

Abstract
Cerebrovascular pathology is pervasive in Alzheimer’s disease (AD), yet it is unknown whether cerebrovascular dysfunction contributes to the progression or etiology of AD. In human subjects and in animal models of AD, cerebral hypoperfusion and hypometabolism are reported to manifest during the early stages of the disease and persist for its duration. Amyloid-β is known to cause cellular injury in both neurons and endothelial cells by inducing the production of reactive oxygen species and disrupting intracellular Ca2+ homeostasis. We present a mechanism for mitochondrial degeneration caused by the production of mitochondrial superoxide, which is driven by increased mitochondrial Ca2+ uptake. We found that persistent superoxide production injures mitochondria and disrupts electron transport in cerebrovascular endothelial cells. These observations provide a mechanism for the mitochondrial deficits that contribute to cerebrovascular dysfunction in patients with AD.
Keywords: Amyloid-β, calcium, mitochondria, superoxide, vascular endothelial cells
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of senile dementia and is characterized by progressive neurodegeneration and cognitive decline. Parenchymal plaques are a hallmark of AD and are formed by the deposition of amyloid-β (Aβ) polymers [1, 2]. The contribution of Aβ deposition to the manifestation and progression of AD has been the focus of investigation since the discovery of the peptide. Remarkably, 90% of patients with AD demonstrate cerebral amyloid angiopathy, a neuropathological disease characterized by the deposition of Aβ on the walls of cerebral vasculature [3–6].
Cerebrovascular comorbidities are common in AD, as many as 92% of AD patients demonstrate ischemic white matter lesions that resemble arteriosclerosis of small vessels. Microvascular degeneration can have obvious detrimental consequences to cerebral tissue. A number of reports show regional hypoperfusion, hypometabolism, and blood-brain barrier hyperpermeability in subjects with AD [7–9]. To date, a large prevalence of patients with AD are at risk for developing severe vascular conditions that include: hemorrhagic stroke, spontaneous cerebral emboli, cerebral microinfarctions, and microhemorrhages [8, 10–12]. A growing body of evidence has emerged documenting cerebrovascular dysfunction preceding cognitive decline in AD patients [13–15], suggesting that vascular dysfunction may play a causative role in the emergence of AD. However, the mechanism(s) by which Aβ exerts it cytotoxic effects on the cerebrovasculature are not yet known.
It is known that bioenergetic deficits occur in neurons exposed to Aβ and that mitochondrial dysfunction precedes the onset of pathology in AD [16, 17]. These observations suggest that mitochondrial dysfunction during the early stages of AD (non-symptomatic) compromise cerebrovascular function resulting in cerebral Aβ retention, vascular degeneration, and hypoperfusion, which together may initiate the cognitive decline and symptomatic phase of AD. Observations of postmortem brain tissue from AD patients have revealed extensive mitochondrial network disruption, presenting as evidence of excessive mitochondrial fission and fragmented cristae [18–21]. In cultured cortical neurons, Aβ exposure results in deficits of synaptic mitochondria and reduced mitochondrial transport, likely contributing to the synaptic dysfunction in AD [22]. Together, these observational studies indicate a pivotal role for mitochondrial injury in the cytotoxicity of Aβ. Cellular production of reactive oxygen species (ROS) has been shown to result in S-nitrosylation of Drp-1 resulting in aberrant mitochondrial fission, synaptic loss, and neuronal damage [23]. Increased production of Aβ-induced ROS has also been demonstrated in cerebrovascular endothelial cells and causes cell dysfunction, blood-brain barrier disruption, and degeneration [6, 24]. The mechanism by which Aβ induces the production of ROS in endothelial cells is thought to involve the activation of the NADPH oxidase membrane-bound subunit, Nox [25, 26]. Genetic ablation of vascular endothelial cell Nox2 or inhibiting NADPH assembly abrogates ROS and vascular dysfunction induced by Aβ [24, 27, 28].
In the present study, we describe a mechanism for Aβ-induced cerebrovascular endothelial cell injury. We provide experimental evidence from primary cultures of vascular endothelial cells and a bEnd.3 cell line exposed to Aβ, which showed increased production of mitochondrial superoxide via direct Aβ1-42 interaction with mitochondria. Aβ induced a dose-dependent increase in mitochondrial superoxide production that paralleled an increase in mitochondrial oxygen consumption and ATP production, suggesting mitochondrial stress. We found that Aβ exerts these effects by increasing mitochondrial Ca2+ concentration, driving hyper-oxidative metabolism, and increasing ROS production by mitochondria. Furthermore, removal of extracellular Ca2+ abrogates the Aβ1-42-induced increase of mitochondrial oxygen consumption, ATP production, Ca2+ accumulation, superoxide production, and cerebrovascular endothelial cell death. These Aβ-induced effects on mitochondrial function may provide a mechanism for the mitochondrial dysfunction and deficits observed in AD and provide evidence for a therapeutic intervention that targets extracellular calcium.
MATERIALS AND METHODS
Study design
All experiments were performed using either primary cerebrovascular endothelial cells from mice or the bEnd.3 [BEND3] (RRID: CVCL_0170, ATCC® CRL-2299) mouse brain capillary endothelial cell line. The brain endothelial cell line is not listed as a commonly misidentified cell line by the International Cell Line Authentication Committee. Initial authentication of the bEnd.3 cell line was performed by the vendor; no further authentication was performed in the laboratory. Cells were cultured with Dulbecco’s modified Eagles medium (DMEM) (Cat. No. SH30022.01 (2019), HyClone GE) supplemented with 10% fetal bovine serum (Cat. No. S12450 (2019), Atlanta Biosciences) and 1% penicillin streptomycin (Cat. No. SV30010 (2019), HyClone GE) (DMEM+). Cells were plated in either 96-well assay plates (1.5x104 cells/well), 24-well culture plates (3.5x104 cells/well), or 10-cm culture dishes (1.5x105 cells/dish) and incubated at 37°C in a humidified incubator under 5% CO2. Primary cerebrovascular endothelial cells were subcultivated every 4 days (ratio = 1 : 2) and bEnd.3 cells every 3 days (ratio = 1 : 8). The first and second passage of primary cell cultures were retained for cryopreservation. For experimental purposes, primary endothelial cell subcultures were utilized beginning at passage 3 up until a maximum passage number of 6. Experiments that used the bEnd.3 cell line were performed on cells at passage 15–20. This study was not pre-registered or blinded. Stratified randomization was employed in all experiments that utilized cultured cells to prevent any experimental condition to be statically positioned across all experiments.
Animal usage
Three-month-old C57BL6 (National Institutes on Aging) pregnant female mice (n = 4) were used to produce primary cerebrovascular endothelial cell cultures for the purposes of this study. Primary cell cultures were prepared from embryos at gestational day 19 from each of the pregnant mice. Primary cells cultures were prepared from each mouse over four separate occasion, around 12 : 00pm eastern time. Pregnant mice produced an average of 6 embryos that were harvested and pooled together to produce a culture of primary cerebrovascular endothelial cells. Therefore, each mouse produced a single culture of primary cells and each of the four instances of prepared primary cultures was used to replicate the experiments described in the study. Power analysis for ANOVA designs indicated a sample size of 3 pregnant mice (power = 0.999) for an effect size of Δ = 1.25. Mice were housed in accordance to IACUC guidelines of West Virginia University (protocol #: 13-0704). Animals were maintained under a light/dark cycle (12 : 12 h) with food and water available ad libitum. For all procedures performed in this study that involved the use of laboratory animals were carried out in accordance with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publication No. 8023, revised 1978) and in compliance with the ARRIVE guidelines.
Preparation of primary cerebrovascular endothelial cell cultures
Primary cerebrovascular endothelial cells were prepared from embryonic mice. At embryonic day 19, pregnant mice were deeply anesthetized with 4% isoflurane diffused into 70% nitrogen and 30% oxygen mixture. After confirming deep anesthetization via tail pinch, mice were euthanized by cervical dislocation. Mice received a vertical incision to the abdomen. Through the incision, each fetus was extracted, removed from the amniotic sack, and decapitated. The collected fetal heads were immediately immersed in ice-cold HBSS buffer (Cat. No. 14025-092 (2019), Gibco). Each brain was extracted through an incision made along the superior sagittal suture followed by the removal of the cerebellum for discard. The remaining tissue was placed into a 50 mL centrifuge tube containing ice-cold DMEM. The brain tissue from each fetus was consolidated into a single 50 mL centrifuge tube containing 20 mL of ice-cold DMEM then centrifuged at 2500 x g for 3 min. The supernatant was decanted and the tissue pellet was resuspended in 1 mL of ice-cold DMEM. The suspended tissue was then transferred to a Dounce homogenizer that was kept on ice. After homogenization, the tissue was transferred into a 50 mL tube containing 1.5 % HEPES 1M (Cat. No. H0887-100ML (2019), Sigma Aldrich) in 20 mL HBSS. The tissue was briefly vortexed followed by centrifugation at 2000 x g for 10 min at 4°C. Supernatant was decanted and the pellet was resuspended in a solution of 18% (w/v) dextran (Cat. No. 31392-50G (2018), Sigma Aldrich) and 1.5% HEPES 1M in 20 mL HBSS. The tube was mixed by inverting then centrifuged at 5000 x g for 10 min at 4°C. Once centrifuged, the supernatant along with the cholesterol interphase was discarded; the pellet was then resuspended in 20 mL of warm DMEM containing 10% FBS and 1% penicillin streptomycin. Vessels were isolated from the suspension by passing the solution through a 50 μm filter, and rinsed with 3 mL of DMEM. Isolated vessels were removed from the filter by immersion in warm DMEM containing 10% FBS and 1% penicillin streptomycin. Collected vessels were centrifuged in a 50 mL tube at 2000 x g for 3 min, resuspended in 1 mL of DMEM and plated in 10 cm culture dishes. Vessels were allowed to reach 65% confluence before passaging and several passages were performed before experimental use. This procedure yields cerebrovascular endothelial cell cultures primarily of capillary in origin.
Mitochondria isolation from cerebrovascular endothelial cells
Mitochondrial isolation was performed on primary cerebrovascular endothelial cells via a previously reported technique [29]. Briefly, cells were cultured to 85% confluence then harvested with a cell scraper and collected into a 50 mL centrifuge tube. Endothelial cells were pelleted by centrifugation at 2000 x g for 3 min, supernatant was discarded and pellet resuspended in 8 mL. With a Dounce homogenizer, cell membranes were fractured, releasing the mitochondria into the medium. The homogenate was transferred into a 15 mL centrifuge tube then aspirated several times using a 10 mL syringe with a 27-gauge needle. The homogenate was centrifuged at 800 x g to pellet the cell debris. After centrifugation, the supernatant was decanted into a new 15 mL tube then distributed into 2 mL microcentrifuge tubes. Microcentrifuge tubes were centrifuged at 10000 x g for 5 min at 5°C to pellet the mitochondria. Once centrifugation completed, the supernatant was discarded and the mitochondrion pellet was resuspended in a solution of 10 mM Tris-MOPS (Cat. No. M-8899 (2019), Sigma Aldrich) at pH 7.4, 1 mM EGTA-Tris (Cat. No. E8145-10G (2019), Sigma Aldrich), and 200 mM sucrose (Cat. No. S5-12 (2018), Fisher Scientific).
Aβ1-42 preparation
Four separate preparations of Aβ1-42 (Cat. No. 03112 (2019), Thermo Fisher Scientific) were performed over the duration of the study. For each preparation, 1 mg of lyophilized Aβ1-42 monomers was suspended in 167 μL of HPLC grade water (Cat. No. AM9937 (2018), Ambion) and incubated at room temperature (RT) for 5 min. The dissolved Aβ1-42 was then diluted to 230 μM by adding 833 μL of Ca2+-free phosphate buffered saline (PBS) (Cat. No. P4417-100TAB (2019), 0.01M, Sigma Aldrich) and incubated for 48 h at 37°C for polymerization. After polymerization, Aβ1-42 was separated into 50 μL aliquots and stored at −80°C. The final product produced 230 μM Aβ1-42 at mixed polymerization states.
Endothelial cell death assay
To assess Aβ1-42-induced cell death, bEnd.3 cells were seeded in 24-well plates at a density of 2.5x104 and allowed to reach 70–85% confluency. Then, vascular endothelial cells were rinsed with pre-warmed DMEM containing 10% FBS and 1% penicillin streptomycin. Aβ1-42 was prepared in 5-, 9-, and 18-μM solutions diluted in DMEM+then added to the wells containing the vascular endothelial cells. Cerebrovascular endothelial cells were exposed to Aβ1-42 for a duration of 24 h. After exposure, cells were rinsed 3 times with 0.1 M PBS, and then fixed with paraformaldehyde (PFA) 4% (Cat. No. 15714 (2018), Electron Microscopy Sciences) for 15 min at room temperature (RT). After fixation, endothelial cells were washed 2 times for 5 min with 0.1 M PBS containing 0.045% Tween-20 (Cat. No. P1379-500ML (2018), Sigma Aldrich). Residual detergent was removed with an additional 2 washes, each for 5 min. Nuclei were stained with 50 nM 4’, 6-diamidino-2-phenylindole (DAPI) (Cat. No. D-21490 (2017), Molecular Probes) counter stain. After nuclear staining, cells were washed 3 times for 5 min with 0.1 M PBS and imaged on an EVOS2 FL Auto microscope (Invitrogen). Cells were imaged at 10x magnification and fluorescence excitation using a mercury arc lamp and a DAPI filter set. Three images were acquired for each well and saved as tiff-formatted images. The number of nuclei in each of the three images were averaged and the average number of nuclei was multiplied by the area of the well floor divided by the microscope frame size, yielding the total number of cells per well. Cell death by chronic exposure to Aβ1-42 was assessed using the live-dead probe (Cat. No. R37609 (2019), Invitrogen). Cells were plated on 96-well assay plates and allowed 24 h of growth before receiving 9 μM Aβ1-42 in DMEM+for 48, 72, 96, and 122 h. After exposure, dead and live cells were labeled and quantified.
Mitochondrial superoxide assay
Mitochondrial superoxide production in response to Aβ1-42 exposure was quantified using MitoSOX Red (Cat. No. M36008 (2019), Molecular Probes) according to the manufacturer’s guidelines. Cerebrovascular endothelial cells were seeded on 96-well plates at a density of 1.5x104 cells per well. Endothelial cells were allowed to proliferate for 24 h ( 85% confluency). Cells were rinsed twice with pre-warmed DMEM and then exposed to 0-, 5-, 9-, and 18-μM Aβ1-42 for 24 h. After exposure to Aβ1-42, cells were rinsed 3 times with warm DMEM+and endothelial cells were loaded with 5 μM MitoSOX Red diluted in DMEM+for 10 min at 37°C. After incubation, cells were rinsed 3 times with warm DMEM+. MitoSOX fluorescence was measured with a spectrophotometer by endpoint kinetic set to an excitation of 510 nm and an emission of 580 nm. Each well was measured in relative fluorescence units (RFU). The RFU measurement from each well per group (n =12) was normalized to the average RFU of the control (0 μM Aβ1-42) group to produce a fold-change value.
Endothelial cell ATP content assay
Endothelial cell ATP content was quantified using the CellTiter-Glo luminescent assay kit (Cat. No. G7571 (2018), Promega) according to the manufacturer’s guidelines. The bEnd.3 cells were seeded on 96-well plates at a density of 1.5x104 cells per well then allowed to proliferate for 24 h ( 85% confluency). Endothelial cells were rinsed twice with warm DMEM+then exposed to 5-, 9-, and 18-μM Aβ1-42 for 24 h. Cells were allowed to equilibrate to RT for 30 min before the ATP assay was implemented. The luciferase-based solution was measured with a spectrophotometer (BioTek) by endpoint kinetic set to measure luminescence. ATP was estimated in nM per well using a standard curve. To prevent ATP degradation, the standard curve was created immediately prior to running the assays. The standard curve was prepared by serial tenfold dilutions of ATP in DMEM+.
Mitochondrial respiration assay
Mitochondrial respiration was measured using a Seahorse XFe (Seahorse Biosciences) bioanalyzer using the MitoStress Kit (Cat. No. 101706-100 (2018), Seahorse Bioscience) according to the manufacturer’s guidelines. Cultured bEnd.3 cells were seeded on 96-well XF Cell Culture Microplates at a density of 1.5x104 cells per well and allowed to proliferate for 24 h. Endothelial cells were exposed to 5-, 9-, and 18-μM Aβ1-42 for 24 h. Three hours prior to running the procedure, the sensor cartridges were hydrated and the instrument parameters set to run the assay. Endothelial cells were rinsed with XF assay medium and placed in the bioanalyzer for calibration. After calibration, each well was measured via the MitoStress Test protocol that used a base measurement of oxygen consumption (pmol/min) to calculate basal respiration, maximal respiration, ATP production, spare capacity, proton leak, and non-mitochondrial respiration.
Cellular calcium assay
Cytosolic and mitochondrial Ca2+ levels were measured with Fluo-4AM (Cat. No. F14201 (2018), Molecular Probes) or Rhod-2AM (Cat. No. R1245MP (2018), Molecular Probes) according to the manufacturer’s guidelines, respectively. Endothelial cells were seeded on either 96-well assay plates at a density of 1.5x104 or glass-bottom culture dishes at a density of 3.5x104 cells. Cells were exposed to 5-, 9, and 18-μM Aβ1-42 for 24 h. Before conducting the experiment, cells were rinsed twice with DMEM+. To enhance dispersion of the Ca2+ indicators, an equal volume of dimethyl sulfoxide (DMSO) (Cat. No. D2650-100ML (2019), Sigma Aldrich) containing 20% pluronic F-127 (Cat. No. P2443-1KG (2018), Sigma Aldrich) was added to the Fluo-4AM or Rhod-2AM stock solution. The Fluo-4AM or Rhod-2AM solution was diluted to 5 μM in DMEM and added to each well. Cells were loaded for 45 min at 37°C. After loading, endothelial cells were washed 3 times with DMEM+and allowed to incubate for an additional 30 min for complete de-esterification. Rhod-2AM was incubated for an additional 24 h to allow for mitochondrial (non-cytosolic) Ca2+ indication. Fluorescence was measured with a spectrophotometer (BioTek) by area scan routine using an excitation of 494 or 552 and an emission of 506 or 581 to measure Fluo-4AM or Rhod-2AM, respectively. Each well was measured in RFU and normalized to the average of the control group to produce a fold change value. For cells that were plated on glass-bottom culture dishes, dynamic Ca2+ indication was measured via confocal microscopy. Cells were imaged over a period of 94 min at 30-s intervals.
Calcium pathway inhibitors
The IP3 R inhibitor, 2-aminoethoxydiphenylborate (2-APB) (Cat. No. 100065-100MG (2020), Sigma Aldrich) was prepared as a 100 mM stock solution in DMSO; subsequent dilution to 50 μM working concentration was prepared in DMEM+. The MCU antagonist, 4-Quinolinecarboxamide (ER-000444793) (Cat. No. 50-193-1971 (2020), Fisher Scientific) was prepared as a 5 mM stock solution in DMSO; working concentration was prepared in DMEM+at 5 μM. The mNCX inhibitor, 2-[4-[(4-nitrophenyl)methoxy]phenyl]ethyl carbamimidothioate (KB-R7943) (Cat. No. 12-441-0 (2020), Fisher Scientific) stock solutions was prepared in DMSO at a concentration of 5 mM; subsequent dilution to 7 μM working concentration was prepared in DMEM+. All compounds used in the following studies were prepared at working concentration immediately before use.
Statistics
All experiments were performed on either primary cerebrovascular endothelial cells from mice or the bEnd.3 cell line. Experiments involving isolated mitochondria were derived from primary cerebrovascular endothelial cells. Quantitative experiments were repeated a minimum of three times. Qualitative experiments were repeated a minimum of two times. Data are reported as the mean value of the experimental group across all replicates plus or minus the standard deviation (mean ± SD). The criteria for sample exclusion consisted of human error or instrument failure. No data was excluded from the statistical analysis of the study. Statistical comparison was performed on GraphPad Prism version 8.0 (RRID: SCR_002798) by one-way ANOVA (Supplementary Table 1) to assess normality of data and with either Bonferroni’s multiple comparison analysis (to compare each group to all other groups), Dunnett’s post hoc analysis (to compare all groups to control group), or linear trend analysis (linear trend to determine dose-dependency) (Supplementary Table 2). All p-values less than 0.05 were considered significant (*p < 0.05; **p < 0.001; ***p < 0.0001).
RESULTS
Acute and chronic exposure to Aβ is cytotoxic to cerebrovascular endothelial cells
Previous studies have shown that, in neuronal cultures, exposure to Aβ1-42 is cytotoxic, causing cellular death at relatively high concentrations [30–32]. However, these experiments describe an unsustainable cellular environment at late stages of AD, when Aβ1-42 is found at significantly elevated concentrations in the brain compared to the early stages of AD. We constructed an experimental paradigm that describes the early changes to cells during the disease progression, when Aβ1-42 is found at lower levels in the brain. To determine the cytotoxicity of acute exposure of Aβ1-42 to cerebrovascular endothelial cells, we used brightfield microscopy to visualize live bEnd.3 cells exposed to 18 μM Aβ1-42. We observed marked changes in cell morphology that resembled apoptosis, characterized by an initial retraction of cellular processes after 120 min that progressed and resulted in a spherical cellular morphology after only 240 min of exposure to 18 μM Aβ1-42 (Fig. 1A). To provide a quantitative measure of the bEnd.3 cell death we observed via live cell imaging, we counted the number of nuclei of cells exposed to 5-, 9-, and 18-μM Aβ1-42 for 24 h and compared this to the number of nuclei of bEnd.3 cells exposed to vehicle control. We found no significant difference in the number of nuclei between bEnd.3 cells that were exposed to Aβ1-42 and cells that were not (Fig. 1B). Although, we found some evidence of cytotoxicity for acute exposure to Aβ1-42, we next evaluated whether prolonged exposure of cerebrovascular endothelial cells to Aβ1-42 could induce cytotoxicity. We assessed this by exposing bEnd.3 cells to 9 μM Aβ1-42 over a period of 48–144 h. In order to account for differences in the number of cells between the latency-groups, we normalized our cell death measurement by counting the total number of nuclei of both dead and live cells to produce a dead-to-live ratio (FD−/F−L), and this value was converted to fold-change from the latency-matched non-exposed control group. We found that cell death caused by 9 μM Aβ1-42 begins after 96 h of exposure. Specifically, cell death was increased by 1.56 ± 0.15 (p < 0.0001)-fold and 1.52 ± 0.21 (p < 0.0001)-fold by 96 h and 120 h of exposure, respectively (Fig. 1C).
Fig. 1.
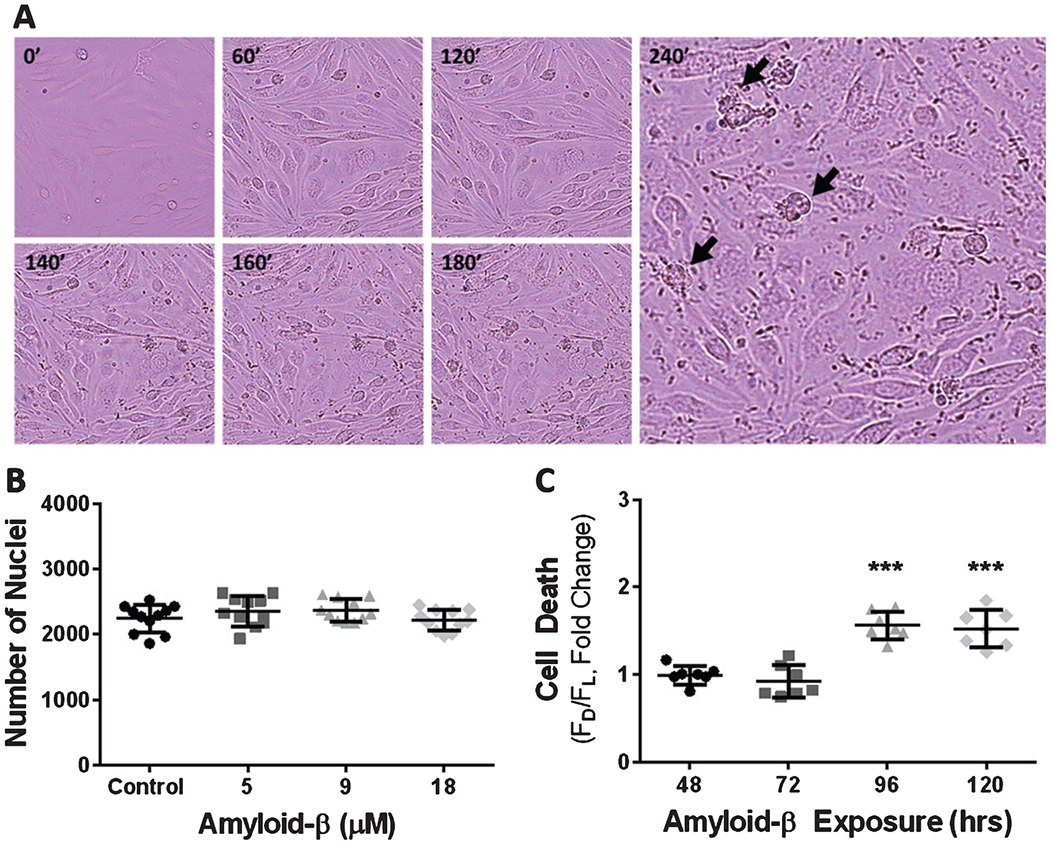
Cerebrovascular endothelial cell exposure to Aβ results in cell death. A) Brightfield micrographs depicting bEnd.3 cell death (black arrows) following exposure to 18 μM Aβ1-42 for 0, 60, 120, 140, 160, 180, and 240 min. B) Scatter plot (mean ± SD) demonstrating the average number of nuclei per well after exposure to vehicle, 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42 (n = 11 wells per group). C) Scatter plot (mean ± SD) depicting endothelial cell death after chronic exposure to 9 μM Aβ1-42 for 48, 72, 96, 120, and 144 h (n = 7 wells per group). One-way ANOVA with Dunnett’s post analysis was used to determine the level of significance between the exposure groups (***p < 0.0001).
Cerebrovascular endothelial cell exposure to Aβ results in mitochondrial dysregulation characterized by accelerated mitochondrial oxidative phosphorylation
Most cells respond to stress by increasing oxidative phosphorylation. For example, cellular damage originating from starvation [33, 34], inflammation [35], membrane rupture [36], thermal limitations [37, 38], and ionic imbalance [39, 40] can result in increased oxidative phosphorylation, which may provide additional metabolic resources that are needed to repair damage incurred. We first assessed mitochondrial oxidative phosphorylation after exposure to Aβ1-42. We used an extracellular flux bioanalyzer XFe 96 to assess changes in mitochondrial oxygen consumption following Aβ1-42 exposure. Oxygen consumption in cultured bEnd.3 cell was measured for 14 min to obtain a baseline metabolic oxygen consumption rate. After 14 min, bEnd.3 cells were exposed to either 9 μM Aβ1-42 or DMEM (control) for a total of 589 min, to determine if Aβ1-42 modulates mitochondrial oxygen consumption. Our analysis revealed a progressive increase in mitochondrial oxygen consumption that began 397 min post-Aβ1-42 exposure, which increased 1.81 ± 1.77-fold (p < 0.001) above the control group after 461 min and continued to increase up to 3.76 ± 1.41-fold (p < 0.001) above the control group over the duration of the experiment (Fig. 2A). These observations were in contrast to bEnd.3 cells maintained in DMEM without Aβ1-42, which demonstrated an essentially unchanged oxygen consumption rate for the duration of the experiment (Fig. 2A, black).
Fig. 2.
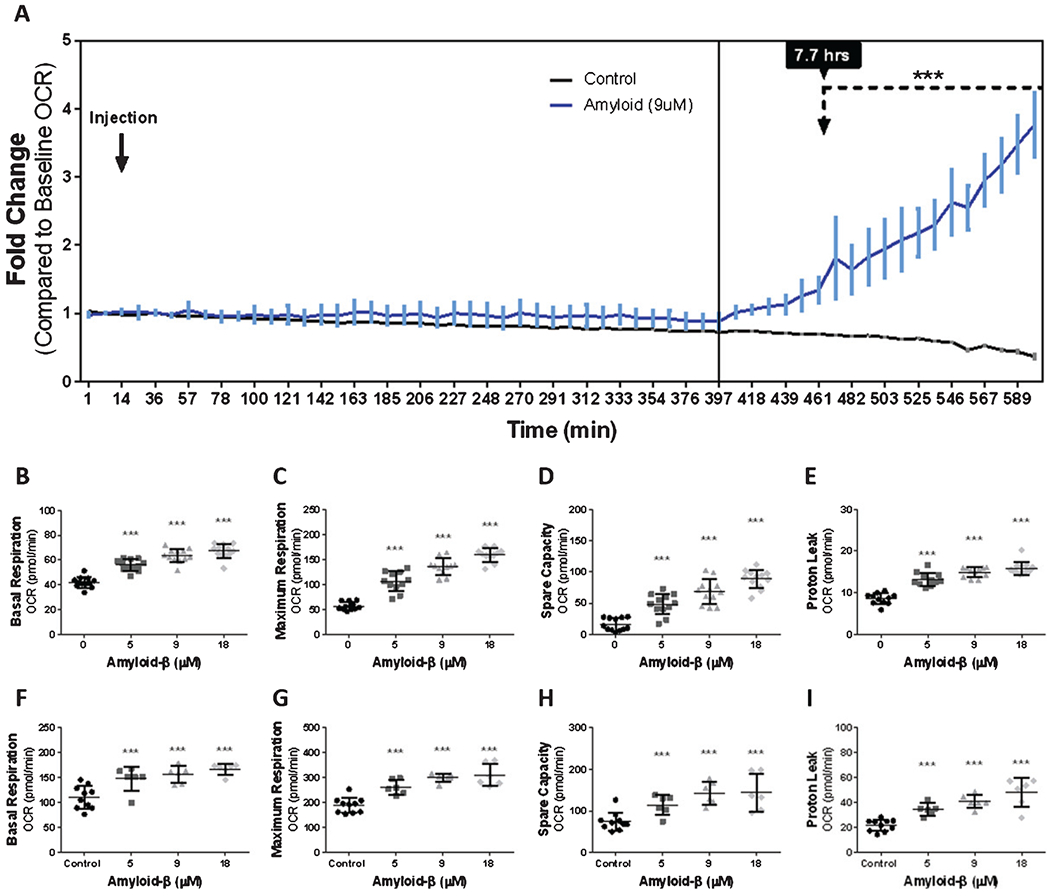
Exposure to Aβ for 24-h results in a dose-dependent increase in mitochondrial oxygen consumption. A) Line graph (mean ± SD) depicting the fold-change of oxygen consumption to baseline as a function of time from bEnd.3 cells exposed to vehicle and 9 μM Aβ1-42. Scatter plot(s) (mean ± SD) demonstrating oxygen consumption (pmol/min) by (B, F) basal respiration, (C, G) maximum respiration, (D, H) spare capacity, and (E, I) proton leak from (B–E) bEnd.3 cells and (F–H) primary cerebrovascular endothelial cells after 24 h exposure to vehicle, 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42. One-way ANOVA with Dunnett’s post analysis and linear trend analysis was used to determine the level of significance between the treatment groups (*p < 0.05; **p < 0.001; ***p < 0.0001), (B–E, n = 12 wells per group; F–I, control = 10, all other conditions = 6 wells per group).
Since our results demonstrated a clear increase in mitochondrial oxygen consumption in response to Aβ1-42 exposure, we characterized specific indices of mitochondrial oxygen consumption. To determine this, we measured cellular basal respiration, maximum respiration, spare capacity, and proton leak after bEnd.3 cells were exposed for 24 h to 5-, 9-, and 18-μM Aβ1-42. We found that Aβ1-42 dose-dependently increased mitochondrial basal respiration, maximal respiration, spare capacity, and proton leak after 24 h of exposure. For all evaluated parameters of oxidative phosphorylation, the maximum effect was measured for bEnd.3 cells exposed to 18 μM Aβ1-42. Specifically, exposure to Aβ1-42 resulted in an increased oxygen consumption to 67 ± 5.6 (p < 0.0001), 159 ± 13.5 (p < 0.0001), 88 ± 14.9 (p < 0.0001), and 15 ± 1.6 (p < 0.0001) pmol/min of oxygen used for basal respiration, maximum respiration, spare capacity, and proton leak, respectively (Fig. 2B–E).
We then repeated this experiment on primary cerebrovascular endothelial cells to determine if Aβ1-42 exposure results in similar mitochondrial bioenergetic hyperactivity. We observed a similar dose-dependent upregulation of mitochondrial oxygen consumption. The maximum effect was measured from cells exposed to 18 μM Aβ1-42, which caused oxygen consumption to increase to 167 ± 11.2 (p < 0.0001), 311 ± 45.3 (p < 0.0001), 144 ± 44.9 (p < 0.001), and 48 ± 11.5 (p < 0.0001) pmol/min of oxygen used for basal respiration, maximum respiration, spare capacity, and proton leak, respectively (Fig. 2F–I). Thus, both primary cerebrovascular endothelial cells and bEnd.3 cells responded to Aβ1-42 by increasing cellular respiration in a dose-dependent manner.
Aβ exposure results in an increased mitochondrial Ca2+ concentration
It is firmly established that mitochondrial activity is modulated by Ca2+. Since mitochondrial oxygen consumption was increased in both primary cerebrovascular endothelial cells and in bEnd.3 cells that were exposed to Aβ1-42, we hypothesized that mitochondrial Ca2+ dyshomeostasis participated in the Aβ1-42-induced mitochondrial dysregulation. We next measured mitochondrial matrix Ca2+ concentration in bEnd.3 cells after exposure to 5-, 9-, and 18-μM of Aβ1-42 for 24 h and compared this to the Ca2+ concentration in cells that were not exposed to Aβ1-42. We found a dose-dependent increase of mitochondrial matrix Ca2+ concentrations after 24 h exposure to Aβ1-42. Compared to vehicle-treated controls (100%), our data demonstrated that Aβ1-42 increased mitochondrial Ca2+ levels to 119 ± 30.1% (p < 0.001) at 9 μM Aβ1-42 and 153 ± 24.7% (p < 0.0001) at 18 μM Aβ1-42 (Fig. 3A).
Fig. 3.
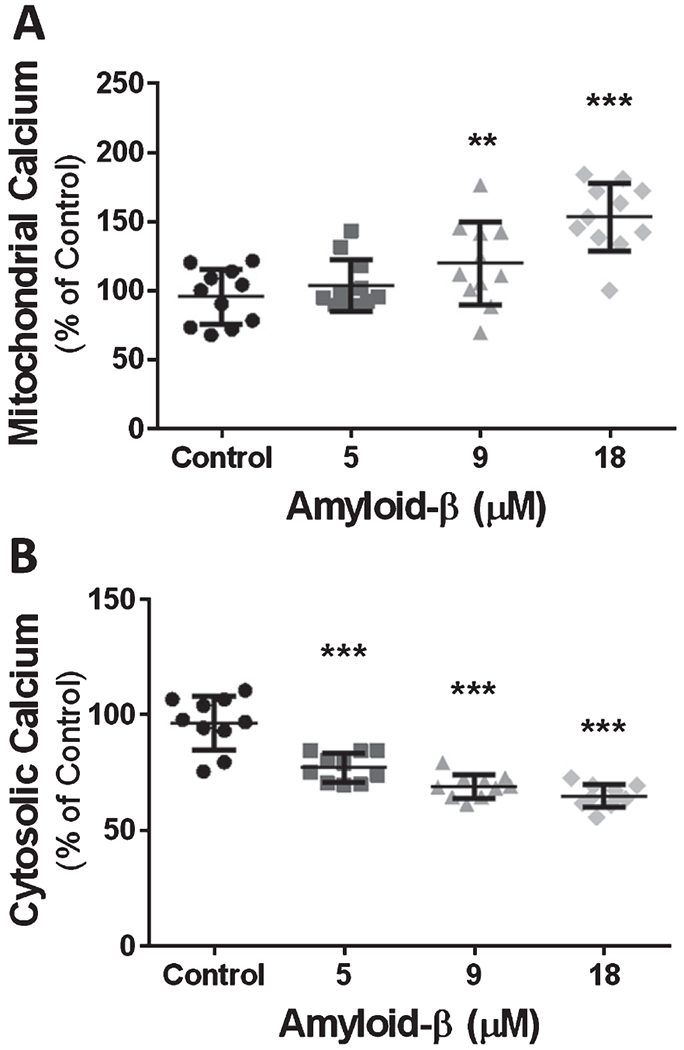
Exposure to Aβ for 24 h causes a dose-dependent increase in mitochondrial Ca2+ content and a dose-dependent decrease of cytosolic Ca2+. A) Scatter plot (mean ± SD) depicting mitochondrial Ca2+ content as percentage of vehicle treated cells, after 24 h exposure to 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42 (n = 11 wells per group). B) Scatter plot (mean ± SD) demonstrating cytosolic Ca2+ content as percentage of vehicle treated cells after 24 h exposure to 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42 (n = 10 wells per group). One-way ANOVA with Dunnett’s post analysis and linear trend analysis was used to determine the level of significance between the treatment groups (**p = 0.001; ***p = 0.0001).
Because mitochondrial uptake of Ca2+ is a critical process required to clear cytosolic Ca2+, we assessed the concentration of cytosolic Ca2+ after exposure to Aβ1-42. Cytosolic Ca2+ was measured viaFluo-4AM fluorescence intensity after exposure to 5-, 9-, and 18-μM Aβ1-42 for 24 h. We found a dose-dependent decrease in cytosolic Ca2+ concentration following Aβ1-42 exposure. In bEnd.3 cells exposed to 18 μM Aβ1-42, cytosolic Ca2+ declined to nearly 64 ± 4.9% (p < 0.0001) of the Ca2+ measured in non-exposed cells (Fig. 3B).
Cellular ATP content increases dose-dependently following exposure to Aβ1-42
Cellular ATP production is stringently coupled to the cell’s ATP requirements. Following the observation, that Aβ1-42 exposure leads to mitochondrial Ca2+ accumulation and because intra-mitochondrial Ca2+ can modulate ATP production, we questioned whether changes to ATP production result from exposure to Aβ1-42. We evaluated mitochondrial ATP production by calculating the oxygen consumption used to produce ATP by summing the oxygen consumed by proton leak and mitochondrial respiration then subtracting this value from the oxygen consumed by the cell’s basal respiration. This produced an estimate for oxygen in pmol/min that was used for ATP production. Our analysis indicated that cells exposed to Aβ1-42 dose-dependently increased ATP production after 24 h of exposure. bEnd.3 cells exposed to 18 μM Aβ1-42 consumed as much as 51 ± 4.6 (p < 0.0001) pmol/min of oxygen for ATP production compared to only 33 ± 3.0 pmol/min of oxygen by non-exposed cells (Fig. 4A).
Fig. 4.
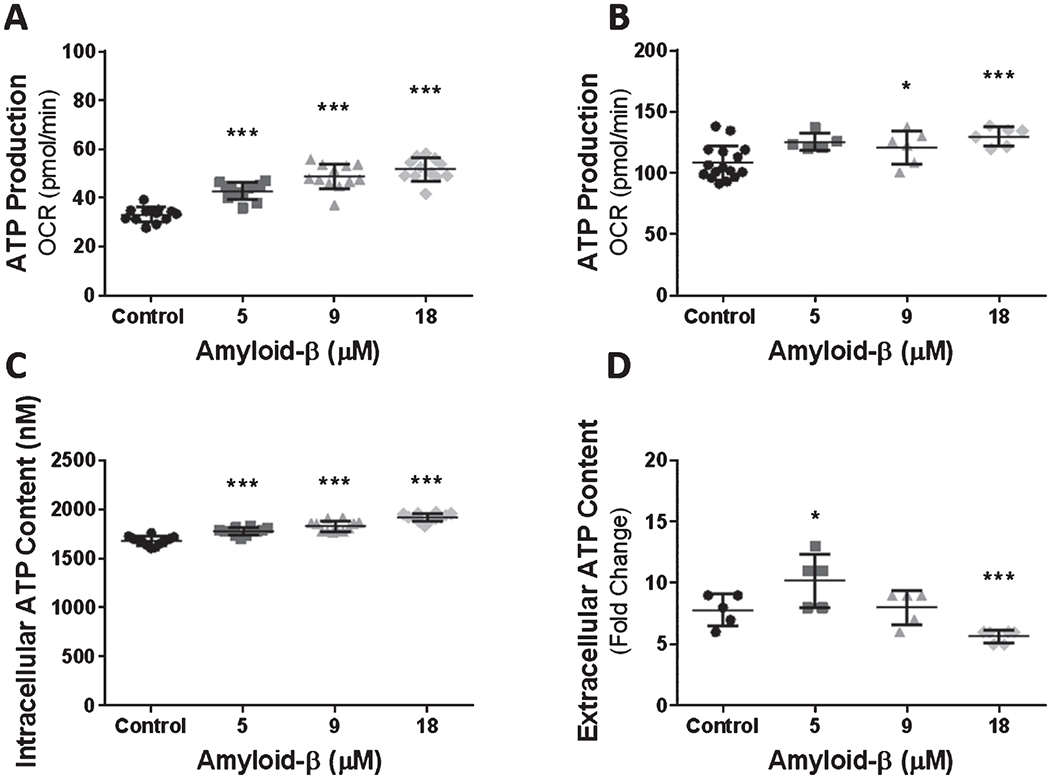
Exposure to Aβ causes a dose-dependent increase in cellular ATP production and cytosolic ATP content. Scatter plot(s) (mean ± SD) depicting mitochondrial oxygen consumption (pmol/min) for ATP production in (A) bEnd.3 (n = 12 wells per group) and (B) primary cerebrovascular endothelial cells after 24 h exposure to vehicle control, 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42 (control = 15, else = 6 wells per group). Scatter plot(s) (mean ± SD) demonstrating the (C) intracellular (n = 12 wells per group) and (D) extracellular (n = 5 wells per group) ATP content (nM) in bEnd.3 cells after exposure to vehicle control, 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42. Statistical analysis was performed by One-way ANOVA with Dunnett’s post analysis and linear trend analysis to determine the level of significance (*p = 0.01; **p = 0.001; ***p = 0.0001).
We next repeated the experiment on primary cerebrovascular endothelial cells to determine if Aβ1-42 mediates similar alterations to oxygen consumption for ATP production. Primary cerebrovascular endothelial cells demonstrated a dose-dependent increase of oxygen consumed for ATP production. Specifically, cells exposed to 9 μM and 18 μM Aβ1-42 consumed 121 ± 13.8 (p < 0.05) and 130 ± 7.6 (p < 0.0001) pmol/min of oxygen, respectively (Fig. 4B). These data indicate that primary cerebrovascular endothelial cells respond to Aβ1-42 exposure similarly to bEnd.3 cells.
An increase in mitochondrial oxidative phosphorylation is often indicative of a cellular ATP deficit. This may result from cellular injury, increased cellular activity, or as a response to cell signaling. Because we observed an increase in mitochondrial oxygen consumption for ATP production, we next measured cellular ATP content in bEnd.3 cells to determine if the changes in oxygen consumption reflect ATP availability. We found that 24 h of exposure to Aβ1-42 resulted in a dose-dependent increase in intracellular ATP content. Exposure to 18 μM Aβ1-42 resulted in an increase in the intracellular ATP concentration to 1922 ± 41.4 (p < 0.0001) nM compared to 1683 ± 46.2 nM in cells that were not exposed to Aβ1-42 (Fig. 4C). Interestingly, the dose-dependent increase of intracellular ATP content occurred alongside a dose-dependent decrease in extracellular ATP. We found that the medium from cell that were not exposed to Aβ1-42 contained 7.8 ± 1.3 nM of ATP which decreased as low as 5.6 ± 0.5 (p < 0.0001) nM of ATP in the medium from cell exposed to 18 μM Aβ1-42 (Fig. 4D). These data suggest that dysregulated mitochondria metabolism is characterized by aberrant ATP production that surpasses ATP utilization.
Mitochondrial superoxide production is driven by Aβ-induced acceleration of mitochondrial oxygen consumption
Mitochondrial production of superoxide is the primary contributor to the oxidative damage during aging and degenerative diseases [41]. We next assessed whether the Aβ1-42-induced increase in mitochondrial oxygen consumption in both primary cerebrovascular endothelial cells and bEnd.3 cells results in elevated mitochondrial superoxide production. Mitochondrial superoxide was probed with a fluorescence indicator after bEnd.3 cells were exposed to Aβ1-42 for 24 h. The fluorescence intensity of the superoxide indicator was converted into fold-change relative to vehicle treated cells. Cultured bEnd.3 cells demonstrated a remarkable dose-dependent increase in mitochondrial superoxide production following 24 h exposure to Aβ1-42. Mitochondrial superoxide production increased 9.2 ± 3.9 (p < 0.0001)-fold and 12.1 ± 3.8 (p < 0.0001)-fold above vehicle treated cells after 24 h exposure to 9 μM and 18 μM Aβ1-42, respectively (Fig. 5D).
Fig. 5.
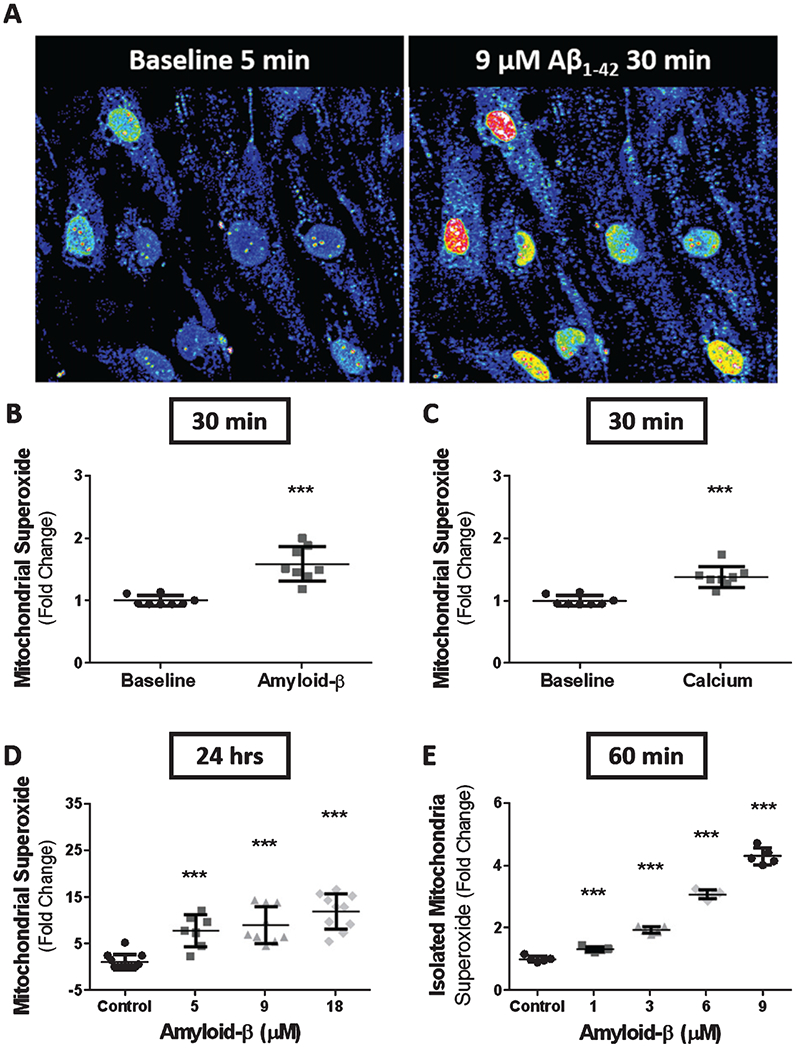
Exposure to Aβ or Ca2+ results in an increase in mitochondrial superoxide production. A) Time-lapse confocal micrographs depicting mitochondrial superoxide production in bEnd.3 cells at baseline (left) and after 30 min of exposure to 9 μM Aβ1-42 (right). Scatter plot(s) (mean ± SD) demonstrating superoxide production from time-lapse images as fold-change from baseline for bEnd.3 cells exposed to (B) 9 μM Aβ1-42 (n = 8 cells) and (C) 5mMCaCl2 (n = 8 cells) for 30 min. D) Scatter plot (mean ± SD) demonstrating mitochondrial superoxide production as fold-change in bEnd.3 cells after 24 h of exposure to vehicle, 5 μM Aβ1-42, 9 μM Aβ1-42, and 18 μM Aβ1-42 (n = 10 wells per group). E) Scatter plot (mean ± SD) depicting superoxide production as fold-change by isolated mitochondria from primary cerebrovascular endothelial cells after 1 h exposure to 0, 1, 3, 6, and 9 μM Aβ1-42 (n = 5 wells per group). Statistical analysis was performed by one-way ANOVA with Dunnett’s post analysis and linear trend analysis to determine the level of significance (***p = 0.0001).
We next assessed the effects of increased extracellular Ca2+ on mitochondrial superoxide production and compared this with superoxide production following Aβ1-42 exposure. To determine this, bEnd.3 cells were probed with a fluorescent superoxide indicator then imaged via confocal microscope after 5 min to obtain a baseline and measurement (Fig. 5A). After baseline measurement, cells were exposed to either 5 mM of Ca2+ or 9 μM of Aβ1-42 and incubated for 30 min before obtaining an outcome measurement. Our time series image analysis revealed a1.6 ± 0.2 (p < 0.0001)-fold increase above baseline in mitochondrial superoxide production after 30 min exposure to 9 μM Aβ1-42 (Fig. 5B) Similarly, cells that were exposed to an additional 5 mM of Ca2+ demonstrated a 1.4 ± 0.1 (p < 0.0001)-fold increase above baseline in superoxide production after 30 min of exposure (Fig. 5C). These data suggest that increased Ca2+ concentration can drive superoxide production in bEnd.3 cells at a magnitude similar to cells exposed to Aβ1-42 for the same amount of time.
We were unsure if direct interaction between Aβ1-42 and mitochondria produce the mitochondrial changes we observed. To answer this question, we isolated the mitochondria form primary cerebrovascular endothelial cells and measured superoxide production following Aβ1-42 exposure for 1 h. Isolated mitochondria exposed to Aβ1-42 for 1 h resulted in a dose-dependent increase in mitochondrial superoxide production. Exposure to 1-, 3-, 6-, and 9-μM Aβ1-42 demonstrated a 1.35 ± 0.07 (p < 0.001), 1.95 ± 0.10 (p < 0.0001), 3.10 ± 0.13 (p < 0.0001), and 4.29 ± 0.27 (p < 0.0001)-fold increase in superoxide production relative to control, respectively (Fig. 5E).
Aβ mediates mitochondrial dysregulation by increasing mitochondrial matrix Ca2+ that accelerates mitochondrial oxidative phosphorylation and superoxide production
Mitochondrial influx of Ca2+ has been shown to accelerate oxidative phosphorylation and ATP production by increasing the activity of mitochondrial dehydrogenase enzymes. Therefore, we hypothesized that the increased oxygen consumption and production of superoxide by cerebrovascular endothelial cell mitochondria following exposure to Aβ1-42 is mediated by the Aβ1-42-induced Ca2+ accumulation in mitochondria. To determine if the increased Ca2+ in the mitochondrial matrix following the exposure of bEnd.3 cells to Aβ1-42 mediates the hyperoxidative state of mitochondria, we reduced the Ca2+ available to mitochondria by chelation. Extracellular Ca2+ was chelated with 1 mM ethylenediaminetetraacetic acid (EDTA) supplemented in cell culture medium with or without exposure to Aβ1-42. We first assessed whether chelation of extracellular Ca2+ results in reduced A-β1-42-induced mitochondrial Ca2+ accumulation. Cultured bEnd.3 cells were plated in 96-well assay plates at a density of 1.5x104 cells per well and grown for 24 h. Cells were then exposed to either vehicle or 9 μM Aβ1-42 with or without 1 mM EDTA for 24 h and then assessed for changes in mitochondrial Ca2+ levels. Our results indicated that 1 mM EDTA effectively abolished the Aβ1-42-induced accumulation of Ca2+ in the mitochondrial matrix (Fig. 6A). Specifically, cells exposed to 9 μM Aβ1-42 without EDTA (9 μM Aβ1-42/0 mM EDTA) increased mitochondrial Ca2+ to 1.8 ± 0.4-fold (versus control, p < 0.0001) above cells exposed to vehicle (control). Whereas, cells exposed to 9 μM Aβ1-42 with 1 mM EDTA (9 μM Aβ1-42/1 mM EDTA) completely abolished the Aβ1-42-induced accumulation of mitochondrial Ca2+ to 0.9 ± 0.4 (mean ± SD)-fold (versus Aβ only, p < 0.0001) compared to control cells (p = 0.65) (Fig. 6A). These data indicate that extracellular Ca2+ chelation with EDTA is effective at reducing mitochondrial Ca2+.
Fig. 6.
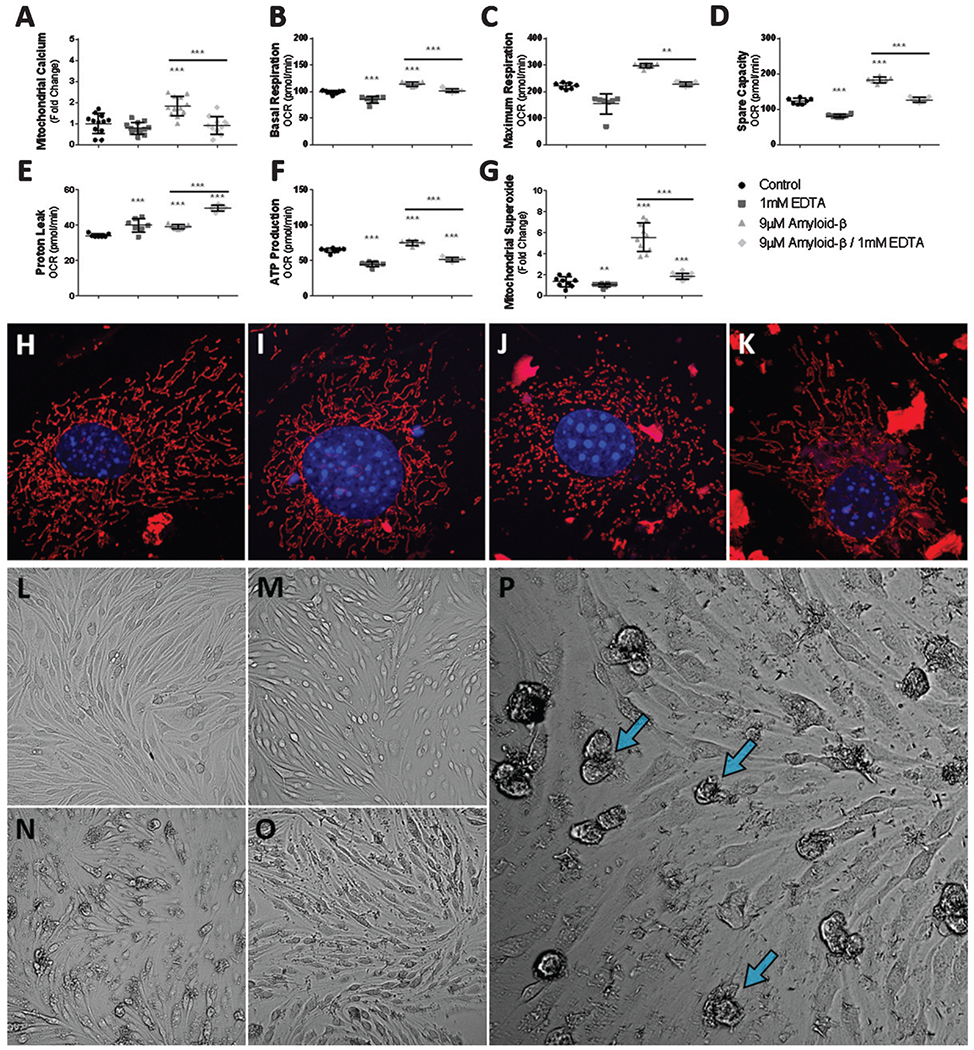
Ca2+ chelation reduces the Aβ induced hyper-oxidative state of mitochondria and superoxide production via reduced mitochondrial Ca2+ uptake. A) Scatter plot (mean ± SD) demonstrating mitochondrial Ca2+ accumulation as fold-change in bEnd.3 cells after 24 h exposure to vehicle control, 1 mM EDTA, 9 μM Aβ1-42, and 9 μM Aβ1-42 with 1 mM EDTA (n = 12 per group). Scatter plot (mean ± SD) demonstrating mitochondrial oxygen consumption (pmol/min) by (B) basal respiration, (C) maximum respiration, (D) spare capacity, (E) proton leak, and (F) ATP production from bEnd.3 cells after 24 h exposure to vehicle control, 1 mM EDTA, 9 μM Aβ1-42, and 9 μM Aβ1-42 with 1 mM EDTA (n = 7 wells per group). G) Scatter plot (mean ± SD) demonstrating mitochondrial superoxide production as fold-change in bEnd.3 cells after 24 h exposure to vehicle control, 1 mM EDTA, 9 μM Aβ1-42, and 9 μM Aβ1-42 with 1 mM EDTA (n = 10 wells per group). Confocal micrographs at 63x magnification depicting mitochondrial fragmentation (red) and nuclei (blue) in bEnd.3 cell exposed to (H) vehicle, (I) 1 mM EDTA, (J) 9 μM Aβ1-42, and (K) 9 μM Aβ1-42 with 1 mM EDTA. Note mitochondria morphology depicted in panel J, demonstrating numerous fragmented and punctated mitochondria while panel(s) H-I, K depict(s) complex and elongated mitochondria. Brightfield micrographs depicting bEnd.3 cell death (blue arrows) following exposure to 9 μM Aβ1-42 after 24 h exposure to (L) vehicle control, (M) 1 mM EDTA, (N, P) 9 μM Aβ1-42, and (O) 9 μM Aβ1-42 with 1 mM EDTA. One-way ANOVA with Dunnett’s post analysis was used to determine significance compared to control while a student’s 2-tailed t-test was used to compare the mean(s) of the 9 μM Aβ1-42 group and the 9 μM Aβ1-42 with 1 mM EDTA experimental group (*p < 0.05; **p < 0.001; ***p < 0.0001).
We next assessed the effectiveness of reducing mitochondrial Ca2+ accumulation with EDTA at ameliorating elevated mitochondrial oxygen consumption after exposure to Aβ1-42. To determine this, we measured the oxygen consumption rate of bEnd.3 cells exposed to either 0 or 9 μM Aβ1-42 with or without 1 mM EDTA. As before, cells exposed to 9 μM Aβ1-42 without EDTA for 24 h demonstrated increased oxygen consumption by basal respiration (114 ± 4.0 pmol/min, p < 0.0001), maximum respiration (296 ± 8.4 pmol/min, p < 0.0001), and spare capacity (182 ± 8.0 pmol/min, p < 0.0001). However, treatment with 1 mM EDTA completely ameliorated the Aβ1-42–induced increase in oxygen consumption. In cells exposed to 9 μM Aβ1-42 with 1 mM EDTA oxygen consumption returned to near vehicle control values for basal respiration (101 ± 3.3 pmol/min, p < 0.02), maximum respiration (227 ± 8.8 pmol/min, p < 0.0001), and spare capacity (126 ± 7.2 pmol/min, p < 0.0001) (Fig. 6B–D). Interestingly, proton leak was the only oxidative phosphorylation parameter that increased further with EDTA treatment. Cells exposed to Aβ1-42 alone consumed 39 ± 1.2 (p < 0.0001) pmol/min of oxygen by proton leak while cells exposed to both Aβ1-42 and EDTA consumed 49 ± 1.6 (p < 0.0001) pmol/min of oxygen (Fig. 6E). In support of our hypothesis, reduction of mitochondrial Ca2+ ameliorates the increased oxygen consumption mediated by mitochondrial Ca2+ accumulation following exposure to Aβ1-42. These data suggest that Aβ1-42 exposure results in mitochondrial dysfunction by increasing mitochondrial Ca2+ levels.
Mitochondrial Ca2+ content is stimulatory to ATP synthesis by the ATP synthase respiratory complex. Therefore, we predicted that by blocking Ca2+ from entering mitochondria, the Aβ1-42-induced upregulation of ATP production should be prevented. To determine the interplay between elevated mitochondrial oxygen consumption and matrix Ca2+ accumulation on the production of ATP, we exposed bEnd.3 cells to 9 μM Aβ1-42 with or without 1 mM EDTA for 24 h and then estimated the production of ATP by mitochondria. Consistently, bEnd.3 cells exposed to 9 μM Aβ1-42 responded by increasing oxygen consumption for ATP production to 75 ± 3.5 (p < 0.0001) pmol/min. Oxygen consumption in cells that were exposed to 9 μM Aβ1-42 - with 1 mM EDTA restored ATP production to near vehicle control values, 51 ± 2.6 (p < 0.0001) (Fig. 6F).
Since mitochondrial Ca2+ accelerates electron transport by increasing the activity of mitochondrial dehydrogenase enzymes, we hypothesized that blocking Aβ1-42-induced mitochondrial Ca2+ accumulation would mitigate the increased production of superoxide by mitochondria following exposure to Aβ1-42. To determine if mitochondrial Ca2+ accumulation mediates the Aβ1-42 -induced increase in mitochondrial superoxide production, we exposed bEnd.3 cells to Aβ1-42 in the presence or absence of EDTA then measured mitochondrial superoxide levels. Superoxide production by bEnd.3 cells exposed to 9 μM Aβ1-42/0mM EDTA increased superoxide production 5.5 ± 1.3 (versus control, p < 0.0001)-fold above control cells, whereas bEnd.3 cells exposed to 9 μM Aβ1-42/1 mM EDTA reduced superoxide production to 1.8 ± 0.2 (versus Aβ only, p < 0.0001)-fold of control cells (Fig. 6G).
Mitochondrial fission-fusion dynamics are a transient process that serves a physiologically important function, i.e., regulation of mitochondrial quality control and metabolic homeostasis. Mitochondrial fragmentation often precedes mitophagy of damaged mitochondria that occurs during states of metabolic and oxidative stress [42]. Mitochondrial fragmentation is well-documented in AD animal models and in human AD patients [43, 44]. It is clear that oxidative stress can cause transient changes to the morphology of mitochondria while prolonged oxidation can result mitochondrial network fragmentation leading to apoptosis [45–47]. Since our results demonstrated that chelation of extracellular Ca2+ was exceptionally effective at ameliorating the Aβ1-42-induced dysregulation of mitochondrial activity, we asked whether the chelation of extracellular Ca2+ can rescue mitochondria from aberrant fragmentation. We answered this by labeling mitochondria in bEnd.3 cells with MitoTracker CMXRos Red and the nuclei with DAPI counterstain following 24 h exposure to 9 μM Aβ1-42- with or without 1 mM EDTA. Indeed, we observed fragmentation of the mitochondrial network in bEnd.3 cells exposed to Aβ1-42 alone and found that by chelation of extracellular Ca2+ completely prevented the morphological changes to the mitochondrial network (Fig. 6H–K). These observations demonstrate the role of Aβ1-42-induced mitochondrial Ca2+ accumulation in the dysregulation of mitochondrial bioenergetics, resulting in mitochondrial fragmentation following oxidative damage.
Our data describes a mechanism in which mitochondrial Ca2+ accumulation following cell exposure to Aβ1-42 mediates cell damage by dysregulating mitochondrial function. We next asked whether preventing the Aβ1-42-induced mitochondrial dysfunction could rescue cerebrovascular endothelial cells from Aβ1-42 mediated death. To assess the cytoprotective effects of blocking Aβ1-42-induced mitochondrial Ca2+ accumulation against Aβ1-42 mediated cell death, we exposed bEnd.3 cells to 9 μM Aβ1-42 with or without 1 mM EDTA for 24 h and then imaged cellular changes in morphology. Exposure to either vehicle control (Fig. 6L) or 1 mM EDTA alone (Fig. 6M) caused no apoptotic cells upon visualization. While, bEnd.3 cells that were exposed to 9 μM Aβ1-42/0mM EDTA (Fig. 6N, P) demonstrated numerous apoptotic cells. However, bEnd.3 cells that were exposed to 9 μM Aβ1-42 with 1 mM EDTA (Fig. 6O) completely prevented the apoptotic cells observed in cultures exposed to Aβ1-42 alone. We observed a reduced number of apoptotic cells in cultures exposed to Aβ1-42 with 1 mM EDTA. We observed no apoptotic cells in cultures that were not exposed to Aβ1-42 with and without 1 mM EDTA.
Aβ associated calcium entry into the mitochondria occurs via multiple intracellular calcium regulation pathways
Since we found that the Aβ1-42-induced elevated mitochondrial calcium could be attenuated by extracellular calcium chelation, we next assessed intracellular calcium regulation pathways involved in calcium uptake by mitochondria. To assess the contribution of intracellular calcium regulation pathways, we used pharmacological inhibitors, which prevent calcium entry into the mitochondria from the endoplasmic reticulum, mitochondrial permeability transition pore (mPTP), and mitochondrial membrane associated calcium exchangers. Mitochondrial calcium was measured with Rhod-2AM as previously stated, then exposed to 9 μM Aβ1-42 or DMEM+control containing 50 μM 2-APB, 5 μM ER-000444793, 7 μM KB-R7943, or vehicle (DMSO) for 24 h. We found that bEnd.3 cells exposed to 9 μM Aβ1-42 -vehicle resulted in a 3.2 ± 0.46 (p < 0.0003)-fold increase mitochondrial calcium above DMEM control 1 ± 0.35-fold. Whereas, treatment with the IP3 receptor antagonist 2-APB (50 μM) attenuated the elevated mitochondrial matrix calcium to 1.2 ± 0.27 (p < 0.00007)-fold. Inhibition of the mPTP with ER-000444973 (5 μM) also reduced the Aβ1-42-induced mitochondrial calcium accumulation to 1.8 ± 0.24 (p < 0.002)-fold. Treatment with an inhibitor of mitochondrial reverse mode Na+/Ca+ exchanger and calcium uniporter, KB-R7943 (7 μM) similarly reduced calcium in the mitochondrial matrix upon exposure to Aβ1-42 to 1.4 ± 0.15 (p < 0.0002) (Fig. 7).
Fig. 7.
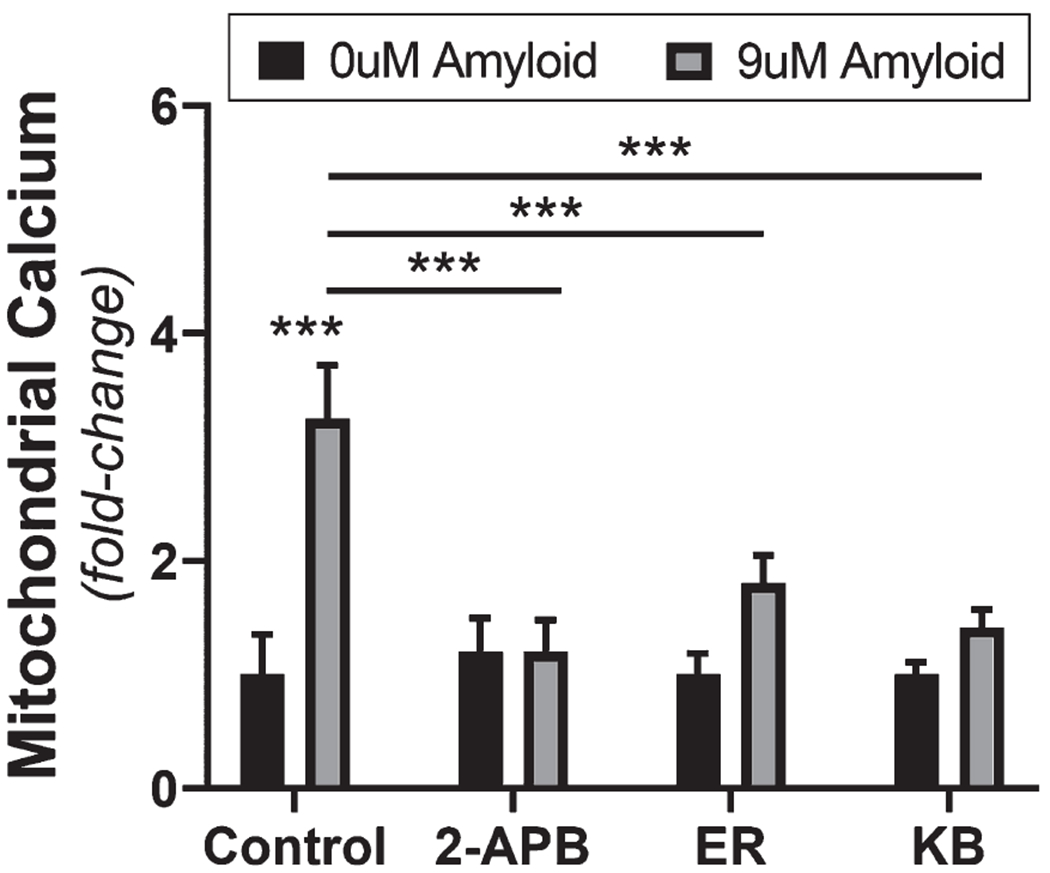
Antagonism of regulatory intracellular calcium pathways attenuates the Aβ-induced mitochondrial calcium influx. Grouped bar graph (mean ± SD) demonstrating mitochondrial Ca2+ accumulation as fold-change in bEnd.3 cells after 24 h exposure to vehicle control, 9 μM Aβ1-42, and 9 μM Aβ1-42 with either 50 μM 2-APB, 5 μM ER-000444793, or 7 μM KB-R7943 (n = 12 per group). Student’s 2-tailed t-test was used to compare the mean(s) of each experimental group (***p < 0.0001).
DISCUSSION
Although Aβ is heavily studied in AD research, its precise role in the disease pathogenesis remains unknown. In AD brains, Aβ plaques are found clustered around regions with damaged and degenerating mitochondria [48, 49]. In culture, Aβ causes ROS production and cell death [6, 23]. The mechanism that drives the Aβ-induced production of ROS is thought to involve the Nox subunit of NADPH oxidase that catalyzes the S-nitrosylation of cellular components [25, 26]. S-nitrosylation of mitochondrial Drp-1 is shown to cause mitochondrial fission and may contribute to the mitochondrial fragmentation found in AD both in humans and in animal models [23]. In addition, Aβ has been shown to cause intracellular Ca2+ dyshomeostasis in culture [50]. However, the interplay between Aβ-induced ROS production, mitochondrial effects, and intracellular Ca2+ levels has not been elucidated previously.
We demonstrate in brain vascular endothelial cells that exposure to Aβ causes a rapid change in cellular bioenergetics, characterized by accelerated mitochondrial oxygen consumption and increased superoxide production. We find that these changes were concurrent with the accumulation of calcium in mitochondria and could be prevented by blocking calcium from entering mitochondria by chelation.
In the present study, we investigate the early changes to endothelial cells following exposure to Aβ that contribute to the cellular death observed after prolong exposure to the peptide. We noticed a rapid change to mitochondrial oxygen consumption after 7 h of exposure to Aβ, and in both bEnd.3 cells and in primary cerebrovascular endothelial cells, Aβ exposure increased basal respiration, maximum respiration, spare capacity, and proton leak by 24 h. Mitochondrial basal respiration is strongly influenced by the turnover of ATP and partially by the oxidation of substrates and by the leakage of protons from the intermembrane space [51, 52]. Therefore, the rate of basal respiration reflects the cellular demand for ATP. In the present study, elevated basal respiration may indicate an increased endothelial demand for ATP in response to the early cellular injury following exposure to Aβ. The spare respiratory capacity of a cell is defined by the ability to utilize substrate and electron transport to meet cellular energy demand before reaching its biological limit [53, 54]. We observed a dose-dependent increase in spare respiratory capacity for both bEnd.3 cells and primary cerebrovascular endothelial cells. Because we observed both an elevated basal respiration and spare respiratory capacity in cells exposed to Aβ, we interpret this to suggest that both an enhanced mitochondrial respiratory competency and an elevated respiratory rate participate in the mechanism of Aβ-induced cellular injury.
It is firmly established that mitochondrial activity is modulated by Ca2+. The influx of Ca2+ to the mitochondrial matrix can exert both stimulatory and inhibitory effects on mitochondrial function. Mitochondrial influx of Ca2+ results in upregulated oxidative phosphorylation by activating mitochondrial dehydrogenase enzymes, glycerophosphate dehydrogenase, isocitrate dehydrogenase, pyruvate dehydrogenase, and oxoglutarate dehydrogenase [55–57]. Activation of these enzymes by Ca2+ increases mitochondrial oxidative phosphorylation and ATP production [58]. We found that Aβ causes increased mitochondrial matrix calcium while decreasing cytosolic calcium in bEnd.3 cells. The stimulatory activity of calcium to resident dehydrogenase enzymes in the mitochondrial matrix may account for the elevated respiration we observed in both bEnd.3 cells and in primary cerebrovascular endothelial cells. In addition, increased concentrations of mitochondrial matrix Ca2+ have been shown to cause the formation of the permeability transition pore (PTP) on the mitochondrial membrane, allowing the release of apoptotic signaling molecules which facilitate mitochondrial-mediated apoptosis [59–61]. Thus, the cytotoxicity we observed following prolong exposure to Aβ may be driven by the PTP following Aβ-induced accumulation of mitochondrial matrix calcium.
Exposure to Aβ in both bEnd.3 cells and in primary endothelial cells resulted in increased oxygen consumption for ATP production and intracellular ATP content. This upregulated ATP synthesis may be an effect of the increased calcium levels in the mitochondrial matrix and its stimulatory role with mitochondrial dehydrogenase enzymes [55–57]. The elevated activity of mitochondrial dehydrogenase enzyme enhances the production of NADH. Catabolism of NADH to NAD+increases the proton-motive force by serving as a proton donor substrate, increasing the throughput of hydrogen ions pumped into the mitochondrial intermembrane space at complex I. The increased hydrogen ions pumped across the mitochondrial inner membrane drives the accelerated ATP synthesis by mechanically powering the coupling of ADP to inorganic phosphate by ATP synthase.
Mitochondrial superoxide production is sensitive to the proton-motive force [41]. High rates of electron transport can cause its reversal, delivering electrons to complex I; these electrons are coupled to molecular oxygen and produce superoxide radicals [62]. Indeed, exposure to Aβ resulted in an increased production of mitochondrial superoxide in intact cells and in isolated mitochondria. These observations indicate that the aberrant elevated mitochondrial respiration following Aβ exposure is inefficient, resulting in increased production of ROS where it can potentially damage the cell and subsequently lead to cell death.
Since mitochondrial calcium is implicated in many of the cellular changes following amyloid-β exposure, we hypothesized that blocking the Aβ-induced mitochondrial calcium accumulation would mitigate the elevated mitochondrial respiration, superoxide production, and cell death following exposure to Aβ. Chelation of extracellular calcium with EDTA was effective at preventing the Aβ-induced accumulation of calcium in the mitochondrial matrix. Thus, we evaluated the role of mitochondrial calcium accumulation as central to the mechanism underlying Aβ mediated endothelial cell injury. We found that by preventing calcium accumulation in the mitochondria, the elevated respiration following Aβ could be mitigated. In addition, blocking the Aβ associated calcium in the mitochondrial matrix, the elevated ATP production and superoxide generation could be halted. These data suggest that calcium influx to mitochondria is a central component to the mechanism mediated by Aβ during the early phase of cellular injury in cerebrovascular endothelial cells.
We further investigated the route by which calcium influx through the plasma membrane enters mitochondria. Specifically, we assessed the contribution of calcium to the mitochondria through the endoplasmic reticulum IP3 R, reverse mode mNCX, and the PTP. Each pathway was evaluated by exposing cells to Aβ-1-42 in the presence of specific antagonists then measuring mitochondrial calcium content.
Our data describe a major role for extracellular calcium in the mechanism mediating the cytotoxicity of Aβ1-42. We found that chelation of extracellular calcium prevented its accumulation in mitochondria (Fig. 6) and the increased oxidative phosphorylation and superoxide production by mitochondria. Recent studies have demonstrated that exposure of the cell membrane to Aβ results in increased permeability of the bilayer, caused by the insertion of Aβ into the membrane, forming ion conductive channels [63, 64]. In principal, these channels would allow the influx of calcium into the cell. Furthermore, we used agents that antagonize mPTP formation, mNCX, and IP3 R to evaluate the contribution of intracellular calcium regulatory pathways in the accumulation of calcium in mitochondria. In our experiments, inhibition of any of these three calcium pathways resulted in attenuated calcium influx into mitochondria. Therefore, these data suggest that exposure to Aβ1-42 causes the cell membrane to become conductive to calcium, the resulting influx of calcium is shunted into mitochondria via multiple calcium regulatory pathways that cause the hyperoxidative state of mitochondria observed in endothelial cell mitochondria.
It is well known that mitochondria possess a sodium/calcium exchanger (mNCX) that functions in the efflux of calcium from mitochondria [65, 66]. It is possible for mNCX to function in reverse mode during conditions of metabolic and calcium dysregulation [67, 68], transporting calcium into the mitochondria. To assess this possibility, we used KB-R7943, an inhibitor of reverse mode mNCX at concentrations previously used in experiments [69], and observed a profound reduction in Aβ1-42-induced mitochondrial calcium influx (Fig. 7).
Mitochondrial calcium uptake is a critical cellular function that maintains low cytosolic calcium concentrations. Calcium influx into the mitochondrial matrix also participates in a regulatory role facilitating mitochondrial dehydrogenase enzyme activity. However, excessive accumulation of calcium by mitochondria may lead to mitochondrial damage via the induction of the mPTP. Calcium-dependent induction of the mPTP is a major mechanism of calcium-induced damage to mitochondria.
Calcium uptake into mitochondria primarily occurs via diffusion across the outer mitochondrial membrane through the voltage-dependent anion channel (VDAC). Translocation of hydrogen by the respiratory chain forms the mitochondrial membrane potential that serves as the driving force to transport calcium across the inner mitochondrial membrane, down its electrochemical gradient. Calcium influx into the mitochondrial matrix is mediated by the mitochondrial calcium uniporter (MCU) at the inner mitochondrial membrane. The MCU channel conducts calcium ions from the outer membrane space into the mitochondrial matrix. Antagonism of MCU substantially reduced Aβ1-42-induced mitochondrial calcium influx (Fig. 7).
The efflux of calcium from the endoplasmic reticulum via IP3 R produce localized microdomains of elevated calcium concentration that are crucial for calcium uptake by the mitochondria [70]. The contact sites that hold mitochondria stationary and within the calcium-rich microdomain occurs at regions termed mitochondrial associated membranes (MAMs). MAMs are sites involved in the exchange of biomolecules, such as lipids, ROS, and calcium between the endoplasmic reticulum and mitochondria. At MAM sites, calcium is conducted into the mitochondrial intermembrane space via a large protein complex involving VDAC. To determine whether the influx of calcium into mitochondria following Aβ1-42 exposure involves the IP3 R pathway, we used 2-APB, an IP3 R antagonist. Inhibition of IP3 R eliminated Aβ1-42-induced mitochondrial calcium influx (Fig. 7).
Together, these data describe a role for calcium in the cytotoxicity exerted by Aβ1-42, where the cell membrane becomes conductive to extracellular calcium, therefrom the influx of calcium is shunted into the mitochondria via multiple calcium regulatory pathways, causing the hyperoxidative state of mitochondria observed following exposure to Aβ1-42.
Consistent with the literature, we observed morphological changes to the mitochondrial network, characterized by punctated mitochondria via excessive fission [23, 43, 71–73]. Mitochondrial fission and fusion is a dynamic process modulated by the energy demand of the cell and participates in the quality control of the mitochondrial network. Damage to mitochondria, such as from ROS can cause mitochondrial fragmentation, similar to our observation in vascular endothelial cells [74–77]. Thus, we interpret these data to suggest that the fragmentation of mitochondria in bEnd.3 cells is mediated by the elevated mitochondrial superoxide production following Aβ exposure. Therefore, because we found that the Aβ associated elevation of mitochondrial superoxide production could be mitigated by preventing mitochondrial matrix calcium. As a result, we expect that the fragmentation of mitochondria could be abrogated. Indeed, we found that by reducing mitochondrial calcium via chelation halted the aberrant mitochondrial fission following Aβ exposure, lending evidence to a mechanism involving aberrant mitochondrial fission driven by the oxidative damage to mitochondria in cerebrovascular endothelial cells exposed to Aβ. Lastly, when we prevented the Aβ-induced accumulation of mitochondrial matrix calcium, the apoptotic morphology of endothelial cells was completely prevented.
Overall, our study provides evidence of early events of cellular injury that involves the elevation of mitochondrial respiration, calcium accumulation in mitochondria, and the production of superoxide. We found that calcium influx into the mitochondria is a central component to the mechanism mediating cell injury. Furthermore, our data indicates that blocking mitochondrial calcium can completely halt the pathological mechanism mediating cell death. In addition, these findings provide a mechanism for the mitochondrial dysfunction and deficits observed in AD and provide evidence for a therapeutic strategy that targets mitochondrial matrix calcium.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH grants P20 GM109098, P01 AG027956, U54 GM104942, T32 AG052375, K01 NS081014, and K01 MH117343. Imaging experiments and image analyses were performed in the West Virginia University Microscope Imaging Facility, which has been supported by the Mary Babb Randolph Cancer Center and NIH grants P20 RR016440, P30 RR032138/GM103488 and P20 RR016477.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0964r2).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-190964.
REFERENCES
- [1].Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- [2].Castellani RJ, Rolston RK, Smith MA (2010) Alzheimer disease. Dis Mon 56, 484–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Janson CG (2015) AD and CAA: Independent risk factors for dementia. Sci Transl Med 7, 318ec214. [Google Scholar]
- [4].DeSimone CV, Graff-Radford J, El-Harasis MA, Rabinstein AA, Asirvatham SJ, Holmes DR (2017) Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J Am Coll Cardiol 70, 1173–1182. [DOI] [PubMed] [Google Scholar]
- [5].Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18, 311–324. [DOI] [PubMed] [Google Scholar]
- [6].Han BH, Zhou M-L, Johnson AW, Singh I, Liao F, Vellimana AK, Nelson JW, Milner E, Cirrito JR, Basak J, Yoo M, Dietrich HH, Holtzman DM, Zipfel GJ (2015) Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci U S A 112, E881–E890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].de la Torre JC (2004) Alzheimer’s disease is a vasocognopathy: A new term to describe its nature. Neurol Res 26, 517–524. [DOI] [PubMed] [Google Scholar]
- [8].Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ (2012) Cerebral microinfarcts: A systematic review of neuropathological studies. J Cereb Blood Flow Metab 32, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brundel M, Heringa SM, de Bresser J, Koek HL, Zwanenburg JJM, Jaap Kappelle L, Luijten PR, Biessels GJ (2012) High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer’s disease. J Alzheimers Dis 31, 259–263. [DOI] [PubMed] [Google Scholar]
- [10].Chi N-F, Chien L-N, Ku H-L, Hu C-J, Chiou H-Y (2013) Alzheimer disease and risk of stroke: A population-based cohort study. Neurology 80, 705–711. [DOI] [PubMed] [Google Scholar]
- [11].Purandare N, Burns A (2009) Cerebral emboli in the genesis of dementia. J Neurol Sci 283, 17–20. [DOI] [PubMed] [Google Scholar]
- [12].Tolppanen A-M, Lavikainen P, Solomon A, Kivipelto M, Soininen H, Hartikainen S (2013) Incidence of stroke in people with Alzheimer disease: A national register-based approach. Neurology 80, 353–358. [DOI] [PubMed] [Google Scholar]
- [13].de la Torre JC (2010) Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev 9, 218–225. [DOI] [PubMed] [Google Scholar]
- [14].Jellinger KA (2010) Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neurodegener Dis 7, 112–115. [DOI] [PubMed] [Google Scholar]
- [15].Kalaria RN (2010) Vascular basis for brain degeneration: Faltering controls and risk factors for dementia. Nutr Rev 68(Suppl 2), S74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106, 14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid p protein toxicity. Cell 77, 817–827. [DOI] [PubMed] [Google Scholar]
- [18].Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Miller SW, Davis RE, Parker WD (2000) Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 162, 37–50. [DOI] [PubMed] [Google Scholar]
- [19].Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21, 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum Mol Genet 20, 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, Torres S, Fujioka H, Xu Y, Perry G, Zhu X (2017) Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26, 4118–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107, 18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Park L, Anrather J, Forster C, Kazama K, Carlson GA, Iadecola C (2004) Ap-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 24, 334–342. [DOI] [PubMed] [Google Scholar]
- [25].Drummond GR, Selemidis S, Griendling KK, Sobey CG (2011) Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10, 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miller AA, Drummond GR, Schmidt HHHW, Sobey CG (2005) NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res 97, 1055–1062. [DOI] [PubMed] [Google Scholar]
- [27].Park L, Anrather J, Girouard H, Zhou P, Iadecola C (2007) Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27, 1908–1918. [DOI] [PubMed] [Google Scholar]
- [28].Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C (2005) NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25, 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lampl T, Crum JA, Davis TA, Milligan C, Del Gaizo Moore V (2015) Isolation and functional analysis of mitochondria from cultured cells and mouse tissue. J Vis Exp, doi: 10.3791/52076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ferreira A, Sinjoanu RC, Nicholson A, Kleinschmidt S (2011) A3 toxicity in primary cultured neurons. Methods Mol Biol 670, 141–153. [DOI] [PubMed] [Google Scholar]
- [31].Takuma H, Tomiyama T, Kuida K, Mori H (2004) Amyloid beta peptide-induced cerebral neuronal loss is mediated by caspase-3 in vivo. J Neuropathol Exp Neurol 63, 255–261. [DOI] [PubMed] [Google Scholar]
- [32].Bastianetto S, Ramassamy C, Dore S, Christen Y, Poirier J, Quirion R (2000) The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur J Neurosci 12, 1882–1890. [DOI] [PubMed] [Google Scholar]
- [33].Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292, 7189–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zeidler JD, Fernandes-Siqueira LO, Carvalho AS, Cararo-Lopes E, Dias MH, Ketzer LA, Galina A, Da Poian AT (2017) Short-term starvation is a strategy to unravel the cellular capacity of oxidizing specific exogenous/endogenous substrates in mitochondria. J Biol Chem 292, 14176–14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brace LE, Vose SC, Stanya K, Gathungu RM, Marur VR, Longchamp A, Treviho-Villarreal H, Mejia P, Vargas D, Inouye K, Bronson RT, Lee C-H, Neilan E, Kristal BS, Mitchell JR (2016) Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis 2, 16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yajima D, Motani H, Hayakawa M, Sato Y, Sato K, Iwase H (2009) The relationship between cell membrane damage and lipid peroxidation under the condition of hypoxia-reoxygenation: Analysis of the mechanism using antioxidants and electron transport inhibitors. Cell Biochem Funct 27, 338–343. [DOI] [PubMed] [Google Scholar]
- [37].Jarmuszkiewicz W, Woyda-Ploszczyca A, Koziel A, Majerczak J, Zoladz JA (2015) Temperature controls oxidative phosphorylation and reactive oxygen species production through uncoupling in rat skeletal muscle mitochondria. Free Radic Biol Med 83, 12–20. [DOI] [PubMed] [Google Scholar]
- [38].Downs CA, Heckathorn SA (1998) The mitochondrial small heat-shock protein protects NADH: Ubiquinone oxidoreductase of the electron transport chain during heat stress in plants. FEBS Lett 430, 246–250. [DOI] [PubMed] [Google Scholar]
- [39].Garlid KD, Paucek P (2003) Mitochondrial potassium transport: The K+ cycle. Biochim Biophys Acta 1606, 23–41. [DOI] [PubMed] [Google Scholar]
- [40].Pilchova I, Klacanova K, Tatarkova Z, Kaplan P, Racay P (2017) The involvement of Mg 2+ in regulation of cellular and mitochondrial functions. Oxid Med Cell Longev 2017, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N (2004) Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37, 755–767. [DOI] [PubMed] [Google Scholar]
- [42].Quintana DD, Garcia JA, Sarkar SN, Jun S, Engler-Chiurazzi EB, Russell AE, Cavendish JZ, Simpkins JW (2019) Hypoxia-reoxygenation of primary astrocytes results in a redistribution of mitochondrial size and mitophagy. Mitochondrion 47, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 105, 19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].DuBoff B, Feany M, Gotz J (2013) Why size matters – balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci 36, 325–335. [DOI] [PubMed] [Google Scholar]
- [45].Qi X, Disatnik M-H, Shen N, Sobel RA, Mochly-Rosen D (2011) Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol Biol Cell 22, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ (2013) Mitochondrial alterations near amyloidplaques in an Alzheimer’s disease mouse model. J Neurosci 33, 17042–17051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B (2007) Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 7, 605–616. [DOI] [PubMed] [Google Scholar]
- [50].LaFerla FM (2002) Calcium dyshomeostasis and intracellular signaling in Alzheimer’s disease. Nat Rev Neurosci 3, 862–872. [DOI] [PubMed] [Google Scholar]
- [51].Brown GC, Lakin-Thomas PL, Brand MD (1990) Control of respiration and oxidative phosphorylation in isolated rat liver cells. Eur J Biochem 192, 355–362. [DOI] [PubMed] [Google Scholar]
- [52].Ainscow EK, Brand MD (1999) Top-down control analysis of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes. Eur J Biochem 263, 671–685. [DOI] [PubMed] [Google Scholar]
- [53].Yadava N, Nicholls DG (2007) Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci 27, 7310–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Choi SW, Gerencser AA, Nicholls DG (2009) Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: Spare respiratory capacity and stochastic mitochondrial failure. J Neurochem 109, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hansford RG, Chappell JB (1967) The effect of Ca2+ on the oxidation of glycerol phosphate by blowfly flight-muscle mitochondria. Biochem Biophys Res Commun 27, 686–692. [DOI] [PubMed] [Google Scholar]
- [56].Denton RM, Randle PJ, Martin BR (1972) Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128, 161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McCormack JG, Denton RM (1979) The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J 180, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McCormack JG, Halestrap AP, Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70, 391–425. [DOI] [PubMed] [Google Scholar]
- [59].Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6, 513–519. [DOI] [PubMed] [Google Scholar]
- [60].Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C (2008) Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299. [DOI] [PubMed] [Google Scholar]
- [61].Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol Cell Biol 4, 552–565. [DOI] [PubMed] [Google Scholar]
- [62].Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab 23, 254–263. [DOI] [PubMed] [Google Scholar]
- [63].Bode DC, Baker MD, Viles JH (2017) Ion channel formation by amyloid-β42 oligomers but not amyloid-β40 in cellular membranes. J Biol Chem 292, 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280, 17294–17300. [DOI] [PubMed] [Google Scholar]
- [65].Hoyt KR, Arden SR, Aizenman E, Reynolds IJ (1998) Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol Pharmacol 53, 742–749. [PubMed] [Google Scholar]
- [66].Kiedrowski L (1999) N-methyl-D-aspartate excitotoxicity: Relationships among plasma membrane potential, Na+/Ca2+ exchange, mitochondrial Ca2+ overload, and cytoplasmic concentrations of Ca2+ H+ and K+. Mol Pharmacol 56, 619–632. [DOI] [PubMed] [Google Scholar]
- [67].Griffiths EJ (1999) Reversal of mitochondrial Na/Ca exchange during metabolic inhibition in rat cardiomyocytes. FEBSLett 453, 400–404. [DOI] [PubMed] [Google Scholar]
- [68].Smets I, Caplanusi A, Despa S, Molnar Z, Radu M, VandeVen M, Ameloot M, Steels P (2004) Ca2+ uptake in mitochondria occurs via the reverse action of the Na+/Ca2+ exchanger in metabolically inhibited MDCK cells. Am J Physiol Renal Physiol 286, F784–F794. [DOI] [PubMed] [Google Scholar]
- [69].Brustovetsky T, Brittain MK, Sheets PL, Cummins TR, Pinelis V, Brustovetsky N (2011) KB-R7943, an inhibitor of the reverse Na+/Ca2+ exchanger, blocks N-methyl-D-aspartate receptor and inhibits mitochondrial complex i. Br J Pharmacol 162, 255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. [DOI] [PubMed] [Google Scholar]
- [71].Bartolome F, de la Cueva M, Pascual C, Antequera D, Fernandez T, Gil C, Martinez A, Carro E (2018) Amyloid 3-induced impairments on mitochondrial dynamics, hippocampal neurogenesis, and memory are restored by phosphodiesterase 7 inhibition. Alzheimers Res Ther 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang L, Trushin S, Christensen TA, Bachmeier BV., Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, Gylys KH, Patterson ER, Parisi JE, Diaz Brinton R, Salisbury JL, Trushina E (2016) Alteredbrain energetics induces mitochondrial fission arrest in Alzheimer’s disease. Sci Rep 6, 18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Almeida A, Medina JM (1998) A rapid method for the isolation of metabolically active mitochondria from rat neurons and astrocytes in primary culture. Brain Res Protoc 2, 209–214. [DOI] [PubMed] [Google Scholar]
- [74].Ježek J, Cooper KF, Strich R (2018) Reactive oxygen species and mitochondrial dynamics: The yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel) 7, E13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wu S, Zhou F, Zhang Z, Xing D (2011) Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 278, 941–954. [DOI] [PubMed] [Google Scholar]
- [76].Willems PHGM, Rossignol R, Dieteren CEJ, Murphy MP, Koopman WJH (2015) Cell metabolism perspective redox homeostasis and mitochondrial dynamics. Cell Metab 22, 207–218. [DOI] [PubMed] [Google Scholar]
- [77].Hung CH-L, Cheng SS-Y, Cheung Y-T, Wuwongse S, Zhang NQ, Ho Y-S, Lee SM-Y, Chang RC-C (2018) A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol 14, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.