

Summary
Third-generation sequencing technologies from companies such as Oxford Nanopore and Pacific Biosciences have paved the way for building more contiguous and potentially gap-free assemblies. The larger effective length of their reads has provided a means to overcome the challenges of short to mid-range repeats. Currently, accurate long read assemblers are computationally expensive, whereas faster methods are not as accurate. Moreover, despite recent advances in third-generation sequencing, researchers still tend to generate accurate short reads for many of the analysis tasks. Here, we present HASLR, a hybrid assembler that uses error-prone long reads together with high-quality short reads to efficiently generate accurate genome assemblies. Our experiments show that HASLR is not only the fastest assembler but also the one with the lowest number of misassemblies on most of the samples, while being on par with other assemblers in terms of contiguity and accuracy.
Subject Areas: Genomics, Bioinformatics, Sequence Analysis
Graphical Abstract
Highlights
-
•
We introduce HASLR, a fast tool for hybrid assembly of short reads and long reads
-
•
HASLR proposes a new data structure called backbone graph
-
•
The backbone graph provides a large-scale map of the whole genome
-
•
Our experiments demonstrate that HASLR generates low number of misassemblies
Genomics; Bioinformatics; Sequence Analysis
Introduction
Long reads (LRs) generated by third-generation sequencing (TGS) technologies such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) have revolutionized the landscape of de novo genome assembly. Although LRs have a higher error rate compared with short reads (SRs) generated by next-generation sequencing (NGS) technologies such as Illumina, they have been shown to result in accurate assemblies given sufficient coverage. Indeed, the length of TGS LRs enables the resolution of many short and mid-range repeats that are problematic when assembling genomes from SRs. Recent advances in sequencing ultra-long ONT reads have moved us closer to the complete reconstruction of entire genomes (including difficult-to-assemble regions such as centromeres and telomeres) than ever before (Miga et al., 2019). Similarly, HiFi PacBio reads have been shown to be capable of improving the contiguity and accuracy in complex regions of the human genome (Vollger et al., 2019). These advances toward more accurate and complete genome assembly could not be achieved without the recent development of assemblers specifically tailored for LRs. These tools assemble LRs either after an error correction step (Koren et al., 2017; Chin et al., 2016) or directly without any prior error correction (Li, 2016; Ruan and Li, 2019; Kolmogorov et al., 2019).
Although LRs are becoming more widely used for de novo genome assembly, using hybrid approaches (that utilize a complementary SR dataset) is still popular for several reasons: (1) SRs have higher accuracy and can be generated by Illumina sequencers at a high throughput for a lower cost; (2) plenty of SR datasets are already publicly available for many genomes; (3) for some basic tasks such as variant calling (SNV and short indel detection), SRs still provide better resolution owing to their high accuracy, which often motivates researchers to generate SRs even when LRs are in hand; and (4) unlike PacBio assemblies whose accuracy increases with the depth of coverage thanks to their unbiased random error model (Myers, 2014), constructing reference quality genomes solely from ONT reads remains challenging owing to biases in base calling, even with a high coverage (Koren et al., 2017; Antipov et al., 2015). As a result, hybrid assembly approaches are still useful (Jaworski et al., 2019; Jiang et al., 2019; Kadobianskyi et al., 2019).
Hybrid approaches for de novo genome assembly can be classified into three groups: (1) methods that first correct raw LRs using SRs and then build contigs using corrected LRs only (e.g., PBcR [Koren et al., 2012] and MaSuRCA [Zimin et al., 2017]). In recent years, many tools have been proposed for hybrid error correction of long reads that can be used toward this goal (see Salmela and Rivals (2014), Haghshenas et al. (2016), and Wang et al. (2018) for examples of such tools); (2) methods that first assemble raw LRs and then correct/polish the resulting draft assembly with SRs using polishing tools such as Pilon (Walker et al., 2014) and Racon (Vaser et al., 2017); and (3) methods that first assemble SRs and then utilize LRs to generate longer contigs (e.g., hybridSPAdes [Antipov et al., 2015], Unicycler [Wick et al., 2017], DBG2OLC [Ye et al., 2016], and Wengan [Di Genova et al., 2019]).
PBcR and MaSuRCA correct LRs using their internal correction algorithm and then employ CABOG (Miller et al., 2008) (Celera Assembler with the Best Overlap Graph) for assembling corrected LRs. hybridSPAdes and Unicycler are similar in design. Both of these tools first use SPAdes (Bankevich et al., 2012), which takes SRs as input and generates an assembly graph, a data structure in which multiple copies of a genome segment are collapsed into a single contig (see Zerbino and Birney (2008) for more details). This data structure also records connections between subsequent contigs such that every region of the genome corresponds to a path in the graph. hybridSPAdes and Unicycler then align LRs to this assembly graph in order to resolve ambiguities and generate longer contigs. On the other hand, DBG2OLC first assembles contigs from SRs and maps them onto raw LRs to get a compressed representation of LRs based on SR contig identifiers and then applies an overlap-layout-consensus (OLC) approach on these compressed LRs to assemble the genome. Since compressed LRs are much shorter compared to raw LRs, building an overlap graph from them is quicker than building it from raw LRs, owing to the faster pairwise alignment. Finally, the more recent tool, Wengan, assembles short reads and then builds multiple synthetic paired-read libraries of different insert sizes from LR sequences. These synthetic paired reads are then aligned to short read contigs and a scaffolding graph is built from the resulting alignments. In the end, the final assembly is generated by traversing proper paths of the scaffolding graph.
Among the above tools, hybridSPAdes and Unicycler have been designed specifically for bacterial and small eukaryotic genomes and do not scale for the assembly of large genomes. PBcR, MaSuRCA, DBG2OLC, and Wengan are the only hybrid assemblers that are capable of assembling large genomes, such as the human genome. However, for mammalian genomes, PBcR and MaSuRCA require a large computational time and cannot be used without a computing cluster. DBG2OLC is faster owing to its use of compressed LRs. Wengan is also a fast assembler and can be used for assembling large genomes in a reasonable time.
In this paper, we introduce HASLR, a fast hybrid assembler that is capable of assembling large genomes. Based on our results, HASLR is the fastest between all the assemblers we tested, while generating the lowest number of mis-assemblies on most datasets. Furthermore, it generates assemblies that are comparable with the best performing tools in terms of contiguity and accuracy. HASLR is also capable of assembling large genomes using less time and memory than other tools.
Results
HASLR's Overview
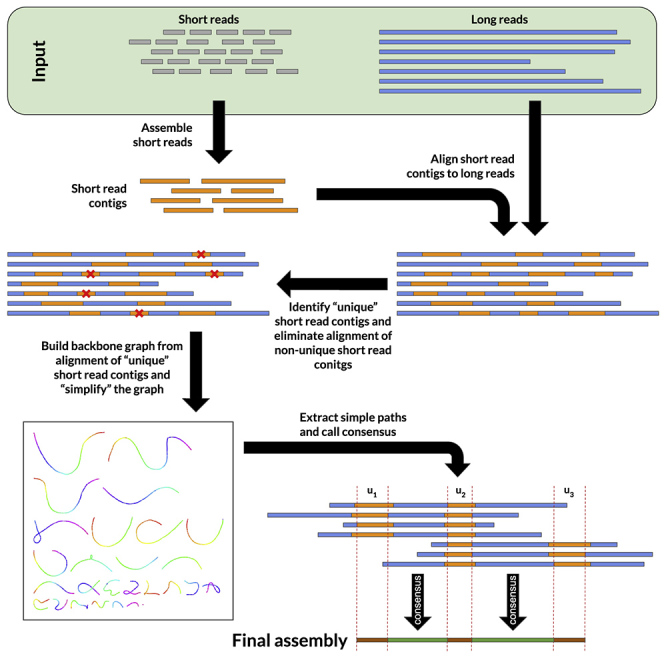
Here, we present an overview of HASLR. See Transparent Methods Section in the Supplemental Information for more detailed description of HASLR. The input to HASLR is a set of long reads (LRs) and a set of short reads (SRs) from the same sample, together with an estimation of the genome size. HASLR performs the assembly using a novel approach that rapidly assembles the genome without performing all-versus-all LR alignments. HASLR, similar to hybridSPAdes, Unicycler, and Wengan, builds SR contigs using a fast SR assembler (i.e., Minia). Then, it uses LRs to put SR contigs in the order of their expected appearance in the genome. This is done by building a novel data structure called backbone graph that models the connections between SR contigs based on their alignments onto LRs. Note that the backbone graph is built only using “unique” SR contigs, those SR contigs that are likely to appear in the genome only once. This is because repetitive SR contigs will cause branching in the backbone graph (see Figure 1 for the backbone graphs built using Yeast dataset utilizing unique versus all SR contigs). Next, the backbone graph is simplified to reduce the effect of wrong SR contig to LR alignments. Finally, a consensus sequence is calculated for each edge that fills the gap between its neighboring SR contigs. The final assembly is generated using all SR contigs and consensus sequences in the simplified backbone graph.
Figure 1.

Two Backbone Graphs Built from a Real PacBio Dataset Sequenced from a Yeast Genome
Each graph is visualized with Bandage (Wick et al., 2015) and colored using its rainbow coloring feature. Each chromosome is colored with a full rainbow spectrum. (Left) The backbone graph built from all SR contigs. (Right) The backbone graph built from unique SR contigs. As it can be seen, using only unique SR contigs for building the backbone graph resolves many of the complexities and ambiguities in the graph. However, it is important to note that excluding non-unique SR contigs could potentially result in a more fragmented graph (some chromosomes are split into multiple paths rather than a single one) and assembly.
It is important to note that the backbone graph is not an assembly graph per se, for two reasons. First, the regions between each pair of connected unique SR contigs are not present in the graph. These missing regions are obtained by calculating the consensus of LR subsequences between each pair of unique SR contigs. Second, unlike assembly graphs, there are some segments of the genome that cannot be translated to a path in the backbone graph. This is due to the potential fragmentation that was mentioned earlier.
Identification of Unique Short Read Contigs
In order to measure the efficacy of our approach for identifying unique SR contigs (see Transparent Methods Section in the Supplemental Information for more details), we conducted a set of experiments as follows. First, we simulated an SR dataset based on six different reference genomes: E. coli, Yeast, C. elegans, A. thaliana, D. melanogaster, and GRCh38 human reference genome. For each genome, we used ART (Huang et al., 2011) to simulate 50× coverage short Illumina reads (2 × 100 bp long, 500 bp insert size mean, and 50 bp insert size deviation) using the Illumina HiSeq 2000 error model. Next, we used Minia to assemble the simulated short reads using k-mer size 49. Finally, to form the ground truth for copy count of each SR contig, we mapped the assembled SR contigs to the reference genome using minimap2 (Li, 2018).
For identification of unique SR contigs, we use the notion of mean k-mer frequency of SR contigs as follows. We calculate the mean and standard deviation of k-mer frequency of 30 longest contigs (favg, fstd). At the end, every SR contig whose mean k-mer frequency is below favg+3fstd is considered as unique contig.
Here, we report the precision and recall of the above-mentioned approach in identifying unique SR contigs. For each dataset, we evaluate the performance of our approach in identifying unique SR contigs that are longer than a threshold. The “length threshold” that is used to discard small contigs in this experiment changes from 100 to 1,000 with a step size of 100.
As it can be seen in Figure 2, the precision of the identified unique SR contigs is always high regardless of the “length threshold.” In addition, in all the experiments a big jump in recall is observed at “length threshold” of 300. The results of this experiment show that the proposed approach for identifying unique SR contigs performs well with high precision and recall.
Figure 2.

Precision and Recall Results in Identification of Unique Short Read Contigs on Six Different Reference Genomes
Precision is shown with blue dots and recall is shown with orange dots. Precision is always high across the different experiments, and in all the experiments a big jump in recall happens at length threshold of 300.
Experimental Setup
We evaluated the performance of HASLR on both simulated and real datasets. We selected five hybrid assemblers: hybridSPAdes (Antipov et al., 2015), Unicycler (Wick et al., 2017), DBG2OLC (Ye et al., 2016), MaSuRCA (Zimin et al., 2017), Wengan (Di Genova et al., 2019); four long read methods: Canu (Koren et al., 2017), Flye (Kolmogorov et al., 2019), wtdbg2 (Ruan and Li, 2019), miniasm (Li, 2016); and two short read methods: Minia (Chikhi and Rizk, 2013), SPAdes (Bankevich et al., 2012). All experiments were performed on isolated nodes of a cluster (i.e., no other simultaneous jobs were allowed on each node). Each node runs CentOS 7 and is equipped with 32 cores (2 threads per core; total of 64 CPUs) Intel(R) Xeon(R) processors (Gold 6130 @ 2.10 GHz) and 720 GB of memory. Each tool was run with their recommended settings. See Table S1 and Supplemental Information for more details about the versions of tools and the employed commands. Note that, for wtdbg2, we used the provided wtdbg2.pl wrapper, which automatically performs a polishing step using the embedded polishing module.
For each experiment, assemblies were evaluated by comparing against their corresponding reference genome using QUAST (Mikheenko et al., 2018). QUAST reports on a wide range of assembly statistics, but we are mostly interested in misassemblies, NGA50, and rate of small errors (mismatch or indel). QUAST detects and reports misassemblies when a contig cannot align to the reference genome as a single continuous piece. Misassemblies indicate structural assembly errors. For computing NGA50, unlike N50 and NG50, only segments of assembled contigs that are aligned to the reference genome are considered. In addition, QUAST breaks contigs with extensive misassemblies before calculation of NGA50. Therefore, NGA50 is a good indicator of the contiguity of the assembly, while taking misassemblies into consideration.
Experiment on Simulated Dataset
We evaluated all the selected methods on four simulated datasets, namely, E. coli, yeast, C. elegans, and human, to provide a wide range of genome sizes and complexity. For each genome, we used ART (Huang et al., 2011) to simulate 50× coverage short Illumina reads (2 × 150 bp long, 500 bp insert size mean, and 50 bp insert size deviation) using the Illumina HiSeq 2000 error model. We also simulated 50× coverage long PacBio reads using PBSIM (Ono et al., 2012). In order to capture the characteristics of real datasets, a set of PacBio reads generated from a human genome (see Supplemental Information for details) with P6-C4 chemistry was passed to PBSIM via option --sample-fastq. This enables PBSIM to sample the read length and error model from the real long reads.
Table 1 shows the QUAST metrics calculated for assemblies generated by different tools. As it can be seen, HASLR generates assemblies with the lowest number of misassemblies in all datasets. It is important to note that since reads are simulated from the same reference used for this assessment, any misassembly reported by QUAST is indeed a structural assembly mistake. In terms of the contiguity, HASLR achieves NGA50 on par with other tools for all datasets except for C. elegans where Canu shows an NGA50 twice larger than others tools. On the human dataset, HASLR generates the most contiguous assembly with an NGA50 of 17.03 Mb and only two extensive misassemblies, although at the price of a lower genome fraction (see Discussion). In addition, HASLR is the fastest assembler across the board. wtdbg2 has a comparable speed but generates lower quality assemblies, both in terms of misassemblies and mismatch/indel rate.
Table 1.
Comparison between Draft Assemblies Obtained by Different Tools on Simulated Data
| Genome | Assembler | Contigs | Genome Fraction | NGA50 | Misassemblies Extensive + Local | Mismatch Rate | Indel Rate | Time | Memory (GB) |
|---|---|---|---|---|---|---|---|---|---|
| E. coli | Canu | 1 | 99.648 | 4,625,313 | 0 + 0 | 0.86 | 15.85 | 30:18 | 4.16 |
| Flye | 1 | 99.937 | 4,639,833 | 0 + 0 | 0.34 | 25.31 | 5:59 | 12.10 | |
| wtdbg2 | 135 | 96.158 | 107,864 | 4 + 79 | 216.99 | 492.12 | 0:46 | 19.36 | |
| miniasm | 4 | 99.470 | 4,178,447 | 0 + 1 | 52.24 | 646.11 | 0:41 | 2.56 | |
| Minia | 162 | 97.713 | 58,763 | 0 + 0 | 0.26 | 0.00 | 0:26 | 3.04 | |
| SPAdes | 79 | 98.333 | 176,163 | 1 + 2 | 1.69 | 0.11 | 6:56 | 113.92 | |
| hybridSPAdes | 1 | 100.000 | 4,641,652 | 0 + 0 | 6.18 | 0.32 | 8:05 | 113.92 | |
| Unicycler | 1 | 99.997 | 4,641,530 | 0 + 0 | 3.12 | 0.45 | 18:43 | 21.56 | |
| DBG2OLC | 2 | 92.497 | 2,647,379 | 0 + 0 | 0.28 | 30.05 | 4:37 | 1.35 | |
| MaSuRCA | 1 | 99.874 | 4,636,209 | 0 + 4 | 0.56 | 0.19 | 5:21 | 32.52 | |
| Wengan | 1 | 100.000 | 4,641,731 | 0 + 0 | 2.54 | 5.36 | 2:21 | 3.19 | |
| HASLR | 1 | 99.999 | 4,643,699 | 0 + 0 | 2.00 | 42.89 | 0:41 | 3.04 | |
| Yeast | Canu | 21 | 98.831 | 910,628 | 0 + 0 | 3.18 | 25.44 | 44:10 | 5.51 |
| Flye | 19 | 99.418 | 916,686 | 6 + 1 | 11.37 | 49.72 | 9:03 | 19.65 | |
| wtdbg2 | 490 | 92.871 | 77,726 | 24 + 191 | 259.00 | 577.63 | 1:58 | 28.35 | |
| miniasm | 18 | 96.637 | 776,254 | 0 + 0 | 54.28 | 709.35 | 1:49 | 6.63 | |
| Minia | 608 | 94.104 | 39,673 | 0 + 0 | 0.46 | 0.04 | 1:03 | 5.05 | |
| SPAdes | 211 | 95.231 | 151,550 | 0 + 0 | 5.62 | 0.69 | 16:16 | 113.93 | |
| hybridSPAdes | 38 | 97.840 | 797,316 | 2 + 12 | 41.54 | 2.12 | 19:41 | 113.93 | |
| Unicycler | 52 | 97.893 | 799,601 | 0 + 1 | 8.81 | 0.44 | 57:47 | 22.99 | |
| DBG2OLC | 18 | 98.492 | 771,063 | 1 + 0 | 5.9 | 85.95 | 13:29 | 1.21 | |
| MaSuRCA | 17 | 99.476 | 919,651 | 0 + 3 | 5.97 | 0.56 | 15:10 | 32.66 | |
| Wengan | 22 | 97.065 | 796,244 | 0 + 0 | 6.14 | 24.48 | 4:14 | 5.55 | |
| HASLR | 18 | 96.597 | 796,649 | 0 + 0 | 5.39 | 76.63 | 1:52 | 10.48 | |
| C. elegans | Canu | 10 | 99.847 | 13,775,238 | 3 + 1 | 5.88 | 67.73 | 5:15:05 | 13.76 |
| Flye | 16 | 99.798 | 15,266,425 | 8 + 0 | 1.10 | 55.35 | 1:01:26 | 89.50 | |
| wtdbg2 | 4,487 | 95.468 | 81,074 | 194 + 506 | 246.33 | 657.89 | 15:57 | 29.45 | |
| miniasm | 37 | 99.696 | 7,468,924 | 3 + 7 | 68.24 | 864.11 | 20:37 | 19.35 | |
| Minia | 13,546 | 86.788 | 10,047 | 13 + 4 | 0.76 | 0.11 | 6:18 | 8.36 | |
| SPAdes | 3,219 | 94.713 | 58,307 | 30 + 62 | 6.42 | 1.36 | 2:45:34 | 114.80 | |
| hybridSPAdes | 340 | 98.643 | 924,797 | 67 + 197 | 73.26 | 9.14 | 3:11:50 | 114.79 | |
| Unicycler | NA | ||||||||
| DBG2OLC | 16 | 99.692 | 6,732,354 | 10 + 7 | 8.55 | 174.21 | 2:04:23 | 7.99 | |
| MaSuRCA | 18 | 99.609 | 4,614,507 | 34 + 123 | 14.89 | 4.56 | 2:07:41 | 33.76 | |
| Wengan | 46 | 98.917 | 2,042,350 | 53 + 20 | 7.26 | 59.81 | 28:21 | 11.18 | |
| HASLR | 25 | 99.182 | 6,455,832 | 0 + 0 | 14.74 | 230.58 | 10:45 | 22.42 | |
| Human | Canu | 1,461 | 97.279 | 15,045,226 | 854 + 99 | 37.7 | 196.78 | 562:14:04 | 58.72 |
| Flye | NA | ||||||||
| wtdbg2 | 122,438 | 92.735 | 87,595 | 3,436 + 13,041 | 224.02 | 598.87 | 10:25:19 | 190.07 | |
| miniasm | 2,528 | 97.170 | 10,294,834 | 374 + 181 | 71.56 | 775.18 | 110:33:23 | 511.16 | |
| Minia | 593,601 | 80.704 | 4,537 | 1,016 + 16 | 1.55 | 0.13 | 3:29:08 | 8.91 | |
| SPAdes | NA | ||||||||
| hybridSPAdes | NA | ||||||||
| Unicycler | NA | ||||||||
| DBG2OLC | 1,906 | 91.013 | 14,385,033 | 221 + 246 | 8.43 | 201.56 | 81:18:15 | 69.53 | |
| MaSuRCA | NA | ||||||||
| Wengan | 1,776 | 94.617 | 11,216,374 | 185 + 70 | 3.84 | 33.5 | 20:12:12 | 38.08 | |
| HASLR | 897 | 91.213 | 17,025,446 | 2 + 5 | 11.32 | 207.88 | 6:06:43 | 58.55 |
Note: Mismatch and indel rates are reported per 100 kbp. Unicycler crashed on C. elegans dataset due to maximum recursion limit. For the human dataset, Flye, SPAdes, hybridSPAdes, and Unicycler failed due to memory limit and MaSuRCA failed due to a segmentation fault.
It is particularly interesting to compare HASLR with hybridSPAdes, Unicycler, and Wengan, since they share similar design in that they connect short read contigs rather than explicitly assembling long reads. In addition, Wengan uses short read contigs generated by Minia, similar to HASLR. hybridSPAdes and Unicycler do not scale for large genomes as they have been designed for small and bacterial genomes. On C. elegans dataset, HASLR gives significantly more contiguous assembly than hybridSPAdes and Wengan without any structural assembly error. For the human dataset, HASLR has a higher NGA50 while generating significantly less misassemblies.
Note that, HASLR does not employ any polishing step either internally or externally. Thus, the indel rate of the draft assemblies generated by HASLR is less than desirable. Since SR contigs generated by Minia do not contain many indels, it is expected that most of these indels are within the consensus sequence calculated by partial order alignment. However, these types of local assembly errors can be easily addressed through a polishing step as shown in Table S4. With a single round of polishing, both indel and mismatches rates match the other tools in two datasets.
Experiment on Real Dataset
To compare the performance of HASLR on real data with other tools, we tested them on four publicly available datasets, E. coli, yeast, C. elegans, and human. Table 2 contains details about these real datasets. Similar to simulated datasets, on real dataset HASLR generates less misassembly compared to other assemblers while remaining the fastest (see Table 3). Compared with other hybrid assemblers, HASLR performs similar or better in terms of contiguity, whereas it stands behind self-assembly tools with a lower NGA50.
Table 2.
Statistics of Real Long Read Datasets
| Dataset | Technology | N50 Length | Estimated Coverage | Total Size (Gb) | Aligned Size (Gb) | Avg. Alignment Identity (%) |
|---|---|---|---|---|---|---|
| E. coli | ONT R9.4 | 63,747 | 1,080 | 5.01 | 4.31 | 85.03 |
| (K-12 MG1655) | Illumina | 2 × 151 | 372 | 1.73 | – | – |
| Yeast | PacBio | 8,561 | 132 | 1.61 | 1.42 | 86.90 |
| (S288C) | Illumina | 2 × 150 | 82 | 1.00 | – | – |
| C. elegans | PacBio | 16,675 | 47 | 4.73 | 4.32 | 87.43 |
| (Bristol) | Illumina | 2 × 100 | 67 | 6.76 | – | – |
| Human | PacBio | 19,960 | 59 | 182.51 | 163.51 | 85.85 |
| (CHM1) | Illumina | 2 × 151 | 41 | 127.76 | – | – |
Note: Alignment statistics were obtained by aligning long reads against their reference genome using lordFAST (Haghshenas et al., 2019).
Table 3.
Comparison between Assemblies Obtained by Different Tools on Real Data
| Dataset | Assembler | Contigs | Genome Fraction | NGA50 | Misassemblies Extensive + Local | Mismatch Rate | Indel Rate | Time | Memory (GB) |
|---|---|---|---|---|---|---|---|---|---|
|
E. coli (ONT) |
Canu | 1 | 99.976 | 3,647,271 | 2 + 6 | 108.85 | 1,254.40 | 702:57:07 | 32.39 |
| Flye | NA | ||||||||
| wtdbg2 | 9 | 79.114 | 141,474 | 38 + 72 | 245.82 | 1,501.74 | 4:57 | 28.05 | |
| miniasm | 3 | 99.992 | 3,106,217 | 4 + 10 | 279.13 | 1,263.23 | 50:00 | 55.56 | |
| Minia | 177 | 97.698 | 57,763 | 0 + 0 | 0.24 | 0.02 | 2:22 | 4.76 | |
| SPAdes | 95 | 98.281 | 133,063 | 0 + 9 | 1.16 | 0.15 | 34:51 | 114.29 | |
| hybridSPAdes | 15 | 99.964 | 3,863,268 | 2 + 7 | 7.16 | 0.50 | 3:38:13 | 114.29 | |
| Unicycler | NA | ||||||||
| DBG2OLC | 1 | 99.950 | 3,539,045 | 3 + 4 | 46.86 | 335.82 | 8:25 | 8.74 | |
| MaSuRCA | 1 | 99.988 | 3,892,134 | 3 + 7 | 2.82 | 0.50 | 30:28 | 32.66 | |
| Wengan | 3 | 99.998 | 3,346,596 | 3 + 2 | 4.74 | 9.24 | 20:02 | 14.37 | |
| HASLR | 2 | 99.992 | 3,970,011 | 2 + 2 | 22.62 | 79.85 | 3:18 | 5.78 | |
| Yeast (PacBio) |
Canu | 23 | 99.724 | 739,932 | 29 + 2 | 8.85 | 7.99 | 1:00:19 | 5.97 |
| Flye | 19 | 99.511 | 566,399 | 28 + 2 | 11.60 | 28.41 | 26:10 | 17.49 | |
| wtdbg2 | 28 | 97.668 | 640,895 | 20 + 3 | 10.65 | 27.17 | 3:04 | 16.26 | |
| miniasm | 88 | 98.292 | 547,238 | 21 + 34 | 31.45 | 381.55 | 5:59 | 15.58 | |
| Minia | 722 | 93.758 | 33,472 | 1 + 1 | 1.67 | 0.81 | 1:18 | 6.36 | |
| SPAdes | 246 | 95.054 | 126,338 | 4 + 2 | 6.44 | 1.47 | 17:11 | 114.09 | |
| hybridSPAdes | 61 | 97.207 | 436,584 | 28 + 20 | 44.77 | 3.71 | 20:58 | 114.09 | |
| Unicycler | 51 | 97.555 | 531,185 | 15 + 5 | 15.13 | 4.22 | 2:09:27 | 36.90 | |
| DBG2OLC | 24 | 63.275 | 229,397 | 25 + 10 | 28.37 | 58.43 | 9:51 | 0.99 | |
| MaSuRCA | 24 | 99.262 | 538,374 | 30 + 8 | 11.83 | 5.85 | 23:15 | 32.69 | |
| Wengan | 29 | 96.258 | 528,763 | 14 + 10 | 11.86 | 34.29 | 6:38 | 8.64 | |
| HASLR | 28 | 95.735 | 530,856 | 11 + 5 | 8.13 | 100.64 | 2:25 | 11.30 | |
|
C. elegans (PacBio) |
Canu | 172 | 99.665 | 561,201 | 723 + 596 | 65.28 | 58.82 | 4:15:23 | 11.62 |
| Flye | 64 | 99.638 | 558,112 | 550 + 450 | 50.50 | 52.89 | 1:08:43 | 31.60 | |
| wtdbg2 | 288 | 98.994 | 561,292 | 329 + 596 | 26.82 | 79.72 | 14:13 | 21.19 | |
| miniasm | 174 | 99.537 | 540,855 | 505 + 432 | 79.10 | 393.94 | 20:12 | 19.95 | |
| Minia | 17,388 | 86.274 | 7,198 | 33 + 27 | 1.34 | 0.99 | 8:05 | 6.61 | |
| SPAdes | 7,234 | 92.003 | 23,152 | 257 + 256 | 11.87 | 4.72 | 2:00:57 | 74.10 | |
| hybridSPAdes | 2,336 | 96.720 | 84,003 | 633 + 638 | 108.04 | 15.96 | 2:47:32 | 74.11 | |
| Unicycler | 858 | 97.102 | 139,992 | 940 + 692 | 58.36 | 45.47 | 23:49:29 | 105.06 | |
| DBG2OLC | 206 | 99.100 | 421,196 | 546 + 383 | 44.75 | 80.61 | 2:34:44 | 11.36 | |
| MaSuRCA | 216 | 97.013 | 471,366 | 368 + 504 | 49.20 | 23.50 | 1:57:49 | 33.48 | |
| Wengan | 270 | 93.341 | 341,861 | 308 + 336 | 35.75 | 121.11 | 45:45 | 8.02 | |
| HASLR | 261 | 97.431 | 453,631 | 259 + 331 | 26.08 | 140.40 | 15:35 | 17.93 | |
| CHM1 (PacBio) |
Canu | 2,110 | 96.084 | 2,329,909 | 6,715 + 7,048 | 145.81 | 120.69 | 689:26:01 | 70.44 |
| Flye | NA | ||||||||
| wtdbg2 | 3,723 | 92.896 | 2,081,842 | 3,535 + 6,286 | 118.45 | 72.54 | 11:35:22 | 202.41 | |
| miniasm | NA | ||||||||
| Minia | 697,240 | 65.977 | 1,823 | 955 + 823 | 87.93 | 13.17 | 3:13:13 | 9.56 | |
| SPAdes | NA | ||||||||
| hybridSPAdes | NA | ||||||||
| Unicycler | NA | ||||||||
| DBG2OLC | 2,118 | 95.547 | 1,599,466 | 3,718 + 8,690 | 116.81 | 116.89 | 78:21:08 | 64.94 | |
| MaSuRCA | 3,781 | 93.782 | 1,761,291 | 4,984 + 7,491 | 180.83 | 57.53 | 350:35:59 | 225.63 | |
| Wengan | 4,474 | 88.948 | 875,489 | 2,771 + 7,577 | 115.65 | 160.71 | 18:19:47 | 112.73 | |
| HASLR | 1,469 | 92.664 | 1,699,092 | 2,097 + 7,661 | 113.06 | 281.74 | 6:32:33 | 60.75 |
Note: Mismatch and indel rates are reported per 100 kbp. Flye, SPAdes, hybridSPAdes, and Unicycler failed on human genome datasets due to memory limit. Unicycler did not finish on E. coli dataset within one month. Flye failed on E. coli with error "No disjointigs were assembled."
For real datasets, we further evaluated the accuracy of assemblies by performing gene completeness analysis using BUSCO (Simão et al., 2015), which quantifies gene completeness using single-copy orthologs. Table 4 shows the results of BUSCO on E. coli, yeast, and C. elegans. We were unable to obtain BUSCO results for the human genome owing to a high run time requirement.
Table 4.
Gene Completeness Analysis
| Dataset | Assembler | Complete (%) | Complete Single Copy (%) |
Complete Duplicate (%) | Fragmented (%) | Missing (%) | Total BUSCO Groups |
|---|---|---|---|---|---|---|---|
|
E. coli (ONT) |
Canu | 4.1 | 4.1 | 0.0 | 16.8 | 79.1 | 440 |
| Flye | NA | ||||||
| wtdbg2 | 1.8 | 1.8 | 0.0 | 9.1 | 89.1 | 440 | |
| miniasm | 3.0 | 3.0 | 0.0 | 18.0 | 79.0 | 440 | |
| minia | 99.8 | 99.3 | 0.5 | 0.2 | 0.0 | 440 | |
| SPAdes | 100.0 | 99.5 | 0.5 | 0.0 | 0.0 | 440 | |
| hybridSPAdes | 100.0 | 99.5 | 0.5 | 0.0 | 0.0 | 440 | |
| Unicycler | NA | ||||||
| DBG2OLC | 35.9 | 35.7 | 0.2 | 33.0 | 31.1 | 440 | |
| MaSuRCA | 99.7 | 98.6 | 1.1 | 0.0 | 0.3 | 440 | |
| Wengan | 100.0 | 99.5 | 0.5 | 0.0 | 0.0 | 440 | |
| HASLR | 97.8 | 97.3 | 0.5 | 1.6 | 0.6 | 440 | |
| Yeast (PacBio) |
Canu | 96.6 | 94.8 | 1.8 | 0.2 | 3.2 | 2,137 |
| Flye | 94.6 | 93.0 | 1.6 | 0.1 | 5.3 | 2,137 | |
| wtdbg2 | 88.4 | 86.8 | 1.6 | 0.8 | 10.8 | 2,137 | |
| miniasm | 25.8 | 25.6 | 0.2 | 5.2 | 69.0 | 2,137 | |
| minia | 96.3 | 94.9 | 1.4 | 0.1 | 3.6 | 2,137 | |
| SPAdes | 96.3 | 94.5 | 1.8 | 0.2 | 3.5 | 2,137 | |
| hybridSPAdes | 96.6 | 94.8 | 1.8 | 0.1 | 3.3 | 2,137 | |
| Unicycler | 96.4 | 94.7 | 1.7 | 0.1 | 3.5 | 2,137 | |
| DBG2OLC | 57.1 | 56.5 | 0.6 | 0.5 | 42.4 | 2,137 | |
| MaSuRCA | 96.3 | 94.1 | 2.2 | 0.1 | 3.6 | 2,137 | |
| Wengan | 96.5 | 94.9 | 1.6 | 0.0 | 3.5 | 2,137 | |
| HASLR | 95.8 | 94.4 | 1.4 | 0.1 | 4.1 | 2,137 | |
|
C. elegans (PacBio) |
Canu | 97.4 | 96.8 | 0.6 | 1.1 | 1.5 | 3,131 |
| Flye | 98.6 | 98.0 | 0.6 | 0.3 | 1.1 | 3,131 | |
| wtdbg2 | 97.1 | 96.5 | 0.6 | 1.3 | 1.6 | 3,131 | |
| miniasm | 83.2 | 82.8 | 0.4 | 6.5 | 10.3 | 3,131 | |
| minia | 80.4 | 79.9 | 0.5 | 9.0 | 10.6 | 3,131 | |
| SPAdes | 91.4 | 90.8 | 0.6 | 4.1 | 4.5 | 3,131 | |
| hybridSPAdes | 96.4 | 95.8 | 0.6 | 1.3 | 2.3 | 3,131 | |
| Unicycler | 97.7 | 97.1 | 0.6 | 0.7 | 1.6 | 3,131 | |
| DBG2OLC | 97.5 | 95.8 | 1.7 | 0.6 | 1.9 | 3,131 | |
| MaSuRCA | 95.5 | 94.1 | 1.4 | 0.4 | 4.1 | 3,131 | |
| Wengan | 91.6 | 91.1 | 0.5 | 0.9 | 7.5 | 3,131 | |
| HASLR | 97.1 | 96.7 | 0.4 | 0.8 | 2.1 | 3,131 |
Note: We used enterobacterales odb10, saccharomycetes odb10, and nematoda odb10 gene sets for assessing gene completeness of E. coli, Yeast, and C. elegans assemblies, respectively. We were not able to obtain the gene completeness results for the human dataset due to time restrictions.
Another observation is that, for some experiments, HASLR does not perform as well as others in terms of genome fraction (see Discussion for more details). However, our gene completeness analysis shows that HASLR is on par with other tools based on BUSCO gene completeness measure (see Table 4). Note that very low gene completeness of Canu, wtdbg2, and DBG2OLC on E. coli dataset could be due to high indel rates of their assemblies. This high indel rate might be caused by the deep coverage of this dataset (>1000×).
We additionally ran RepeatMasker (Smit et al., 2013-2015) on CHM1 assembly generated by HASLR and discovered 1,519,699 SINEs, 922,706 LINEs, and 485,530 LTRs, spanning 13.22%, 21.73%, and 9.21% of the assembly, respectively. In addition, there are 2,275 microsatellites, 659,551 simple repeats, and 97,783 low complexity regions, covering 0.26%, 1.36%, and 0.22% of the assembly, respectively. Further investigation showed that these repeats have a wide range of sizes (see Figure 3 for distribution of identified repeats). This suggests that similar to other long read assemblers, HASLR is capable of resolving large repeats.
Figure 3.

Distribution of Repeats in HASLR's Assembly of CHM1 Dataset Identified Using RepeatMasker
Long Read Coverage Analysis
In order to investigate the required coverage for de novo assembly using HASLR, we assessed its performance on different values for long read coverage. Although HASLR requires only three long reads (minSupp = 3) connecting two unique SR contigs to have a corresponding edge in the backbone graph, in practice a higher coverage is required. We subsampled reads from each simulated and real dataset to 5×, 10×, 15×, 20×, 25×, 30×, 35×, 40×, and 45× coverage. After assembling the subsampled datasets, we measured the NGA50 and Genome fraction using QUAST for each obtained assembly.
As depicted in Figure 4, higher coverage of long reads results in a better assembly. It is interesting that, in most cases, starting from 15× coverage, the genome fraction does not improve significantly. Although the continuity of assemblies keeps improving with increasing coverage, the biggest jump in NGA50 happens between 20× and 30× coverage. Changes in NGA50 above 30× coverage is not significant.
Figure 4.

Performance of HASLR in Assembling Different Datasets on Subsampled Coverage
Discussion
HASLR introduces the notion of backbone graph for hybrid genome assembly. This enables HASLR to keep up with increasing throughput of LR sequencing technologies while remaining time and memory efficient. The high speed of HASLR is due to two reasons: (1) HASLR uses the fast SPOA consensus module rather than normal POA implementation and (2) HASLR uses only the longest 25× coverage of LRs for assembly. Assemblies generated by HASLR are similar to those generated by best-performing tools in terms of contiguity while having the lowest number of misassemblies. In other words, we prefer to remain conservative in resolving ambiguous regions without strong signal rather than aggressively resolving them to generate longer contigs and possibly generating misassemblies. However, the conservative nature of HASLR does not imply that it compromises on assembling complex regions. Every complex region that is covered by a sufficient number of LRs, together with its flanking unique SR contigs, would be resolved. In fact, based on our manual inspections, there are regions that HASLR assembles properly but all other tools either misassemble or generate fragmented assembly (see Figures S1–S10 for visual examples of such cases).
There are a number of future directions that are planned for future releases of HASLR. First, compared with other tools, HASLR usually has a higher indel rate. Note that most of the small local assembly mistakes (including mismatch and indel errors) can be fixed by further polishing. But since a large portion of the assembled genome is built from SR contigs, a polishing module could be specifically designed for HASLR that only polishes the regions between unique SR contigs which have been generated using SPOA. This would enable a faster polishing phase.
An important factor in the contiguity of assemblies generated by HASLR is the length of reads. Obviously, longer reads would generate a more connected and resolved backbone graph. With the recent advancements in the Nanopore technology and the introduction of ultra-long Nanopore reads (whose length can go beyond 1 Mbp), one can expect to get much more contiguous assemblies. Therefore, supporting ultra-long ONT reads is an important feature to address in the future.
HASLR sometimes generates assemblies with relatively lower genome fraction and/or NGA50 compared with other tools. This is clearer when we compare it against Canu, especially on a large and complex genome like the human genome. The main reason is that the connectivity of the backbone graph depends on the existence of unique SR contigs. Therefore, the lack of unique SR contigs in a large region results in multiple connected components rather than a single connected component in the backbone graph. However, that region as a whole (considering all SR contigs aligned to that region) might be different from any other region in the genome because of the order of aligned SR contigs. This means that such region can be resolved using overlap-based assembly approaches. This limitation could be mitigated by extracting unused LRs and assembling them in an OLC fashion (e.g., using miniasm [Li, 2016]). Note that only a small portion of LRs is unused compared to the original input dataset. As a result, using an OLC approach for such a small set of LRs should not affect the total running time significantly.
One of the main bottlenecks of OLC-based assembly approach in terms of speed is that they require to find all overlaps between input reads. Recent LR assemblers have tried to speed up this process by using minimizers (Li, 2016; Koren et al., 2017) or compressed representation of LRs (Ruan and Li, 2019) techniques. However, an all-versus-all alignment is still required in order to generate such a graph. In fact, OLC-based assemblers can use HASLR (or the idea of backbone graph assembly) as a first step before performing the computationally expensive all-versus-all alignment step.
Finally, phased assembly of diploid genomes is an active area of research (see Garg et al. (2018) for an example). Toward this goal, there are two directions of future work: (1) heterozygosity-aware consensus calling of subreads falling between two unique SR contigs is one of our main future directions; this would be possible via clustering of subreads that fall between consecutive unique SR contigs into two groups and performing consensus calling for each group separately. (2) Resolving highly heterozygous regions; we observed that some of the regions with high heterozygosity are not resolved by HASLR. This is because the short read contigs (produced by Minia) for these regions are fragmented. Thus, they are filtered by HASLR, which requires a minimum length for short read contig to long read alignments (controlled via “--aln-block” option; default 500). As a result, regions with high heterozygosity are more likely to be absent from the backbone graph, which makes the backbone graph more fragmented. This means that HASLR might generate more fragmented assembly that has a lower NGA50 (see Tables S2 and S3 which contain results of assemblies for HG002 human dataset). One of the future directions is to explore how a short read assembler like Minia can be adapted for high heterozygosity regions (e.g., by collapsing heterozygous events to generate longer contigs). Although some heterozygosity information might be lost as a result of this modification, a post-assembly step can be used to retrieve this information (e.g., via mapping long reads to the assembled contigs).
Limitations of the Study
The current version of HASLR does not generate a phased assembly for diploid genomes. In addition, the assemblies generated by HASLR might be fragmented owing to high heterozygosity regions or repetitive regions that are not spanned by long reads. We refer the reader to the Discussion section for more details about these limitations.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Faraz Hach (faraz.hach@ubc.ca).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The instructions to generate simulated data used in this article can be found in Supplemental Information. Nanopore reads for E.coli were downloaded from http://lab.loman.net/2017/03/09/ultrareads-for-nanopore and the corresponding Illumina data were downloaded from ftp://webdata:webdata@ussd-ftp.illumina.com/Data/SequencingRuns/MG1655/MiSeq_Ecoli_MG1655_110721_PF_R1.fastq.gz and ftp://webdata:webdata@ussd-ftp.illumina.com/Data/SequencingRuns/MG1655/MiSeq_Ecoli_MG1655_110721_PF_R2.fastq.gz. The yeast PacBio dataset was obtained via accessions ERX1725434, ERX1725435, and ERX1725441, whereas yeast Illumina reads are accessible via ERX1943903. PacBio reads for C.elegans are available at https://github.com/PacificBiosciences/DevNet/wiki/C.-elegans-data-set and the accession ID for the corresponding Illumina is SRR065390. For the CHM1 sample, PacBio reads can be obtained at https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP044331 and Illumina dataset is available via accession ID SRX652547. HASLR is an open source tool implemented in C++ and Python. Its source code is publicly available at https://github.com/vpc-ccg/haslr. HASLR can be installed via Bioconda package manager as well.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Baraa Orabi for providing feedback on the original manuscript. This work is funded in part by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grants to F.H. (RGPIN-05952) and C.C. (RGPIN-03986) and NSERC CREATE program to E.H..
Author Contributions
E.H., C.C., and F.H. developed underlying methodology with feedback from J.S.; E.H. implemented HASLR and performed evaluation of methods; H.A. performed parameter tuning; E.H. and F.H. wrote the manuscript with the help and feedback from C.C. and J.S.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101389.
Supplemental Information
References
- Antipov D., Korobeynikov A., McLean J.S., hybridspades P.A. Pevzner. An algorithm for hybrid assembly of short and long reads. Bioinformatics. 2015;32:1009–1015. doi: 10.1093/bioinformatics/btv688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R., Rizk G. Space-efficient and exact de bruijn graph representation based on a bloom filter. Algorithms Mol. Biol. 2013;8:22. doi: 10.1186/1748-7188-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C.-S., Peluso P., Sedlazeck F.J., Nattestad M., Concepcion G.T., Clum A., Dunn C., O’Malley R., Figueroa-Balderas R., Morales-Cruz A. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods. 2016;13:1050. doi: 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Genova A., Buena-Atienza E., Ossowski S., Wengan M.-F.S. Efficient and high quality hybrid de novo assembly of human genomes. BioRxiv. 2019:840447. doi: 10.1038/s41587-020-00747-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S., Rautiainen M., Novak A.M., Garrison E., Durbin R., Marschall T. A graph-based approach to diploid genome assembly. Bioinformatics. 2018;34:i105–i114. doi: 10.1093/bioinformatics/bty279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghshenas E., Hach F., Sahinalp S.C., Chauve C. Colormap: correcting long reads by mapping short reads. Bioinformatics. 2016;32:i545–i551. doi: 10.1093/bioinformatics/btw463. [DOI] [PubMed] [Google Scholar]
- Haghshenas E., Sahinalp S.C., Hach F. lordFAST: sensitive and Fast Alignment Search Tool for LOng noisy Read sequencing Data. Bioinformatics. 2019;35:20–27. doi: 10.1093/bioinformatics/bty544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Li L., Myers J.R., Marth G.T. Art: a next-generation sequencing read simulator. Bioinformatics. 2011;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski C.C., Allan C.W., Matzkin L.M. Chromosome-level hybrid de novo genome assemblies as an attainable option for non-model organisms. BioRxiv. 2019:748228. doi: 10.1111/1755-0998.13176. [DOI] [PubMed] [Google Scholar]
- Jiang J.B., Quattrini A.M., Francis W.R., Ryan J.F., Rodríguez E., McFadden C.S. A hybrid de novo assembly of the sea pansy (Renilla muelleri) genome. GigaScience. 2019;8:giz026. doi: 10.1093/gigascience/giz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadobianskyi M., Schulze L., Schuelke M., Judkewitz B. Hybrid genome assembly and annotation of Danionella translucida. BioRxiv. 2019:539692. doi: 10.1038/s41597-019-0161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M., Yuan J., Lin Y., Pevzner P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019;37:540. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Koren S., Schatz M.C., Walenz B.P., Martin J., Howard J.T., Ganapathy G., Wang Z., Rasko D.A., McCombie W.R., Jarvis E.D. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012;30:693. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics. 2016;32:2103–2110. doi: 10.1093/bioinformatics/btw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga K.H., Koren S., Rhie A., Vollger M.R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G.A. Telomere-to-telomere assembly of a complete human x chromosome. BioRxiv. 2019:735928. doi: 10.1038/s41586-020-2547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A., Prjibelski A., Saveliev V., Antipov D., Gurevich A. Versatile genome assembly evaluation with quast-lg. Bioinformatics. 2018;34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.R., Delcher A.L., Koren S., Venter E., Walenz B.P., Brownley A., Johnson J., Li K., Mobarry C., Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers G. Efficient local alignment discovery amongst noisy long reads. In: Brown D., Morgenstern B., editors. International Workshop on Algorithms in Bioinformatics. Springer; 2014. pp. 52–67. [Google Scholar]
- Ono Y., Asai K., Hamada M. Pbsim: pacbio reads simulator toward accurate genome assembly. Bioinformatics. 2012;29:119–121. doi: 10.1093/bioinformatics/bts649. [DOI] [PubMed] [Google Scholar]
- Ruan J., Li H. Fast and accurate long-read assembly with wtdbg2. BioRxiv. 2019:530972. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmela L., Rivals E. Lordec: accurate and efficient long read error correction. Bioinformatics. 2014;30:3506–3514. doi: 10.1093/bioinformatics/btu538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. Busco: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Smit A., Hubley R., Green P. 2013-2015. RepeatMasker.http://repeatmasker.org [Google Scholar]
- Vaser R., Sović I., Nagarajan N., Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger M.R., Logsdon G.A., Audano P.A., Sulovari A., Porubsky D., Peluso P., Concepcion G.T., Munson K.M., Baker C., Sanders A.D. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. BioRxiv. 2019:635037. doi: 10.1111/ahg.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.R., Holt J., McMillan L., Jones C.D. Fmlrc: Hybrid long read error correction using an fm-index. BMC Bioinformatics. 2018;19:50. doi: 10.1186/s12859-018-2051-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick R.R., Schultz M.B., Zobel J., Holt K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3350–3352. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick R.R., Judd L.M., Gorrie C.L., Holt K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C., Hill C.M., Wu S., Ruan J., Ma Z.S. Dbg2olc: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016;6:31900. doi: 10.1038/srep31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. Velvet: algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A.V., Puiu D., Luo M.-C., Zhu T., Koren S., Marçais G., Yorke J.A., Dvořák J., Salzberg S.L. Hybrid assembly of the large and highly repetitive genome of aegilops tauschii, a progenitor of bread wheat, with the masurca mega-reads algorithm. Genome Res. 2017;27:787–792. doi: 10.1101/gr.213405.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The instructions to generate simulated data used in this article can be found in Supplemental Information. Nanopore reads for E.coli were downloaded from http://lab.loman.net/2017/03/09/ultrareads-for-nanopore and the corresponding Illumina data were downloaded from ftp://webdata:webdata@ussd-ftp.illumina.com/Data/SequencingRuns/MG1655/MiSeq_Ecoli_MG1655_110721_PF_R1.fastq.gz and ftp://webdata:webdata@ussd-ftp.illumina.com/Data/SequencingRuns/MG1655/MiSeq_Ecoli_MG1655_110721_PF_R2.fastq.gz. The yeast PacBio dataset was obtained via accessions ERX1725434, ERX1725435, and ERX1725441, whereas yeast Illumina reads are accessible via ERX1943903. PacBio reads for C.elegans are available at https://github.com/PacificBiosciences/DevNet/wiki/C.-elegans-data-set and the accession ID for the corresponding Illumina is SRR065390. For the CHM1 sample, PacBio reads can be obtained at https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP044331 and Illumina dataset is available via accession ID SRX652547. HASLR is an open source tool implemented in C++ and Python. Its source code is publicly available at https://github.com/vpc-ccg/haslr. HASLR can be installed via Bioconda package manager as well.