

Key Points
CH is frequent in the bone marrow and the blood of carefully selected healthy volunteers.
The detection of CH in the bone marrow must be interpreted with caution, even in the presence of an unexplained cytopenia.
Abstract
Clonal hematopoiesis (CH) of indeterminate potential has been described in blood samples from large series of patients. Its prevalence and consequences are still not well understood because sequencing methods vary and because most studies were performed in cohorts comprising individuals with nonhematologic diseases. Here, we investigated the frequency of CH in 82 paired bone marrow and blood samples from carefully selected healthy adult volunteers. Forty-one genes known to be mutated in myeloid malignancies were sequenced with a 1% threshold of detection. In bone marrow samples, clones were found in almost 40% of healthy volunteers more than 50 years old. The most frequent mutations were found in DNMT3A and TET2, with 1 individual carrying 3 variants. Variant allele frequencies were highly concordant between blood and bone marrow samples. Blood parameters were normal except for those in 2 individuals: 1 had a mild macrocytosis and 1 had a mild thrombocytosis. Furthermore, no morphologic abnormalities or dysplasia were detected when bone marrow smears were carefully evaluated. Individuals with CH differed from others by age (62.8 vs 38.6 years; P < .0001) and platelet count (294 vs 241 ×109/L; P = .0208), the latter being no more significant when removing the 2 individuals who carried the JAK2 p.V617F mutation. These results confirm that CH is a very common condition in healthy adults over 50 years old. Consequently, the detection of driver myeloid mutations should be interpreted with caution in the absence of cytologic abnormalities in the blood and/or the bone marrow.
Visual Abstract
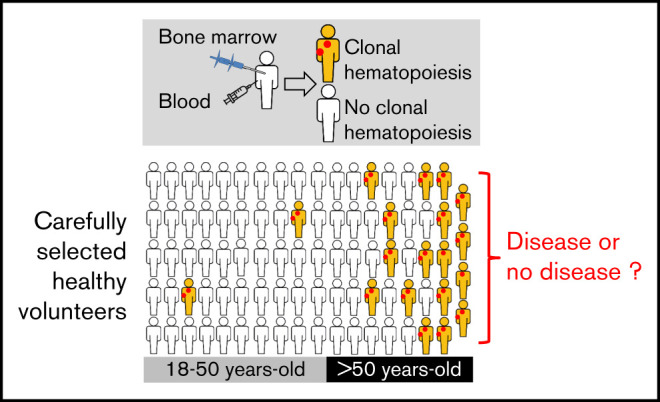
Introduction
Clonal hematopoiesis of indeterminate potential (CHIP) is defined as the presence of a clone at more than 2% of variant allele frequency (VAF) in peripheral blood (PB) of individuals with no evidence of hematologic disease.1,2 In 2014, 3 studies identified clonal hematopoiesis (CH) by analyzing PB samples from thousands of individuals with no history of hematologic disease. However, these cohorts included both healthy people and patients with solid cancers, diabetes, cardiovascular diseases, psychiatric diseases, or other medical conditions.3-5 Thus, these cohorts may not be fully representative of healthy individuals. The pioneer studies used whole-exome sequencing (WES) to detect CH in 1% of individuals younger than 40 years old and up to 20% in the elderly. More recently, targeted sequencing techniques allowed the discovery of a higher prevalence of CH, prefacing the observation that virtually all healthy adults have detectable clones, provided that a highly sensitive method of detection is used.6,7 CHIP has been described as a risk factor for blood cancer.5,8 However, the diagnosis of most blood cancers relies on morphologic and genetic examination of the bone marrow (BM), and CH has never been evaluated in BM samples from healthy individuals. To address this issue, we studied CH in paired PB and BM samples from an unprecedented cohort of rigorously selected healthy volunteers.
Methods
Healthy volunteers
In all, 102 healthy volunteers aged 18 to 81 years were enrolled in the HEALTHOX clinical trial (ClinicalTrials.gov identifier: NCT02789839) of Tours University Hospital (Tours, France), and all provided written informed consent. This study was approved by the ethics committees of the Hospital (CPP [Comité de protection des personnes] Tours and AFSSAPS [Agence française de sécurité sanitaire des produits de santé] identifier ID-RCB: 2011-A00262-39; CPP Ile-de-France III: 2753). All volunteers were given an initial complete medical examination. Exclusion criteria for this study included previous hematologic or tumoral disease, exposure to chemotherapy or radiotherapy, major infectious diseases (ie, bacterial or fungal invasive infection or chronic viral infection), inflammatory diseases, and anti-agregant and anticoagulant treatments. Because this trial also investigated oxidative metabolism, an antioxidant diet that included ascorbic acid or N-acetylcysteine was another exclusion criteria.
Sample collection and morphologic analyses
BM samples were collected through sternal puncture using the classical diagnostic procedure that is standard in France, and PB samples were taken at the same time. No BM biopsy was performed. Cytomorphologic analyses were performed independently by 3 expert cytopathologists at Tours University Hospital. Standard 2016 World Health Organization (WHO) criteria for myeloid diseases were used. These cytomorphologic analyses of the BM included the quantification of the hematopoietic lineages and their maturation as well as the assessment of dysplasia, defined as more than 10% of a lineage with dysplasia.
DNA extraction and next-generation sequencing analyses
PB cells and mononuclear BM cells were frozen and stored for analysis. Both PB and BM samples were analyzed for 82 volunteers, but only PB samples were available for 8 individuals and only BM samples were available for 7 individuals. DNA extraction was performed using QIAsymphony DNA kits (Qiagen, Hilden, Germany). Libraries and sequencing of a panel of 41 genes commonly mutated in myeloid malignancies (supplemental Table 1) were carried out as previously described.9,10 Read alignment and variant calling were performed using Sophia DDM software v5.0.12 (Sophia Genetics, Lausanne, Switzerland) with a sensitivity of 1%. CH was defined as the presence of at least 10 reads carrying 1 somatic variant with a VAF >1%. All variants were checked using IGV software v2.3 and were confirmed on both PB and BM samples as true somatic mutations.11
Statistical analyses
Statistical analyses were performed using Mann-Whitney U, χ2, Fisher’s exact, and Wilcoxon matched-pairs tests or Spearman correlation with GraphPad Prism 6 software (GraphPad Software, San Diego, CA).
Results
Description of the cohort
A total of 102 volunteers were initially included. A full medical evaluation was performed to ensure that only healthy adults were included. Thirteen volunteers were excluded from the study (BM DNA was not available or the patient withdrew consent), so samples from 89 volunteers were included in the final analyses (Figure 1). Included volunteers were aged from 18.6 to 80.1 years (median, 43.6 years). Twenty-six were younger than 30, 24 were between 31 and 50, and 39 were over 50 years old (Figure 2A). Complete blood and BM parameters for all volunteers are shown in Table 1 and in supplemental Tables 2 and 3. These parameters did not reveal any major abnormalities. Complete blood counts, BM counts, and cell morphology did not reveal any cytopenia or dysplasia.
Figure 1.

Flowchart of the study.
Figure 2.

Clonal hematopoiesis in 89 healthy volunteers. (A) Frequency of mutations in each age class, detailing single, double, or triple mutations per individual. (B) Nature and frequency of all the somatic mutations found in the BM of individuals with CH. (C) VAFs of the 25 mutations found in the BM and PB of the 19 volunteers with CH.
Table 1.
Demographic and biological characteristics of individuals with and without CH
| Characteristic | Total (N = 89) | CH (n = 19) | No CH (n = 70) | P | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | Median | Range | No. | % | Median | Range | No. | % | Median | Range | ||
| Age, y | 43.6 | 18.6-80.1 | 62.8 | 28.3-72.4 | 36.8 | 18.6-80.1 | <.0001 | ||||||
| Sex | |||||||||||||
| Male | 36 | 40.4 | 4 | 11.1 | 32 | 88.9 | .0667 | ||||||
| Female | 53 | 59.6 | 15 | 28.3 | 38 | 71.7 | |||||||
| Hemoglobin, g/L | 142 | 118-165 | 139 | 123-161 | 142 | 118-165 | .2059 | ||||||
| Mean globular volume, fL | 89.8 | 77.6-101.4 | 91.3 | 86.1-101.4 | 89.6 | 77.6-98.4 | .0611 | ||||||
| Red cell distribution width, % | 13.4 | 12.1-15.7 | 13.6 | 12.3-15.0 | 13.4 | 12.1-15.7 | .5031 | ||||||
| Thrombocytes, ×109/L | 245 | 128-439 | 294 | 213-439 | 241 | 128-404 | .0208 | ||||||
| Leukocytes, ×109/L | 5.8 | 3.6-10.5 | 5.7 | 4.5-7.0 | 5.9 | 3.6-10.5 | .4690 | ||||||
| Neutrophils, ×109/L | 3.3 | 1.3-7.6 | 3.3 | 2.3-4.6 | 3.2 | 1.3-7.6 | .8058 | ||||||
| Monocytes, ×109/L | 0.5 | 0.1-1.0 | 0.5 | 0.1-0.8 | 0.5 | 0.3-1.0 | .3770 | ||||||
| Gamma glutamyl transferase, IU/L | 22 | 7-182 | 31 | 10-80 | 21 | 7-182 | .1055 | ||||||
| C-reactive protein, mg/L | 1.2 | 0.3-26.2 | 1.8 | 0.3-5.8 | 1.1 | 0.3-26.2 | .1653 | ||||||
| Iron, µmol/L | 18 | 5-43 | 17 | 11-26 | 18 | 5-43 | .8404 | ||||||
| Ferritin, µg/L | 101 | 6-531 | 130 | 29-531 | 92 | 6-455 | .0962 | ||||||
CH in the BM
We evaluated the frequency of CH in the BM of the 89 volunteers (Figure 2A-B). Overall 25 mutations with VAFs greater than 1% and depths ranging from 291 to 2769 reads were detected in 19 individuals (21.3%) (Table 2). Among the volunteers, 14 harbored 1 variant, 4 carried 2 mutations, and 1 had 3 mutations (Figure 2A,C). The most frequently mutated genes were DNMT3A (12 mutations in 9 volunteers), TET2 (n = 7), ASXL1 (n = 2), and JAK2 (n = 2) (Figure 2C). VAFs (mean, 6.0%; range, 1%-49.4%) were not correlated with age (P = .2505). When setting the VAF threshold to the usual value of 2% for CHIP detection, only 4 individuals (age 28, 52, 66, and 67 years, respectively) moved to the no CH group (Table 2). Consequently, the corrected CH frequencies were 0% before the age of 40 years, 30.8% in the 51- to 60-year-old group, and 40% in volunteers aged 61 to 70 years.
Table 2.
Mutations and VAF found in the BM and PB samples from healthy volunteers with CH
| UPN | Age, y | Gene | Mutation | VAF BM, % | VAF PB, % |
|---|---|---|---|---|---|
| #33 | 28 | TET2 | p.Lys1877Glu | 1.1 | 0.6 |
| #36 | 47 | JAK2 | p.Val617Phe | 3.1 | 1.3 |
| #84 | 52 | DNMT3A | p.Arg771* | 1.2 | 1.8 |
| #05 | 53 | TET2 | p.Ala295*fs*1 | 1 | 0.5 |
| DNMT3A | p.? Splice | 2.1 | 1.8 | ||
| #68 | 54 | DNMT3A | p.Ile310_Val328del | 5.6 | 5.9 |
| #101 | 54 | TET2 | p.His1904Arg | 4.7 | 4.5 |
| #13 | 59 | DNMT3A | p.? Splice | 15 | 13 |
| TET2 | p.Gly1154Asp | 14 | 11.2 | ||
| #43 | 61 | JAK2 | p.Val617Phe | 3.2 | 4.2 |
| #99 | 61 | TET2 | p.Cys484Leufs*2 | 2.2 | 2 |
| #49 | 63 | DNMT3A | p.Tyr735Ser | 3.4 | 1.4 |
| #30 | 63 | DNMT3A | p.Arg736Cys | 3.2 | 1.5 |
| #44 | 63 | DNMT3A | p.Ile310Ser | 5.2 | 4.2 |
| #45 | 66 | SRSF2 | p.Tyr44His | 1.4 | 1.7 |
| #47 | 66 | RAD21 | p.Leu393Pro | 2.7 | ND |
| #72 | 67 | TET2 | p.Cys1289Tyr | 1 | 0.5 |
| #38 | 69 | ASXL1 | p.Cys1527* | 49.4 | 50.8 |
| TET2 | p.Leu446* | 4.9 | 4.2 | ||
| #53 | 70 | DNMT3A | p.Leu347Gln | 4.3 | ND |
| DNMT3A | p.Ser714Cys | 4.2 | ND | ||
| DNMT3A | p.Arg882His | 1.8 | ND | ||
| #78 | 71 | ASXL1 | p.Gly649* | 9 | 11 |
| #29 | 72 | DNMT3A | p.Glu578Gly | 5.1 | 4.5 |
| DNMT3A | p.R882H | 1.6 | 1.6 |
ND, no data; UPN, uniform patient number.
Comparison of individuals with and without CH
When comparing individuals with CH to those without CH (Table 1; Figure 3), we found that only 2 parameters were significantly different: platelet counts (294 vs 241 ×109/L, respectively; P = .0208) and age (62.8 vs 36.8 years, respectively; P < .0001), with only 2 (4.0%) of 50 individuals under 50 years old and 17 (43.6%) of 39 individuals over 50 years old having a clone. There was a trend for sex ratio, mean corpuscular volume, and ferritin to differ when the 2 groups were compared. In individuals with mutations, BM examination did not show any abnormality in any lineage or any dysplasia, and BM erythroblasts and granulocyte maturation did not differ between the 2 groups (Figures 3B-C and 4).
Figure 3.
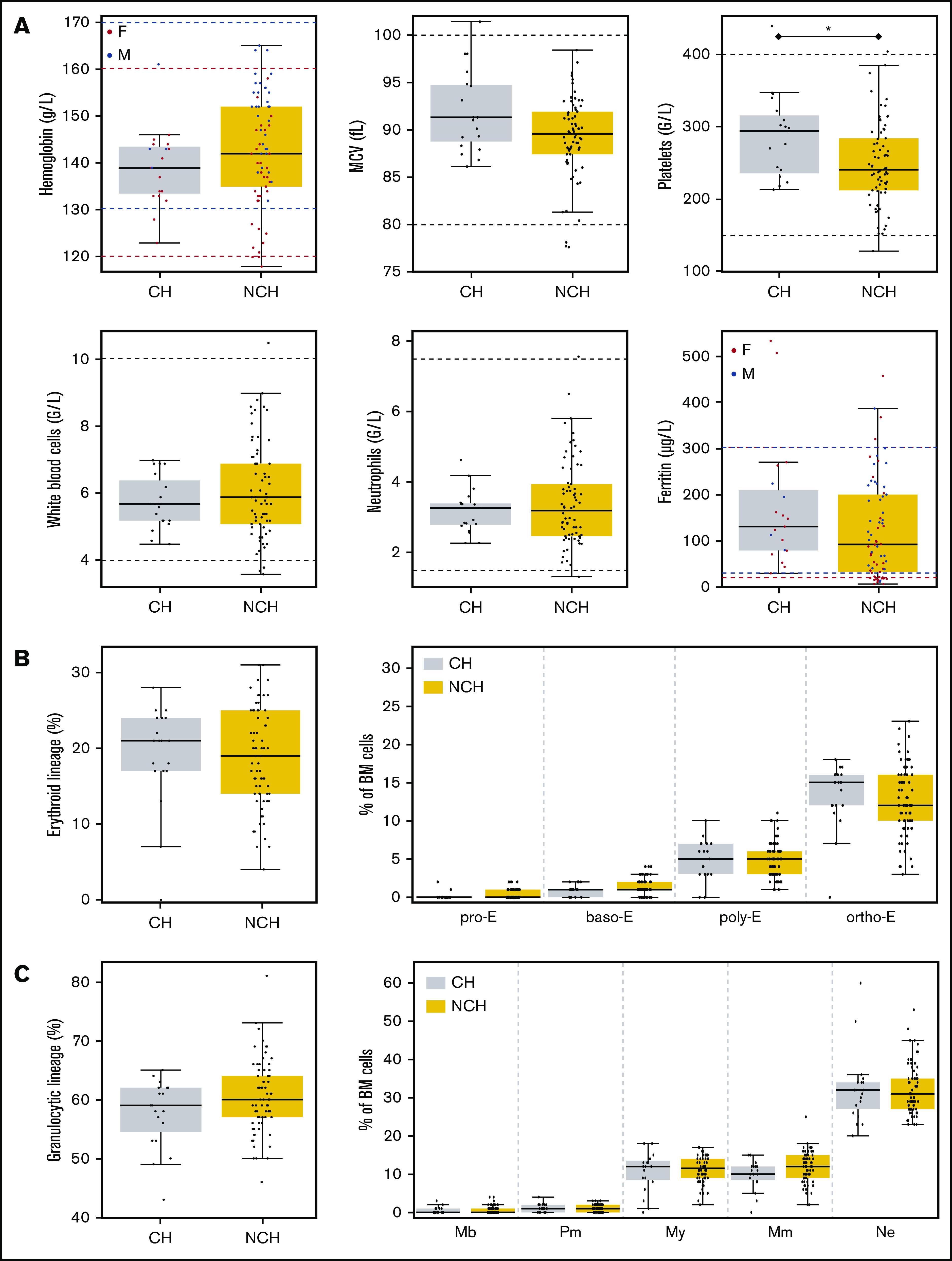
PB and BM parameters according to the presence of CH or absence of CH (NCH). (A) Six PB parameters are shown in the boxplot. Horizontal dashed lines indicate usual values. For parameters impacted by sex, values for women are in red and values for men are in blue. (B) Percentages of BM cells from the erythroid lineage (left panel) and maturation of the erythroid lineage (right panel) in the BM of volunteers with CH or without CH (NCH) are shown in boxplots. (C) Percentages of BM cells from the granulocytic lineage (left panel) and maturation of the granulocytic lineage (right panel) in the BM of volunteers with or without CH are shown in boxplots. baso-E, basophilic erythroblasts; F, female; M, male; Mb, myeloblasts; MCV, mean corpuscular volume; Mm, metamyelocytes; My, myelocytes; ortho-E, orthochromatic erythroblasts; Pm, promyelocytes; poly-E, polychromatic erythroblasts; pro-E, proerythroblasts.
Figure 4.

Cytomorphologic aspects of the BM of individuals with or without CH. BM photos from 2 volunteers without CH (#98 and #102) and 6 volunteers with CH (#13, #36, #38, #45, #53, #78). Representative cells from the megakaryocytic, granulocytic, and erythroid lineages are shown. Mutated genes are indicated at top of each panel. Scale bars represent 50 µm.
The analysis of PB also did not reveal any abnormalities, except for patient #29, who had a mild isolated macrocytosis (101.4 fL) with 2 DNMT3A mutations, and patient #43, who had a mild thrombocytosis (439 ×109/L) and a JAK2 p.V617F mutation. It is worth noting that the other volunteer with the JAK2 mutation (patient #36) had a normal platelet count (347 ×109/L). The 2 volunteers with the JAK2 V617F mutation had platelet counts below 450 ×109/L; therefore, they did not meet the first criterium required by the WHO classification for a diagnosis of essential thrombocythemia. However, it cannot be assumed that these 2 individuals could be at the early preclinical phase of a myeloproliferative neoplasm. BM biopsies were not performed, and no follow-up was planned in the study to support this hypothesis. Thus a second analysis was performed that excluded these 2 individuals with JAK2 p.V617F mutations. Platelets count tended to be different between the CH and no CH groups, without reaching statistical significance (276 vs 241 ×109/L, respectively; P = .0805) (data not shown).
Comparison of BM and PB for CH detection
We then compared the detection of CH in the blood to that in the BM from the 82 volunteers with paired samples available (Table 2). Variants were called in PB using the same detection algorithm with a sensitivity of 1% (see “Methods”). Three volunteers had variants that were detected only in the PB and 6 had variants only detected in the BM with the algorithms. Systematic IGV checking of these variants confirmed their presence in both samples. Failure of the automated tool to detect variants were a result of either having VAFs less than 1% or having insufficient numbers of mutated reads (<10). When comparing the VAFs obtained from blood samples to those from paired BM samples, no statistical difference was found (P = .1116). VAFs measured from the 2 sources were correlated (R = .8877; P < .0001), with a trend toward higher values in BM than in blood samples (supplemental Figures 1 and 2).
Discussion
In 2014, there were 3 studies that described CH in large groups of individuals who did not have hematologic malignancies.3-5 WES allowed the detection of mutations within the coding sequence of all genes, but with a low sensitivity. Subsequently, a 2% cutoff VAF was proposed for the validation of CHIP. By using this threshold in the PB, sequencing of genes mutated in myeloid cancer highlighted CH in 13.7% of 2530 women older than age 55 without hematologic malignancies.12 Next, Young et al6 used error-corrected sequencing and digital droplet polymerase chain reaction to quantify small circulating clones (0.03% VAF). They identified CH in PB samples from 19 of 20 healthy women aged 50 to 60 years. In our series, PB and BM samples from healthy volunteers were concomitantly obtained and studied for the first time. With a 1% threshold for variant detection, our data confirm that CH is a very frequent condition after the age of 50 years in healthy adults.
Recent studies support the hypothesis that CH mutations may be the cause of a delay in differentiation.13 Because CH mutations occur in hematopoietic stem cell (HSC) compartments, we expect that BM VAF may be higher compared with PB VAF. In our study, we did not sort HSCs because we chose to identify CH frequency using a panel-based sequencing that is also used for hematologic diagnosis. Only a slight difference was observed between the 2 values with a trend toward a higher VAF in BM compared with VAF in PB samples. Despite a potential increase in self-renewal because of somatic CHIP mutations, HSCs are rare in the BM in comparison with their mature progeny.13 It is noteworthy that the maturation pyramids of late progenitors (erythroblasts and granulocytes) did not differ between the 2 groups. This could explain why next-generation sequencing with a sensitivity of 1% in bulk DNA does not highlight any VAF difference between PB and BM.
The diagnosis of myelodysplastic syndromes (MDSs) still relies on a set of clinical and biological arguments that allow the clone and the disease to be linked in most patients. This is not the case in the diagnostic context of clonal cytopenia of undetermined significance (CCUS), which is defined as the association of CH and 1 or more cytopenias without any criteria for a myeloid neoplasm.14-16 A recent study in individuals with CCUS showed that distinction could be drawn between patients with MDS-like symptoms that have a high risk of evolution and others on the basis of mutation profiles. Isolated mutation of DNMT3A, TET2, and ASXL1 (also known as DTA when the 3 genes are grouped together) may not be predictive for hematologic malignancy. In contrast, mutations in spliceosome genes (such as SF3B1 or SRSF2), JAK2 mutations, comutations, or large clone size may be present in individuals with MDS-like symptoms who have survival characteristics or risk of disease progression similar to those of patients with MDS.17,18 In our series, 3 volunteers harbored isolated non-DTA mutation. Yet none of them revealed any PB parameter abnormality or any dysplasia.
Our cohort of healthy individuals showed a high frequency of CH with normal BM cytomorphology and normal blood count. Thus, the causal link between an eventual cytopenia and a clonal mutation, including non-DTA mutations, should be carefully assessed. To that end, a BM examination is mandatory for diagnosing a possible BM dysplasia. Moreover, in the absence of documented BM dysplasia, this causal link might still be highly questionable, and all alternative diagnoses should be investigated before declaring a diagnosis of CCUS.
To conclude, we find a high prevalence of CH after 50 years of age by studying for the first time both BM and PB from healthy volunteers for whom we have assessed the normality of PB and BM morphology. In light of these results, the medical follow-up of individuals with CH can be carefully managed. We think that marrow examination and medical follow-up are essential when a patient presents with the combination of a cytopenia and CH in a blood sample to avoid making a wrong diagnosis of CCUS at some point. Further prospective studies that include patients with cytopenia and medical follow-up are required to evaluate the evolutionary potential of such clones in healthy individuals.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported by the French National Cancer Institute (INCA), the French Ministry of Solidarity and Health, and INSERM (plancancer-THE 201606, INCA-DGOS-Inserm_12560 “SiRIC CURAMUS”, and INCA-DGOS 2017-1-RT-03 “MIF_AML”), La Ligue Nationale Contre le Cancer, CANCEN, Les Sapins de l’Espoir Contre le Cancer, the Club of Blois of International Rotary, and Sociétés d’Accélération du Transfert de Technologie Grand Centre.
Footnotes
Requests for data should be sent by e-mail to Pierre Hirsch (pierre.hirsch@aphp.fr) or Olivier Herault (olivier.herault@univ-tours.fr).
Authorship
Contribution: P.H. and C.D. performed next-generation sequencing experiments and interpreted the results; N.R., A.F., E.R., and S.L. collected blood and bone marrow samples and analyzed their biological parameters; P.H., H.G., O.H., and F.D. interpreted the results and wrote the manuscript with the help of J.-A.M., E.S., J.B., and L.S; O.H. was the main investigator of the HEALTHOX clinical trial, and managed the collection of samples with the help of E.G., N.G., and V.G.; and F.D., O.H., and P.H. designed the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: François Delhommeau, Hôpital Saint-Antoine, 184 Rue du Faubourg Saint-Antoine, F-75012 Paris, France; e-mail: francois.delhommeau@aphp.fr; Olivier Herault, Hôpital Bretonneau, 2 Boulevard Tonnellé, F-37000 Tours, France; e-mail: olivier.herault@univ-tours.fr; and Pierre Hirsch, Hôpital Saint-Antoine, 184 Rue du Faubourg Saint-Antoine, F-75012 Paris, France; e-mail: pierre.hirsch@aphp.fr.
References
- 1.Calvillo-Argüelles O, Jaiswal S, Shlush LI, et al. . Connections between clonal hematopoiesis, cardiovascular disease, and cancer: a review. JAMA Cardiol. 2019;4(4):380-387. [DOI] [PubMed] [Google Scholar]
- 2.Shlush LI. Age-related clonal hematopoiesis. Blood. 2018;131(5):496-504. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal S, Fontanillas P, Flannick J, et al. . Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie M, Lu C, Wang J, et al. . Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Genovese G, Kähler AK, Handsaker RE, et al. . Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7(1):12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zink F, Stacey SN, Norddahl GL, et al. . Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018;2(22):3404-3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirsch P, Zhang Y, Tang R, et al. . Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat Commun. 2016;7(1):12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Gerstung M, Bullinger L, et al. . Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. . Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buscarlet M, Provost S, Zada YF, et al. . DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130(6):753-762. [DOI] [PubMed] [Google Scholar]
- 13.Eudy E, Loberg M, Bell R, Stearns T, Trowbridge J. Dnmt3a mutation confers a selective advantage specifically to cells within the long-term hematopoietic stem cell (LT-HSC) compartment [abstract]. Blood 2019;134(suppl 1). Abstract 448.
- 14.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 15.Kwok B, Hall JM, Witte JS, et al. . MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126(21):2355-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cargo CA, Rowbotham N, Evans PA, et al. . Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362-2365. [DOI] [PubMed] [Google Scholar]
- 17.Malcovati L, Gallì A, Travaglino E, et al. . Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abelson S, Collord G, Ng SWK, et al. . Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.