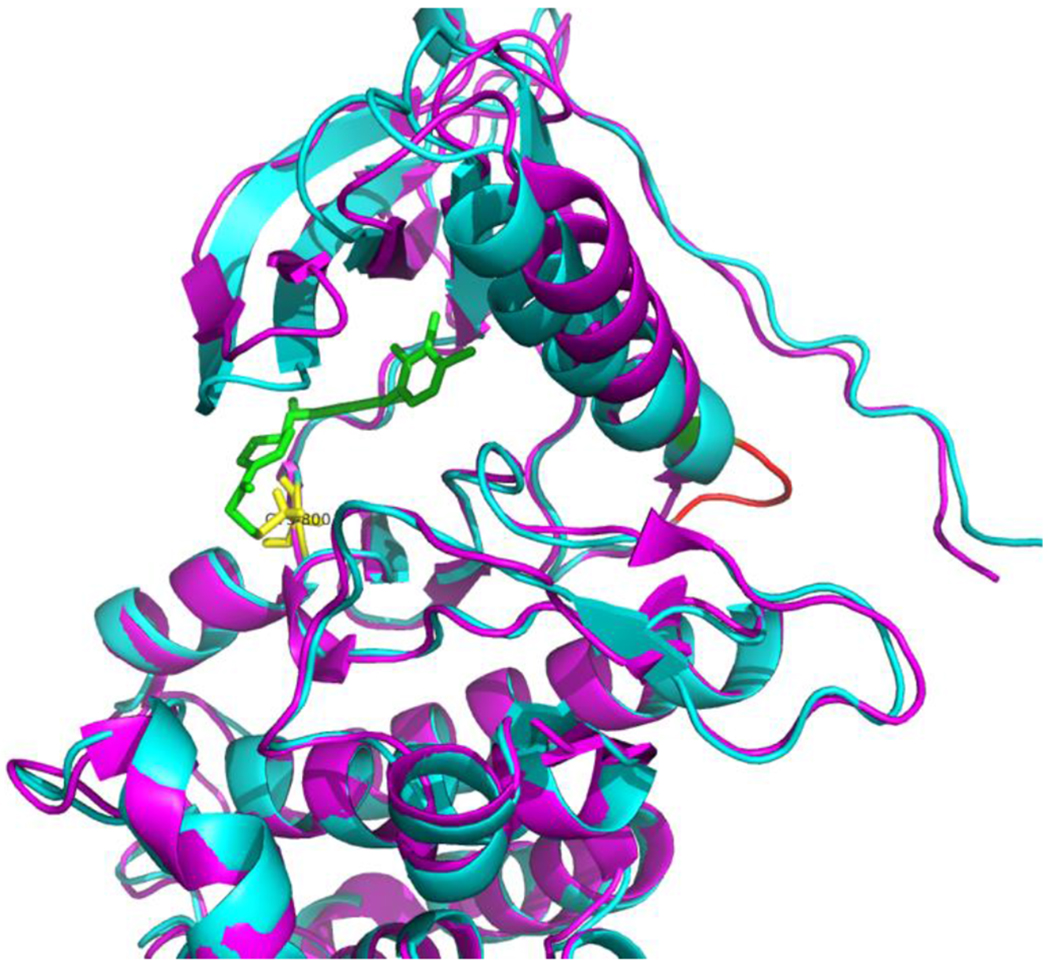
The insertion increases the rigidity of the α-helix of EGFR, leading to a smaller and more rigid binding pocket. Poziotinib could fit into the pocket and forms covalent bond with Cys800. Magenta: EGFR with T790M; Cyan: EGFR with Exon 20; Green: Poziotinib; Yellow: Cys 800; Red: EGFR SVD insertion.
Previously we also successfully repurposed vismodegib, a Hedgehog inhibitor primarily used to treat basal cell carcinoma (BCC), to for NSCLC therapy [165]. In this discovery, a novel SMO P641A mutation was discovered in a NSCLC patient at MD Anderson Cancer Center. This patient did not respond to neither chemotherapy (cisplatin/etoposide) nor targeted therapy (erlotinib/MK2206). Using computational modeling and computational alanine scan, we hypothesized that this germline mutation impairs SMO/PTCH1 interactions and this decreases SMO inhibition. Based on our observations, the patient was treated with vismodegib, a Hedgehog inhibitor, and maintained response for six months. Another example is to repurpose mebendazole as a novel TNIK inhibitor[166]. We constructed CoMSIA and variable-selection k-nearest neighbor models and employed them to screen 1,448 FDA-approved small molecule drugs. Finally, we identified mebendazole, an approved anthelmintic drug, could selectively inhibit TNIK kinase activity with a dissociation constant Kd = ~1 μM. As mentioned, numerous drug repurposing projects are summarized in Table 3 and the drug properties are in the supplementary Table 3.1.
It is worth mentioning that several national and international collaborative initiatives are being developed for drug repurposing. For instance, Repurposing Drugs in Oncology (ReDO) is an international collaborative efforts towards bringing less toxic cancer treatments to patients in short amount of time[167]. ReDO offers a database of non-cancer drugs that have shown some anticancer activity in preclinical and clinical settings. Another such initiative is ReFRAMEdb with a collection of about 14,000 FDA approved and experimental drugs with associated bioactivity data. Through collaboration of US, UK and Germany, the team is leveraging ReFRAMEdb resource to repurpose drug for COVID-19 treatment[168]. Additionally, drug repurposing is one of the main strategies employed to tackle the recent COVID19 pandemic. According to clinicaltrials.gov, 915 clinical trials are reported by WHO International Clinical Trials Registry Platform (https://clinicaltrials.gov/ct2/who_table); and 16 federally-funded clinical trials began in USA alone (https://clinicaltrials.gov/ct2/results?cond=COVID-19&fund=0&fund=1) as of April 25, 2020, including the well-known remdesvir and hydroxychloroquine to be repurposed for COVID-19 treatment (Table 3).
7.3. Rational Design of Multi-target Directed Ligands (MTDLs)
Traditional concept of drug design involves identification of magic bullet that can target specific receptor and modulates the disease condition. Given the promiscuous nature of drugs and involvement of multiple targets in complex diseases, it is of great significance to develop drugs that can target multiple pre-selected targets for better efficacy against the disease with minimal side effects (Figure. 1A). To achieve these goals, multiple strategies have been suggested and implemented with great successes[3, 18, 19, 20, 21, 22, 169, 170, 171]. For instance, Lim et al. used rational approach to identify multi-target PDE/kinase inhibitor using structural systems pharmacology for precision anti-cancer therapy[172]. The first step of rational multi-targeting drug development involves identification of specific targets. This is a challenging task and requires great understanding of disease mechanisms. One of the straightforward ways is to identify multiple targets from a single pathway that is directly involved in disease. Tan et al. reviewed structural systems pharmacology concepts to identify and prioritize genome wide target profiles[173]. Tolcher et al. discussed novel strategies for combination therapies targeting the MAP kinase pathway to treat solid tumors[174]. Another way for target selection is based on clinical observations. Using the patient response and genetic markup, physicians routinely identify various targets to obtain maximal effects. For example, antipsychotics and anticancer drugs are regularly prescribed as a combination therapy to treat some patients[6, 175]. A variety of in silico methods have been developed to predict target combinations using systems biology/pharmacology data[56, 149, 176, 177, 178, 179].
In the second step, multiple frameworks from known compounds targeting pre-selected targets are taken into consideration and they are either used in combination, linked, fused or merged together to develop multi-targeting compounds (Figure 1 B–E). In combination approach, two or more drugs are rationally selected against pre-selected target combinations (Figure 1B). For instance, Somayaji et al. recently published a study on optimizing drug combinations for multiple targets to modulate the primary and secondary brain damage cascades[180]. To this end, the authors developed multi-scale models covering aspects such as whole-body physiology, primary and secondary injury mechanisms, drug metabolisms and pre-clinical to human extrapolation methods. In a linked approach, two pharmacophores or drug molecules are linked by a spacer to act against multiple targets (Figure 1C). For example, Fancellu et al. recently described multi-targeted tacrine-benzofuran hybrid molecules for the treatment of Alzheimer’s disease[181]. Another example is PROTAC molecules which gained large attention due to their ability to facilitate ubiquitination of the targeted proteins resulting in protein degradation. Zeng et al. discussed PROTAC strategies targeting oncogenic KRASG12C protein in their recent article[182]. They developed novel PROTAC series based on CRBN degrader that could degrade KRASG12C in reporter assays. In fused molecules, two pharmacophores or drug molecules are fused together without a linker (Figure 1D). For instance, Jerabek et al. developed Tacrin-resveratrol fused compound as a multi-targeting agent for Alzheimer’s disease[183]. With the merged molecule approach, two pharmacophore or drug molecules are merged usually on a common substructure to develop small MW and simpler molecules (Figure 1E). Sheng and co-workers designed a merged molecule by combining part of deferiprone and amino-propoxyphenyl scaffold from a H3R antagonist[184]. The authors showed that the merged molecule exhibited excellent activity against β-amyloid aggregation, selective H3R antagonism, metal ion chelation, and radical scavenging. Recently, Ferraro et al. have shown that the results from extended MD simulation trajectories of multi-targeted dopamine D3 modulators can be employed to study the ligand flexibility and interactions with multiple receptors[169]. In a similar study, Xue et al. predicted the binding mode and affinities of triple reuptake inhibitor amitifadine in complex with various human monoamine transporters (MATs)[170]. The authors demonstrated that such approaches could facilitate rational optimization of MTDLs.
7.3. Polypharmacology in Epigenetics
Epigenetics is one of the rapidly growing fields with high therapeutic potentials. Several drugs targeting epigenetic enzymes such as DNA methylation inhibitors, histone methyl transferase inhibitors, bromodomain inhibitors have been approved or in clinical trials for cancer treatments[185]. In 2016, de Lera et al. published a comprehensive review article on epigenetic polypharmacology covering topics from combination therapy to multi-targeted approach[186]. Singh et al. published a computational study of EZH2, HDAC and HKMT inhibitors, and predicted that these modulators can be used as multi-targeted agents against the Hedgehog pathway for cancer treatments[187].
A recent paper by Tomaselli et al. used multi-target-directed ligands (MTDLs), for targeting multiple epigenetic targets[188]. In the context of epigenetic polypharmacology, such MTDLs could act on an epigenetic target with one component and on either a non-epigenetic target or another epigenetic target with another component. These different components could act on different pathways as well, decreasing the chance of acquired resistance due to use of another pathway to compensate for the targeted one. Most of the epigenetic MTDLs developed so far are hybrid molecules consisting of a histone deacetylase inhibitor (HDACi) linked to another targeted drug, usually a kinase inhibitor[189, 190].
7.4. Polypharmacology in Natural Products
Natural products with polypharmacological profiles have demonstrated great promise as novel therapeutics for various complex diseases including cancer. In a review study, Fang et al. constructed polypharmacological and systems immunology profiles of five natural compounds, then summarized the different resources available for reconstructing drug-target networks of natural targets[38]. They also reviewed computational tools for the prediction of drug-target interactions and provided an example of application to precision oncology within a systems biology framework through the integration of polypharmacology of natural products and large-scale cancer genomics data. The article highlighted the promising immunotherapies and combination therapies that harness the immunological and inflammatory side effects of natural products to target the tumor microenvironment. In another study, Grisoni et al. developed a virtual-screening protocol based on machine learning and identified mimetics of the natural product galantamine against its targets[191]. The eight compounds they identified had different polypharmacological profiles, and two had activity against multiple targets associated with Alzheimer’s disease and were suitable for further drug development. This supports the use of chemically informed machine-learning models in virtual screening protocols for polypharmacology design.
8. Expert Opinion
Key Findings and Potentials:
It would be ideal if we could experimentally identify all possible targets for a given compound in order to design multi-targeting strategies and maximally reduce potential toxicities. Apparently, this is practically impossible due to the tremendous number of compounds and targets. As we updated here, a variety of strategies have been implemented to predict toxicities, repurpose drugs, and rationally design multi-targeting agents. In particular, several large national and international collaborative initiatives for drug repurposing have been carried out with high expectations including the latest efforts to address COVID-19 pandemic[167, 168, 192][193, 194]. Encouragingly, the number of successful clinical trials show that both private and public sectors recognized the value of drug repurposing and ignited their significant interests. As polypharmacology can predict drug effects, both on-target and off-target, it could provide a better picture of targeting diseases. Therefore, the rational polypharmacological drug design holds a great promise and potential to future drug discovery. However, to achieve such ambitious goals and ultimately translate the knowledge to effective patient therapies, we must realize that there is still a long way to go and we need to overcome a variety of weaknesses and challenges.
Weakness and Challenges:
One of the major challenges in developing polypharmacology methods is lack of the experimentally derived activities of compounds for a wide range of targets. An ideal dataset would require activities of diverse chemotypes against large number of targets. Multiple reports have shown that publicly available databases are skewed towards certain drug targets such as kinases and GPCRs with small molecules with limited chemotypes[195, 196, 197]. As a matter of fact, for many diseases, we only understand the pathways/mechanism partially, if not minimally, at the molecular level. It is of great issue to derive the polypharmacological network using such incomplete data and such effort usually leads to biased models with limited usages. The accompanying problem is the lack of negative data. Frequently computer-generated, so called random negative datasets are utilized for method development. For instance, there are essentially no large negative datasets with true experimentally confirmed non-interacting compound-target pairs. In most of the cases, there are only a few confirmed interacting targets for a given compound; for many other compounds and targets, we just do not have the data. Indeed, some negative data can be mined from large databases such as Pubchem[198]. Wassermann et al. complied a dataset of “Dark Chemical Matter” (DCM)[199]. However, even these compounds are not tested against all of the protein targets. What we need is a large number of compounds with experimentally determined activity against as many targets as possible, no matter active or inactive. Therefore, in addition to developing efficient HTS assays for compound-target interactions, we should also encourage publications of negative data as they are critical for methodology development and modeling building.
Another challenge is that there is a great demand of sophisticated assay systems for multi-targeting compound characterization. Most of the current assays are designed to test activity of the drug against one target. Therefore, new assay systems need to be developed so that they can accurately detect binding to multiple targets or take readouts of multiple functional activities simultaneously. Recently, a few methods such as Kinobeads [200], drug affinity responsive target stability (DARTS)[201], and cellular thermal shift assays (CETSA)[202] have shown promises in detecting multi-target binding. An issue of these assays is that they lack the complexities to address the disease states and thus do not provide the overall picture of the effects of multi-targeted ligands on the body. To this end, in vivo animal models can be developed to simulate complex disease environments. Some recent studies assessed combinations of sEH inhibitors and PPARγ agonists in a cross-breeding spontaneously hypertensive obese (SHROB) rat[203] model to reveal synergistic effects through complex read-outs of the whole range of metabolic parameters[204, 205]. However, more complex parameters and models are needed for a realistic animal disease representation useful for multiple targeting in other diseases, and in some cases advanced species (e.g., primate) models may be required[206].
Current status and future perspective:
It is well understood that most drugs exert their effects well beyond their on-targets, and drug off-targets are poorly understood or largely unknown, in most cases. It is not possible to exhaustively evaluate their target space with wet lab experiments. Computational polypharmacology has advanced to the stage where tangible hypothesis can be developed using prediction results to guide the wet lab experiments.
In the next few years, we anticipate standardized datasets, open source libraries, rigorous method validation strategies, and accurate polypharmacology prediction algorithms will be implemented and used routinely in the drug discovery pipeline. In addition, most of the current methods are implemented either as web servers or standalone packages. Since community efforts become more crucial, it will be important to develop portable programming libraries which can be employed by the community developers to modify existing or implement new toolkits. In terms of experimental assays, more cell-free, cell-based, and animal models are needed to simultaneously assess the effects of compounds on multiple targets or functions. As animal models are usually complex, costly and time-consuming, use of simple organisms such as C. elegan and zebrafish may be a more economical way for proof-of-concept studies[207]. On the one hand, polypharmacology studies are important for understanding possible adverse effects of new drugs in the pipeline of development. On the other hand, it is particularly helpful for drug repurposing where new indications of existing drugs/agents can be identified. Rational multi-targeting drug design such as Da et al [208] and Proschak et al.[43] provides a great promise to routine application of polypharmacology methods in drug discovery.
Along the same line, advances in computational sciences will further enable polypharmacology modeling. Especially, AI will be able to take the unstructured big data and look for complex relationships, as demonstrated by the IBM cognitive computing platform Watson (https://www.ibm.com/watson) and DeepMind AlphaFold and AlphaGo [150, 209]. With development of new AI algorithms, combined with plethora of multi-omics big data, we have been equipped with potentially game-changing toolboxes. AI will be particularly powerful for the coming age of big data analysis in pharmaceutical research and ultimately personalized precision medicine although it is a long haul with a lot of hurdles[154]. Down the road in the next few years, we anticipate that more sophisticated, robust, and accurate polypharmacology approaches will boom and rational design of more potent but less toxic multi-targeting agents may become possible. However, it needs to be realized that such tasks remain extremely challenging at the current stage.
Supplementary Material
Article Highlights.
This review article covers the updated computational aspects of polypharmacology, focusing on reports published after 2016, including the efforts addressing the current COVID-19 pandemic.
Many new technologies and transformative concepts have been developed for multi-targeting agents with clinical successes.
High-level data curation/integration and novel computational approaches have been developed, but more are needed for robust and accurate polypharmacology prediction.
More experimental assays and models are in great demand to study the multi-targeting and multi-functional effects of compounds for different diseases.
Deep learning and AI technologies, in combination with multi-omics big data, are being adopted for retrieving hidden and complex relations between biological targets and chemical entities.
Significant challenges still lie ahead for polypharmacology studies, in particular for rational design of effective multi-targeting agents.
Acknowledgements:
The authors give special thanks to the HPC resources from Texas Advanced Computing Center (TACC) and the University of Texas M.D. Anderson Cancer Center
Funding:
S Zhang is partially supported by the Institutional Research Grant (IRG) Program at The University of Texas MD Anderson Cancer Center, CPRIT RP170333, and NIH/NCI grants 1R01CA225955-01 and P30CA016672.
Footnotes
Declaration of Interest:
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- 1.Ramsay RR, Popovic-Nikolic MR, Nikolic K, et al. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018. January 17;7(1):3. doi: 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saginc G, Voellmy F, Linding R. Cancer systems biology: Harnessing off-target effects. Nat Chem Biol. 2017. November 21;13(12):1204–1205. doi: 10.1038/nchembio.2519. [DOI] [PubMed] [Google Scholar]
- 3.***.Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017. December 1;358(6367). doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]; A thorough analysis using chemical proteomics of the target spectrum of clinically evaluated kinase drugs and their unknown targets, offering a perspective on the “druggable” kinome, (non)kinase off-targets, and their potential therapeutic applications.
- 4.Kroschinsky F, Stolzel F, von Bonin S, et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit Care. 2017. April 14;21(1):89. doi: 10.1186/s13054-017-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.***.Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014. October 9;57(19):7874–87. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]; An excellent review on polypharmacology that describes advantages and disadvantages of combination therapy, multi-targeted drugs, and drug repurposing. It also describes applications of polypharmacology in cancer therapy and CNS diseases.
- 6.Bayat Mokhtari R, Homayouni TS, Baluch N, et al. Combination therapy in combating cancer. Oncotarget. 2017. June 6;8(23):38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.***.Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018. May;24(5):638–646. doi: 10.1038/s41591-018-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; A great example of successful precision oncology with repurposing a shelved drug to treat a subpopulation of patients with a specific protein mutation.
- 8.*.Kuenzi BM, Remsing Rix LL, Stewart PA, et al. Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat Chem Biol. 2017. December;13(12):1222–1231. doi: 10.1038/nchembio.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]; Another example of succcessful drug repurposing based on drug polypharmacology.
- 9.***.Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 2020. January 13. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; A latest comprehensive review on the PROTAC technology.
- 10.Khan S, Zhang X, Lv D, et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019. December;25(12):1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schapira M, Calabrese MF, Bullock AN, et al. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019. December;18(12):949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- 12.Farnaby W, Koegl M, Roy MJ, et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019. July;15(7):672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.**.Labrijn AF, Janmaat ML, Reichert JM, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019. August;18(8):585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]; A great review on bispecific antibody development.
- 14.Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2019. December 17. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.**.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019. September;25(9):1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]; A great review on CAR-T development.
- 16.Priest BT, Bell IM, Garcia ML. Role of hERG potassium channel assays in drug development. Channels (Austin). 2008. Mar-Apr;2(2):87–93. doi: 10.4161/chan.2.2.6004. [DOI] [PubMed] [Google Scholar]
- 17.Du-Cuny L, Chen L, Zhang S. A critical assessment of combined ligand- and structure-based approaches to HERG channel blocker modeling. J Chem Inf Model. 2011. November 28;51(11):2948–60. doi: 10.1021/ci200271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.*.Castillo-Quan JI, Tain LS, Kinghorn KJ, et al. A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc Natl Acad Sci U S A. 2019. October 15;116(42):20817–20819. doi: 10.1073/pnas.1913212116. [DOI] [PMC free article] [PubMed] [Google Scholar]; An example to demonstrate how to use a triple drug combination targeting.
- 19.Yu JX, Craig AJ, Duffy ME, et al. Phenotype-Based Screens with Conformation-Specific Inhibitors Reveal p38 Gamma and Delta as Targets for HCC Polypharmacology. Mol Cancer Ther. 2019. September;18(9):1506–1519. doi: 10.1158/1535-7163.MCT-18-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.***.Peng Y, McCorvy JD, Harpsoe K, et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell. 2018. February 8;172(4):719–730 e14. doi: 10.1016/j.cell.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; A great example of investigating the structural basis of polypharmacology at canonical GPCRs and illustrating how such understanding may ultimately facilitate drug design for multiple GPCRs.
- 21.Chan JD, Cupit PM, Gunaratne GS, et al. The anthelmintic praziquantel is a human serotoninergic G-protein-coupled receptor ligand. Nat Commun. 2017. December 5;8(1):1910. doi: 10.1038/s41467-017-02084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammam K, Saez-Ayala M, Rebuffet E, et al. Dual protein kinase and nucleoside kinase modulators for rationally designed polypharmacology. Nat Commun. 2017. November 10;8(1):1420. doi: 10.1038/s41467-017-01582-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinzi L, Rastelli G. Identification of Target Associations for Polypharmacology from Analysis of Crystallographic Ligands of the Protein Data Bank. J Chem Inf Model. 2020. January 27;60(1):372–390. doi: 10.1021/acs.jcim.9b00821. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Li Z, Wu X, et al. Deep Learning Enhancing Kinome-Wide Polypharmacology Profiling: Model Construction and Experiment Validation. J Med Chem. 2019. August 15. doi: 10.1021/acs.jmedchem.9b00855. [DOI] [PubMed] [Google Scholar]
- 25.**.Da C, Zhang D, Stashko M, et al. Data-Driven Construction of Antitumor Agents with Controlled Polypharmacology. J Am Chem Soc. 2019. October 2;141(39):15700–15709. doi: 10.1021/jacs.9b08660. [DOI] [PMC free article] [PubMed] [Google Scholar]; An interesting data-driven strategy achieving restricted polypharmacology in the design of antitumor agents selectively targeting multiple targets.
- 26.Stork C, Chen Y, Sicho M, et al. Hit Dexter 2.0: Machine-Learning Models for the Prediction of Frequent Hitters. J Chem Inf Model. 2019. March 25;59(3):1030–1043. doi: 10.1021/acs.jcim.8b00677. [DOI] [PubMed] [Google Scholar]
- 27.Li Z, Li X, Liu X, et al. KinomeX: a web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics. 2019. December 15;35(24):5354–5356. doi: 10.1093/bioinformatics/btz519. [DOI] [PubMed] [Google Scholar]
- 28.Miljkovic F, Vogt M, Bajorath J. Systematic computational identification of promiscuity cliff pathways formed by inhibitors of the human kinome. J Comput Aided Mol Des. 2019. June;33(6):559–572. doi: 10.1007/s10822-019-00198-9. [DOI] [PubMed] [Google Scholar]
- 29.Jasial S, Gilberg E, Blaschke T, et al. Machine Learning Distinguishes with High Accuracy between Pan-Assay Interference Compounds That Are Promiscuous or Represent Dark Chemical Matter. J Med Chem. 2018. November 21;61(22):10255–10264. doi: 10.1021/acs.jmedchem.8b01404. [DOI] [PubMed] [Google Scholar]
- 30.Schneider P, Rothlisberger M, Reker D, et al. Spotting and designing promiscuous ligands for drug discovery. Chem Commun (Camb). 2016. January 21;52(6):1135–8. doi: 10.1039/c5cc07506h. [DOI] [PubMed] [Google Scholar]
- 31.Yang JJ, Ursu O, Lipinski CA, et al. Badapple: promiscuity patterns from noisy evidence. J Cheminform. 2016;8:29. doi: 10.1186/s13321-016-0137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez-Nueno VI, Karaboga AS, Souchet M, et al. GES polypharmacology fingerprints: A novel approach for drug repositioning. Journal of Chemical Information and Modeling. 2014;54:720–734. doi: 10.1021/ci4006723. [DOI] [PubMed] [Google Scholar]
- 33.***.Chaudhari R, Tan Z, Huang B, et al. Computational polypharmacology: a new paradigm for drug discovery. Expert Opin Drug Discov. 2017. March;12(3):279–291. doi: 10.1080/17460441.2017.1280024. [DOI] [PMC free article] [PubMed] [Google Scholar]; The current update is based on this review, with details on databases and methods used in polypharmacology modeling.
- 34.Tan Z, Chaudhai R, Zhang S. Polypharmacology in Drug Development: A Minireview of Current Technologies. ChemMedChem. 2016. June 20;11(12):1211–8. doi: 10.1002/cmdc.201600067. [DOI] [PubMed] [Google Scholar]
- 35.Amelio I, Lisitsa A, Knight RA, et al. Polypharmacology of Approved Anticancer Drugs. Curr Drug Targets. 2017;18(5):534–543. doi: 10.2174/1389450117666160301095233. [DOI] [PubMed] [Google Scholar]
- 36.Bolognesi ML. Harnessing Polypharmacology with Medicinal Chemistry. ACS Med Chem Lett. 2019. March 14;10(3):273–275. doi: 10.1021/acsmedchemlett.9b00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerisier N, Petitjean M, Regad L, et al. High Impact: The Role of Promiscuous Binding Sites in Polypharmacology. Molecules. 2019. July 10;24(14). doi: 10.3390/molecules24142529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang J, Liu C, Wang Q, et al. In silico polypharmacology of natural products. Brief Bioinform. 2018. November 27;19(6):1153–1171. doi: 10.1093/bib/bbx045. [DOI] [PubMed] [Google Scholar]
- 39.Karuppasamy R, Veerappapillai S, Maiti S, et al. Current progress and future perspectives of polypharmacology : From the view of non-small cell lung cancer. Seminars in Cancer Biology. 2019. 10.1016/j.semcancer.2019.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyers J, Chessum NEA, Ali S, et al. Privileged Structures and Polypharmacology within and between Protein Families. ACS Med Chem Lett. 2018. December 13;9(12):1199–1204. doi: 10.1021/acsmedchemlett.8b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moya-Garcia A, Adeyelu T, Kruger FA, et al. Structural and Functional View of Polypharmacology. Sci Rep. 2017. August 31;7(1):10102. doi: 10.1038/s41598-017-10012-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinzi L, Caporuscio F, Rastelli G. Selection of protein conformations for structure-based polypharmacology studies. Drug Discov Today. 2018. November;23(11):1889–1896. doi: 10.1016/j.drudis.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 43.***.Proschak E, Stark H, Merk D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J Med Chem. 2019. January 24;62(2):420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]; Reccomended article to gain comprehensive overview of multi-targeted drug design from a medicinal chemist’s perspective.
- 44.Ravikumar B, Aittokallio T. Improving the efficacy-safety balance of polypharmacology in multi-target drug discovery. Expert Opin Drug Discov. 2018. February;13(2):179–192. doi: 10.1080/17460441.2018.1413089. [DOI] [PubMed] [Google Scholar]
- 45.Allen WJ, Balius TE, Mukherjee S, et al. DOCK 6: Impact of new features and current docking performance. Journal of computational chemistry. 2015;36:1132–56. doi: 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YZ, Ung CY. Computer automated prediction of potential therapeutic and toxicity protein targets of bioactive compounds from Chinese medicinal plants. Am J Chin Med. 2002;30(1):139–54. doi: 10.1142/S0192415X02000156. [DOI] [PubMed] [Google Scholar]
- 47.Wang JC, Chu PY, Chen CM, et al. idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res. 2012. July;40(Web Server issue):W393–9. doi: 10.1093/nar/gks496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo H, Chen J, Shi L, et al. DRAR-CPI: a server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011. July;39(Web Server issue):W492–8. doi: 10.1093/nar/gkr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Gao Z, Kang L, et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006. July 1;34(Web Server issue):W219–24. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friesner Ra, Banks JL, Murphy RB, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of Medicinal Chemistry. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 51.McGann M FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011. March 28;51(3):578–96. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 52.Pereira JC, Caffarena ER, Dos Santos CN. Boosting Docking-Based Virtual Screening with Deep Learning. J Chem Inf Model. 2016. December 27;56(12):2495–2506. doi: 10.1021/acs.jcim.6b00355. [DOI] [PubMed] [Google Scholar]
- 53.Shulman-Peleg A, Nussinov R, Wolfson HJ. SiteEngines: recognition and comparison of binding sites and protein-protein interfaces. Nucleic Acids Res. 2005. July 1;33(Web Server issue):W337–41. doi: 10.1093/nar/gki482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jambon M, Imberty A, Deleage G, et al. A new bioinformatic approach to detect common 3D sites in protein structures. Proteins. 2003. August 1;52(2):137–45. doi: 10.1002/prot.10339. [DOI] [PubMed] [Google Scholar]
- 55.Chartier M, Adriansen E, Najmanovich R. IsoMIF Finder: online detection of binding site molecular interaction field similarities. Bioinformatics. 2016. February 15;32(4):621–3. doi: 10.1093/bioinformatics/btv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siragusa L, Cross S, Baroni M, et al. BioGPS: navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins. 2015. March;83(3):517–32. doi: 10.1002/prot.24753. [DOI] [PubMed] [Google Scholar]
- 57.Yeturu K, Chandra N. PocketMatch: a new algorithm to compare binding sites in protein structures. BMC Bioinformatics. 2008;9:543. doi: 10.1186/1471-2105-9-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.ProBiS-database: Precalculated binding site similarities and local pairwise alignments of PDB structures. 2012. [DOI] [PMC free article] [PubMed]
- 59.Gao M, Skolnick J. APoc: large-scale identification of similar protein pockets. Bioinformatics. 2013. March 1;29(5):597–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krotzky T, Fober T, Hullermeier E, et al. Extended Graph-Based Models for Enhanced Similarity Search in Cavbase. IEEE/ACM Trans Comput Biol Bioinform. 2014. Sep-Oct;11(5):878–90. doi: 10.1109/TCBB.2014.2325020. [DOI] [PubMed] [Google Scholar]
- 61.Hoffmann B, Zaslavskiy M, Vert JP, et al. A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction. BMC Bioinformatics. 2010;11:99. doi: 10.1186/1471-2105-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aung Z, Tong JC. BSAlign: a rapid graph-based algorithm for detecting ligand-binding sites in protein structures. Genome Inform. 2008;21:65–76. [PubMed] [Google Scholar]
- 63.Liu X, Ouyang S, Yu B, et al. PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010. July;38(Web Server issue):W609–14. doi: 10.1093/nar/gkq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stank A, Kokh DB, Horn M, et al. TRAPP webserver: predicting protein binding site flexibility and detecting transient binding pockets. Nucleic Acids Res. 2017. July 3;45(W1):W325–W330. doi: 10.1093/nar/gkx277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Z, Li X, Liu X, et al. KinomeX: a web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics. 2019. June 22. doi: 10.1093/bioinformatics/btz519. [DOI] [PubMed] [Google Scholar]
- 66.Dey F, Caflisch A. Fragment-based de novo ligand design by multiobjective evolutionary optimization. J Chem Inf Model. 2008. March;48(3):679–90. doi: 10.1021/ci700424b. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Zhao Z, Liu Z, et al. AutoT&T v.2: An Efficient and Versatile Tool for Lead Structure Generation and Optimization. J Chem Inf Model. 2016. February 22;56(2):435–53. doi: 10.1021/acs.jcim.5b00691. [DOI] [PubMed] [Google Scholar]
- 68.Maass P, Schulz-Gasch T, Stahl M, et al. Recore: a fast and versatile method for scaffold hopping based on small molecule crystal structure conformations. J Chem Inf Model. 2007. Mar-Apr;47(2):390–9. doi: 10.1021/ci060094h. [DOI] [PubMed] [Google Scholar]
- 69.Durrant JD, Lindert S, McCammon JA. AutoGrow 3.0: an improved algorithm for chemically tractable, semi-automated protein inhibitor design. J Mol Graph Model. 2013. July;44:104–12. doi: 10.1016/j.jmgm.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keiser MJ, Roth BL, Armbruster BN, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007. February;25(2):197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 71.Peon A, Li H, Ghislat G, et al. MolTarPred: A web tool for comprehensive target prediction with reliability estimation. Chem Biol Drug Des. 2019. July;94(1):1390–1401. doi: 10.1111/cbdd.13516. [DOI] [PubMed] [Google Scholar]
- 72.Nickel J, Gohlke BO, Erehman J, et al. SuperPred: update on drug classification and target prediction. Nucleic Acids Res. 2014. July;42(Web Server issue):W26–31. doi: 10.1093/nar/gku477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gfeller D, Grosdidier A, Wirth M, et al. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014. July;42(Web Server issue):W32–8. doi: 10.1093/nar/gku293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L, Ma C, Wipf P, et al. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013. April;15(2):395–406. doi: 10.1208/s12248-012-9449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu TP, Hsieh YY, Chou CJ, et al. Systematic polypharmacology and drug repurposing via an integrated L1000-based Connectivity Map database mining. R Soc Open Sci. 2018. November;5(11):181321. doi: 10.1098/rsos.181321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016. January 4;44(D1):D380–4. doi: 10.1093/nar/gkv1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duan Q, Flynn C, Niepel M, et al. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 2014. July;42(Web Server issue):W449–60. doi: 10.1093/nar/gku476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012. May;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee K, Lee M, Kim D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target-fishing server. BMC Bioinformatics. 2017. December 28;18(Suppl 16):567. doi: 10.1186/s12859-017-1960-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de la Vega de Leon A, Chen B, Gillet VJ. Effect of missing data on multitask prediction methods. J Cheminform. 2018. May 22;10(1):26. doi: 10.1186/s13321-018-0281-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allaway RJ, La Rosa S, Guinney J, et al. Probing the chemical-biological relationship space with the Drug Target Explorer. J Cheminform. 2018. August 20;10(1):41. doi: 10.1186/s13321-018-0297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Awale M, Reymond JL. Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J Chem Inf Model. 2019. January 28;59(1):10–17. doi: 10.1021/acs.jcim.8b00524. [DOI] [PubMed] [Google Scholar]
- 83.Chen C, Wu M, Cen S, et al. MTLD, a Database of Multiple Target Ligands, the Updated Version. Molecules. 2017. September 6;22(9). doi: 10.3390/molecules22091375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun J, Jeliazkova N, Chupakin V, et al. ExCAPE-DB: an integrated large scale dataset facilitating Big Data analysis in chemogenomics. J Cheminform. 2017;9:17. doi: 10.1186/s13321-017-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim S, Thiessen PA, Bolton EE, et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016. January 4;44(D1):D1202–13. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Research. 2012;40. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gilson MK, Liu T, Baitaluk M, et al. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016. January 4;44(D1):D1045–53. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014. January;42(Database issue):D1091–7. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Zhang S, Li F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2019. November 6. doi: 10.1093/nar/gkz981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gunther S, Kuhn M, Dunkel M, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008. January;36(Database issue):D919–22. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reddy AS, Tan Z, Zhang S. Curation and analysis of multitargeting agents for polypharmacological modeling. J Chem Inf Model. 2014. September 22;54(9):2536–43. doi: 10.1021/ci500092j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu Z, Chang YC, Wang Y, et al. VisANT 4.0: Integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res. 2013. July;41(Web Server issue):W225–31. doi: 10.1093/nar/gkt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kringelum J, Kjaerulff SK, Brunak S, et al. ChemProt-3.0: a global chemical biology diseases mapping. Database (Oxford). 2016;2016. doi: 10.1093/database/bav123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu QN, Deng Z, Tu W, et al. VNP: Interactive Visual Network Pharmacology of Diseases, Targets, and Drugs. CPT Pharmacometrics Syst Pharmacol. 2014. March 12;3:e105. doi: 10.1038/psp.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Z, Li J, Dang R, et al. PhIN: A Protein Pharmacology Interaction Network Database. CPT Pharmacometrics Syst Pharmacol. 2015. March;4(3):e00025. doi: 10.1002/psp4.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.von Eichborn J, Murgueitio MS, Dunkel M, et al. PROMISCUOUS: a database for network-based drug-repositioning. Nucleic Acids Res. 2011. January;39(Database issue):D1060–6. doi: 10.1093/nar/gkq1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Betts KS. Tox21 to date: steps toward modernizing human hazard characterization. Environ Health Perspect. 2013. July;121(7):A228. doi: 10.1289/ehp.121-a228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Papatheodorou I, Moreno P, Manning J, et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2019. October 30. doi: 10.1093/nar/gkz947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics. 2016. June 20;54:1 30 1–1 30 33. doi: 10.1002/cpbi.5. [DOI] [PubMed] [Google Scholar]
- 100.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002. January 1;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome. Nucleic acids research. 2004;32:D277–80. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hess JL. The Cancer Genome Anatomy Project: power tools for cancer biologists. Cancer Invest. 2003. April;21(2):325–6. doi: 10.1081/cnv-120016428. [DOI] [PubMed] [Google Scholar]
- 103.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013. October;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015. October 1;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu H, Zhang W, Zou B, et al. DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res. 2020. January 8;48(D1):D871–D881. doi: 10.1093/nar/gkz1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coker EA, Mitsopoulos C, Tym JE, et al. canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. 2019. January 8;47(D1):D917–D922. doi: 10.1093/nar/gky1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schneider P, Schneider G. Privileged Structures Revisited. Angew Chem Int Ed Engl. 2017. June 26;56(27):7971–7974. doi: 10.1002/anie.201702816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Monteleone S, Fuchs JE, Liedl KR. Molecular Connectivity Predefines Polypharmacology: Aliphatic Rings, Chirality, and sp(3) Centers Enhance Target Selectivity. Front Pharmacol. 2017;8:552. doi: 10.3389/fphar.2017.00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duran-Frigola M, Siragusa L, Ruppin E, et al. Detecting similar binding pockets to enable systems polypharmacology. PLoS Comput Biol. 2017. June;13(6):e1005522. doi: 10.1371/journal.pcbi.1005522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ehrt C, Brinkjost T, Koch O. Binding site characterization - similarity, promiscuity, and druggability. Medchemcomm. 2019. July 1;10(7):1145–1159. doi: 10.1039/c9md00102f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Naderi M, Lemoine JM, Govindaraj RG, et al. Binding site matching in rational drug design: algorithms and applications. Brief Bioinform. 2018. August 31. doi: 10.1093/bib/bby078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gilberg E, Gutschow M, Bajorath J. Promiscuous Ligands from Experimentally Determined Structures, Binding Conformations, and Protein Family-Dependent Interaction Hotspots. ACS Omega. 2019. January 31;4(1):1729–1737. doi: 10.1021/acsomega.8b03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lim H, Xie L. Omics Data Integration and Analysis for Systems Pharmacology. Methods Mol Biol. 2019;1939:199–214. doi: 10.1007/978-1-4939-9089-4_11. [DOI] [PubMed] [Google Scholar]
- 114.Moya-García AA, Ranea JAG. Insights into polypharmacology from drug-domain associations. Bioinformatics. 2013;29:1934–1937. doi: 10.1093/bioinformatics/btt321. [DOI] [PubMed] [Google Scholar]
- 115.Wu Z, Li W, Liu G, et al. Network-Based Methods for Prediction of Drug-Target Interactions. Front Pharmacol. 2018;9:1134. doi: 10.3389/fphar.2018.01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheng F, Kovacs IA, Barabasi AL. Network-based prediction of drug combinations. Nat Commun. 2019. March 13;10(1):1197. doi: 10.1038/s41467-019-09186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hafner M, Mills CE, Subramanian K, et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol. 2019. August 15;26(8):1067–1080 e8. doi: 10.1016/j.chembiol.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011. October 30;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 119.Sakamoto KM, Kim KB, Kumagai A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001. July 17;98(15):8554–9. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Konstantinidou M, Li J, Zhang B, et al. PROTACs- a game-changing technology. Expert Opin Drug Discov. 2019. December;14(12):1255–1268. doi: 10.1080/17460441.2019.1659242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bondeson DP, Smith BE, Burslem GM, et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol. 2018. January 18;25(1):78–87 e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang HT, Dobrovolsky D, Paulk J, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol. 2018. January 18;25(1):88–99 e6. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vij R, Nath R, Afar DEH, et al. First-in-Human Phase 1 Study of ABBV-838, an Antibody-Drug Conjugate Targeting SLAMF7/CS1 in Patients With Relapsed and Refractory Multiple Myeloma. Clin Cancer Res. 2020. January 22. doi: 10.1158/1078-0432.CCR-19-1431. [DOI] [PubMed] [Google Scholar]
- 124.Gerber DE, Infante JR, Gordon MS, et al. Phase Ia Study of Anti-NaPi2b Antibody-Drug Conjugate Lifastuzumab Vedotin DNIB0600A in Patients with Non-Small Cell Lung Cancer and Platinum-Resistant Ovarian Cancer. Clin Cancer Res. 2020. January 15;26(2):364–372. doi: 10.1158/1078-0432.CCR-18-3965. [DOI] [PubMed] [Google Scholar]
- 125.Narayan R, Blonquist TM, Emadi A, et al. A phase 1 study of the antibody-drug conjugate brentuximab vedotin with re-induction chemotherapy in patients with CD30-expressing relapsed/refractory acute myeloid leukemia. Cancer. 2019. December 20. doi: 10.1002/cncr.32657. [DOI] [PubMed] [Google Scholar]
- 126.Danila DC, Szmulewitz RZ, Vaishampayan U, et al. Phase I Study of DSTP3086S, an Antibody-Drug Conjugate Targeting Six-Transmembrane Epithelial Antigen of Prostate 1, in Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2019. December 20;37(36):3518–3527. doi: 10.1200/JCO.19.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kahl BS, Hamadani M, Radford J, et al. A Phase I Study of ADCT-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody-Drug Conjugate, in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Clin Cancer Res. 2019. December 1;25(23):6986–6994. doi: 10.1158/1078-0432.CCR-19-0711. [DOI] [PubMed] [Google Scholar]
- 128.Kopp LM, Malempati S, Krailo M, et al. Phase II trial of the glycoprotein non-metastatic B-targeted antibody-drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. Eur J Cancer. 2019. November;121:177–183. doi: 10.1016/j.ejca.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim SB, Meric-Bernstam F, Kalyan A, et al. First-in-Human Phase I Study of Aprutumab Ixadotin, a Fibroblast Growth Factor Receptor 2 Antibody-Drug Conjugate (BAY 1187982) in Patients with Advanced Cancer. Target Oncol. 2019. October;14(5):591–601. doi: 10.1007/s11523-019-00670-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sandhu S, McNeil CM, LoRusso P, et al. Phase I study of the anti-endothelin B receptor antibody-drug conjugate DEDN6526A in patients with metastatic or unresectable cutaneous, mucosal, or uveal melanoma. Invest New Drugs. 2019. August 5. doi: 10.1007/s10637-019-00832-1. [DOI] [PubMed] [Google Scholar]
- 131.Massard C, Soria JC, Krauss J, et al. First-in-human study to assess safety, tolerability, pharmacokinetics, and pharmacodynamics of the anti-CD27L antibody-drug conjugate AMG 172 in patients with relapsed/refractory renal cell carcinoma. Cancer Chemother Pharmacol. 2019. June;83(6):1057–1063. doi: 10.1007/s00280-019-03796-4. [DOI] [PubMed] [Google Scholar]
- 132.Collins DM, Bossenmaier B, Kollmorgen G, et al. Acquired Resistance to Antibody-Drug Conjugates. Cancers (Basel). 2019. March 20;11(3). doi: 10.3390/cancers11030394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, et al. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J Med Biotechnol. 2019. Jan-Mar;11(1):3–23. [PMC free article] [PubMed] [Google Scholar]
- 134.Lamb YN. Inotuzumab Ozogamicin: First Global Approval. Drugs. 2017. September;77(14):1603–1610. doi: 10.1007/s40265-017-0802-5. [DOI] [PubMed] [Google Scholar]
- 135.Traynor K Ado-trastuzumab emtansine approved for advanced breast cancer. Am J Health Syst Pharm. 2013. April 1;70(7):562. doi: 10.2146/news130023. [DOI] [PubMed] [Google Scholar]
- 136.Teicher BA, Doroshow JH. The promise of antibody-drug conjugates. N Engl J Med. 2012. November 8;367(19):1847–8. doi: 10.1056/NEJMe1211736. [DOI] [PubMed] [Google Scholar]
- 137.Fan G, Wang Z, Hao M, et al. Bispecific antibodies and their applications. J Hematol Oncol. 2015. December 21;8:130. doi: 10.1186/s13045-015-0227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Muller D, Kontermann RE. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs. 2010. April 1;24(2):89–98. doi: 10.2165/11530960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 139.Jensen TI, Axelgaard E, Bak RO. Therapeutic gene editing in haematological disorders with CRISPR/Cas9. Br J Haematol. 2019. June;185(5):821–835. doi: 10.1111/bjh.15851. [DOI] [PubMed] [Google Scholar]
- 140.Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015. August;36(8):494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ledford H Engineered cell therapy for cancer gets thumbs up from FDA advisers. Nature. 2017. July 12;547(7663):270. doi: 10.1038/nature.2017.22304. [DOI] [PubMed] [Google Scholar]
- 142.Xin Yu J, Hubbard-Lucey VM, Tang J. The global pipeline of cell therapies for cancer. Nat Rev Drug Discov. 2019. October;18(11):821–822. doi: 10.1038/d41573-019-00090-z. [DOI] [PubMed] [Google Scholar]
- 143.Avram S, Curpan R, Bora A, et al. Enhancing Molecular Promiscuity Evaluation Through Assay Profiles. Pharm Res. 2018. October 18;35(11):240. doi: 10.1007/s11095-018-2523-1. [DOI] [PubMed] [Google Scholar]
- 144.Hu Y, Bajorath J. Polypharmacology directed compound data mining: identification of promiscuous chemotypes with different activity profiles and comparison to approved drugs. J Chem Inf Model. 2010. December 27;50(12):2112–8. doi: 10.1021/ci1003637. [DOI] [PubMed] [Google Scholar]
- 145.Hu Y, Bajorath J. Molecular scaffolds with high propensity to form multi-target activity cliffs. J Chem Inf Model. 2010. April 26;50(4):500–10. doi: 10.1021/ci100059q. [DOI] [PubMed] [Google Scholar]
- 146.Zhang J, Mucs D, Norinder U, et al. LightGBM: An Effective and Scalable Algorithm for Prediction of Chemical Toxicity-Application to the Tox21 and Mutagenicity Data Sets. J Chem Inf Model. 2019. October 28;59(10):4150–4158. doi: 10.1021/acs.jcim.9b00633. [DOI] [PubMed] [Google Scholar]
- 147.Rendleman MC, Buatti JM, Braun TA, et al. Machine learning with the TCGA-HNSC dataset: improving usability by addressing inconsistency, sparsity, and high-dimensionality. BMC Bioinformatics. 2019. June 17;20(1):339. doi: 10.1186/s12859-019-2929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jones D, Bopaiah J, Alghamedy F, et al. Polypharmacology Within the Full Kinome: a Machine Learning Approach. AMIA Joint Summits on Translational Science. 2018:98–107. [PMC free article] [PubMed] [Google Scholar]
- 149.Chua HE, Bhowmick SS, Tucker-Kellogg L. Synergistic target combination prediction from curated signaling networks: Machine learning meets systems biology and pharmacology. Methods. 2017. October 1;129:60–80. doi: 10.1016/j.ymeth.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 150.**.Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020. January 15. doi: 10.1038/s41586-019-1923-7. . [DOI] [PubMed] [Google Scholar]; The latest breakthrough in protein structure prediction and drug design using AI.
- 151.Mysinger MM, Carchia M, Irwin JJ, et al. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012. July 26;55(14):6582–94. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li H, Sze K-H, Lu G, et al. Machine-learning scoring functions for structure-based drug lead optimization. WIREs Computational Molecular Science.n/a(n/a):e1465. doi: 10.1002/wcms.1465. [DOI] [Google Scholar]
- 153.Shen C, Ding J, Wang Z, et al. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. WIREs Computational Molecular Science. 2020;10(1):e1429. doi: 10.1002/wcms.1429. [DOI] [Google Scholar]
- 154.*.Strickland E IBM Watson, heal thyself: How IBM overpromised and underdelivered on AI health care. IEEE Spectrum. 2019;56(4):24–31. doi: 10.1109/MSPEC.2019.8678513. [DOI] [Google Scholar]; A report on real world applications and failures of AI to patient treatment.
- 155.LaBute MX, Zhang X, Lenderman J, et al. Adverse drug reaction prediction using scores produced by large-scale drug-protein target docking on high-performance computing machines. PLoS One. 2014;9(9):e106298. doi: 10.1371/journal.pone.0106298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ji ZL, Wang Y, Yu L, et al. In silico search of putative adverse drug reaction related proteins as a potential tool for facilitating drug adverse effect prediction. Toxicol Lett. 2006. July 1;164(2):104–12. doi: 10.1016/j.toxlet.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 157.de Anda-Jauregui G, Guo K, Hur J. Network-Based Assessment of Adverse Drug Reaction Risk in Polypharmacy Using High-Throughput Screening Data. Int J Mol Sci. 2019. January 17;20(2). doi: 10.3390/ijms20020386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zitnik M, Agrawal M, Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018. July 1;34(13):i457–i466. doi: 10.1093/bioinformatics/bty294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou H, Gao M, Skolnick J. Comprehensive prediction of drug-protein interactions and side effects for the human proteome. Sci Rep. 2015. June 9;5:11090. doi: 10.1038/srep11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Uner OC, Gokberk Cinbis R, Tastan O, et al. DeepSide: A Deep Learning Framework for Drug Side Effect Prediction. bioRxiv. 2019:843029. doi: 10.1101/843029. [DOI] [Google Scholar]
- 161.Kim D, Lee J, Lee S, et al. Predicting unintended effects of drugs based on off-target tissue effects. Biochem Biophys Res Commun. 2016. January 15;469(3):399–404. doi: 10.1016/j.bbrc.2015.11.095. [DOI] [PubMed] [Google Scholar]
- 162.Le P, Kunold E, Macsics R, et al. Repurposing human kinase inhibitors to create an antibiotic active against drug-resistant Staphylococcus aureus, persisters and biofilms. Nat Chem. 2019. December 16. doi: 10.1038/s41557-019-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Noronha V, Choughule A, Patil VM, et al. Epidermal growth factor receptor exon 20 mutation in lung cancer: types, incidence, clinical features and impact on treatment. Onco Targets Ther. 2017;10:2903–2908. doi: 10.2147/OTT.S133245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tu HY, Ke EE, Yang JJ, et al. A comprehensive review of uncommon EGFR mutations in patients with non-small cell lung cancer. Lung Cancer. 2017. December;114:96–102. doi: 10.1016/j.lungcan.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 165.Tsao AS, Wistuba I, Xia D, et al. Germline and Somatic Smoothened Mutations in Non–Small-Cell Lung Cancer Are Potentially Responsive to Hedgehog Inhibitor Vismodegib. JCO Precision Oncology. 2017. (1):1–10. doi: 10.1200/po.17.00149. [DOI] [PubMed] [Google Scholar]
- 166.Tan Z, Chen L, Zhang S. Comprehensive Modeling and Discovery of Mebendazole as a Novel TRAF2- and NCK-interacting Kinase Inhibitor. Sci Rep. 2016. September 21;6:33534. doi: 10.1038/srep33534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pantziarka P, Bouche G, Meheus L, et al. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience. 2014;8:442. doi: 10.3332/ecancer.2014.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Janes J, Young ME, Chen E, et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc Natl Acad Sci U S A. 2018. October 16;115(42):10750–10755. doi: 10.1073/pnas.1810137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ferraro M, Decherchi S, De Simone A, et al. Multi-target dopamine D3 receptor modulators: Actionable knowledge for drug design from molecular dynamics and machine learning. Eur J Med Chem. 2020. February 15;188:111975. doi: 10.1016/j.ejmech.2019.111975. [DOI] [PubMed] [Google Scholar]
- 170.Xue W, Wang P, Tu G, et al. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys Chem Chem Phys. 2018. February 28;20(9):6606–6616. doi: 10.1039/c7cp07869b. [DOI] [PubMed] [Google Scholar]
- 171.*.Zhou J, Jiang X, He S, et al. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J Med Chem. 2019. October 24;62(20):8881–8914. doi: 10.1021/acs.jmedchem.9b00017. [DOI] [PubMed] [Google Scholar]; Another great review article on MTDL.
- 172.*.Lim H, He D, Qiu Y, et al. Rational discovery of dual-indication multi-target PDE/Kinase inhibitor for precision anti-cancer therapy using structural systems pharmacology. PLoS Comput Biol. 2019. June;15(6):e1006619. doi: 10.1371/journal.pcbi.1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]; An example of rational design of multi-targeting agents.
- 173.Tan H, Ge X, Xie L. Structural systems pharmacology: a new frontier in discovering novel drug targets. Curr Drug Targets. 2013. August;14(9):952–8. doi: 10.2174/1389450111314090003. [DOI] [PubMed] [Google Scholar]
- 174.Tolcher AW, Peng W, Calvo E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol Cancer Ther. 2018. January;17(1):3–16. doi: 10.1158/1535-7163.MCT-17-0349. [DOI] [PubMed] [Google Scholar]
- 175.Ortiz-Orendain J, Castiello-de Obeso S, Colunga-Lozano LE, et al. Antipsychotic combinations for schizophrenia. Cochrane Database Syst Rev. 2017. June 28;6:CD009005. doi: 10.1002/14651858.CD009005.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sheng Z, Sun Y, Yin Z, et al. Advances in computational approaches in identifying synergistic drug combinations. Brief Bioinform. 2018. November 27;19(6):1172–1182. doi: 10.1093/bib/bbx047. [DOI] [PubMed] [Google Scholar]
- 177.Ryall KA, Tan AC. Systems biology approaches for advancing the discovery of effective drug combinations. J Cheminform. 2015;7:7. doi: 10.1186/s13321-015-0055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Identification of similar binding sites to detect distant polypharmacology, (2013). [DOI] [PubMed]
- 179.Al-Ali H, Lee DH, Danzi MC, et al. Rational Polypharmacology: Systematically Identifying and Engaging Multiple Drug Targets To Promote Axon Growth. ACS Chem Biol. 2015. August 21;10(8):1939–51. doi: 10.1021/acschembio.5b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Somayaji MR, Przekwas AJ, Gupta RK. Combination Therapy for Multi-Target Manipulation of Secondary Brain Injury Mechanisms. Curr Neuropharmacol. 2018;16(4):484–504. doi: 10.2174/1570159X15666170828165711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fancellu G, Chand K, Tomas D, et al. Novel tacrine-benzofuran hybrids as potential multi-target drug candidates for the treatment of Alzheimer’s Disease. J Enzyme Inhib Med Chem. 2020. December;35(1):211–226. doi: 10.1080/14756366.2019.1689237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zeng M, Xiong Y, Safaee N, et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem Biol. 2020. January 16;27(1):19–31 e6. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 183.Jerabek J, Uliassi E, Guidotti L, et al. Tacrine-resveratrol fused hybrids as multi-target-directed ligands against Alzheimer’s disease. Eur J Med Chem. 2017. February 15;127:250–262. doi: 10.1016/j.ejmech.2016.12.048. [DOI] [PubMed] [Google Scholar]
- 184.Sheng R, Tang L, Jiang L, et al. Novel 1-Phenyl-3-hydroxy-4-pyridinone Derivatives as Multifunctional Agents for the Therapy of Alzheimer’s Disease. ACS Chem Neurosci. 2016. January 20;7(1):69–81. doi: 10.1021/acschemneuro.5b00224. [DOI] [PubMed] [Google Scholar]
- 185.Wu J, Brown M. Chapter 2 - Epigenetics and Epigenomics In: Hoffman R, Benz EJ, Silberstein LE, et al. , editors. Hematology (Seventh Edition): Elsevier; 2018. p. 17–24. [Google Scholar]
- 186.de Lera AR, Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin Epigenetics. 2016;8:105. doi: 10.1186/s13148-016-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Singh AN, Sharma N. Epigenetic Modulators as Potential Multi-targeted Drugs Against Hedgehog Pathway for Treatment of Cancer. The protein journal. 2019;38(5):537–550. doi: 10.1007/s10930-019-09832-9. [DOI] [PubMed] [Google Scholar]
- 188.Tomaselli D, Lucidi A, Rotili D, et al. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med Res Rev. 2020. January;40(1):190–244. doi: 10.1002/med.21600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cuadrado-Tejedor M, Garcia-Barroso C, Sanchez-Arias JA, et al. A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice. Neuropsychopharmacology. 2017. January;42(2):524–539. doi: 10.1038/npp.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.De Simone A, Milelli A. Histone Deacetylase Inhibitors as Multitarget Ligands: New Players in Alzheimer’s Disease Drug Discovery? ChemMedChem. 2019. June 5;14(11):1067–1073. doi: 10.1002/cmdc.201900174. [DOI] [PubMed] [Google Scholar]
- 191.Grisoni F, Merk D, Friedrich L, et al. Design of Natural-Product-Inspired Multitarget Ligands by Machine Learning. ChemMedChem. 2019. June 18;14(12):1129–1134. doi: 10.1002/cmdc.201900097. [DOI] [PubMed] [Google Scholar]
- 192.Allison M NCATS launches drug repurposing program. Nat Biotechnol. 2012. July 10;30(7):571–2. doi: 10.1038/nbt0712-571a. [DOI] [PubMed] [Google Scholar]
- 193.Chiotos K, Hayes M, Kimberlin DW, et al. Multicenter initial guidance on use of antivirals for children with COVID-19/SARS-CoV-2. J Pediatric Infect Dis Soc. 2020. April 22. doi: 10.1093/jpids/piaa045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.**.Harrison C Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. 2020. April;38(4):379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]; a latest demonstration of using drug repurposing to address COVID-19 pandemic.
- 195.Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017. January;16(1):19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hu Y, Bajorath J. Growth of ligand-target interaction data in ChEMBL is associated with increasing and activity measurement-dependent compound promiscuity. J Chem Inf Model. 2012. October 22;52(10):2550–8. doi: 10.1021/ci3003304. [DOI] [PubMed] [Google Scholar]
- 197.Southan C, Sitzmann M, Muresan S. Comparing the Chemical Structure and Protein Content of ChEMBL, DrugBank, Human Metabolome Database and the Therapeutic Target Database. Mol Inform. 2013. December;32(11–12):881–897. doi: 10.1002/minf.201300103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kim S Getting the most out of PubChem for virtual screening. Expert Opin Drug Discov. 2016. September;11(9):843–55. doi: 10.1080/17460441.2016.1216967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wassermann AM, Lounkine E, Hoepfner D, et al. Dark chemical matter as a promising starting point for drug lead discovery. Nat Chem Biol. 2015. December;11(12):958–66. doi: 10.1038/nchembio.1936. [DOI] [PubMed] [Google Scholar]
- 200.Bantscheff M, Eberhard D, Abraham Y, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007. September;25(9):1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 201.Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A. 2009. December 22;106(51):21984–9. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013. July 5;341(6141):84–7. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 203.Ernsberger P, Koletsky RJ, Kline DD, et al. The SHROB model of syndrome X: effects of excess dietary sucrose. Ann N Y Acad Sci. 1999. November 18;892:315–8. doi: 10.1111/j.1749-6632.1999.tb07806.x. [DOI] [PubMed] [Google Scholar]
- 204.Hye Khan MA, Kolb L, Skibba M, et al. A novel dual PPAR-gamma agonist/sEH inhibitor treats diabetic complications in a rat model of type 2 diabetes. Diabetologia. 2018. October;61(10):2235–2246. doi: 10.1007/s00125-018-4685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Imig JD, Walsh KA, Hye Khan MA, et al. Soluble epoxide hydrolase inhibition and peroxisome proliferator activated receptor gamma agonist improve vascular function and decrease renal injury in hypertensive obese rats. Exp Biol Med (Maywood). 2012. December;237(12):1402–12. doi: 10.1258/ebm.2012.012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bezard E, Imbert C, Gross CE. Experimental models of Parkinson’s disease: from the static to the dynamic. Rev Neurosci. 1998;9(2):71–90. doi: 10.1515/revneuro.1998.9.2.71. [DOI] [PubMed] [Google Scholar]
- 207.Schlegel A, Stainier DY. Lessons from “lower” organisms: what worms, flies, and zebrafish can teach us about human energy metabolism. PLoS Genet. 2007. November;3(11):e199. doi: 10.1371/journal.pgen.0030199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Da C, Zhang D, Stashko M, et al. Data-Driven Construction of Antitumor Agents with Controlled Polypharmacology. Journal of the American Chemical Society. 2019;141(39):15700–15709. doi: 10.1021/jacs.9b08660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Silver D, Schrittwieser J, Simonyan K, et al. Mastering the game of Go without human knowledge. Nature. 2017. October 18;550(7676):354–359. doi: 10.1038/nature24270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.