

Abstract
The gene encoding the proto-oncogene GTPase RAS is frequently mutated in human cancers. Mutated RAS proteins trigger antiapoptotic and cell-proliferative signals and lead to oncogenesis. However, RAS also induces apoptosis and senescence, which may contribute to the eradication of cells with RAS mutations. We previously reported that Ras association domain family member 6 (RASSF6) binds MDM2 and stabilizes the tumor suppressor p53 and that the active form of KRAS promotes the interaction between RASSF6 and MDM2. We also reported that Unc-119 lipid-binding chaperone (UNC119A), a chaperone of myristoylated proteins, interacts with RASSF6 and regulates RASSF6-mediated apoptosis. In this study, using several human cancer cell lines, quantitative RT-PCR, RNAi-based gene silencing, and immunoprecipitation/-fluorescence and cell biology assays, we report that UNC119A interacts with the active form of KRAS and that the C-terminal modification of KRAS is required for this interaction. We also noted that the hydrophobic pocket of UNC119A, which binds the myristoylated peptides, is not involved in the interaction. We observed that UNC119A promotes the binding of KRAS to RASSF6, enhances the interaction between RASSF6 and MDM2, and induces apoptosis. Conversely, UNC119A silencing promoted soft-agar colony formation, migration, and invasiveness in KRAS-mutated cancer cells. We conclude that UNC119A promotes KRAS-mediated p53-dependent apoptosis via RASSF6 and may play a tumor-suppressive role in cells with KRAS mutations.
Keywords: apoptosis, KRAS, TP53, tumor suppressor, Ras association domain family member 6 (RASSF6), human retina gene 4 (HRG4), cancer, cell signaling, cell proliferation, GTPase, tumor suppressor gene, p53
Humans have two homologues of unc-119 of Caenorhabditis elegans, UNC119 and UNC119B (1, 2). UNC119 was first identified as a retina-enriched gene and was named human retina gene 4 (HRG4) (3). UNC119 is conventionally called UNC119A and has two splicing variants, UNC119Aa and UNC119Ab, which have different C-terminal sequences (4). The middle portion of UNC119A has a structure similar to that of the hydrophobic pockets of cGMP phosphodiesterase (PDEδ) and Rho GDP-dissociation inhibitor (RhoGDI), which bind lipid-modified proteins, such as RAS, RAB3, and RHO (5–8). However, unlike PDEδ and RhoGDI, UNC119A interacts with myristoylated proteins, including SRC, FYN, LCK, ABL1, ABL2, and GNAT (transducin-α) (9–13). UNC119A also binds RIBEYE, encoded by CTBP2, a component of ribbon synapses, and ARL2/3, the GTP-binding proteins (2, 5, 14). A series of studies have revealed that UNC119A plays versatile roles in ribbon synapse formation, T cell activation/differentiation, myoblast differentiation, cilia formation, endocytosis, cytokinesis, SRC activation, and ABL inhibition (2, 9, 11, 14–19).
UNC119A is reported to promote hepatocellular carcinoma through WNT signaling (20). A small molecule that interferes with the interaction between UNC119A and SRC has attracted attention as a potential anticancer drug (21). However, in certain cancers, a low expression of UNC119A is associated with poor patient prognosis (22). We identified UNC119A as a binding protein of RASSF6, one of the members of tumor suppressor RASSF proteins (22). RASSF6 has a Ras association (RA) domain, binds the GTP-bound form of RAS, and is involved in RAS-mediated apoptosis and senescence (23). RASSF6 also has a coiled-coil motif, named the SARAH domain, and interacts with mammalian Ste20-like kinases, the core kinases of the tumor suppressor Hippo pathway (24). In a previous study, we revealed that UNC119A promotes the binding of RASSF6 to MDM2, inhibits the MDM2-mediated p53 degradation, and functions as a tumor suppressor (22). In this study, we investigated the implication of RAS in the UNC119A-RASSF6-MDM2-p53 axis. We found that UNC119A binds KRAS in a nucleotide-dependent manner. Mutation in the C-terminal cysteine of KRAS abrogates the interaction; however, UNC119A with mutations in the hydrophobic pocket still binds KRAS, suggesting that farnesylated KRAS binds to UNC119A in a different manner than that of the myristoylated proteins. We also demonstrated that UNC119A enhances the interaction between RASSF6 and MDM2 via KRAS and promotes the KRAS-induced p53-mediated apoptosis.
Results
UNC119A interacts with KRAS
PDEδ, a KRAS-binding protein, has a hydrophobic pocket into which the farnesylated peptide is inserted. Although PDEδ and UNC119A have similar structures, the residues forming the hydrophobic pockets of PDEδ and UNC119A are only partially conserved. Accordingly, a previous study using fluorescent synthetic peptides revealed that UNC119A binds only the myristoylated peptides and does not interact with the prenylated peptides (25). However, when we performed immunoprecipitation with anti-UNC119 antibody using the lysates of SW480 cells harboring KRASB G12V, KRAS was detected in the immunoprecipitates (Fig. 1A). To further characterize the interaction, we exogenously expressed various FLAG-UNC119 proteins and Myc-KRASB G12V in HEK293FT cells and immunoprecipitated Myc-KRASB G12V. UNC119Aa, UNC119Ab, and UNC119B coimmunoprecipitated with KRASB G12V (Fig. 1B). As the interaction with UNC119Aa was weaker than that with UNC119Ab, we focused on UNC119Ab. UNC119B, which was previously demonstrated not to bind RASSF6, interacted with KRASB G12V (22). Therefore, it is unlikely that RASSF6 is involved in the interaction between KRASB G12V and UNC119A. Neither RASSF6 silencing nor RASSF6 coexpression had any effects on the interaction between UNC119Ab and KRASB G12V (Fig. S1A and B). Compared with the WT KRASA and KRASB, the active mutants, KRASA G12V and KRASB G12V, interacted with UNC119Ab more efficiently, but the negative mutant, KRASB S17N, did not (Fig. 1C). This is in stark contrast to PDEδ, which does not recognize the GTPase switch region and interacts with RAS, independent of the GTP/GDP-bound state (6, 26, 27). Among RAS-related proteins, HRAS and NRAS also interacted with UNC119Ab, whereas RAP1B did not (Fig. 1D).
Figure 1.

Interaction between UNC119A and RAS proteins. A, UNC119A was immunoprecipitated (IP) from SW480 cells and immunoblotted (IB) with anti-UNC119 and anti-KRAS antibodies. Single and double asterisks indicate IgG heavy and light chains, respectively. UNC119A and KRAS are indicated with arrows. B, C, and D, Myc-tagged proteins were coexpressed with FLAG-tagged proteins in HEK293FT cells, and 48 h after transfection, immunoprecipitation was performed with anti-Myc antibody. B, pCIneoMyc-KRASB G12V, pCIneoFH-UNC119Aa (FLAG-UNC119Aa), pCIneoFHF-UNC119Ab (FLAG-UNC119Ab), and pCIneoFHF-UNC119B (FLAG-UNC119B) were used. UNC119Aa, UNC119Ab, and UNC119B were coimmunoprecipitated with KRASB G12V. C, pCIneoMyc-KRASA WT, pCIneoMyc-KRASA G12V, pCIneoMyc-KRASB WT, pCIneoMyc-KRASB G12V, pCIneoMyc-KRASB S17N, and pCIneoFHF-UNC119Ab (FLAG-UNC119Ab) were used. KRASA G12V and KRASB G12V more efficiently trapped UNC119Ab than KRASA WT and KRASB WT. KRASB S17N did not bind UNC119Ab (the last lane). D, pCIneoMyc2-KRASA WT, pCIneoMyc2-KRASB WT, pCIneoMyc2-HRAS WT, pCIneoMyc2-NRAS WT, pCIneoMyc2-RAP1B, and pCIneoFHF UNC119Ab (FLAG-UNC119Ab) were used. KRASA and NRAS efficiently bound UNC119Ab compared with KRASB and HRAS. The interaction between UNC119Ab and RAP1B was not detected. Five experiments for panels A and D and three experiments for panels B and C were performed by two members.
Molecular requirements of KRAS to interact with UNC119Ab
To characterize the difference between KRASB and RAP1B, we prepared chimeric constructs (Fig. 2A). The chimeric proteins, KRASB G12V(1–63)/RAP1B(64–184), KRASB G12V(1–76)/RAP1B(77–184) and KRASB G12V(1–86)/RAP1B(87–184), could bind to UNC119Ab, whereas KRASB G12V(1–60)/RAP1B(61–184) did not (Fig. 2B). This result implies that the N-terminal region of KRASB covering 63 residues is involved in the interaction. In this regard, UNC119Ab is different from PDEδ, which binds only the C-terminal region of KRAS (28). The C-terminal region of KRASB undergoes farnesylation at Cys185, followed by the removal of 3 amino acids and carboxymethylation (29). KRASA is similarly modified but is additionally palmitoylated at Cys180 (30). As we expected that UNC119Ab would bind KRAS independently of the C-terminal modification, we examined the interaction of UNC119Ab with KRASB G12V C185S, which does not undergo farnesylation and subsequent modification (Fig. 2A). Surprisingly, KRASB G12V C185S did not bind to UNC119Ab (Fig. 2C). Similarly, KRASA G12V C186S did not bind to UNC119Ab, whereas KRASA G12V C180S did (Fig. 2D). Moreover, atorvastatin, which suppresses farnesylation, attenuated the binding of KRAS G12V to UNC119Ab (Fig. 2E). Hence, the C-terminal modification of KRASB is required for the interaction with UNC119Ab. To exclude the possibility that UNC119Ab binds KRASB via PDEδ, we knocked down PDE6D and confirmed that the interaction between UNC119Ab and KRASB was not suppressed (Fig. S1C).
Figure 2.

Molecular requirement of KRAS for the interaction with UNC119Ab. A, schematic diagram of KRAS (dark gray) and RAP1B (light gray) constructs. C, G, S, and V stand for cysteine, glycine, serine, and valine, respectively. Numbers indicate amino acid residue numbers. B, C, D, and E, Myc-tagged proteins were coexpressed with FLAG-UNC119Ab in HEK293FT cells, and 48 h after transfection, immunoprecipitation was performed with anti-Myc antibody. B and C, pCIneoMyc-KRASB G12V, pCIneoMyc-2 KRASB G12V, pCIneoMyc2-KRASB G12V 60/RAP1B, pCIneoMyc2-KRASB G12V 63/RAP1B, pCIneoMyc2-KRASB G12V 76/RAP1B, pCIneoMyc2-KRASB G12V 86/RAP1B, pCIneoMyc-KRASB G12V C185S, and pCIneoFHF-UNC119Ab (FLAG-UNC119Ab) were used. KRASB G12V 63/RAP1B bound to UNC119Ab, but KRASB G12V 60/RAP1B did not. KRASB G12V C185S did not bind UNC119Ab either. D, pCIneoMyc-KRASA G12V, pCIneoMyc-KRASA G12V C180S, pCIneoMyc-KRASA C186S, pCIneoMyc-KRASA G12V C180/186S, and pCIneoFHF-UNC119Ab (FLAG-UNC119Ab) were used. KRASA lacking C186 did not interact with UNC119Ab. E, HEK293FT cells were pretreated with the indicated concentration of atorvastatin for 18 h, and then transfection was performed. Cells were cultured in the medium containing the same concentration of atorvastatin until immunoprecipitation. The signals were measured by use of ImageJ (numbers under the image). Three experiments for panels B, D, and E and five experiments for panel C were performed by two members.
Molecular requirement of UNC119Ab to interact with KRASB G12V
We next analyzed the KRAS-binding region of UNC119Ab. UNC119A has 2 beta sheets composed of 9 beta strands (12). Similar to KRAS, ARL3 interacts with UNC119A depending on the GTP-GDP state. A previous study suggested that the switch I region of ARL3 binds to Lys92 and Arg94, whereas the interswitch region and switch II region bind to the residues between Phe179 and Asp195, which cover the 7th beta strand. We mutated Lys92/Arg94 to alanine and Phe177/Phe179/Phe181 to aspartic acid in UNC119Ab (UNC119Ab K92/R94A and UNC119Ab F177/F179/F181D). The latter mutation decreased the interaction, whereas the former had no effect (Fig. 3A and B, arrow). We also mutated Tyr137/Tyr194/Phe196 to aspartic acid (UNC119Ab Y137/Y194/F196D). Although these residues are important in forming the hydrophobic pocket, the mutant could bind KRASB G12V (Fig. 3C). To exclude the possibility that these mutations have undesired consequences, we confirmed that there was no change in the stabilities of UNC119Ab F177/F179/F181D and UNC119Ab Y137/Y194/F198D and that these proteins interacted with RASSF6 as well as the WT of UNC119Ab (Fig. S2). UNC119A interacts with myristoylated tyrosine kinases, such as ABL1 and SRC. During this study, we found that UNC119Ab is tyrosine phosphorylated by ABL1 and SRC. UNC119Ab has 5 tyrosine residues. To determine which residue is phosphorylated, we prepared 5 mutants in which each tyrosine was mutated to phenylalanine. We found that only UNC119Ab Y194F was not phosphorylated (Fig. S3A, arrow). We hypothesized that if the hydrophobic pocket is important for the interaction, phosphorylation of Tyr194 should attenuate the interaction. We coexpressed FLAG-tagged UNC119Ab and Myc-tagged KRASB G12V with N terminus-truncated ABL1b (ABL1b del N), which is more active than the full-length ABL1b, and performed immunoprecipitation analysis. ABL1b-del N did not weaken but rather strengthened the interaction (Fig. S3B, left). SRC similarly enhanced the interaction (Fig. S3B, right). The immunoblotting with anti-phosphotyrosine antibody confirmed that tyrosine-phosphorylated UNC119Ab coimmunoprecipitated (Fig. S3B, P-Tyr, arrows).
Figure 3.

Molecular requirement of UNC119Ab for the interaction with KRASB. A, B, and C, various FLAG-tagged UNC119Ab proteins were coexpressed with GFP- or Myc-KRASB G12V in HEK293FT cells. pCIneoFHF-UNC119Ab, pCIneoFHF-UNC119Ab K92/R94A, pCIneoFHF-UNC119Ab F177/F179/F181D, pCIneoFHF-UNC119Ab Y137/Y194/F196D, pCIneoGFP-KRASB G12V, and pCIneoMyc-KRASB G12V were used. KRASB G12V bound to UNC119Ab K92/R94A (A) and UNC119Ab Y137/Y194/F196D (C) but not UNC119Ab F177F179F181D (B) (arrow). The interaction of KRASB G12V with UNC119Ab Y137/Y194/F196D, in which Y194 was mutated to aspartic acid, was slightly high. Three experiments for panels A, B, and C were performed by two members.
Localization of KRASB on the plasma membrane may be important for the interaction
The above findings support the notion that the C terminus of KRASB is involved in the interaction but binds to a region other than the hydrophobic pocket of UNC119Ab. Because of the observed requirement of the C-terminal modification of KRASB, we considered the possibility that the membrane anchoring of KRASB is a prerequisite for the interaction with UNC119Ab. To address this question, we fused the N-terminal sequence of PSD-95, which is palmitoylated, to KRASB G12V C185S (Palm-Myc-KRASB G12V C185S) (31). We also prepared Myr-GFP-KRASB G12V C185S with the mouse Lck-derived myristoylation signal sequence at the N terminus. Both Palm-Myc-KRASB G12V C185S and Myr-GFP-KRASB G12V C185S were recovered in the membrane fraction, whereas Myc-KRASB G12V C185S was exclusively detected in the soluble fraction (Fig. S4, A and B). However, as shown by the immunofluorescence analysis, Myr-GFP-KRASB G12V C185S and Palm-Myc-KRASB G12V C185S were differentially distributed. The former was accumulated on the plasma membrane, whereas the latter was not (Fig. S4C). As shown in the immunoprecipitation analysis, UNC119Ab could bind Myr-GFP-KRASB G12V C185S but not Palm-Myc-KRASB G12V C185S (Fig. 4, A and B, arrow). Control myristoylated-GFP did not bind to UNC119Ab. Myristoylated-GFP-KRASB G12V C185S could bind to UNC119Ab Y137/Y194/F196D (Fig. 4C). Hence, the possibility that the myristoylated N terminus artificially fits into the hydrophobic pocket of UNC119Ab can be excluded. These findings suggest that the plasma membrane localization is important for the interaction between KRAS and UNC119Ab.
Figure 4.
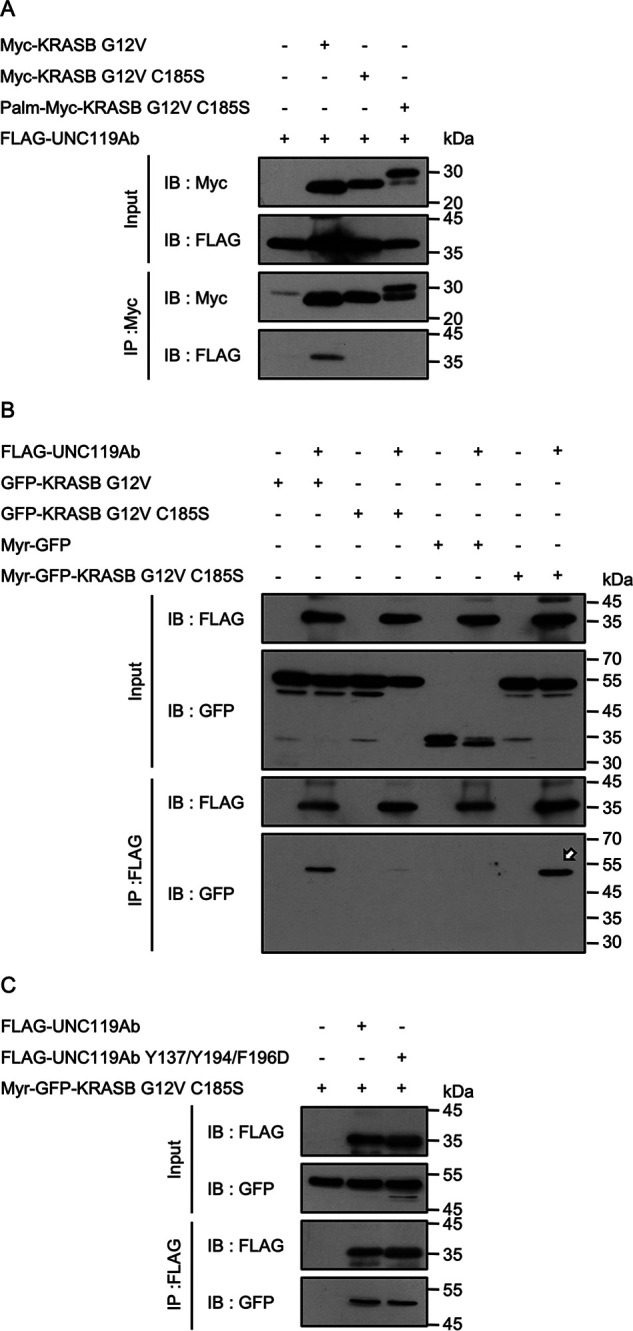
The effect of lipid modification on the interaction between KRASB and UNC119Ab. A, Myc-KRASB G12V, Myc-KRASB G12V C185S, and Palm-Myc-KRASB G12V C185S were coexpressed with FLAG-UNC119Ab in HEK293FT cells. Immunoprecipitation was performed with anti-Myc antibody. UNC119Ab coimmunoprecipitated with neither Myc-KRASB G12V C185S nor Palm-Myc-KRASB G12V C185S. B, FLAG-UNC119Ab was coexpressed with GFP-KRASB G12V, GFP-KRASB G12V C185S, Myr-GFP, and Myr-GFP-KRASB G12V C185S in HEK293FT cells and was immunoprecipitated. GFP-KRASB G12V and Myr-GFP-KRASB G12V C185S coimmunoprecipitated with FLAG-UNC119Ab (arrow), whereas GFP-KRASB G12V C185S or Myr-GFP was not. C, Myr-GFP-KRASB G12V C185S coimmunoprecipitated with FLAG-UNC119Ab Y137/Y194/F196D, which excludes the possibility that the artificial myristoylated sequence binds to the hydrophobic pocket. Four experiments for panel A and three experiments for panels B and C were performed by two members.
UNC119Ab enhances the interaction between RASSF6 and KRASB
As shown in Fig. S1, RASSF6 had no effect on the interaction between UNC119Ab and KRASB G12V. As a reverse experiment, we examined the effect of UNC119A silencing and UNC119Ab coexpression on the interaction between RASSF6 and KRASB G12V. UNC119A silencing attenuated the interaction (Fig. 5A, arrows), whereas UNC119Ab coexpression enhanced it (Fig. 5B, arrow). UNC119A silencing also abolished the interaction between KRASB WT and RASSF6 (Fig. S5A). UNC119Ab coexpression augmented the interaction between RASSF6 and KRASB WT (Fig. S5B). However, UNC119A silencing had no effect on the interaction between KRASB G12V and RAF1 (Fig. S5C). As shown in Fig. S3, ABL1 and SRC enhanced the binding of KRASB G12V to UNC119Ab. Consistent with this, KRASB G12V did not bind to UNC119Ab Y194F as efficiently as UNC119Ab (Fig. S6A). UNC119Ab Y194F did not enhance the interaction between RASSF6 and KRAS G12V as much as UNC119Ab, although UNC119Ab and UNC119Ab Y194F interacted with RASSF6 with similar affinity (Fig. S6B and C). This means that when KRASB does not bind to UNC119A, the interaction between KRASB and RASSF6 is compromised. We next tested the possibility that RASSF6 indirectly associates with KRAS via UNC119A. We depleted UNC119A in SW480 cells using CRISPR/Cas9. KRAS coimmunoprecipitated with RASSF6 from UNC119A-depleted cells, supporting the idea that although UNC119A enhances the interaction between RASSF6 and KRAS, RASSF6 can interact with KRAS independently of UNC119A (Fig. S7).
Figure 5.

UNC119Ab enhances the interactions between RASSF6 and KRASB G12V and between RASSF6 and MDM2. A, B, and C, Myc-tagged, FLAG-tagged, and HA-tagged proteins were expressed in HEK293FT cells as indicated. D, mCherry-MDM2 was expressed in SW480 cells. A, HEK293FT cells were plated at 8 × 105 cells/well in a 6-well plate and transfected with control siRNA (siCont.) or UNC119A siRNA (siUNC119A#1 and #2). 24 h later, cells were transfected with pCIneoMyc-KRASB G12V and pCIneoFHF-RASSF6 (FLAG-RASSF6). 48 h after transfection, immunoprecipitation was performed. UNC119A silencing, which suppressed UNC119A expression (arrowhead), abolished the interaction between RASSF6 and KRASB G12V (white arrows). B, pCIneoMyc-KRASB G12V, pCIneoFHF-RASSF6 (FLAG-RASSF6), and pCIneoFHF-UNC119Ab (FLAG-UNC119Ab) were used. In this experiment, immunoprecipitation was performed 24 h, not 48 h, after transfection. The interaction between RASSF6 and KRASB G12V was barely detectable without UNC119Ab but visible with UNC119Ab (white arrow). The signals were measured by use of ImageJ (numbers under the image). HEK293FT cells (C) and SW480 cells (D) were transfected with control siRNA (siCont.) or KRAS siRNA (siKRAS). 24 h later, transfection was performed with pCIneoFHF-RASSF6 (FLAG-RASSF6), pCIneoMyc-UNC119Ab (Myc-UNC119Ab), pCIneoHAHA-MDM2 (HA-MDM2), or iPS-mCherry-MDM2 (mCherry-MDM2). 48 h after transfection, FLAG-RASSF6 was immunoprecipitated to evaluate the coimmunoprecipitation of HA-MDM2 or mCherry-MDM2. UNC119Ab enhanced the interaction between RASSF6 and MDM2, but KRAS silencing, which suppressed KRAS expression (black arrow), abolished the effect (white arrow). Single asterisks indicate IgG light chains. Three experiments for panels A, B, and C were performed by two members. Three experiments were performed for panel D.
UNC119Ab promotes the binding of RASSF6 to MDM2 via KRAS
We previously reported that UNC119A promotes the binding of RASSF6 to MDM2 (22). We also reported that the RA domain of RASSF6 binds to MDM2 and triggers apoptosis by stabilizing p53 (32). The binding of the RA domain to MDM2 is intramolecularly inhibited by the SARAH domain, and KRASB eliminates this inhibition (32). Therefore, we surmised that UNC119Ab promotes the binding of RASSF6 to MDM2 via KRAS. As expected, UNC119Ab failed to enhance RASSF6-MDM2 interaction under KRAS silencing in HEK293FT cells (Fig. 5C, white arrow). We also performed the same experiment in SW480 cells harboring KRASB G12V and obtained similar results (Fig. 5D). These findings imply that UNC119Ab promotes the binding of KRASB G12V to RASSF6, subsequently augmenting the binding of RASSF6 to MDM2.
MDM2 is recruited to the cytoplasm in the presence of active KRASB, which depends on UNC119A and RASSF6
The nuclear export of MDM2 is required for the nuclear export of p53, as shown in HeLa cells (33). In osteosarcoma SJSA, breast cancer MCF-7, and neuroblastoma cells, leptomycin B treatment or the masking of the nuclear export signal (NES) of p53 stabilized p53, indicating that p53 is not degraded unless it is exported to the cytoplasm (34, 35). Consistent with this, the NES mutant of MDM2, which cannot export p53 to the cytoplasm, could not induce p53 degradation (33). This implies that MDM2 export antagonizes p53 signaling in these cells. However, it has also been shown that the NES mutant of MDM2 induces ubiquitination and degradation of p53 inside the nucleus in U2OS cells (36). In this case, MDM2 nuclear export segregated MDM2 from p53 and enhanced the p53 signaling. Consistent with this, the clinical study of human breast cancers concludes that the cytoplasmic MDM2-positive cases show better prognosis than the nuclear MDM2-positive cases (37). These controversial reports indicate that the effect of MDM2 export on p53 signaling is cell dependent. In HEK293FT cells, MDM2 was mainly recovered in the nucleus even when overexpressed (22), whereas in the p53-null H1299 cells, GFP-MDM2 was also detected in the cytoplasm. Notably, when mCherry-RASSF6 was coexpressed, MDM2 shifted to the cytoplasm (Fig. 6A). The mCherry-KRASB G12V mutant showed a similar effect (Fig. 6B). KRASB G12V-mediated shift was suppressed by the silencing of RASSF6 or UNC119A, indicating that KRASB G12V induces the shift of MDM2 depending on both RASSF6 and UNC119A in H1299 cells (Fig. 6C).
Figure 6.
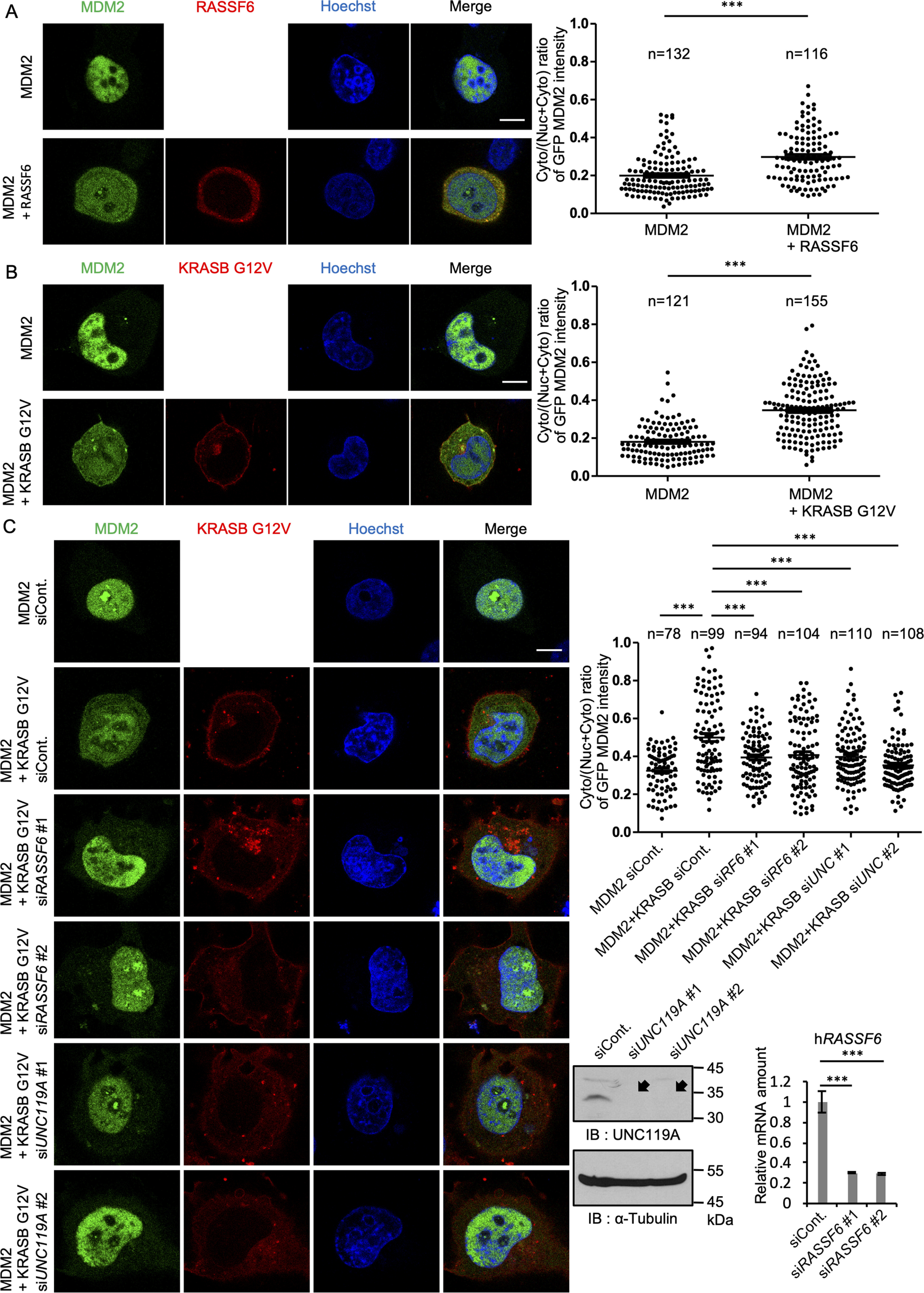
KRASB G12V induces the cytoplasmic shift of MDM2 depending on RASSF6 and UNC119A. A, B, and C, H1299 cells were transfected with pCIneoGFP-MDM2, pCIneomCherry-RASSF6, and pCIneomCherry-KRASB G12V as indicated. C, cells were first transfected with control siRNA, UNC119A siRNAs (siUNC119A#1 and #2), or RASSF6 siRNAs (siRASSF6#1 and #2). The efficiency of knockdown was evaluated for UNC119A by Western blotting and for RASSF6 by quantitative RT-PCR. 24 h later, the cells were replated in a 12-well plate and transfection was performed. A, B, and C, 24 h after transfection, cells were treated with 30 μm MG132 for 6 h and fixed. The nuclei were visualized with Hoechst 33342 (Hoechst). GFP signals in cytoplasm and nucleus were measured as described in Experimental procedures, and the ratio was calculated for each cell. Dot plots exhibit the ratios of cytoplasmic signal intensity over cytoplasmic plus nuclear signal intensity for each cell. Numbers indicate numbers of counted cells. Horizontal bars show the averages. ***, p < 0.001. Bar, 10 μm. Three experiments were performed. The data are means with S.E. Numbers of samples are shown. Statistical analyses were performed with one-way ANOVA with Tukey's test. Arcsine transformation was applied for panels A, B, and C.
UNC119Ab and KRAS cooperatively induce apoptosis, which depends on RASSF6
UNC119Ab regulates the RASSF6-MDM2-p53 axis to induce cell cycle arrest and apoptosis (22). RASSF6 synergizes with KRASB and induces p53-mediated apoptosis (23). Consistent with this, we observed that KRASB triggered the interaction between RASSF6 and MDM2 (32). As we found that UNC119Ab promotes the binding of KRASB to RASSF6, we predicted that UNC119Ab enhances KRASB-mediated apoptosis via RASSF6. We first confirmed the effect of RASSF6 on KRASB-induced apoptosis in HCT116 cells (Fig. S8). We next examined whether UNC119Ab also cooperates with KRASB to induce apoptosis. The p53-positive HCT116 cells expressing UNC119Ab exhibited nuclear condensation, cytochrome c release, and apoptosis-inducing factor release (Fig. 7A, first and second bars). Although KRASB expression did not significantly induce apoptosis, KRASB enhanced the UNC119A-induced apoptosis (Fig. 7A, second and fourth bars). RASSF6 silencing attenuated the synergistic effect of UNC119Ab and KRASB (Fig. 7B). UNC119Ab expression increased the nuclear p53 expression, which was blocked by KRAS silencing or RASSF6 silencing (Fig. 8A). KRASB WT augmented the effect of UNC119Ab on p53 expression, and RASSF6 silencing abolished the effect (Fig. 8B). As shown in the immunoblotting data, UNC119Ab increased the protein expression levels of p53, p21, and BAX, whereas silencing of KRAS and RASSF6 reduced those levels (Fig. 8C). These findings are consistent with the model that UNC119Ab promotes the binding of KRAS to RASSF6 and activates the KRAS–RASSF6–p53 axis.
Figure 7.
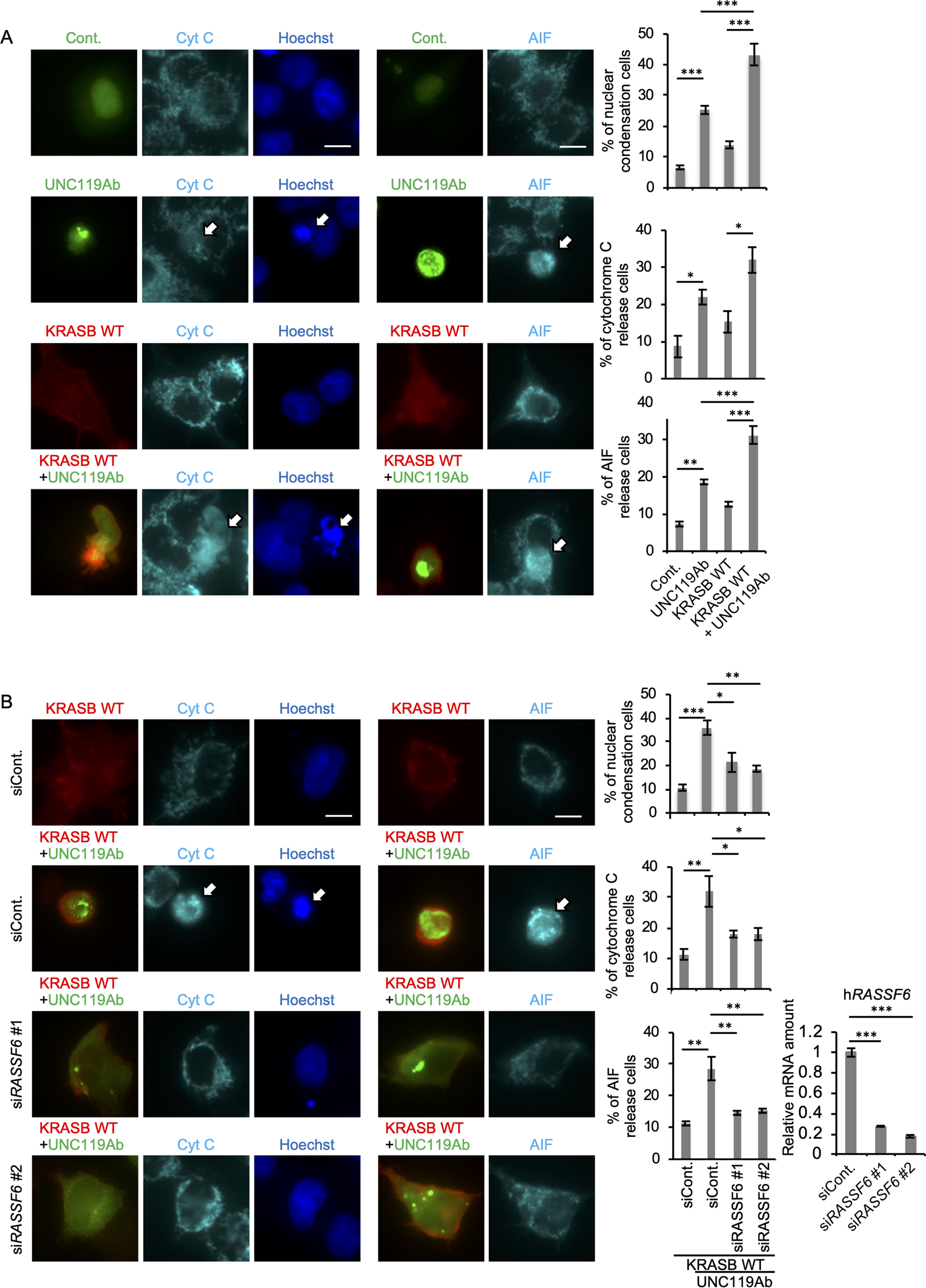
UNC119Ab and KRASB WT synergistically induce apoptosis, which depends on RASSF6. A and B, HCT116 cells were transfected with pBudCGFP-SUMO (Cont), pCIneoGFP-UNC119Ab (UNC119Ab), and pCIneoMyc2-KRASB WT (KRASB WT). B, cells were transfected with control siRNA (siCont) or RASSF6 siRNA (siRASSF6#1 and #2). 24 h later, cells were replated in a 12-well plate and transfection was performed. 24 h after transfection, cells were immunostained with anti-cytochrome c (Cyt C) and anti-apoptosis-inducing factor (AIF) antibodies. Nuclei were visualized with Hoechst 33342. 50 GFP-positive cells were observed. Ratios of cells with nuclear condensation, cytochrome c release, and AIF release were calculated. The validation of RASSF6 silencing was performed by quantitative RT-PCR. Data are means with S.E. *, p < 0.05; **, p < 0.01; and ***, p < 0.001. Bar, 10 μm. Three experiments were performed. Fifty cells were evaluated for each condition. Statistical analyses were performed with one-way ANOVA with Tukey's test.
Figure 8.
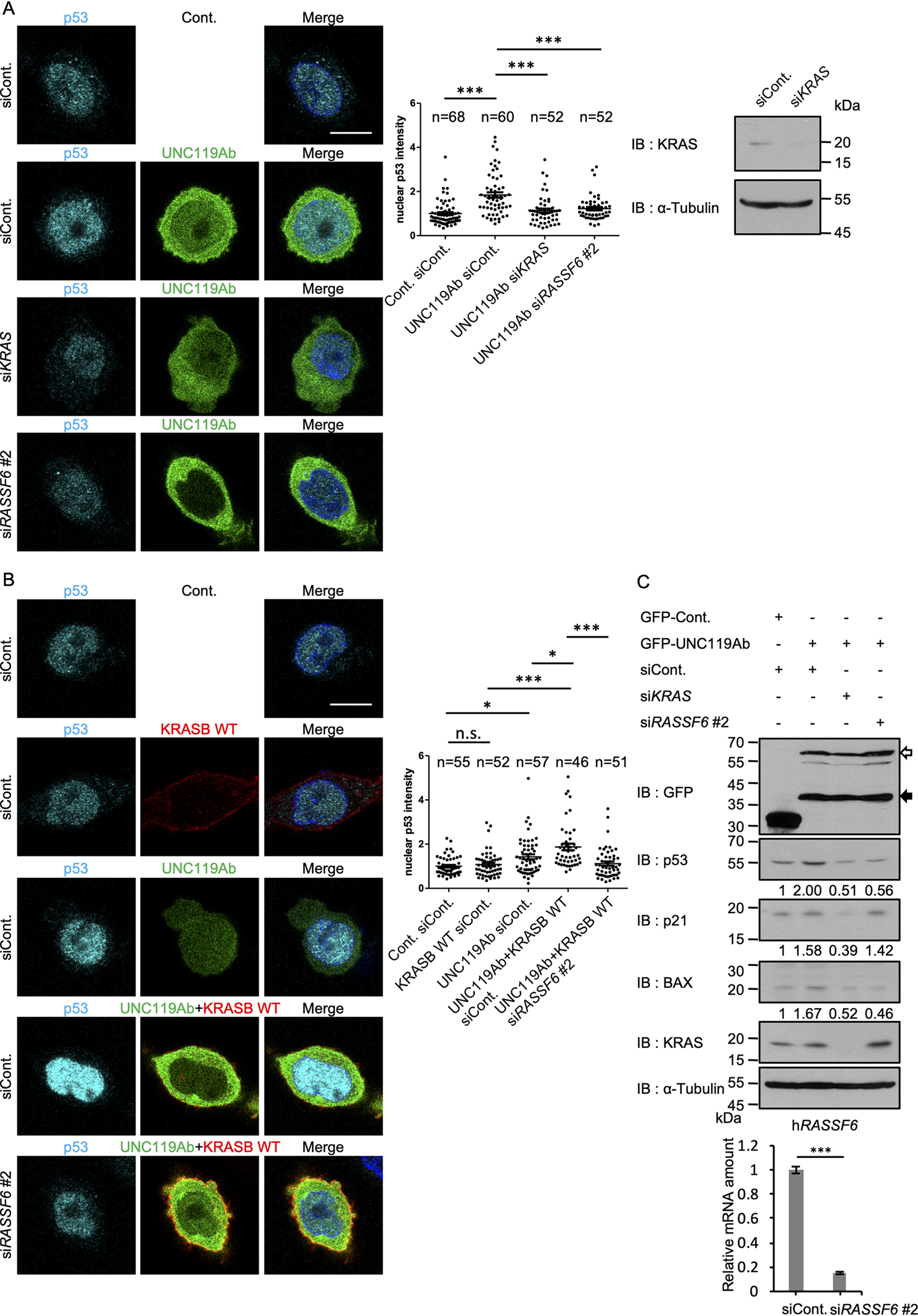
UNC119Ab and KRASB WT synergistically enhance p53 expression, which depends on RASSF6. KRAS silencing and RASSF6 silencing were performed in HCT116 cells as described for Fig. 7. GFP-UNC119Ab and Myc2-KRASB WT were expressed, and endogenous p53 was immunostained. Nuclear p53 signal was measured by ImageJ in the indicated number of cells. A, UNC119Ab augmented the signal of p53 in the nucleus (siCont), but the silencing of KRAS and RASSF6 reduced it. B, KRASB WT further enhanced the nuclear p53 signal in UNC119Ab-expressing cells (UNC119Ab+KRASB WT, siCont), whereas RASSF6 silencing abolished the effect. C, HCT116 cells were infected with retrovirus vectors harboring GFP or GFP-UNC119Ab and cultured in the medium containing 1 mg/liter of puromycin. 2 days later, the silencing of KRAS and RASSF6 was performed. 72 h later, whole-cell lysates were immunoblotted with the indicated antibodies. White and black arrows indicate GFP-UNC119Ab and its degraded product. The efficiency of knockdown was evaluated for KRAS by Western blotting and for RASSF6 by quantitative RT-PCR. The data are means with S.E. *, p < 0.05; ***, p < 0.001; n.s, not significant. Bar, 10 μm. Three experiments were performed. Numbers of samples are shown. Statistical analyses were performed with one-way ANOVA with Tukey's test.
UNC119A antagonizes oncogenic potency of KRAS
Based on the above data, we hypothesized that UNC119A plays a tumor-suppressive role in cells with KRAS mutations. To test this idea, we expressed KRASB G12V in mouse NIH3T3 cells to induce transformation and evaluated the effect of UNC119A silencing. NIH3T3 cells expressing KRASB G12V formed colonies in soft-agar culture, and UNC119A silencing significantly enhanced colony formation (Fig. 9A). We next knocked down UNC119A in human lung cancer A549 cells expressing KRASB G12S. UNC119A silencing promoted colony formation in soft-agar culture and sphere formation, and it enhanced migration and invasion in a transwell assay (Fig. 9B, C, and D). The additional silencing of KRAS abolished the effect of UNC119A silencing (Fig. 9B, C, and D). We also suppressed UNC119A and KRAS in SW480 cells and obtained similar results (Fig. S9A, B, and C). To exclude the off-target effects, we reintroduced RNAi-resistant mutants derived from UNC119A (UNC119A-R-#1, -#2) and UNC119A F177/F179/F181D (UNC119A-FD-R-#1, #2) in A549 cells (Fig. S10). UNC119A-R-#1 and -#2 suppressed the effect of UNC119A silencing with siUNC119A#1 and #2, respectively, although UNC119A-FD-R-#1 or -#2 had no effect (Fig. S11).
Figure 9.

UNC119A silencing malignant transformation in NIH3T3 cells expressing KRASB G12V and human lung cancer A549 cells. A, control and Myc-KRASB G12V-expressing NIH3T3 cells were transfected with control siRNA (siCont) or UNC119A siRNA (siUNC119A#1). Soft-agar colony formation assay was performed. Colony area was evaluated in 15 independent fields. Dot plots exhibit each value. Horizontal bars show the averages. ***, p < 0.001. The immunoblottings demonstrate the expression of Myc-KRASB G12V and the suppression of UNC119A. B, C, and D, A549 cells were transfected with control siRNA (siCont), UNC119A siRNA (siUNC119A#1), and KRAS siRNA (siKRAS). Soft-agar colony formation, sphere formation, and transwell (migration and invasion) assays were performed. The immunoblottings demonstrate the suppression of UNC119A and KRAS. B, colony formation was evaluated as described for panel A. C, cell aggregates larger than 50 µm were defined as spheres, and the number of spheres was counted in 5 independent fields. D, cells were cultured on transwell inserts without or with Matrigel. 36 h later, the membranes were fixed and the cells on the lower surface were stained with crystal violet. The intensity of staining was quantified by ImageJ in 5 independent fields. ***, p < 0.001. Scale bars, 100 μm (A, B, and C) and 200 µm (D). Three experiments were performed. The data are means with S.E. Numbers of samples are shown. Statistical analyses were performed with one-way ANOVA with Tukey's test.
Frequency of KRAS mutation is lower in cancer cells with UNC119A-high expression
To discuss the physiological relevance of our observations, we finally examined whether UNC119A contributes to the eradication of cells with KRAS mutations using data from The Cancer Genome Atlas (TCGA). We selected pancreatic, colon, and lung adenocarcinoma, which are frequently associated with KRAS mutations. In pancreatic and lung adenocarcinoma, the ratio of cancer cells with KRAS mutations was lower in cells expressing UNC119A at high levels (Table S1). In colon adenocarcinoma, although the deviation was not statistically significant, the ratio of mutations was relatively low in cells with UNC119A-high expression. In contrast, the frequencies of TP53 mutations in pancreatic adenocarcinoma and APC mutations in colon adenocarcinoma were not altered based on the UNC119A expression level. The ratio of mutations of LRP1B, which is the second most frequently mutated gene in TCGA lung adenocarcinoma, is higher in cancers with UNC119A-high expression. These observations are consistent with the assumption that UNC119A promotes apoptosis in cells with KRAS mutations.
Discussion
Although PDEδ, a chaperone for farnesylated proteins, and UNC119A have similar structures, UNC119A is a chaperone of the myristoylated proteins (38, 39). Previous studies revealed how myristoylated peptides fit into the hydrophobic pocket, whereas farnesylated peptides fail to bind the hydrophobic pocket (25). However, UNC119A also interacts with proteins that are not modified by myristoylation. RIBEYE recruits UNC119A to ribbon synapses (14). Small GTP-binding proteins, ARL2 and ARL3, which do not have myristoylated signals, also bind to UNC119A (2, 5, 40). We reported the interaction between UNC119A and RASSF6 (22). RASSF6 has an RA domain that interacts with the GTP-bound RAS (23). RAS promotes the binding of RASSF6 to MDM2 (32). RAS presumably induces a conformational change in the RA domain, which subsequently binds to the RING-finger E3 ligase region of MDM2 and blocks the MDM2-mediated p53 degradation (32). Consequently, RAS triggers p53-mediated apoptosis through the RASSF6-MDM2-p53 axis (32). In this way, RASSF6 potentially antagonizes RAS signaling and plays a tumor-suppressive role in cells with RAS mutations. We also found that UNC119A promotes the interaction between RASSF6 and MDM2 and induces apoptosis via p53 (22). Therefore, we wanted to examine whether and how the KRAS-RASSF6-MDM2-p53 axis and UNC119A-RASSF6-MDM2-p53 axis are related to each other.
We first addressed the question of whether UNC119A interacts with KRAS. We detected the coimmunoprecipitation of endogenous UNC119A and KRAS in colon cancer SW480 cells. We attempted to rule out the possibility that UNC119A and KRASB interact with each other through a certain third molecule. To this end, we confirmed that the silencing of RASSF6 or PDEδ had no effect on the interaction between UNC119A and KRASB G12V. UNC119B, which does not interact with RASSF6, also binds KRASB (22). UNC119A interacts with KRAS depending on the GTP-GDP state, whereas PDEδ binds both the GTP-bound and GDP-bound KRAS (6). These findings also support that neither RASSF6 nor PDEδ is involved in the interaction between UNC119A and KRASB.
To further confirm the interaction between UNC119A and KRAS, we mapped the molecular determinants of the interaction. Unexpectedly, KRAS mutants lacking farnesylated cysteine failed to bind to UNC119A. Nevertheless, the mutations in the hydrophobic pocket of UNC119A had no effect on the interaction, which implies that the farnesylated C-terminal region binds to the other region of UNC119A. The replacement of Phe177, Phe179, and Phe181 by aspartic acid abolished the interaction. We speculate that the sequence including these residues is involved in the interaction with KRASB. Notably, these residues are conserved in UNC119B. The interaction between UNC119Aa and KRASB is weaker than that between UNC119Ab and KRASB. A possible explanation for this observation is that the longer C-terminal region of UNC119Aa interferes with the access of KRASB to the RAS-binding region of UNC119A. Not only KRAS but also HRAS and NRAS bind to UNC119A, whereas RAP1B does not. Although RAP1B has a geranyl-geranylated cysteine, the failure to bind is not because of the difference in lipid modification, as the chimeric proteins composed of the N-terminal KRASB and C-terminal RAP1B bind to UNC119A. The N-terminal 60 amino acids of KRAS are not sufficient, and 63 amino acids must be included in the chimeric protein. Gln61 is one of the frequently mutated residues in KRAS and is involved in GTP hydrolysis (41). As UNC119A binds the GTP-bound KRAS, the region around Gln61 may be important for the interaction with UNC119A.
The interaction between RASSF proteins other than NORE1/RASSF5 and RAS proteins is weak and is still a matter of debate. However, as RASSF6 and KRAS coimmunoprecipitate from the lysates of UNC119A-depleted SW480 cells, we could conclude that RASSF6 can interact with KRAS independent of UNC119A, and UNC119A enhances the basal interaction between RASSF6 and KRAS. Interestingly, ABL1 and SRC strengthen the binding of KRASB G12V to UNC119Ab, whereas UNC119Ab Y194F does not enhance the interaction between KRASB and RASSF6 as efficiently as UNC119Ab. This finding suggests that tyrosine phosphorylation at Y194 has a regulatory role in the UNC119A–KRAS–RASSF6–MDM2–p53 axis.
The expression of UNC119A is regarded as a biomarker for predicting both good and poor prognosis in cancer patients. UNC119A promotes cell proliferation and migration in hepatocellular cancer cell lines (20). UNC119A regulates the distribution and activation of SRC (9). A small molecule targeting the interaction of UNC119A with myristoylated proteins suppresses cell proliferation in SRC-dependent cancer cells (21). On the contrary, we reported earlier that UNC119A regulates p53 through the interaction with RASSF6 and acts as a tumor suppressor in colon cancer cells (22). We have extended our observations in this study. UNC119A enhances the response of the RASSF6-MDM2-p53 axis in the presence of KRAS. Hence, UNC119A contributes to the tumor-suppressive role of RASSF6 in cancer cells with KRAS mutations. UNC119A silencing in cells expressing KRASB G12V promotes malignant transformation. The analysis of the human cancer database supports the assumption that UNC119A plays a tumor-suppressive role in cells expressing mutated KRAS proteins. In contrast, UNC119A is neutral against the mutations of TP53 and APC. The frequency of mutations of LRP1B, which is regarded as an oncogene in lung adenocarcinoma, is associated with a high expression of UNC119A (42). Therefore, to target UNC119A in cancer therapy, it is important to evaluate whether UNC119A plays an oncogenic or a tumor-suppressive role in each cancer type.
Experimental procedures
DNA constructs and virus production
pCIneoMyc, pCIneoGFP, pCIneoFHF, pCIneomCherry, pCIneoFHF-RASSF6, pCIneoFH-UNC119Aa, pCIneoFHF-UNC119Ab, pCIneoFHF-UNC119B, pCIneoGFP-MDM2, pCIneoHAHA-MDM2, pCIneomCherry-MDM2, pBudCGFP-SUMO, and pQCXIP EF were described previously (22, 32, 43–46). Oligonucleotides are listed in Table S2. Oligonucleotides (H4222 and H4223) were phosphorylated by T4 kinase, annealed, and ligated into EcoRI/MluI sites of pCIneoMyc to generate pCIneoMyc2, in which the N-terminal Myc-tag is followed by seven glycine residues. Human cDNAs of KRASA, KRASB, HRAS, NRAS, RAP1B, and RAF1 were obtained by PCR from a human lung and kidney cDNA library (Clontech) with the indicated primers (Table S2). Products were digested with MluI/Sall and ligated into the same sites of pCIneoMyc2, pCIneoFHF, and pCIneomCherry to generate pCIneoMyc2-KRASA WT, pCIneoMyc2-KRASB WT, pCIneoMyc2-HRAS, pCIneoMyc2-NRAS, pCIneoMyc2-RAP1B, pCIneoFHF-RAF1, and pCIneomCherry-C-SRC. KRASA G12V, KRASB G12V, and KRASB S17N were generated by a two-step PCR method with the indicated primers. For instance, the first PCR was performed with the primers H4065/H4069 and H4068/H4066 on pCIneoMyc2-KRASB WT. The products were isolated, mixed, and used as the template for the second PCR with the primers H4065/H4066 to generate KRASB G12V, which was ligated into MluI/Sall sites of pCIneoMyc. Other constructs with mutations were prepared in a similar manner. To generate KRASA G12V C186S and KRASB G12V C185S, PCR was conducted by using primers H4065/H4074 and H4065/H4067 on KRASA G12V and KRASB G12V, respectively. KRASA G12V C180/186S was generated from KRASA G12V C180S. Chimeric constructs of KRASB/RAP1B were also prepared by a two-step PCR method. For instance, the first PCR was performed with H4065/H4385 on KRASB G12V and H4384/H4251 on RAP1B, and the mixture of products was amplified with H4065/H4251 to generate KRASB G12V 60/RAP1B. Five UNC119Ab mutants, in which each tyrosine is replaced by alanine, were prepared by using the PrimeSTAR mutation basal kit (Takara Bio Inc.) on pCIneoFHF-UNC119Ab with the indicated primers. The other three UNC119Ab mutants, UNC119Ab K92/R94A, F177/F179/F181D, and Y137/Y194/F196D, were prepared by a two-step PCR method. For UNC119Ab Y137/Y194/F196D, Tyr137 and Tyr194/Phe196 were mutated sequentially. pLenti-CRISPR2-UNC119A CR1 was generated by ligating the indicated primers to lentiCRISPR ver2 (Addgene, 52961, a gift from Feng Zhang) (47). RNAi-resistant constructs were generated with PCR using the indicated primers to introduce three mutations in the target sequences in either UNC119Ab or UNC119Ab Y177/179/181D. The products were subcloned into pCIneoGFP and then into pQCXIP. cDNA of human ABL1b was obtained from Owen N. Witte (University of California, Los Angeles) through Yoshiro Maru (Tokyo Women's Medical University School of Medicine). PCR was performed with primers H3818/H3819 to generate ABL1b-delN, which lacks the myristoylated N terminus. To generate Palm-Myc-KRASB G12V C185S, the first PCR was performed by using H4540/H4544 on pCMV rat PSD-95 (44). The product was mixed with pCIneoMyc2-KRASB G12V C185S. The second PCR was performed with H4540/H4520. The second product was digested with NheI/NotI and ligated into the same sites of pCIneo. To generate Myr-GFP, the first PCR was performed by using H2962/H543 on pCIneoGFP. The product was ligated into NheI/NotI sites of pCIneo to generate pCIneoMyr-GFP. The MluI/Sall fragment from pCIneoMyc2 KRASB G12V C185S was ligated into the same sites of pCIneoMyr-GFP to generate pCIneoMyr-GFP-KRASB G12V C185S. NheI/NotI fragments from pCIneoGFP, pCIneoGFP-UNC119Ab, and pCIneoMyc KRASB G12V were ligated into XbaI/NotI sites of pQCXIP to generate pQCXIP-GFP, pQCXIP-GFP-UNC119Ab, and pQCXIP-Myc-KRASB G12V. The NheI/NotI fragment from pCIneomCherry-MDM2 was ligated into XbaI/NotI sites of iPSC hNanog (System Biosciences) to generate piPS-mCherry-MDM2.
Antibodies and reagents
UNC119 antibody was described previously (22). Other antibodies and reagents were obtained from commercial sources. The primary antibodies used are listed in Table S3. Other antibodies and reagents are Hoechst 33342 and epidermal growth factor (E9644) (Sigma-Aldrich, St. Louis, Dallas, TX, USA); MG-132 (09494-64) and cycloheximide (06741-04) (Nacalai Tesque, Tokyo, Japan); anti-DYKDDDDK-tag beads (016-22784), 4-amidinophenylmethanesulfonyl fluoride (APMSF), leupeptin (334-40414), basic fibroblast growth factor (064-04541), and fibronectin solution from human plasma (063-00591) (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan); Matrigel matrix basement membrane growth factor reduced (354230) (Corning Inc.); protein G-Sepharose 4 fast flow (GE Healthcare); peroxidase-conjugated anti-rabbit (0855676) secondary antibodies (MP Biomedicals, Santa Ana, CA, USA); Alexa Fluor® 488 goat anti-rabbit IgG (A11034), Alexa Fluor® 568 goat anti-rabbit IgG (A11036), and Alexa Fluor® 647 goat anti-mouse IgG (A21235) (Thermo Fisher Scientific, Waltham, MA, USA); and atorvastatin (A2476) (Tokyo Medical Industry, Tokyo, Japan).
Cell cultures and transfection
HEK293FT, HCT116, H1299, SW480, A549, and NIH3T3 cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS) and 10 mm Hepes-NaOH, pH 7.4, under 5% CO2 at 37 °C. Lipofectamine 2000 (ThermoFisher Scientific) was used for transfection. SW480 cells were infected with lentivirus vector (pLenti-CRSIRP2-UNC119A-CR1) and cultured with 1 mg/liter puromycin for 4 days. Clones were isolated and UNC119A knockout clones were selected.
Viral infection
HEK293FT cells were used for generating retrovirus and lentivirus vectors. The medium was collected and used for the infection.
Quantitative RT-PCR
Cells were harvested with TRI Reagent (Molecular Research Center, Inc.), and mRNA was extracted according to the manufacturer's protocol. cDNA was prepared from mRNA using ReverTra Ace (TOYOBO). Quantitative RT-PCR analysis was performed by using SYBR Green (Roche) and an ABI7500 real-time PCR system (Applied Biosystems). PCR conditions were 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The primers used are human GAPDH, 5′-CCACTCCTCCACCTTTGAC-3′ and 5′-ACCCTGTTGCTGTAGCCA-3′; human RASSF6, 5′-ACGTCTTCTCCAGCAAAGGA-3′ and 5′-CAGAGCTGCTTCACTCATGG-3′; and human PDE6D, 5′-ATCCTGAGGGGCTTCAAACTA-3′ and 5′-TGCACTTGAGGATTTTCTTGG-3′.
RNAi
RNA interference was performed by use of Lipofectamine RNAiMAX (Thermo Fisher Scientific). The small interfering RNAs (siRNAs) used are Silencer Select Negative Control No. 2 (4390846), human UNC119A#1 (s17350), human UNC119A#2 (s17351), human RASSF6#1 (s46639), and human RASSF6#2 (s46640) (Thermo Fisher Scientific), and human KRAS (sc-35731), mouse UNC119A (sc-75763), and human PDEδ (sc-61309) (Santa Cruz Biotechnology). The validity of the knockdown was confirmed by quantitative RT-PCR (RASSF6 and PDEδ) or immunoblotting (UNC119A and KRAS).
Western blotting
Samples were separated on 8–12% acrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% (w/v) milk in TBS–Tween buffer (50 mm Tris, 138 mm NaCl, 2.7 mm KCl, 0.1% [v/v] Tween 20, pH 7.4) and incubated overnight at 4 °C with the indicated antibodies. Membranes were washed in TBS–Tween buffer, incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature, and washed again in TBS–Tween buffer. Signals were detected using ECL (GE Healthcare) reagent and X-ray films.
Immunoprecipitation of UNC119A and RASSF6 from SW480 cells
SW480 cells at 50 to 60% confluence in two 100-mm dishes were suspended in buffer A (50 mm Tris-HCl [pH 8.0], 50 mm NaCl, 0.5% [w/v] CHAPS, 7.5 mm MgCl2, 5 mm EDTA) containing 50 μm APMSF and 10 mg/liter leupeptin and lysed by sonication at high power 6 times for 10 s each time with 1-min intervals. The lysates were centrifuged at 20,000 × g for 10 min at 4 °C. The supernatant was incubated with mouse anti-UNC119 antibody (Abnova) or control mouse IgG for 4 h at 4 °C and further incubated with protein G-Sepharose 4 fast flow beads (GE Healthcare) for 1 h at 4 °C. The beads were washed four times with buffer A, and the precipitates were analyzed by SDS-PAGE and immunoblotting with anti-UNC119 and anti-KRAS antibodies. The immunoprecipitation of RASSF6 from UNC119A knockout SW480 cells was conducted in a similar manner.
Coimmunoprecipitation from HEK293FT and SW480 cells
To detect the interaction of exogenously expressed proteins, HEK293FT cells were plated at 8 × 105 cells/well in a 6-well plate, and 24 h later, the indicated plasmids were transfected with Lipofectamine 2000 to express various tagged proteins. 48 h later, cells were lysed in buffer D (25 mm Tris-HCl [pH 8.0], 100 mm NaCl, 1% [w/v] Triton X-100, 7.5 mm MgCl2, 5 mm EDTA, 50 μm APMSF, and 10 mg/liter leupeptin). Lysates were centrifuged at 20,000 × g for 10 min at 4 °C. To immunoprecipitate FLAG-tagged proteins from the supernatant (input), anti-DYKDDDDK tag beads were used. For Myc-tagged proteins, anti-Myc antibody was added to the supernatant and incubated for 2 h. Protein G-Sepharose 4 fast flow beads then were added. After 30 min of incubation, beads were washed four times with buffer D. Proteins in the inputs and in the immunoprecipitates were detected with appropriate antibodies. In the experiments to evaluate the coimmunoprecipitation of MDM2, cells were pretreated with 30 μm MG132 for 6 h and then lysed in buffer D containing 30 μm MG132. Beads were washed four times with buffer D containing 500 mm NaCl. In the experiments including ABL1 and SRC, cells were lysed in buffer D containing 50 mm NaF, 25 mm β-glycerophosphate, and 2 mm Na3VO4, and beads were rinsed with the same buffer. SW480 cells were plated at 1.1 × 106 cells in a 6-cm dish, and two dishes were used for each immunoprecipitation in the same manner, except that cells were lysed by sonication at high power 3 times for 10 s each time with 1-min intervals.
Subcellular fractionation
Cells were rinsed with ice-cold PBS, harvested by scraping, and centrifuged at 600 × g at 4 °C for 3 min. The cells were resuspended with 560 µl of the hypotonic buffer (20 mm Hepes-NaOH [pH 7.4], 10 mm KCl, 2 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 50 μm APMSF, 10 mg/liter leupeptin, 1 mm DTT), kept at 4 °C for 15 min, lysed by sonication at high power 3 times for 10 s each time with 10-s intervals, and kept at 4 °C for 20 min. 60 µl of the lysate was saved as the whole-cell lysate, and the remaining lysate was centrifuged at 100,000 × g at 4 °C for 60 min. The supernatant was collected as the cytoplasmic fraction. The pellet was rinsed twice with the hypotonic buffer, suspended with 500 µl of the hypotonic buffer, and used as the membrane fraction.
Immunofluorescence
HCT116 cells were fixed with 3.7% (w/v) formaldehyde in PBS at room temperature for 15 min and quenched with 50 mm glycine in PBS. The samples were incubated with 0.2% (v/v) Triton X-100 in PBS for 15 min and subsequently with PBS containing 1% (w/v) BSA and 0.1% (v/v) Triton X-100. The samples then were incubated with various primary antibodies and secondary antibodies and were counterstained with Hoechst 33342 (Sigma-Aldrich) to visualize cell nuclei. The images were obtained with Keyence BZ-X700 (Keyence Cooperation, Osaka, Japan) or Leica TCS SP8 (Leica Biosystems, Wetzlar, Germany).
Analysis of MDM2 distribution
GFP-MDM2 was expressed in H1299 cells. GFP fluorescence intensity in each well was measured with ArrayScan VTI (Cellomics). An algorithm for data acquisition was designed by using vHCS Scan software. In brief, Hoechst 33342 staining was used to identify the nucleus. Each cell was identified within the field, and the nuclear boundary was drawn. The nuclear area was defined as the area 2 pixels inside the nuclear boundary to minimize the cytoplasmic contamination. The cytoplasmic area was defined as the ring with a width of 5 pixels around the nuclear boundary. The intensity of GFP fluorescence was averaged over the measured nuclear and cytoplasmic area. The ratio of the cytoplasmic GFP to the nuclear GFP was calculated by using BioApplication Compartment Analysis V4 (Thermo Fisher Scientific). 50 to 150 cells were analyzed for each sample. The images were obtained with a Leica TCS SP8 (Leica Biosystems, Wetzlar, Germany).
Soft-agar colony formation assay
1 ml melted agar solution containing DMEM, 10% (v/v) FBS, and 0.5% (w/v) agarose (STAR agarose) (RSV-AGRP) (RIKAKEN, Tokyo, Japan) was plated in one well of a 12-well plate. 500 µl DMEM containing 1 × 104 NIH3T3, A549, or SW480 cells, 10% (v/v) FBS, and 0.3% (w/v) agarose then was overlaid and kept for 30 min at room temperature. After agarose solidified, 400 µl DMEM containing 10% (v/v) FBS was further overlaid and cultured for 7 days. The images were obtained with a fluorescence microscope BZ-X700 (Keyence, Osaka, Japan) and analyzed by the BZ-H3C Hybrid Cell Count Module. The area of cells was measured in 15 independent fields.
Sphere formation assay
A549 and SW480 cells were plated at 1,000 cells/well in a 96-well microplate with a cell-repellent surface (655970) (Greiner Bio-One International) and cultured in serum-free DMEM–F12 containing 0.4% (w/v) BSA, 10 ng/ml basic fibroblast growth factor, 20 ng/ml epidermal growth factor, 5 μg/ml insulin, and 1% (w/v) methylcellulose for 10 and 7 days, respectively. Cell aggregates larger than 50 μm were defined as spheres.
Transwell migration and invasion assays
Transwell assays were performed according to the protocol by Corning, Inc. Briefly, transwell inserts (Falcon® permeable support for 24-well plate with 8.0-μm transparent PET membrane) (353097) (Corning Inc.) were coated with 10 µg/cm2 fibronectin solution. The inserts used for invasion assay were further coated with 30 µg Matrigel in 100 µl of 10 mm Tris-HCl (pH 8.0) containing 0.7% (w/v) NaCl. 4 × 104 A549 cells/200 μl serum-free DMEM were seeded on the inserts. 750 µl DMEM with 10% FBS was added. 36 h later, nonmigrating or noninvading cells on the upper surface were removed with a cotton swab. Migrating or invading cells on the lower surface were fixed with methanol and stained with 0.2% (w/v) crystal violet. SW480 cells were used in a similar manner, except that cells were seeded at 1 × 105 cells/200 µl and fixed after 144 h. The area of migrating or invading cells was quantified using ImageJ in 5 independent fields.
Human cancer database analyses
Datasets of pancreatic adenocarcinoma, colon adenocarcinoma, and lung adenocarcinoma were obtained from TCGA and PanCancer-Atlas through cBioPortal for Cancer Genomics (RRID:SCR_014555) (48). After the samples lacking the information regarding KRAS gene status were omitted, pancreatic adenocarcinoma, colon adenocarcinoma, and lung adenocarcinoma contained 172, 532, and 510 samples, respectively. The samples were stratified into three groups depending on UNC119A gene expression level. The numbers of samples with or without KRAS mutations were determined in each group. As references, the frequency of mutations of TP53, APC, and LRP1B were evaluated in pancreatic, colon, and lung adenocarcinomas, respectively.
Statistical analysis
Statistical analyses were performed with Student's t test for comparison between two samples and analysis of variance (ANOVA) with Tukey's test for multiple comparison using GraphPad Prism 5.0 (GraphPad Software). Chi-square analysis was performed by using Microsoft Excel.
Data availability
All data are contained within the manuscript.
Supplementary Material
Acknowledgments
We thank T. Sawada and Y. Morimoto for their help. We are especially grateful to Susumu Hirabayashi (Imperial College London) for instructions about human cancer database analysis. We appreciate Feng Zhang (Broad Institute) and Koji Okamoto and Hitoshi Nakagama (National Cancer Center Research Institute) for plasmids and Editage for English language editing.
This article contains supporting information.
Author contributions—T. S. and Y. H. conceptualization; T. S. data curation; T. S., T. Nakamura, and Y. H. investigation; T. S. and Y. H. writing-original draft; H. Inaba, H. Iwasa, T. Nakata, and Y. H. supervision; H. Inaba methodology; H. Iwasa and Y. H. funding acquisition; H. Iwasa, J. M., K. A.-M., T. Nakata, H. N., and Y. H. writing-review and editing; H. N. resources; Y. H. project administration.
Funding and additional information—This work was funded by the Japan Society for the Promotion of Science (JSPS) (26460359, 26293061, 17K08624, and 19H03414) and the Mitsubishi Foundation (26138).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RA
- Ras association
- NES
- nuclear export signal
- TCGA
- The Cancer Genome Atlas
- APMSF
- 4-amidinophenylmethanesulfonyl fluoride
- ANOVA
- analysis of variance.
References
- 1. Kobayashi A., Higashide T., Hamasaki D., Kubota S., Sakuma H., An W., Fujimaki T., McLaren M. J., Weleber R. G., and Inana G. (2000) HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest. Ophthalmol. Vis. Sci. 41, 3268–3277 [PubMed] [Google Scholar]
- 2. Wright K. J., Baye L. M., Olivier-Mason A., Mukhopadhyay S., Sang L., Kwong M., Wang W., Pretorius P. R., Sheffield V. C., Sengupta P., Slusarski D. C., and Jackson P. K. (2011) An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 25, 2347–2360 10.1101/gad.173443.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Higashide T., Murakami A., McLaren M. J., and Inana G. (1996) Cloning of the cDNA for a novel photoreceptor protein. J. Biol. Chem. 271, 1797–1804 10.1074/jbc.271.3.1797 [DOI] [PubMed] [Google Scholar]
- 4. Swanson D. A., Chang J. T., Campochiaro P. A., Zack D. J., and Valle D. (1998) Mammalian orthologs of C. elegans unc-119 highly expressed in photoreceptors. Invest. Ophthalmol. Vis. Sci. 39, 2085–2094 [PubMed] [Google Scholar]
- 5. Kobayashi A., Kubota S., Mori N., McLaren M. J., and Inana G. (2003) Photoreceptor synaptic protein HRG4 (UNC119) interacts with ARL2 via a putative conserved domain. FEBS Lett. 534, 26–32 10.1016/S0014-5793(02)03766-3 [DOI] [PubMed] [Google Scholar]
- 6. Nancy V., Callebaut I., El Marjou A., and de Gunzburg J. (2002) The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J. Biol. Chem. 277, 15076–15084 10.1074/jbc.M109983200 [DOI] [PubMed] [Google Scholar]
- 7. Sasaki T., Kikuchi A., Araki S., Hata Y., Isomura M., Kuroda S., and Takai Y. (1990) Purification and characterization from bovine brain cytosol of a protein that inhibits the dissociation of GDP from and the subsequent binding of GTP to smg p25A, a ras p21-like GTP-binding protein. J. Biol. Chem. 265, 2333–2337 [PubMed] [Google Scholar]
- 8. Ueda T., Kikuchi A., Ohga N., Yamamoto J., and Takai Y. (1990) Purification and characterization from bovine brain cytosol of a novel regulatory protein inhibiting the dissociation of GDP from and the subsequent binding of GTP to rhoB p20, a ras p21-like GTP-binding protein. J. Biol. Chem. 265, 9373–9380 [PubMed] [Google Scholar]
- 9. Cen O., Gorska M. M., Stafford S. J., Sur S., and Alam R. (2003) Identification of UNC119 as a novel activator of SRC-type tyrosine kinases. J. Biol. Chem. 278, 8837–8845 10.1074/jbc.M208261200 [DOI] [PubMed] [Google Scholar]
- 10. Palacios E. H., and Weiss A. (2004) Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000 10.1038/sj.onc.1208074 [DOI] [PubMed] [Google Scholar]
- 11. Vepachedu R., Karim Z., Patel O., Goplen N., and Alam R. (2009) Unc119 protects from Shigella infection by inhibiting the Abl family kinases. PLoS ONE 4, e5211 10.1371/journal.pone.0005211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang H., Constantine R., Vorobiev S., Chen Y., Seetharaman J., Huang Y. J., Xiao R., Montelione G. T., Gerstner C. D., Davis M. W., Inana G., Whitby F. G., Jorgensen E. M., Hill C. P., Tong L., et al. (2011) UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci. 14, 874–880 10.1038/nn.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gopalakrishna K. N., Doddapuneni K., Boyd K. K., Masuho I., Martemyanov K. A., and Artemyev N. O. (2011) Interaction of transducin with uncoordinated 119 protein (UNC119): implications for the model of transducin trafficking in rod photoreceptors. J. Biol. Chem. 286, 28954–28962 10.1074/jbc.M111.268821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alpadi K., Magupalli V. G., Käppel S., Köblitz L., Schwarz K., Seigel G. M., Sung C. H., and Schmitz F. (2008) RIBEYE recruits Munc119, a mammalian ortholog of the Caenorhabditis elegans protein unc119, to synaptic ribbons of photoreceptor synapses. J. Biol. Chem. 283, 26461–26467 10.1074/jbc.M801625200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorska M. M., Stafford S. J., Cen O., Sur S., and Alam R. (2004) Unc119, a novel activator of Lck/Fyn, is essential for T cell activation. J. Exp. Med. 199, 369–379 10.1084/jem.20030589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vepachedu R., Gorska M. M., Singhania N., Cosgrove G. P., Brown K. K., and Alam R. (2007) Unc119 regulates myofibroblast differentiation through the activation of Fyn and the p38 MAPK pathway. J. Immunol. 179, 682–690 10.4049/jimmunol.179.1.682 [DOI] [PubMed] [Google Scholar]
- 17. Gorska M. M., Goplen N., Liang Q., and Alam R. (2010) Uncoordinated 119 preferentially induces Th2 differentiation and promotes the development of asthma. J. Immunol. 184, 4488–4496 10.4049/jimmunol.0903115 [DOI] [PubMed] [Google Scholar]
- 18. Karim Z., Vepachedu R., Gorska M., and Alam R. (2010) UNC119 inhibits dynamin and dynamin-dependent endocytic processes. Cell Signal. 22, 128–137 10.1016/j.cellsig.2009.09.022 [DOI] [PubMed] [Google Scholar]
- 19. Lee Y., Chung S., Baek I. K., Lee T. H., Paik S. Y., and Lee J. (2013) UNC119a bridges the transmission of Fyn signals to Rab11, leading to the completion of cytokinesis. Cell Cycle 12, 1303–1315 10.4161/cc.24404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lei B., Chai W., Wang Z., and Liu R. (2015) Highly expressed UNC119 promotes hepatocellular carcinoma cell proliferation through Wnt/β-catenin signaling and predicts a poor prognosis. Am. J. Cancer Res. 5, 3123–3134 [PMC free article] [PubMed] [Google Scholar]
- 21. Garivet G., Hofer W., Konitsiotis A., Klein C., Kaiser N., Mejuch T., Fansa E., Alsaabi R., Wittinghofer A., Bastiaens P. I. H., and Waldmann H. (2019) Small-molecule inhibition of the UNC-Src interaction impairs dynamic Src localization in cells. Cell Chem. Biol. 26, 842–851 10.1016/j.chembiol.2019.02.019 [DOI] [PubMed] [Google Scholar]
- 22. Iwasa H., Sarkar A., Shimizu T., Sawada T., Hossain S., Xu X., Maruyama J., Arimoto-Matsuzaki K., Withanage K., Nakagawa K., Kurihara H., Kuroyanagi H., and Hata Y. (2018) UNC119 is a binding partner of tumor suppressor Ras-association domain family 6 and induces apoptosis and cell cycle arrest by MDM2 and p53. Cancer Sci. 109, 2767–2780 10.1111/cas.13706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allen N. P., Donninger H., Vos M. D., Eckfeld K., Hesson L., Gordon L., Birrer M. J., Latif F., and Clark G. J. (2007) RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene 26, 6203–6211 10.1038/sj.onc.1210440 [DOI] [PubMed] [Google Scholar]
- 24. Ikeda M., Kawata A., Nishikawa M., Tateishi Y., Yamaguchi M., Nakagawa K., Hirabayashi S., Bao Y., Hidaka S., Hirata Y., and Hata Y. (2009) Hippo pathway-dependent and -independent roles of RASSF6. Sci. Signal. 2, ra59 10.1126/scisignal.2000300 [DOI] [PubMed] [Google Scholar]
- 25. Mejuch T., van Hattum H., Triola G., Jaiswal M., and Waldmann H. (2015) Specificity of lipoprotein chaperones for the characteristic lipidated structural motifs of their cognate lipoproteins. Chembiochem 16, 2460–2465 10.1002/cbic.201500355 [DOI] [PubMed] [Google Scholar]
- 26. Hanzal-Bayer M., Renault L., Roversi P., Wittinghofer A., and Hillig R. C. (2002) The complex of Arl2-GTP and PDE delta: from structure to function. EMBO J. 21, 2095–2106 10.1093/emboj/21.9.2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ismail S. A., Chen Y. X., Rusinova A., Chandra A., Bierbaum M., Gremer L., Triola G., Waldmann H., Bastiaens P. I., and Wittinghofer A. (2011) Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 7, 942–949 10.1038/nchembio.686 [DOI] [PubMed] [Google Scholar]
- 28. Dharmaiah S., Bindu L., Tran T. H., Gillette W. K., Frank P. H., Ghirlando R., Nissley D. V., Esposito D., McCormick F., Stephen A. G., and Simanshu D. K. (2016) Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U S A 113, E6766–E6775 10.1073/pnas.1615316113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hancock J. F., Cadwallader K., and Marshall C. J. (1991) Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 10, 641–646 10.1002/j.1460-2075.1991.tb07992.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaplan J. B., and Sass P. M. (1991) Post-translational processing of purified human K-ras in Xenopus oocytes. Cancer Commun. 3, 383–388 10.3727/095535491820873704 [DOI] [PubMed] [Google Scholar]
- 31. Topinka J. R., and Bredt D. S. (1998) N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20, 125–134 10.1016/S0896-6273(00)80440-7 [DOI] [PubMed] [Google Scholar]
- 32. Sarkar A., Iwasa H., Hossain S., Xu X., Sawada T., Shimizu T., Maruyama J., Arimoto-Matsuzaki K., and Hata Y. (2017) Domain analysis of Ras-association domain family member 6 upon interaction with MDM2. FEBS Lett. 591, 260–272 10.1002/1873-3468.12551 [DOI] [PubMed] [Google Scholar]
- 33. Roth J., Dobbelstein M., Freedman D. A., Shenk T., and Levine A. J. (1998) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 17, 554–564 10.1093/emboj/17.2.554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freedman D. A., and Levine A. J. (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 18, 7288–7293 10.1128/mcb.18.12.7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stommel J. M., Marchenko N. D., Jimenez G. S., Moll U. M., Hope T. J., and Wahl G. M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18, 1660–1672 10.1093/emboj/18.6.1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geyer R. K., Yu Z. K., and Maki C. G. (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2, 569–573 10.1038/35023507 [DOI] [PubMed] [Google Scholar]
- 37. Park H. S., Park J. M., Park S., Cho J., Kim S. I., and Park B. W. (2014) Subcellular localization of Mdm2 expression and prognosis of breast cancer. Int. J. Clin. Oncol. 19, 842–851 10.1007/s10147-013-0639-1 [DOI] [PubMed] [Google Scholar]
- 38. Ismail S. A., Chen Y. X., Miertzschke M., Vetter I. R., Koerner C., and Wittinghofer A. (2012) Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J. 31, 4085–4094 10.1038/emboj.2012.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Constantine R., Zhang H., Gerstner C. D., Frederick J. M., and Baehr W. (2012) Uncoordinated (UNC)119: coordinating the trafficking of myristoylated proteins. Vision Res. 75, 26–32 10.1016/j.visres.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bologna G., Yvon C., Duvaud S., and Veuthey A. L. (2004) N-terminal myristoylation predictions by ensembles of neural networks. Proteomics 4, 1626–1632 10.1002/pmic.200300783 [DOI] [PubMed] [Google Scholar]
- 41. Privé G. G., Milburn M. V., Tong L., de Vos A. M., Yamaizumi Z., Nishimura S., and Kim S. H. (1992) X-ray crystal structures of transforming p21 ras mutants suggest a transition-state stabilization mechanism for GTP hydrolysis. Proc. Natl. Acad. Sci. U S A 89, 3649–3653 10.1073/pnas.89.8.3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding L., Getz G., Wheeler D. A., Mardis E. R., McLellan M. D., Cibulskis K., Sougnez C., Greulich H., Muzny D. M., Morgan M. B., Fulton L., Fulton R. S., Zhang Q., Wendl M. C., Lawrence M. S., et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 10.1038/nature07423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hirao K., Hata Y., Ide N., Takeuchi M., Irie M., Yao I., Deguchi M., Toyoda A., Sudhof T. C., and Takai Y. (1998) A novel multiple PDZ domain-containing molecule interacting with N-methyl-D-aspartate receptors and neuronal cell adhesion proteins. J. Biol. Chem. 273, 21105–21110 10.1074/jbc.273.33.21105 [DOI] [PubMed] [Google Scholar]
- 44. Irie M., Hata Y., Takeuchi M., Ichtchenko K., Toyoda A., Hirao K., Takai Y., Rosahl T. W., and Südhof T. C. (1997) Binding of neuroligins to PSD-95. Science 277, 1511–1515 10.1126/science.277.5331.1511 [DOI] [PubMed] [Google Scholar]
- 45. Ikeda M., Hirabayashi S., Fujiwara N., Mori H., Kawata A., Iida J., Bao Y., Sato Y., Iida T., Sugimura H., and Hata Y. (2007) Ras-association domain family protein 6 induces apoptosis via both caspase-dependent and caspase-independent pathways. Exp. Cell Res. 313, 1484–1495 10.1016/j.yexcr.2007.02.013 [DOI] [PubMed] [Google Scholar]
- 46. Yang Z., Nakagawa K., Sarkar A., Maruyama J., Iwasa H., Bao Y., Ishigami-Yuasa M., Ito S., Kagechika H., Hata S., Nishina H., Abe S., Kitagawa M., and Hata Y. (2014) Screening with a novel cell-based assay for TAZ activators identifies a compound that enhances myogenesis in C2C12 cells and facilitates muscle repair in the muscle injury model. Mol. Cell. Biol. 34, 1607–1621 10.1128/MCB.01346-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoadley K. A., Yau C., Hinoue T., Wolf D. M., Lazar A. J., Drill E., Shen R., Taylor A. M., Cherniack A. D., Thorsson V., Akbani R., Bowlby R., Wong C. K., Wiznerowicz M., Sanchez-Vega F., Cancer Genome Atlas Network (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e296 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.