

Abstract
Little is known about the role of epithelial membrane protein-2 (EMP2) in breast cancer development or progression. In this study, we tested the hypothesis that EMP2 may regulate the formation or self-renewal of breast cancer stem cells (BCSC) in the tumor microenvironment. In silico analysis of gene expression data demonstrated a correlation of EMP2 expression with known metastasis related genes and markers of cancer stem cells (CSC) including aldehyde dehydrogenase (ALDH). In breast cancer cell lines, EMP2 overexpression increased and EMP2 knockdown decreased the proportion of stem-like cells as assessed by the expression of the CSC markers CD44+/CD24-, ALDH activity, or by tumor sphere formation. In vivo, upregulation of EMP2 promoted tumor growth while knockdown reduced the ALDHhigh CSC population as well as retarded tumor growth. Mechanistically, EMP2 functionally regulated the response to hypoxia through the upregulation of HIF-1αa transcription factor previously shown to regulate the self-renewal of ALDHhigh CSC. Furthermore, in syngeneic mouse models and primary human tumor xenografts, mAbs directed against EMP2 effectively targeted CSC, reducing the ALDH+ population and blocking their tumor initiating capacity when implanted into secondary untreated mice. Collectively, our results show that EMP2 increases the proportion of tumor initiating cells providing a rationale for the continued development of EMP2 targeting agents.
Keywords: Epithelial membrane protein-2, breast cancer, antibody therapy, cancer stem cells
Introduction
Breast cancer is a leading cause of death among women with over 90% of these deaths due to the development of metastasis(1,2). Despite considerable progress in basic research leading to an increased understanding of breast cancer biology and advances in the treatment of early stage disease, metastatic breast cancer remains largely incurable(3). Substantial evidence suggests that one source of treatment resistance is the existence of cells that display “stem like” properties. These cancer stem cells (CSC) are endowed with intrinsic properties that contribute to treatment resistance including increased DNA repair, expression of drug transporter proteins, anti-oxidant defenses, and slow cell cycle kinetics(4,5). In addition, their phenotypic plasticity facilitates their epigenetic reprogramming rendering them resistant to multiple treatments including molecularly targeted agents(6).
A number of cell surface markers including CD44+/CD24- and enzymatic activities including aldehyde dehydrogenase (ALDH) are associated with cells that display increased tumor initiating capacity in mouse xenograft models(7,8). These cells also display increased invasive capacity and may play a fundamental role in mediating tumor metastasis(8,9). Furthermore, the expression of these CSC markers correlates with poor prognosis in patients across a spectrum of tumor types including breast cancer(10–12). These data highlight the importance of developing therapeutic strategies capable of successfully targeting the CSC population.
One strategy to accomplish this is to identify and target CSC regulatory proteins. Epithelial membrane protein-2 (EMP2) is a tetraspan protein, which has been reported to be overexpressed in a number of cancers including its expression in 63% of invasive breast cancers where EMP2 levels correlate with disease progression(13). Additionally, its expression has been reported in breast metastases as well as in circulating tumor cells(13–15). However, little is known about its role in disease etiology.
In this study, the role EMP2 in breast cancer progression and in the regulation of breast cancer stem cells (BCSC) were investigated using cell lines and xenograft models. The clinical relevance of these studies was supported by in silico analysis of TGCA datasets demonstrating a correlation of EMP2 expression with markers of metastasis in breast cancers across a spectrum of breast cancer subtypes. Using gene knockdown, we validated EMP2 as a CSC regulatory target. We demonstrate that a monoclonal antibody (mAb) directed against EMP2 reduces CSC in vitro and blocks tumor initiating capacity and tumor growth in mouse xenograft models. Collectively, these studies support the continued development of anti-EMP2 therapeutics to target the CSC population which mediates tumor metastasis and treatment resistance.
Methods and Materials
Cell Culture.
The human breast cancer cell lines MDA-MB-231, MDA-MB-468, HS578T, BT474, BT-20, MDA-MB-157, MCF7, ZR-75–30, and SK-BR-3 (American Type Culture Collection; ATCC, Manassas, VA) were cultured in DMEM with 10% fetal calf serum (FCS; Hyclone Laboratories, Logan, UT), 1% sodium pyruvate, 1% penicillin-streptomycin, and 1% L-glutamine (Life Technologies, Carlsbad, CA) at 37C in a humidified 5% CO2. HCC1806, HCC202, MDA-MB-453, HCC1419 (all ATCC) and JIMT-1 (German Tissue Repository; DSMZ) were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mmol/L glutamine, and 1% penicillin G-streptomycin-fungizone solution (PSF, Irvine Scientific). MDA-MB-436, UACC812 and UACC893 (all ATCC) were grown in L15 medium supplemented with 15% heat-inactivated FBS, 2 mmol/L glutamine, and 1% PSF. UACC732 (ATCC), UACC- 732 cells were maintained in MEM supplemented with 10% heat-inactivated FBS, 2 mmol/L glutamine, 1% PSF, 1% MEM nonessential amino acids, and 1 μmol/L sodium pyruvate. SUM190 (University of Michigan) cells were grown in DMEM-F12 medium supplemented with 10% heat-inactivated FBS, 1% PSF, 10 μg/mL insulin, and 1 μg/mL hydrocortisone. MCF10A and MCF12A cells (kind gift of Jaydutt Vadgama, Charles Drew University) were grown in Dulbecco’s Modified Eagle’s/F12 medium (DMEM/F12, 1:1) containing 5% equine serum (ES), 20 ng/mL epidermal growth factor, 0.5 μg/mL hydrocortisone, 0.1 μg/mL cholera toxin, and 10 μg/mL insulin. Cells were passaged weekly and tested periodically for mycoplasma contamination using MycoAlert Kit (Lonza, Hayward, CA). SUM149PT cells was maintained in Ham’s F12K media supplemented with 5% FBS, 5μg/ml insulin, 1μg/ml hydrocortisone and 4 μg/ml gentamicin in addition to the supplements listed above. Finally, 4T1 cells, a spontaneous mammary tumor syngeneic in BALB/c mice, were obtained from ATCC. 4T1 cells were maintained in RPMI medium supplemented as above. Both 4T1 and MDA-MB-231 cells were stably infected with firefly luciferase (FLUC) as previously described (16). Cell lines were used within 3 months after resuscitation of frozen aliquots and were authenticated based on viability, recovery, growth, morphology, and isoenzymology by the supplier. Modulation of EMP2 expression in cell lines is described in the Supplemental Methods.
Production of the anti-EMP2 mAb.
Anti-EMP2 mAb is currently produced in bulk by Lake Pharma on a contractual basis according to their standard practices. The variable sequences used to generate the mAb are provided below:
HC-hmAb:
QVQLVQSGGGVVQPGRSLRLSCAASGFTFSSYAMHWVRQAPGKGLEWVAVISYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARDRRGRKSAGIDYWGQGTLVTVSS
LC-hKappa:
DIQMTQSPSSLSASVGDRVTITCQASQDISNYLNWYQQKPGKAPKLLIYAASSLQSGVPSRFS
GSGSGTDFTLTISSLQPEDFATYYCLQDYNGWTFGQGTKVDIK
The affinity of the antibody is verified against a specific and scrambled EMP2 peptide using ForteBio Octet with an affinity between 5–8 nM considered acceptable.
TCGA Data.
The expression of EMP2 in breast cancer was obtained through the CBioPortal for Cancer Genomics (http://cbioportal.org)(17). 1105 provisional cases of invasive breast cancer were evaluated for EMP2 expression (RNA Seq V2 RSEM; z-score threshold± 1.1). A minimum criteria of at least 20 (out of 814) cases was set. From the dataset, the genes that co-expressed with EMP2 were selected and used to generate a ranked list (from high to low correlation). This ranked list was then analyzed using Gene Set Enrichment Analysis (GSEA; http://software.broadinstitute.org/gsea/index.jsp), which identified 61 gene sets that had an FDR q-val < 0.5.
Immunohistochemistry
Patient samples
Approval from the institution review board at UCLA was obtained before initiating the study. EMP2 and ALDH1 expression was measured in 62 patients with breast cancer. The patients represent the population of women who had undergone breast surgery at King/Drew Medical Center from 1999 to 2005, and profile as 57% African American, 37% Latina, 1% Asian and 4% Caucasian. Samples were stained as described in the Supplemental Methods section.
Treatment and analysis using mouse xenograft model.
Animal studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Animal Research Committee at the University of California, Los Angeles or the University of Michigan approved all procedures. All efforts were made to minimize animal suffering. The Supplemental Methods section describes the models created.
Information about SDS-PAGE/Western blot analysis, RNA sequencing, flow cytometry, tumorspheres and statistical analyses are described in the Supplemental Methods.
Results
EMP2 is expressed in human breast cancers and is associated with metastasis.
In a previous study, EMP2 protein was shown to be upregulated in all subsets of breast cancer(13), but little is known about its role in the etiology or clinical course of this disease. To address this, EMP2 expression was queried using data generated by the TCGA Research Network: http://cancergenome.nih.gov/. Concordant with previous publications on EMP2 protein expression, EMP2 mRNA expression was significantly increased in women with breast cancer, with 28.4% of ductal carcinomas and 20.3% of lobular breast cancers showing increased expression (Fig S1A). Mixed ductal and lobular carcinomas also showed increased EMP2 mRNA expression in 29.6% of patients. To next elucidate gene expression changes associated with EMP2 mRNA upregulation, gene set enrichment analysis (GSEA) was performed on a ranked list of genes co-expressed with EMP2. EMP2 was co-expressed with genes enriched in all breast cancer classes, with hypoxia, and gene sets associated with breast cancer metastasis and disease relapse (Fig S1B; Table S1).
EMP2 affects tumor growth in mouse xenografts
Initially, the expression of EMP2 was analyzed in cell lines representing luminal, basal, mesenchymal and HER2 subtypes of disease as well as in two normal mammary cell lines (MCF10A and MCF12A; Table S2)(18,19). As demonstrated in Fig 1A, low levels of EMP2 were detectable in both MCF10A and MCF12A. However, significantly higher levels of the protein were present in all breast cancer cell lines with the exception of HS578t, a breast carcinosarcoma cell line. To investigate its functional role within these cells, we overexpressed or knocked down EMP2 utilizing specific shRNAs in cell lines representing major breast cancer subtypes. Overexpression of EMP2 accelerated and knockdown significantly inhibited the growth of both hormone receptor positive and TNBC cells lines represented by BT474 and MDA-MB-231 (Fig 1B-C). In the case of SUM149, whereas knockdown of EMP2 significantly inhibited tumor growth compared to the vector control, less pronounced changes were observed upon overexpression (Fig 1D). As all three cell lines exhibited reduced tumor load concomitant to lower EMP2 levels, the effect of EMP2 knockdown was further investigated.
Figure 1: EMP2 expression promotes tumor growth.
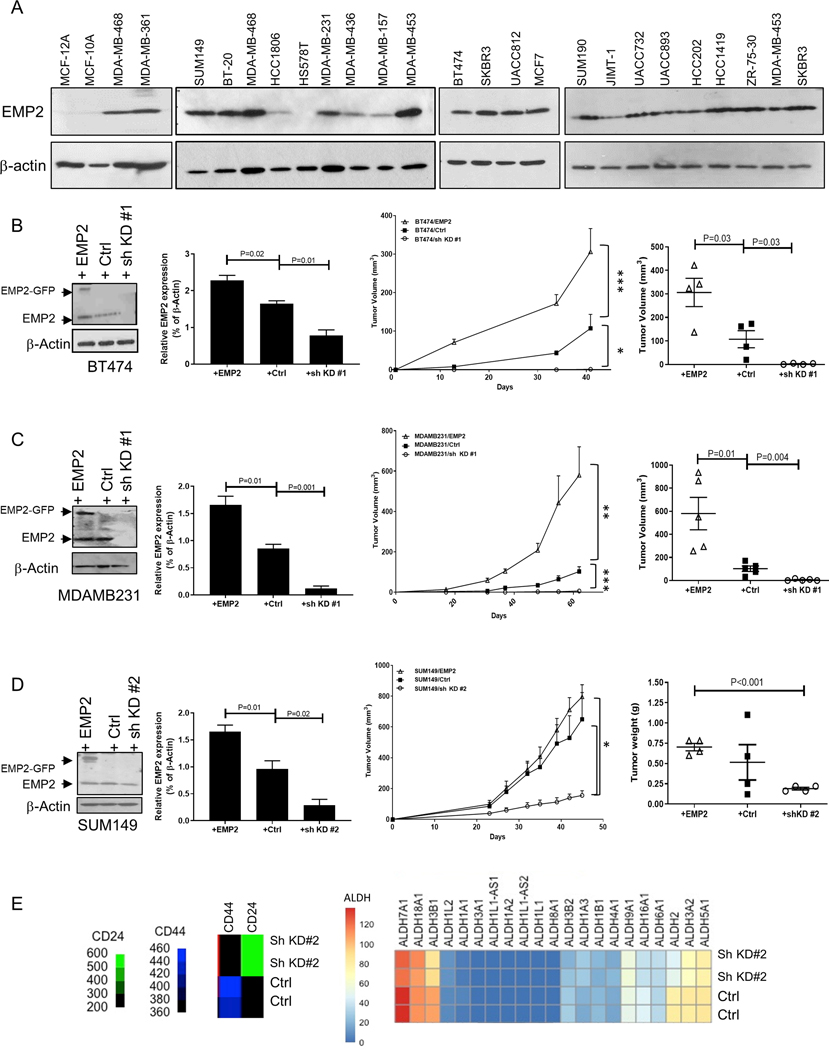
A. A panel of normal mammary cell lines and breast cancer cell lines (MCF10A and MCF12A) representing the major molecular subtypes were tested for EMP2 expression with β-Actin used as a loading control. B. To determine the role of EMP2 in tumorigenesis, BT474 breast cancer cells with high EMP2, endogenous, or low levels (downregulation through sh KD #1) were injected in Balb/c nude mice. Quantitation of EMP2 expression in the different cell lines is depicted on the left. The growth of the cells over 40 days is shown in the middle, with final tumor volumes illustrated on the right. N=4. Significance was determined using 2 way ANOVA, p<0.0001, and results from Bonferroni’s multiple comparisons test shown on the figure. C. MDA-MB-231 cells with modified EMP2 levels as above were injected into Balb/c nude mice and measured over 62 days. Significance using a 2-way ANOVA, p=0.0004. Bonferroni’s multiple comparisons was used tested post hoc with results shown on the figure. N=5. Right, Final tumor volumes are highlighted. D. EMP2 expression in SUM149 cells were modified to overexpress or reduce EMP2 levels (sh KD#2) compared to a vector control. Cells were injected into Balb/c nude mice, and tumor load monitored over 45 days. Significance using a 2-way ANOVA, p=0.009. Post hoc analysis using Bonferroni’s multiple comparisons test is shown on the figure. N=4. Right, Graph illustrates final tumor weights. E. Transcriptomic data showing differential expression in SUM149 cell line infected with a vector control or shRNA lentiviral vector (sh KD #2). TMM normalized read counts per million were utilized to generate a heatmap based on differential expression of CD44, CD24, and various ALDH isoforms. EMP2 significantly altered the expression of multiple ALDH isoforms, and it was associated with increased CD24 and reduced CD44. *, indicates p<0.05.
EMP2 knockdown affects expression of genes associated with cancer “stemness”
In order to elucidate pathways regulated by EMP2, we determined the effect of EMP2 knockdown on gene expression in SUM149 cells via RNA sequencing (RNAseq). Comparison of the vector control versus shRNA knockdown revealed 332 genes with differential expression between the two groups. Gene set enrichment analysis (GSEA) revealed a significant enrichment, relative to EMP2 levels, for the response to hypoxia, similar to the enrichment seen using TCGA data as above (Table S3).
Substantial evidence now exists that many cancers including breast cancer are hierarchically organized and contain subpopulations of cells that display stem cell properties. These cells play putative roles in treatment resistance and metastasis(20,21). Several markers including CD44+/CD24- and ALDH activity have been shown to enrich for tumor cells displaying “stemlike” properties. As hypoxia is a known regulator of CSC(5,22), we interrogated the RNAseq data to elucidate the effect of EMP2 knockdown on CSC marker expression. As shown in Fig 1E, EMP2 knockdown increased CD24 expression while decreasing expression of CD44 as well as a number of ALDH isoforms. These results suggested that EMP2 may regulate proteins associated with “stemness”.
EMP2 promotes tumorsphere formation.
One recognized property of CSC is their ability to survive in anchorage-independent conditions, and as such, tumorsphere assays can be used to identify cells with stem-like characteristics(23,24). To determine if EMP2 can alter the tumorsphere forming efficiency of breast cancer cells, MDA-MB-231 and BT474 cells with varying levels of EMP2 were plated under low adherence conditions, and after two weeks, tumorspheres were enumerated. In both MDA-MB-231 and BT474, overexpression of EMP2 correlated with higher sphere forming efficiency than the vector control, and conversely, reduction in EMP2 levels significantly prevented tumorsphere formation (Fig 2A).
Figure 2. EMP2 expression promotes tumorsphere formation and markers associated with stemness.
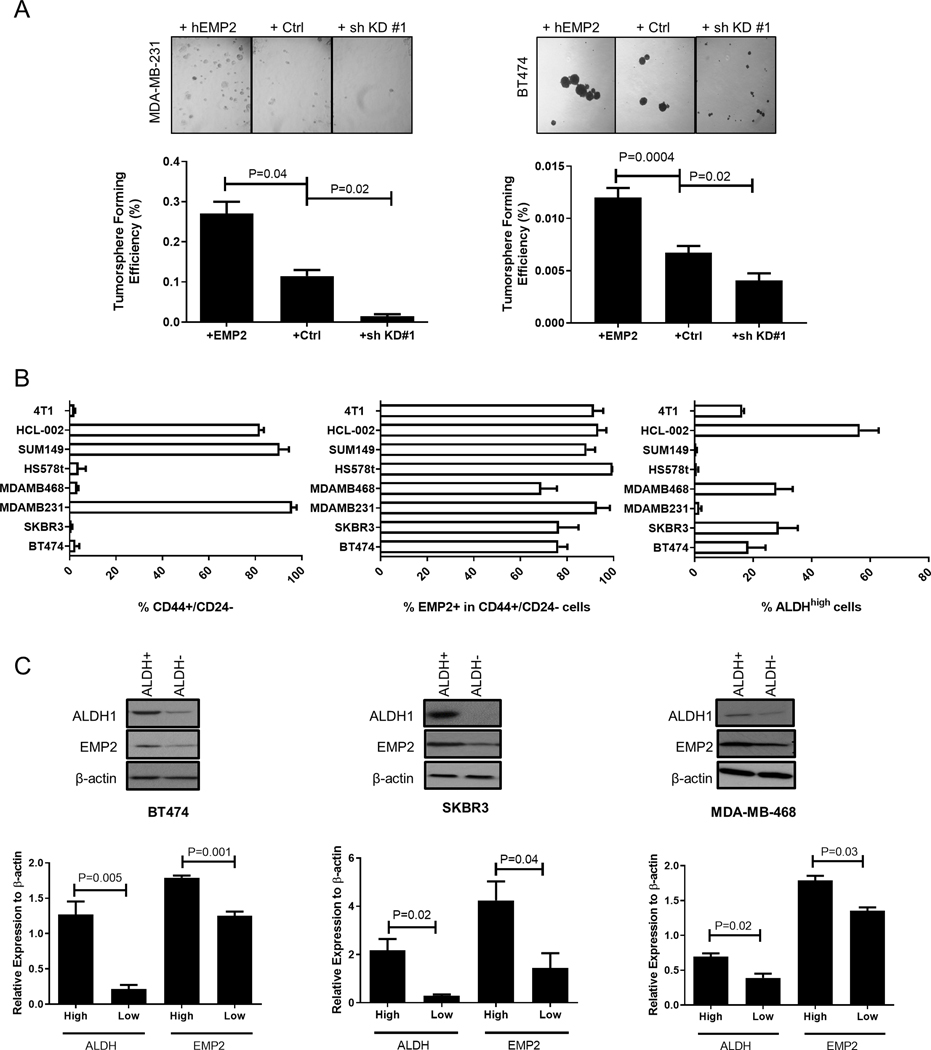
A. The effects of EMP2 expression on mammosphere formation was evaluated in MDA-MB-231 and BT474. EMP2 levels directly correlated with increased mammosphere formation. N=4; significance was determined using Student’s t test. B. Breast cancer cells express varying levels of CD44+/CD24- or ALDH activity as determined by the ALDEFLUOR assay. Left, The percentage of CD44+/CD24- cells observed in a panel of breast cancer cell lines as determined by flow cytometry. Middle Panel, Percentage of EMP2+ cells was determined in cells gated on CD44+/CD24- using flow cytometry. Right, Assessment of ALDH activity in a panel of breast cancer cell lines as measured by the ALDEFLUOR assay. N=3 with results expressed as the mean± SEM. C. High levels of ALDH1 correlated with high levels of EMP2 expression. Cells were sorted into ALDHhigh or ALDHlow as determined using the ALDEFLUOR assay and subsequently analyzed for EMP2 and ALDH1 expression by western blot analysis. Results were normalized to β-Actin levels and tabulated as the mean± SEM, with significance determined using Student’s t test. N=3.
As these results suggested that EMP2 may enhance properties associated with CSC, we utilized the tumorsphere assay as a means to calculate the frequency of tumor initiating cells based on EMP2 expression. 50, 500, and 5000 BT474 cells with modified EMP2 levels were cultured on low adhesion plates, and spheres enumerated after two weeks. Upregulation of EMP2 increased the tumor initiating frequency within the population. In contrast, reduction in EMP2 expression reduced the tumor initiating frequency, or stemness, of these cells (Table S4).
EMP2 is expressed on CD44+ and ALDH+ cells.
A number of previous studies have characterized CSC using markers such as CD44, CD24, as well as ALDHhigh activity(8,9,25). CSC maintain plasticity, and studies have shown that these states can be identified based on CD44+/CD24– staining or via ALDHhigh activity. These respective markers reflect differing mesenchymal or epithelial CSC states, respectively (26), and it is believed that cells displaying either sets of markers are capable of initiating tumors in NOD/SCID mice(27,28). To determine if EMP2 is present in cells expressing these CSC markers, we utilized flow cytometry on a panel of seven breast cancer cell lines as well as the primary tumorgraft HCl-002(29). Consistent with previous reports, the expression of CD44+/CD24- and ALDHhigh activity varied significantly among these different cells (8) (Fig 2B). Next, the expression of EMP2 within CD44+/CD24- or ALDHhigh populations was determined, and within both states, EMP2 was highly expressed. EMP2+ expression significantly overlapped with 65–99% of CD44+/CD24- cells in all cell lines (Fig S1C), and its expression was enriched in cells displaying an ALDHhigh phenotype. ALDHhigh and ALDHlow cells were sorted and analyzed for ALDH1 and EMP2 expression using western blot analysis (Fig S1D). In all three cell lines tested, ALDH activity as assessed by the ALDEFLUOR assay correlated with ALDH1 protein expression, and similarly, levels of EMP2 were higher in ALDHhigh compared to ALDHlow populations (Fig 2C). To validate this finding, the overlap between ALDH activity and EMP2 levels was determined using flow cytometry using Diethylaminobenzaldehyde or DEAB, a specific inhibitor of ALDH, as a negative control. All ALDHhigh cells showed high expression of EMP2, with results from representative cell lines provided (Fig S1E).
Correlation of HIF-1α, ALDH and EMP2 expression.
To further our understanding of the relationship between EMP2 and ALDH1, the effects of EMP2 knockdown on ALDH activity were analyzed using DEAB as a negative control. In these cells, knockdown of EMP2 significantly reduced ALDH activity as assessed by the ALDEFLUOR assay (Fig 3A). To verify the dependence of ALDH1 expression on EMP2, cells were next transiently transfected with an EMP2 siRNA or scrambled control. Similar to effects observed with the shRNA knockdown, transient knockdown of EMP2 using a pooled set of four siRNA significantly reduced ALDH1 expression (Fig 3B). These results validate the correlation of EMP2 expression with ALDH1 expression in vitro.
Figure 3. ALDH activity changes relative to EMP2 expression.
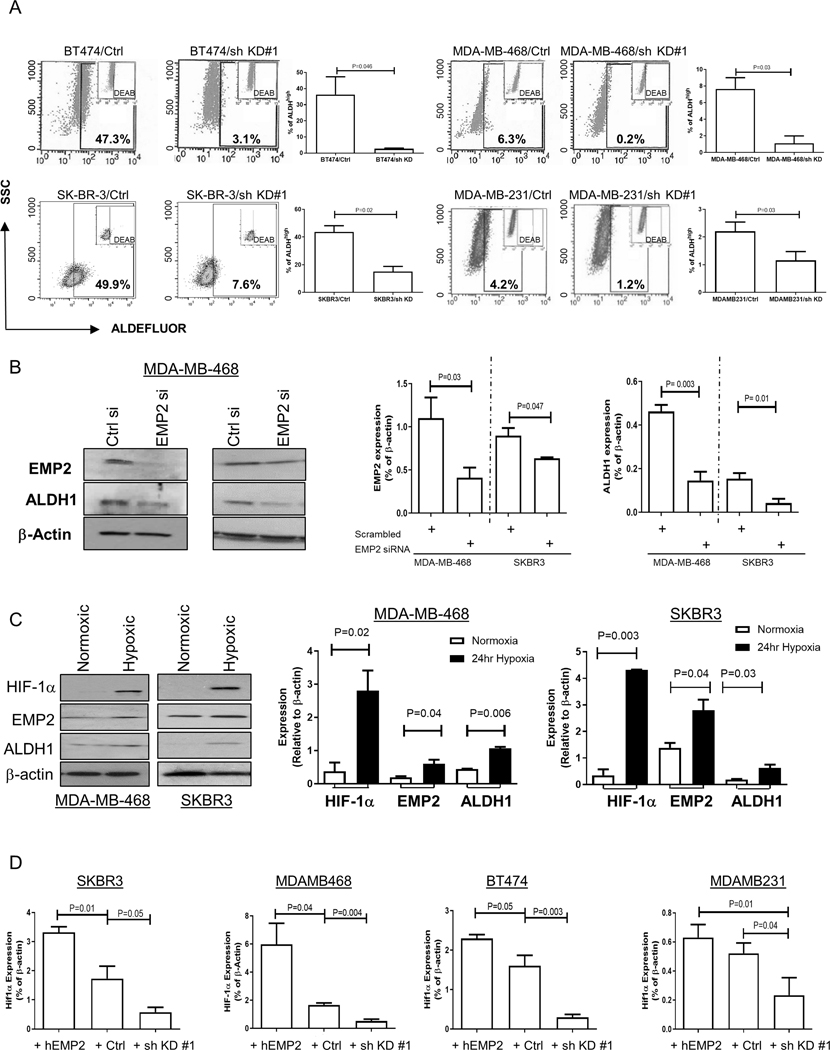
A. As a functional readout of ALDH activity, breast cancer cells with endogenous or shRNA knockdown of EMP2 were tested using the ALDEFLUOR assay. A specific inhibitor of ALDH, diethylaminobenzaldehyde or DEAB, was used as a negative control. Higher EMP2 levels correlated with an increase in the number of ALDHhigh cells. B. To independently confirm the regulation of ALDH1 by EMP2, cells were transiently transfected with an EMP2 or scrambled siRNA. Knockdown of EMP2 levels significantly reduced ALDH1 expression in two representative cell lines. N=3, with results tabulated as the mean ± SEM. C. Hypoxia increases ALDH1 and EMP2 levels. MDA-MB-468 and SKBR3 were plated under both normoxic and 0.1% O2 hypoxic conditions. After 24 hours, ALDH1 and EMP2 levels were measured relative to β-Actin. A representative western blot is shown on the left, and results tabulated from three independent experiments shown on the right. Data is expressed as the mean ± SEM. D. SKBR3, MDA-MB-468, BT474 and MDA-MB-231 cells were placed in 0.1% O2 hypoxic chamber. HIF-1α expression was monitored in response to changes in EMP2. All experiments were repeated 3 times, with results expressed as the mean ± SEM. P values show significant differences between groups using Student’s t test.
Previous studies have shown links between EMP2 and hypoxia(30) as well as hypoxia and the formation of BCSC(31,32). In order to determine if exposure to hypoxia alters EMP2 and ALDH1 expression, two representative cell lines were placed in a hypoxic chamber for 24 hours and EMP2 and ALDH1 levels analyzed by western blot analysis. In response to hypoxia exposure, expression of HIF-1α, ALDH1, and EMP2 were all significantly increased (Fig 3C). To extend upon this observation, cells with varying levels of EMP2 were placed in hypoxic conditions for 24 hours. In all 4 cell lines, HIF-1α expression directly correlated with EMP2 levels (Fig 3D; Fig S1F), suggesting that EMP2 helps regulate the hypoxic response of breast cancer cells.
Clinical correlation of ALDH and EMP2 expression.
To investigate the clinical relevance of these findings, we utilized immunohistochemistry to examine the relationship between EMP2 and ALDH1 expression in breast cancer biopsies. The patient cohort consisted of 24 women with TNBC, 4 women with HER2+, hormone receptor negative (HR-) disease, 23 women with luminal A (ER+ and/or PR+/HER2-) disease, and 11 women with luminal B (ER+ and/or PR+/HER2+) disease. Although all women analyzed presented with invasive ductal carcinoma (IDC), foci of ductal carcinoma in situ (DCIS) were also observed.
High levels of EMP2 were observed in both DCIS and IDC spots independent of hormone status compared to histologically “normal” lobules adjacent to the tumor (Fig 4A; Table 1). No statistical differences were observed in the mean expression of EMP2 in DCIS foci and IDC (1.84±0.22 and 1.86±0.08, respectively), and both were higher than the relative expression observed in normal breast tissue (p=0.002, Kriskal Wallis). Interestingly, in 68% of patients, adjacent “normal” tissue showed some low levels (0.72±0.11) of EMP2 as well. Fig S2A illustrates staining between normal, DCIS and IDC spots. While our results suggest that EMP2 may be expressed at pre -invasive stages of breast cancer progression, it is also possible that the similar expression observed in DCIS and IDC may be attributable to “field effects” (33).
Figure 4. ALDH1 and EMP2 are co-expressed.
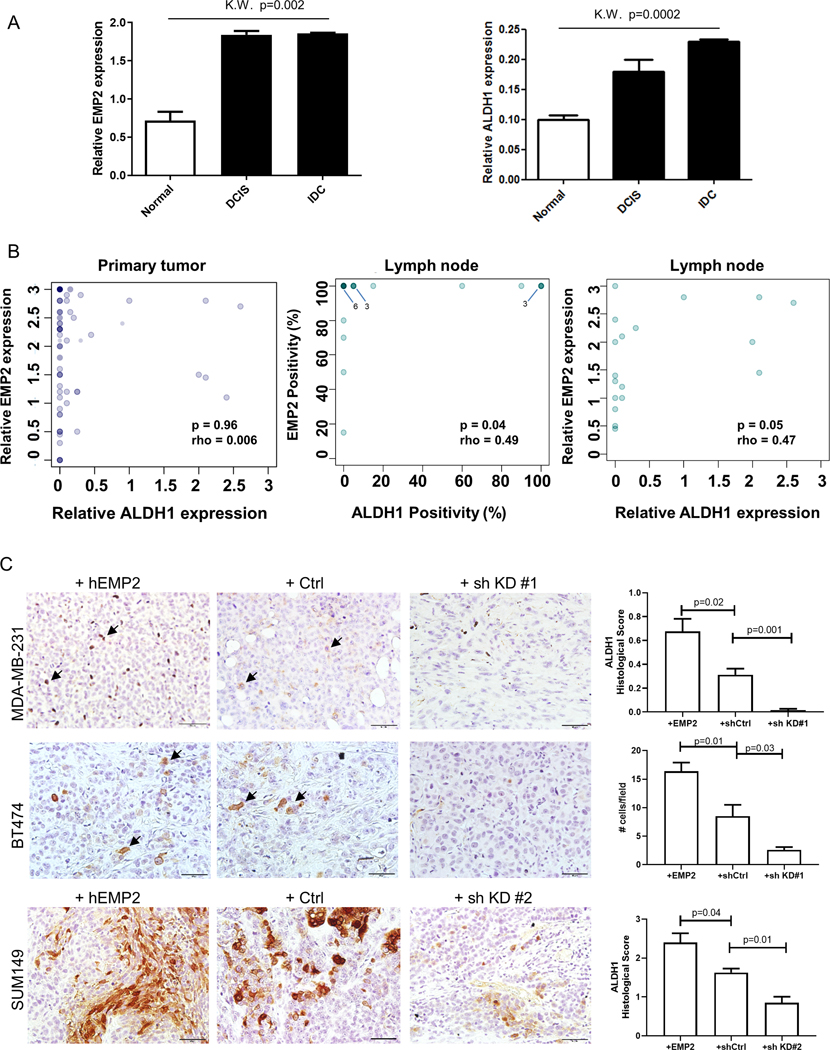
A. EMP2 and ALDH1 expression were measured in a tissue microarray from 62 women with invasive ductal carcinoma. Relative expression measures both the percentage of positive cells and the intensity of their staining, with a maximum score of 3. EMP2 and ALDH1 expression were significantly upregulated in cancer compared to adjacent normal tissue (p=0.002 and p=0.0002, respectively; Kruskal Wallis). Within the same patient, no significant differences were observed in DCIS relative to IDC for both ALDH1 and EMP2. B. Correlation of EMP2 and ALDH1 in patient samples. While all ALDH1 positive tumors were EMP2 positive, the relative expression of EMP2 did not correlate with that of ALDH1 in the primary tumor (N=47). Middle, A significant correlation between the percentage of ALDH1 and EMP2 cells occurred within lymph node metastasis (p=0.04; N=19). Right, This correlation was also significant when expression intensity was included (p=0.05). In all graphs, the intensity of color (darkness) is proportional to the number of samples at a given location. C. ALDH1 expression (brown) was measured in MDA-MB-231, BT474, and SUM149 xenografts that overexpress EMP2, express a vector control or express EMP2 shRNA knockdown tumors. Representative images are shown at a 400X magnification, with some ALDH1 positive cells highlighted with arrows. Scale bar = 50μM. N=4 with results shown as the mean ± SEM.
Table 1. Characterization of 62 patients with breast cancer stained for EMP2 or ALDH1.
An adjusted histological score of 0.4 was used to set the threshold for positivity based on staining in a positive and negative control tissue. EMP2 and ALDH1 are present in all breast cancer subtypes.
| Core tissue sample comparisons |
| Variables | n | adj EMP2>0 |
|---|---|---|
| Histology | ||
| normal* | 55 | 46 |
| DCIS | 19 | 19 |
| IDC | 195 | 180 |
| LN metastasis | 63 | 59 |
| Grade | ||
| 1 | 29 | 26 |
| 2 | 92 | 84 |
| 3 | 88 | 84 |
| Variables | n | ALDH>0 |
|---|---|---|
| Histology | 66 | 7 |
| normal* | 19 | 9 |
| DCIS | 201 | 75 |
| IDC | 65 | 28 |
| LN metastasis | ||
| Grade | ||
| 1 | 36 | 11 |
| 2 | 92 | 24 |
| 3 | 87 | 46 |
| patient overall histology comparisons |
| subtype by IHC | n | adj EMP2>0 |
|---|---|---|
| luminal A | 23 | 22 |
| luminal B | 11 | 11 |
| HER2+ | 4 | 3 |
| TNBC | 24 | 23 |
| subtype by IHC | n | adj EMP2>0 |
| luminal A | 23 | 22 |
| luminal B | 11 | 11 |
| HER2+ | 4 | 3 |
| TNBC | 24 | 23 |
n = tumor core samples with adequate marker information
tumor adjacent
Sections were also stained for ALDH1. Consistent with previous reports, ALDH1 displayed a heterogenous staining pattern within select patients with breast cancer, which was shared across molecular breast cancer subtypes (Table 1). Within the tumor, ALDH1 expression was often observed both within the parenchyma as well as within the tumor stroma (Fig S2B)(34). 26 of 47 patients (55.3%) expressed ALDH1 within the tumor parenchyma albeit at low levels, and its expression was higher in cancer compared to normal epithelia (p = 0.0002, Kruskal Wallis; Fig 4A). Similar to EMP2, no significant differences were observed in the relative expression of ALDH1 in DCIS and IDC from these patients (0.18±0.08 and 0.23±0.04, respectively). In normal breast tissue, minimal expression of ALDH1 was observed within epithelia (0.1±0.01; Fig S2C).
Correlation of EMP2 and ALDH1 expression showed that all patients with ALDH1+ cells were EMP2+ as well. However, when the intensity of staining was taken into account, there was no direct correlation between the relative expression of EMP2 and ALDH1 within the primary tumor (Fig 4B). This likely reflects the low overall staining observed with ALDH1 relative to EMP2. In addition to the primary tumor, the tissue array also included lymph nodes with metastatic disease from a subset of patients, allowing for the analysis of protein expression in these lesions. A significant correlation between EMP2 and ALDH1 occurred within lymph node metastasis (Fig 4B). The trend of EMP2 and ALDH1 positivity within lymph nodes was significant both based by percent positivity and intensity of expression. To confirm these results, a limited number (n=10) of full section metastatic lesions or IDC samples were independently double stained for both markers. Similar to the results obtained above, all ALDH1 positive tumor cells (visualized as brown) co-expressed EMP2 as well (visualized as red). This occurred in both representative metastatic lesions (Fig S2D) as well as in patients with IDC (Fig S2E), although notably, many IDC samples did not express detectable ALDH1 (Fig S3F). Together these results suggest that EMP2, ALDH1 co-expressing cells may represent a subset of the EMP2+ primary tumor with increased metastatic capacity. Further studies testing if EMP2 regulates metastatic potential will be required to test this hypothesis.
To extend this observation, the expression of EMP2 and ALDH1 was next examined in the xenograft models created from MDA-MB-231, BT474 and SUM149 described above. All three cell lines in vivo showed increased numbers of ALDH1 positive cells with increased EMP2 expression. In contrast, low to moderate levels of ALDH1 were observed in the vector control samples, while knockdown of EMP2 produced a statistically significant reduction in its levels (Fig 4C). These results suggest that knockdown of EMP2 may sufficiently target this population of cells.
Anti-EMP2 mAb reduces the proportion of BCSC’s in vitro.
As the above data suggested that downregulation of EMP2 reduced markers and activities associated with BCSC, we next determined whether an anti-EMP2 mAb(13) could produce a similar effect. Fig S3A and S3B provide validation of the integrity and affinity of the anti-EMP2 mAb, respectively. To test the effects of the mAb, MDA-MB-468 cells were treated with control human mAbs or anti-EMP2 mAbs for 1–5 days, and EMP2 and ALDH1 levels relative to β-actin were quantitated. Relative to the control, anti-EMP2 mAbs significantly reduced EMP2 (p=0.02, day 5) and ALDH1 (p=0.0002, day 5) levels (Fig 5A).
Figure 5. Anti-EMP2 mAbs reduce markers associated with stemness.
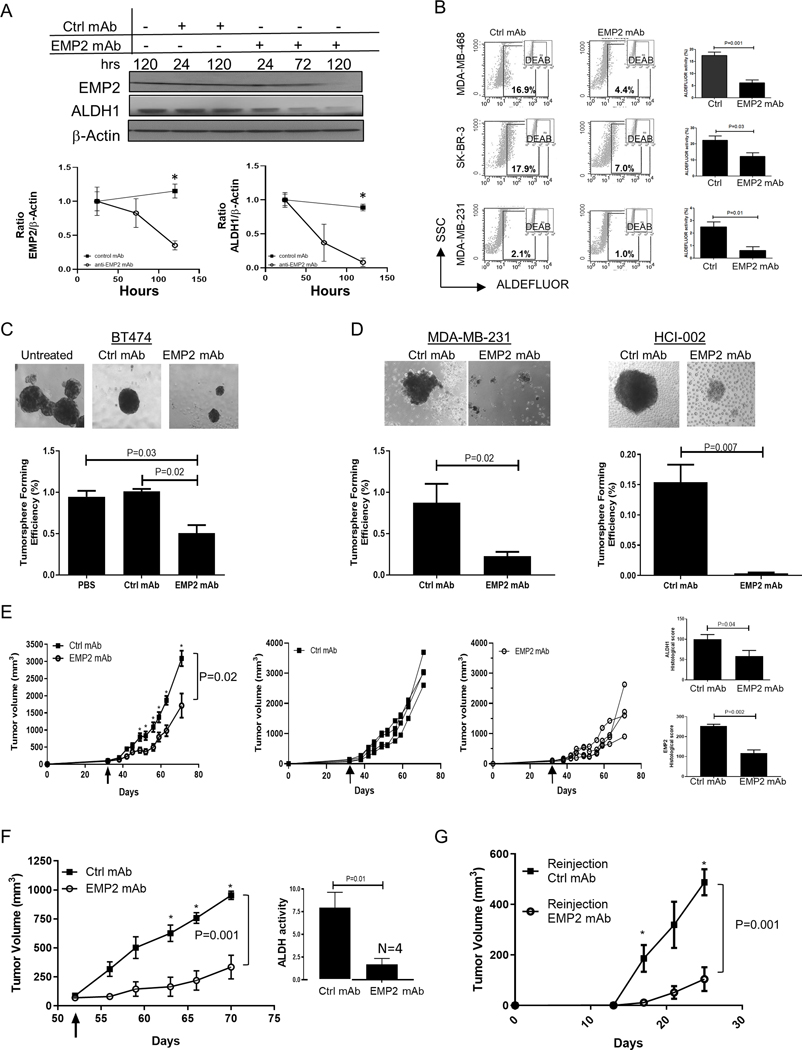
A. MDA-MB-468 cells were treated with 50μg/ml of anti-EMP2 or control human mAbs, and cell lysates were probed by Western blot analysis for EMP2 or ALDH1 expression for up to 120 hrs. Lower panels: Densiometric analysis of Western blots for expression of EMP2 or ALDH1 relative to β-actin. Samples were normalized to the 24 hours controls. N=3. *, p<0.05 by Student’s t test. B. MDA-MB-468, SKBR3, MDA-MB-231 cells were treated with 50μg/ml anti-EMP2 or control mAbs (anti-CD20) for 24 hours to monitor ALDH activity via the ALDEFLUOR assay. Diethylaminobenzaldehyde or DEAB, a specific inhibitor of ALDH, was used as a negative control. The graphs represent the average ± SEM of ALDH activity following treatment with control or anti-EMP2 mAbs from three independent experiments. C. Left panel, 5000 BT474 cells were left untreated, treated with 50 μg/ml control human or treated with anti-EMP2 mAbs and cultured for two weeks under low adherence conditions. Tumorspheres were enumerated, with representative examples shown. Below, quantitation of three independent experiments showing the average tumorsphere forming efficiency ± SEM, with significance determined using Student’s t test. Magnification=100X. D. MDA-MB-231 or HCI-002 cells were isolated from xenografts created in nude or NOD-SCID animals, respectively. Isolated cells (5000) were cultured in triplicate and treated as above with control human mAbs or anti-EMP2 mAbs. The graphs below represent the averaged results ± SEM from triplicate samples prepared from two independent experiments. Magnification=100X. E. The triple negative primary tumorgraft HCI-002 was implanted into the mammary fat pad of NOD-SCID animals. Grouped and individual data are provided for animals treated with 10mg/kg twice a week with either control mAbs or an anti-EMP2 mAb. N=4. The arrow denotes when tumor treatment was initiated. Two way ANOVA, p=0.02. *, significance by Bonferroni’s multiple comparisons test. Right panels, At day 71, tumors were harvested, fixed, and stained for ALDH1 or EMP2 expression using standard immunohistochemistry. The graphs show the average immunohistochemical intensity of each protein, ± SEM. N=4. Significance was determined using Student’s t test. F. 1×106 MDA-MB-231 cells were implanted into the mammary fat pad of Balb/c nude mice. When tumors averaged 80–100mm3, they were grouped and treated with human control or anti-EMP2 mAbs at 10mg/kg twice a week. N=4. Two way ANOVA, p=0.001. *, significance by Bonferroni’s multiple comparisons test. Right, Following treatment, tumors were dissociated, and some of the cells monitored for ALDH activity via the ALDEFLUOR assy. Results are expressed as the average ± SEM. N=4. G. A single cell suspension of the resected primary tumor initially treated with control or anti-EMP2 mAbs was reinjected into secondary animals at either 500, 5000, or 50,000 cells. No additional treatments were given. In animals reinjected with 50,000 cells, tumor load was monitored and the mean± SEM illustrated. N=4. Two way ANOVA, p=0.001. *, significance by Bonferroni’s multiple comparisons test.
To corroborate these findings, changes in ALDH activity were next determined using the ALDEFLUOR assay. MDA-MB-468, SKBR3, and MDA-MB-231 cell lines were incubated with anti-EMP2 or control mAbs for 24 hours, and changes in ALDH activity measured relative to the DEAB inhibitor. In all three cell lines, anti-EMP2 mAbs significantly reduced ALDH activity by 50–80% compared to those treated with control mAbs (Fig 5B).
We next tested the ability of the anti-EMP2 mAbs to reduce the self-renewal capability of CSC using a tumorsphere assay. Initially, 5000 BT474 cells were treated with saline or 50 μg/ml control human or anti-EMP2 mAbs and cultured for two weeks under low adherence conditions. Anti-EMP2 mAbs significantly reduced tumorsphere forming efficiency compared to vehicle and mAb controls (Fig 5C). To extend upon these observations, we tested the ability of the anti-EMP2 mAbs to reduce the self-renewal capability of CSC using a single cell tumorsphere assay. 1, 5, or 10 live cells were incubated with 10μg/ml of control or anti-EMP2 mAb, and viable tumorspheres enumerated after two weeks (Table 2). Treatment with anti-EMP2 mAbs significantly inhibited tumorsphere formation in the multiple cell lines tested.
Table 2. Tumorsphere formation of breast cancer cells treated with Anti-EMP2 mAb.
Either 1, 10 or 50 live cells were FACS sorted into 50uL of Enhanced serum-free media (+/− 10ug/mL Anti-EMP2 mAb) per well in non-treated 384-well plates. Spheres were counted 2 weeks later. The frequency of tumorsphere-initiating cells (TIC) was calculated using the online Extreme Limiting Dilution Analysis (ELDA) calculator [http://bioinf.wehi.edu.au/software/elda/index.html]. P-values were calculated by testing for inequality in frequency between multiple groups.
| # spheres / wells | Treatment | 1 cell / well | 10 cells / well | 50 cells / well | 1 / TIC frequency (95 CI) | P-value |
|---|---|---|---|---|---|---|
| BT474 | Control | 49/128 | 113/128 | 126/128 | 1 / 7.62 cells (6.82 – 8.5) | 5.05e-15 |
| 40/128 | 96/128 | 118/128 | ||||
| 22/128 | 105/128 | 120/128 | ||||
| Anti-EMP2 mAb | 30/128 | 96/128 | 113/128 | 1 / 12.99 cells (11.69 – 14.4) | ||
| 27/128 | 84/128 | 108/128 | ||||
| 40/128 | 94/128 | 112/128 | ||||
| SKBR3 | Control | 31/120 | 109/120 | 144/144 | 1 / 3.31 cells (2.93 – 3.73) | 2.70e-32 |
| 32/120 | 118/120 | 144/144 | ||||
| 24/120 | 118/120 | 144/144 | ||||
| Anti-EMP2 mAb | 12/120 | 66/120 | 143/144 | 1 / 8.97 cells (8.02 – 10.03) | ||
| 17/120 | 83/120 | 144/144 | ||||
| 15/120 | 87/120 | 144/144 | ||||
| SUM149 | Control | 171/384 | ND | ND | 1 / 1.98 cells (1.81 – 2.18) | 5.1e-05 |
| 144/384 | ND | ND | ||||
| 141/384 | ND | ND | ||||
| Anti-EMP2 mAb | 126/384 | ND | ND | 1 / 2.64 cells (2.38 – 2.93) | ||
| 127/384 | ND | ND | ||||
| 110/384 | ND | ND | ||||
Several groups have shown that CSC subpopulations can arise during tumor progression(35). To study the emergence of CSC subpopulations during this process in vivo, we next tested the ability of the anti-EMP2 mAbs to target xenograft derived cells. MDA-MB-231 and primary HCI-002 xenografts were created, and cells were isolated from harvested tumors and plated as above. In both cases, anti-EMP2 mAbs significantly inhibited tumorsphere formation compared to control mAbs (Fig 5D).
Anti-EMP2 mAb reduces tumor growth and tumor initiating potential
Our results thus far suggested that anti-EMP2 mAbs may successfully reduce markers and features associated with BCSC. To translate this observation, we utilized the models tested above to determine the effects of anti-EMP2 mAbs in vivo. Initially, primary HCI-002 tumor pieces were implanted into NOD/SCID animals, and when tumors reached ~50–75mm3, treatment with 10mg/kg anti-EMP2 or an isotype matched control antibody was initiated. Anti-EMP2 mAb significantly reduced tumor growth of the primary tumorgrafts compared to control mAbs (Fig 5E, Two way ANOVA, p=0.02). We next determined the effects of treatment on ALDH1 and EMP2 expression using standard immunohistochemistry (Fig S3C). A quantitative reduction in the percentage and intensity of EMP2 and ALDH1 expressions were observed post anti-EMP2 mAb treatment compared to the control as scored by a pathologist (Y.E.) masked to the treatment groups (p=0.002 and p=.04, respectively; Fig 5E, right).
To extend upon these observations, a second model was created using MDA-MB-231 cells. In two independent experiments, treatment with anti-EMP2 mAb significantly reduced tumor load compared to treatment with control human mAbs (Fig 5F; Fig S3D-E). As the “gold standard” assay for CSC is their ability to initiate tumors in secondary mice, two assays were performed on remnant cells of the tumor to determine if anti-EMP2 mAbs were sufficient to prevent tumor re-initiation. After tumor cell disassociation, cells were either analyzed for ALDH activity or reinjected into naïve animals to monitor secondary tumor formation. Treatment with anti-EMP2 mAb produced a marked reduction in ALDH activity as measured by the ALDEFLUOR assay compared to those treated with human control mAbs (Fig 5F, right). To further assess the ability of remnant cells to initiate tumor formation, we injected serial limiting dilutions (50,000, 5000 or 500 cells) into nude mice and monitored tumor formation without any further treatment. Fig 5G follows tumor re-initiation from cells injected with 50,000 cells, and results indicate that anti-EMP2 mAbs dampen the efficiency of tumor re-initiation. Similarly, at all three doses of cells, a significant inhibition in secondary tumor formation was observed post anti-EMP2 treatment compared to the control suggesting that anti-EMP2 mAbs effectively target a significant portion of BCSC within this tumor model (Table S5).
Anti-EMP2 mAb reduces tumor metastasis
It has been suggested that CSC dictate the fate and behavior of the tumor(35). As in silico analysis suggested an enrichment of genes involved in metastasis with higher EMP2 levels, we determined whether anti-EMP2 mAbs could inhibit metastasis in an experimental mouse xenograft model. Luciferase labelled MDA-MB-231 cells were injected into the left ventricle of Balb/c nude mice, and animals were treated twice weekly with 10mg/kg control or anti-EMP2 mAbs. Tumor metastasis was monitored using bioluminescence imaging. As shown in Fig 6A, anti-EMP2 treatment selectively produced a quantitative reduction in tumor metastasis.
Figure 6. Anti-EMP2 therapy reduces tumor growth and metastasis.
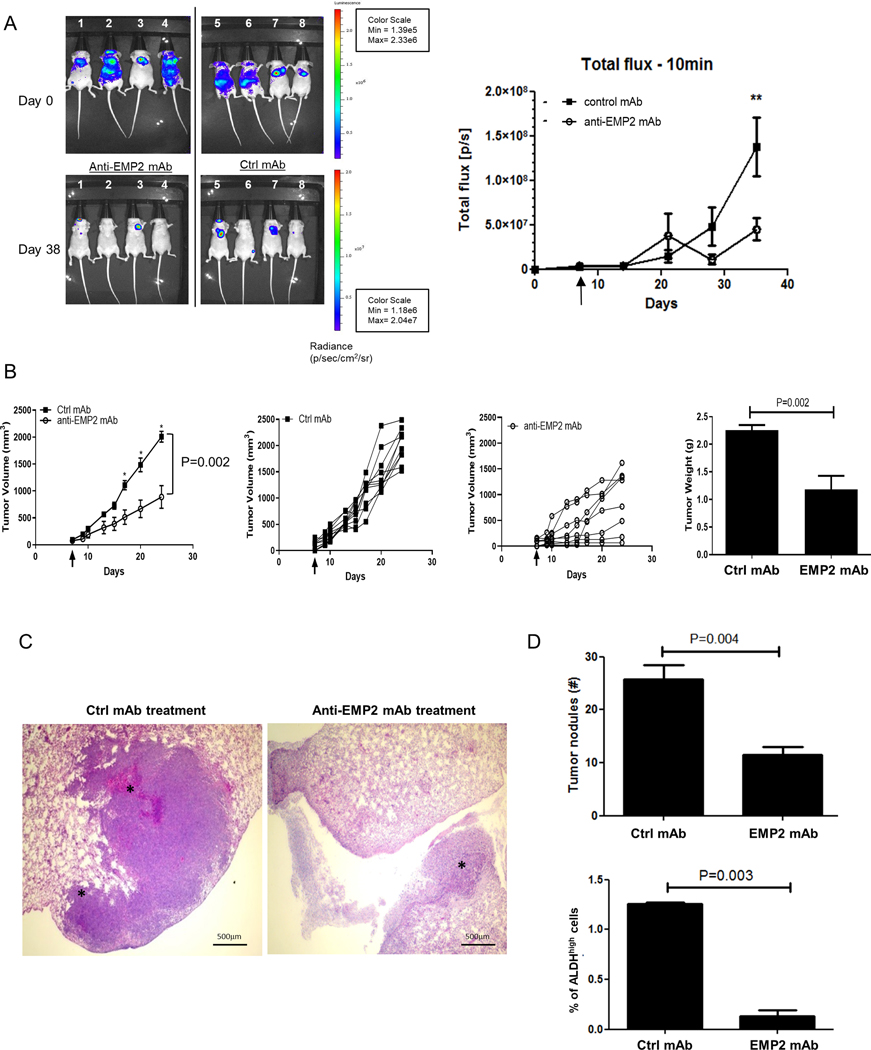
A. In order to create a model of metastasis, MDA-MB-231/Luc+ cells were injected into the left ventricle of Balb/c nude mice, and tumor load was determined using bioiluminescence. Mice were treated systemically, beginning at day 5, with 10mg/kg anti-EMP2 IgG1 or control human IgG. Data was analyzed using the maximum photon flux emission (photons/second) throughout the whole animal. N=4. *, p<0.05 as determined by Student’s t test. B. 5×104 4T1-FLUC cells were implanted into the mammary fat pad of Balb/c immunocompetent mice and treated twice a week with 10mg/kg control human mAbs or anti-EMP2 mAb starting on day 7 when tumors approached 75–100mm3. The arrow demarks treatment initiation, with grouped and individual data shown. N=8. Two way ANOVA, p=0.002. *, significant by Bonferroni post test. Right, Animals were sacrificed at day 24, and tumors weighed. Mean tumor weight for the control mAb treated group was 1.5±0.3 while the mean tumor weight for the anti-EMP2 mAb was 0.95± 0.1 (p=0.04, Mann Whitney test). N=8. C. In order to assess pulmonary metastases, lungs were removed and stained with hematoxylin and eosin. Representative images are shown. Magnification=400X. D. Top, Enumeration of tumor nodules per 4x field. p=0.004, Student’s t test. Below, For half of the animals, ALDH activity was measured using the ALDEFLUOR assay in the remaining primary tumor. Average results are shown from 4 animals ± SEM, with significance determined using Student’s t test.
Previous studies have shown that anti-human EMP2 mAbs cross react with murine EMP2(13), and we thus hypothesized that these mAbs may reduce the metastatic burden in a 4T1 syngeneic model. Given that these animals have an intact immune system, they may also be more clinically relevant. 4T1 cells labelled with firefly luciferase (FLUC) were injected into the mammary fat pad of Balb/c immunocompetent mice, and treatment was initiated when tumors reached 75–100mm3. Treatment with anti-EMP2 mAbs significantly reduced primary tumor load compared to those treated with control mAbs (Fig 6B; Two way ANOVA, p=0.002), and a reduction in tumor weight was also computed following anti-EMP2 mAb treatment (right panel; p=0.002). To further interrogate these results, metastatic burden was assessed using the ALDEFLUOR assay on the primary tumor, and lung nodules were enumerated manually by counting 4X fields from 5 tumors (Fig 6C-D). Treatment with anti-EMP2 mAbs resulted in a marked reduction in lung metastasis, and representative images of lung metastasis post treatment with either the Ctrl or anti-EMP2 mAbs are shown in Fig 6C. Enumeration of nodules showed a greater than 2-fold reduction in tumor lesions in the lung (p=0.004) with an average of 25.8±2.7 nodules counted following Ctrl mAb treatment compared to 11.5±1.6 nodules following anti-EMP2 mAb treatment (Fig 6D, top). We next correlated ALDH activity using the ALDEFLUOR assay with the number of lung nodules. Anti-EMP2 mAbs produced a significant reduction in ALDH activity, supporting previous studies that ALDH activity serves as a surrogate for metastatic potential (8,36) (p=0.003; Fig 6D bottom). Collectively, our results identify EMP2 as an attractive therapeutic target and demonstrate that anti-EMP2 mAbs effectively inhibit the BCSC population reducing tumor initiation, growth and metastasis across a spectrum of breast cancer models.
Discussion
Over the last decade, significant developments have furthered the understanding of intra- and inter-lesional tumor cell heterogeneity. Contributing to this heterogeneity are cellular hierarchies which resemble those established during normal development and organogenesis. At the apex of these hierarchies are “stem like” cells that may drive tumor initiation and metastasis. These cells may also contribute to the resistance of tumors to a variety of cancer therapeutic agents(4,6,35), highlighting the need to identify novel therapeutic approaches capable of targeting this cell population. In these studies, we identify EMP2 as an important regulator of BCSC and demonstrate that EMP2 genetic knockdown or inhibition with a mAb reduces the proportion of CSC in vitro and in vivo using mouse xenograft models. Although the mechanism of EMP2 regulation is not known, this study supports and extends the observation that EMP2 may help regulate HIF-1α expression(16), a transcription factor critical for the cellular response to hypoxia(22). We have recently demonstrated that HIF-1 regulates ALDH+ BCSC’s through an AMPK dependent pathway(37). Although our data demonstrates that EMP2 is upstream of HIF-1α and helps stabilize and/or promote its expression, it does not establish a clear causal relationship between the two.
Within breast cancer, EMP2 was predominantly expressed on the membrane and cytoplasm of the tumor parenchyma with low levels of protein observed in histologically “normal” adjacent mammary tissue. In disease progression, no differences in EMP2 expression were observed in patients with IDC who also had foci diagnosed as DCIS. This is not completely unexpected as several studies have shown that the regions up to a 1cm from the margin of the tumor display abnormalities relative to normal, non-tumor bearing tissue(33). These results suggest that overexpression of EMP2 may be an early event in carcinogenesis, or it may simply reflect the similarities of high grade DCIS with IDC.
ALDH1 expression, in contrast, displayed a heterogeneous staining pattern within the tumor parenchyma, with tumor nests visualized in some patients. Although all ALDH1 positive tumor cells were EMP2 positive, no clear correlation between EMP2 and ALDH1 expression intensity occurred within patients’ primary tumors. In contrast to this, EMP2 and ALDH1 expression correlated within metastatic lymph nodes of patients. Several possibilities may account for this apparent discrepancy. First, we have reported that while ALDH1 is a marker of epithelial proliferative BCSC, more mesenchymal BCSC are characterized by CD44+/CD24- expression(21). BCSC’s maintain the plasticity to transition between these states in a process regulated by the tumor microenvironment(26). Thus, it may simply be that the tumors analyzed were captured in a mesenchymal state. A second possibility is that ALDH1 expression is insufficient to independently characterize CSC. Historically, ALDH1A1 and ALDH1A3 have both been associated in BCSC identification(38), although in recent years, many other family members such as ALDH2, ALDH3A1, ALDH5A1, and ALDH7A1 have been implicated as well(39). Consistent with this possibility, in SUM149, differential mRNA expression was observed in 10 different ALDH isoforms. A final possibility may involve temporal regulation for ALDH1 in disease progression, with its induction occurring after the upregulation of EMP2. This possibility would be consistent with reports that ALDH1 expression correlates with larger tumor size(40).
One defining feature of BCSC is therapy resistance, and in this regard, data presented here and in the literature suggest that EMP2 may play a significant role in this process. Several studies have documented EMP2 expression in endocrine resistant(41), Adriamycin resistant(42), as well as dasatinib resistant tumors(43). Moreover, this study furthers these associations as GSEA suggests an association between clinical upregulation of EMP2 and invasion, stemness, metastasis. Critically, anti-EMP2 mAbs inhibited many of the defining features associated with these cells. Anti-EMP2 therapy significantly reduced a number of in vitro markers associated with BCSC including tumorsphere formation and ALDH activity. Moreover, anti-EMP2 mAbs significantly reduced primary tumor formation in both primary tumorgrafts and cultured cell lines. As these mAbs cross react with murine EMP2(13), syngeneic systems were also created to determine if the immune system can enhance or inhibit these effects. Similar to the results observed in immunocompromised models, anti-EMP2 mAbs inhibited tumor growth and specifically suppressed markers associated with stem-ness including ALDH1. Finally, anti-EMP2 mAbs inhibited tumor metastasis as a significant reduction in pulmonary metastasis were observed. Collectively, given the lack of toxicity observed to date with anti-EMP2 mAbs in animal models, these results support the continued development of EMP2 inhibitors in breast cancer.
In conclusion, our data supports the role of EMP2 in BCSC formation. Additional experiments will be required to elucidate the mechanisms that regulate EMP2 expression as well as how it regulates BCSC. Nevertheless, our results suggest that EMP2 may serve as novel therapeutic target and that anti-EMP2 therapeutics may provide a novel means for BCSC elimination.
Supplementary Material
Acknowledgments
We are very thankful for the continuous support of Dr. Jason Lee in the Crump Imaging Core. This work was generously supported by Charles Drew University/UCLA NIH U54-CA-143931 (Y.E.; Y.W.; J.V.), the NIH/National Center for Advancing Translational Sciences (NCATS) UL1TR000124, NIH/NCI P50-CA211015 (M.W.), NCI R01 CA163971 (M. W.), and by grants R35 CA197585 from the NIH, Breast Cancer Research Foundation (BCRF) and Forbes Institute for Cancer Discovery to M.S.W. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30 CA016042 and 5P30 AI028697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office, and the UCLA Vice Chancellor’s Office of Research.
Footnotes
Conflict of interest
M.W., L.K.G, and J.B. are inventors on the University of California patents related to the anti- EMP2 mAb antibody presented in this work. M.W. was a scientific advisor to Oncoresponse. MSW has financial holdings and is a scientific advisor for OncoMed Pharmaceuticals, Verastem, and MedImmune and receives research support from Dompe Pharmaceuticals and MedImmune. No other authors have competing interests.
References
ADDIN EN.REFLIST
- 1.Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Seminars in cancer biology 2012;22(3):234–49 doi 10.1016/j.semcancer.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Seyfried TN, Huysentruyt LC. On the Origin of Cancer Metastasis. Crit Rev Oncog 2013;18(1–2):43–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med 2013;274(2):113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Seminars in cancer biology 2018;53:156–67 doi 10.1016/j.semcancer.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. The EMBO Journal 2017;36(3):252–9 doi 10.15252/embj.201695204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem cells international 2018;2018:5416923 doi 10.1155/2018/5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde Dehydrogenase 1–Positive Cancer Stem Cells Mediate Metastasis and Poor Clinical Outcome in Inflammatory Breast Cancer. Clinical Cancer Research 2010;16(1):45–55 doi 10.1158/1078-0432.ccr-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Scientific reports 2017;7(1):13856 doi 10.1038/s41598-017-14364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabinovich I, Sebastiao APM, Lima RS, Urban CA, Junior ES, Anselmi KF, et al. Cancer stem cell markers ALDH1 and CD44+/CD24- phenotype and their prognosis impact in invasive ductal carcinoma. European journal of histochemistry : EJH 2018;62(3) doi 10.4081/ejh.2018.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer research 2009;69(4):1302–13 doi 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu Y, Pu T, Guo P, Wei B, Zhang Z, Zhang H, et al. ALDH+/CD44+ cells in breast cancer are associated with worse prognosis and poor clinical outcome. Experimental and Molecular Pathology 2016;100(1):145–50 doi 10.1016/j.yexmp.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 12.Batlle E, Clevers H. Cancer stem cells revisited. Nature Medicine 2017;23:1124 doi 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 13.Fu M, Maresh EL, Helguera GF, Kiyohara M, Qin Y, Ashki N, et al. Rationale and preclinical efficacy of a novel anti-EMP2 antibody for the treatment of invasive breast cancer. Mol Cancer Ther 2014;13(4):902–15 doi 10.1158/1535-7163.MCT-13-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, Yao L, Burner D, Minev B, Lu L, Wang M, et al. Epithelial membrane protein 2: a novel biomarker for circulating tumor cell recovery in breast cancer. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 2019;21(4):433–42 doi 10.1007/s12094-018-1941-1. [DOI] [PubMed] [Google Scholar]
- 15.Obermayr E, Sanchez-Cabo F, Tea MK, Singer CF, Krainer M, Fischer MB, et al. Assessment of a six gene panel for the molecular detection of circulating tumor cells in the blood of female cancer patients. BMC Cancer 2010;10:666 doi 10.1186/1471-2407-10-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiyohara MH, Dillard C, Tsui J, Kim SR, Lu J, Sachdev D, et al. EMP2 is a novel therapeutic target for endometrial cancer stem cells. Oncogene 2017;36(42):5793–807 doi 10.1038/onc.2017.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Research 2011;13(4):215 doi 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121(7):2750–67 doi 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell stem cell 2015;17(3):260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast Cancer Cell Lines Contain Functional Cancer Stem Cells with Metastatic Capacity and a Distinct Molecular Signature. Cancer Research 2009;69(4):1302–13 doi 10.1158/0008-5472.can-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2016;1863(3):382–91 doi 10.1016/j.bbamcr.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YC, Ingram PN, Fouladdel S, McDermott SP, Azizi E, Wicha MS, et al. High-Throughput Single-Cell Derived Sphere Formation for Cancer Stem-Like Cell Identification and Analysis. Scientific reports 2016;6:27301 doi 10.1038/srep27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardo Y, de Giorgio A, Coombes CR, Stebbing J, Castellano L. Mammosphere formation assay from human breast cancer tissues and cell lines. Journal of visualized experiments : JoVE 2015(97) doi 10.3791/52671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem cell reports 2014;2(1):78–91 doi 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem cell reports 2014;2(1):78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al Hajj M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ginestier C, Hur M, Charafe-Jauffret E, Monville F, Dutcher J, Brown M. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007;1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeRose YS, Wang G, Lin Y-C, Bernard PS, Buys SS, Ebbert MTW, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17(11):1514–20 doi http://www.nature.com/nm/journal/v17/n11/abs/nm.2454.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon LK, Kiyohara M, Fu M, Braun J, Dhawan P, Chan A, et al. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene 2013;32(46):5369–76 doi Doi 10.1038/Onc.2012.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proceedings of the National Academy of Sciences 2014. doi 10.1073/pnas.1421438111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Kazi AA, Shah P, Schech A, Sabnis G, Chumsri S, Brodie A. Inhibiiton of non-hypoxic HIF-1 expression in letrozole-resistant breast cancer cells reduces their cancer stem cell characteristics. Cancer Research 2013;73(8 Supplement):95. [Google Scholar]
- 33.Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nature Communications 2017;8(1):1077 doi 10.1038/s41467-017-01027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collina F, Di Bonito M, Li Bergolis V, De Laurentiis M, Vitagliano C, Cerrone M, et al. Prognostic Value of Cancer Stem Cells Markers in Triple-Negative Breast Cancer. BioMed research international 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. Journal of biomedical science 2018;25(1):20– doi 10.1186/s12929-018-0426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clinical & experimental metastasis 2016;33(1):97–113 doi 10.1007/s10585-015-9755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell metabolism 2018;28(1):69–86 e6 doi 10.1016/j.cmet.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016;7(10):11018–32 doi 10.18632/oncotarget.6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res 2010;70(12):5163–73 doi 10.1158/0008-5472.can-09-3806. [DOI] [PubMed] [Google Scholar]
- 40.Kida K, Ishikawa T, Yamada A, Shimada K, Narui K, Sugae S, et al. Effect of ALDH1 on prognosis and chemoresistance by breast cancer subtype. Breast Cancer Research and Treatment 2016;156(2):261–9 doi 10.1007/s10549-016-3738-7. [DOI] [PubMed] [Google Scholar]
- 41.Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, et al. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Research 2008;68(18):7493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Györffy B, Surowiak P, Kiesslich O, Denkert C, Schäfer R, Dietel M, et al. Gene expression profiling of 30 cancer cell lines predicts resistance towards 11 anticancer drugs at clinically achieved concentrations. International Journal of Cancer 2006;118(7):1699–712 doi 10.1002/ijc.21570. [DOI] [PubMed] [Google Scholar]
- 43.Huang F, Reeves K, Han X, Fairchild C, Platero S, Wong TW, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Research 2007;67(5):2226–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.