

Abstract
We previously found that inhibition of p66Shc confers protection against hepatic stellate cell (HSC) activation during liver fibrosis. However, the effect of p66Shc on HSC proliferation, as well as the mechanism by which p66Shc is modulated, remains unknown. Here, we elucidated the effect of p66Shc on HSC proliferation and evaluated microRNA (miRNA)-p66Shc-mediated reactive oxidative species (ROS) generation in liver fibrosis. An in vivo model of carbon tetrachloride (CCl4)-induced liver fibrosis in rats and an LX-2 cell model were developed. p66Shc expression was significantly upregulated in rats with CCl4-induced liver fibrosis and in human fibrotic livers. Additionally, p66Shc knockdown in vitro attenuated mitochondrial ROS generation and HSC proliferation. Interestingly, p66Shc promoted HSC proliferation via β-catenin dephosphorylation in vitro. MicroRNA (miR)-203a-3p, which was identified by microarray and bioinformatics analyses, directly inhibited p66Shc translation and attenuated HSC proliferation in vitro. Importantly, p66Shc was found to play an indispensable role in the protective effect of miR-203a-3p. Furthermore, carnosic acid (CA), the major antioxidant compound extracted from rosemary leaves, protected against CCl4-induced liver fibrosis through the miR-203a-3p/p66Shc axis. Collectively, these results suggest that p66Shc, which is directly suppressed by miR-203a-3p, is a key regulator of liver fibrosis. This finding may lead to the development of therapeutic targets for liver fibrosis.
Graphical Abstract
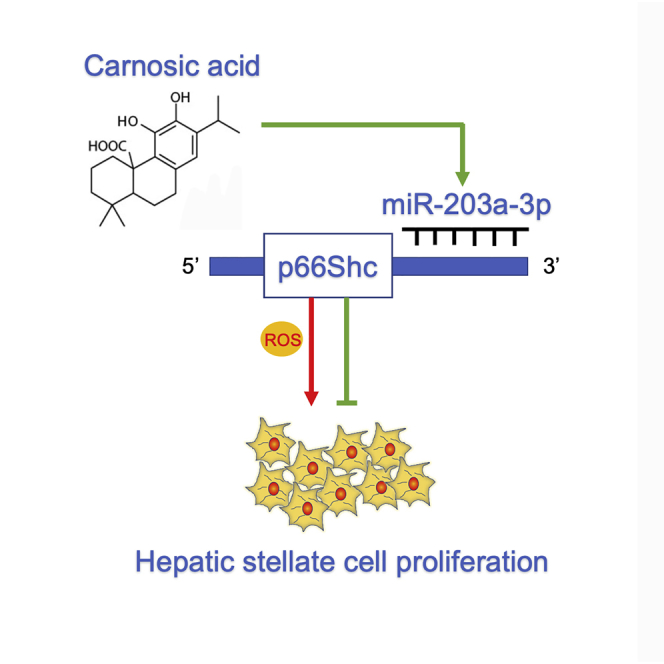
Wang et al. elucidated the effect of p66Shc on HSC proliferation and evaluated miR-203a-3p/p66Shc-mediated reactive oxidative species (ROS) generation during liver fibrosis both in vivo and in vitro. The authors also reported that carnosic acid (CA) protected liver fibrosis through the miR-203a-3p/p66Shc axis.
Introduction
Liver fibrosis is an inevitable stage in the progression of chronic liver disease to cirrhosis.1,2 Liver fibrosis development is characterized by excessive deposition of extracellular matrix (ECM), which is primarily produced by activated hepatic stellate cells (HSCs).3 Accumulating evidence indicates that reactive oxidative species (ROS) play an indispensable role in HSC activation and proliferation.4 Furthermore, oxidative stress markers have been detected in biopsy samples, in serum from experimental liver fibrosis/cirrhosis animal models, and in liver cirrhosis patients.5 Accordingly, the decreasing of ROS production would be an effective means to attenuate the progression of liver fibrosis. However, the source of ROS and the mechanism underlying ROS production in liver fibrosis require further investigation.
Recently, an important pathway for ROS regulation mediated by the adaptor protein p66Shc was discovered. p66Shc, which regulates cellular redox states, metabolism, and lifespan, is a critical mediator of oxidative signal transduction.6,7 p66Shc gene knockout mice exhibited significantly reduced oxidative stress injury in a variety of models, such as high-fat diet, calcium overload, and myocardial ischemia/reperfusion (I/R) models.8, 9, 10 Interestingly, p66Shc acts not only in the cytosol but also as a specific redox enzyme in mitochondria, generating hydrogen peroxide.11 Mitochondrial ROS represent a major source of cellular ROS production, particularly during sustained dysfunction.12 p66Shc induces mitochondrial ROS generation by reducing equivalents of the mitochondrial electron transfer chain and oxidation of cytochrome c.13 Our recent studies have demonstrated that high expression of p66Shc promotes HSC activation in liver fibrosis by inducing mitochondrial ROS generation.14 Nevertheless, the role of p66Shc in HSC proliferation and the mechanism by which p66Shc is modulated have not yet been explored.
Recent studies have shown that the expression of p66Shc can be suppressed by microRNAs (miRNAs, or miRs) in different diseases.15,16 miRNAs are small noncoding RNA molecules that are approximately 21–25 nt in length, which play essential regulatory roles in plants and animals by inhibiting transcript translation or promoting degradation.17, 18, 19 Although the functions of miRNAs have been widely studied in regard to apoptosis, development, and differentiation,20,21 only a few studies have focused on miRNAs in oxidative signaling regulation, especially in the regulation of mitochondrial ROS. The examination of the function of miRNAs in regulating mitochondrial ROS generation might shed new light on the machinery that underlies mitochondrial ROS regulation and might promote effective therapy for liver fibrosis. Based on the above findings regarding p66Shc, we hypothesize that miRNA modulation may decrease mitochondrial ROS production by targeting p66Shc, thereby alleviating liver fibrosis.
Carnosic acid (CA), the major antioxidant compound found in Rosmarinus officinalis, has a variety of biological activities, such as anti-oxidant and anti-apoptotic activities.22, 23, 24 CA can alleviate liver I/R injury and lipopolysaccharide (LPS)-induced liver injury by inhibiting ROS production.25, 26, 27 Our recent studies demonstrated that CA could reduce dimethylnitrosamine (DMN)-induced and bile duct ligation (BDL)-induced liver fibrosis through the SIRT1/SMAD3 and miR-29b-3p/HMGB1 signaling pathways.28,29 However, carbon tetrachloride (CCl4)-induced liver fibrosis is more closely related to ROS production.30 Whether CA can alleviate CCl4-induced liver fibrosis, as well as the underlying mechanism, remains unknown.
In this study, our aims were as follows: (1) to determine the potential role of p66Shc-mediated ROS generation during HSC proliferation in liver fibrosis; (2) to identify miRNAs that potentially bind to p66Shc mRNA through bioinformatic analysis and then screen their expression in liver fibrosis; and (3) to test whether CA can alleviate CCl4-induced liver fibrosis through the identified miRNA/p66Shc axis.
Results
CA Attenuates CCl4-Induced Liver Fibrosis
To investigate whether CA could attenuate CCl4-induced rat liver fibrosis, the protein levels of α-SMA and COL1A2 and the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined, and hematoxylin and eosin (H&E) and Masson staining were performed. α-SMA and COL1A2 protein expression and the serum levels of ALT and AST were significantly increased in response to CCl4 treatment compared with those in the control group (Figures 1A and 1B), whereas CA treatment significantly decreased the expression of α-SMA and COL1A2 and the ALT and AST levels in a dose-dependent manner, which suggested that CA is an effective, protective agent against CCl4-induced liver fibrosis in rats. According to H&E and Masson staining, CCl4-induced hepatic steatosis, necrosis, and collagen deposition in rat liver tissues were attenuated by CA (Figures 1C and 1D). These results demonstrated that CA protects against CCl4-induced liver fibrosis in rats.
Figure 1.

CA Attenuates CCl4-Induced Liver Fibrosis
Rats were intraperitoneally injected with olive oil or CCl4 dissolved in olive oil combined with or without CA treatment. (A) α-SMA and COL1A2 protein expression in livers; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the CCl4 group; ##p < 0.01 versus the CCl4 group. (B) Serum ALT and AST levels; n = 8. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the CCl4 group; ##p < 0.01 versus the CCl4 group. (C) Liver tissue sections stained with H&E (×200); n = 8. (D) Liver tissue sections stained with Masson (×200); n = 8.
p66Shc Plays an Important Role in CA Liver Fibrosis Regulation
To assess the role of p66Shc in liver fibrosis, we first determined the expression of p66Shc in CCl4-induced rat liver fibrosis. As expected (Figure 2A), p66Shc expression was significantly increased under CCl4 treatment compared with control treatment. However, CA treatment abrogated the increase in p66Shc.
Figure 2.

p66Shc Contributes to ECM Production during Liver Fibrosis and Is Modulated by CA
(A) p66Shc protein expression in rat livers; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the CCl4 group; ##p < 0.01 versus the CCl4 group. (B) p66Shc, α-SMA, and COL1A2 protein expression in LX-2 cells. LX-2 cells were treated with CA, PDGF-BB, SHC1 siRNA, or nothing; n = 3. ∗p < 0.05 versus the control group; ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group; ##p < 0.01 versus the PDGF-BB group. (C) Immunofluorescence images (×200) showing p66Shc (red) and α-SMA (green) expression in LX-2 cells. DAPI was used as a counterstain.
HSC proliferation plays an essential role in promoting ECM production and liver fibrosis.3 To assess whether p66Shc is required for the proliferation of HSCs in vitro, LX-2 cells were transfected with p66Shc small interfering RNA (siRNA) and then treated with platelet-derived growth factor-BB (PDGF-BB). As shown in Figure 2B, p66Shc expression was significantly increased with PDGF-BB treatment, but CA treatment abrogated the increase in p66Shc. Additionally, after transfection with p66Shc siRNA, the α-SMA and COL1A2 protein levels were decreased compared with those in the model group. Furthermore, double immunofluorescence staining showed that p66Shc localized with α-SMA-positive cells, and the increase in p66Shc and α-SMA induced by PDGF-BB was reduced by p66Shc siRNA (Figure 2C). These results indicate that p66Shc contributes to ECM production in vitro. Furthermore, to investigate the mechanisms by which p66Shc exerts its effects on fibrogenesis, flow cytometry, Cell Counting Kit 8 (CCK8), and cyclin D1 expression analyses were performed to determine whether p66Shc could promote HSC proliferation.31, 32, 33 As shown in Figures 3A and B, the percentage of cells in G1 phase was increased by transfection with p66Shc siRNA. In addition, p66Shc siRNA attenuated cell proliferation and the expression of cyclin D1 and p21 (Figures 3C and 3D). These results suggest that p66Shc contributes to HSC proliferation in vitro. Additionally, mitochondrial ROS levels were alleviated by transfection with p66Shc siRNA (Figure 3E). Thus, p66Shc, which contributes to HSC proliferation, plays an essential role in regulating liver fibrosis, and CA-mediated protection against liver fibrosis is related to p66Shc regulation.
Figure 3.

p66Shc Contributes to HSC Proliferation during Liver Fibrosis and Is Modulated by CA
(A and B) The percentage of LX-2 cells in the G1 phase in different groups; n = 3. ∗∗p < 0.01 versus the control group; ##p < 0.01 versus the PDGF-BB group. (C) CCK8 assays were used to examine cell proliferation at the indicated time points; n = 6. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group; ##p < 0.01 versus the PDGF-BB group. (D) Cyclin D1 and p21 protein expression in LX-2 cells; n = 3. ∗∗p < 0.01 versus the control group; ##p < 0.01 versus the PDGF-BB group in different groups. (E) Immunofluorescence images (×200) showing mitochondrial ROS (red) production in LX-2 cells. Hoechst was used as a counterstain.
p66Shc Promotes HSC Proliferation via β-Catenin Dephosphorylation
β-catenin activation plays an important role in HSC proliferation.34, 35, 36 Recent research has demonstrated that p66Shc knockdown inhibits Wnt3a-stimulated β-catenin dephosphorylation and accumulation.37 Thus, we next analyzed the potential role of p66Shc-mediated ROS in β-catenin activation. We first examined β-catenin expression in vivo. In response to CCl4, the phosphorylated β-catenin level was decreased compared with that in the control group (Figure 4A). However, CA treatment abrogated the decrease in phosphorylated β-catenin. Consistently, in vitro, p66Shc siRNA or CA increased the expression of phosphorylated β-catenin in LX-2 cells treated with PDGF compared with the model group (Figure 4B). The qRT-PCR results showed that p66Shc siRNA or CA decreased the expression of the β-catenin downstream targets C-MYC (cellular-myelocytomatosis viral oncogene) and cyclin D1, suggesting that p66Shc contributes to HSC proliferation through β-catenin (Figure 4C). In addition, the antioxidants N-acetyl cysteine (NAC) and polyethylene glycol-catalase (CAT) substantially inhibited the β-catenin dephosphorylation induced by p66Shc overexpression (Figure 4D). Furthermore, we obtained similar results in LX-2 cells treated with H2O2 (Figures 4E–4G). Collectively, these results suggest that p66Shc-mediated ROS generation promotes HSC proliferation, which is related to β-catenin activation.
Figure 4.

p66Shc Promotes HSC Proliferation via β-Catenin Dephosphorylation
(A) Phosphorylated β-catenin and β-catenin protein expression in rat livers; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the CCl4 group; ##p < 0.01 versus the CCl4 group. (B) Phosphorylated β-catenin and β-catenin protein expression in LX-2 cells in different groups; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group; ##p < 0.01 versus the PDGF-BB group. (C) Cyclin D1 and C-MYC mRNA levels in LX-2 cells. ∗∗p < 0.01 versus the control group; ##p < 0.01 versus the PDGF-BB group. (D) Phosphorylated β-catenin and β-catenin protein expression in LX-2 cells. LX-2 cells were treated with pcDNA-p66Shc, N-acetyl cysteine (NAC), polyethylene glycol-catalase (CAT), or nothing; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the pcDNA-p66Shc group; ##p < 0.01 versus the pcDNA-p66Shc group. (E–G) p66Shc (E), phosphorylated β-catenin and β-catenin (F), and α-SMA and COL1A2 (G) protein expression in LX-2 cells from different groups. LX-2 cells were treated with CA, H2O2, SHC1 siRNA, or nothing; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the H2O2 group; ##p < 0.01 versus the H2O2 group.
Selection of miRNAs Targeting p66Shc in Liver Fibrosis
To investigate the molecular mechanism underlying the effects of p66Shc upregulation on liver fibrosis, we assessed the regulation of p66Shc by miRNA. We referred to the miRNA array data from Gene Expression Omnibus (GEO): GSE77271 and GSE66278 and a reference38 to determine the downregulated miRNAs (Table 1). Among the 57 downregulated miRNAs, 8 were predicted to bind the 3′ UTR of SHC1. Furthermore, only miR-203a-3p and miR-96-5p were predicted to be conserved among species (Figure 5A). Therefore, we used qRT-PCR to analyze further the expression of the two miRNAs, revealing that the miR-203a-3p level was significantly decreased in the model group, whereas miR-96-5p was not significantly decreased (Figure 5B). In addition, miR-203a-3p is highly conserved across humans, rats, and mice, according to bioinformatics analysis (Figure 6A). Furthermore, the miR-203a-3p level was significantly decreased in the model group both in vivo and in vitro, and CA treatment ameliorated this decrease (Figures 6B and 6C).
Table 1.
miRNAs in the GEO: GSE77271 and GSE66278 Datasets and a Reference Dataset That Are Reportedly Downregulated in a CCl4 Liver Fibrosis Model
| Downregulation of miRNAs | ||||||
|---|---|---|---|---|---|---|
| Decreased miRNAs | miR-434-3p | miR-183 | miR-22 | miR-29b | miR-206 | miR-877 |
| miR-340-3p | miR-705 | miR-29a | miR-174 | miR-341 | miR-29c | |
| miR-202-3p | miR-101a | miR-30c | miR-365 | miR-677 | miR-96 | |
| miR-378a-3p | miR-148a | miR-704 | miR-193 | miR-378b | miR-30b | |
| miR-212-3p | miR-1904 | miR-let-7 | miR-7a | miR-378d | miR-152 | |
| miR-126-5p | miR-27b | miR-30d | miR-361 | miR-26b | miR-98 | |
| miR-542-5p | miR-103 | miR-185 | miR-26a | miR-23b | miR-107 | |
| miR-345-5p | miR-10b | miR-210 | miR-802 | miR-194 | miR-16 | |
| miR-345-3p | miR-203 | miR-192 | miR-30a | miR-429 | miR-200a | |
| miR-126-3p | miR-15a | miR-872 | ||||
| TargetScan | miR-212-3p | miR-365 | miR-203 | miR-96 | miR-429 | miR-7 |
| miR-let-7 | miR-200a | |||||
| Conservation | miR-203a-3p | miR-96-5p | ||||
Based on the human TargetScan database, we predicted that certain miRNAs may bind with p66Shc.
Figure 5.

Selection of miRNAs Targeting p66Shc in Liver Fibrosis
(A) The process of screening for miRNAs involved in liver fibrosis. (B) miR-96-5p and miR-203a-3p expression in rat livers; n = 3. ∗∗p < 0.01 versus the control group.
Figure 6.

CA Downregulates p66Shc through miR-203a-3p
(A) miR-203a-3p has a predicted binding site in p66Shc from humans, mice, and rats. (B) miR-203a-3p expression in rat livers; n = 3. ∗p < 0.05 versus the control group; ∗∗p < 0.01 versus the control group; #p < 0.05 versus the CCl4 group; ##p < 0.01 versus the CCl4 group. (C) miR-203a-3p expression in LX-2 cells; n = 3. ∗p < 0.05 versus the control group; ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group. (D) CA modulates p66Shc via miR-203a-3p. LX-2 cells were transfected with wild-type or mutant SHC1-3′ UTR luciferase constructs and with antago-miR-203a-3p control or antago-miR-203a-3p. After 24 h, the cells were exposed to 20 μM CA for 6 h or left untreated. ∗p < 0.05 versus the antago-miR-203a-3p control group; ∗∗p < 0.01 versus the antago-miR-203a-3p control group; #p < 0.05 versus the antago-miR-203a-3p group. (E–G) miR-203a-3p (E), p66Shc (F), and phosphorylated β-catenin and β-catenin (G) expression in LX-2 cells. LX-2 cells were treated with CA, antago-miR-203a-3p control, antago-miR-203a-3p, or nothing; n = 3. ∗p < 0.05 versus the antago-miR-203a-3p control group; ∗∗p < 0.01 versus the antago-miR-203a-3p control group; #p < 0.05 versus the antago-miR-203a-3p group.
CA Downregulates p66Shc through miR-203a-3p
To investigate further whether miR-203a-3p targets the 3′ UTR of p66Shc, we transfected LX-2 cells with a dual-luciferase construct containing either the wild-type or mutated p66Shc 3′ UTR in the presence or absence of CA. As shown in Figure 6D, antago-miR-203a-3p clearly increased the luciferase activity of the wild-type reporter. Compared with the antago-miR-203a-3p group, the CA pretreatment group exhibited decreased luciferase activity. However, this effect was not observed for the mutated p66Shc 3′ UTR. We next investigated whether the expression of p66Shc is suppressed by miR-203a-3p. As shown in Figures 6E–6G, p66Shc expression was significantly increased after transfection with antago-203a-3p, but CA markedly decreased the expression of p66Shc. Taken together, these results confirm that p66Shc is a direct target of miR-203a-3p and that CA can decrease p66Shc expression through miR-203a-3p.
miR-203a-3p Inhibits HSC Proliferation by Targeting p66Shc
To explore whether miR-203a-3p can regulate ECM production, LX-2 cells were transfected with ago-miR-203a-3p or the control. As shown in Figure 7A, agomir (ago)-miR-203a-3p increased the miRNA level of miR-203a-3p after PDGF-BB treatment, and a similar effect was observed after CA treatment. In addition, the evaluation of the effect of ago-miR-203a-3p was based on the expression of p66Shc, β-catenin, α-SMA, and COL1A2 (Figures 7B–7D). Ago-miR-203a-3p attenuated the p66Shc upregulation and the phosphorylated β-catenin downregulation induced by PDGF-BB. α-SMA and COL1A2, generally recognized as biomarkers of liver fibrosis, were markedly decreased in the ago-miR-203a-3p group treated with PDGF-BB. The above results indicate that ago-miR-203a-3p downregulated p66Shc, α-SMA, and COL1A2 expression in LX-2 cells.
Figure 7.

miR-203a-3p Inhibits HSC Proliferation by Targeting p66Shc
(A–D) miR-203a-3p (A), p66Shc (B), phosphorylated β-catenin and β-catenin (C), and α-SMA and COL1A2 (D) expression in LX-2 cells from different groups. LX-2 cells were treated with CA, PDGF-BB, ango-miR-203a-3p control, ango-miR-203a-3p, or nothing; n = 3. ∗∗p < 0.01 versus the control group; ##p < 0.01 versus the PDGF-BB group. (E–H) CCK8 assay results (E) and the p66Shc and cyclin D1 (F), phosphorylated β-catenin and β-catenin (G), and α-SMA and COL1A2 (H) protein expression levels in LX-2 cells. LX-2 cells were treated with PDGF-BB, ango-miR-203a-3p control, ango-miR-203a-3p, target protector-p66Shc, or nothing; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group; ##p < 0.01 versus the PDGF-BB group; &p < 0.05 versus the ango-miR-203a-3p group; &&p < 0.01 versus the ango-miR-203a-3p group. (I and J) The percentage of LX-2 cells in the G1 phase; n = 3. ∗∗p < 0.01 versus the control group; #p < 0.05 versus the PDGF-BB group; &p < 0.05 versus the ango-miR-203a-3p group.
We then investigated whether p66Shc plays an essential role in the protective effects of miR-203a-3p on HSC proliferation. We employed target protector technology, in which a target protector is able to disrupt the specific interaction of miRNA-mRNA pairs. To this end, we produced a p66Shc target protector and observed that p66Shc expression was increased in the presence of the p66Shc target protector upon PDGF-BB treatment and transfection with ago-miR-203a-3p (Figure 7F). In addition, the target protector of p66Shc resulted in decreased phosphorylated β-catenin protein levels and increased α-SMA and COL1A2 expression (Figures 7G and 7H). Furthermore, as shown in Figures 7E, 7F, 7I, and 7J, the decrease in HSC proliferation resulting from transfection with ago-miR-203a-3p was reversed by the p66Shc target protector. These results indicated that miR-203a-3p exerts its protective effects on HSC proliferation by modulating p66Shc expression.
Silencing p66Shc Attenuates Liver Fibrosis in Mice
The p66Shc expression in mice was silenced with a p66Shc-specific short hairpin RNA (shRNA), followed by CCl4 administration. When challenged with CCl4, mice deficient for p66Shc exhibited reduced histological liver damage and decreased collagen accumulation in the liver (Figures 8A and 8B). Consistently, the serum levels of ALT and AST were significantly decreased in response to p66Shc silencing (Figure 8C). Furthermore, p66Shc silencing increased phosphorylated β-catenin expression and markedly decreased p66Shc, α-SMA, and COL1A2 expression (Figures 8D–8F). These results indicated that silencing p66Shc attenuates CCl4-induced liver fibrosis.
Figure 8.

Silencing p66Shc Attenuates Liver Fibrosis in Mice
Mice were intraperitoneally injected with olive oil or CCl4 dissolved in olive oil combined with or without p66Shc-specific shRNA. (A) Liver tissue sections stained with H&E (×200). (B) Liver tissue sections stained with Masson (×200). (C) Serum ALT and AST levels; n = 8. ∗∗p < 0.01 versus the LV (lentivirus)-control group; ##p < 0.01 versus the CCl4 group. (D-F) p66Shc, phosphorylated β-catenin, β-catenin, α-SMA and COL1A2 protein expression in the liver; n=3. ∗∗p < 0.01 versus the LV-control group; ##p < 0.01 versus the CCl4 group.
miR-203a-3p Ameliorates Liver Fibrosis in Mice
Mice were administered ago-negative control (NC) or miR-203a-3p ago-miR (ago-203) intravenously and subjected to CCl4 treatment. Compared to those in the ago-NC group, animals administered ago-203 exhibited substantially alleviated liver injury and fibrosis that was induced by CCl4 stimulation. The morphological changes were visualized by H&E and Masson staining (Figures 9A and 9B). Consistently, the serum levels of ALT and AST were significantly decreased in response to ago-203 (Figure 9C). Furthermore, ago-203 increased miR-203a-3p and phosphorylated β-catenin expression and markedly decreased p66Shc, α-SMA, and COL1A2 expression (Figures 9D–9G). These findings thus support that miR-203a-3p effectively inhibits liver fibrosis.
Figure 9.

miR-203a-3p Ameliorates Liver Fibrosis in Mice
Mice were intraperitoneally injected with olive oil or CCl4 dissolved in olive oil combined with Ago-203 or Ago-NC. (A) Liver tissue sections stained with H&E (×200); n = 8. (B) Liver tissue sections stained with Masson (×200); n = 8. (C) Serum ALT and AST levels; n = 8. ∗∗p < 0.01 versus the Ago-NC group; ##p < 0.01 versus the CCl4 group. (D) miR-203a-3p expression in the liver; n = 3. ∗∗p < 0.01 versus the Ago-NC group; ##p < 0.01 versus the CCl4 group. (E-G) p66Shc, phosphorylated β-catenin, β-catenin, α-SMA and COL1A2 protein expression in the liver; n=3. ∗∗p < 0.01 versus the LV-control group; ##p < 0.01 versus the CCl4 group.
miR-203a-3p, p66Shc, and β-Catenin Expression Is Correlated with Liver Fibrosis in Patients
To identify the biological relevance of miR-203a-3p, p66Shc, and β-catenin in liver fibrosis in clinical situations, the expression of miR-203a-3p, p66Shc, and β-catenin was compared between healthy controls and fibrosis patients. As shown in Figures 10A–10C, p66Shc expression was increased and was negatively correlated with the expression of miR-203a-3p and phosphorylated β-catenin in patients with liver fibrosis. These results demonstrate that miR-203a-3p downregulation and p66Shc upregulation are involved in human liver fibrosis.
Figure 10.

miR-203a-3p, p66Shc, and β-Catenin Expression Is Correlated with Liver Fibrosis in Patients
(A–C) miR-203a-3p (A), p66Shc (B), and phosphorylated β-catenin and β-catenin (C) expression in healthy human livers and fibrotic human livers; n = 3. ∗p < 0.05 versus the healthy control group; ∗∗p < 0.01 versus the healthy control group.
Discussion
Liver fibrosis is widely recognized as a contributor to clinical morbidity, and there are no effective treatments for this disease. Therefore, it is essential to propose therapeutic interventions for liver fibrosis. In this study, we provide the first evidence of the following findings: (1) p66Shc plays an essential role in regulating HSC proliferation during liver fibrosis; (2) miR-203a-3p inhibits HSC proliferation by targeting p66Shc; and (3) CA has the potential to regulate the miR-203a-3p/p66Shc pathway to alleviate liver fibrosis.
Excessive ROS production is widely recognized as a key mediator of liver disease.39 Accumulating evidence links p66Shc to oxidative stress because this adaptor protein functions as an ROS producer.7 In a previous study, we showed that liver I/R activated p66Shc via the sirt1-dependent pathway, leading to liver oxidative stress.40 In addition, we observed that p66Shc is associated with hepatocyte oxidative stress in chronic alcoholic liver injury.41 These observations underscore the potential contribution of p66Shc to liver disease. In this study, we found that p66Shc expression was significantly upregulated in rats with CCl4-induced liver fibrosis and in human fibrotic livers, and silencing p66Shc markedly attenuated CCl4-induced liver fibrosis. Furthermore, p66Shc knockdown attenuated mitochondrial ROS accumulation in vitro. More importantly, upregulation of α-SMA and COL1A2 induced by H2O2 was prevented by p66Shc knockdown, suggesting that p66Shc-mediated ROS generation is responsible for ECM production. Thus, these data demonstrate that p66Shc-mediated ROS play crucial roles in liver fibrosis.
HSC proliferation is a critical event in liver fibrosis.4 When exposed to profibrogenic factors, HSCs are activated and transdifferentiate into myofibroblasts (MFs) that proliferate. Then, ECM protein synthesis and deposition by MFs are increased, contributing to the progression of liver fibrosis.2 In this study, we used PDGF-BB, the most potent HSC proliferation stimulator,42 to further explore the effects of p66Shc on HSC proliferation. We found that knockdown of p66Shc in vitro significantly decreased the upregulation of α-SMA and COL1A2 induced by PDGF-BB, which indicates that inhibition of p66Shc can attenuate ECM production. In addition, p66Shc knockdown decreased the proliferation of HSCs, as indicated by decreased cell viability and cyclin D1 expression and increased G1-phase cell numbers and p21 expression. Taken together, the above findings show that p66Shc promotes HSC proliferation, which contributes to liver fibrosis.
Recent studies have shown that miRNAs regulate liver fibrosis progression, and aberrant miRNA expression in the liver is related to liver fibrosis disease.43,44 However, only a few miRNAs have been found to be involved in HSC proliferation. To clarify thoroughly the effects of miRNA on HSC proliferation during liver fibrosis, we utilized miRNA array data from GEO: GSE77271 and GSE66278 and from the study by Roderburg et al.38 to identify 57 miRNAs that are downregulated in the liver after CCl4 treatment. Based on the role of p66Shc in HSC proliferation, bioinformatics analysis was used to investigate whether miR-203a-3p targets p66Shc. The binding sequences of miR-203a-3p in the p66Shc 3′ UTR are highly conserved across humans, rats, and mice. Therefore, miR-203a-3p was determined to be the optimal miRNA screen targeting p66Shc in liver fibrosis. Furthermore, luciferase assays confirmed that p66Shc is a target of miR-203a-3p in LX-2 cells. Our results also demonstrated that miR-203a-3p attenuated HSC proliferation in vitro and ameliorated liver fibrosis progression in vivo. Taken together, our findings reveal that miR-203a-3p inhibits HSC proliferation during liver fibrosis, which is associated with the regulation of p66Shc expression.
It has been reported that a single miRNA targets approximately 200 transcripts.45 Therefore, perhaps other miR-203a-3p targets exist that can regulate the progression of liver fibrosis. With the consideration of the results regarding miR-203a-3p targeting p66Shc, it is crucial to investigate whether p66Shc is indispensable for miR-203a-3p activity during liver fibrosis. In this study, we found that a p66Shc target protector abrogated the effect of ago-miR-203a-3p on HSC proliferation in vitro. Thus, the effect of miR-203a-3p on HSC proliferation may be primarily dependent on p66Shc regulation. Furthermore, our study demonstrated that p66Shc-mediated ROS generation contributes to β-catenin activation in LX-2 cells. β-catenin activation contributes to ECM accumulation and HSC proliferation.35, 36, 37 We found that phosphorylated β-catenin was upregulated when miR-203a-3p was activated to inhibit p66Shc-mediated HSC proliferation. These data suggest that miR-203a-3p inhibits HSC proliferation and attenuates liver fibrosis by modulating the p66Shc/β-catenin pathway.
CA, a well-known antiadipogenic and antioxidant agent,29,46 shows protective effects against liver I/R injury, chronic alcoholic liver injury, and nonalcoholic fatty liver disease.40,41,47 Here, for the first time, we provide evidence that CA attenuates PDGF-BB-induced LX-2 cell proliferation and CCl4-induced liver fibrosis in rats. We recently reported that CA effectively inhibits p66Shc expression.40,41,47 Likewise, in our current study, CA treatment protected against liver fibrosis in association with inhibition of p66Shc. Additionally, the inhibition of p66Shc was closely associated with the upregulation of miR-203a-3p induced by CA. Furthermore, we also discovered that 12-O-methylcarnosic acid, a main CA active metabolite in hepatocytes,48 significantly decreased the expression of α-SMA and COL1A2 in vitro. These data show that CA, as well as its main active metabolite, has protective effects against liver fibrosis via the miR-203a-3p/p66Shc axis.
Above all, our study demonstrated that inhibition of p66Shc-mediated oxidative signaling via CA-induced upregulation of miR-203a-3p alleviates liver fibrosis progression, potentially representing a new therapeutic target for liver fibrosis.
Materials and Methods
Animals and Treatments
Male Sprague-Dawley (180 to 220 g) rats were obtained from the Experimental Animal Center of Dalian Medical University (Dalian, China). CA (98% purity) was obtained from Shanghai Winherb Medical Science (Shanghai, China) and dissolved in olive oil. CCl4 (0.5 mL/kg) was also dissolved in olive oil at a proportion of 1:10. The experimental rats were randomly divided into five groups: (1) control, (2) control + CA (40 mg/kg), (3) CCl4, (4) CCl4 + CA (20 mg/kg), and (5) CCl4 + CA (40 mg/kg). The rats were gavaged with CA and olive oil every day, and CCl4 and olive oil were intraperitoneally injected twice a week. After 4 weeks, the animals were euthanized, and blood and liver tissue samples were collected for analysis. These procedures were performed according to the guidelines of the Use Committee of Dalian Medical University and Institutional Animal Care and were approved by the Institutional Ethics Committee of Dalian Medical University.
Male C57BL/6 mice (6–8 weeks old) were obtained from the Animal Center of Dalian Medical University (Dalian, China). CCl4 (2 mL/kg) was intraperitoneally injected twice a week for 4 weeks (eight total injections), and additional control group mice were injected with olive oil. The mice were injected intravenously only once with an shRNA-p66Shc or NC lentivirus (1 × 109 viral particles, 100 μL/mouse) before the first CCl4 injection. In addition, mice received intravenous injections of ago-203 or ago-NC at a dose of 50 μg twice per week in addition to CCl4 for 4 weeks. The lentivirus and ago-miR were purchased from GenePharma (Shanghai, China). Blood and liver tissue samples were collected at 48 h after the last CCl4 injection. All procedures involving animals were performed according to animal-use protocols approved by the Ethics Committee of Dalian Medical University in compliance with the Guide for the Care and Use of Laboratory Animals.
Cell Culture and Treatment
The human LX-2 cell line was purchased from the China Cell Culture Center (Shanghai, China) and cultivated in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C and 5% CO2. DMEM and FBS were both purchased from Life Biotechnologies (Gibco-BRL, USA). PDGF-BB was obtained from Sigma-Aldrich (St. Louis, MO, USA). In vitro, LX-2 cells were treated with 20 μM CA or 10 μM 12-O-methylcarnosic acid (BioBioPha, Yunnan, China) for 6 h and then with 20 ng/mL PDGF-BB for 24 h before being processed for total RNA and protein extraction. For NAC and CAT pretreatment, cells were incubated with 3 mM NAC and 1,000 μg/mL CAT for 1 h before total RNA and protein extraction.
Cell Proliferation Assay
CCK8 (Bimake, China) was used to measure cell proliferation according to the manufacturer’s recommendations.
Transfection with Agomirs, Antagomirs, siRNA, pcDNA, and Target Protector
Agomirs, antagomirs, siRNA targeting SHC1, pcDNA-SHC1, and the corresponding NCs were purchased from GenePharma. The SHC1 miScript target protector and the corresponding NC were purchased from QIAGEN. The manufacturer’s protocols were used to guide the transfection procedures.
Cell-Cycle Analysis
A Cell Cycle and Apoptosis Analysis Kit (Beyotime, China) was used to examine the cell cycle. A FACSCalibur flow cytometer (Becton Dickinson [BD], USA) was used to perform the fluorescence analysis. CellQuest Pro software was used to determine the percentage of cells in different cell-cycle phases.
Serum ALT/AST Activities
Commercial kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) were used to determine serum ALT and AST levels according to the manufacturer’s instructions.
Liver Histological Observation
For H&E and Masson staining, liver tissue samples were embedded in paraffin and then cut into 5 mm-thick sections. The sections were viewed under a light microscope.
qRT-PCR
Total RNA was isolated from liver tissues or LX-2 cells using TRIzol reagent (TaKaRa, China). A Transcript All-in-one SuperMix qPCR Kit (TransGen, Beijing, China) was used to extract miRNA. Mature miRNA was quantified via real-time PCR with a SYBR Real-time PCR Kit (TransGen, Beijing, China) and an Applied Biosystems 7300 system (Applied Biosystems, Foster City, CA, USA). mRNA or miRNA expression was normalized to that of endogenous RNA β-actin or U6 small nuclear 2 (RNU6B).
Western Blotting
Briefly, 10%–15% SDS-PAGE gels were used to separate equal amounts of protein, which was then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). After blocking, primary antibodies specific for p66Shc (Abcam, USA), β-catenin, α-SMA, COL1A2, cyclin D1 (Proteintech, China), p21 (Proteintech, China), phosphorylated (p)-β-catenin (Bioss, China), and β-actin (ZSGB-BIO) were used to immunoblot the membranes. After washing, the appropriate secondary antibodies were incubated with the membranes. Enhanced chemiluminescence-plus reagents (Beyotime Institute of Biotechnology, Hangzhou, China) were used to develop the membranes. The emitted light was captured by a BioSpectrum 410 multispectral imaging system with a Chemi 410 high-resolution (HR) camera, and the results were analyzed using Gel-Pro Analyzer Version 4.0 software (Media Cybernetics, MD, USA).
Luciferase Activity Assay
Plasmids containing the miR-203a-3p-p66Shc 3′ UTR response element (3′ UTR-wild type [WT]) and the corresponding mutant (3′ UTR-mut) were purchased from GenePharma. LX-2 cells were cotransfected with the antagomir or NC, along with the plasmid DNA. When appropriate, the cells were incubated with or without (control) 20 μL of CA for 24 h after transfection. Reporter assays were conducted at 36 h after transfection. Luciferase activity was determined with a Dual-Luciferase Reporter Assay Kit (TransGen, Beijing, China) using a Dual-Light Chemiluminescent Reporter Gene Assay System (Berthold, Germany), and the results were normalized to Renilla luciferase activity.
Immunofluorescence
LX-2 cells were fixed and incubated with p66Shc or an α-SMA antibody and then with a secondary antibody conjugated to Alexa Fluor 594 or 488 (Proteintech, China) or fixed and incubated with MitoSOX Red Mitochondrial Superoxide Indicator (Invitrogen Life Technologies, Carlsbad, CA, USA). Then, 4,6-diamidino-2-phenylindole (DAPI; Beyotime, China) and Hoechst (Beyotime, China) were added, and a Leica DM 4000B microscope was used to examine staining.
Patients
The study was approved by the Institutional Ethics Committee of Dalian Medical University (Dalian, China), and patient samples were collected after informed written consent was obtained. Six normal tissues and six liver fibrosis tissues were obtained from the Second Affiliated Hospital of Dalian Medical University (Dalian, China). Biopsies of livers from healthy donors who were unsuitable for liver transplantation for nonhepatic reasons served as the normal samples. Biopsies of livers from patients with liver fibrosis served as the liver fibrosis samples. The tissue samples were immediately snap frozen for western blot and qRT-PCR analyses.
Statistical Analyses
The results are expressed as the mean ± SD. Statistical analyses were performed using GraphPad Prism software (version 5.0; GraphPad Prism Software, La Jolla, CA, USA). The data were analyzed with a two-tailed unpaired Student’s t test or one-way analysis of variance to determine the statistical significance of differences between the groups. p values <0.05 were considered to indicate statistical significance.
Author Contributions
Z.W., Y.Z., X.T., and J.Y. designed the study. Z.W., Y.Z., H.Z., D.F., F.T., and Y.L. performed the study. Z.W., Y.Z., and Z.C. analyzed the data. Z.W., Y.Z., J.Z., L.L., X.M., X.T., and J.Y. drafted the manuscript. J.Y. and X.T. and all authors collaborated in the collection and interpretation of the data and contributed to the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (nos. 81773799, 81871547, 81473266, and 81500406).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.013.
Contributor Information
Xiaofeng Tian, Email: txfdl@dmu.edu.cn.
Jihong Yao, Email: yaojihong65@hotmail.com.
Supplemental Information
References
- 1.Schuppan D., Kim Y.O. Evolving therapies for liver fibrosis. J. Clin. Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez-Gea V., Friedman S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 3.Wei J., Feng L., Li Z., Xu G., Fan X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. 2013;67:387–392. doi: 10.1016/j.biopha.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Crosas-Molist E., Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015;6:106–111. doi: 10.1016/j.redox.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yadav D., Hertan H.I., Schweitzer P., Norkus E.P., Pitchumoni C.S. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am. J. Gastroenterol. 2002;97:2634–2639. doi: 10.1111/j.1572-0241.2002.06041.x. [DOI] [PubMed] [Google Scholar]
- 6.Migliaccio E., Giorgio M., Mele S., Pelicci G., Reboldi P., Pandolfi P.P., Lanfrancone L., Pelicci P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 7.Luzi L., Confalonieri S., Di Fiore P.P., Pelicci P.G. Evolution of Shc functions from nematode to human. Curr. Opin. Genet. Dev. 2000;10:668–674. doi: 10.1016/s0959-437x(00)00146-5. [DOI] [PubMed] [Google Scholar]
- 8.Bellisario V., Berry A., Capoccia S., Raggi C., Panetta P., Branchi I., Piccaro G., Giorgio M., Pelicci P.G., Cirulli F. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc-/-) mouse. Front. Behav. Neurosci. 2014;8:285. doi: 10.3389/fnbeh.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrussi L., Giommoni N., Pellegrini M., Gamberucci A., Baldari C.T. p66Shc-dependent apoptosis requires Lck and CamKII activity. Apoptosis. 2012;17:174–186. doi: 10.1007/s10495-011-0663-4. [DOI] [PubMed] [Google Scholar]
- 10.Di Lisa F., Giorgio M., Ferdinandy P., Schulz R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 2017;174:1690–1703. doi: 10.1111/bph.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra M., Duraisamy A.J., Bhattacharjee S., Kowluru R.A. Adaptor Protein p66Shc: A Link Between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2019;30:1621–1634. doi: 10.1089/ars.2018.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang J.W., Hong J.M., Lee S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016;60:383–393. doi: 10.1111/jpi.12319. [DOI] [PubMed] [Google Scholar]
- 13.Giorgio M., Migliaccio E., Orsini F., Paolucci D., Moroni M., Contursi C., Pelliccia G., Luzi L., Minucci S., Marcaccio M. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y., Wang Z., Feng D., Zhao H., Lin M., Hu Y., Zhang N., Lv L., Gao Z., Zhai X. p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics. 2019;9:1510–1522. doi: 10.7150/thno.29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu F., Pang L., Cai X., Liu X., Yuan S., Fan X., Jiang B., Zhang X., Dou Y., Gorospe M., Wang W. let-7-repressesed Shc translation delays replicative senescence. Aging Cell. 2014;13:185–192. doi: 10.1111/acel.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J.M., Tao J., Chen D.D., Cai J.J., Irani K., Wang Q., Yuan H., Chen A.F. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2014;34:99–109. doi: 10.1161/ATVBAHA.113.302104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim V.N. MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 18.Neilson J.R., Sharp P.A. Small RNA regulators of gene expression. Cell. 2008;134:899–902. doi: 10.1016/j.cell.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–1786. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 20.Jia Y.J., Liu Z.B., Wang W.G., Sun C.B., Wei P., Yang Y.L., You M.J., Yu B.H., Li X.Q., Zhou X.Y. HDAC6 regulates microRNA-27b that suppresses proliferation, promotes apoptosis and target MET in diffuse large B-cell lymphoma. Leukemia. 2018;32:703–711. doi: 10.1038/leu.2017.299. [DOI] [PubMed] [Google Scholar]
- 21.Wang L.Q., Yu P., Li B., Guo Y.H., Liang Z.R., Zheng L.L., Yang J.H., Xu H., Liu S., Zheng L.S. miR-372 and miR-373 enhance the stemness of colorectal cancer cells by repressing differentiation signaling pathways. Mol. Oncol. 2018;12:1949–1964. doi: 10.1002/1878-0261.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordán M.J., Lax V., Rota M.C., Lorán S., Sotomayor J.A. Relevance of carnosic acid, carnosol, and rosmarinic acid concentrations in the in vitro antioxidant and antimicrobial activities of Rosmarinus officinalis (L.) methanolic extracts. J. Agric. Food Chem. 2012;60:9603–9608. doi: 10.1021/jf302881t. [DOI] [PubMed] [Google Scholar]
- 23.Park M.Y., Mun S.T. Dietary carnosic acid suppresses hepatic steatosis formation via regulation of hepatic fatty acid metabolism in high-fat diet-fed mice. Nutr. Res. Pract. 2013;7:294–301. doi: 10.4162/nrp.2013.7.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petiwala S.M., Berhe S., Li G., Puthenveetil A.G., Rahman O., Nonn L., Johnson J.J. Rosemary (Rosmarinus officinalis) extract modulates CHOP/GADD153 to promote androgen receptor degradation and decreases xenograft tumor growth. PLoS ONE. 2014;9:e89772. doi: 10.1371/journal.pone.0089772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aruoma O.I., Halliwell B., Aeschbach R., Löligers J. Antioxidant and pro-oxidant properties of active rosemary constituents: carnosol and carnosic acid. Xenobiotica. 1992;22:257–268. doi: 10.3109/00498259209046624. [DOI] [PubMed] [Google Scholar]
- 26.Romo Vaquero M., Yáñez-Gascón M.J., García Villalba R., Larrosa M., Fromentin E., Ibarra A., Roller M., Tomás-Barberán F., Espín de Gea J.C., García-Conesa M.T. Inhibition of gastric lipase as a mechanism for body weight and plasma lipids reduction in Zucker rats fed a rosemary extract rich in carnosic acid. PLoS ONE. 2012;7:e39773. doi: 10.1371/journal.pone.0039773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiang Q., Liu Z., Wang Y., Xiao H., Wu W., Xiao C., Liu X. Carnosic acid attenuates lipopolysaccharide-induced liver injury in rats via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2013;53:1–9. doi: 10.1016/j.fct.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y., Shi X., Ding C., Feng D., Li Y., Hu Y., Wang L., Gao D., Tian X., Yao J. Carnosic acid prevents COL1A2 transcription through the reduction of Smad3 acetylation via the AMPKα1/SIRT1 pathway. Toxicol. Appl. Pharmacol. 2018;339:172–180. doi: 10.1016/j.taap.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S., Wang Z., Zhu J., Xu T., Zhao Y., Zhao H., Tang F., Li Z., Zhou J., Gao D. Carnosic Acid Alleviates BDL-Induced Liver Fibrosis through miR-29b-3p-Mediated Inhibition of the High-Mobility Group Box 1/Toll-Like Receptor 4 Signaling Pathway in Rats. Front. Pharmacol. 2018;8:976. doi: 10.3389/fphar.2017.00976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X., Wang X., Lv Y., Xu L., Lin J., Diao Y. Protection effect of kallistatin on carbon tetrachloride-induced liver fibrosis in rats via antioxidative stress. PLoS ONE. 2014;9:e88498. doi: 10.1371/journal.pone.0088498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilhelm A., Aldridge V., Haldar D., Naylor A.J., Weston C.J., Hedegaard D., Garg A., Fear J., Reynolds G.M., Croft A.P. CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF-regulated mechanism. Gut. 2016;65:1175–1185. doi: 10.1136/gutjnl-2014-308325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abramovitch S., Dahan-Bachar L., Sharvit E., Weisman Y., Ben Tov A., Brazowski E., Reif S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut. 2011;60:1728–1737. doi: 10.1136/gut.2010.234666. [DOI] [PubMed] [Google Scholar]
- 33.Nevzorova Y.A., Bangen J.M., Hu W., Haas U., Weiskirchen R., Gassler N., Huss S., Tacke F., Sicinski P., Trautwein C., Liedtke C. Cyclin E1 controls proliferation of hepatic stellate cells and is essential for liver fibrogenesis in mice. Hepatology. 2012;56:1140–1149. doi: 10.1002/hep.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng J.H., She H., Han Y.P., Wang J., Xiong S., Asahina K., Tsukamoto H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G39–G49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 35.Kordes C., Sawitza I., Häussinger D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2008;367:116–123. doi: 10.1016/j.bbrc.2007.12.085. [DOI] [PubMed] [Google Scholar]
- 36.Jiang F., Parsons C.J., Stefanovic B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 2006;45:401–409. doi: 10.1016/j.jhep.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Vikram A., Kim Y.R., Kumar S., Naqvi A., Hoffman T.A., Kumar A., Miller F.J., Jr., Kim C.S., Irani K. Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2014;34:2301–2309. doi: 10.1161/ATVBAHA.114.304338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roderburg C., Urban G.W., Bettermann K., Vucur M., Zimmermann H., Schmidt S., Janssen J., Koppe C., Knolle P., Castoldi M. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 39.Mansouri A., Gattolliat C.H., Asselah T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology. 2018;155:629–647. doi: 10.1053/j.gastro.2018.06.083. [DOI] [PubMed] [Google Scholar]
- 40.Yan H., Jihong Y., Feng Z., Xiaomei X., Xiaohan Z., Guangzhi W., Zhenhai M., Dongyan G., Xiaochi M., Qing F. Sirtuin 1-mediated inhibition of p66shc expression alleviates liver ischemia/reperfusion injury. Crit. Care Med. 2014;42:e373–e381. doi: 10.1097/CCM.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 41.Gao L., Shan W., Zeng W., Hu Y., Wang G., Tian X., Zhang N., Shi X., Zhao Y., Ding C. Carnosic acid alleviates chronic alcoholic liver injury by regulating the SIRT1/ChREBP and SIRT1/p66shc pathways in rats. Mol. Nutr. Food Res. 2016;60:1902–1911. doi: 10.1002/mnfr.201500878. [DOI] [PubMed] [Google Scholar]
- 42.van Dijk F., Olinga P., Poelstra K., Beljaars L. Targeted Therapies in Liver Fibrosis: Combining the Best Parts of Platelet-Derived Growth Factor BB and Interferon Gamma. Front. Med. (Lausanne) 2015;2:72. doi: 10.3389/fmed.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernández-Ramos D., Fernández-Tussy P., Lopitz-Otsoa F., Gutiérrez-de-Juan V., Navasa N., Barbier-Torres L., Zubiete-Franco I., Simón J., Fernández A.F., Arbelaiz A. MiR-873-5p acts as an epigenetic regulator in early stages of liver fibrosis and cirrhosis. Cell Death Dis. 2018;9:958. doi: 10.1038/s41419-018-1014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao L., Xue D., Shen D., Ma W., Zhang J., Wang X., Zhang W., Wu L., Pan K., Yang Y. MicroRNA-942 mediates hepatic stellate cell activation by regulating BAMBI expression in human liver fibrosis. Arch. Toxicol. 2018;92:2935–2946. doi: 10.1007/s00204-018-2278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krek A., Grün D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., Rajewsky N. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 46.Rašković A., Milanović I., Pavlović N., Ćebović T., Vukmirović S., Mikov M. Antioxidant activity of rosemary (Rosmarinus officinalis L.) essential oil and its hepatoprotective potential. BMC Complement. Altern. Med. 2014;14:225. doi: 10.1186/1472-6882-14-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shan W., Gao L., Zeng W., Hu Y., Wang G., Li M., Zhou J., Ma X., Tian X., Yao J. Activation of the SIRT1/p66shc antiapoptosis pathway via carnosic acid-induced inhibition of miR-34a protects rats against nonalcoholic fatty liver disease. Cell Death Dis. 2015;6:e1833. doi: 10.1038/cddis.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romo Vaquero M., García Villalba R., Larrosa M., Yáñez-Gascón M.J., Fromentin E., Flanagan J., Roller M., Tomás-Barberán F.A., Espín J.C., García-Conesa M.T. Bioavailability of the major bioactive diterpenoids in a rosemary extract: metabolic profile in the intestine, liver, plasma, and brain of Zucker rats. Mol. Nutr. Food Res. 2013;57:1834–1846. doi: 10.1002/mnfr.201300052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.