

Abstract
Effective treatment of advanced prostate cancer persists as a significant clinical need as only 30% of patients with distant disease survive to 5 years after diagnosis. Targeting signaling and tumor cell-immune cell interactions in the tumor microenvironment has led to the development of powerful immunotherapeutic agents, however the prostate tumor milieu remains impermeable to these strategies highlighting the need for novel therapeutic targets. In this study, we provide compelling evidence to support the role of the RON receptor tyrosine kinase as a major regulator of macrophages in the prostate tumor microenvironment. We show that loss of RON selectively in prostate epithelial cells leads to significantly reduced prostate tumor growth and metastasis and is associated with increased intratumor infiltration of macrophages. We further demonstrate that prostate epithelial RON loss induces transcriptional reprogramming of macrophages to support expression of classical M1 markers and suppress expression of alternative M2 markers. Interestingly, our results show epithelial RON activation drives upregulation of RON expression in macrophages as a positive feed-forward mechanism to support prostate tumor growth. Using 3D co-culture assays, we provide additional evidence that epithelial RON expression coordinates interactions between prostate tumor cells and macrophages to promote macrophage-mediated tumor cell growth. Taken together, our results suggest that RON receptor signaling in prostate tumor cells directs the functions of macrophages in the prostate tumor microenvironment to promote prostate cancer.
Keywords: RON receptor tyrosine kinase, macrophages, macrophage activation, tumor microenvironment, prostate cancer
Introduction
Prostate cancer is the second most common cancer and the second leading cause of cancer-related deaths among men in the United States (1). Although the majority of cases are clinically localized disease which responds well to mainstay treatments, such as surgery, radiation and androgen deprivation, advanced prostate cancer remains a significant challenge to effectively treat as the 5-year survival rate is only 30% in these patients (1). Tyrosine kinase inhibitors and immunotherapy agents have emerged as promising therapeutic strategies, however both have shown limited clinical efficacy against prostate cancer (1–3). In clinical trials, tyrosine kinase inhibitors did not demonstrate significant improvement in overall survival in prostate cancer patients and were associated with high discontinuation rates due to adverse events (2). Immunotherapy agents have had overwhelming success in several cancers, such as melanoma and lung cancer. However, these therapies have generated limited responses in prostate cancer, in part due to the markedly low immunogenicity of prostate tumors. Thus, it is imperative to investigate the underlying mechanisms that define the pathophysiology of prostate cancer to refine the current strategies and develop more effective treatments for advanced disease.
Tumor-associated macrophages (TAMs) represent a vital arm of the antitumor immune response and are known to regulate tumor growth and progression based on their activation status (4). M1 macrophages kill microorganisms and tumor cells and produce pro-inflammatory molecules, whereas M2 macrophages promote tissue repair and tumorigenesis and tune the inflammatory response (5). In prostate cancer, increased recruitment of M1 macrophages was observed in organ-confined disease, whereas M2 macrophages were dominant in invasive prostate tumors, suggesting that increased recruitment of M1 macrophages may be protective against prostate cancer progression (6). Furthermore, a recent study found that high infiltration of M2 macrophages in the prostate tumor microenvironment was associated with increased odds of lethal prostate cancer (7). Thus, regulation of macrophage activation could be exploited to enhance immunotherapeutic targeting of prostate cancer.
The RON receptor (MST1R) is a member of the Met family of receptor tyrosine kinases and is preferentially expressed on epithelial cells and macrophages (8, 9). RON is overexpressed in >90% of human prostate cancers, and high RON expression correlates with poor prognosis and progression to therapy-resistant disease (9–11). Our laboratory has established that the RON receptor plays a critical role in regulating prostate cancer growth and the tumor microenvironment. We found that ubiquitous ablation of RON signaling in the Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) murine model significantly reduces tumor growth, tumor cell survival, angiogenesis, and metastasis (12). We further demonstrated that RON signaling selectively in prostate epithelial cells is necessary and sufficient to drive prostate cancer (10, 13, 14).
Additional studies from our laboratory demonstrated that whole-body loss of RON signaling led to significantly increased TAM infiltration into prostate and breast tumors (15, 16). These tumors were associated with decreased intratumor staining of inducible nitric oxide synthase (iNOS), a marker of M1 macrophages, and increased staining of Arginase-1, characteristic of M2 macrophages, suggesting that RON modulates TAM functions to support tumor growth. RON signaling in macrophages has been established as an important negative regulator of inflammation, in part by promoting M2 macrophage activation, and our previous study found RON signaling in TAMs promotes macrophage Arginase-1 expression and drives prostate tumor growth (17–19). However, the role of epithelial-specific RON signaling in regulating immune cells in the tumor microenvironment remains unexplored.
Herein, we show that RON signaling in prostate epithelial cells is critical for prostate tumor growth and progression. Selective loss of RON signaling in prostate epithelial cells leads to significant reductions in tumor cell proliferation and survival, angiogenesis, and metastatic outgrowth in TRAMP mice. Additionally, epithelial RON loss leads to enhanced infiltration of TAMs and upregulated expression of M1 macrophage activation markers. Further, we show that prostate epithelial RON expression supports a feed-forward upregulation of RON expression in macrophages which in turn promotes macrophage-dependent tumor cell growth. Our findings present prostate epithelial RON as a crucial regulator of the macrophage antitumor immune response in prostate cancer and suggests RON as a promising target to combat advanced prostate cancer.
Materials and Methods
Mice.
Mice were maintained under specific pathogen-free conditions according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. Experimental protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee. The C57BL/6 transgenic adenocarcinoma of the mouse prostate (TRAMP) model was purchased from The Jackson Laboratory (20, 21). C57BL/6 mice with loxP sites flanking the tyrosine kinase domain of RON (exons 13 through 18) (RONF/F) have been previously characterized (22). C57BL/6 Probasin Cre mice (PbCre) (23) were crossed into RONF/F mice to generate mice with a prostate epithelial-specific deletion of the RON receptor (RONΔEpi). For study animals, RONF/F mice were crossed with TRAMP mice to generate the control group (RONF/F/TRAMP). RONF/F/TRAMP female mice were then crossed with RONΔEpi male mice to generate the experimental group (RONΔEpi/TRAMP males). Data were collected from male study mice that survived to 30 weeks of age. For tumor measurements, the prostate was removed and weighed. For proliferation studies, mice were injected with 150 mg/kg 5-Bromo-2-deoxyuridine (BrdU) 2 hours prior to sacrifice. Genotyping of transgenic mice was performed by PCR analysis (See Supplemental Methods). Primer sets for identification of RONF/F and RONΔEpi mice are listed in Supplemental Table 1.
Histopathology and Immunohistochemistry.
Mouse prostate and distal organs were fixed in 10% formalin, paraffin embedded, and cut into 4 μm sections. Sections were stained with hematoxylin and eosin, BrdU (BrdU Staining Kit, Invitrogen), TUNEL (In Situ Apoptosis Detection Kit, EMD Millipore), F4/80 (14-4801-85, eBiosciences), iNOS (610328, BD Transduction Labs), and CD31 (557355, BD Pharmingen) as previously described (16). For histopathology, images of at least 3 fields per slide were captured for each prostate tumor, and prostate glands in each field were categorized as previously described (13, 24). At least 10 total glands were evaluated per prostate.
Metastasis.
For metastatic analysis, mice were examined upon necropsy for gross metastatic foci, and the mesenteric lymph node and lung were removed, serially sectioned (3–5 serial sections per organ, 100 μm apart), and examined by hematoxylin and eosin staining.
Tumor Associated Macrophage (TAM) Isolation.
Prostate tumors taken from 25–40 week old RONF/F/TRAMP and RONΔEpi/TRAMP mice were mechanically dissociated then digested and filtered to obtain single-cell suspensions as previously described (19, 25). TAMs were enriched from the suspension using CD11b-coated magnetic beads (Miltenyi Biotech) per manufacturer’s instructions.
Cells and Reagents.
Cells were maintained in DMEM with 10% Cosmic Calf Serum and 0.2% gentamicin at 37°C and 5% CO2 unless noted. Murine TRAMP C2RE3 cells were obtained from Dr. Zhongyun Dong as previously described (19, 26). shRON and shNT TRAMP C2RE3 cells were generated by infection with murine RON shRNA lentivirus (TRCN0000023547, Sigma-Alrdich) or empty vector (pLKO.1), respectively. Cells were cultured biweekly and stocks of cells were passaged at least twice before use in experiments. Macrophage colony stimulating factor (M-CSF)-containing culture supernatants from murine CMG14–12 cells were obtained from Dr Yi Zheng (27) as previously described (25, 28). Supernatants were ccollected and used for bone marrow-derived macrophage (BMDM) differentiation. To generate BMDMs, bone marrow cells were isolated from wild-type C57BL/6 mice as previously described (25). M-CSF was added at a 1:10 dilution for at least 6 days to generate differentiated BMDM. After differentiation, BMDMs were harvested using Accutase (Innovative Cell Technologies) and used for experiments. All murine lines were obtained in 2012 from the sources noted with cells expanded and initial stocks generated following 1–3 initial passages. Cells were most recently confirmed negative for mycoplasma through PCR on 2/26/2020 at Cincinnati Children’s Hospital Medical Center. Authentication of the murine cell lines was not performed.
Quantitative Real-Time PCR (qRT-PCR).
RNA was extracted using TRIzol (Invitrogen) per manufacturer’s instructions. Extraction of RNA from macrophages was performed using Direct-zol RNA Mini-Prep Kit (Zymo Research). cDNA from 0.5–1 μg of RNA per sample was prepared using the High-Capacity cDNA Reverse Transcriptase kit (Applied Biosystems). Data were normalized to 18S reference gene and analyzed by ΔΔCT (29). Primer sequences are listed in Supplemental Table 1.
Western Blot Analyses.
Cells were homogenized in RIPA buffer as previously described (30). Antibodies for analyses included: RON (1:200, sc-322, Santa Cruz Biotechnology), Arginase-1 (1:1000, 610708, BD Transduction Labs), p-STAT3 (Y705) (1:800, 9145S, Cell Signaling Technology), STAT3 (1:500, 9139S, Cell Signaling Technology), and C4-actin (ACTIN) (1:40,000, Cincinnati Children’s Hospital Medical Center; 1:5000, sc-47778, Santa Cruz Biotechnology).
Flow Cytometry Analyses.
Flow cytometry analysis for proliferation or apoptosis was performed on TRAMP C2RE3 cells cultured in complete media or cultured in serum free media for 48 hours. For proliferation, cells were labeled with 10μM BrdU for 4 hours, stained with BrdU-APC (17-5071-41, eBiosciences) following the manufacturer’s instructions. For apoptosis, cells were stained with Annexin V-APC (550474, BD Biosciences) and 50 ng Propidium Iodide (556463, BD Biosciences) per manufacturer’s instructions. Flow cytometry was performed and analyzed using the LSRFortessa and FACS Diva software (BD Biosciences).
Transwell Co-culture Assay.
0.5×106 TRAMP C2RE3 cells were seeded in complete media onto Transwell inserts with 0.4 μm pore size (Corning) in 6-well plates. Four hours later, media was replaced with 1.5 mL of DMEM supplemented with 0.2% gentamicin (serum free media), and inserts were transferred to plates containing 0.25×106 BMDM per well in 2.6 mL serum free media. BMDMs plated in serum free media alone served as a control for transcriptional analyses. Cells were co-cultured up to 18 hours, then BMDMs were harvested for RNA analyses.
Conditioned Media.
1×106 TRAMP C2RE3 cells were seeded in a 6-well plate and incubated in 1.5mL of serum free media for 24 hours. Following incubation, the culture supernatants were collected, centrifuged to remove cells, and diluted 1:1 with serum free media prior to being placed on BMDMs.
3D Cell Culture Assays.
3D cell culture assays were performed as described previously (31) using a 1:1 ratio of macrophages to tumor cells based on previous 3D co-culture models (32, 33). Briefly, 2×104 TRAMP C2RE3 cells alone or with 2×104 BMDMs were plated on top of 1.0% agarose in duplicate wells of 6 well plates in 3mL of serum-containing media. After 10 to 14 days, images of spheres were taken using a Zeiss Axiovert S100TV inverted microscope (Carl Zeiss Microscopy) and spheres > 25 μm in diameter were measured using ImageJ software. The 25μm threshold was established based on the average sphere size obtained for the control cells.
Statistical Analysis.
Incidence of metastasis was analyzed using Fisher’s exact test. All other data are expressed as mean ± standard error of the mean (SEM). Unless noted, all data represent mean values from at least 3 independent experiments. Statistical significance was determined by performing Student’s t-test for unpaired samples or ANOVA for comparison of multiple groups using GraphPad Prism software. Welch’s correction was used for samples with unequal variances as determined by the F test (GraphPad Software). Significance was set at P < 0.05.
Results
RON loss in prostate epithelial cells significantly reduces prostate tumor burden and progression in TRAMP mice
We have previously shown that whole-body loss of RON signaling abrogates prostate tumor growth and metastasis in TRAMP mice (12). These studies were the first to establish RON in the development and progression of prostate cancer. We next sought to interrogate the mechanistic contributions of prostate epithelial-specific RON signaling to prostate cancer by generating RONF/F/TRAMP and RONΔEpi/TRAMP mice. Of note, no appreciable histological differences were observed in the prostates of TRAMP-negative RONF/F and RONΔEpi mice, suggesting prostate epithelial Ron loss does not lead to overt abnormalities in normal prostate development (Supplemental Figure S1). Prostate tumors were harvested from RONF/F/TRAMP and RONΔEpi/TRAMP mice at 30 weeks of age to examine tumor burden. RONΔEpi/TRAMP mice exhibited significantly reduced prostate tumor growth compared to controls (Figure 1A). Prostates from several RONΔEpi/TRAMP mice (n= 6) that were over 30 weeks of age (mean of 34.5 weeks, with a range of 32–40 weeks) continued to exhibit reduced tumor growth (mean weight, 0.24g) compared to control mice at 30 weeks (mean weight, 0.38g, P=0.05). mRNA analysis confirms significantly reduced RON expression in prostate tumors from RONΔEpi/TRAMP mice compared to control tumors (Figure 1B).
Figure 1. RON loss selectively in the prostate epithelium of TRAMP mice reduces prostate tumor burden and metastasis.
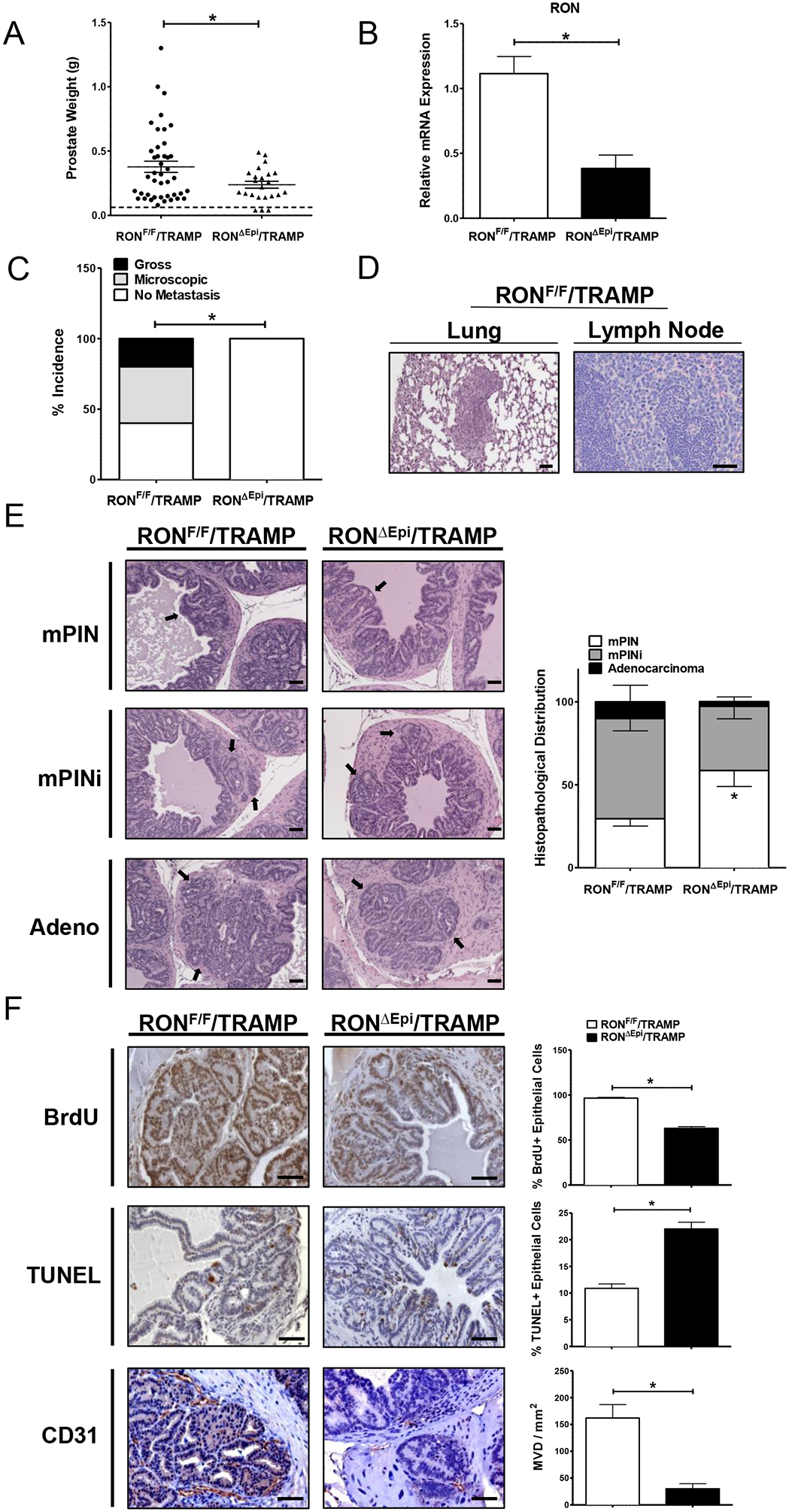
A) Prostate weights collected at 30 weeks from RONF/F/TRAMP (n=42) and RONΔEpi/TRAMP mice (n=23). Each dot represents a single prostate. Dotted horizontal line represents the average weight of prostates from TRAMP-negative mice. B) qRT-PCR analysis for RON mRNA expression in prostate tumors from RONF/F/TRAMP (n=9) and RONΔEpi/TRAMP mice (n=7). C) Percent incidence of metastasis in mice from each genotype. D) Representative images of hematoxylin and eosin (H&E) staining of lungs from RONF/F/TRAMP and RONΔEpi/TRAMP mice. E) Representative images of H&E staining of prostate tumors from RONF/F/TRAMP and RONΔEpi/TRAMP mice with quantification of histopathological distribution (n=4–6 mice per genotype). mPIN=mouse prostate intraepithelial neoplasia, mPINi=mPIN with invasion, or Adenocarcinoma. Arrows indicate characteristics of the assigned histopathological grade. F) Representative images and quantification of immunohistochemical staining of prostate tumors from RONF/F/TRAMP and RONΔEpi/TRAMP mice for BrdU, TUNEL, and CD31 (n=3 mice per genotype). Scale bars= 100 μm. *P < 0.05.
To assess the effect of prostate epithelial RON loss on metastatic burden, we determined the incidence of gross or microscopic metastatic outgrowth in lymph nodes and lungs from RONF/F/TRAMP and RONΔEpi/TRAMP mice in a cohort of 10 mice per group with similar prostate tumor weights (Supplemental Figure S2). Metastases were observed in 60% of RONF/F/TRAMP mice wherein 20% exhibited gross metastatic lesions (Figure 1C). Strikingly, no metastases were detected in tumor size-matched RONΔEpi/TRAMP mice. Representative images of metastases from the lung and lymph node of RONF/F/TRAMP mice are depicted in Figure 1D.
To determine the contribution of epithelial RON expression to prostate tumor progression, we characterized histopathology in prostate tumors from RONF/F/TRAMP and RONΔEpi/TRAMP mice and found a significant difference in the presence of prostate intraepithelial neoplasia (mPIN) in tumors from RONΔEpi/TRAMP mice compared to control tumors (Figure 1E). Correspondingly, we observed reduced local invasion (mPINi) in tumors from RONΔEpi/TRAMP mice compared to controls suggesting that epithelial RON loss leads to a less invasive phenotype in prostate tumors. We examined tumor cell proliferation and death and found significantly decreased proliferation and increased apoptosis in prostate tumor cells in RONΔEpi/TRAMP mice compared to control mice (Figure 1F). Interestingly, epithelial RON loss leads to a 2-fold increase in apoptosis compared to a 66% reduction in proliferation, suggesting RON in tumor cells is a major promoter of prostate tumor cell survival. Consistent with our previous work showing RON in prostate cancer cells is critical for microvessel formation and angiogenic chemokine production to promote prostate tumor growth (10), we observed a striking decrease in microvessel density of over 5-fold in tumors from RONΔEpi/TRAMP mice compared to control mice as measured by CD31 staining (Figure 1F).
Loss of RON expression in prostate epithelial cells promotes the influx and M1 activation of macrophages in the tumor microenvironment
Given that ubiquitous loss of RON expression or loss of RON expression selectively in macrophage/myeloid cells leads to changes in the tumor microenvironment (14–16, 18, 19), we next sought to examine the effects of prostate epithelial RON loss on macrophages in the prostate tumor microenvironment of TRAMP mice. We observed a significant increase in F4/80+ macrophage infiltration into the prostate tumors of RONΔEpi/TRAMP mice compared to controls, and this was associated with an increase in intratumor staining of the M1 macrophage marker iNOS (Figure 2A). To verify the effect of epithelial RON loss on macrophage activation in vivo, we isolated CD11b+ tumor-associated macrophages (TAMs) from RONF/F/TRAMP and RONΔEpi/TRAMP mice and analyzed the expression of a panel of well-established markers of M1 (CXCL9, INOS, IL-12B, IFNγ, TNFα, VEGFa) and M2 macrophages (ARG1, IL-10, TGFβ) (Figure 2B) (5). Indeed, we observed increased expression of multiple M1 markers and decreased expression of M2 markers in TAMs from RONΔEpi/TRAMP mice compared to control mice. Overall, our data suggests that prostate epithelial RON loss leads to increased infiltration of M1 TAMs in the prostate tumor microenvironment.
Figure 2. Epithelial RON loss leads to increased infiltration and M1 activation of macrophages in the tumor microenvironment.
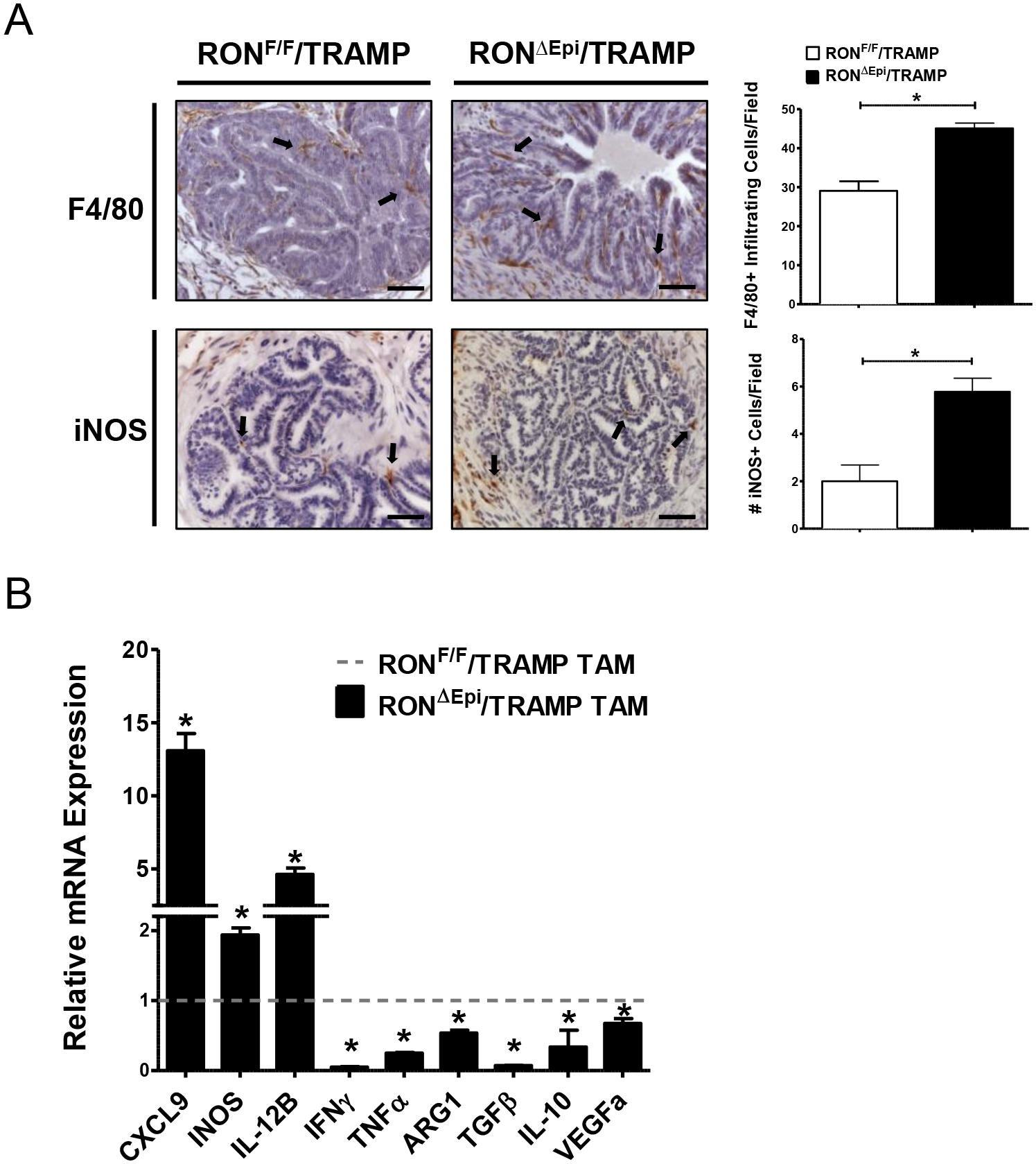
A) Representative images and quantification of immunohistochemical staining of prostate tumors from RONF/F/TRAMP and RONΔEpi/TRAMP mice for F4/80 and iNOS (n=3 mice per genotype). Scale bars= 100 μm. B) qRT-PCR analyses for CXCL9, INOS, IL-12B, IFNγ, TNFα, ARG1, TGFβ, IL-10, and VEGFa mRNA expression in TAMs from RONF/F/TRAMP and RONΔEpi/TRAMP mice. Fold change in RONΔEpi/TRAMP TAMs over controls is shown. Dotted horizontal line represents the values of RONF/F/TRAMP TAMs, which were set to 1. Data represent mean values of 3 independent isolations of TAMs from pooled prostate tumors from each genotype. *P < 0.05.
Prostate epithelial RON loss induces M1 marker expression in macrophages in vitro through secretion of soluble mediators
We next sought to interrogate the functional consequences of prostate epithelial RON loss on macrophage activation marker expression in vitro. We utilized the TRAMP C2RE3 prostate cancer cell line and used stable shRNA knockdown to decrease RON expression at the transcript (Figure 3A) and protein level (Figure 3B). We employed an in vitro transwell co-culture system to examine the interaction between Control or shRON TRAMP C2RE3 cells and bone marrow-derived macrophages (BMDM) isolated from syngeneic wild type mice. Consistent with our in vivo data, our results show RON depletion in tumor cells is sufficient to induce the expression of M1 macrophage markers and suppress expression of M2 macrophage markers compared to BMDM incubated with Control cells (Figure 3C–D). Conditioned media from Control and shRON TRAMP C2RE3 cells produced similar results when cultured with BMDMs (Figure 3E), demonstrating epithelial RON expression regulates macrophage activation through secretion of one or more soluble mediators from the tumor cells.
Figure 3. Epithelial RON loss induces M1 marker expression and suppresses M2 marker expression in macrophages through the production of tumor cell-derived soluble mediators.
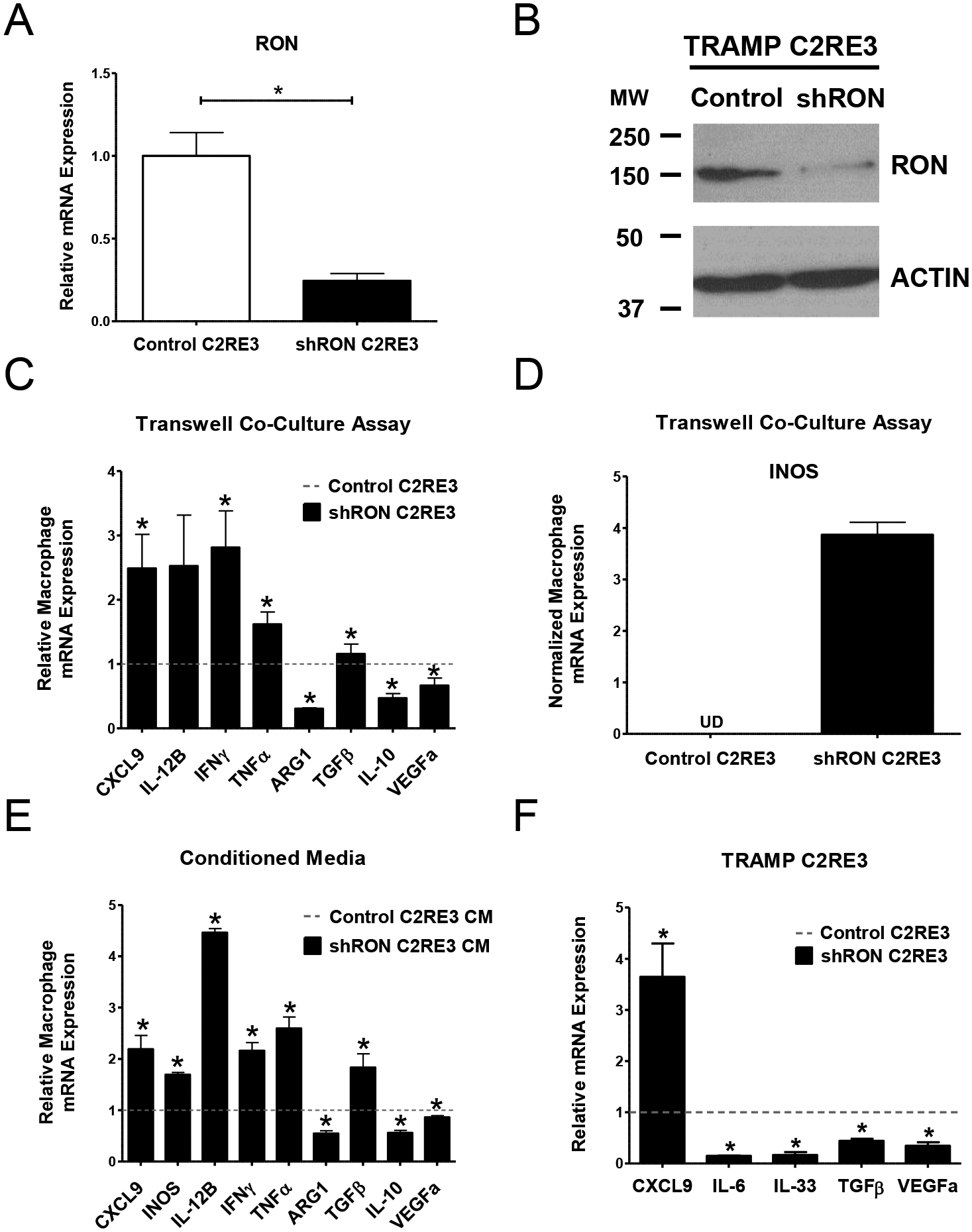
A) qRT-PCR analyses for RON mRNA expression in Control TRAMP C2RE3 and shRON TRAMP C2RE3 cells. B) Western blot images showing the expression of RON in Control and shRON TRAMP C2RE3 cells. ACTIN is used as a loading control. MW, molecular weight (kDa). C-E) qRT-PCR analyses for CXCL9, INOS, IL-12B, IFNγ, TNFα, ARG1, TGFβ, IL-10, and VEGFa mRNA expression in BMDM either co-cultured with Control or shRON TRAMP C2RE3 cells (C-D) or cultured in conditioned media from Control or shRON TRAMP C2RE3 cells (E). The fold change in BMDMs co-cultured with or treated with conditioned media from shRON TRAMP C2RE3 cells over Control TRAMP C2RE3 cells is shown in C and in E. Dotted horizontal lines represent the values of macrophages co-cultured with or treated with conditioned media from Control TRAMP C2RE3 cells, which were set to 1. UD= undetectable. F) qRT-PCR analyses for CXCL9, IL-6, IL-33, TGFβ, and VEGFa mRNA expression in Control and shRON TRAMP C2RE3 cells. Fold change in shRON TRAMP C2RE3 cells over Control cells is shown. Dotted horizontal line represents the values of Control TRAMP C2RE3 cells, which were set to 1. *P < 0.05.
Given the significant transcriptional changes reflected in macrophages following co-culture with or conditioned media from RON modulated TRAMP C2RE3 cells, we next examined Control and shRON TRAMP C2RE3 cells for the expression of several important cytokines known for modulating macrophage recruitment and activation. As depicted in Figure 3F, we observed increased CXCL9 expression and decreased expression of IL-6, IL-33, VEGFa, and TGFβ in shRON TRAMP C2RE3 cells compared to Control cells, suggesting these molecules as potential soluble effectors which may be involved in recruiting and transcriptionally reprogramming macrophages in this experimental setting.
RON expression in prostate epithelial cells upregulates macrophage RON expression in a paracrine manner
Unexpectedly, we found significant induction of RON gene expression in macrophages co-cultured with or treated with conditioned media from RON-proficient tumor cells compared to macrophages exposed to RON-deficient tumor cells (Figure 4A). Gene expression levels of RON were similar between macrophages exposed to RON-deficient tumor cells and untreated macrophages (data not shown). To corroborate these findings in vivo, we performed Western blot analyses of TAMs isolated from RONF/F/TRAMP and RONΔEpi/TRAMP mice. We observed markedly higher expression of RON in TAMs from control mice, coupled with higher Arginase-1 protein expression (Figure 4B). A change in status of STAT3, a common marker of M2 macrophage activation, in TAMs from RONΔEpi/TRAMP mice, was also observed (Supplementary Figure S3) (5, 34–39).
Figure 4. Prostate epithelial RON expression promotes RON expression in macrophages.
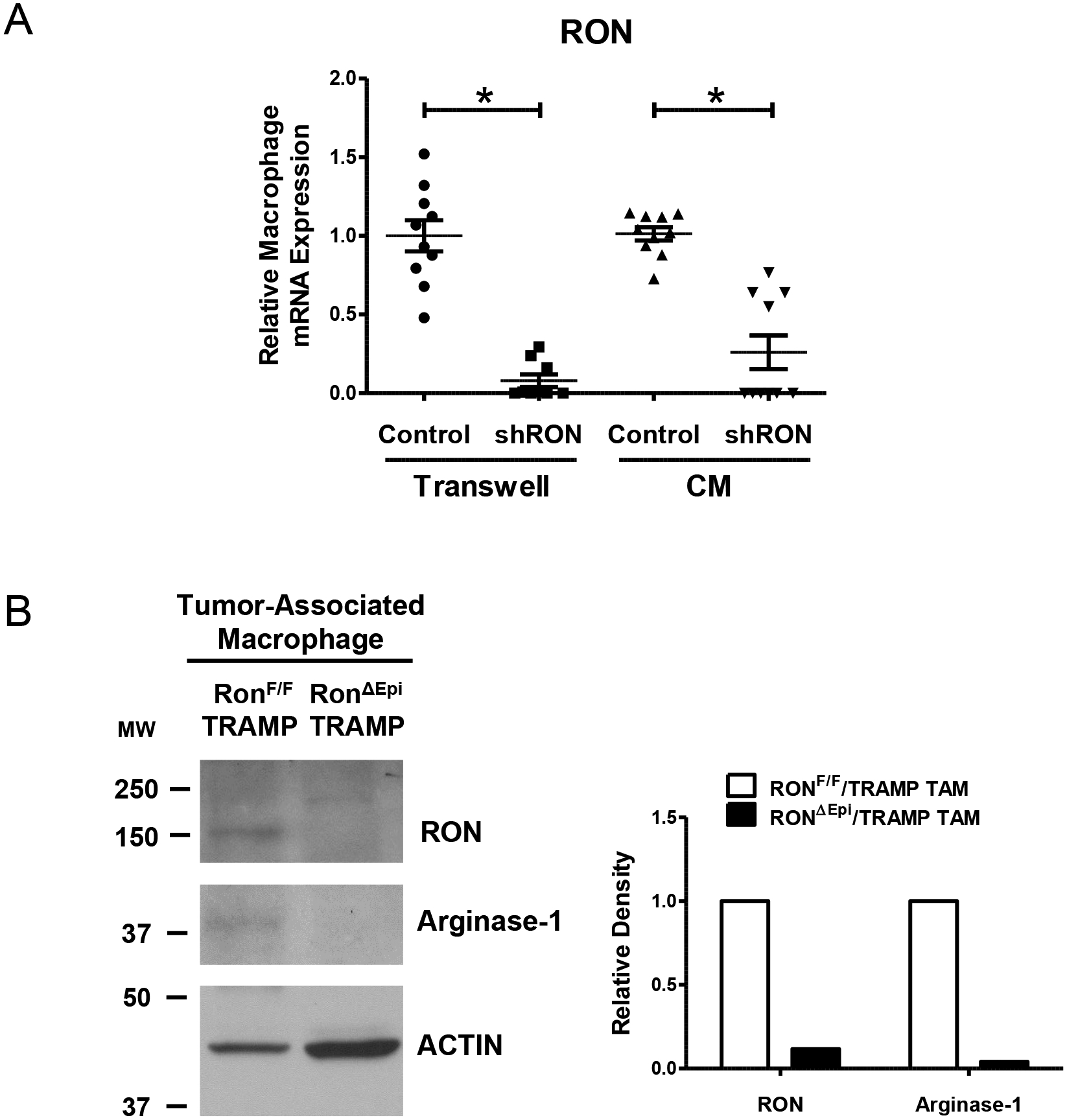
A) qRT-PCR analyses for RON mRNA expression in BMDM either co-cultured with Control or shRON TRAMP C2RE3 cells or cultured in conditioned media (CM) from Control or shRON TRAMP C2RE3 cells. *P < 0.05. B) Western blot images and densitometric quantitation of the expression of RON and Arginase-1 in lysates of TAMs isolated from pooled prostate tumors (n= 2–4 mice per genotype). ACTIN is used as a loading control. Quantitation of RON and Arginase-1 expression was normalized to ACTIN. MW, molecular weight (kDa).
Prostate epithelial RON expression is required for macrophages to promote tumor cell growth in 3D co-cultures in vitro
Due to the significant differences in BrdU and TUNEL tumor cell staining observed in the prostate tissue from RONF/F/TRAMP and RONΔEpi/TRAMP mice, we sought to examine the role of prostate epithelial RON expression in tumor cell proliferation and apoptosis. Following RON knockdown, no appreciable effect on proliferation or cell death was observed between Control and shRON TRAMP C2RE3 cells in vitro when grown in 2D culture under serum-containing (COMPLETE) or serum-starved (SFM) conditions (Figure 5A–B).
Figure 5. Prostate epithelial RON expression leads to increased 3D tumor cell growth in co-culture with macrophages.
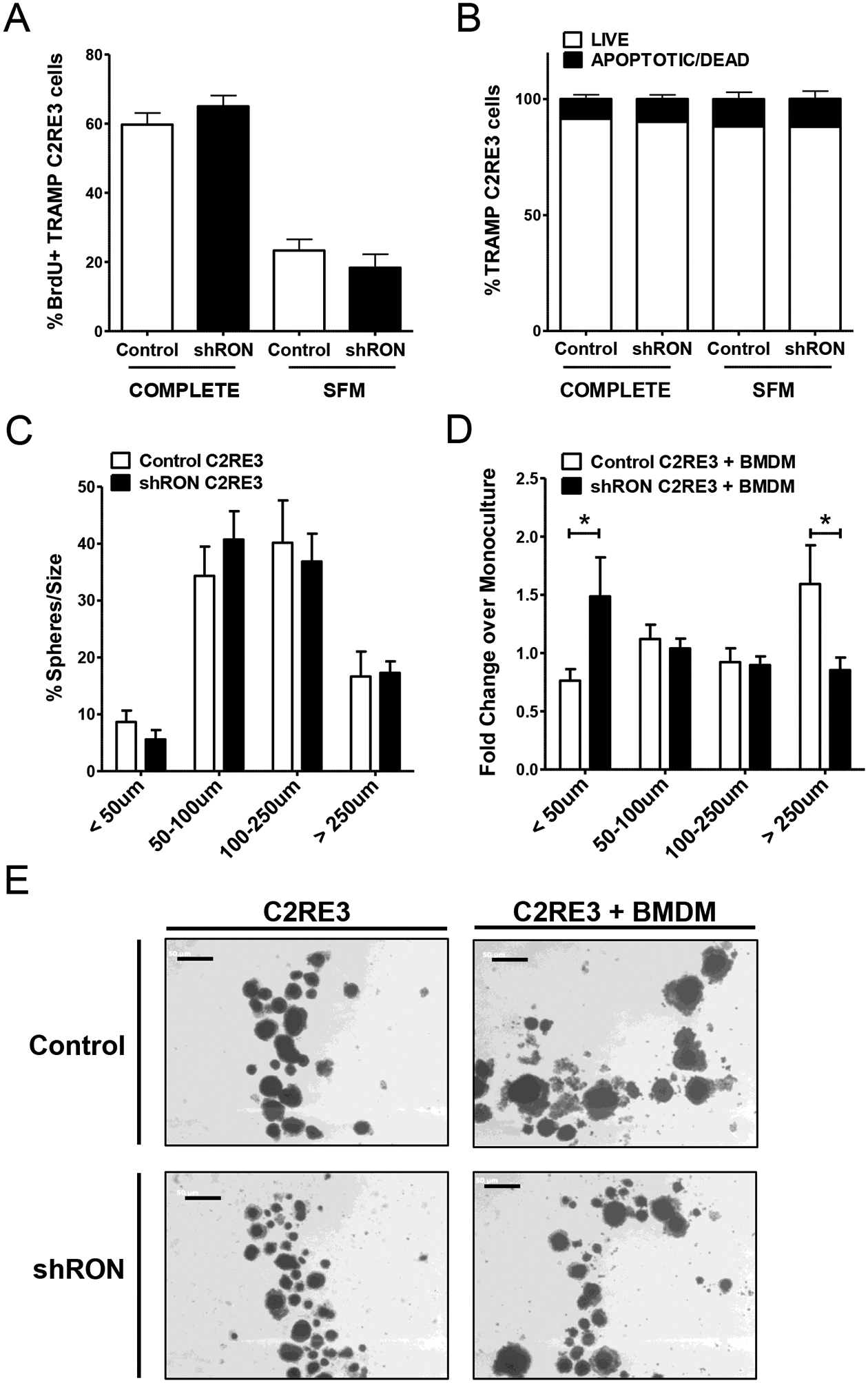
A) Percentage of BrdU positive TRAMP C2RE3 cells as determined by flow cytometry from cells cultured in COMPLETE or serum free media (SFM) in 2D cell culture conditions. B) Percentage of TRAMP C2RE3 cells cultured in complete or serum free media (SFM) in 2D cell culture conditions subjected to Annexin V/Propidium Iodide (PI) staining; Annexin V-/PI- (LIVE), Annexin V+/PI-, Annexin V+/PI+, and Annexin V-/PI+ (APOPTOTIC/DEAD). C) Percent of TRAMP C2RE3 spheres in each size classification. D) Fold change in the percent of spheres in each size classification in co-cultures of BMDM and Control or shRON TRAMP C2RE3 cells over the respective monocultures. *P < 0.05. E) Representative images of TRAMP C2RE3 spheres in monoculture or co-culture with BMDM (scale bar=500 μm).
Based on our data showing prostate epithelial RON promotes M2 marker expression in macrophages and that M2 macrophages are known to promote tumor cell proliferation (40), we tested the effect of macrophages on TRAMP C2RE3 cell growth using 3D cell culture assays to mimic in vivo tumor conditions and to facilitate cell-cell interactions. While there was no difference in the size distribution of spheres between Control or shRON TRAMP C2RE3 cells alone in 3D (Figure 5C), significantly more large spheres (> 250 μm) were found when Control cells were co-cultured with macrophages compared to shRON TRAMP C2RE3 cell co-cultures (Figure 5D–E). Conversely, significantly more small spheres (< 50 μm) were found in shRON TRAMP C2RE3 cell co-cultures compared to Control co-cultures, suggesting that epithelial RON expression is critical for promoting tumor cell interactions with macrophages that lead to enhanced growth. Overall, our findings support a working model wherein RON expression in prostate epithelial cells induces macrophage RON expression and supports M2 macrophage activation to promote prostate cancer (Figure 6).
Figure 6. Working model depicting the mechanism by which epithelial RON signaling regulates macrophages in the tumor microenvironment to promote prostate cancer.

Schematic of the tumor cell-macrophage communication network wherein RON signaling in prostate epithelial cells promotes RON expression and M2 activation in macrophages through the production of RON-dependent tumor cell-secreted soluble factors. Consequently, the RON-educated macrophages help support prostate tumor growth. The net effect of this feed-forward loop directs tumor cell metastasis and angiogenesis and supports an immunosuppressive tumor microenvironment.
Discussion
The RON receptor is a central regulator of the prostate tumor microenvironment yet the epithelial-specific roles of RON in prostate cancer remain poorly understood. In this study, we interrogated the consequences of prostate epithelial RON loss in the TRAMP mouse model. Similar to our previous study involving whole-body deletion of RON in TRAMP mice (12), our new data indicate that loss of prostate epithelial-specific RON signaling attenuates tumor growth and is critical for prostate cancer metastasis (Figure 1). Overexpression of RON in prostate epithelial cells has been shown to be sufficient to induce the development of advanced histopathology in the prostate, leading to prostate adenocarcinoma (13). Consistent with this data, our current studies show that loss of prostate epithelial RON in a spontaneous prostate cancer mouse model significantly reduces the presence of advanced prostate histopathology (Figure 1). Combined, these data support a crucial role for RON in mediating prostate cancer aggressiveness.
While no changes in tumor cell proliferation were observed in TRAMP mice with whole-body loss of either RON or its ligand, HGFL (12, 15), our data showed significantly decreased proliferation in mice with prostate epithelial RON loss compared to control mice (Figure 1). These findings suggest RON signaling in other cell types may contribute to the regulation of prostate tumor cell proliferation, reinforcing the importance of dissecting the cell type-specific roles for RON in the prostate tumor microenvironment. Interestingly, our data revealed marked increases in tumor cell apoptosis and significantly reduced tumor angiogenesis in TRAMP mice with prostate epithelial RON loss (Figure 1). These results are similar to those observed in the context of whole-body HGFL or RON loss (12, 15), and thus provide evidence to support the role of prostate epithelial RON as a key promoter of tumor cell survival and neovascularization in prostate cancer.
Targeting the complex network of cell-cell communications in the tumor microenvironment has emerged as one of the frontiers of cancer research. TAMs coordinate a majority of these communications as they govern many processes that influence tumor growth and metastasis, such as angiogenesis, immune cell recruitment and activation, and tumor cell migration and invasion, depending on their activation status (4, 5, 40, 41). Therefore, identifying and targeting regulators of TAM activation holds significant potential for gaining more effective control of the tumor microenvironment to direct it against tumor growth and progression. In this report, we demonstrate the novel role of epithelial-specific RON receptor signaling in regulating TAM infiltration and activation. Loss of epithelial RON leads to increased infiltration of F4/80-positive macrophages into the stroma directly adjacent to the prostate epithelial cells, whereas macrophages were mostly localized to the stroma surrounding the glands in control tumors (Figure 2). Similar results were observed in tumors from TRAMP mice with whole-body HGFL loss (15).
In addition to increased intratumor infiltration of macrophages, we show that prostate epithelial RON loss modulates macrophage activation (Figure 2–3). Interestingly, our studies show that prostate epithelial RON loss leads to an upregulation of CXCL9 in macrophages, although the extent of this increase varied with the use of TAMs versus BMDM in our studies. Paradoxically, we found downregulation of M2 marker TGFβ in TAMs from epithelial RON-deficient mice, but this was not observed in BMDMs exposed to RON-deficient tumor cells. Similarly, epithelial RON loss leads to increased expression of IFNγ and TNFα in macrophages in vitro yet opposite trends were observed in TAMs. The differences observed between TAMs and BMDM could be partially attributed to inherent differences in the origins of the two macrophage populations. Recent evidence demonstrates that TAMs derive from embryonic-derived tissue resident macrophages and monocyte precursors recruited from the bone marrow, can exhibit distinct cytokine production profiles and varying capacities to be activated to an M1 or M2 phenotype based on marker gene expression (42–44). TAMs are highly plastic cells that respond to diverse cues from numerous cell types within the tumor microenvironment, thus macrophage activation occurs over a continuum in vivo and TAMs can express markers of both activation states (5, 41). Our in vitro experiments do not fully recapitulate the heterogeneous network of cell types and signals in the prostate tumor microenvironment, and instead, represent a purified system testing the direct communication between tumor cells and macrophages. Therefore, we anticipate that epithelial RON may enlist multiple cell types to modulate TAM cytokine production and ultimately promote M2 activation in vivo.
Overall, our data show that prostate epithelial RON loss leads to increased M1 marker expression and suppressed M2 marker expression in macrophages through a paracrine interaction. These findings are analogous with previous literature that has established macrophage-intrinsic RON expression as a key regulator of macrophage activation (17, 18). Similar to these studies on RON-deficient macrophages, we found that epithelial RON loss induces increased iNOS expression in macrophages and attenuates macrophage Arginase-1 expression at the transcript and protein level (Figure 3). Thus, our data demonstrate that RON expression in prostate epithelial cells is necessary to support M2 macrophage activation.
We also noted tumor cell autonomous changes in cytokine expression associated with RON loss including increased CXCL9 and decreased IL-6, IL-33, and TGFβ expression (Figure 3). These molecules have documented roles in directing infiltration and activation of macrophages (5, 45). Recent literature also points to a role for CXCL9 in prostate cancer progression (46), although the upregulation of CXCL9 observed in both tumor cells and macrophages upon epithelial RON loss suggests this chemokine as a potential mediator of heightened M1 tumor-killing macrophage infiltration in prostate tumors lacking epithelial RON signaling. IL-33, a member of the IL-1 signaling family and a promoter of Th2 immune responses, has emerged as an attractive anti-tumor target. Inhibition of IL-33 has recently been shown to abrogate M2 activation of TAMs leading to reduced tumor growth in a preclinical lung cancer model (47). Thus, our data present these immunomodulatory cytokines as potential soluble mediators of macrophage recruitment and activation regulated by prostate epithelial RON and warrant further investigation into their specific contributions in prostate cancer.
In regards to angiogenesis, our results show that epithelial RON loss leads to reduced VEGFa expression in both tumor cells and macrophages (Figure 2–3). VEGFa is a marker of M2 activation as well as a key cytokine produced by TAMs to promote angiogenesis (4, 5, 40, 41). Our data show that the difference in VEGFa expression in much greater (at least 2-fold) in RON-deficient tumor cells compared to that observed in macrophages conditioned by RON-deficient tumor cells, suggesting that loss of epithelial RON is more detrimental to angiogenic chemokine production in prostate tumor cells than in macrophages. These data support our previous study which demonstrated that RON in prostate cancer cells positively regulates angiogenic chemokines and recruits endothelial cells to promote prostate tumor growth and vascularization (10). In contrast, loss of myeloid-specific RON expression had no appreciable effect on microvessel density in prostate tumors (19). Taken together, these findings lead us to speculate that RON signaling in tumor cells, rather than macrophages, is a major driver of angiogenesis in prostate cancer.
Notably, our experiments revealed a novel discovery that prostate epithelial RON is required for the corresponding upregulation of RON expression in macrophages (Figure 4). Prior work from our laboratory demonstrated that loss of RON signaling in TAMs led to significantly increased macrophage tumor infiltration, increased iNOS expression and reduced Arginase-1 expression in macrophages, and prostate tumor growth inhibition (19). Therefore, we posit that RON signaling in prostate epithelial cells promotes M2 macrophage polarization in part by enhancing macrophage RON expression, leading to reduced antitumor immune responses and the promotion of prostate tumor growth. Consistent with our findings, a recent study showed reciprocal HGFL-RON-signaling in pancreatic cancer was associated with altered macrophage polarization and increased tumor growth (48), thus lending support to the functional importance of a RON-driven positive regulatory loop across multiple epithelial cancers (Figure 6).
Loss of RON expression in TRAMP C2RE3 cells did not affect tumor cell proliferation or apoptosis in vitro, however co-culture with BMDMs induced growth of Control TRAMP C2RE3 spheres while diminishing growth of shRON TRAMP C2RE3 spheres (Figure 5). Combined with our results showing epithelial RON modulates macrophage activation, these findings suggest that prostate epithelial RON educates macrophages to directly promote tumor growth in part through M2 macrophage activation. This is the first report to show that macrophages help support tumor cell growth in a manner that is dependent on epithelial RON expression. Given the critical role of RON in driving castration-resistant prostate cancer (CRPC) (31), the importance of RON-regulated macrophage functions in CRPC certainly warrants exploration.
Collectively, the results of our study implicate RON in a novel feed-forward regulatory mechanism within the interaction between prostate tumor cells and TAMs that ultimately coordinates an immunosuppressive and tumor-supporting prostate tumor microenvironment (Figure 6). We provide novel insights into the role of epithelial RON in prostate cancer and the utility of RON as a novel immunotherapeutic target. Increased infiltration and antitumor activation of macrophages observed with epithelial RON inhibition could be harnessed to overcome the markedly low immune infiltrate characteristic of prostate cancer. Furthermore, tumor infiltration of M1/M2 macrophages represents a potential biomarker for responsiveness to RON targeting and could be used in clinical trials to monitor efficacy of anti-RON therapy. Thus, prostate epithelial RON inhibition represents an attractive novel therapeutic strategy for multifaceted reprogramming of the prostate tumor microenvironment to attack prostate tumor growth and progression. Targeting RON in prostate epithelial cells holds significant potential for enhancing the efficacy of current therapies to effectively treat advanced prostate cancer.
Supplementary Material
Implications:
Epithelial RON is a novel immunotherapeutic target that is responsible for directing the macrophage antitumor immune response to support prostate tumor growth and progression.
Acknowledgements
This work was supported in part by the National Institutes of Health Grants T32CA117846 (SEW, CS, NEB, JV), R01CA125379 (SEW), F31CA228425 (CS, SEW), R01CA239697 (SEW) and F31CA200390 (NEB, SEW); United States Department of Veterans Affairs research grant 1IOBX000803 (SEW); and DOD PC101840 (JV, SEW). We would like to thank Lauren Beckmeyer, Brian Hunt, Glenn Doerman, Delia Miller, Andrew Paluch, and Drs. Vidjaya Premkumar Letchoumy and Jennifer Bourn for assistance with this project.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Society AC. Prostate Cancer: American Cancer Society; 2019. [Available from: https://www.cancer.org/cancer/prostate-cancer.html.
- 2.Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J Clin Oncol. 2016;34(25):3005–13. [DOI] [PubMed] [Google Scholar]
- 3.Graff JN, Chamberlain ED. Sipuleucel-T in the treatment of prostate cancer: an evidence-based review of its place in therapy. Core Evid. 2014;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Frontiers in oncology. 2018;8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33(19):2423–31. [DOI] [PubMed] [Google Scholar]
- 7.Erlandsson A, Carlsson J, Lundholm M, Fält A, Andersson S-O, Andrén O, et al. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate. 2019;79(4):363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonis MA, Thobe MN, Waltz SE. Ron-receptor tyrosine kinase in tumorigenesis and metastasis. Future Oncol. 2007;3(4):441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thobe MN, Gurusamy D, Pathrose P, Waltz SE. The Ron receptor tyrosine kinase positively regulates angiogenic chemokine production in prostate cancer cells. Oncogene. 2010;29(2):214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Toole JM, Rabenau KE, Burns K, Lu D, Mangalampalli V, Balderes P, et al. Therapeutic implications of a human neutralizing antibody to the macrophage-stimulating protein receptor tyrosine kinase (RON), a c-MET family member. Cancer Res. 2006;66(18):9162–70. [DOI] [PubMed] [Google Scholar]
- 12.Thobe MN, Gray JK, Gurusamy D, Paluch AM, Wagh PK, Pathrose P, et al. The Ron receptor promotes prostate tumor growth in the TRAMP mouse model. Oncogene. 2011;30(50):4990–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray JK, Paluch AM, Stuart WD, Waltz SE. Ron receptor overexpression in the murine prostate induces prostate intraepithelial neoplasia. Cancer Lett. 2012;314(1):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown NE, Sullivan C, Waltz SE. Therapeutic Considerations for Ron Receptor Expression in Prostate Cancer. EMS Cancer Sci J. 2018;1(1). [PMC free article] [PubMed] [Google Scholar]
- 15.Vasiliauskas J, Nashu MA, Pathrose P, Starnes SL, Waltz SE. Hepatocyte growth factor-like protein is required for prostate tumor growth in the TRAMP mouse model. Oncotarget. 2014;5(14):5547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benight NM, Wagh PK, Zinser GM, Peace BE, Stuart WD, Vasiliauskas J, et al. HGFL supports mammary tumorigenesis by enhancing tumor cell intrinsic survival and influencing macrophage and T-cell responses. Oncotarget. 2015;6(19):17445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Correll P, Morrison A, Lutz M. Receptor tyrosine kinases and the regulation of macrophage activation. Journal of leukocyte biology. 2004;75(5):731–7. [DOI] [PubMed] [Google Scholar]
- 18.Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol. 2011;187(5):2181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurusamy D, Gray JK, Pathrose P, Kulkarni RM, Finkleman FD, Waltz SE. Myeloid-specific expression of Ron receptor kinase promotes prostate tumor growth. Cancer Res. 2013;73(6):1752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg N, DeMayo F, Finegold M, Medina D, Tilley W, Aspinall J, et al. Prostate cancer in a transgenic mouse. Proceedings of the National Academy of Sciences. 1995;92(8):3439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol. 2001;Chapter 20:20.5. [DOI] [PubMed] [Google Scholar]
- 22.Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J Clin Invest. 2001;108(4):567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maddison LA, Nahm H, DeMayo F, Greenberg NM. Prostate specific expression of Cre recombinase in transgenic mice. genesis. 2000;26(2):154–6. [DOI] [PubMed] [Google Scholar]
- 24.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer research. 2013;73(9):2718–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni RM, Stuart WD, Gurusamy D, Waltz SE. Ron receptor signaling is protective against DSS-induced colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2014;306(12):G1065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olson M, Lee J, Zhang F, Wang A, Dong Z. Inducible nitric oxide synthase activity is essential for inhibition of prostatic tumor growth by interferon-β gene therapy. Cancer gene therapy. 2006;13(7):676. [DOI] [PubMed] [Google Scholar]
- 27.Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J, et al. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J Clin Invest. 2010;120(6):1981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15(8):1477–88. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. methods. 2001;25(4):402–8. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Torres SJ, Benight NM, Karns RA, Lower EE, Guan JL, Waltz SE. HGFL-mediated RON signaling supports breast cancer stem cell phenotypes via activation of non-canonical beta-catenin signaling. Oncotarget. 2017;8(35):58918–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown NE, Paluch AM, Nashu MA, Komurov K, Waltz SE. Tumor Cell Autonomous RON Receptor Expression Promotes Prostate Cancer Growth Under Conditions of Androgen Deprivation. Neoplasia. 2018;20(9):917–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza-Alvarez P, et al. 3D-3-culture: A tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials. 2018;163:185–97. [DOI] [PubMed] [Google Scholar]
- 33.Raghavan S, Mehta P, Xie Y, Lei YL, Mehta G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. Journal for ImmunoTherapy of Cancer. 2019;7(1):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gironella M, Calvo C, Fernández A, Closa D, Iovanna JL, Rosello-Catafau J, et al. Reg3β deficiency impairs pancreatic tumor growth by skewing macrophage polarization. Cancer research. 2013;73(18):5682–94. [DOI] [PubMed] [Google Scholar]
- 35.Xu M, Zhang S, Jia L, Wang S, Liu J, Ma X, et al. E-M, an Engineered Endostatin with High ATPase Activity, Inhibits the Recruitment and Alternative Activation of Macrophages in Non-small Cell Lung Cancer. Front Pharmacol. 2017;8:532-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers J-WJ, et al. STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. Journal of Biological Chemistry. 1996;271(22):13221–7. [DOI] [PubMed] [Google Scholar]
- 37.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18(5):2545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499–513. [DOI] [PubMed] [Google Scholar]
- 39.Christine R, Sylvie R, Erik B, Geneviève P, Amélie R, Gérard R, et al. Implication of STAT3 Signaling in Human Colonic Cancer Cells during Intestinal Trefoil Factor 3 (TFF3) – and Vascular Endothelial Growth Factor–Mediated Cellular Invasion and Tumor Growth. Cancer Research. 2005;65(1):195. [PubMed] [Google Scholar]
- 40.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer research. 2006;66(2):605–12. [DOI] [PubMed] [Google Scholar]
- 41.Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer research. 2018;78(19):5492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laviron M, Boissonnas A. Ontogeny of Tumor-Associated Macrophages. Front Immunol. 2019;10:1799-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y-L, Tian P-X, Han F, Zheng J, Xia X-X, Xue W-J, et al. Comparison of the characteristics of macrophages derived from murine spleen, peritoneal cavity, and bone marrow. J Zhejiang Univ Sci B. 2017;18(12):1055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kermanizadeh A, Brown DM, Stone V. The variances in cytokine production profiles from non-or activated THP-1, Kupffer cell and human blood derived primary macrophages following exposure to either alcohol or a panel of engineered nanomaterials. PloS one. 2019;14(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janatpour MJ, Hudak S, Sathe M, Sedgwick JD, McEvoy LM. Tumor necrosis factor–dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. The Journal of experimental medicine. 2001;194(9):1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan S, Wang K, Sun F, Li Y, Gao Y. CXCL9 promotes prostate cancer progression through inhibition of cytokines from T cells. Molecular medicine reports. 2018;18(2):1305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang K, Shan S, Yang Z, Gu X, Wang Y, Wang C, et al. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget. 2017;8(40):68571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Babicky ML, Harper MM, Chakedis J, Cazes A, Mose ES, Jaquish DV, et al. MST1R kinase accelerates pancreatic cancer progression via effects on both epithelial cells and macrophages. Oncogene. 2019;38(28):5599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.