

Abstract
Although accumulation of myeloid-derived suppressor cells (MDSC) is a hallmark of cancer, the underlying mechanism of this accumulation within the tumor microenvironment remains incompletely understood. We report here that TNFα-RIP1-mediated necroptosis regulates accumulation of MDSCs. In tumor-bearing mice, pharmacological inhibition of DNMT with the DNA methyltransferease inhibitor decitabine (DAC) decreased MDSC accumulation and increased activation of antigen-specific cytotoxic T lymphocytes (CTL). DAC-induced decreases in MDSC accumulation correlated with increased expression of the myeloid cell lineage-specific transcription factor IRF8 in MDSCs. However, DAC also suppressed MDSC-like cell accumulation in IRF8-deficient mice, indicating that DNA methylation may regulate MDSC survival through an IRF8-independent mechanism. Instead, DAC decreased MDSC accumulation by increasing cell death via disrupting DNA methylation of RIP1-dependent targets of necroptosis. Genome-wide DNA bisulfite sequencing revealed that the Tnf promoter was hypermethylated in tumor-induced MDSCs in vivo. DAC treatment dramatically increased TNFα levels in MDSC in vitro, and neutralizing TNFα significantly increased MDSC accumulation and tumor growth in tumor-bearing mice in vivo. Recombinant TNFα induced MDSC cell death in a dose- and RIP1-dependent manner. IL6 was abundantly expressed in MDSCs in tumor-bearing mice and human colorectal cancer patients. In vitro, IL6 treatment of MDSC-like cells activated STAT3, increased expression of DNMT1 and DNMT3b, and enhanced survival. Overall, our findings reveal that MDSCs establish a STAT3-DNMT epigenetic axis, regulated by autocrine IL6, to silence TNFα expression. This results in decreased TNFα-induced and RIP1-dependent necroptosis to sustain survival and accumulation.
Keywords: MDSCs, RIP1, Necroptosis, IL6, STAT3, DNMT, TNFα
Introduction
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that are induced under various pathological conditions, including cancer (1). In mice, MDSCs are defined as CD11b+Gr1+ immature myeloid cells that can be further divided into monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) (2). In humans, the phenotype of MDSCs is less defined, but a phenotype of CD11b+CD33+HLA-DR− in combination with other markers is often used to identify MDSCs in human cancer patients (2). A key function of MDSCs is immune suppression. MDSCs use various mechanisms to suppress activation and function of T cells and NK cells to enable tumor immune escape (3). Furthermore, MDSCs also promote tumor growth through secreting cytokines and growth factors to modulate tumor angiogenesis and to silence tumor suppressors by an epigenetic mechanism (4,5). Therefore, MDSCs are key targets in cancer immunotherapy.
Immune cell homeostasis is controlled by the balance of cell lineage differentiation and cell death rate. MDSCs are induced from myeloid progenitor cells by proinflammatory mediators in the tumor microenvironment (6). It was also well-documented that transcription factor IRF8 regulates lineage-specific differentiation of CD11b+Gr1+ immature cells from myeloid progenitor cells (7). Regulation of MDSC turnover is not well understood. The fact that MDSCs massively accumulate in tumor-bearing host suggests that MDSCs could have a decreased cell death rate (8-11). Indeed, several cellular pathways have been shown to play important roles in MDSC survival and accumulation (8-10). The TNFR2 signaling up-regulates c-FLIP and inhibits caspase 8 activation to promote MDSC survival (12). The IL4Ra signaling is also pivotal for MDSC survival and accumulation (13). Moreover, MDSCs tends to downregulate the death receptor Fas and downstream apoptosis-regulatory mediators Bax and Bcl-xL to escape from CTL-mediated cytotoxicity (14,15). Autophagy plays opposing role in MDSC survival. A ceramide mimetic has been shown to activate lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress MDSCs (16), but autophagy also promote MDSC survival and function (8,17). On the other hand, MDSCs have increased levels of death receptor TRAIL-R as compared to mature myeloid cells such as neutrophils and monocytes (9), and TRAIL-R2 agonist antibody immunotherapy eliminated MDSCs in tumor-bearing mice and human cancer patients (18). We report here the identification of DNA methylation as a mechanism of MDSC survival and accumulation. Specifically, we determined that DNA methylation sustains MDSC accumulation through protecting MDSCs from necroptosis at the post lineage differentiation phase in vivo. Mechanistically, we determined that MDSC autocrine IL6 activates the STAT3-DNMT intrinsic signaling pathway to hypermethylate the Tnf promoter to silence Tnf expression, resulting in impaired RIP1-mediated necroptosis to promote MDSC survival and accumulation.
Materials and Methods
Mice and human specimens:
IRF8 KO and IRF8-GFP mice were as described (7,19). C57BL/6 and BALB/c mice were obtained from Jackson Laboratory (Bar Harbor, ME). Use of mice was performed according to approved protocols by institutional animal use and care committee of Augusta University (Protocol# 2008-0162). Peripheral blood specimens from healthy donors were provided by Shepeard Community Blood Bank (Augusta, GA). Peripheral blood specimens of colorectal cancer patients were collected from Georgia Cancer Center and Charlie Norwood VA Medical Center according to approved protocols by Augusta University Institutional Review Board (Protocol# 1354508-1) and Charlie Norwood VA Medical Center Institutional Review Board (Protocol # 1314554-4).
Mouse tumor models.
Colon carcinoma cell line CT26 were obtained directly from ATCC (Manassas, VA). ATCC has characterized this cell line by morphology, immunology, DNA fingerprint, and cytogenetics. J774M cells were sorted from J774 cells and established as CD11b+Gr1+ cell line. J774M cells were phenotypically and functionally characterized as previously described (20). The AT3 cell line was derived from C57BL/6 mice and was kindly provided by Dr. Scott Abrams (Roswell Park Cancer Institute, NY) and was characterized as previously described (21). All cell lines were stored in aliquots in liquid nitrogen and all cell lines were used in less than 30 passages after obtaining them. These cell lines were not further authenticated by the authors. All cell lines were tested for mycoplasma every two monthly and all cells used in this study were negative for mycoplasma. AT3 cells were injected to the mammary gland of female C57BL/6 to establish orthotopic tumor. CT26 cells were injected to the right flank of BALB/c mice (2x105 cells/mouse) to establish subcutaneous tumor or surgically injected to cecal wall of BALB/c mice (1x104 cells/mouse) to establish orthotopic colon tumor.
Reagents.
Decitabine (DAC), Necrostatin-1 (Nec-1), and Ferrostostatin-1 (Fer-1) were obtained from Sigma-Aldrich (St. Luis, MO). Necrosulfonamide (NSA) was obtained from Calbiochem (Burlington, MA). Z-DEVD-FMK was obtained from BD Pharmingen. Recombinant mouse TNFα was purchased from R & D Systems (Minneapolis, MN). Mouse TNFα neutralization mAb (clone XT3.11) was obtained from Bio X cell (West Lebanon, NH). Anti-mouse CD11b, Gr1, CD11c, CD4, CD8, CD19, NK1.1, F4/80, CD45.2, TNFα mAbs, Annexin V, Zobbie Violet, and 4’, 6-Diamidino-2-Phenylindole (DAPI) were obtained from Biolegend (San Diego, CA). Anti-pSTAT3 was obtained from Cell Signaling. Anti-STAT3 was obtained from BD Biosciemces. Propidium iodide (PI) was obtained from MP Biomedicals (Santa Ana, CA).
Flow cytometry.
Tissue collection and analysis by flow cytometry was performed as previously described (5,22). Flow cytometry analysis was done on the LSRFortessa for multicolor panels, on the BD FACSCalibur for two color panels, and on the BD Accuri C6 flow cytometer for three color panels.
Targeted CpG site bisulfite sequencing of the Irf8 DNA promoter region in MDSCs.
Genomic DNA samples were bisulfite converted using the EZ DNA Methylation- Lightning Kit (Zymo Research) according to the manufacturer’s instructions. Eight PCR amplicons were designed to cover the 2000 bp upstream of the mouse Irf8 gene according to the coordinates for mm10 from NCBI. The covered sequence was slightly extended into the first exon to cover the full CpG island. The resulting amplicons were pooled for harvesting and subsequent barcoding. After barcoding, samples were purified and then prepared for massively parallel sequencing using a MiSeq V2 300bp Reagent Kit (Illumina)and paired-end sequencing protocol according to the manufacturer’s guidelines. Sequence reads were identified using standard Illumina base-calling software and then analyzed using a Zymo Research proprietary analysis pipeline by Zymo Research. Low quality nucleotides and adapter sequences were trimmed off. Sequence reads were aligned back to the reference genome using Bismark (Galaxy tool version: 0.22.1). The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. The genomic coordinates, The methylated CpG count and total CpG count for CpG sites were then calculated and tracked to the genomic coordinates using UCSC genomic browser.
Genome-wide bisulfite DNA methylation sequencing.
CD11b+Gr1+ MDSCs were purified from spleens of AT3 tumor-bearing mice using CD11b MicroBeads and MACS separation column according to the manufacturer’s instructions (Miltenyi Biotec Inc. San Diego, CA). The purity of CD11b+Gr1+ cells is greater than 95%. Genomic DNA was extracted from the purified MDSCs using Quick-DNA Miniprep Plus Kit (Zymo Research, Irvine, CA). The genomic DNA was then fragmented, ligated with an adaptor, subjected to bisulfite conversion, size selected for library construction. The library was sequenced using the Illumina next generation high-through sequencing. The bisulfite sequences alignment and methylation call were carried out using Galaxy/Europe Server (usegalaxy.eu). The raw reads were first analyzed by FastQC (Galaxy tool version: 0.72) program to obtain basic statistics of the sequencing runs. The raw reads were then trimmed with Trimmomatic (Galaxy tool version: 0.36.5) to filter out adaptor contents and low-quality reads. The cleaned reads were mapped to the mouse genome (mm10) using Bismark Mapper. PCR duplicates were removed using Bismark Deduplicate (Galaxy tool version: 0.22.1). The methylation call was performed using MethylDeckel (Galaxy tool version: 0.3.0.2). The circos plot was generated using R package circlize (Version 0.4.6). Heatmaps were plotted using R package EnrichedHeatmap (version 1.14.0). The entire dataset is deposited in GEO database (accession # GSE144649).
TCGA database genomic data mining:
IL6 expression datasets in human colon carcinoma and melanoma were extracted from TCGA Colon Cancer (COAD) and melanoma (Melanoma) ploy A+ IlluminaHiSeq pancan normalized RNA seq dataset using UCSC Xena Cancer Genomics Browser.
Induction of MDSCs from bone marrow.
Bone marrow (BM) cells (5×105 cells/ml) were cultured with RPMI 1640 medium containing 10% (v/v) FCS and supplemented with either 20 ng/ml recombinant mouse GM-CSF or 50% (v/v) of tumor cell–conditioned medium for 5 days.
IL6 protein measurement.
Cell culture medium was collected and analyzed for IL6 protein levels using multiplex LEGENDplex Mouse Inflammation Panel (BioLegend) according to the manufacturer’s instructions. Data were collected on FACSCalibur two-laser flow cytometer (BD Biosciences) and analyzed using LEGENDplex Data Analysis Software (BioLegend).
Statistical analysis.
Statistical analysis was performed using Student’s t test. A P <0.05 was taken as statistically significant.
Results
IRF8 is selectively silenced in MDSCs among immune cells in tumor-bearing mice.
IRF8 is a transcriptional factor that controls lineage differentiation of monocytes and dendritic cells in humans. Global deletion of Irf8 in mice leads to accumulation of CD11b+Gr1+ immature myeloid cells (7) that phenotypically and functionally resemble tumor-induced MDSCs (21). To determine IRF8 expression profiles in immune cells in tumor-bearing host, we made use of the IRF8-GFP reporter mice (19). As expected, CD11b+Gr1+ MDSCs massively accumulate in spleen, blood, and tumor in tumor-bearing mice (Fig. 1A-D and Figs. S1 & 2). IRF8 expression level is significantly decreased in MDSCs and F4/80 macrophage cells, but not in other subset of myeloid cells, T cells, B cells and NK cells (Fig. S1A-H & S2A-H).
Figure 1. DNA methylation sustains tumor-induced MDSC accumulation in vivo.
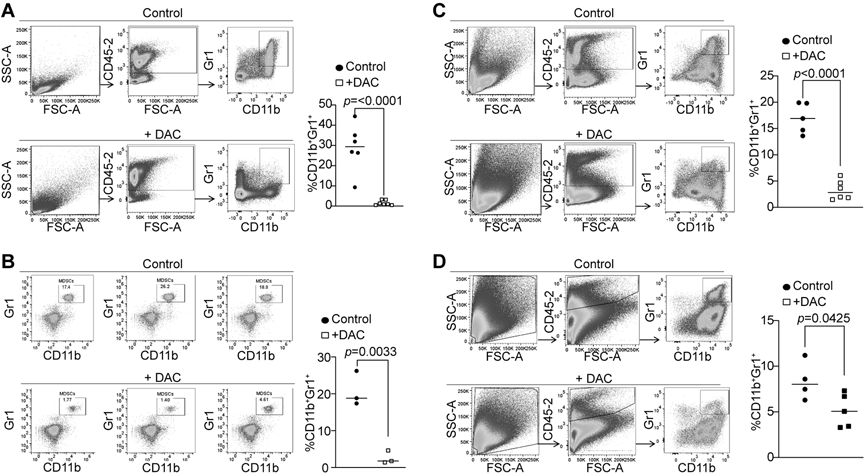
A. AT3 tumor cells (2.5x105 cells/mouse) were injected into the mammary gland of C57BL/6 mice. Tumor-bearing mice were treated with saline (control, n=6) and DAC (2 mg/kg body weight, n=7) by i.v. injection at days 21-23 after tumor cell injection once every 2 days for 2 times. Mice were sacrificed 1 day after the last treatment. Spleens cells were stained with CD45.2−, CD11b−, and Gr1-specific antibodies. All CD45.2+ cells were gated and analyzed for CD11b+Gr1+ cells. Shown are gating strategy (left panel) and quantification of CD11b+Gr1+ cells (right panel). B. AT3 tumor-bearing mice were treated with saline (control, n=3) and DAC (2 mg/kg body weight, n=3) as in A. The peripheral blood was collected and processed for white blood cells. The cells were then stained and analyzed as in A. C. AT3 tumor-bearing mice were treated with saline (control, n=5) and DAC (2 mg/kg body weight, n=6) as in A. Tumor tissues were collected, digested with collagenase to make single cells. The cells were then stained with CD45.2−, CD11b−, and Gr1-specific antibodies. All CD45.2+ cells were gated and analyzed for CD11b+Gr1+ cells. Shown are gating strategy (left panel) and quantification of CD11b+Gr1+ cells (right panel). D. CT26 cells (2x105) were injected s.c. to BALB/c mice. The tumor-bearing mice were treated with saline (control, n=4) and DAC (2 mg/kg body weight, n=5) at days 21 and 23 after tumor cell injection. Mice were sacrificed at day 24. Tumor tissues were collected, digested with collagenase to make single cells. The cells were then stained with CD45.2−, CD11b−, and Gr1-specific antibodies. All CD45.2+ cells were gated and analyzed for CD11b+Gr1+ cells. Shown are gating strategy (left panel) and quantification of CD11b+Gr1+ cells (right panel).
Inhibition of DNA methylation reduces MDSC accumulation and increased antigen-specific CTL activation in vivo.
IRF8 is an essential regulator of myeloid cell lineage differentiation from hematopoietic stem and myeloid progenitor cells (7). DNA methylation is a key regulator of myeloid cell differentiation from hematopoietic stem and progenitor cells (23) and IRF8 is regulated by its promoter DNA methylation in myeloid cells (24). We therefore aimed at testing the hypothesis that tumor cells use DNA methylation to silence IRF8 expression to induce MDSC accumulation. The chemotherapeutic agent DAC was used as a DNMT inhibitor here. Tumor-bearing mice were treated with DAC and analyzed for MDSC accumulation. DAC treatment at a dose of 1 mg/kg body weight significantly decreased MDSC levels in spleens but not in the tumors in the tumor-bearing mice (Fig. S3A & B). However, increasing DAC dose to 2 mg/kg body weight significantly decreased MDSC levels in spleens, peripheral blood, and tumors in the mammary carcinoma AT3 tumor-bearing mice (Fig. 1A-C). The colon carcinoma CT26 tumor model was then used as a complementary model in mice that were then treated with DAC. As observed in the AT3 tumor-bearing mice, DAC therapy at a dose of 2 mg/kg body weight also significantly decreased MDSC accumulation in the tumor (Fig. 1D). These findings indicate that DNA methylation maintains MDSC accumulation in tumor-bearing mice.
The CT26 tumor-bearing mice were then analyzed for T cell activation. DAC therapy significantly increased tumor-infiltrating CD8+ T cells (Fig. S4A). CT26 tumor cells harbor the viral gp70 protein which may serve as an antigen to generate antigen-specific T cells. The tumors were analyzed with a tetramer that is specific for an epitope (AH1) of the gp70 protein. It is clear that DAC therapy significantly and dramatically increased the tumor-infiltrating antigen-specific CTLs in the tumor-bearing mice (Fig. S4A & B). These findings indicate that DAC therapy is effective in suppressing MDSC accumulation and increasing antigen-specific CTL activation.
DNA methylation regulates MDSC accumulation at the post differentiation phase.
To determine whether inhibition of DNA methylation activates IRF8 expression to suppress MDSC differentiation, we analyzed the Irf8 promoter DNA methylation. The mouse Irf8 promoter contains a CpG island (Fig. 2A). Bisulfite DNA sequencing of tumor-induced MDSCs revealed that the CpG island region and a region upstream of the Irf8 transcription start site are methylated (Fig. 2A) and DAC treatment induced the Irf8 promoter demethylation (Fig. 2B). Consistent with the methylation status of tumor-induced MDSCs, DAC treatment significantly increased IRF8+ MDSC cells in the tumor microenvironment (Fig. 2C). We then treated IRF8 KO mice with DAC. The rationale is that if DAC targets MDSC accumulation at the differentiation stage through up-regulating IRF8, then DAC therapy should not affect the CD11b+Gr1+ immature myeloid cell accumulation in IRF8 KO mice since IRF8 expression and function is lost in IRF8 KO mice. However, DAC therapy still significantly decreased MDSC accumulation in IRF8 KO mice (Fig. 2D). These findings indicate that DNA methylation regulates MDSC accumulation post MDSC lineage differentiation.
Figure 2. The Irf8 promoter is hypermethylated in MDSCs.
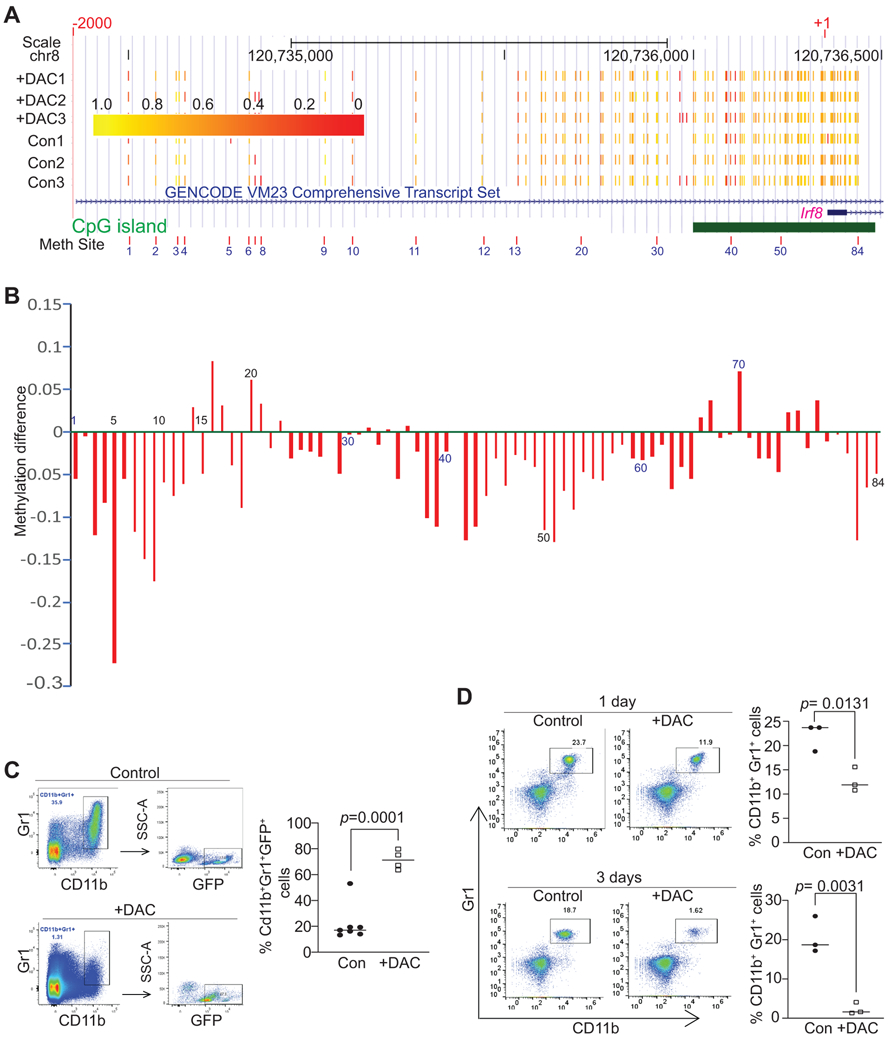
A. CD11b+Gr1+ MDSCs were purified from spleens of control (n=3) and DAC-treated (n=3) AT3 tumor-bearing mice as described in C below. Genomic DNA was purified, bisulfite converted and subjected to the Irf8 promoter (−2000 to +1) targeted bisulfite Sequencing as described in the materials and methods section. Shown is the UCSC genomic browser track of methylation of CpG sites. The number 1-84 correspond to coordinates for each of the detected CpG sites at the Irf8 promoter region. The predicted CpG island is indicated by the green bar. Yellow corresponds to higher methylation levels, while red corresponds to lower methylation levels. B. Methylation difference is calculated by the formula: methylation ratio of the DAC-treated MDSCs (mean of the three samples) – methylation ratio of the control group (mean of the three samples) at the specified site as indicated (#1-84). Methylation ratio was calculated as the measured number of methylated cytosines divided by total number of cytosines covered at that site. The numbers under or above the bar indicate the genomic coordinates as in A. C. AT3 tumor cells (2.5x105 cells/mouse) were injected s.c. to IRF8-GFP reporter mice. The tumor-bearing mice were treated with saline (control, n=7) and DAC (2 mg/kg body weight, n=4) by i.v. injection at days 21 and 23 after tumor cell injection. Mice were sacrificed at day 24. Spleens were collected and stained with CD45.2−, CD11b−, and Gr1-specific mAbs plus DAPI. The live CD45.2+ cells were dated and analyzed for CD11b+Gr1+ cells. The CD11b+Gr1+ cells were then gated for GFP+ cells. The % GFP+ cells were quantified and presented at the right. D. IRF8 KO mice were treated with saline (control, n=3) and DAC (2 mg/kg body weight, n=3) by i.v. injection once every 2 days for 2 times. Blood samples were collected from the mice at day 1 and 3 after the 1st DAC treatment. White blood cells were processed and stained with CD11b−, and Gr1-specific mAbs. Left panel shows dot plots of the CD11b+Gr1+ cells, and right panel shows quantification of CD11b+Gr1+ cells.
DNA methylation maintains tumor-induced MDSC survival in vivo.
The above finding that DNA methylation regulates MDSC accumulation at the post lineage differentiation stage suggests that DNA methylation might regulate MDSC survival. To test this hypothesis, the AT3 tumor-bearing and CT26 tumor-bearing mice were treated with DAC. Spleen cells from the tumor-bearing mice were then analyzed for cell death. Cell death was measured by % PI+ or DAPI+ and % Annexin V+ cells of the gated CD11b+Gr1+ cells. DAC therapy significantly increased % DAPI+, % Annexin V+DAPI−, and % Annexin V+DAPI+ cells in the CD11b+Gr1+ spleen cells in both AT3 tumor-bearing (Fig. 3A & B) and CT26 tumor-bearing (Fig. 3C & D) mice. The effect of DAC on tumor-infiltrating MDSC cell death was not measured due to technical difficulties. These observations indicate that DNA methylation regulates MDSC accumulation through enhancing MDSC survival.
Figure 3. DNA methylation sustains MDSC accumulation through maintaining cellular survival in tumor-bearing mice.
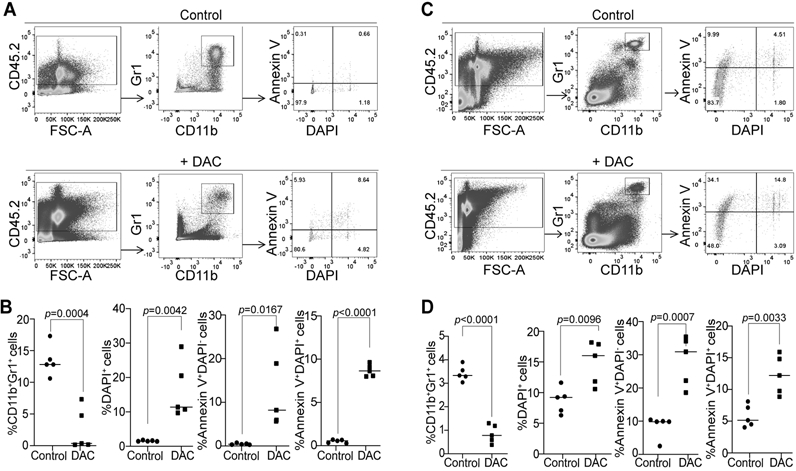
A. AT3 tumor-bearing mice were treated with saline (control, n=5) and DAC (2 mg/kg body weight, n=5), respectively, at day 19 after tumor cell injection once every 2 days for 2 times. Mice were sacrificed one day after the last treatment. Spleens were collected and processed in cold PBS. Cells were then stained with CD45.2−, CD11b, and Gr1-specific antibodies plus Annexin V and DAPI, and analyzed by flow cytometry. The CD45.2+ cells were gated and analyzed for CD11b+Gr1+ cells. The CD11b+Gr1+ cells were then analyzed for Annexin V+ and DAPI+ cells. B. Quantification of CD11b+Gr1+, CD11b+Gr1+DAPI+, CD11b+Gr1+Annexin V+DAPI, and CD11b+Gr1+Annexin V+DAPI+ cells as gated in A. C. CT26 tumor-bearing were treated with saline (control, n=5) and DAC (2 mg/kg body weight, n=5) at day 19 after tumor cell injection once every 2 days for 2 times. Mice were sacrificed I day after the last treatment. Spleens were collected and processed in cold PBS. The spleen cells were then analyzed as in A. D. Quantification of CD11b+Gr1+, CD11b+Gr1+DAPI+, CD11b+Gr1+Annexin V+DAPI, and CD11b+Gr1+Annexin V+DAPI+ cells as gated in C.
DNA methylation impairs necroptosis to promote MDSC survival.
To determine which cell death pathway underlies DNA methylation-dependent MDSC survival, we made use of the MDSC-like J774M cells. J774M cells have a CD11b+Gr1+ phenotype and exhibit potent suppressive activity against CTL activation and proliferation (20), and thus phenotypically and functionally resemble MDSCs. Treatment of J774M cells with DNA methylation inhibitor DAC induced cell death in a dose-dependent manner in vitro (Fig. 4A & B). To determine which cell death pathways are activated by DAC, we treated cells with DAC in the presence of cell death pathway-selective inhibitors. Only RIP1-specific inhibitor significantly decreased DAC-induced cell death in J774M cells (Fig. 4C & D). This observation indicates that DAC induces J774M cell death through RIP1-mediated necroptosis in vitro, suggesting that DNA methylation regulates MDSC necroptosis (Fig. 4C & D).
Figure 4. DNA methylation impairs necroptosis to promote MDSC survival in vitro.
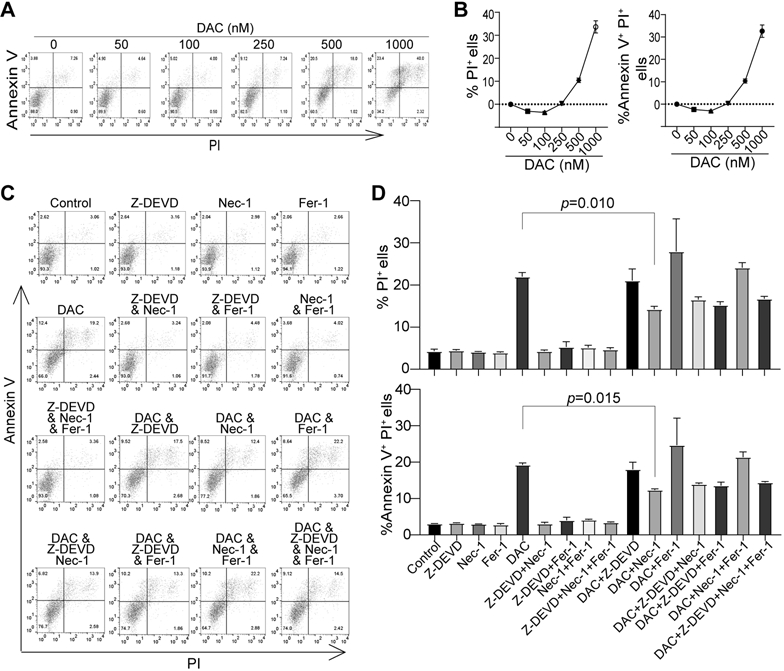
A. J774M cells were plated in 24-well plates (8.3x104 cells/well in 1 ml medium) and treated with DAC at the indicated doses for 3 days. The floating and adherent cells were collected, stained with Annexin V and PI, and analyzed by flow cytometry. Shown are dot plots of representative results. B. The % PI+, and % Annexin V+PI+ cells were quantified as expressed as % PI+ cells in the treatment group - % PI+ cells of control group, and %Annexin V+PI+ cells of treatment group - % Annexin V+PI+ cells of control group, respectively. C. J774M cells were seeded as in A and treated with the solvent (control), Caspase 3 inhibitor Z-DEVD (10 μM), Necroptosis inhibitor Necropstain-1 (Nec-1, 10 μM), Ferroptosis inhibitor Ferrostain-1 (Fer-1, 10 μM), and DAC (1 μM), either alone or in combination as indicated, for 3 days. Floating and adherent cells were collected and analyzed as in A. D. The % PI+, and % Annexin V+PI+ cells were quantified as in B. Shown are results of one of two experiments. Column: mean, Bar: SD.
DNA methylation and necroptosis in tumor-induced MDSCs.
We next performed genome-wide DNA methylation analysis of tumor-induced MDSCs. Purified MDSCs from spleens of AT3 tumor-bearing mice were used for genomic DNA preparation that was subjected to bisulfite conversion and genome-wide high throughout DNA sequencing. DNA hypermethylation occurs throughout the entire genome in tumor-induced MDSCs (Fig. 5A). The promoter regions of several genes with known functions in cell death were hypermethylated (Fig. 5B). Particularly, the promoters of Tnf, Ripk1, and Ripk3 were hypermethylated (Fig. 5B). Analysis of the promoter DNA sequences revealed that the Tnf promoter lacks a CpG island, whereas both Ripk1 and Ripk3 promoters contains CpG islands (Fig. 5C). However, all three promoters are hypermethylated (Fig. 5D).
Figure 5. The promoter DNA of Tnf, Ripk1 and Ripk3 are methylated in tumor-induced MDSCs in vivo.
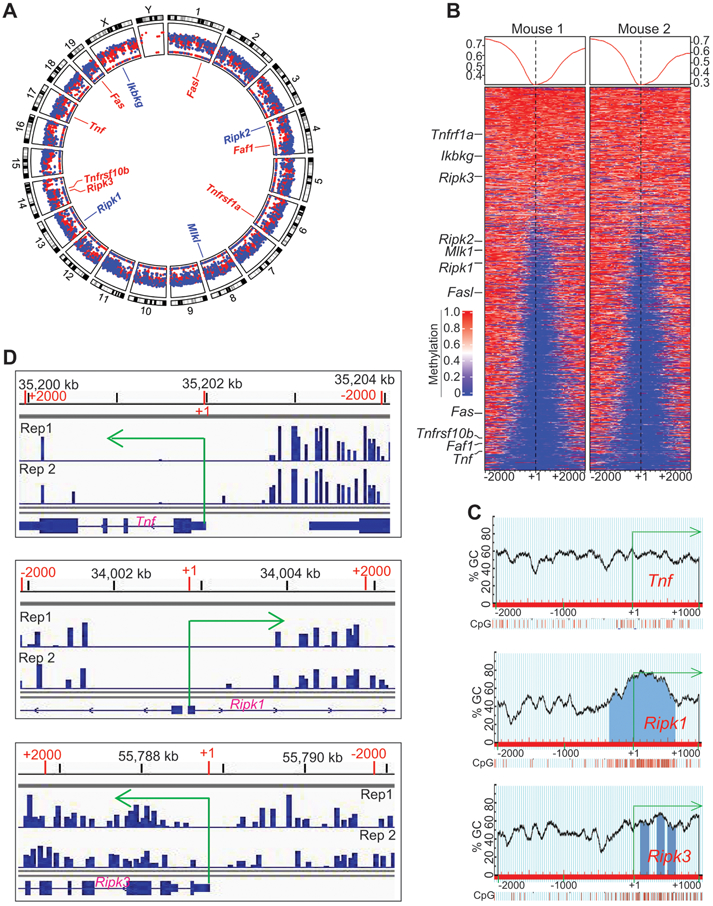
A. Spleens were collected from AT3 tumor-bearing mice as in Fig. 1A. The genomic DNA from purified CD11b+Gr1+ cells were then analyzed by bisulfite conversion and Illumina NGS sequencing. The sequencing data were transformed to raw reads, and the raw reads were further analyzed by Bismark program to align bisulfite-treated reads to reference genome. The gene annotation files and methylation level were then determined. Shown is the Circos plot of the genome-wide distribution of hyper and hypomethylated regions in the tumor-induced MDSC genome by chromosome. The mean methylation levels within a 2000 bp windows starting TSS to +2000 bp upstream are displayed. The promoters with methylation levels >= 0.33 are displayed in red and those < 0.3 are displayed in blue. Several genes with known functions in cell death pathways are highlighted in the inner circle. Shown is one of the two representative experiments. B. Promoter-specific heatmap of methylation at DMR loci identified in the MDSCs from two tumor-bearing mice. The methylation signals within the ±2000 of TSS was calculated using 50 bp windows and plotted in a heatmap. The color gradient indicates with methylation from 0 to 1, corresponding to 0% to 100% methylation. Several genes with known functions in cell death pathways are indicated at the left. C. The Tnf, Ripk1, and Ripk3 promoter DNA sequences (−2000 to +1000 relative to the transcription start site) were extracted from the mouse genome database using ensemble genome browser. The DNA sequences were uploaded to the MethPrimer for predication CpG islands. Shown are the predicted CpG islands and CpG content of their indicated gene promoters. D. CD11b+Gr1+ cells were purified from spleens of AT3 tumor-bearing mice using the CD11b microbeads and LS columns (Miltenyi Biotech). Genomic DNA was prepared from the purified cells and subjected to bisulfite-sequencing. The methylation reads were loaded to IGV genome browser. Shown is snapshots of the DNA methylation profiles at the TNf, Ripk1, and Ripk3 promoter regions.
DNA methylation sustains MDSC survival through impairing the TNFα-RIP1 necroptosis pathway.
The above findings suggest that DNA methylation maintain MDSC survival at least in part through silencing the RIP1-dependent necroptosis pathway. TNFα is a potent necroptosis inducer (25) and its promoter DNA is hypermethylated in tumor-induced MDSCs (Fig. 5D). We then analyzed TNFα expression in J774M cells. Consistent with the observation that the Tnf promoter is hypermethylated (Fig. 5B & D), DAC treatment significantly increased TNFα expression level in J774M cells (Fig. 6A). Furthermore, TNFα protein induced J774M cell death in a dose-dependent manner (Fig. 6B), and neutralizing TNFα diminished DAC-induced cell death (Fig. 6C). To determine whether this in vitro finding can be extended to in vivo MDSC survival regulation, tumor-bearing mice with treated with IgG control and TNFα neutralization mAb, respectively. Neutralizing TNFα significantly increased tumor growth (Fig. S5A) and increased MDSC accumulation in the tumor (Fig. S5B & C). Although the promoters of Ripk1 and Ripk3 are hypermethylated (Fig. 5B-D), DAC treatment failed to increase RIP1 expression levels in J774M cells (Fig. S6A-B), suggesting that DNA methylation is not a major mechanism underlying RIP1 expression regulation in MDSCs.
Figure 6. DNA methylation-mediated TNFα silencing impairs necroptosis in MDSC-like cells in vitro.
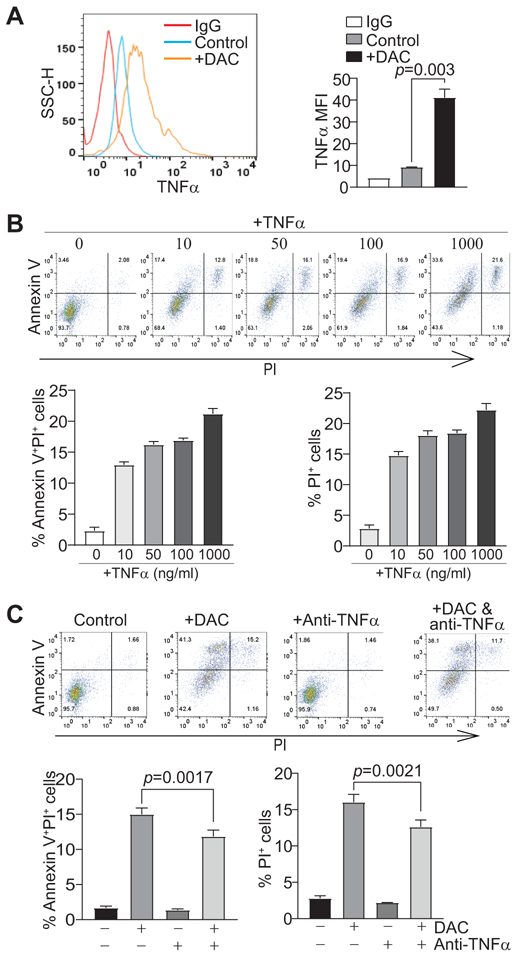
A. J744M cells were treated with DAC (1 μM) for 3 days, intracellularly stained with TNFα-specific mAb, and analyzed by flow cytometry. Left panel shows TNFα levels in untreated and treated cells, and right panel shows quantification of TNFα MFI. B. J774M cells were treated with recombinant TNFα at the indicated doses for 3 days. Floating and adherent cells were collected, stained with Annexin V and PI, and analyzed by flow cytometry. The top panel shows representative results of dot plots. The % PI+, and % Annexin V+PI+ cells were quantified as expressed as % PI+ cells in the treatment group - % PI+ cells of control group, and %Annexin V+PI+ cells of treatment group - % Annexin V+PI+ cells of control group, respectively, and shown at the bottom panel. Column: Mean; Bar: SD. C. J774M cells were treated with DAC (1 μM) in the presence of IgG or anti-mouse TNFα (1 mg/ml) for 3 days. Floating and adherent cells were collected and analyzed by flow cytometry as in B. Shown is representative results of one of two experiments.
IL6 intrinsic signaling silences the TNFα-RIP1 necroptosis pathway.
DNMT1 and DNMT3b are transcriptionally regulated by activated STAT3 (5,26). IL6 is one of the STAT3 inducers (27,28). It is therefore likely that MDSCs may express IL6 to activate DNMTs to hypermethylate TNFα. To extend these findings to myeloid cells, we analyzed IL6 expression levels in tumor-infiltrating MDSCs in vivo. Although only weak IL6 expression was detected in tumor cells, the tumor-infiltrating MDSCs express relatively high level of IL6 (Fig. 7A & B). To determine the human relevance, we collected peripheral blood specimens from healthy donors and colorectal cancer patients (Table S1 & 2). The CD11b+CD33+HLA-DR− MDSCs were gated and analyzed for IL6 level. As expected, human colorectal cancer patients have a significantly higher level of MDSC accumulation than healthy donors (Fig. 7C, D & F). IL6 protein level is significantly higher in MDSCs from colorectal cancer patients as compared to health donors (Fig. 7C, E and G). Analysis of the human colon cancer dataset revealed that IL6 expression level is significantly higher in human colon carcinomas than in the normal colon tissues (Fig. S7A). No enough metastatic colon carcinoma data is available from the TCGA database, but analysis of human melanoma IL6 expression level indicates that there is no significant difference in IL6 expression between primary and metastatic human melanoma (Fig. S7B).
Figure 7. Autocrine IL6 activates STAT3 to up-regulates DNMT1 and DNMT3b in MDSCs.
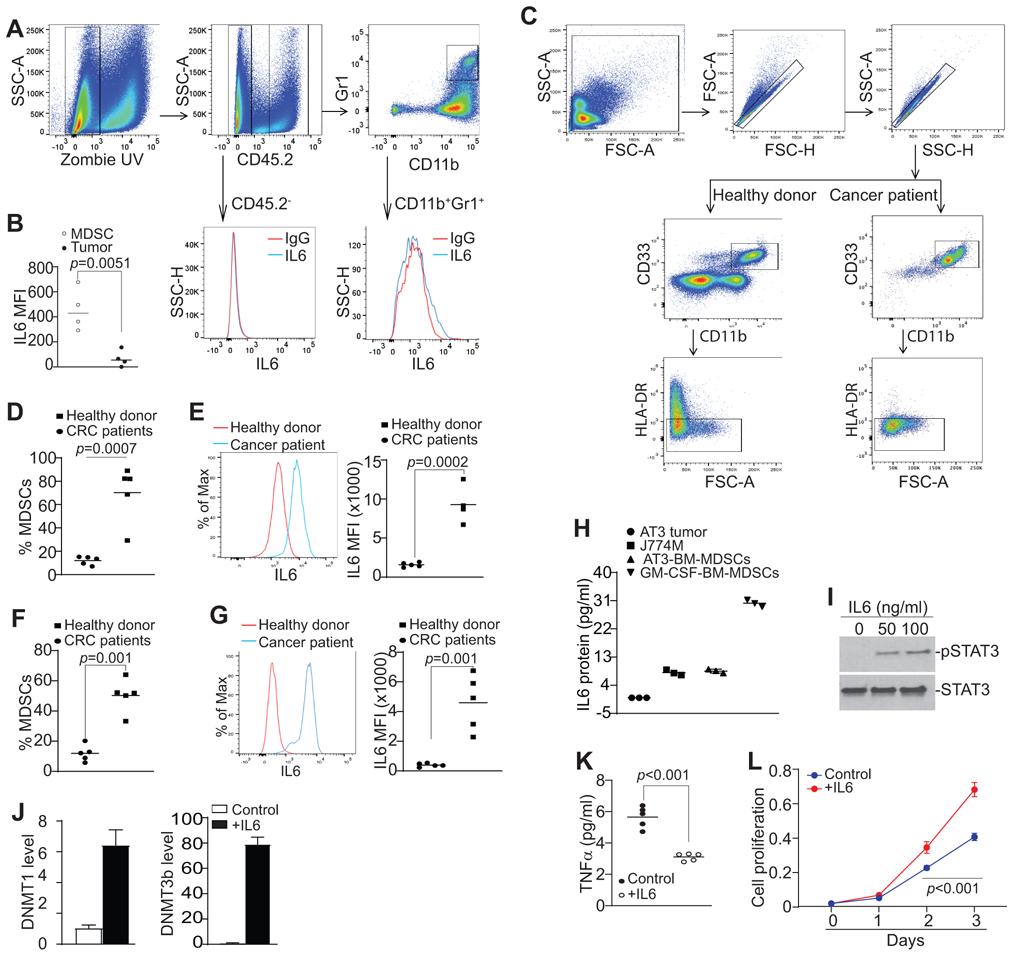
A. CT26 cells were orthotopically transplanted to cecal wall of BALB/c mice (n=4). The tumor tissues were collected, digested in collagenase solution to make single cells. The single cell mixtures were then stained with IgG isotype, CD45.2−, CD11b−, Gr1−, and IL6-specific mAbs plus Zombie UV, and analyzed by flow cytometry. The gating strategy is shown. The CD45.2− tumor cells and CD11b+Gr1+ cells were gated and analyzed for IL6 protein MFI, respectively. Shown are representative results from one of four mice. B. Quantification of IL6 protein level (MFI) in tumor cells and tumor-infiltrating MDSCs. IL6 MFI is calculated as IL6 MFI of the anti-IL6 mAb-stained sample – IgG MFI of the same sample. C. Peripheral blood specimens were collected from healthy donors (n=5) and colorectal cancer patients (n=5). Buffy coat was processed, stained with CD11b−, CD33−, HLA-DR−, and IL6-specific antibodies, and analyzed by flow cytometry. The CD11b+CD33+HLA-DR− cells were gated as shown (top two panels). D. Quantification of CD11b+CD33+HLA-DR− MDSCs as shown in C. Each dot represents % MDSCs from one donor or patient as described in Table S1. E. The representative overlay of IL6 MFI of MDSCs of one healthy donor and one colorectal cancer patient. Quantification of MFIs were shown at the right panel. Each dot represents % MDSCs from one donor or patient as described in Table S1. F & G. Peripheral blood specimens were collected from five more healthy donors (n=5) and colon cancer patients (n=5) as described in Table S2 and repeated the analysis of IL6 in MDSCs as in C-E. H. AT3 tumor cells and J774M cells were cultured for 24h and the culture medium was collected and analyzed for IL6 protein. To induce MDSC from BM cells, BM cells were isolated from C57BL/6 mice and cultured with AT3 tumor cell-conditioned medium or GM-CSF for 5 days. The culture supernatants were then collected and analyzed for IL6 protein level. I. J774M cells were treated with recombinant IL6 protein at the indicated doses for 1.5h and analyzed by Western blotting analysis for the indicated proteins. J. J774M cells were treated with recombinant IL6 protein (100 ng/ml) for 24h and analyzed for DNMT1 and DNMT3b expression by qPCR with Rpl13a as an internal control. K. J774M cells (1x105/well) were seeded in a 96-well plate and cultured with or without 100 ng/mL IL-6 (R&D). Supernatants were collected 24h later and measured for TNFα by ELISA. F. J774M cells (1x103 cells/well) were seeded in a 96 well plate and treated with 100 ng/mL IL-6. Cell proliferation at indicated time points was measured using AqeousOne reagent according to manufacturer’s instructions (Promega).
Analysis of culture supernatant revealed that the MDSC-like J774M cells secrete abundant IL6 protein under in vitro culture conditions, whereas the in vitro cultured tumor cells have undetectable IL6 protein under in vitro culture conditions (Fig. 7H). However, bone marrow-derived MDSCs induced tumor cell-conditioned medium and GM-CSF also secrete abundant IL6 protein in vitro (Fig. 7H). IL6 is a potent activator of STAT3 (29), and STAT3 is known to up-regulate DNMT1 and DNMT3b expression (5,26-28). The high IL6 protein level in MDSCs suggest that MDSCs may use autocrine IL6 to activate STAT3 to upregulate DNMT1 and DNMT3b. To test this hypothesis, J774M cells were treated with IL6 and analyzed STAT3 activation and expression of DNMT1 and DNMT3b. Indeed, IL6 treatment induced STAT3 activation (Fig. 7I) and significantly increased DNMT1 and DNMT3b expression in J774M cells (Fig. 7J). At the same time, IL6 treatment also decreased TNFα production in J774M cells (Fig. 7K) and increased J774M cell proliferation (Fig. 7L). Taken together, our data indicate that MDSC autocrine IL6-STAT3-DNMT-TNFa-RIP1 pathway promotes MDSC survival and accumulation.
Discussion
IRF8 is an essential lineage-specific transcription factor for myeloid cell differentiation and maturation. Loss of IRF8 expression or function leads to interrupted myelopoiesis in humans (30) and accumulation of immature myeloid cells (IMC) in mice with a CD11b+Gr1+ phenotype (7). IRF8 therefore functions as a suppressor of CD11b+Gr1+ immature myeloid cells under physiological conditions (7). One phenotype of tumor-bearing mice is also the accumulation of CD11b+Gr1+ MDSCs (31) and IRF8 is silenced in the tumor-induced MDSCs (21,32). IRF8 therefore also acts as a suppressor of CD11b+Gr1+ MDSC differentiation under pathological conditions (21). In addition, IRF8 regulates expression of apoptosis regulatory factors, such as Caspase 3, Bcl-2, Bcl-xL, Bax, and Fas, in myeloid cells and MDSCs to promote myeloid cells/MDSC apoptosis (14,32-34). IRF8 therefore functions in regulation of both myeloid cell lineage differentiation and apoptosis. In this study, we determined that the Irf8 promoter DNA is hypermethylated in MDSCs and DAC treatment decreased the Irf8 promoter DNA methylation in tumor-bearing mice in vivo. However, inhibition of DNA also diminished CD11b+Gr1+ cell accumulation in IRF8 KO mice in which IRF8 function is lost. Our findings thus suggest that DNA methylation regulates MDSC accumulation not at the stage of MDSC differentiation from myeloid progenitor cells but rather at the stage post MDSC lineage differentiation. Our findings also indicate that DNA methylation regulates MDSC death through an IRF8-independent mechanism.
Cellular turnover plays a key role in MDSC accumulation and function (8,9,11,12,35). Although apoptosis and autophagy are known to be involved in MDSC survival (9,14,32), these cell death mechanisms involve extrinsic ligands such as TRAIL and FasL (9,14). In this study, we extended the regulation of MDSC cell death to an epigenetic mechanism. We determined that DNA methylation is essential for MDSC survival and accumulation in tumor-bearing mice. Furthermore, we extended the cell death pathways to necroptosis in MDSCs. We determined that MDSCs can be induced to die in a RIP1-dependent manner. It is known that TNFα is a potent inducer of RIP1-dependent necroptosis (36,37). Consistent with this phenomenon, we observed that inhibition of DNA methylation significantly increased TNFα expression in MDSCs and MDSC-produced TNFα induces MDSC necroptosis through an autocrine mechanism. We should emphasize that the CD11b+Gr1+ MDSCs are still sensitive to cell death induction, such as to TRAIL- and FasL-induced apoptosis (9,18) and TNFα-induced necroptosis as observed in this study. However, CD11b+Gr1+ myeloid cells in tumor-bearing hosts and in IRF8 KO mice exhibit significantly less spontaneous apoptosis in vivo and are less sensitive to FasL-induced apoptosis as compared to CD11b+Gr1+ myeloid cells in tumor-free and WT mice (32). This relative decrease in sensitivity to cell death induction may have a significant impact on MDSC accumulation. Several cell death pathways regulate MDSC turnover (8-16). The TNFα-induced necroptosis is therefore one of the mechanisms that regulate MDSC turnover. The relative contribution of this TNFα-RIP1 necroptosis pathway in overall MDSC turnover in the tumor microenvironment requires further study.
Apoptosis and necroptosis attenuate each other in mammalian cell death regulation (38). In this study, we observed that inhibition of DNA methylation induced Annexin V+ cells in MDSCs and RIP1-selective inhibitor necropstain-1 also decreased this Annexin V+ population in DAC-treated cells. It was recently proposed that cells can die through RIP1-dependent apoptosis (RDA) (39). It is therefore possible that this Annexin V+ cells might undergo RDA, which also require further study.
IL6 is well known to promote MDSC differentiation (6) and tumor cell-produced IL6 exhibits immune suppressive function and promotes tumor stemness (40-42). Here, we observed that MDSCs is the major producer of IL6 and express much higher level of IL6 than tumor cells in tumor-bearing mice. Furthermore, IL6 expression level is significantly elevated in MDSCs from cancer patients. Although STAT3 can be activated by several cytokines (43,44), it is known that IL6 activates STAT3 and the activated STAT3 activates both histone methyltransferase and DNA methyltransferases to induce an epigenetic silencing program (27-29,45-47). We extended this phenomenon to MDSCs. We determined that IL6 is dramatically up-regulated in MDSCs and autocrine IL6 intrinsic signaling activates STAT3 to up-regulated DNMT1 and DNMT3b in MDSCs. Therefore, we conclude that the IL6-STAT3-DNMT1/3b epigenetic pathway silencing the TNFα-RIP1 necroptosis pathway to increase MDSC survival to maintain MDSC accumulation in the tumor-bearing host.
A recent genome-wide CRISPR/Cas9 screen uncovered the TNFα level is low in tumor and TNF antitumor activity is only limited in tumors at baseline and in immune checkpoint blockade immunotherapy (ICB) non-responders (48). It is possible that TNFα is silenced by its promoter DNA methylation in the tumor microenvironment. Pharmacologic sensitization of tumor cells to the TNFα-RIP1 pathway significantly increased the efficacy of ICB immunotherapy (48). Furthermore, a multi-center phase III clinical trial revealed that DAC induced suppression of MDSC-like neutrophils (49). Pathologically activated neutrophils are a subset of PMN-MDSCs that contribute to the failure of cancer therapies and are associated with poor clinical outcomes (50). Therefore, DAC-mediated epigenetic re-activation of TNFα may have dual efficacies: activate the MDSC intrinsic TNFα-RIP1 pathway to suppress MDSC accumulation and enhance the tumor intrinsic TNFα signaling pathway to augment the sensitivity of tumor cells to ICB immunotherapy.
Supplementary Material
Significance: Findings demonstrate that targeting IL6 expression or function represent potentially effective approaches to suppress MDSC survival and accumulation in the tumor microenvironment.
Acknowledgement:
We thank Dr. Roni J. Bollag at the Georgia Cancer Center Biorepository for advice and for providing human blood specimens. We also thank Dr. Rafal Pacholczyk for advice and assistance in cytokine analysis. Grant support from US Department of Veterans Affairs Merit Review Award (I01 CX001364, to K.L.) and National Institutes of Health (R01 CA133085, R01 CA182518, and R01 CA227433 to K.L., 1F30CA236436, to J.D.K., and 1F31 AI120487, to A.V.P)
Footnotes
Conflict of Interest: The authors declares no conflict of interest
References
- 1.Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol 2019;10:1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008;8:618–31 [DOI] [PubMed] [Google Scholar]
- 5.Ibrahim ML, Klement JD, Lu C, Redd PS, Xiao W, Yang D, et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep 2018;25:3036–46 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010;32:790–802 [DOI] [PubMed] [Google Scholar]
- 7.Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 1996;87:307–17 [DOI] [PubMed] [Google Scholar]
- 8.Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA. Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest 2014;124:2626–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker KH, Horn LA, Ostrand-Rosenberg S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J Leukoc Biol 2016;100:463–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 2014;41:947–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 2012;122:4094–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth F, De La Fuente AC, Vella JL, Zoso A, Inverardi L, Serafini P. Aptamer-mediated blockade of IL4Ralpha triggers apoptosis of MDSCs and limits tumor progression. Cancer Res 2012;72:1373–83 [DOI] [PubMed] [Google Scholar]
- 14.Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 2011;117:5381–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chornoguz O, Grmai L, Sinha P, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Proteomic pathway analysis reveals inflammation increases myeloid-derived suppressor cell resistance to apoptosis. Mol Cell Proteomics 2011;10:M110 002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest 2018;128:3840–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin Cancer Res 2017;23:2942–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Yan M, Sun J, Jain S, Yoshimi R, Abolfath SM, et al. A Reporter Mouse Reveals Lineage-Specific and Heterogeneous Expression of IRF8 during Lymphoid and Myeloid Cell Differentiation. J Immunol 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016;5:e1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest 2013;123:4464–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redd PS, Ibrahim M, Klement JD, Sharman SK, Paschall AV, Yang D, et al. SETD1B activates iNOS expression in myeloid-derived suppressor cells. Cancer Res 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunimoto H, McKenney AS, Meydan C, Shank K, Nazir A, Rapaport F, et al. Aid is a key regulator of myeloid/erythroid differentiation and DNA methylation in hematopoietic stem/progenitor cells. Blood 2017;129:1779–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu X, Yang D, Zimmerman M, Liu F, Yang J, Kannan S, et al. IRF8 regulates acid ceramidase expression to mediate apoptosis and suppresses myelogeneous leukemia. Cancer Res 2011;71:2882–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 2015;6:e1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS, et al. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer 2015;136:547–59 [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Deuring J, Peppelenbosch MP, Kuipers EJ, de Haar C, van der Woude CJ. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis 2012;33:1889–96 [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A 2005;102:6948–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009;15:103–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011;365:127–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Bardhan K, Paschall AV, Yang D, Waller JL, Park MA, et al. Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J Biol Chem 2013;288:19103–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabriele L, Phung J, Fukumoto J, Segal D, Wang IM, Giannakakou P, et al. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J Exp Med 1999;190:411–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burchert A, Cai D, Hofbauer LC, Samuelsson MK, Slater EP, Duyster J, et al. Interferon consensus sequence binding protein (ICSBP; IRF-8) antagonizes BCR/ABL and down-regulates bcl-2. Blood 2004;103:3480–9 [DOI] [PubMed] [Google Scholar]
- 35.Ostrand-Rosenberg S, Sinha P, Chornoguz O, Ecker C. Regulating the suppressors: apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol Immunother 2012;61:1319–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007;12:445–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011;477:335–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 2013;14:727–36 [DOI] [PubMed] [Google Scholar]
- 39.Amin P, Florez M, Najafov A, Pan H, Geng J, Ofengeim D, et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc Natl Acad Sci U S A 2018;115:E5944–E53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Zong X, Mitra S, Mitra AK, Matei D, Nephew KP. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018;67:320–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin Cancer Res 2019;25:3643–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumida K, Ohno Y, Ohtake J, Kaneumi S, Kishikawa T, Takahashi N, et al. IL-11 induces differentiation of myeloid-derived suppressor cells through activation of STAT3 signalling pathway. Scientific Reports 2015;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eichten A, Su J, Adler AP, Zhang L, Ioffe E, Parveen AA, et al. Resistance to Anti-VEGF Therapy Mediated by Autocrine IL6/STAT3 Signaling and Overcome by IL6 Blockade. Cancer Res 2016;76:2327–39 [DOI] [PubMed] [Google Scholar]
- 45.Ozes AR, Pulliam N, Ertosun MG, Yilmaz O, Tang J, Copuroglu E, et al. Protein kinase A-mediated phosphorylation regulates STAT3 activation and oncogenic EZH2 activity. Oncogene 2018;37:3589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quan Z, He Y, Luo C, Xia Y, Zhao Y, Liu N, et al. Interleukin 6 induces cell proliferation of clear cell renal cell carcinoma by suppressing hepaCAM via the STAT3-dependent upregulation of DNMT1 or DNMT3b. Cell Signal 2017;32:48–58 [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood 2006;108:1058–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019;178:585–99 e15 [DOI] [PubMed] [Google Scholar]
- 49.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019;569:73–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.