

Abstract
Obesity is an established risk factor for many cancers and has recently been found to alter the efficacy of T cell–based immunotherapies. Currently, however, the effects of obesity on immunometabolism remain unclear. Understanding these associations is critical, given the fact that T cell metabolism is tightly linked to effector function. Thus, any obesity-associated changes in T cell bioenergetics are likely to drive functional changes at the cellular level, alter the metabolome and cytokine/chemokine milieu, and impact cancer immunotherapy outcomes. Here, we provide a brief overview of T cell metabolism in the presence and absence of solid tumor growth and summarize current literature regarding obesity-associated changes in T cell function and bioenergetics. We also discuss recent findings related to the impact of host obesity on cancer immunotherapy outcomes and present potential mechanisms by which T cell metabolism may influence therapeutic efficacy. Finally, we describe promising pharmaceutical therapies that are being investigated for their ability to improve CD8 T cell metabolism and enhance cancer immunotherapy outcomes in patients, regardless of their obesity status.
Keywords: immunotherapy, obesity, T cell metabolism, tumor immunology
1 |. INTRODUCTION
As of 2016, approximately two thirds of adults in the United States were defined as either overweight (body mass index [BMI] of 25–29.9 kg/m2) or obese (BMI ≥ 30 kg/m2), a trend that shows no sign of slowing.1 Obesity is associated with a chronic, low level of inflammation that is physiologically detrimental and is driven, in part, by adipocyte dysfunction and alterations in the composition and function of leukocytes residing in white adipose tissue.2,3 It is therefore not surprising that obesity is linked to numerous health problems including an increased risk for diabetes, heart disease, and stroke,4,5 and increased complication rates and death following influenza infection.6 Central to this review, epidemiologic data have shown that obesity increases the risk of developing 13 different types of cancers including pancreatic, endometrial, liver, postmenopausal breast, and renal cell carcinoma (RCC).7 Interestingly, for reasons that are incompletely understood, many prevalent cancers, such as cutaneous melanoma and bladder cancer, do not demonstrate an increased prevalence in individuals with obesity.7 Even for non–obesity-associated tumors, host obesity can impair outcomes and increase the risk of recurrence, as has been seen for breast cancer.8
Clinical management of patients with advanced cancers increasingly includes the use of immune-stimulatory therapies. As a result, cancer immunotherapy has become the “fourth pillar” of cancer treatment, alongside the more traditional approaches of surgery, radiation, and chemotherapy.9 Therefore, as the incidence of obesity in the United States continues to rise, it is likely that a growing number of cancer patients with obesity will be treated with immunotherapies. For this reason, understanding the ways in which host obesity subverts normal immune function, in the presence and absence of immune-stimulatory therapies, is a critical issue in the field. Here, we summarize current literature regarding the broad effects of obesity on anti-tumor immunity and cancer immunotherapy outcomes with a focus on understanding how obesity-associated alterations in immunometabolism might drive these changes. Because many of the cancer immunotherapies currently in use or under development work by improving the ability of CD8 T lymphocytes to maintain effector function and kill tumor targets, we will primarily discuss advances in our understanding of how host obesity and the immunosuppressive tumor microenvironment, independently and together, modulate CD8 T cell metabolism. However, as this is a relatively new area of research, we will highlight key findings regarding other leukocyte populations, as needed, to present mechanisms that may also regulate CD8 T cell metabolic fitness and function in the obese tumor microenvironment.
2 |. OVERVIEW OF GLYCOLYSIS
Metabolic pathways are exquisitely regulated to match both the energy demands (ie, requirements for adenosine triphosphate or “ATP”) of cells and the production of numerous biosynthetic intermediates that are required to sustain cellular proliferation, function, and survival. These interconnected biochemical pathways regulate the synthesis (or anabolism), maintenance, and breakdown (or catabolism) of biomolecules through glycolysis, the tricarboxylic acid (TCA) or Krebs cycle, and the pentose phosphate pathway, among others. A comprehensive review of cellular metabolism as related to normal immune function is beyond the scope of this review, but multiple excellent publications have previously covered this topic.10,11
T cells, as with many other types of cells, use glucose to produce ATP, in addition to other substrates such as fatty acids and glutamine. Glycolysis is a multi-step process that breaks glucose molecules down into two molecules of pyruvate, a process that can occur with or without oxygen.12 In many differentiated cells that have a limited proliferative capacity, pyruvate is utilized in one of two ways: (a) It can be oxidized via the TCA cycle to create NADH and FADH2, which are then used in the mitochondria to drive oxidative phosphorylation (OXPHOS), an oxygen-dependent process that produces 36 molecules of ATP per one molecule of glucose, or (b) it can be fermented into lactic acid in the cytosol, an oxygen-independent process called anaerobic glycolysis that yields just two molecules of ATP per one molecule of glucose. A frequently used method for evaluating cellular metabolism is to determine the relative reliance of cells on OXPHOS vs glycolysis for ATP production, as these pathways can dictate ancillary metabolic processes and provide insight into the overall metabolic state of a given cell population. Although OXPHOS robustly generates large amounts of ATP, it also generates reactive oxygen species (ROS), which can be detrimental. Many proliferative cells, like recently activated T cells undergoing clonal expansion, shift instead toward a reliance upon aerobic glycolysis, a process that produces less ATP per glucose molecule but also generates metabolic intermediates needed for the pentose phosphate pathway and TCA cycle.12–16 In doing so, aerobic glycolysis increases the synthesis of lipids, nucleotides, and amino acids, which are key biomolecules needed to sustain rapid cellular proliferation.
Because obesity is frequently associated with the acquisition of insulin resistance and therefore chronically elevated levels of blood glucose, the consideration of glucose metabolism is central to the topic of this review. However, glucose metabolism is not the sole method of providing energy and biomolecules for cells with proliferative needs. Lipid metabolism is also an important contributor to cellular energetics (reviewed in references10,17) and its role as a regulator of T cell metabolism in the context of host obesity has become increasingly scrutinized due to the systemic increases in free fatty acids that are also present in individuals with obesity.18 In addition, amino acids can be used to meet cellular energy demands via processes such as glutamine catabolism. Here, we will focus primarily on glucose metabolism in CD8 T cells and how it is altered by the tumor environment or obesity. We also include a discussion of recent advances in our understanding of fatty acid oxidation as a critical regulator of CD8 T cell metabolic fitness and anti-tumor immunity.
3 |. INTRODUCTION TO T CELL METABOLISM
The ability of a CD8 T cell to mount an anti-tumor response is determined by its metabolic state, which is driven by both intrinsic and extrinsic signals.19–21 Naive CD8 T cells must maintain ion homeostasis, membrane integrity, and actin cytoskeleton rearrangement demands as they surveil for cognate antigens. Because naive T cells have limited proliferation needs, their metabolic profile is largely catabolic and they rely primarily on OXPHOS for ATP production (Figure 1). Activation of naive T cells requires three signals: (a) engagement of the T cell receptor (TCR) by peptide/major histocompatibility complexes; (b) binding of co-stimulatory receptors such as CD28 or inducible T cell co-stimulator (ICOS) on the T cell surface to their respective ligands (ie, co-stimulatory ligands CD80 and CD86, and ICOSL, respectively) on antigen-presenting cells (APCs); and (c) cytokine interaction with receptors on T cells (reviewed in reference22). TCR and co-stimulatory receptor engagement on naive T cells orchestrates rapid transcriptional reprogramming that drives their differentiation into actively proliferating effector cells. Because T cell activation and proliferation require large amounts of biomass and energy indicative of anabolic growth, the metabolic profile of activated T cells also changes, resulting in a greater utilization of aerobic glycolysis10,17 (Figure 1). To fill the need for increased glucose, TCR ligation results in the enhanced expression of glucose transporter (GLUT) proteins.23 Similarly, co-stimulatory receptor ligation and cytokine signaling, especially via IL-2,24 activate phosphoinositide-3 kinase (PI3K) and its downstream target protein kinase B (Akt), which further increase GLUT protein expression on the T cell surface and upregulate glycolytic enzyme activity.25,26 Similar trends are also present in CD4 T cells; it was recently shown that following TCR plus CD28 ligation, naive CD4 T cells rely upon Akt and STAT5 to drive their switch from a relatively quiescent metabolic state to one characterized by increased rates of both glycolysis and OXPHOS.27 Thus, provision of signals 2 and 3 during T cell activation reinforces metabolic changes induced via TCR ligation. In addition to a reliance on glucose metabolism, activated T cells require specific amino acids (eg, leucine, glutamine, arginine, serine, tryptophan) to serve as both building blocks for protein synthesis and as metabolic intermediates for the TCA cycle.11 Overall, metabolic reprogramming from OXPHOS toward aerobic glycolysis increases the rate of ATP production, reduces the production of damaging metabolic byproducts such as ROS in the mitochondria, and builds biomass by providing metabolic intermediates for the pentose phosphate pathway and TCA cycle. Simultaneously, this metabolic shift reduces effector T cell reliance on oxygen for energy production, which enables these cells to retain cytokine production and cytolytic activity after migrating into oxygen-poor environments, such as hypoxic regions within solid tumors.
FIGURE 1.
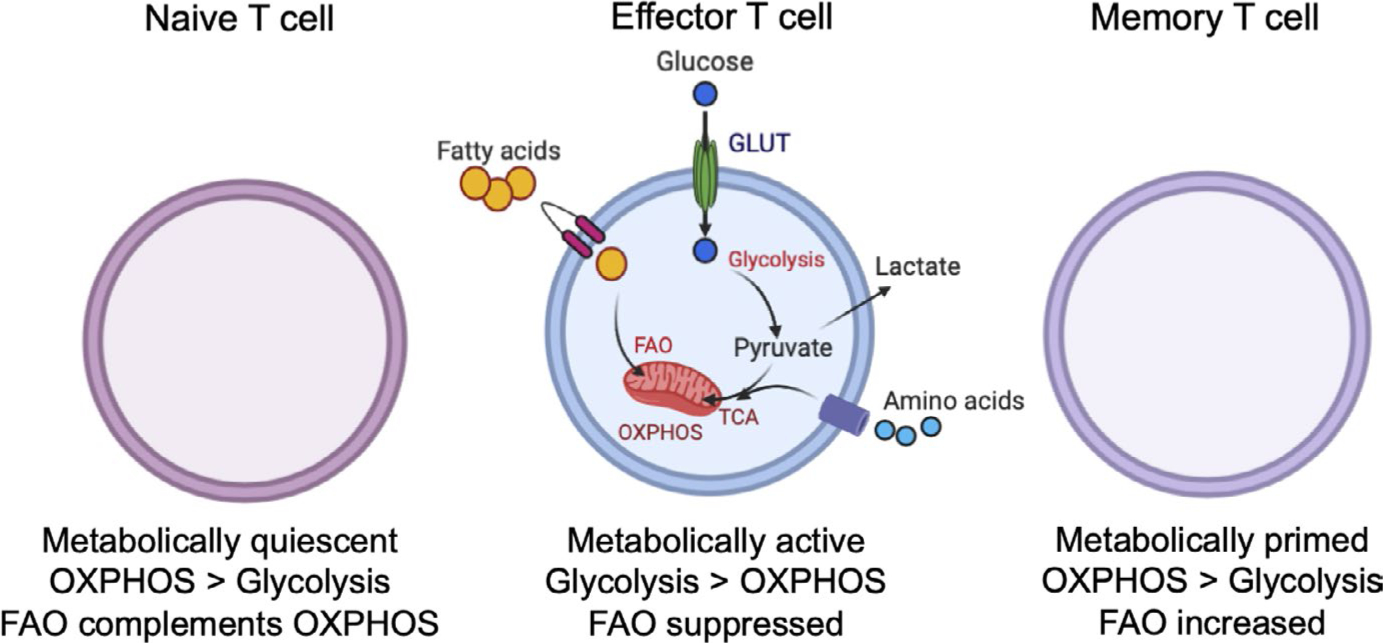
T cell metabolic profiles change with activation status. Naive T cells are metabolically quiescent and tend to use mitochondrial oxidative phosphorylation (OXPHOS) to a greater extent than glycolysis. Following activation, effector T cells exhibit greater metabolic activity, which is supported by increased uptake of glucose and fatty acids through glucose transporters (GLUTs) and fatty acid transporters, respectively. Effector T cells tend to use aerobic glycolysis to a greater extent than OXPHOS. Memory T cells are metabolically primed and ready to respond rapidly to antigen encounter; these cells tend to rely more heavily on fatty acid oxidation to fuel metabolic demands. Figure created using BioRender
In response to both TCR engagement and depletion of ATP reserves, effector T cells utilize nutrient-sensing pathways to regulate proliferative bursts and functional capacity. TCR ligation increases glucose and amino acid uptake by T cells, which activates the nutrient-sensing mammalian target of rapamycin (mTOR) kinase.28 mTOR signaling skews T cell metabolism toward anabolic, biomass- and energy-producing pathways, like aerobic glycolysis, that promote T cell differentiation and enhance the secretion of effector cytokines. In contrast, when energy stores are low, 5′ adenosine monophosphate-activated protein kinase (AMPK) becomes activated and subsequently inhibits mTOR signaling to promote increased OXPHOS and increase ATP availability.29,30
The resolution of ongoing CD8 T cell responses involves a contraction phase in which a majority of effector T cells undergo apoptosis and a minority of cells become effector memory cells, a process that is heavily influenced by metabolic cues.31 In contrast to naive and effector T cells, long-lived memory T cells possess a unique metabolic phenotype with decreased mTOR signaling, a reduced reliance on glycolysis, and a switch to mitochondrial fatty acid oxidation32 (Figure 1). Recent studies have indicated that central memory T cells, which reside preferentially in secondary lymphoid organs, and tissue-resident memory T cells, which reside in non-lymphoid peripheral tissues, have different metabolic profiles. A study by Pan et al33 determined that tissue-resident memory T cells have increased requirements for fatty acid oxidation relative to central memory T cells. However, the generalization that memory T cell has a greater reliance on fatty acid oxidation relative to naive or effector T cells appears to remain well-supported by the literature.
4 |. OBESITY-ASSOCIATED ALTERATIONS IN CD8 T CELL FUNCTION AND METABOLIC FITNESS
The metabolic programming of naive, activated, effector, and memory CD8 T cells is highly sensitive to extrinsic factors. Thus, obesity-induced alterations in CD8 T cell metabolic fitness could greatly impact their differentiation, effector function, and transition to memory cells. To date, most of the research done in this area has supported a scenario in which host obesity is detrimental to immune responses at both the cellular and metabolic levels. However, as the majority of these studies have been conducted in rodent models, it remains to be determined whether similar effects of comparable magnitude are seen in humans with obesity. Here, we will summarize what is known about the ways in which host obesity impacts CD8 T cell metabolic fitness and effector function.
4.1 |. Obesity drives a pro-inflammatory host environment
Healthy adipose tissue is comprised of a mixture of adipocytes and resident leukocyte populations associated with anti-inflammatory immune responses (eg, T-helper [Th]2 cells, T regulatory [Treg] cells, anti-inflammatory or “M2” macrophages, group 2 innate lymphoid cells [ILC2s], and eosinophils) that strive to maintain homeostasis and buffer against metabolic dysfunction and insulin resistance.34–37 In response to a high-fat diet (HFD) or in genetically obese murine models, adipocytes in white adipose tissue undergo hyperplasia and hypertrophy to compensate for increased energy intake. Prolonged positive energy intake (ie, taking in more energy than the body requires) and dysfunctional adipose tissue remodeling result in an environment characterized by a shift in adipokine production from the anti-inflammatory adiponectin to the pro-inflammatory leptin coincident with elevated local concentrations of pro-inflammatory mediators such as TNFα, CRP, IL-6, and MCP-1. These changes are due, in part, to the chronic generation of hypoxia-related genes and ROS. Production of pro-inflammatory cytokines and chemokines coincides with an influx of immune populations associated with pro-inflammatory cellular immune responses (eg, Th1 cells, CD8 T cells, M1 macrophages, group 1 innate lymphoid cells [ILC1s], and invariant natural killer T [iNKT] cells) into adipose tissue34,38–40 and a simultaneous loss of anti-inflammatory immune cells.35,41 If increased adiposity and positive energy balance remain unresolved, these leukocyte populations contribute to further metabolic dysregulation and the long-term systemic inflammation that is a hallmark of obesity. The critical role of CD8 T cells in the initiation and propagation of adipose tissue inflammation and remodeling with obesity is becoming clear and appears to involve intricate crosstalk between T cells, adipocytes, and M1 macrophages via the production of inflammatory cytokines like IFNγ, TNFα, and IL-6.42–45 Therefore, in response to HFD administration, an early influx of CD8 T cells into white adipose tissue contributes to the development of classic, systemic hallmarks of obesity.
One note of caution in interpreting murine studies on the physiological and immunological effects of diet-induced obesity (DIO) is the fact that strain differences in the immunometabolic phenotype and response to chronic HFD administration have been reported between commonly used C57BL/6 and BALB/c mice,46,47 as the latter are much more resistant to developing obesity and metabolic disease. In response to chronic HFD administration, mice of the C57BL/6 background exhibit greater weight gain, glucose intolerance, and insulin resistance than mice of the BALB/c strain. Thus, C57BL/6 mice display a host environment where obesity is coupled with naturally occurring type 2 diabetes. In contrast, BALB/c mice frequently exhibit obesity without the complications of insulin resistance. An additional concern when examining preclinical data from HFD studies is the potential cofounding effects of altered diet composition. Studies often compare chow-fed control mice to mice administered a 45%−60% Kcal from fat diet. Data from our laboratory demonstrates that diet-matching is possible in BALB/c mice since a portion of mice fed a HFD fail to gain weight and are metabolically similar to chow-fed, lean controls.48 We refer to these HFD-fed lean mice as “obese-resistant”. Therefore, strain-specific metabolic differences and potential diet effects should be considered when performing or interpreting studies examining the effects of murine DIO on immune function.
4.2 |. The complex effects of host obesity on T cell effector function
At an organismal level, obesity induces widespread alterations in glucose metabolism that lead to insulin resistance and metabolic syndrome. Multiple prior studies have shown that obesity in mice triggers detrimental changes in the T cell compartment, which range from reduced numbers of T cell progenitors in the thymus and bone marrow to accelerated thymic involution, diminished thymic lymphopoiesis, and restricted TCR diversity.43,49 It is therefore not surprising that obesity is associated with diminished CD8 T cell responses to influenza vaccination,50 decreased frequencies of antigen-specific CD8 T cells in the peripheral blood of C57BL/6 DIO mice following peptide-pulsed dendritic cell immunization,51 and an increased prevalence of programmed cell death 1 (PD-1)+ exhausted CD8 T cells in the livers52 and adipose tissue53 of C57BL/6 DIO mice. Simultaneously, however, obesity can activate mTOR and hypoxia inducible factor-1 (HIF1) pathways to not only regulate T cell trafficking but also support sustained glycolysis and effector function in CD8 T cells.54 Thus, obesity may have both beneficial and detrimental effects on T cell immunity, both of which are discussed below. At this time, it is not clear how signals from multiple obesity-associated factors (eg, general nutrient excess, elevated glucose, increased lipid availability, and elevated plasma leptin) interact to ultimately dictate net positive or negative outcomes on T cell function.
As mentioned, obesity is associated with elevated systemic levels of both insulin and the pro-inflammatory adipokine leptin.55,56 T cells express surface receptors for leptin, making them susceptible to obesity-induced alterations in this protein throughout the entirety of T cell differentiation, trafficking, effector function, and transition to memory cells. Much evidence exists to indicate that leptin is beneficial for T cell immunity, with some reports suggesting that leptin can poise CD8 T cells to engage more efficiently in host immune responses.57–60 Leptin may also activate the nutrient-sensing PI3K/Akt/mTOR pathway in effector T cells to promote glycolysis.61 In addition, signaling through the leptin receptor can upregulate GLUT1 in activated T cells to support increased glucose demands in these metabolically reprogrammed cells.62 With regard to CD4 T cell subset differentiation, leptin promotes the polarization of CD4 T cells to favor expansion of the Th1 subset while suppressing Th2 differentiation, thereby increasing pro-inflammatory secretion of TNFα, IL-6, and IL-12.63,64 Conversely, leptin deficiency in mice and humans skews CD4 T cells toward the Th2 phenotype, and fasting-induced reductions in circulating leptin can result in blunted glucose uptake, impaired glycolysis, and defective CD4 T cell activation.62–65 As cytotoxic CD8 T cells have been described as existing in synonymous pro-inflammatory, IFNγ+Tc1, and anti-inflammatory, IL-4+Tc2, subsets,66,67 it is possible that obesity-associated leptin may also skew CD8 effector T cell differentiation toward a pro-inflammatory profile, although this idea remains to be tested. However, not all effects of leptin are beneficial, as recent evidence suggests that leptin can impair CD8 T cell immunity via a STAT3-dependent signaling pathway that culminates in elevated PD-1 expression and suppression of T cell proliferation, leading to functional exhaustion.68
With obesity, adipocytes fail to properly store fatty acids, resulting in increased free fatty acids in the plasma of humans and mice.18 However, many cell types, including T lymphocytes, have a limited capacity to process lipids, so chronically elevated intracellular concentrations of these substances can induce lipotoxicity, which manifests as cellular damage, dysfunction, and eventually death (reviewed in reference17). In particular, an excess of free fatty acids can diminish mitochondrial membrane potential, increase expression of phosphatidylserine on the outer leaflet of the plasma membrane, and increase caspase activation.17 Studies are beginning to elucidate how specific lipid metabolites can impact T cell signaling cascades to alter polarization and effector function. For example, following chronic HFD administration, an overabundance of the fatty acid palmitate was shown to increase the oxygen consumption rate of naive CD4 T cells during stimulation with IL-7, indicating an elevated OXPHOS response.69 In this study, excess palmitate during CD4 T cell priming biased polarization toward an effector memory-like phenotype via enhanced activation of the PI3K-p110δ-Akt-dependent pathway. The resulting effector memory-like T cells showed preferential trafficking into sites of inflammation.69 Although not shown to be directly triggered by elevated free fatty acid concentrations, shifts in PI3K/Akt/mTOR signaling in CD8 T cells also alter cellular function and trafficking. For example, increased Akt signaling can downregulate CD62L and CCR7 expression on CD8 T cells, induce preferential trafficking into sites of inflammation, and increase transcriptional programming to direct CD8 effector function independent of changes in cell metabolism.70
As mentioned previously, upon activation, T cells undergo extensive metabolic programming, shifting from OXPHOS to glycolytic metabolism, making them highly sensitive to alterations in glucose homeostasis. Glucose deprivation, which will be discussed in more detail in the context of the tumor microenvironment, is a potent inducer of CD8 T cell dysfunction that is characterized by reduced IFNγ, granzyme, and perforin production.71–73 Interestingly, excessive glucose also has modest negative effects on CD8 T cell proliferation and cytokine production in vitro. The in vivo effects of elevated glucose remain in question. For example, in mice with increased blood sugar levels due to chemically induced diabetes, CD8 T cell proliferation, IFNγ production, and cytolytic killing remain intact.74 Thus, elevated blood sugar alone may not be sufficient in vivo to drive the obesity-associated alterations that have been detected in CD8 T cell function.
4.3 |. Obesity alters T cell metabolism
Currently, the extent to which obesity alters CD8 T cell metabolism is incompletely understood. Metabolic profiling of CD4 and CD8 T cells in the context of combined obesity and influenza infection was recently reported by Beck and colleagues.75 In this setting, age-matched DIO or chow-fed control C57BL/6 mice were given a primary infection with X-31 influenza. Following resolution of the primary infection, mice received a secondary PR8 influenza challenge, and CD4 and CD8 T cells were examined ex vivo for metabolic parameters. Prior to secondary influenza infection, splenic CD4 memory T cells from obese mice were found to possess significant increases in both basal oxygen consumption (an indicator of OXPHOS) and extracellular acidification rates (an indicator of glycolysis). In contrast, CD8 memory T cells from DIO mice at this time point had no increase in basal oxygen consumption but did exhibit an increase in basal glycolysis. At day 7 postsecondary influenza infection, splenic CD4 T cells from obese mice exhibited a robust, significant increase in basal oxygen consumption rate and an overall shift toward OXPHOS vs glycolysis, which is atypical for effector T cells. Of note, effector CD8 T cells from influenza-infected DIO mice displayed a comparable shift toward OXPHOS and away from glycolysis. In the lungs of mice with secondary influenza infection, obesity was associated with reduced IFNγ secretion and CD69 expression by CD8 memory T cells.75 Surprisingly, obesity-induced T cell metabolic perturbations were not reversed by weight loss in this model, suggesting that the obese host environment may imprint physiologic changes on both CD4 and CD8 T cells that are not easily reversed.
To determine whether obesity-associated changes were present in splenic T cells from obese BALB/c mice, we made use of our diet-matched DIO and obese-resistant (OB-Res) mice (eg, all mice fed HFD). Splenic CD8 T cells were isolated from age-matched, unmanipulated BALB/c mice that had been on HFD for 20 weeks and were subjected to mitochondrial and glycolysis measurements using an Agilent Seahorse Analyzer. Although basal mitochondrial respiration and ATP production were comparable in CD8 T cells from DIO and obese-resistant mice, CD8 T cells from DIO mice showed a trending increase in their spare respiratory capacity (Figure 2A), suggesting that these cells may be better able to overcome oxidative stress. Similarly, when the glycolytic capacity of these CD8 T cells was examined, the glycolytic reserve of T cells from DIO mice again showed a trending increase (Figure 2B), implying that T cells from DIO mice have a heightened ability to generate ATP to fuel energetic needs. Interestingly, these results suggest that CD8 T cells derived from an obese host environment may have adaptations that support their metabolic fitness (Figure 3). More research in this area is needed to determine the effects of obesity on primary effector vs memory T cell metabolism and the contributions of differing mouse genetic backgrounds. It will also be important for researchers to examine CD4 T cell metabolism in BALB/c DIO mice, which display obesity without insulin resistance, and also investigate the potential impact of dietary composition on both CD8 and CD4 T cells across the spectrum of activation and differentiation.
FIGURE 2.
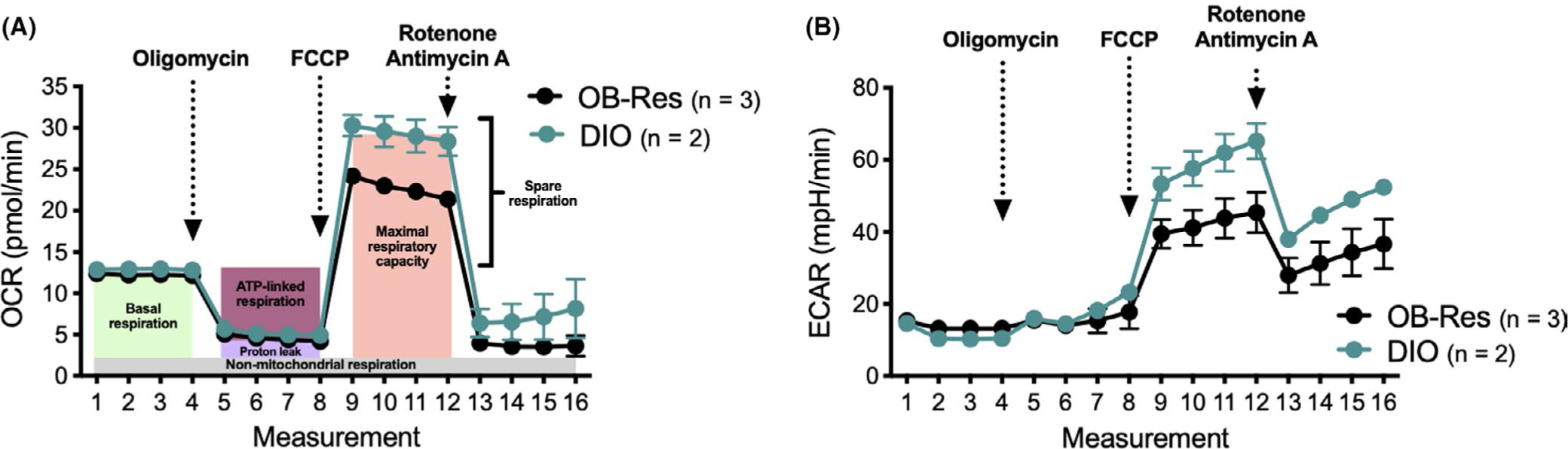
Splenic CD8 T cells from diet-induced obese (DIO) mice have increased spare respiratory capacity and greater glycolytic response relative to T cells from age- and diet-matched obese-resistant (OB-Res) mice. (A) Oxygen consumption rate (OCR, an indicator of OXPHOS) and (B) extracellular acidification rate (ECAR, an indicator of glycolysis) were assessed via examination of isolated CD8 T cells from naive OB-Res (n = 3) and DIO (n = 2) mice
FIGURE 3.
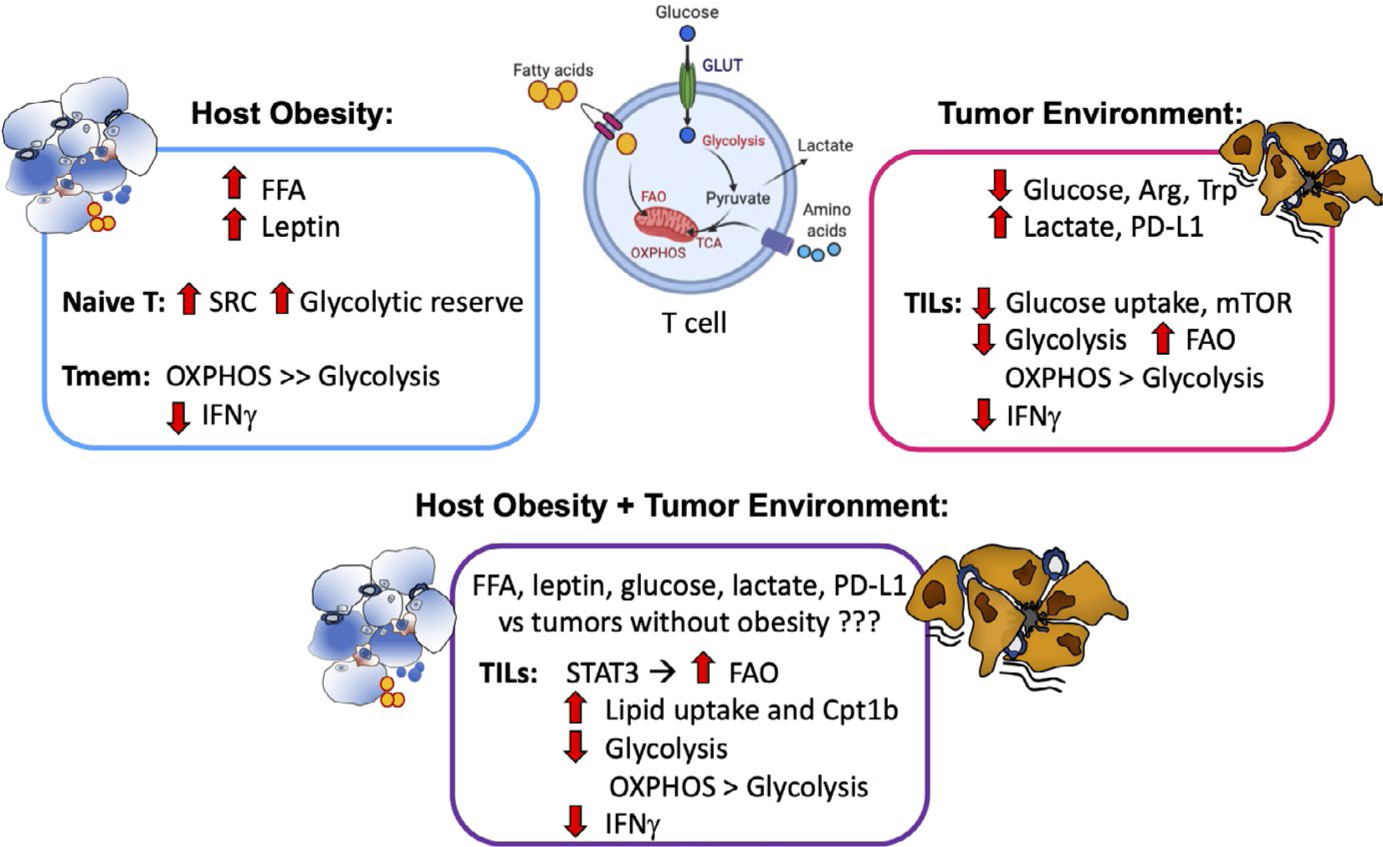
Host obesity and solid tumor microenvironments alter T cell metabolism. Host obesity is associated with systemic increases in free fatty acids (FFA) and leptin. Analysis of splenic T cells from naive mice shows trends toward increased spare respiratory capacity (SRC) and glycolytic reserves in CD8 T cells. Obesity has been described as impairing T cell effector function, as evidenced by decreased memory T cell secretion of IFNγ in response to influenza re-challenge. The underlying metabolic impairments remain poorly understood but appear to include a greater dependence on mitochondrial oxidative phosphorylation (OXPHOS) vs glycolysis; potential alterations to fatty acid oxidation (FAO) have not been examined. Tumors are nutrient and oxygen-poor environments characterized by reduced glucose, arginine (Arg), and tryptophan (Trp), but increased lactate and programmed cell death-ligand 1 (PD-L1); all of these changes are detrimental to tumor-infiltrating lymphocytes (TILs), as indicated by reduced IFNγ. Not surprisingly, tumors also cause metabolic dysregulation in TILs, illustrated by decreased glucose uptake, mTOR activation, and glycolysis. As a result, TILs utilize OXPHOS and FAO to a greater extent than glycolysis. Combined host obesity and tumor growth appear to increase FAO and decrease glycolysis relative to tumors from lean mice, the net result was reduced IFNγ secretion in TILs. Figure created using BioRender
5 |. TUMOR CELL METABOLISM AS A DRIVER OF TUMOR-INFILTRATING LYMPHOCYTE (TIL) DYSFUNCTION
5.1 |. Highly glycolytic tumor cells rely upon Warburg metabolism
Tumor cells are highly proliferative and they sustain continued cell division through numerous mechanisms, including evasion of growth suppressor signaling, upregulation of cell cycle control proteins (eg, cyclins, cyclin-dependent kinases), and resistance to apoptotic cell death.14 Constant, dysregulated cell cycle progression has broad influences on cancer cell biology, as it triggers alterations in tumor-induced secretory factors and promotes an enhanced metastatic potential that favors tumor cell survival. Notably, these hallmarks of malignancy require ample amounts of biomaterials and energy which are provided at the metabolic level by a predominance of aerobic glycolysis, in a manner similar to that discussed for effector T cells, above. This reliance of malignant cells on glucose and their metabolic shift away from OXPHOS and toward the less efficient glycolysis— even in the presence of oxygen–is referred to as “Warburg” metabolism, after Otto Warburg, the Nobel Laureate who discovered this phenomenon.76 In tumor cells, Warburg metabolic reprogramming tends to result in substrate inflexibility and dependence on a sustained influx of glucose carbons through glycolysis,16,77,78 factors that are now being exploited to develop therapeutic strategies to limit tumor progression.79
Tumor cells have multiple mechanisms to compensate for this increased glucose demand, including the upregulation of GLUTs and glycolytic enzymes, or downregulation of enzymes that promote the opposing gluconeogenesis pathway. For example, in clear cell RCC, induction of HIF-1 is a defining characteristic of this tumor type, even under normoxic conditions.80 HIF-1 activation promotes aerobic glycolysis by increasing the expression of several enzymes involved in this process, including phosphoglycerate kinase (PGK1) and lactate dehydrogenase A (LDHA).81 Equally important as a driver of RCC glycolysis is the finding by Li et al82 that nearly 100% of clear cell RCC tumor biospecimens examined had suppressed expression of fructose-1,6-bisphosphatase 1 (FBP1), a key driver of gluconeogenesis, the metabolic pathway that opposes glycolysis. Of note, elevated expression of GLUT proteins in solid tumors may also act as an indicator of patient outcomes. A meta-analysis by Zhao et al83 summarized data from 41 studies involving 4794 cancer patients to assess the prognostic significance of GLUT1 on survival across various cancers. High GLUT1 expression was significantly associated with lower disease-free and overall survival rates in all cancers examined. Subgroup analysis demonstrated a significant GLUT1 expression association in gastric, urinary, ovarian, pancreatic, colorectal, gallbladder, and lung cancers, as well as oral and esophageal squamous cell carcinomas. Clinical data also demonstrate that increased expression of glycolysis-related genes in squamous cancer cells corresponds with reduced frequencies of tumor-infiltrating CD8 T cells.20 Thus, metabolic shifts toward aerobic glycolysis are critical mediators of malignant transformation and also appear to influence disease prognosis and local T cell immunity.
5.2 |. The tumor microenvironment promotes CD8 TIL metabolic dysfunction and decreases effector function
A positive association exists between the accumulation of tumor-infiltrating CD8 T cells in the treatment-naive setting and favorable clinical outcomes in patients with most types of solid tumors,84 RCC being a notable exception.85 However, it is widely recognized that the tumor microenvironment suppresses CD8 T cell effector function through a variety of mechanisms that have been thoroughly reviewed by others.14,86–88 At the cellular level, tumor-secreted factors orchestrate extrinsic changes within local tissues (eg, the induction of cancer-associated-fibroblasts89) and promote the expansion and accumulation of immunosuppressive tumor-infiltrating myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, and Tregs,90 all of which generate a feed-forward pro-inflammatory milieu. In addition to this accumulation of immunosuppressive cells, solid tumors can negatively impact CD8 TILs via tumor cell and leukocyte expression of inhibitory immune checkpoint molecules,91 nutrient deprivation (eg, depletion of glucose, lipids, and amino acids15,92–99), and the release of hypoxia-related factors and immunosuppressive molecules (eg, adenosine).100
The robust glycolytic nature of tumor cells has been shown to provide competition for the limiting amounts of glucose that are present in the tumor environment. One study by Pearce et al illustrated that in the presence of highly glycolytic tumor cells, CD8 TILs resorted to a state of diminished glucose uptake, and had reduced mTOR activity and glycolysis, and impaired IFNγ secretion and anti-tumor activity.101 The authors further determined that CD8 TILs in progressively growing tumors exhibited an abnormal metabolic state, with increased OXPHOS relative to glycolysis. This work supports earlier studies showing that low glucose concentrations within tumors impede Akt signaling in T cells, thereby suppressing glycolysis and inhibiting the differentiation of naive T cells into fully functioning effectors.102 Similar defects in glucose uptake and glycolytic capacity in CD8 TILs are seen in humans. Siska et al103 identified metabolic defects in CD8 TILs from human renal tumors that included diminished glucose uptake, impaired glycolytic capacity, and fragmented mitochondria; these changes coincided with a reduced ability of TILs to proliferate in the tumor microenvironment (Figure 3). The authors additionally reported that metabolic defects in TILs could be overcome by addition of exogenous pyruvate to circumvent glycolytic defects or by reduction of mitochondria-produced ROS via the addition of the mitochondrial antioxidant, MitoQ.
In addition to creating a situation where competition for glucose is fierce, the robust glycolytic nature of tumor cells means that they secrete metabolites such as lactic acid into the tumor microenvironment, a phenomenon that directly inhibits TIL proliferation and cytokine production.104–106 Given this, a 2019 study by Renner et al107 demonstrated that in human melanoma and pancreatic tumor cells, the anti-inflammatory drug diclofenac reduced lactate secretion by tumor cells via inhibition of lactate transporters (monocarboxylate transporter [MCT]1 and MCT4). Importantly, while MCT inhibition by diclofenac reduced the glycolytic activity of T cells, effector functions and viability were maintained by shifting glucose flux into TCA metabolites and enhanced respiration.107 In a complementary study, Delgoffe et al reported that CD8 TILs have reduced mitochondrial content and impaired glucose uptake.108 They subsequently determined that forced expression of leptin in T cells reversed these metabolic defects and improved T cell–mediated tumor clearance.109 Notably, the latter approach was only successful in lean mice, as the leptin resistance present in DIO animals negated its beneficial effects.
Intra-tumoral deprivation of nutrients other than glucose can also impair the metabolic fitness and effector functions of CD8 TILs. Tumor cells and tumor-infiltrating immunosuppressive cells deplete nutrients such as L-arginine, L-cysteine, and L-tryptophan from the environment via consumption and sequestration. This leads to impaired function of both APCs that are needed to stimulate naive tumor antigen-specific T cells and previously differentiated effector T cells.94,96 For example, depletion of L-arginine downregulates the zeta (ζ)-chain of the TCR complex, which impedes normal TCR signal transduction pathways and results in the proliferative arrest of antigen-activated T cells.93,94,96 A study by Geiger et al110 suggests that reversing metabolic dysfunction in TILs can be accomplished by modulating intracellular arginine levels. Using a metabolomics approach, these authors determined that during T cell activation, intracellular arginine concentrations decrease, forcing increased arginine expression in activated T cells, promoting OXPHOS at the expense of glycolysis, and leading to enhanced anti-tumor activity in mice.110 Thus, although the tumor microenvironment induces numerous metabolic defects in TILs, it is possible to reverse these dysfunctional states through multiple approaches, at least in preclinical murine or ex vivo human studies. The translational potential of these approaches remains to be determined.
6 |. OBESITY PROMOTES CANCER PROGRESSION WHILE LIMITING PROTECTIVE T CELL IMMUNITY
As stated above, the reported effects of obesity on CD8 T cell effector function in mice are overwhelmingly negative. Given the critical role of CD8 T cells in controlling nascent or established tumors, it is plausible to assume that obesity would promote tumor progression. Indeed, a multitude of publications support this idea. Both genetic models of obesity (eg, leptin receptor-deficient db/db mice) and models of DIO have demonstrated accelerated primary and/or metastatic tumor growth relative to chow-fed controls, particularly in preclinical models of breast cancer.111–113 In humans, obesity is associated with an increased risk for developing 13 different types of cancer, including renal and postmenopausal breast,114 but it is not known whether these trends are due to obesity-associated decreases in immune surveillance by T cells and other cytotoxic leukocytes, such as natural killer (NK) cells. Another possible pathway correlating obesity with cancer development is genetic instability through oxidative stress-induced DNA damage and impaired DNA repair pathways.115 Multiple preclinical studies show that obesity causes DNA damage in multiple organs, including brain, liver, colon, and testes.116 Obesity may directly impact oncogenic mutation rates to rewire metabolic pathways and enhance the proliferative capacity of malignant cells to promote their survival.115,117,118 However, a lack of longitudinal, mechanistic studies makes it difficult to draw direct links. Additionally, obesity can promote glycolytic cancer cell metabolism through changes in sex hormone levels, inflammatory signals, metabolic signals (eg, insulin and IGF-1), and/or adipokine hormone levels. Regarding the latter, a recent meta-analysis reported that the concentrations of plasma leptin levels increase with disease severity in breast cancer patients.119 The causes and consequences of elevated leptin in breast cancer patients remain questions for future investigations. Nevertheless, because elevated leptin concentrations have been shown to promote T cell exhaustion in the presence and absence of melanoma challenge in C57BL/6 DIO mice,52 it is possible that increased leptin detected in breast cancer patients may be acting to suppress protective CD8 T cell anti-tumor immunity even in the absence of host obesity. In this section, we will briefly summarize known effects of the tumor microenvironment on T cell metabolic fitness, and then, we will discuss the current understanding in the field regarding combined effects of host obesity and solid tumor growth on T cell metabolism and function. Because this is an emerging area of study, we will highlight recent reports that have investigated the net effect of these factors on the metabolic fitness of CD8 T cells as well as other leukocyte populations, as doing so may reveal findings relevant to TIL biology.
6.1 |. Host obesity impedes protective T cell responses to solid tumors
A developing area of research within the broader field of cancer immunology is to determine whether obesity has a net positive or negative impact on specific facets of anti-tumor immunity, then discern the mechanisms by which obesity either diminishes or promotes protective anti-tumor mechanisms and tumor-promoting/ immunosuppressive factors.120–122 We have found that in a preclinical model of orthotopic renal cancer, obesity is associated with an increased accumulation of regulatory dendritic cells within tumors.123 When sort-purified, these CD11b+ dendritic cells were unable to activate naive CD8 T cells and induce their proliferation. Instead, tumor-infiltrating dendritic cells used an arginase-dependent pathway to actively suppress CD8 T cell proliferation when co-cultured with naive antigen-specific T cells and stimulatory, peptide-pulsed dendritic cells from the spleens of tumor-free mice. Thus, renal tumor-infiltrating dendritic cells from DIO mice acquired the regulatory features we had previously identified in a model of mammary carcinoma.124 Renal tumors from obese mice also displayed a heightened accumulation of MDSCs, likely in response to elevated intra-tumoral CCL2 concentrations, and tumor-infiltrating MDSC from both obese and lean mice exerted robust ex vivo suppression of CD8 T cell proliferation.125 Thus, obesity in this preclinical renal cancer model is associated with an exaggerated accumulation of myeloid-lineage suppressor cells, leading to impaired CD8 T cell immunity as evidenced by reduced frequencies of IFNγ+ CD8 T cells in renal tumors. Our findings are in agreement with preclinical studies from other laboratories that show obesity drives the accumulation of tumor-promoting macrophages,111 MDSCs,112,126 and neutrophils113 in preclinical breast cancer, resulting in diminished T cell function and accelerated primary and/or metastatic tumor progression. Clinically, the accumulation of MDSCs correlates positively with disease stage and severity in breast, pancreatic, esophageal, and gastric tumors and often coincides with a concurrent reduction in intra-tumoral CD8 T cells.127–129 Thus, the obesity-associated accumulation of myeloid-lineage suppressor cells in tumors appears to be a recurrent feature that is consistent across human and murine tumors.
6.2 |. Host obesity exacerbates metabolic dysfunction in TILs
At this time, the combined effects of host obesity and a suppressive tumor microenvironment on CD8 TIL metabolic fitness have not been thoroughly studied. However, a 2019 paper by Zhang et al130 have provided a major advance in our understanding of this topic. Using a spontaneously arising model of breast cancer, the authors demonstrated that obesity is associated with more aggressive tumor outgrowth and weak CD8 TIL effector function, similar to what has been reported in other models. With obesity, CD8 TILs had robust STAT3 signaling, high expression of carnitine palmitoyltransferase 1 (“Cpt1b,” a requisite enzyme in the fatty acid oxidation pathway), elevated fatty acid oxidation, and decreased glycolysis, all of which contributed to the inability of CD8 T cells to control tumor growth (Figure 3). These T cell features could be reversed by inhibiting STAT3 signaling in T cells or by inhibiting fatty acid oxidation with etomoxir; in response, T cells were able to mediate tumor regression. This study therefore illustrates that obesity can trigger lipid uptake in CD8 T cells, which forces a metabolic shift toward fatty acid oxidation and away from glycolysis, leading to impaired anti-tumor activity.
This theme of obesity-associated lipid uptake triggering immune dysfunction has also been illustrated in other models. In one study, Michelet et al131 conducted a thorough evaluation of NK cell metabolic fitness and anti-tumor activities in C57BL/6 DIO mice. The authors found that changes at the transcriptomic level were present after just one week of HFD administration. After eight weeks on diet, nearly 3000 genes were differentially expressed in NK cells from DIO vs chow-fed lean mice. Among the most highly altered pathways were those related to peroxisome proliferator-activated receptor (PPAR) signaling, glutathione metabolism, fructose metabolism, and glycolysis. Thus, HFD-induced obesity triggered marked changes in NK cell metabolic profiles. These transcriptomic changes were accompanied by an accumulation of lipids within NK cells that was found to be dependent upon PPAR signaling. PPARs are a family of transcriptional regulators that control cellular metabolism132 and PPAR signaling in tumor cells can promote CD36-dependent lipid accumulation that supports tumor progression.133 In NK cells, elevated CD36 expression and intracellular lipid accumulation were associated with impaired IFNγ secretion, diminished cytolytic capacity, and NK failure to control a melanoma tumor challenge.131 These detrimental effects of the obese host environment were linked specifically to elevated levels of free fatty acids, as administration of palmitate or PPAR agonists impaired glucose uptake and both glycolysis and OXPHOS in splenic NK cells examined ex vivo. Of note, NK metabolic and effector dysfunction induced by palmitate could be reversed by administration of etomoxir, an inhibitor of Cpt1b and fatty acid oxidation.
A second group evaluated the combined effects of obesity and mammary tumor growth on local adipose tissue macrophages.134 These authors demonstrated that with HFD administration in C57BL/6 mice, macrophages within mammary tissues adopted a pro-inflammatory but metabolically distinct profile (referred to as “MMe”) from that previously reported for classic M1 macrophages. MMe macrophages can be induced by culturing bone marrow precursors with insulin, glucose, or the fatty acid palmitate, and they rely upon PPARγ expression to drive lipid metabolism.135 In mice with mammary tumors, MMe adipose tissue macrophages promoted stemness in breast cancer cells and were defined by their increased expression of CD36 and Plin2, genes that encode proteins associated with lipid uptake, along with heightened expression of genes encoding the pro-inflammatory cytokines IL6, IL-1β, and TNFα. In this model system, as with the Michelet et al study described above, ex vivo administration of the fatty acid palmitate was sufficient to drive adipose tissue macrophages to acquire MMe phenotypic and functional characteristics, which supported tumor progression.134
In addition to these studies, earlier work by Herber et al136 demonstrated that tumor-infiltrating dendritic cells, even in lean mice fed a low-fat chow diet, had enhanced intracellular lipid accumulation, which inhibited their normal capacity to stimulate naive CD8 T cell proliferation and promote T cell–mediated tumor clearance. Similar lipid accumulations were present in dendritic cells isolated from subjects with head and neck cancer. Thus, obesity-driven intracellular lipid accumulation in leukocytes is emerging as a conserved mechanism by which normal anti-tumor effector functions are inhibited in T cells and other leukocytes.
7 |. DECIPHERING THE IMPLICATIONS OF OBESITY- AND TUMOR-DERIVED TIL METABOLIC DEFECTS ON CANCER IMMUNOTHERAPY OUTCOMES
Obesity is a confirmed risk factor for numerous cancers; however, the interplay between obesity, obesity-induced alterations in CD8 T cell metabolic fitness, and response to cancer therapy is less clear. The relationship between obesity-induced impairments in CD8 T cells and clinical responses to cancer therapies likely depends on the mechanism of action of the specific therapeutic approach, and the reliance of this therapy on a CD8 T cell response. Additional research is needed to investigate the local and systemic effects of obesity-induced dysregulation on CD8 T cell metabolism, determine whether there is a context in which this dysregulation is beneficial, and identify potential targets or treatment strategies to boost effector CD8 T cell metabolic fitness to further improve clinical responses.
7.1 |. Immune checkpoint blockade and other cancer immunotherapies
In addition to surgical interventions, current cancer treatments include chemotherapies, radiotherapies, targeted therapies, and immunotherapies. Based on recent advancements in the fields of immunology and molecular biology, immunotherapy has become a promising treatment for advanced cancer patients.137–142 Cancer immunotherapies comprise multiple strategies, including cytokine therapies such as high-dose IL-2,143 targeted antibodies,144 adoptive transfers of ex vivo-activated autologous T cells or genetically engineered chimeric antigen receptor (CAR) T cells,145 cancer vaccines,146 genetically engineered oncolytic viruses,147 and immune checkpoint blockade, described in more detail below.148 Immune checkpoint blockade-based therapies are becoming widely utilized in the clinical setting and are currently FDA-approved as treatment options in patients with many types of advanced cancers.149–151 The immune checkpoint blockade approach to immunotherapy involves using antibodies to maintain T cell activation by either binding and agonizing co-stimulatory receptors or binding and antagonizing inhibitory receptors.139,152
Despite the many reported successes of immunotherapies in advanced cancer patients, less than half of patients who receive immunotherapies experience objective, durable responses.153 For this reason, identifying factors that contribute to either improved or diminished treatment response rates is of great interest. Several tumor-intrinsic and extrinsic factors impact treatment efficacy, including tumor mutational burden, the number and functional status of tumor-infiltrating CD8 T cells, and increased expression of inhibitory ligands in the tumor microenvironment.154–158 However, the full extent to which modifiable host factors, like obesity, alter immune responses to immunotherapies and clinical outcomes remain largely unknown.122
7.2 |. Metabolic responses to PD-1 ligation in T cells
In normal physiology, inhibitory immune checkpoints protect against immune activation targeted toward self-antigens (ie, autoimmunity) and help return active immune responses to a basal level following the clearance of foreign agents.137 Numerous inhibitory cell surface markers have been identified, including PD-1, cytotoxic T lymphocyte–associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), and T cell immunoglobulin and mucin-domain containing-3 (TIM-3), among others.91,139,159 The mechanisms by which these various receptors inhibit T cell responses is being elucidated, but all appear to rely at least partially on blunting the metabolic pathways needed for sustained T cell activation. For example, upon TCR ligation, intracellular calcium influx can induce Pdcd1 transcription (the gene encoding PD-1) via nuclear factor of activated T cells (NFAT) c1.160 When PD-1 is engaged by programmed cell death-ligand 1 (PD-L1], an inhibitory signaling is initiated wherein the phosphatases SHP1 and SHP2 are recruited to the intracellular domain of PD-1. This leads subsequently to inhibition of ZAP70 activation at the intracellular domain of the TCR and inhibition of PI3K activation at the intracellular domain of the co-stimulatory receptor CD28, which in turn blocks Akt activation and glycolysis (reviewed in reference161). PD-1 impairment of glycolysis therefore forces a shift in metabolism toward fatty acid oxidation, creating cells with the metabolic phenotype of memory T cells. Evidence for PD-1 regulation of T cell metabolism has also been documented in human CD4 T cells. Here, the metabolic consequences of PD-1 inhibition are decreases in GLUT1 expression, glucose uptake, and glycolysis; with simultaneous increases in CPT1a expression, fatty acid oxidation, and lipolysis.162 Thus, ligation of PD-1 leads to metabolic changes characterized by decreased glycolysis and a shift toward fatty acid oxidation in effector T cells, which ultimately suppresses tumoricidal effector functions.
7.3 |. Obesity paradox paradigms in cancer therapy outcomes
Understanding the effects of energy balance and increased adiposity on cancer therapy outcomes is an emerging area of research and retrospective analyses are beginning to shed light on these complex relationships. Existing data strongly demonstrate a negative impact of cachexia on cancer treatment efficacy and overall patient outcomes (reviewed in references163,164), likely related to reductions in lean body mass and functional energy reserves that become further exacerbated in the face of cancer and/or therapy-induced challenges. Conversely, multiple studies have reported a beneficial association between obesity and the efficacy of multiple types of cancer therapy, reflecting prior findings of “obesity paradox” paradigms in other disease states.165,166 For example, Albiges et al167 determined that elevated BMI (>25 kg/m2) was associated with improved overall survival (OS) and progression-free survival (PFS) following administration of targeted therapies, including VEGF and mTOR inhibitors, in two independent cohorts of advanced renal cancer patients. These favorable responses were found to be associated with low expression of fatty acid synthase (FASN, a multi-unit enzyme that participates in fatty acid synthesis) in individuals with high BMI. More recently, McQuade et al168 reported that obesity (BMI ≥ 30 kg/m2) at treatment initiation in melanoma patients was associated with improved outcomes following administration of targeted therapies, chemotherapies, or immune checkpoint blockade. Notably, the latter association was true only for men. As this was a retrospective study, mechanisms for improved outcomes with obesity were not identified, but the authors did attempt to control for cachexia in their lean cohort as a confounding factor that may have contributed to reduced survival in these individuals. A complementary study by Wang et al52 provided mechanistic insight into this obesity paradox phenomenon in the context of anti-PD-1 administration in melanoma. As mentioned earlier, these authors found that obesity-associated increases in systemic leptin were responsible for promoting CD8 T cell exhaustion, as evidenced by elevated surface expression of PD-1 on CD8 TILs and a loss of cytokine secretion and cytolytic activity. In this study, anti-PD-1 administration was not efficacious in lean mice, likely due to the low expression of PD-1 on CD8 T cells. In contrast, anti-PD-1 was able to delay B16 melanoma outgrowth in DIO mice. The translatability of these findings was demonstrated through a retrospective study of cancer patients with melanoma and other tumor types, wherein anti-PD-1 checkpoint blockade again produced improved OS and PFS in patients with obesity. Thus, the majority of published retrospective studies in cancer subjects have provided evidence in support of obesity paradox paradigms. Although fewer in number, conflicting findings or null effects of obesity have also been reported. One example is a study by Fukumura et al, which shows that VEGF-targeted therapy is less effective in the context of obesity, due to increased myeloid cell expression of IL-6 and FGF.169
8 |. NOVEL THERAPEUTIC APPROACHES FOCUSED ON IMPROVING TIL METABOLIC FITNESS
8.1 |. Immune checkpoint blockade-based strategies to improve TIL metabolic fitness and function
Given the widespread evidence that both host obesity and the solid tumor microenvironment negatively impact TIL metabolic fitness and effector function, the beneficial effects of host obesity on cancer therapy outcomes is surprising. Furthermore, how can our understanding of T cell metabolism be leveraged to improve TIL function in cancer patients and achieve improved efficacy of cancer immunotherapies in patients regardless of BMI status?
One intriguing possibility is that immune checkpoint blockade therapeutics like anti-PD-1 can improve the metabolic fitness of CD8 TILs directly or indirectly by altering the tumor microenvironment. Immune checkpoint blockade strategies can negatively impact tumor metabolism while simultaneously improving CD8 T cell metabolic fitness. As shown by Pearce et al, blocking PD-L1 dampens tumor-specific glycolysis by inhibiting mTOR activity and reducing the expression of glycolytic enzymes in tumor cells.101 Simultaneously, anti-PD-L1 therapy induces selective expansion of tumor-infiltrating CD8 T cells that co-express the activating marker ICOS and inhibitory markers LAG-3 and PD-1,170 suggesting that immune checkpoint blockade can target tumor cells to disrupt their metabolism while simultaneously expanding effector populations. In addition, the Pearce study demonstrated that combinatorial immune checkpoint blockade (anti-CTLA-4, anti-PD-1, and anti-PD-L1) may protect intra-tumoral CD8 T cells from the negative effects of reduced glucose availability within the tumor microenvironment by promoting T cell glycolytic capacity, which in turn, enhances IFNγ production.101
Alternatively, combinatorial strategies targeting metabolic pathways concurrently with immune checkpoint blockade may improve therapeutic efficacy. For example, data from Pedicord et al171 show that rapamycin, an mTOR inhibitor, in combination with anti-CTLA-4 immune checkpoint blockade increases mitochondrial biogenesis and spare respiratory capacity in memory CD8 T cells, suggesting that targeting mTOR may be a beneficial combinatorial strategy to improve the metabolic programming of CD8 T cells and improve therapeutic outcomes. Lastly, therapies that target co-stimulatory molecules on CD8 T cells, like 4–1BB, can enhance mitochondrial capacity in CD8 T cells and improve the efficacy of anti-PD-1 immune checkpoint blockade.172
Therapy-induced metabolic changes are not exclusive to alterations in glucose metabolism but can also be observed in fatty acid metabolism. Data from Zhang et al173 demonstrate that tumor-infiltrating CD8 T cells can enhance PPARα signaling, mitochondria activation, and the catabolism of fatty acids to overcome tumor-associated hypoglycemia and hypoxia and partially preserve effector functions. Further promoting fatty acid catabolism with fenofibrate, a drug that activates PPARα signaling, improved anti-tumor activity and synergized with anti-PD-1 immune checkpoint blockade to achieve superior efficacy in a murine model of melanoma.173 These findings highlight our currently incomplete understanding of how and when specific metabolic pathways, like fatty acid oxidation, can be engaged to promote, rather than inhibit, CD8 TIL effector functions.
8.2 |. Improving the metabolic fitness of adoptively transferred T cells
Intense efforts are underway to improve the metabolic fitness and function of expanded or engineered T cells used in adoptive cell therapy-based immunotherapy. Adoptive cell transfer therapies use ex vivo expanded autologous effector cells to target tumor cells upon re-injection. Increased expression of glycolysis-related proteins in the tumor microenvironment can reduce the efficacy of adoptive cell therapy.174 In one study, the overexpression of glycolysis-related molecules was associated with an increase in the immunosuppressive metabolic intermediate lactate and a reduction in immunostimulatory molecules, IRF1, and CXCL10, resulting in reduced therapeutic efficacy of adoptive cell therapy.175 Targeting lactate via a lactate dehydrogenase A inhibitor improved the anti-tumor effect of adoptive cell therapy, suggesting that targeting tumor glycolysis may be a successful strategy for improving T cell–mediated anti-tumor activity.
CAR or TCR-based strategies genetically alter T cells to recognize specific proteins to improve their ability to clear tumor cells after re-injection. Studies are attempting to refine the architecture of CAR and TCR constructs to improve the metabolic fitness of cytotoxic cells prior to their infusion into cancer patients. Proof of principle studies are providing mechanistic insight into potential alterations needed to improve the metabolic fitness of engineered T cells to promote potent effector function and maintain long-lived memory populations. For example, the inclusion of 4–1BB (CD137), an inducible co-stimulatory receptor, in the CAR architecture promotes the outgrowth of CD8 central memory T cells with enhanced respiratory capacity, increased fatty acid oxidation, and enhanced mitochondrial biogenesis.176 In contrast, inclusion of CD28, a co-stimulatory receptor required for providing signal 2 during T cell activation, results in effector memory cells with an enhanced capacity for glycolysis.176 Expansion conditions are also ongoing to determine the optimal culture conditions in which to grow tumor-reactive effector cells. Data from Sabatino, et al177 demonstrate that activating naive (CD8+CD62L+CD45RA+) T cells engineered to express a CD19-specific CAR with CD3/CD28 in the presence of IL-7, IL-21, and the glycogen-synthase-3b inhibitor TWS119, enhances metabolic fitness and mediates a robust, long-lasting anti-tumor response against systemic acute lymphoblastic leukemia xenografts compared to current CAR T cell expansion protocols. Interestingly, inhibition of proviral integration site for Moloney murine leukemia virus (PIM) kinases, a family of serine/threonine kinases that participate in T cell glucose metabolism, during CD8 T cell expansion can enhance mTORC1, improve the uptake of glucose by CD8 T cells, and increase anti-tumor effector function.178 Data from Klebanoff, et al179 demonstrate that Akt signaling inhibition is compatible with CAR and TCR retroviral transduction of human T cells and promotes a CD62L-expressing central memory phenotype. In that study, cell yield was not compromised and T cells had preserved mitogen-activated protein kinase (MAPK) activation, promotion of the intra-nuclear localization of forkhead box O1 (FOXO1), a transcriptional regulator of T cell memory, and superior anti-tumor efficacy.
8.3 |. Using anti-diabetic agents to enhance T cell metabolic fitness and anti-tumor activity
Multiple clinical and preclinical studies are examining whether repur-posing commonly prescribed pharmaceutical agents that target metabolic pathways, such as FDA-approved anti-diabetic drugs, can be used to improve response rates to T cell–dependent cancer therapies and if so, whether these benefits are mediated through enhancements in CD8 T cell metabolism. However, since both tumor cells and activated CD8 T cells rely upon aerobic glycolysis, there is the potential that any glucose-limiting intervention that inhibits tumor cell metabolism could also negatively impact CD8 TIL function.
A prime example of this approach is illustrated by studies into the use of metformin, a biguanide that is commonly prescribed for the treatment of type 2 diabetes. Metformin has multiple mechanisms of action, including inhibition of mTOR and Complex I of the mitochondrial respiratory chain.180 Because it is effective at lowering systemic glucose availability, metformin is being examined as an adjuvant to promote CD8 T cell–mediated tumor clearance. Prior studies demonstrate that metformin not only reduces the risk for developing some cancers,181 but can also improve responses to chemotherapy and immune-based therapies. Metformin can increase the abundance of tumor-infiltrating CD8 T cells and promote their secretion of effector cytokines, like IL-2, TNFα, and IFNγ.182,183 In a combinatorial strategy, metformin was found to inhibit tumor cell OXPHOS and glycolytic metabolic pathways, yet simultaneously improve endogenous CD8 T cell OXPHOS and cytokine production; the net result was increased efficacy of anti-PD-1 immune checkpoint blockade.184 The mechanistic drivers of these divergent metabolic outcomes in tumor cells vs T cells are not yet fully understood. Similarly, the combination of metformin and anti-CTLA-4 immune checkpoint blockade was found to increase the abundance of tumor-infiltrating CD8 T cells that produced granzyme B and IFNγ and concurrently reduce tumor burden and improve survival in models of breast, melanoma, and colon cancer.185 However, the positive benefits of metformin may not translate to all types of immunotherapy, as metformin was also found to inhibit CD19-CAR T cell cytotoxicity in vivo.186 These studies emphasize the complexity of the tumor microenvironment, but also the potential benefits of combining an anti-diabetic agent with broad therapeutic benefits, like metformin, with immune checkpoint blockade. Additional studies are needed to identify ways to reduce potential detriments to the metabolic fitness and function of CD8 TILs and determine whether obesity-induced alterations in inflammatory and metabolic mediators provide positive or negative benefits for the adjuvant use of anti-glycemic agents in the setting of cancer immunotherapy administration.
9 |. SUMMARY AND FUTURE DIRECTIONS FOR INVESTIGATION
Obesity is a major health concern across the globe. As the prevalence of obesity continues to rise, so too will the numbers of cancer patients who have obesity at the time of their diagnosis or treatment initiation. Currently, the effects of obesity on T cell metabolism and anti-tumor activity remain poorly understood. Much of the existing data suggest that obesity, even in tumor-free mice or humans, impairs normal immune function. When combined with the presence of a progressing malignancy, which are well-documented to be immunosuppressive, current studies indicate that the net effect should be dire for the normal metabolic health of effector T cells that are needed to mediate tumor control.
Instead, retrospective studies in cancer patients have shown that host obesity is associated with improved immune checkpoint blockade outcomes. The reasons for this are not clear, but several possibilities arise from the studies summarized here. For example, from the perspective of T cell metabolism, it is possible that the combination of obesity-driven increases in circulating leptin, which are generally thought to have positive effects on TIL metabolism and function, plus anti-PD-1 checkpoint blockade is supporting CD8 TIL glycolysis at a level sufficient to cause it to dominate over the less beneficial fatty acid oxidation. The net result would be enhanced CD8 TIL metabolic fitness and promotion of robust cytolytic activity and T cell survival, even in the glucose and nutrient-deprived environment within tumors. As yet, this idea has not been tested. It is also possible that the nutrient excess associated with obesity leads to heightened availability of glucose and other key metabolic substrates in the tumor environment, as compared to the tumors of lean individuals, leading to improved CD8 TIL metabolic fitness even prior to the administration of immunotherapy. To address this possibility, studies similar to the one conducted by the Rathmell laboratories,103 wherein the metabolic profiles of CD8 TILs from human renal tumors were examined, should be conducted with biospecimens segregated according to the BMI status of the donor across a variety of tumor types. In addition, future preclinical studies also will need to examine the individual vs combined impact of various facets of host obesity on the metabolic profiles and effector functions of CD8 TILs, as in the exogenous palmitate addition studies we summarized above.69,131 These studies will again need to be conducted using tumors that arise in different anatomic locations, in the presence vs absence of immune checkpoint inhibitors or other therapies of interest. Performing comparable studies with human specimens will be challenging but is also necessary. Lastly, it is also possible that heightened PD-1 expression on TILs from patients with obesity simply provides an increased number of targets for engagement of anti-PD-1 antibodies, as posited by Wang et al52
Investigations into the intersection of host obesity, solid tumor growth, cancer immunotherapies, and T cell metabolism are truly just beginning. The next several years should provide a wealth of mechanistic information that can be used to tailor combinatorial cancer immunotherapies to the needs of patients with advanced disease, both in the presence and absence of obesity, so they can live longer, healthier lives.
ACKNOWLEDGEMENTS
WJT is supported by The Research Training Program in Basic and Translational Oncology (T32CA183926), The University of Alabama at Birmingham (UAB). National Cancer Institute R01CA181088 and R21CA223126 (to LAN).
Footnotes
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Hales CM, Fryar CD, Carroll MD, Freedman DS, Ogden CL. Trends in obesity and severe obesity prevalence in US youth and adults by sex and age, 2007–2008 to 2015–2016. JAMA. 2018;319(16):1723–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox AJ, West NP, Cripps AW. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015;3(3):207–215. [DOI] [PubMed] [Google Scholar]
- 3.Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. 2017;8:1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verma S, Hussain ME. Obesity and diabetes: an update. Diabetes Metab Syndr. 2017;11(1):73–79. [DOI] [PubMed] [Google Scholar]
- 5.Piché M-E, Poirier P, Lemieux I, Després J-P. Overview of epidemiology and contribution of obesity and body fat distribution to cardiovascular disease: an update. Prog Cardiovasc Dis. 2018;61(2):103–113. [DOI] [PubMed] [Google Scholar]
- 6.Honce R, Schultz-Cherry S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front Immunol. 2019;10:1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer-viewpoint of the IARC Working Group. N Engl J Med. 2016;375(8):794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J Clin. 2017;67(5):378–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCune JS. Rapid advances in immunotherapy to treat cancer. Clin Pharmacol Ther. 2018;103(4):540–544. [DOI] [PubMed] [Google Scholar]
- 10.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 15.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol. 2017;8:1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obesity Boden G. and free fatty acids. Endocrinol Metab Clin North Am. 2008;37(3):635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottensmeier CH, Perry KL, Harden EL, et al. Upregulated glucose metabolism correlates inversely with CD8+ T-cell infiltration and survival in squamous cell carcinoma. Cancer Res. 2016;76(14):4136–4148. [DOI] [PubMed] [Google Scholar]
- 21.Munford H, Dimeloe S. Intrinsic and extrinsic determinants of T cell metabolism in health and disease. Front Mol Biosci. 2019;6:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cretenet G, Clerc I, Matias M, et al. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci Rep. 2016;6:24129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. [DOI] [PubMed] [Google Scholar]
- 25.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5(11):844–852. [DOI] [PubMed] [Google Scholar]
- 26.Jones N, Vincent EE, Cronin JG, et al. Akt and STAT5 mediate naïve human CD4+ T-cell early metabolic response to TCR stimulation. Nat Commun. 2019;10(1):2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones N, Vincent EE, Cronin JG, et al. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat Commun. 2019;10(1):2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmond RJ. mTOR regulation of glycolytic metabolism in T Cells. Front Cell Dev Biol. 2018;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang K, Chi H. AMPK helps T cells survive nutrient starvation. Immunity. 2015;42(1):4–6. [DOI] [PubMed] [Google Scholar]
- 30.Chaube B, Bhat MK. AMPK, a key regulator of metabolic/energy homeostasis and mitochondrial biogenesis in cancer cells. Cell Death Dis. 2016;7(1):e2044–e2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15(12):1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Windt GJ, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan Y, Tian T, Park CO, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One. 2011;6(1):e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molofsky AB, Nussbaum JC, Liang HE, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brestoff JR, Kim BS, Saenz SA, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519(7542):242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond). 2008;32(3): 451–463. [DOI] [PubMed] [Google Scholar]
- 39.Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity. 2010;18(10):1918–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park J, Huh JY, Oh J, et al. Activation of invariant natural killer T cells stimulates adipose tissue remodeling via adipocyte death and birth in obesity. Genes Dev. 2019;33(23–24):1657–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc Res. 2017;113(9):1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Youm YH, Vandanmagsar B, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. 2010;185(3):1836–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schipper HS, Prakken B, Kalkhoven E, Boes M. Adipose tissue-resident immune cells: key players in immunometabolism. Trends Endocrinol Metab. 2012;23(8):407–415. [DOI] [PubMed] [Google Scholar]
- 45.Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol. 2019;10:1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montgomery MK, Hallahan NL, Brown SH, et al. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia. 2013;56(5):1129–1139. [DOI] [PubMed] [Google Scholar]
- 47.Jovicic N, Jeftic I, Jovanovic I, et al. Differential immunometabolic phenotype in Th1 and Th2 dominant mouse strains in response to high-fat feeding. PLoS One. 2015;10(7):e0134089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boi SK, Buchta CM, Pearson NA, et al. Obesity alters immune and metabolic profiles: New insight from obese-resistant mice on high-fat diet. Obesity. 2016;24(10):2140–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H, Youm YH, Vandanmagsar B, et al. Obesity accelerates thymic aging. Blood. 2009;114(18):3803–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheridan PA, Paich HA, Handy J, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes. 2012;36(8):1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan SH, Hemann EA, Legge KL, Norian LA, Badovinac VP. Diet-induced obesity does not impact the generation and maintenance of primary memory CD8 T cells. J Immunol. 2014;193(12):5873–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Aguilar EG, Luna JI, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25(1):141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shirakawa K, Yan X, Shinmura K, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. 2016;126(12):4626–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finlay DK, Rosenzweig E, Sinclair LV, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209(13):2441–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–2556. [DOI] [PubMed] [Google Scholar]
- 56.Hursting SD, Berger NA. Energy balance, host-related factors, and cancer progression. J Clin Oncol. 2010;28(26):4058–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loffreda S, Yang S, Lin H, et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998;12(1):57–65. [PubMed] [Google Scholar]
- 58.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897. [DOI] [PubMed] [Google Scholar]
- 59.Gerriets VA, Danzaki K, Kishton RJ, et al. Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol. 2016;46(8):1970–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naylor C, Petri WA Jr. Leptin regulation of immune responses. Trends Mol Med. 2016;22(2):88–98. [DOI] [PubMed] [Google Scholar]
- 61.Procaccini C, De Rosa V, Galgani M, et al. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. J Immunol. 2012;189(6):2941–2953. [DOI] [PubMed] [Google Scholar]
- 62.Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192(1):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Batra A, Okur B, Glauben R, et al. Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology. 2010;151(1):56–62. [DOI] [PubMed] [Google Scholar]
- 64.Fernández-Riejos P, Najib S, Santos-Alvarez J, et al. Role of leptin in the activation of immune cells. Mediators Inflamm. 2010;2010:568343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med. 2012;33(1):35–45. [DOI] [PubMed] [Google Scholar]
- 66.Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135(3):626–635. [DOI] [PubMed] [Google Scholar]
- 67.Yu Y, Cho HI, Wang D, et al. Adoptive transfer of Tc1 or Tc17 cells elicits antitumor immunity against established melanoma through distinct mechanisms. J Immunol. 2013;190(4):1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Ping FF, Bakht S, Ling J, Hassan W. Immunometabolism features of metabolic deregulation and cancer. J Cell Mol Med. 2019;23(2):694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mauro C, Smith J, Cucchi D, et al. Obesity-induced metabolic stress leads to biased effector memory CD4(+) T cell differentiation via PI3K p110delta-Akt-mediated signals. Cell Metab. 2017;25(3):593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macintyre AN, Finlay D, Preston G, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34(2):224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174(8):4670–4677. [DOI] [PubMed] [Google Scholar]
- 72.Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38(9):2438–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobs SR, Herman CE, MacIver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Recino A, Barkan K, Wong FS, Ladds G, Cooke A, Wallberg M. Hyperglycaemia does not affect antigen-specific activation and cytolytic killing by CD8(+) T cells in vivo. Biosci Rep. 2017;37(4):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rebeles J, Green WD, Alwarawrah Y, et al. Obesity-induced changes in T-cell metabolism are associated with impaired memory T-cell response to influenza and are not reversed with weight loss. J Infect Dis. 2019;219(10):1652–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy. J Exp Clin Cancer Res. 2019;38(1):403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490. [DOI] [PubMed] [Google Scholar]
- 81.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li B, Qiu B, Lee DS, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513(7517):251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao ZX, Lu LW, Qiu J, et al. Glucose transporter-1 as an independent prognostic marker for cancer: a meta-analysis. Oncotarget. 2018;9(2):2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Linette GP, Carreno BM. Tumor-infiltrating lymphocytes in the checkpoint inhibitor era. Curr Hematol Malignancy Rep. 2019;14(4):286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geissler K, Fornara P, Lautenschlager C, Holzhausen HJ, Seliger B, Riemann D. Immune signature of tumor infiltrating immune cells in renal cancer. Oncoimmunology. 2015;4(1):e985082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. [DOI] [PubMed] [Google Scholar]
- 87.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–5596. [DOI] [PubMed] [Google Scholar]
- 88.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40. [DOI] [PubMed] [Google Scholar]
- 89.Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 2015;361(2):155–163. [DOI] [PubMed] [Google Scholar]
- 90.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. [DOI] [PubMed] [Google Scholar]
- 91.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. [DOI] [PubMed] [Google Scholar]
- 92.Indoleamine Mellor A. 2,3 dioxygenase and regulation of T cell immunity. Biochem Biophys Res Commun. 2005;338(1):20–24. [DOI] [PubMed] [Google Scholar]
- 93.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109(4):1568–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84(4):949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–2623. [DOI] [PubMed] [Google Scholar]
- 98.Delgoffe GM, Powell JD. Sugar, fat, and protein: new insights into what T cells crave. Curr Opin Immunol. 2015;33:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy. 2017;9(1):83–97. [DOI] [PubMed] [Google Scholar]
- 100.Fernandez-Ramos AA, Poindessous V, Marchetti-Laurent C, Pallet N, Loriot MA. The effect of immunosuppressive molecules on T-cell metabolic reprogramming. Biochimie. 2016;127:23–36. [DOI] [PubMed] [Google Scholar]
- 101.Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siska PJ, Beckermann KE, Mason FM, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2(12):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brand A, Singer K, Koehl GE, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance By T and NK cells. Cell Metab. 2016;24(5):657–671. [DOI] [PubMed] [Google Scholar]
- 105.Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. [DOI] [PubMed] [Google Scholar]
- 106.Haas R, Smith J, Rocher-Ros V, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015;13(7):e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Renner K, Bruss C, Schnell A, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 2019;29(1):135–150.e139. [DOI] [PubMed] [Google Scholar]
- 108.Scharping NE, Menk AV, Moreci RS, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intra-tumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45(2):374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rivadeneira DB, DePeaux K, Wang Y, et al. Oncolytic viruses engineered to enforce leptin expression reprogram tumor-infiltrating T cell metabolism and promote tumor clearance. Immunity. 2019;51(3): 548–560.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Geiger R, Rieckmann JC, Wolf T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3): 829–842.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arendt LM, McCready J, Keller PJ, et al. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013;73(19):6080–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kolb R, Phan L, Borcherding N, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. 2016;7:13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Quail DF, Olson OC, Bhardwaj P, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. 2017;19(8):974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer—viewpoint of the IARC Working Group. N Engl J Med. 2016;375(8):794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kompella P, Vasquez KM. Obesity and cancer: A mechanistic overview of metabolic changes in obesity that impact genetic instability. Mol Carcinog. 2019;58(9):1531–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Setayesh T, Nersesyan A, Mišík M, et al. Impact of obesity and overweight on DNA stability: Few facts and many hypotheses. Mutat Res. 2018;777:64–91. [DOI] [PubMed] [Google Scholar]
- 117.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doerstling SS, O’Flanagan CH, Hursting SD. Obesity and cancer metabolism: a perspective on interacting tumor-intrinsic and extrinsic factors. Front Oncol. 2017;7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Niu J, Jiang L, Guo W, Shao L, Liu Y, Wang L. The Association between leptin level and breast cancer: a meta-analysis. PLoS ONE. 2013;8(6):e67349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cozzo AJ, Fuller AM, Makowski L. Contribution of adipose tissue to development of cancer. Compr Physiol. 2017;8(1):237–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Donohoe CL, Lysaght J, O’Sullivan J, Reynolds JV. Emerging concepts linking obesity with the hallmarks of cancer. Trends Endocrinol Metab. 2017;28(1):46–62. [DOI] [PubMed] [Google Scholar]
- 122.Dyck L, Lynch L. Cancer, obesity and immunometabolism - connecting the dots. Cancer Lett. 2018;417:11–20. [DOI] [PubMed] [Google Scholar]
- 123.James BR, Tomanek-Chalkley A, Askeland EJ, Kucaba T, Griffith TS, Norian LA. Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J Immunol. 2012;189(3):1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Norian LA, Rodriguez PC, O’Mara LA, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. 2009;69(7):3086–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hale M, Itani F, Buchta CM, Wald G, Bing M, Norian LA. Obesity triggers enhanced MDSC accumulation in murine renal tumors via elevated local production of CCL2. PLoS ONE. 2015;10(3):e0118784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline science: high fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. 2018;103(3):395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nagaraj S, Youn J-I, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol. 2013;191(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang C, Yue C, Herrmann A, et al. STAT3 activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 2020;31(1):148–161.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Michelet X, Dyck L, Hogan A, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19(12):1330–1340. [DOI] [PubMed] [Google Scholar]
- 132.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812(8):1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pascual G, Avgustinova A, Mejetta S, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41–45. [DOI] [PubMed] [Google Scholar]
- 134.Tiwari P, Blank A, Cui C, et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J Exp Med. 2019;216(6):1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kratz M, Coats BR, Hisert KB, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20(4):614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Herber DL, Cao W, Nefedova Y, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Couzin-Frankel J Cancer immunotherapy. Science. 2013;342(6165):1432–1433. [DOI] [PubMed] [Google Scholar]
- 142.Trapani JA, Darcy PK. Immunotherapy of cancer. Aust Fam Physician. 2017;46(4):194–199. [PubMed] [Google Scholar]
- 143.Choudhry H, Helmi N, Abdulaal WH, et al. Prospects of IL-2 in Cancer Immunotherapy. BioMed Res Int. 2018;2018:9056173–9056173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. 2012;148(6):1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. Journal Immunother Cancer. 2018;6(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. [DOI] [PubMed] [Google Scholar]
- 149.Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Network Open. 2019;2(5):e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Toor SM, Sasidharan Nair V, Decock J, Elkord E. Immune checkpoints in the tumor microenvironment. Semin Cancer Biol. 2019. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 152.Sanchez K, Page D, McArthur HL. Immunotherapy in breast cancer: an overview of modern checkpoint blockade strategies and vaccines. Curr Probl Cancer. 2016;40(2–4):151–162. [DOI] [PubMed] [Google Scholar]
- 153.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pitt Jonathan M, Vétizou M, Daillère R, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44(6):1255–1269. [DOI] [PubMed] [Google Scholar]
- 156.Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed Versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol. 2017;1036:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rieth J, Subramanian S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int J Mol Sci. 2018;19(5):1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–1218. [DOI] [PubMed] [Google Scholar]
- 161.Arasanz H, Gato-Cañas M, Zuazo M, et al. PD1 signal transduction pathways in T cells. Oncotarget. 2017;8(31):51936–51945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers. 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 164.Tobert CM, Hamilton-Reeves JM, Norian LA, et al. Emerging impact of malnutrition on surgical patients: literature review and potential implications for cystectomy in bladder cancer. J Urol. 2017;198(3):511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carson KR, Bartlett NL, McDonald JR, et al. Increased body mass index is associated with improved survival in United States veterans with diffuse large B-cell lymphoma. J Clin Oncol. 2012;30(26):3217–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xu H, Cao D, He A, Ge W. The prognostic role of obesity is independent of sex in cancer patients treated with immune checkpoint inhibitors: A pooled analysis of 4090 cancer patients. Int Immunopharmacol. 2019;74:105745. [DOI] [PubMed] [Google Scholar]
- 167.Albiges L, Hakimi AA, Xie W, et al. Body mass index and metastatic renal cell carcinoma: clinical and biological correlations. J Clin Oncol. 2016;34(30):3655–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McQuade JL, Daniel CR, Hess KR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 2018;19(3):310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Incio J, Liu H, Suboj P, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6(8):852–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beyrend G, van der Gracht E, Yilmaz A, et al. PD-L1 blockade engages tumor-infiltrating lymphocytes to co-express targetable activating and inhibitory receptors. J Immunother Cancer. 2019;7(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pedicord VA, Cross JR, Montalvo-Ortiz W, Miller ML, Allison JP. Friends not foes: CTLA-4 blockade and mTOR inhibition cooperate during CD8+ T cell priming to promote memory formation and metabolic readiness. J Immunol. 2015;194(5):2089–2098. [DOI] [PubMed] [Google Scholar]
- 172.Menk AV, Scharping NE, Rivadeneira DB, et al. 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215(4):1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang Y, Kurupati R, Liu L, et al. Enhancing CD8(+) T Cell Fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–391 e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017;26(1):94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cascone T, McKenzie JA, Mbofung RM, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977–987.e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. [DOI] [PubMed] [Google Scholar]
- 177.Sabatino M, Hu J, Sommariva M, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chatterjee S, Chakraborty P, Daenthanasanmak A, et al. Targeting PIM kinase with PD1 inhibition improves immunotherapeutic anti-tumor T-cell response. Clin Cancer Res. 2019;25(3):1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Klebanoff CA, Crompton JG, Leonardi AJ, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. 2013;37(3):207–218. [DOI] [PubMed] [Google Scholar]
- 182.Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kunisada Y, Eikawa S, Tomonobu N, et al. Attenuation of CD4(+) CD25(+) regulatory T cells in the tumor microenvironment by metformin, a Type 2 diabetes drug. EBioMedicine. 2017;25:154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cha JH, Yang WH, Xia W, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71(4):606–620.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mu Q, Jiang M, Zhang Y, et al. Metformin inhibits proliferation and cytotoxicity and induces apoptosis via AMPK pathway in CD19-chimeric antigen receptor-modified T cells. Onco Targets Ther. 2018;11:1767–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]