

Abstract
Estrogenicity-directed fractionation of a methanol extract of the strobiles of Humulus lupulus L. cv. Nugget that had been extracted previously with supercritical CO2, known as “spent hops,” led to the isolation and identification of 22 compounds including 12 prenylated chalcones (1-8, 10-13), five prenylflavanones (14-17), 4-hydroxybenzaldehyde (18), sitosterol-3-O-β-glucopyranoside (19), humulinone (20), and cohumulinone (21). In addition, the prenylated chalcone xanthohumol C (9a) was obtained as a 6:1 mixture along with its 1″,2″-dihydro derivative (9b). Three new chalcones (4, 11, 12) and four previously unreported constituents of hops (5, 6, 9b, 13) are reported. The structures of the new compounds were determined through a combination of spectrometric techniques including 1D and 2D NMR, HRESIMS and ESI-MS-MS. Full 1H NMR spin system analyses were performed to characterize the higher-order glucopyranosyl, prenyl and chalcone B-ring spectra of the isolates. The principle estrogen 8-prenylnaringenin (15) from hops is an artifact formed along with its positional isomer 6-prenylnaringenin (16) through the spontaneous isomerization of the pro-estrogenic chalcone DMX (7).
Keywords: Humulus lupulus, spent hops, chalcone, flavanone, phytoestrogen, prenyl, para-disubstituted benzene, sitosterol glucopyranoside, spin system, proton NMR
In addition to their use in the brewing industry1,2 and as a mild sedative in phytomedicine,3–5 hops have been investigated for their estrogenic6–11 and, more recently, potential cancer chemopreventive12–14 activities. As part of a collaborative, multidisciplinary approach to the investigation of botanicals with potential benefits to women’s health, our long-term goal is to evaluate an extract of hops for its ability to alleviate symptoms related to menopause. The specific purpose of the work presented here was to develop the foundations necessary to produce a standardized estrogenic extract of hops, namely, to isolate and characterize standards for chemical and biological assays, and to identify compounds that may serve as chemical markers for the formulated product.
An estrogen-dependent human endometrial adenocarcinoma epithelial cell line known as the “Ishikawa” cell line was employed to direct the fractionation. Alkaline phosphatase activity in these “Ishikawa cells” correlates with administration of estrogens, but not of other hormones, and this activity is blocked by administration of antiestrogens.15 These cells provide a convenient assay for estrogens and anti-estrogens in which a chromogenic phosphate monoester substrate is hydrolyzed by alkaline phosphatase, and the enzyme’s expression is readily quantified and correlated with estrogenic activity.
Results and Discussion
Compounds present in estrogenic fractions that showed a color reaction with FeCl3 were isolated and chemically characterized as described below. The choice of this phenol-specific spray reagent,16 was made after a literature search revealed all natural estrogen receptor ligands of any significant potency contain at least one phenolic hydroxyl group.17
A majority of the isolates in this work can be considered either direct derivatives or biosynthetic analogues of 1 (see Supplementary Data page S1 for structures). An intact prenyl unit and a para-disubstituted benzene ring are two commonly occurring natural product structural elements present in this molecule. An X-ray crystal structure was obtained for xanthohumol (1), the major hop chalcone (Figure 1). Since the X-ray structure provides absolute proof of connectivity, an extensive analysis of the NMR spectra of 1 can be useful in the interpretation not only of the isolates presented herein, but also of other natural products that contain similar spin systems. After the structure elucidation process was facilitated by the X-ray data, a full spin system analysis was carried out. The first approach used was to simulate the observed spectrum, using the NUTS simulation utility, by adjusting the parameters (δ, J values) until the simulated and experimental spectra were superimposable. Such an approach allowed for all δ and long-range J values in the higher-order systems to be defined to at least 0.2 Hz precision. In a second approach, using the PERCH NMR software, the spectrum was first predicted and simulated based on a molecular model of the compound. Then, using the PERCHit spectra iteration tool, spin system parameters were approximated by iteration of the simulated spectrum until it converged with the experimental data. Although δ/J prediction in PERCH was reasonable, it was necessary before iteration to adjust the predicted δ/J values to parameters that were determined using the first approach discussed above. As a result, the δ/J values were determined with high precision, allowing the underlying higher order spin mechanics and complex long-range J pattern to be fully explained (see Table 1). Provided that the sweep width (SW) and time domain (TD) are optimized, e.g. TD = 32K for SW = 10 ppm, and data sets appended by zero-filling and/or linear prediction, NMR spectra allow for a digital resolution on the order of 0.0001 ppm (0.03 Hz at 300 MHz). Therefore with tools such as PERCH, chemical shifts and coupling constants can accurately be determined to a precision of 0.0001 ppm and 0.01 Hz or better, respectively.
Figure 1.
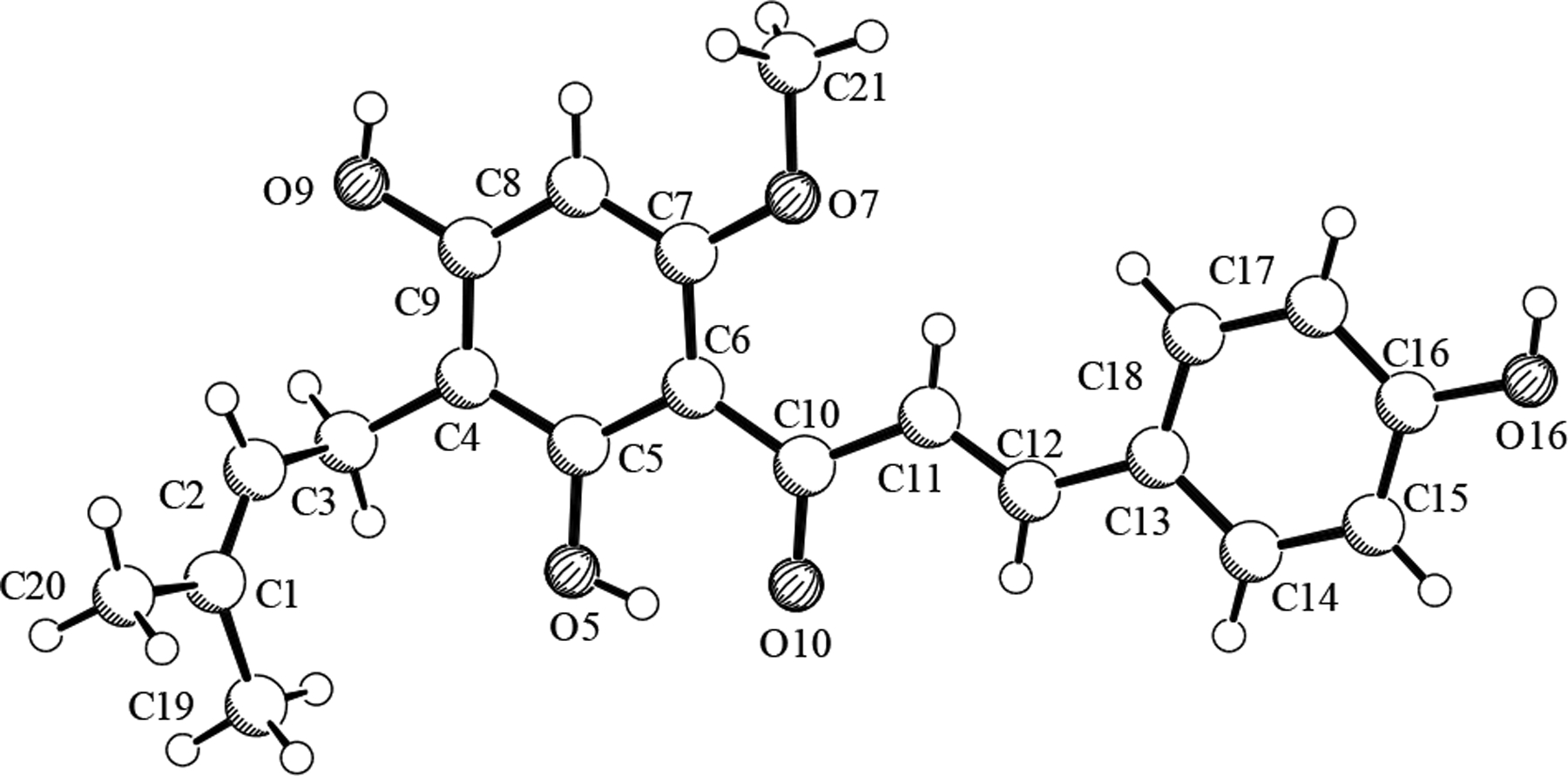
X-ray crystal structure of xanthohumol (1).
Table 1.
1H NMR spin system analyses for reference compounds 1and 10and for new compound 5.
| position | ppmd | multe | spin system | J [Hz] (→ H)f | |
|---|---|---|---|---|---|
| 1a | α | 7.7879 | d | ABMM’XX’ | 15.53 (Hβ) |
| β | 7.6649 | dtt | ABMM’XX’ | 15.53 (Hα), 0.52 (H2,6), 0.34 (H3,5) | |
| 2,6 | 7.4946 | dddd | ABMM’XX’ | 8.51 (ortho), 2.61 (meta), 0.52 (Hβ), 0.34 (para) | |
| 3,5 | 6.8232 | dddd | ABMM’XX’ | 8.51 (ortho), 2.34 (meta), 0.34 (para), 0.34 (Hβ) | |
| 5’ | 6.0146 | s | - | - | |
| 1″-CH2 | 3.2244 | dqq | AM2X3Y3 | 7.21 (H2″), 1.15 (Me-5″), 0.49 (Me-4″) | |
| 2″ | 5.1967 | tqq | AM2X3Y3 | 7.21 (CH2-1″), 1.40 (Me-4″), 1.36 (Me-5″) | |
| 4″-Me | 1.7562 | dtq | AM2X3Y3 | 1.40 (H2″), 0.49 (CH2-1″), 0.36 (Me-5″) | |
| 5″-Me | 1.6494 | dtq | AM2X3Y3 | 1.36 (H2″), 1.15 (CH2-1″), 0.36 (Me-4″) | |
| 6’-OMe | 3.8950 | s | - | - | |
| 5b | α | 7.8031 | d | ABMM’XX’ | 15.54 (Hβ) |
| β | 7.6742 | dtt | ABMM’XX’ | 15.54 (Hα), 0.45 (H2,6), 0.22 (H3,5) | |
| 2,6 | 7.5040 | dddd | ABMM’XX’ | 8.53 (ortho), 2.57 (meta), 0.45 (Hβ), 0.25 (para) | |
| 3,5 | 6.8261 | dddd | ABMM’XX’ | 8.53 (ortho), 2.53 (meta), 0.25 (para), 0.22 (Hβ) | |
| 5’ | 6.0259 | s | - | - | |
| 1″-CH2 | 2.6196 | ddd | AA’MM’ | 16.00 (gem.), 11.79 (trans), 4.88 (gauche) | |
| 2″-CH2 | 1.6375 | ddd | AA’MM’ | 16.00 (gem.), 11.79 trans), 4.88 (gauche) | |
| 4″,5″-Me | 1.1719 | s | - | - | |
| 6’-OMe | 3.9064 | s | - | - | |
| 10c | α | 7.8145 | d | ABMM’XX’ | 15.53 (Hβ) |
| β | 7.7030 | dtt | ABMM’XX’ | 15.53 (Hα), 0.59 (H2,6), 0.46 (H3,5) | |
| 2,6 | 7.5067 | dddd | ABMM’XX’ | 8.46 (ortho), 2.54 (meta), 0.59 (Hβ), 0.37 (para) | |
| 3,5 | 6.8271 | dddd | ABMM’XX’ | 8.46 (ortho), 2.12 (meta), 0.46 (Hβ), 0.37 (para) | |
| 5’ | 5.9896 | s | - | - | |
| 1″-a | 2.8433 | dd | AMX | 16.81 (H1″b), 5.34 (H2″) | |
| 1″-b | 2.5237 | dd | AMX | 16.81 (H1″a), 6.95 (H2″) | |
| 2″ | 3.7648 | dd | AMX | 6.95 (H1″b), 5.34 (H2″) | |
| 4″-Me | 1.3469 | s | - | - | |
| 5″-Me | 1.2939 | s | - | - | |
| 6’-OMe | 3.9024 | s | - | - |
500 MHz spectrum with digital resolution (DR) = 0.084 Hz (0.00017 ppm).
360 MHz spectrum with DR = 0.09 Hz (0.00025 ppm).
300 MHz spectrum with DR = 0.055 Hz (0.00018 ppm).
Coupling constants and chemical shifts were precisely determined by PERCH analysis and are reported to 0.01 Hz precision. The digit shown in smaller font is not precisely certain, but is necessary to fully convey the experimentally observed spin system.
Multiplicity given under first order spin approximation.
Coupling constants and chemical shifts were precisely determined by PERCH analysis and are reported to 0.01 Hz precision. The digit shown in smaller font is not precisely certain, but is necessary to fully convey the experimentally observed spin system.
In 1H NMR, an intact prenyl (isopentenyl) unit is a 9-spin system that is annotated as either an AM2X3Y3 (for chalcones and 6-prenylated flavanones) or AMNX3Y3 system (for 8-prenylated flavanones). In Figure 2, experimental and simulated spectra of the prenyl unit spin system are shown for the chalcone 1. The H2″ olefin proton signal is often reported as a triplet, but already a 300 MHz spectrum clearly reveals its nature as something more appropriately called a tqq or ddqq under nuclei first-order assumptions. While the methyl signals are typically reported as singlets, they each have a unique shape due to extensive long-range coupling. Each methyl group can be approximated as a dtq or dddq and, due to stronger 5J coupling with each of the H1″ protons, the trans-5″-methyl signal is more stout than that of the cis-4″-methyl. The signal corresponding to the methylene 1″ protons appears as one broad isochronic (2H) doublet with J = 7.2 Hz. One reason for the relatively unresolved nature of this peak is that thermal rotation of the 1″-methylene group relative to the aromatic A-ring causes the Larmor frequency (v) of these protons to oscillate, such that after Fourier transformation the frequency-averaged spectrum appears as a broad Gaussian peak.18 However, when the chalcone 1 isomerizes to the flavanone 17, the induced chirality results in chemical non-equivalence of these CH2-1″ protons and, as will be shown below, significantly more structural information can be extracted from the corresponding signal. Spin system analyses carried out on spectra of compounds 2, 7, 8, 14–17 containing this moiety revealed that, whereas the chemical shifts varied widely, coupling constants in the intact prenyl units in all cases were within 0.1 Hz of the values shown for 1 in Figure 2.
Figure 2.
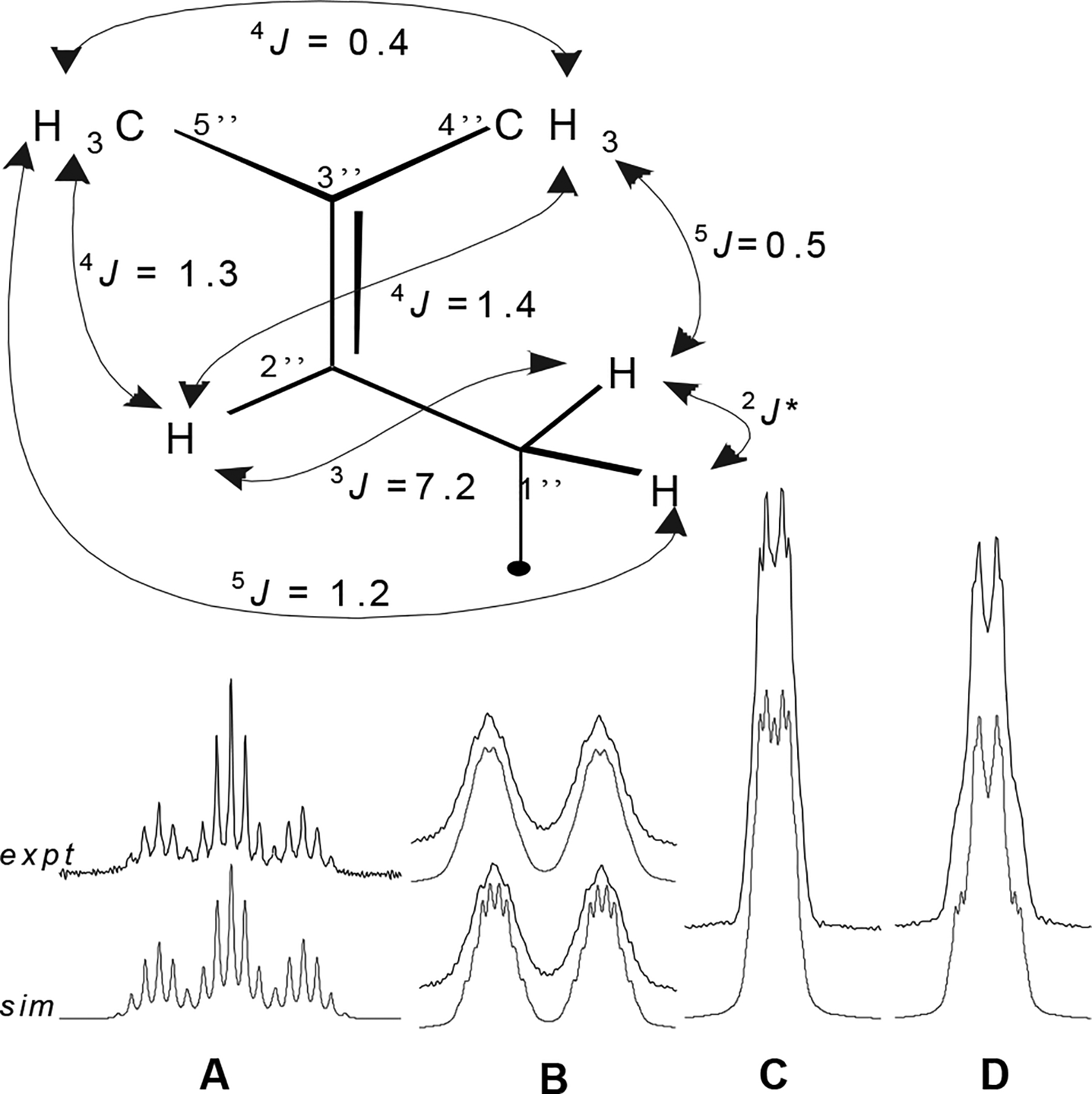
The 1H NMR spin system of the prenyl unit in xanthohumol (1) exhibits multiple long-range coupling. Simulated (sim) and experimental (expt) 500 MHz 1H NMR spectra of the prenyl signals in 1 are shown. Precise δ and J values determined for 1 are given in Table 1. A H2″, B CH2-1″ (simulation linewidth above 0.8 Hz; below 0.5 Hz), C Me-4″, D Me-5″. Linewidth in simulated spectra shown for signals A, C, and D is 0.5 Hz. * 2J coupling not observed; geminal coupling between the 1″ methylene protons was observed only in the 8-prenylated flavanones, 15 and 17 (see Figure 4B). The coupling constants are within 0.1 Hz of those determined for compounds containing this same moiety (2, 7, 8, and 14-17).
The prenylated flavanone, (±)-8-prenylnaringenin 15, is an artifact formed along with (±)-6-prenylnaringenin (16) by spontaneous isomerization of its parent natural product chalcone desmethylxanthohumol (7).8 In their 1H NMR spectra, as illustrated in Figure 3, the H4″-(Z)-methyl protons on the prenyl unit in all three 8-prenylated flavanones (14, 15, 17) were shielded up to 0.5 ppm relative to isomeric chalcones or 6-prenylflavanones. In addition to this Z-methyl shielding in 8-prenylflavanones, another potentially useful observation for placement of intact prenyl units on the flavanone nucleus concerns the methylene H1″ signal. Anisochronicity (Δδ) between the H1″ geminal methylene protons was clearly evident in the 8-prenylated flavanones (15, 17), especially at a higher spectrometer frequency as evidenced in Figure 4B. However, neither chemical nor magnetic non-equivalence between these protons could be demonstrated either for the 6-prenylated flavanone (16) or for any chalcone, wherein they appeared as a broad doublet as shown in Figure 2 for the chalcone 1. The chirality induced when the chalcone isomerizes to the flavanone results in H1″ signals that are significantly more complex and informative.
Figure 3.
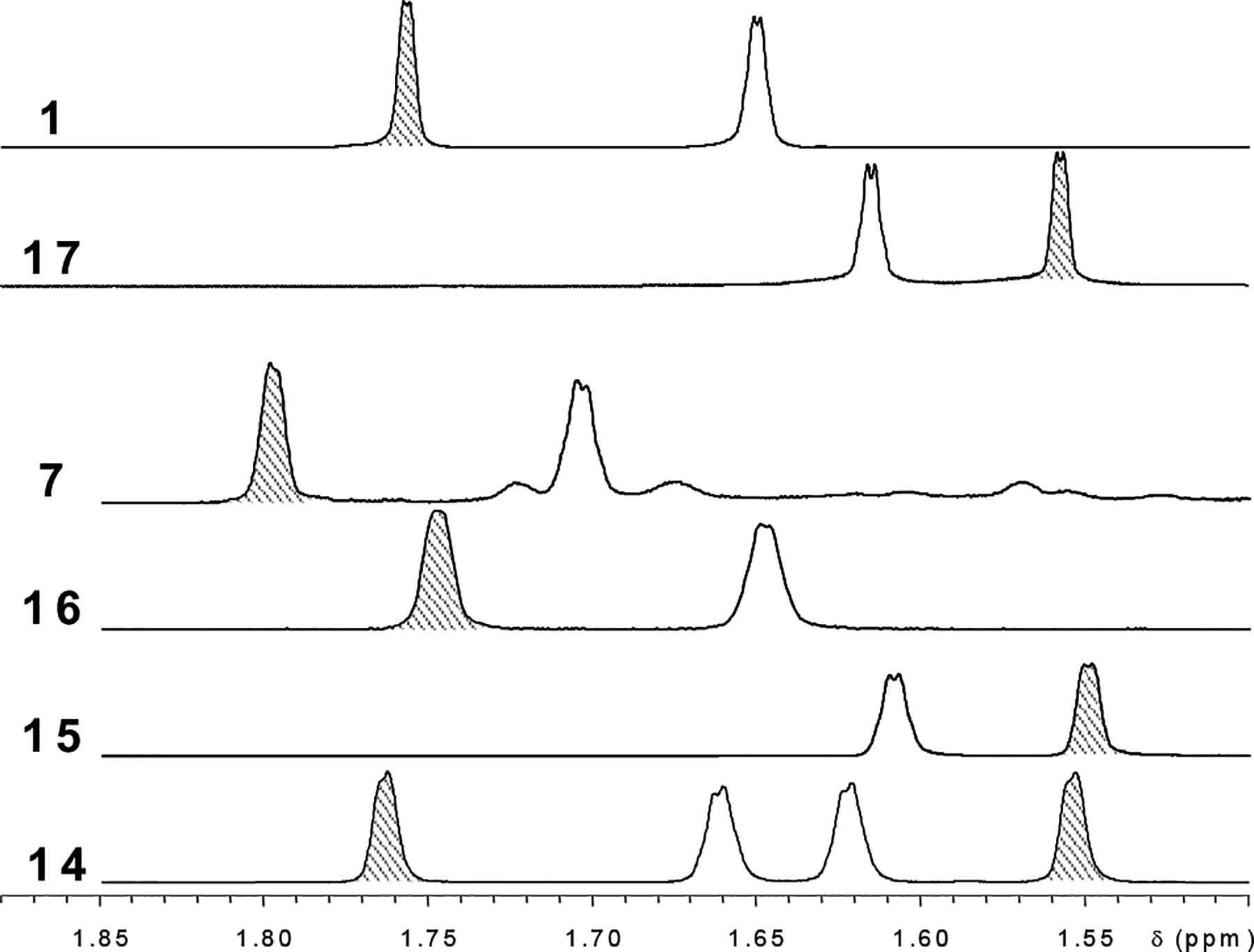
Z-Methyl shielding in the prenyl moiety of 8-prenylated flavanones. The 4″-(Z) prenyl methyl signals are shaded in the above spectra of the methyl region of chalcones and flavanones from Humulus lupulus containing intact prenyl units. The 8-prenylated flavanones 15 (8-prenylnaringenin) and 17 (isoxanthohumol) are clearly distinguished from the 6-prenylated flavanone 16 (6-prenylnaringenin), which has a similar pattern as the chalcones (1, 7). Consistent with this trend, the 4″-Z methyl protons in the 8-prenyl unit of 6,8-diprenylnaringenin (14) are strongly shielded relative to those in the 6-prenyl moiety (Δδ = 0.21 ppm), much more so than the 5″-E-methyl protons (Δδ = 0.06 ppm). These shift rules reflect the underlying difference in chemical environment, and allow assignment of flavanone partial structures based on δ values of methyl protons.
Figure 4.
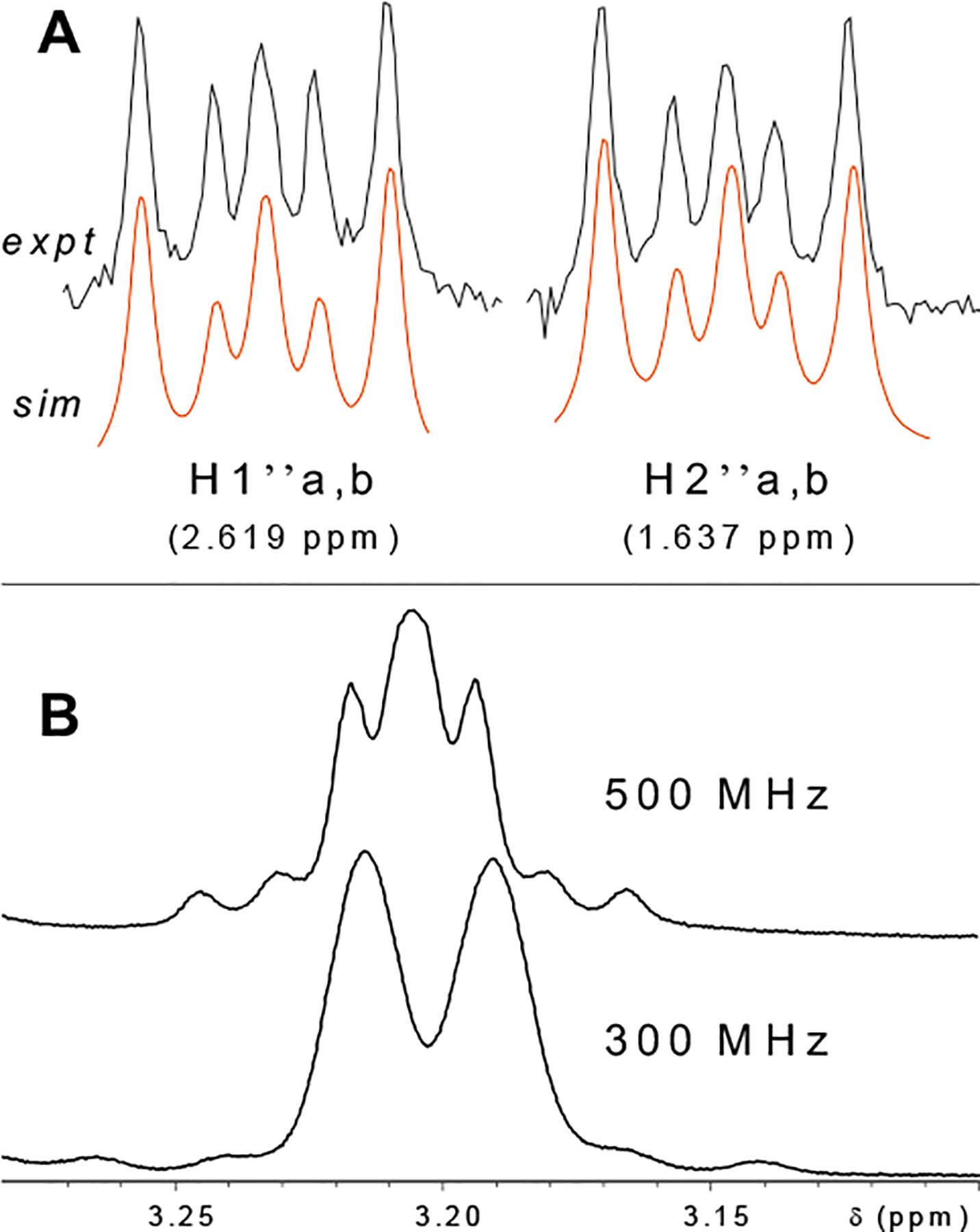
Unique signals diagnostic of partial structures of flavanoids from Humulus lupulus. Above (A) are signals for the AA′XX′ spin system comprised of the prenyl-methylene groups in xanthohumol H (5). The magnetic non-equivalence between the isochronic protons, indicating that this is not a freely-rotating system in MeOH solution at room temperature, is evidenced by the observation of geminal coupling. 2J = 16.00 Hz, 3Jgauche = 4.88 Hz, 3Jtrans=11.79 Hz. In B, the effect of spectrometer frequency on spin multiplicity of the H1″ prenyl-unit methylene signal is illustrated for the 8-prenylated flavanone, isoxanthohumol (17). At 300 MHz, Δv = 6 Hz for the two nuclei, whereas at 500 MHz Δv = 10 Hz. Since the geminal coupling constant of 13.8 Hz is independent of field strength, as spectrometer frequency decreases, the ratio J/Δv increases, resulting in a higher order spectrum. The same higher order effect was observed for the 5-O-demethyl derivative of 17, 8-prenylnaringenin (15). These data cannot be reproduced in tables without first conducting full spin system analyses.
Another structural element of 1 commonly encountered in natural products is the para-disubstituted benzene ring. These aromatic “B-ring” protons are annotated in this case as MM’XX’ indicating two pairs of chemically equivalent, magnetically non-equivalent protons. An additional fine splitting of each half of the Hβ doublet was evident, and the observation that each of the doublet peaks of Hβ are more stout than those of Hα indicated that the aromatic B-ring and the adjacent α- and β-protons together comprise a single spin system (Figure 5). The very long-range 5J coupling between Hβ and H3/H5 is 0.34 Hz, comparable in magnitude with the 0.52 Hz 4J coupling between Hβ and H2/H6. Precise coupling constants and chemical shifts in this ABMM’XX’ spin system shown for 1 in Table 1 are within 0.15 Hz of those determined for the other isolated 4-hydroxychalcones (3-14), allowing for unambiguous dereplication of this moiety in the isolates discussed below. Whereas the B-ring 1H NMR signals are often reported as individual (2H) doublets, they may be considered dddd under first-order assumptions.
Figure 5.
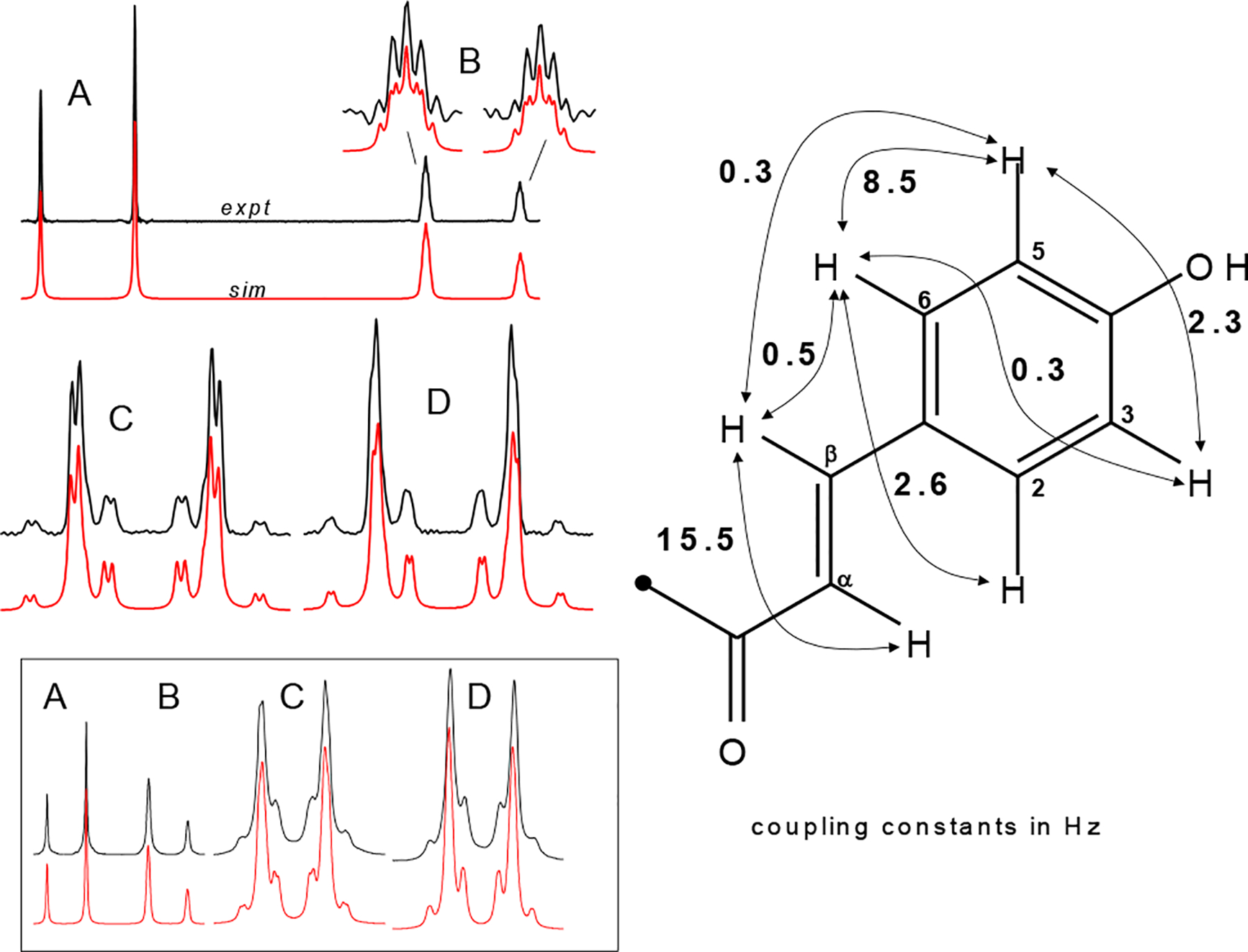
The ABMM’XX’ spin system in chalcones 1–13 shows a characteristic set of signals corresponding to the B-ring and the olefinic α and β protons. Experimental (top) and simulated (bottom) 500 MHz 1H NMR signals of Hα (A), Hβ (B; enlargement at linewidth 0.25 Hz; all other simulated spectra are shown at 0.5 Hz linewidth), H2,6 (C), and H3,5 (D) are shown on the left for xanthohumol (1). Each of the peaks (A-D) are generally reported as individual doublets. Long range coupling results in a finer structure visible even at 300 MHz (inset below). The coupling constants (in Hz) shown on the right are within 0.15 Hz of those determined for 4-hydroxychalcones 1-13. Precise values determined for this spin system in 1, 5, and 10 are given in Table 1.
The HRESIMS [M-H]− m/z value for 4 of 387.1451 provided the molecular formula of C21H24O7 (calcd for 387.1449). From the α,β, and B-ring 1H NMR signals (Table 2) and from the UV spectrum, by analogy to 1, 4 could be identified as a 4-hydroxychalcone. Abundant fragments at m/z 119 (B fragment) and 267 (A fragment) in the MS2 spectrum indicated that, relative to 1, the two additional oxygen atoms and two additional protons were associated with the A-ring. Combining the information obtained from UV, MS and 1H NMR, it was clear that compound 4 was a derivative of 1 with a saturated dihydroxyprenyl unit. With two isochronous and uncoupled methyl groups on the prenyl unit (6H singlet at δ 1.25), one of the hydroxyl groups must be at the 3″-position, as placement anywhere else would result in methyl signals that were either anisochronic or split into doublets. An HMBC correlation between aromatic A-ring carbons and each of the methylene protons placed the methylene group at the 1″-position, and the second hydroxyl group must therefore be placed at the 2″-position. Combining the above information, the structure of 4 was determined to be (±)-(2E)-1-[2,4-dihydroxy-3-(2,3-dihydroxy-3-methylbutyl)-6-methoxyphenyl]-3-(4-hydroxyphenyl)-2-propen-1-one. This marks the first report of this structure, and in keeping with a convention established by Tabata et al. with xanthohumol B (10)19 and continued by Deinzer et al.,20,21 the trivial name xanthohumol G is proposed for 4 (Figure 6).
Table 2.
1H and 13C NMR assignments for chalcones 4,6,9b,11.
| compound | 4 | 6 | 9b | 11 | ||||
|---|---|---|---|---|---|---|---|---|
| position | δH | δC | δH | δC | δH | δC | δH | δC |
| C=O | 194.8 | 194.4 | 194.2 | |||||
| α | 7.807 d (15.5) | 125.9 | 7.79 d (15.5) | 7.821 d (15.6) | 125.6 | 7.935 d (15.6) | 126.1 | |
| β | 7.688, d* (15.5) | 143.3 | 7.67 d* (15.5) | 7.697 d* (15.6) | 143.7 | 7.629 d* (15.6) | 143.0 | |
| 1 | 128.5 | 128.4 | 128.5 | |||||
| 2,6 | 7.507* | 131.3 | 7.50* | 131.4 | 7.50e | 131.5 | 7.502* | 131.2 |
| 3,5 | 6.828* | 116.9 | 6.82* | 117.1 | 6.83e | 116.1 | 6.829* | 116.9 |
| 4 | 161.5 | 162.0 | 161.1 | |||||
| 1’ | 108.6 | 106.2 | ||||||
| 2’ | 163.1 | 162.8b | 163.1 | 166.7 | ||||
| 3’ | 6.071 s | 93.1 | 6.02 s | 5.950 s | 92.8 | 5.930 s | 96.2 | |
| 4’ | 167.5 | 166.2b | 161.6 | 164.3 | ||||
| 5’ | 107.0 | 103.6 | 100.9 | |||||
| 6’ | 167.5 | 166.2b | 161.3 | 157.2 | ||||
| 1″ | 2.603 dd (14.3, 10.0) | 25.3 | 3.31c | 2.591, 2H, t (6.8) | 17.1 | 2.848 dd (16.9, 5.5) | 26.8 | |
| 3.029 dd (14.3, 2.6) | 2.513 dd (16.9, 6.9) | |||||||
| 2″ | 3.555 dd (10.0, 2.6) | 80.6 | 5.49 m# | 125.8b | 1.806, 2H, t (6.8) | 33.1 | 3.790 dd (6.9, 5.5) | 69.5 |
| 3″ | 74.1 | 135.5b | 77.1 | 79.7 | ||||
| 4″ | 1.242 s | 25.9 | 1.81, 3H, br-s# | 13.9 | 1.337 s | 27.0 | 1.387 s | 21.5 |
| 5″ | 1.242 s | 25.9 | 3.89, 2H, br-s# | 69.4 | 1.337 s | 27.0 | 1.447 s | 25.9 |
| O-Me | 3.919 s | 56.2 | 3.90 s | 55.4a | 3.896 s | 56.3 | - | - |
see Figure 5 for B-ring spin multiplicities
insufficient S/N for full multiplicity analysis
assignment based on HMQC correlation
assignment based on HMBC correlation
signal overlapped by solvent was assigned from COSY correlation
signal obscured by corresponding signal in 9a.
Figure 6.
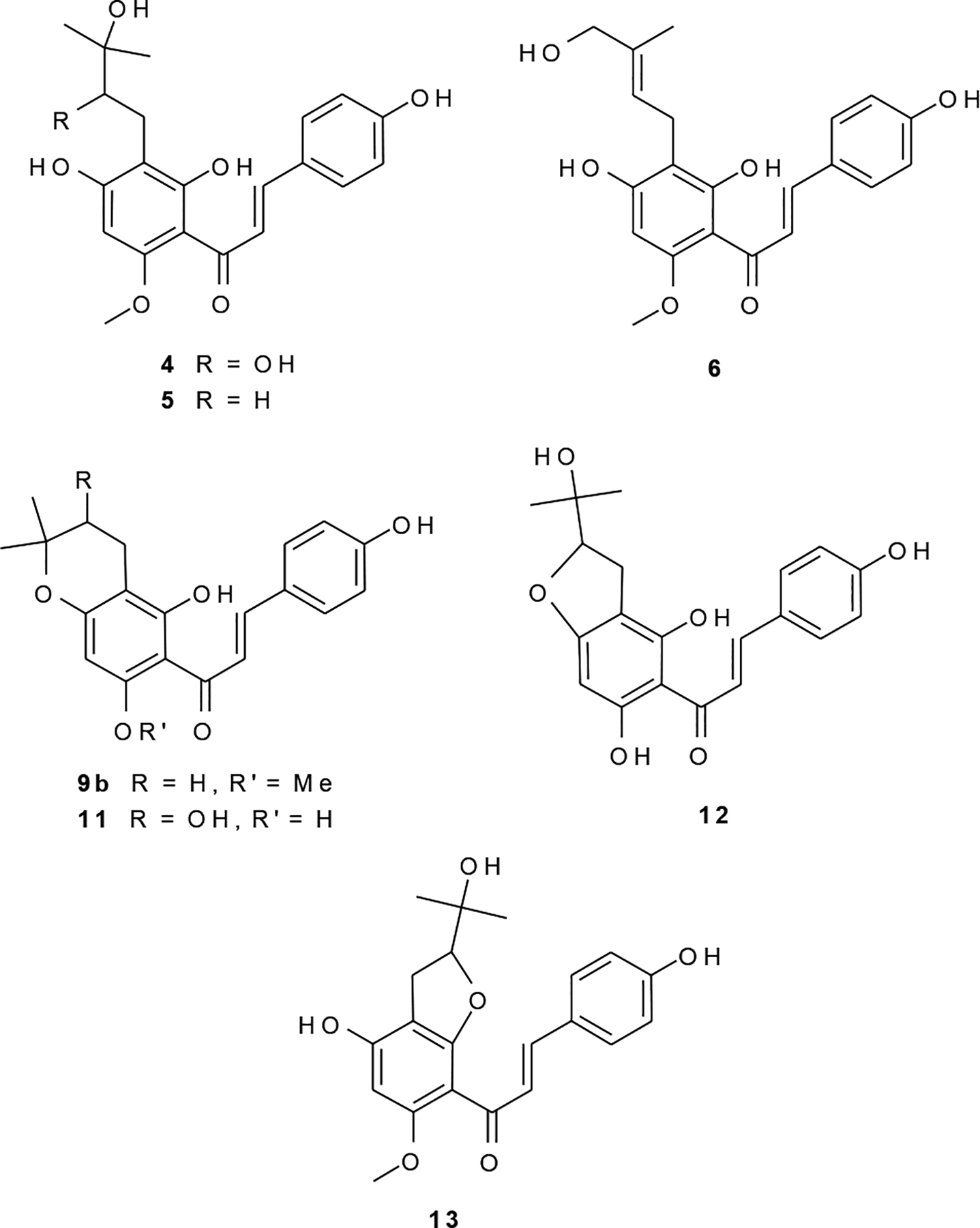
New chalcones (4,6,11,12) and previously unreported constituents (5,9b,13) of Humulus lupulus L.
The molecular formula for 5 of C21H24O6 (calcd for 371.1500) was provided by the HRESIMS [M-H]− m/z value of 371.1509, and by comparing the 1H NMR spectrum with 1, 5 was evidently a hydrated derivative of xanthohumol. This notion was supported by the UV spectrum, which was typical for a chalcone with a 6′ and/or 2′-hydroxyl group.22 Abundant fragments at m/z 119 (B fragment) and 251 (A fragment) in the ESI fragmentation spectrum were consistent with such a proposal and also indicated that the site of hydration was in the A fragment. Considering the 1H NMR spectrum, site of hydroxylation could be localized in the prenyl unit by comparing with that of 1, and given that both methyl groups on the prenyl unit resonated as isochronic singlets (6H singlet at δ 1.24), the hydroxyl group was placed at the 3″-position. Spin simulation experiments were carried out in order to interpret the NMR data, and simulated and experimental spectra for the unique methylene signals for this isolate are shown in Figure 4A. Interestingly, geminal coupling (16.0 Hz) was observed in the 1″ and 2″ methylene groups, contrary to what may be expected for methylene units that supposedly are freely rotating. The structure of 5 was therefore assigned as (2E)-1-[2,4-dihydroxy-3-(3-hydroxy-3-methylbutyl)-6-methoxyphenyl]-3-(4-hydroxyphenyl)-2-propen-1-one. This structure was recently reported as a xanthohumol metabolite in rat feces.23 Although the NMR data were collected in different solvents, they were sufficiently similar to determine that 5 is identical to the xanthohumol metabolite. This marks the first report of this compound as a constituent of hops, and the trivial name xanthohumol H is proposed for 5 (Figure 6).
The HRESIMS [M-H]− m/z value of 369.1350 for 6 provided a molecular formula of C21H22O6 (calcd for 369.1344) consistent with an oxidized derivative of 1. Likewise, the UV spectrum indicated that 6 was a chalcone. A metabolite of xanthohumol (1) produced by human liver microsomes had an identical ESI-MS-MS spectrum as 6, and this was considered proof that 6 was a hydroxylated derivative of XH (1).24 Abundant MS2 fragments at m/z 119 (B fragment) and 249 (A fragment) were respectively consistent with a B-ring hydroxylated chalcone containing an oxidized prenyl in the A-ring. Comparing the 1H NMR spectra of isolates containing intact prenyl units (1, 7, 8, 14–17) and that of compound 6, it was apparent that the prenyl unit in 6 was modified by the addition of a hydroxyl group at either the 4″ or 5″ position. In the mass-limited COSY spectrum, wherein the S/N for all prenyl unit signals was very low, the prenyl unit crosspeaks observed, in order of decreasing S/N, were: H2″/CH2-1″, H2″/Me-4’, CH2-1″/Me-4″, and CH2-1″/ CH2OH-5″. No crosspeak was observed between H2″ and the CH2OH, consistent with a trans arrangement with the hydroxylated methyl(ene) group H1″. Due to very limited sample quantity, the S/N even of a 512 scan 500 MHz 1H NMR spectrum was insufficient to perform a full spin analysis, and precise δ/J values remain incomplete. Thus, only partial 13C assignments could be obtained. Therefore the H2″ peak was labeled “m” in Table 2. Clearly, however, 6 was either the 4″(Z)- or 5″(E)-hydroxyl derivative of 1. Attempts were made to prove the placement of the hydroxyl group with NOE experiments, e.g., to show a correlation between the intact methyl and the H1″ methylene protons or, conversely between H2″ and the hydroxylated methyl. Such an observation was complicated by the fact that the hydroxymethylene signal overlaps the 5-O-methyl in MeOH-d4. While the sample was insoluble in pure CDCl3, these two signals were resolved in a 1H spectrum taken in a mixture of MeOH-d4 and CDCl3 (ca. 1:10). However, due mainly to insufficient sample quantity, no such NOE correlations could be observed. In all likelihood both isoforms exist in hops, i.e. 4″- and 5″-hydroxyxanthohumol. Because the E-olefins are generally regarded as being thermodynamically more stable, it is rational that the corresponding isoform should be more abundant. No chalcone of greater abundance than 6 remained unidentified in related fractions. The presently available spectral evidence is most compatible with the structure of 5″-(E)-hydroxyxanthohumol (Figure 6). Available data do not allow the 4″-(Z) isomer to be ruled out.
It should be emphasized that, particularly for this class of compounds, inverse 13C detection methods, specifically HMBC spectra, provide the same information as 13C direct detection, plus much more, in a shorter amount of spectrometer time. For example, after 45,000 scans (36 hr) at 125 MHz on ca. 200 μg 6, only four carbon peaks were visible, namely, the B-ring 2′,6′ and 3′,5′ signals, and the prenyl C4″ and C5″ signals. These same resonances, as well as cross peaks with C2′, C4′, C6′, C2″, and C3″ were visualized in the HMBC spectrum after only 12 hr spectrometer time. The sensitivity could be enhanced further by 2- to 16-fold f1 zero-filling and/or forward linear prediction prior to Fourier transformation.25
We have twice isolated the mixture 9a:9b (ca. 6:1), from both whole strobiles, and from spent hops. The major component 9a was identified as the known compound, xanthohumol C.21 The UV spectrum of 9b was obtained by analytical HPLC separation of the two congeners using PDA detection, and was essentially the same as that of 1, indicating a free 2′- or 6′-hydroxy. The HRESMS (m/z 353.1438 [M-H]− calcd for 353.1394), MS2, UV, and NMR (Table 2) spectra were all consistent with the 1″,2″-dihydro derivative of 9a. The structure of 9b was therefore assigned as (2E)-1-(3,4-dihydro-5-hydroxy-7-methoxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxyphenyl)-2-propen-1-one, or 1″,2″-dihydroxanthohumol C (Figure 6). This compound has recently been reported as a xanthohumol metabolite in rat feces,23 and the NMR data presented were identical to those reported for 9b.
The HRESIMS [M-H]− m/z value of 355.1182 for 11 corresponded to a molecular formula of C20H20O6 (calcd for 355.1187). An abundant B-ring fragment (m/z 119) and A-ring fragment at m/z 235 were observed in the MS2 spectrum, as was the loss of H2O at m/z 337. The 1H NMR signals for the prenyl unit protons in 11 (Table 2) were very similar to those for 10, which was identified as the known compound, xanthohumol B, by comparison with literature data.19–21,26,27 Following previous reports, the chemical shifts of the 2″ methine proton in furano compounds such as 12 and 13 were much higher than what is observed for 10 and 11 and reported19–21,27,28 for related pyrano compounds (ca. 4.8 ppm vs. ca. 3.8 ppm). This led to the assignment of 11 as the O-demethyl derivative of 10, or (±)-(2E)-1-(3,4-dihydro-3,5,7-trihydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxyphenyl)-2-propen-1-one. The trivial name desmethylxanthohumol B is proposed for 11 (Figure 6) in order to reflect its relationship to the compounds previously named desmethylxanthohumol8 and xanthohumol B.19
For the mass-limited isolate 12 the HRESIMS [M-H]− m/z value of 355.1201 corresponded to a molecular formula of C20H20O6 (calcd for 355.1187), consistent with an isomer of 11 and another oxidized derivative of DMX (7). The UV spectrum also indicated a chalcone. Abundant fragments at m/z 119 (B fragment), 235 (A fragment), and a peak that was weak, but clearly present, at m/z 337 (loss of H2O) in the MS2 spectrum, together indicated that the site of oxidation was in the prenyl unit. The signal for the prenyl unit methine proton H2″ resonated at a relatively low field compared to that observed for the pyrano isomer 11 (δ 4.83 compared with δ 3.79) and for this reason the prenyl side chain was determined to be of a furano constitution. The question remaining was if the cyclization had occurred with the 4′ or the 2′-hydroxyl group. The rationale ultimately used to rule out the O-demethyl derivative of 13 were as follows: The 1H NMR spectra of prenyl unit signals were virtually identical when comparing compounds 10 and 11, and it would therefore be expected that the prenyl signals for the demethyl derivative of 13 would be similar to those for 13 (XH-I). The prenyl unit δ and J-values were notably different comparing 12 and 13, much more so than for the 10/11 pair (XH/DMX-B). By analogy, it did not seem logical that 12/13 constitute the XH/DMX-I pair. Also, the dramatic deshielding of the α proton (δ 8.06) was more pronounced in 12 than for any other isolate. Given the deshielding effect due to H-bonding of free 6′ or 2′-hydroxy groups with the ketone, it is rational to assert that free hydroxyl groups at both 6′ and 2′- would result in the greatest deshielding of the α-proton, as was observed for 12, and indicates that cyclization of the prenyl unit had occurred with the 4′-OH rather than the 2′-OH. The 5-O-demethyl derivative of 13 cannot be entirely ruled out based on the available data. However, all of these observations support the structure assigned to 12, namely, (±)-(2E)-1-[2,3-dihydro-4,6-dihydroxy-2-(1-hydroxy-1-methylethyl)-7-benzofuranyl]-3-(4-hydroxyphenyl)-2-propen-1-one. This structure has not been reported to date, and the trivial name desmethylxanthohumol J is proposed for 12 (Figure 6).
The HRESIMS [M-H]− m/z value of 369.1359 for 13 provided a molecular formula of C21H22O6 (calcd for 369.1344) consistent with an oxidized derivative of 1, while the UV spectrum of 13 was distinct from any other isolate, with λmax 345 nm. In the MS2 spectrum, an abundant fragment at m/z 119 (B fragment) was consistent with a B-ring hydroxylated chalcone, and a weak, but clearly present, fragment at m/z 249 (A fragment) implied the site of oxidation was in the A ring. Also observed in the MS2 spectrum was a fragment corresponding to the neutral loss of acetone (m/z 311). Whereas such a fragment may be characteristic of 13 and related 2-(1-hydroxy-1-methylethyl)furano compounds, it should be noted that fragments corresponding to a neutral loss of 58 were also observed for compounds in which the loss of C3H6O could not readily be visualized (i.e. loss of 58 also observed for 1, 2, 8, 11, 14), and therefore such a product ion cannot presently be regarded as being diagnostic for any particular arrangement of a prenyl unit. Based on the UV spectrum with λmax 345 nm, as opposed to ca. 370 nm for chalcones containing a hydroxyl group(s) at the 2′ and/or 6′ position capable of H-bonding with the ketone, as well as the strongly shielded chemical shift observed for the α proton (δ 7.13, compared with 7.8–8.0 for chalcones containing a free 6′ or 2′-hydroxyl group), it was apparent that the cyclization of the prenyl unit occurred with the 2′- rather than the 4′-hydroxyl group. The remaining question was whether 13 was a furan or a pyran derivative. Comparing the 1H NMR spectrum of 13 with those of 10 and 11 and with literature data for related compounds,19–21,27,28 the pyrano derivative could be ruled out because the δ value of the H2″ methine proton would be significantly lower than what was observed for 13. The structure proposed for 13 has recently been reported as a microbial transformation product of xanthohumol,29 and with the exception of slight chemical shift differences due to solvent, and the revised B-ring coupling constants presented in Figure 5, the spectroscopic data for 13 otherwise agreed with previously reported values29 and allowed for the conclusion that 13 was identical to the XH transformation product, (±)-(2E)-1-[2,3-dihydro-4-hydroxy-2-(1-hydroxy-1-methylethyl)-6-methoxy-5-benzofuranyl]-3-(4-hydroxyphenyl)-2-propen-1-one (Figure 6). This is the second report of this compound, and the first report as a constituent of hops. Since no trivial name was proposed when this compound was first reported,29 and because this marks the first report of 13 as a plant constituent, the common name xanthohumol I is proposed. This chalcone is unique among all presented herein, in that isomerization to the corresponding flavanone is prevented by substitution at both the 2′and 6′ positions. The abundant B-ring fragment at m/z 119 (base peak) in the MS2 spectrum indicates that an alternative mechanism must exist to a previously proposed pathway20 involving isomerization to the flavanone, for 1 and congeners from hops.
Compound 19 was obtained as a colorless, amorphous solid from a relatively polar silica gel fraction (Table 3). The 1H NMR spectrum, with seven signals of sugar protons, including the anomeric resonance at δ 5.07 ppm, was typical of steroidal glycopyranoside with a Δ5,6-stigmastene skeleton.30,31 Due to higher order effects and overlap, complete coupling constants for the spin system of the sugar moiety could only be determined with spin simulation experiments (Figure 7). These experiments allowed the unambiguous identification of the sugar moiety as a β-glucopyranoside residue.32 Interestingly, all attempts to dereplicate 19 by comparison of 1H NMR data with those published for known triterpenoid- or steroidal glucopyranosides were unsuccessful. The experimental chemical shifts combined with the observed J values for the protons (other than H1′) on the sugar moiety, were not consistent with any previously reported compound. The 13C NMR data, however, were identical to those published for sitosterol-3-O-β-D-glucopyranoside.33 The J pattern presented in Figure 7, therefore, represents the first complete 1H NMR analysis of the of the sugar moiety in sitosterol-3-O-β-glucopyranoside (19).30,31,33
Table 3.
Chromatographic behavior on silica gel vacuum-liquid chromatography (VLC), countercurrent chromatography (CCC) partition coefficients (Kp), and overall isolation yields of compounds from spent hops.
| a | b | c | d | e | f | g | h | (%) |
|---|---|---|---|---|---|---|---|---|
| 40 | 1.5 | 0.59 | 1.09 | 1.24 | 38.6 | 0.4 | ||
| 40 | 1.5 | 1.13 | 38.8 | 0.00048 | ||||
| 40 | 2.0 | 0.23 | 0.45 | 33.6 | 0.0019 | |||
| 40 | 3.0 | 0.40 | 31.6 | 0.00036 | ||||
| 40 | 3.0 | 0.40 | 33.7 | 0.00026 | ||||
| 40 | 3.0 | 0.40 | 33.5 | 0.00015 | ||||
| 40 | 3.0 | 0.16 | 0.24 | 37.9 | 0.00056 | |||
| 25 | 0.5 | 0.72 | 1.17 | 46.0 | 0.0019 | |||
| 25 | 0.5 | 0.30 | 0.58 | 44.0 | 0.0016 | |||
| 25 | 0.5 | 0.30 | 0.58 | 43.7 | 0.0002 | |||
| 40 | 2.0 | 0.29 | 0.54 | 35.0 | 0.00085 | |||
| 40 | 3.0 | 0.37 | 29.1 | 0.00051 | ||||
| 40 | 3.0 | 0.40 | 30.9 | 0.00034 | ||||
| 40 | 3.0 | 0.09 | 23.6 | 0.00051 | ||||
| 25 | 0.5 | 0.27 | 0.52 | 43.5 | 0.0016 | |||
| 40 | 2.0 | 0.56 | 1.02 | 0.91 | 34.3 | 0.00034 | ||
| 40 | 2.0 | 0.82 | 1.46 | 38.4 | 0.0039 | |||
| 50 | 5.0 | 0.08 | 0.33 | 32.1 | 0.0011 | |||
| 40 | 1.0 | 0.18 | 0.00093 | |||||
| 2* | 9.0 | 0.0031 | ||||||
| 50 | 4.0 | 0.5 | 0.00087 | |||||
| 21 | 50 | 5.0 | 0.6 | 0.00058 |
VLC elution, % Petr. ether in EtOAc (*except for 19, where percentage indicates % MeOH in EtOAc)
VLC % CHCl3 in MeOH
Partition coefficients determined for CCC solvent systems:
hexanes-EtOAc-MeOH-H2O (HEMWat) 4-1-4-1
HEMWat 3-1-3-1
HEMWat 6-4-6-4
petr. ether-EtOAc-MeOH-H2O 5-5-6-4
HEMWat 5-5-5-5
column Supelco Discovery C18, 250×4.6 mm, 5mm particle size; flow 1.0 mL/min; gradient: A=0.1% HOAc; B=MeOH; 40%B from 0 to 2 min; linear gradient (1.5%/min) to 42 min; hold 100%B.
Figure 7.
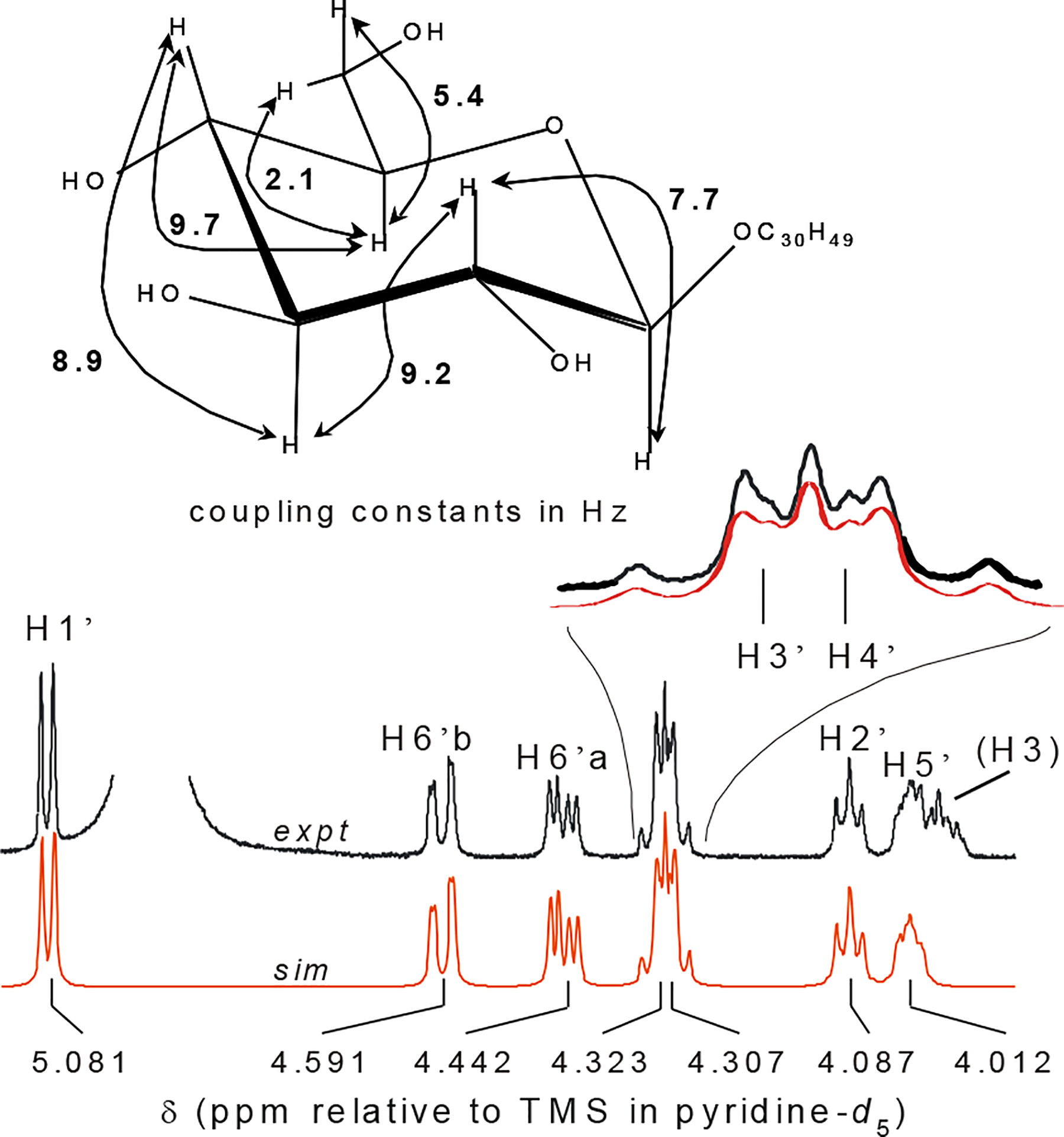
Higher-order spin system of the sugar moiety in sitosterol-3-O-β-glucopyranoside (19). The relative stereochemistry at the 3′, 4′, and 5′ positions, which was determined using spin simulation tools to elucidate the J pattern of these higher order signals, corresponded to an all trans-diaxial arrangement and the identification of glucose as the sugar moiety.
This is the first report on the chemical constituents of spent hops. The bitter acids and volatile oils present in hops, well known for the organoleptic properties that they impart to beer, are effectively separated from the chalcones and flavanones with CO2 extraction. The result is a potentially beneficial situation, whereby what is regarded as an agricultural waste product can essentially be seen for the purposes outlined in the introduction, as pre-purified starting material. After having tested all the isolated compounds individually for estrogenic activity, it can now be stated that the known estrogen (±)-8-prenylnaringenin (15; 8PN) is by far the most potent estrogen present in hops.8,10 This compound is an artifact formed from the chemical isomerization of 3′-prenylnaringenin chalcone, synonymous with desmethylxanthohumol (7; DMX).8,34 In addition, the flavanone isoxanthohumol (17; IX), is a much weaker estrogen that may be of significance due to its greater abundance. In terms of standardizing such a product for potential use in phytoestrogen therapy, 8PN is clearly an important biological marker, and the ratio of DMX/8PN could be used to measure chemical stability. All compounds with a chiral center were found to be optically inactive, indicating that they may be auto-oxidation and/or hydration products of XH or DMX formed by an SN1-type mechanism. Those with chiral centers on the prenyl unit can all be envisioned to arise from a theoretical prenyl-epoxide intermediate. In addition to the compounds detailed above, the known hop constituents α,β-dihydroxanthohumol (2),35 xanthohumol D (3),21 desmethylxanthohumol (7),8 5′-prenylxanthohumol (8),20 6,8-diprenylnaringenin (14), 8-prenylnaringenin (15),8,20,21,26,34,36,37 6-prenylnaringenin (16),8,20,21,26,34,37 isoxanthohumol (17),8,20,21,26,34,38,39 humulinone (20),40,41 and cohumulinone (21)40,41 were also isolated from spent hops and identified by comparison with literature data.
Experimental Section
General Experimental Procedures.
The X-ray data set for 1 was collected with an Enraf Nonius CAD4 diffractometer. Programs used: data collection EXPRESS (Nonius B.V., 1994), data reduction MolEN (K. Fair, Enraf-Nonius B.V., 1990), structure solution SHELXS-97 (G.M. Sheldrick, Acta Cryst. 1990, A46, 467–473), structure refinement SHELXL-97 (G.M. Sheldrick, Universität Göttingen, 1997), graphics SCHAKAL (E. Keller, Universität Freiburg, 1997). Nominal mass, exact mass, and LC-MS data were collected with Waters Micromass Q-Tof2 high resolution hybrid orthogonal angle-Time of Flight tandem LC mass spectrometer equipped with electrospray ionization (ESI) and featuring a quadrupole mass filter and collision cell for high resolution MS/MS analyses. The spectrometer was operated in negative ion mode. Tandem mass spectra were acquired at a collision energy of 25 eV using argon as the collision gas at a pressure of 2.0 × 10−5 mbar. 1H NMR and 13C NMR data were measured on Bruker Avance-500, Avance-360, and DPX-300 instruments, as indicated, with MeOH-d4 as a solvent and tetramethylsilane (TMS) as an internal standard. Spin system analyses were conducted with the PERCH NMR software (PERCH Solutions Ltd., Kuopio, Finland; www.perchsolutions.com) using total-line-shape mode. Offline FID processing and simulation experiments were conducted with the Nuts NMR Utility Transform Software (Acorn NMR, Inc.; www.acornnmr.com). Line resolution of experimental data was enhanced by Lorentz-Gauss (LG) transformation using individually determined parameters (WV mode). [α]D values were recorded on a Perkin Elmer 241 polarimeter at 20 °C. HPLC was carried out with a Waters Delta 600 system equipped with a Waters 996 photodiode array (PDA) detector, Waters 717 plus autosampler, and Millennium32 Chromatography Manager (Waters Corp.) using Supelco (Discovery C18, 250×4.6 mm, 5mm particle size), and a MeOH-H2O solvent system with 0.5% HOAc added to the aqueous phase. UV λmax values were determined from the PDA chromatograms. IR spectra were recorded as a neat film on a Ge ATR crystal with a JASCO FT/IR-410. Vacuum liquid chromatography (VLC) was performed using a modified version of the procedure described by Coll and Bowden,42 in that higher packing heights were used. Countercurrent chromatography was carried out using a J-type HSCCC instrument (Model CCC-1000; Pharma-Tech Research Corporation, Baltimore, MD) containing a self-balancing three-coil centrifuge rotor equipped with 125- or 320 mL columns (1/16 or 1/8″ ID Teflon tubing, respectively), solvent pump (Shimadzu LC-610), dual wavelength UV detector (Shimadzu SPD-10A UV) and a Foxy Jr. fraction collector (Isco, Inc.). Partition coefficients given in Table 3 were calculated using the marker method, where unretained components present in each injected fraction were defined as the marker (Kp≡0).43 To monitor the preparative separations, analytical thin layer chromatography (TLC) was performed at room temperature on precoated 0.25 mm thick silica gel 60 F254 aluminum plates (20 × 20 cm; Merck, Darmstadt, Germany). All TLC chromatograms shown in the Supplementary Data pages were scanned at 150 dpi. For a majority of the TLC chromatograms, experiments were carried out in duplicate: One copy was sprayed with the general purpose reagent p-anisaldehyde-H2SO4-HOAc (1:1:48); the other with a phenol-specific spray reagent FeCl3 (3% in dry EtOH).
Plant Material.
A sample of spent Nugget hop pellets (Humulus lupulus L. cv. Nugget; plant material remaining after supercritical CO2 extraction of palletized strobiles) was provided by Yakima Chief Inc., Sunnyside, WA (Lot # PE-MANU004).
Extraction and Isolation.
In order to investigate potential alternative sources of hop estrogens, an extract of spent hop pellets was prepared by maceration of the plant material (642 g) in MeOH (1.5 L) overnight, and removal of the now twice-spent plant material by gravity filtration. The filtrate from this first maceration was set aside, and the plant material was macerated a second time with MeOH (1.5 L) overnight. The combined filtrates were evaporated to a volume of ca. 800 mL and partitioned by adding H2O (200 mL) and extracting with petroleum ether (600 mL × 1; 400 mL × 2). A small portion of the crude MeOH extract was evaporated to dryness for subsequent bioassay and chemical analyses. The dried MeOH extract (E1) had a shiny black appearance with the consistency of hard taffy and had a faint, musty odor. The petroleum ether partition (P2), after removal of solvent by rotary evaporation was a thick, black liquid with an earthy aroma. The aqueous phase after petroleum ether extraction was subsequently extracted with CHCl3 (500 mL × 1; 400 mL × 2). After thorough removal of solvent and grinding in a mortal and pestle, the CHCl3 partition (P3) was a golden-orange powder, nearly odorless, freely soluble in MeOH, and only partially soluble in CHCl3.
The crude MeOH extract E1 and partitions P2 and P3 were tested for estrogenicity. In terms of Ishikawa EC50 values (determined as described by Liu et al.),44 the CHCl3 partition P3 was ten-fold more potent than E1 and 100-fold more potent than P2 and was therefore selected for bioassay-directed fractionation. The TLC, LC-UV, and LC-MS chromatograms all indicated that XH (1) was the major component of P3. In addition, it was clear from the TLC and LC analyses of P3 that several related chalcones and/or flavanones were also present. The partition P3 was first fractionated by VLC using a petr. ether → EtOAc gradient and all subfractions were combined into three pooled fractions, namely, F1 (least polar), F2 (moderately polar), and F3 (most polar) (see page S4 for TLC analysis). Each of the fractions F1-F3 was then fractionated again by VLC, but with a different solvent gradient (CHCl3 → MeOH) (pages S5–S7). Silica gel fractions were further fractionated by countercurrent chromatography and some isolates were further purified by HPLC. The chromatographic behavior of the isolates is summarized in Table 3.
X-ray crystal structure analysis of xanthohumol (1): formula C21H22O5 * ½ C4H8O2, M = 398.44, yellow-orange crystal 0.35 × 0.15 × 0.05 mm, a = 31.155(2), b = 15.541(1), c = 8.688(2) Å, = 90.42(1)°, V = 4206.4(10) Å3, ρcalc = 1.258 g cm−3, μ = 7.43 cm−1, empirical absorption correction via ψ scan data (0.781 T 0.964), Z = 8, monoclinic, space group P21/c (No. 14), λ= 1.54178 Å, T = 223 K, ω/2θ scans, 9177 reflections collected (h, +k, +l), [(sinθ)/λ] = 0.62 Å−1, 8577 independent (Rint = 0.023) and 4927 observed reflections [I ≥ 2 (I)], 538 refined parameters, R = 0.054, wR2 = 0.155, max. residual electron density 0.27 (−0.35) e Å−3, hydrogen atoms calculated and refined riding.
Xanthohumol
G (±)-{(2E)-1-[2,4-dihydroxy-3-(2,3-dihydroxy-3-methylbutyl)-6-methoxyphenyl]-3-(4-hydroxyphenyl)-2-propen-1-one} (4): yellow-orange powder, [α]D20 0° (0.3 mg/mL), UV (LC-PDA) λmax 369 nm, IR (film) 3270, 2957, 1616, 1336, 1228, 1168, 1141, 1106 cm−1, 1H NMR (360 MHz, MeOH-d4) see Table 2, HRESIMS [M-H]− 387.1451 m/z calcd for C21H24O7 (1.9 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 267 (100) B-fragment 119 (70) other product ions 311, 164.
Xanthohumol H
{(2E)-1-[2,4-dihydroxy-3-(3-hydroxy-3-methylbutyl)-6-methoxyphenyl]-3-(4-hydroxyphenyl)-2-propen-1-one} (5): yellow-orange solid, UV (LC-PDA) λmax 373 nm, IR (film) 3300, 2964, 2923, 1611, 1229, 1168, 1137, 1102 cm−1, 1H NMR, see Table 1, HRESIMS [M-H]− m/z 371.1495 calcd for C21H24O6 (3.8 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 251 (80) B-fragment 119 (50) other fragment 297.
Trans-hydroxyxanthohumol
{1-[2,4-dihydroxy-3-(4-hydroxy-3-methyl-2-butenyl)-6-methoxyphenyl] −3-(4-hydroxyphenyl)-2-propen-1-one} (6): yellow-orange solid, UV (LC-PDA) λmax 370 nm, IR (film) 3300, 2917, 2846, 1605, 1227, 1166, 1140, 1101 cm−1 , NMR (500 MHz, MeOH-d4), see Table 2, HRESIMS [M-H]− 369.1350 m/z calcd for C21H22O6 (3.2 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 249 (50) B-fragment 119 (100) other product ions 295, 219, 181.
1″,2″-Dihydroxanthohumol
C {(2E)- 1-(3,4-dihydro-5-hydroxy-7-methoxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxyphenyl)-2-propen-1-one} (9b): orange solid, UV (LC-PDA) λmax 370 nm, NMR (360 MHz, MeOH-d4), see Table 2, HRESIMS [M-H]− 353.1158 m/z calcd for C21H22O5 (4.5 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 233 (2) B-fragment 119 (100).
Desmethylxanthohumol
B (DMX-B) (±)-{(2E)-1-(3,4-dihydro-3,5,7-trihydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxyphenyl)- 2-propen-1-one} (11): yellow-orange solid, [α]D20 0° (0.5 mg/mL), UV (LC-PDA) λmax 372 nm, IR (film) 3300, 2976, 2923, 1605, 1507, 1348, 1226, 1166, 1132, 830 cm−1 , NMR (360 MHz, MeOH-d4), see Table 2, HRESIMS [M-H]− 355.1188 m/z calcd for C20H20O6 (1.8 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 235 (8), B-fragment 119 (40), other product ions 297, 283, 163.
Desmethylxanthohumol
J (DMX-J) {(2E)-1-[2,3-dihydro-4,6-dihydroxy-2-(1-hydroxy-1-methylethyl)-7-benzofuranyl]-3-(4-hydroxyphenyl)-2-propen-1-one} (12): yellow-orange solid, [α]D20 0° (0.2 mg/mL), UV (LC-PDA) λmax 370 nm, IR (film) 3305, 2970, 2923, 1605, 1353, 1225, 1166, 1026, 982, 832 cm−1, 1H NMR (360 MHz, MeOH-d4) δ 8.06 (1H, d, 15.5, Hα) 7.73 (1H, d*, 15.5, Hβ), 7.54 (2H, m*, H2,6), 6.81 (2H, m*, H3,5), 5.83 (1H, s, H5’), 4.83 (1H, dd, 9.6, 8.7, H2″), 3.04 (1H, dd, 16.0, 8.7, H1″a), 3.03 (1H, dd, 16.0, 9.6, H1″b) 1.41 (3H, s, Me-4″), 1.30 (3H, s, Me-5″); * = see Figure 5 for multiplicity assignments in the chalcone B-ring, HRESIMS [M-H]− 355.1201 m/z calcd for C20H20O6 (5.4 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 235 (65) B-fragment m/z (80) other product ions m/z 297, 177.
Xanthohumol
I (XH-I) {(2E)-1-[2,3-dihydro-4-hydroxy-2-(1-hydroxy-1-methylethyl)-6-methoxy-5-benzofuranyl]-3-(4-hydroxyphenyl)-2-propen-1-one} (13): orange powder, [α]D20 0° (0.3 mg/mL), UV (LC-PDA) λmax 346 nm, IR (film) 3260, 2979, 2929, 1605, 1290, 1217, 1169, 1145, 1101 cm−1, HRESIMS [M-H]− 369.1359 m/z calcd for C21H22O6 (5.6 ppm) ESIMSMS product ions m/z (% base peak) A-fragment 249 (2) B-fragment 119 (100) other product ions 191, 311.
Supplementary Material
Acknowledgment.
The authors gratefully acknowledge Mr. Jim Boyd from Yakima Chief, Inc. (Sunnyside, WA, www.yakimachief.com) for providing the plant material used in this investigation. We also thank the National Center for Complementary and Alternative Medicine (NCCAM; nccam.nih.gov), National Institutes of Health (NIH), the National Institute of General Medical Sciences, and the U.S. Department of Health and Human Services (grant P50 AT00155), for funding this work. We are indebted to Dr. Matthias Niemitz of the University of Kuopio for his valuable PERCH support.
Footnotes
Supplementary Files
Accompanying this manuscript are the following files:
Pauli_hops_revision2.doc = digital version of this manuscript
Pauli_supplementary_data_v2.pdf = supplementary data; processed spectral and preparative chromatographic data (intended as supplementary information to be available through the Journal’s website.
References and Notes
- (1).Burgess A Hops: Botany, Cultivation and Utilization; Leonard Hill: London, 1964. [Google Scholar]
- (2).Neve RA Hops; Chapman and Hall: New York, 1991. [Google Scholar]
- (3).Wohlfart R; Hänsel R; Schmidt H Planta Med. 1983, 48, 120–123. [PubMed] [Google Scholar]
- (4).Fussel A; Wolf A; Brattstrom A Eur. J. Med. Res 2000, 5, 385–390. [PubMed] [Google Scholar]
- (5).Schulz V; Hänsel R; Tyler VE In Rational Phytotherapy; 4th ed.; Springer-Verlag: Berlin, Germany, 2001; pp 98–99. [Google Scholar]
- (6).Koch W; Heim G Estrogens in hops and beer 1953, 95, 845–845. [PubMed] [Google Scholar]
- (7).Nastainczyk W, Untersuchung über die östrogene Wirkung des Hopfens und des Bieres, Ph.D. Dissertation, Universität Saarbrücken, 1972. [Google Scholar]
- (8).Hänsel RV; Schulz J Arch. Pharm. Weinheim 1988, 321, 37–40. [Google Scholar]
- (9).De Keukeleire D; Milligan SR; De Cooman L; Heyerick A Pharm. Pharmacol. Lett 1997, 7, 83–86. [Google Scholar]
- (10).Milligan SR; Kalita JC; Heyerick A; Rong H; De Cooman L; De Keukeleire DJ Clin. Endocrinol. Metab 1999, 84, 2249–2252. [DOI] [PubMed] [Google Scholar]
- (11).Chadwick LR; Pauli GF; Farnsworth NR Phytomedicine 2004, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miranda CL; Stevens JF; Helmrich A; Henderson MC; Rodriguez RJ; Yang YH; Deinzer ML; Barnes DW; Buhler DR Food Chem. Toxicol 1999, 37, 271–285. [DOI] [PubMed] [Google Scholar]
- (13).Miranda CL; Aponso GLM; Stevens JF; Deinzer ML; Buhler DR Cancer Lett. 2000, 149, 21–29. [DOI] [PubMed] [Google Scholar]
- (14).Gerhäuser C; Alt A; Klimo K; Heiss E; Gamal-Eldeen A; Neumann I; Knauft J; Scherf H; Frank N; Bartsch H; Becker H Proc. Am. Assoc. Cancer Res 2001, 42, 18. [Google Scholar]
- (15).Holinka C; Hata H; Kuramoto H; Gurpide E Cancer Res. 1986, 46, 2771–2774. [PubMed] [Google Scholar]
- (16).Wagner H; Bladt S Plant drug analysis: a thin layer chromatography atlas; 2nd ed.; Springer: Berlin, 1996. [Google Scholar]
- (17).Farnsworth NR; Ed. NAPRALERT®: A Database of World Literature on Natural Products Available through STN or directly through the College of Pharmacy, University of Illinois at Chicago [Google Scholar]
- (18).Günther H NMR Spectroscopy; 2nd ed.; John Wiley & Sons Ltd.: West Sussex, England, 1998. [Google Scholar]
- (19).Tabata N; Ito M; Tomoda H; Omura S Phytochemistry 1997, 46, 683–687. [DOI] [PubMed] [Google Scholar]
- (20).Stevens JF; Ivancic M; Hsu V; Deinzer M Phytochemistry 1997, 44, 1575–1585. [Google Scholar]
- (21).Stevens JF; Taylor AW; Nickerson GB; Ivancic M; Henning J; Haunold A; Deinzer ML Phytochemistry 2000, 53, 759–775. [DOI] [PubMed] [Google Scholar]
- (22).Chadwick LR, Estrogens and congeners from spent hops, Ph.D. dissertation, University of Illinois at Chicago, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nookandeh A; Frank N; Steiner F; Ellinger R; Schneider B; Gerhauser C; Becker H Phytochemistry 2004, 65, 561–570. [DOI] [PubMed] [Google Scholar]
- (24).Nikolic D; Li Y; Chadwick LR; van Breemen RB Metabolism of xanthohumol and isoxanthohumol, prenylated flavonoids from hops (Humulus lupulus L.), by human liver microsomes. 52nd ASMS Conference on Mass Spectrometry and Allied Topics, Nashville, Tennessee May 23–27, 2004. [DOI] [PubMed] [Google Scholar]
- (25).Reynolds WF; Enriquez RG J. Nat. Prod 2002, 65, 221–244. [DOI] [PubMed] [Google Scholar]
- (26).Stevens JF; Miranda CL; Buhler DR; Deinzer ML J. Am. Soc. Brew. Chem 1998, 56, 136–145. [Google Scholar]
- (27).Stevens JF; Taylor AW; Clawson JE; Deinzer ML J. Agric. Food Chem 1999, 47, 2421–2428. [DOI] [PubMed] [Google Scholar]
- (28).Seo EK; Silva GL; Chai HB; Chagwedera TE; Farnsworth NR; Cordell GA; Pezzuto JM; Kinghorn AD Phytochemistry 1997, 45, 509–515. [DOI] [PubMed] [Google Scholar]
- (29).Herath WH; Ferreira D; Khan IA Phytochemistry 2003, 62, 673–677. [DOI] [PubMed] [Google Scholar]
- (30).Kojima H; Sato N; Hatano A; Ogura H Phytochemistry 1990, 29, 2351–2355. [Google Scholar]
- (31).Agrawal PK Phytochemistry 1992, 31, 3307–3330. [DOI] [PubMed] [Google Scholar]
- (32).Pauli GF J. Nat. Prod 1995, 58, 483–494. [DOI] [PubMed] [Google Scholar]
- (33).Faizi S; Ali M; Saleem R; Irfanullah; Bibi S Mag. Res. Chem 2001, 39, 399–405. [Google Scholar]
- (34).Stevens JF; Taylor AW; Deinzer ML J. Chromatogr. A 1999, 832, 97–107. [DOI] [PubMed] [Google Scholar]
- (35).Etteldorf N; Etteldorf N; Becker HZ Naturforsch. C (J. Biosci.) 1999, 54, 610–612. [Google Scholar]
- (36).Tahara S; Katagiri Y; Ingham JL; Mizutani J Phytochemistry 1994, 36, 1261–1271. [DOI] [PubMed] [Google Scholar]
- (37).Mizobuchi S; Sato Y Agric. Biol. Chem 1994, 48, 2771–2775. [Google Scholar]
- (38).Sun S; Watanabe S; Saito T Phytochemistry 1989, 28, 1776–1777. [Google Scholar]
- (39).Sun S; Watanabe S; Saito T Kawasaki Med. J 1990, 16, 117–125. [Google Scholar]
- (40).Shoolery JN; Verzele M; Alderweireldt F Tetrahedron 1960, 9, 271–274. [Google Scholar]
- (41).Verzele M; de Keukeleire D Chemistry and Analysis of Hop and Beer Bitter Acids; Elsevier: Amsterdam, 1991; Vol. 27 in Developments in Food Science. [Google Scholar]
- (42).Coll JC; Bowden BF J. Nat. Prod 1989, 49, 934–936. [Google Scholar]
- (43).Ito Y; Conway WD, Eds. High-Speed Countercurrent Chromatography; Wiley: New York, N. Y., 1996; Vol. 132 in Chemical Analysis: A series of monographs on analytical chemistry and its applications. [Google Scholar]
- (44).Liu J; Burdette JE; Xu H; Gu C; van Breemen RB; Bhat KP; Booth N; Constantinou AI; Pezzuto JM; Fong HH; Farnsworth NR; Bolton JL J. Agric. Food. Chem 2001, 49, 2472–2479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.