

Abstract
Human viruses possess very complex supramolecular structures. Both icosahedral and enveloped viruses typically display an array of viral-encoded protein antigens at varied spatial densities on the viral particle surface. The viral nucleic acid genome, on the other hand, is encapsulated inside the viral particle. Although both the surface antigen and the interior nucleic acids could independently produce immunological responses, how B cells integrate these two types of signals and respond to a typical virus particle to initiate activation is not well understood at a molecular level. The study of these fundamental biological processes would benefit from the development of viral structural mimics that are well constructed to incorporate both quantitative and qualitative viral features for presentation to B cells. These novel tools would enable researchers to systematically dissect the underlying processes. Here we report the development of such particulate antigens based on liposomes engineered to display a model protein antigen, hen egg lysozyme (HEL). We developed methods to overexpress and purify various affinity mutants of HEL from E. coli. We conjugated the purified recombinant HEL proteins onto the surface of a virion-sized liposome in an orientation-specific manner at defined spatial densities and also encapsulated nucleic acid molecules into the interior of the liposome. Both the chemical conjugation of the HEL antigen on liposome surfaces and the encapsulation of nucleic acids were stable under physiologically relevant conditions. These liposomes elicited antigen-specific B-cell responses in vitro, which validate these supramolecular structures as a novel and effective approach to mimic and systematically isolate the role of essential viral features in directing the B-cell response to particulate antigens.
INTRODUCTION
The human immune system robustly mounts effective antibody responses against a plethora of viral pathogens. This property of the immune system forms the biological basis for many of the vaccine products that have been licensed for immunization and distribution in the United States.1 However, at a fundamental level, how B cells respond to a viral particulate antigen and initiate the cascade of events that lead to the production of potent neutralizing antibodies and long-lived memory B cells is incompletely understood. From B-cell recognition of relevant antigens to B-cell presentation of antigens to CD4+ T cells and recruitment of CD4+ T cell help to sustain the activation of B cells, our understanding of these processes mostly stems from studies of soluble antigens (for a recent review see 2). Although many fundamental pathways in B-cell activation have been well established, how these pathways may be modulated by the essential and unique quantitative and qualitative features of a viral particulate antigen represents a key gap in our knowledge that needs to be addressed in order to inform rational vaccine design in the future.
It is well known that individual viruses have surface antigens that are displayed at specific densities. It is noteworthy that the spatial densities of these viral surface antigens vary over several orders of magnitude in different naturally occurring viruses.1 Although little is known about how this varied spatial density of viral surface antigens may contribute to the immunogenicity of the viruses, it is well established that antigen organization determines B-cell responses.3 Quantitative differences in epitope density can easily produce quantitative or qualitative differences in antibody responses in mouse models of immunization.3–6 Specifically, a high-density display of an antigen on viral-like particles can elicit strong B-cell antibody responses and even break B-cell tolerance.3,7 In fact, it has been shown recently that a threshold density of epitopes on liposomes can elicit class-switched and autoreactive IgG, demonstrating that epitope density can serve as a stand-alone signal in eliciting B-cell class switching and secretion of autoreactive antibodies.8 Collectively, these studies strongly suggest that particulate antigens can induce effects on B cells that are qualitatively different from those produced by conventional soluble antigen, and thus, studies of such complex particulate antigens are necessary in order to understand B-cell responses to biological viruses.
Model antigens, such as the hapten (4-hydroxy-3-nitrophenyl) acetyl (NP) and various polymers based on this hapten,4 have been indispensable to understand B-cell responses to immunogens,9–13 but model antigens that facilitate the study of viral features on B-cell responses have been limited. An ideal model antigen that mimics natural viruses for the study of B-cell responses to viral infection and vaccine antigens must possess two essential qualities. (1) The spatial density of the surface epitope must be “programmable” to mimic that of naturally occurring viruses. Herein, the “spatial density” of an epitope refers to the number of immunogenic epitopes that is averaged over a single particle, as previously defined by us.14 (2) The interior of the particle should be able to accommodate nucleic acid molecules to at least mimic this essential feature of any virion. Nucleic acids are highly immunostimulatory molecules that can be recognized by both endosomal and cytosolic sensors in B cells and can trigger B-cell activation both independently of and synergistically with B-cell antigen receptor (BCR) engagement.15 Importantly, differences in the site and context in which nucleic acids are sensed by B cells can have profoundly divergent effects on B-cell responses.16,17 Therefore, in order to accurately model the immune responses of B cells toward particulate antigens such as naturally occurring viruses, it is critical to produce synthetic particles that mimic both of these essential features of a viral particle. In order to study these aspects in isolation and collectively, it is ideal for these features to be independently built into a model antigen. Such a construct will enable us to dissect the individual contributions of epitope density and encapsulated nucleic acids on B-cell responses and reveal how they function either independently or synergistically to regulate B-cell activation and immune responses.
Liposome-based nanoparticles have unique advantages in this regard because (1) liposomes themselves are not immunogenic, which offers a clear baseline to understand the role of other components that are incorporated into these particles; (2) a plethora of chemical conjugation techniques has been developed for conjugation of protein molecules onto the surface of liposomes,18 and (3) the interior of the liposome can be used for encapsulation of molecules such as nucleic acids that mimic the viral genome. Lastly, it is important to mention the differences between synthetic particles and viral-like particles that are biologically synthesized using cell-based systems. The compositions of the latter are usually not well or easily defined. Liposome-based synthetic particles, on the other hand, are assembled from purified components. The molecular basis of immunogenicity is well defined, which allows one to dissect the contributions of each feature on B-cell responses and isolate the independent effects of epitope density and encapsulated nucleic acids.
Previously we have developed a set of analytical methods to quantitate the average spatial density of proteins conjugated on the surface of liposomes.14 By quantitating epitope densities on liposomes with time, we uncovered an intrinsic issue associated with the widely used Ni-chelating liposomes and suggest that alternative methods other than the Ni-chelation chemistry is needed for stable conjugation of epitopes onto liposomes for biological studies.14 Our results from that work suggest a general strategy that can be used to precisely regulate the epitope density on liposomes for the study of B-cell responses. In the current study, we report a method that is potentially generic and allows one to conjugate epitopes of interest in an orientation-specific manner to unilamellar liposomes using maleimide chemistry. The epitope densities generated by this method could be regulated, and the synthetic liposomes were stable under physiological conditions. We also report a method to encapsulate nucleic acid molecules into these liposomes, so that the resulting liposomal nanoparticles can mimic both the quantitative and the qualitative features of naturally occurring viral particles (Figure 1).
Figure 1.
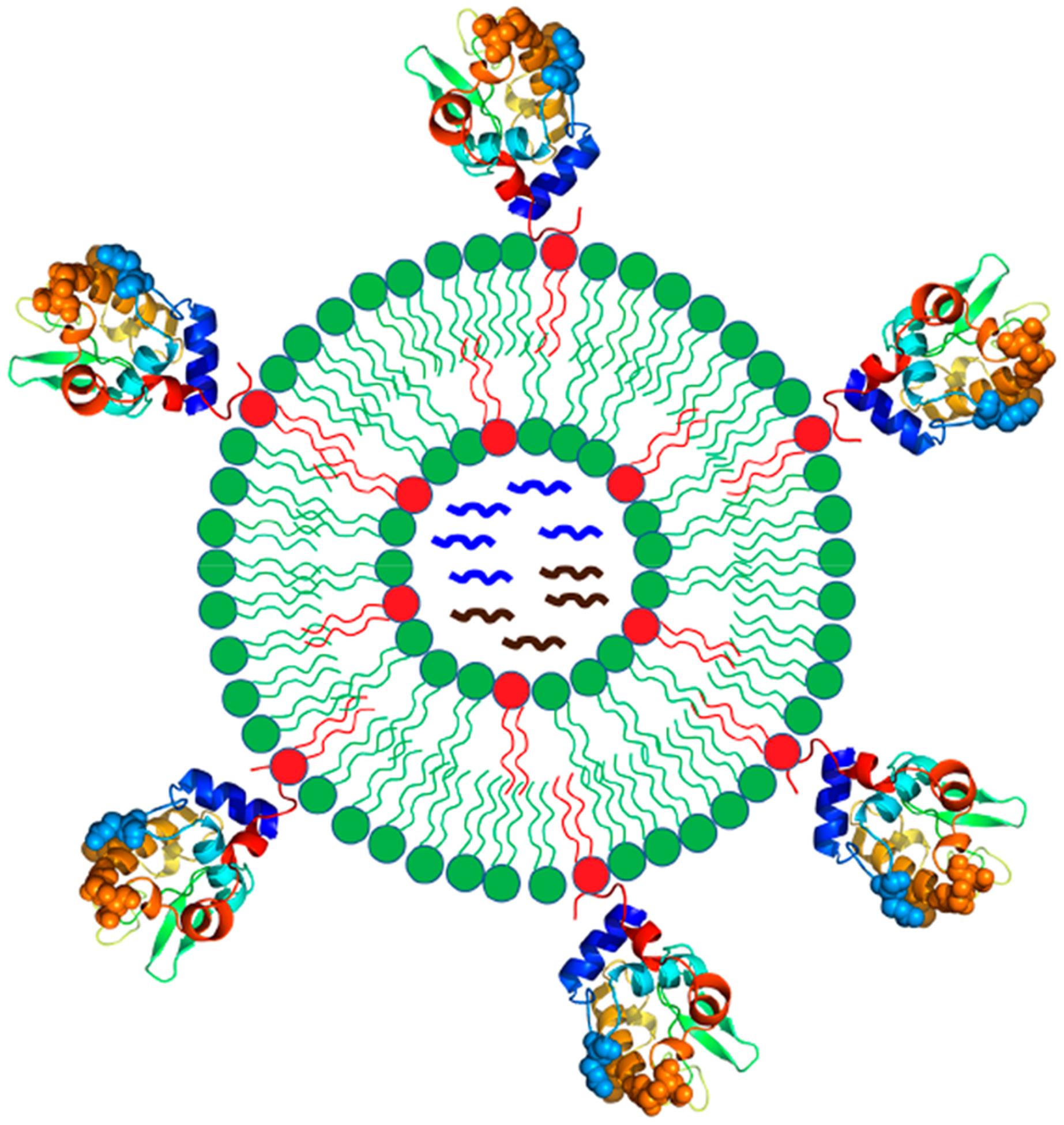
Schematic of functional liposomal nanoparticles that quantitatively incorporate various features of enveloped viruses. Particle surface is conjugated with protein antigen at regulated average spatial densities. Internal space of the liposome is loaded with nucleic acids and other molecules to mimic the internal structure of a typical biological virus.
RESULTS
Choice of Protein Antigen and Protein Expression System.
The model protein antigens that we choose in this study for conjugation onto the surface of liposomes is hen egg lysozyme (HEL). The major reason to choose this protein is that this protein along with its affinity mutants has been used in many classic and recent immunological studies to understand the basic principles of B and T cell responses.19–24 Transgenic mice that carry HEL-specific B cells have been developed, which allows one to use these existing tools for study of immune responses to complex antigens such as particulate antigens of interest here. Because HEL is so well characterized as an immunogen in mouse, much is known about how a soluble antigen of HEL is processed and presented by B cells in mice of various genetic background and how HEL antigen peptides are recognized by MHC class II molecules.19,20 Therefore, particulate antigens based on HEL could potentially build on the existing knowledge about this soluble antigen and further extend to the realm of particulate antigens.
HEL is one of the first few proteins whose three-dimensional structures were solved by X-ray crystallography.25 The relative abundance and availability of this protein from hen egg white has propelled much of the early research on the biochemistry of this protein.26 For our applications where we want to conjugate this protein in a specific orientation onto the surface of liposomes in a stable manner, one solution among others is to engineer free accessible cysteines into the sequence of HEL and use thiol maleimide chemistry to achieve the orientation-specific conjugation.
Wild-type HEL has eight cysteine residues that form four specific pairs of disulfide bonds, which are all important for HEL folding and stability.27–30 To engineer free accessible cysteines for chemistry of our interest without disruption of the native protein structure, it is necessary to purify this protein in native form from its overexpression host.31 Otherwise, the four disulfide bonds present in the native protein may not form correctly after refolding of the denatured proteins that carry additional engineered cysteines.
Site-directed mutagenesis has been extensively conducted on HEL proteins. Various mutants have been purified and studied.29,32,33 However, almost all of these methods employ purification of HEL in denatured form from E. coli followed by refolding of the protein. This approach will not work for our purpose. There are reports in the literature that have expressed HEL in E. coli in native forms, and issues were reported that the cells were lysed.34 However, in that study, HEL was fused with OmpA signal peptide that targeted this protein to periplasmic space, which would unleash its lytic activity. Alternatively, to suppress the lytic activity of lysozyme, one report used tandem expression of human lysozyme with a tight-binding inhibitor Ivy in E. coli.35 This method did work for producing wild-type HEL in native form from E. coli in our hands. However, expression of HEL mutants under the same conditions was very low, which might be due to competition with Ivy expression that was controlled under a separate T7 promoter. For the study of B-cell responses, it is necessary to produce in good quantities a set of HEL mutants that have varied affinities toward the antigen-specific B cells, because affinity between an antigen and the B-cell antigen receptors will modulate B-cell responses. To mimic the biological situation of B-cell encounter of viruses, the initial affinity between the viral antigen and B-cell antigen receptors is likely to be very low. Moreover, the serious drawback of the HEL–Ivy overexpression system was that HEL and Ivy were very close in their molecular masses, so that it was very challenging to resolve the two proteins clearly in a single gel, which created difficulty for assessment of protein purity.
Methods have been reported in the literature to purify HEL mutants in native forms using the yeast expression system Pichia pastoris.36 However, both cloning and selection of transformants were not as convenient as in E. coli. In this work, we report, to the best of our knowledge, the first system in E. coli that allows the overexpression and purification of HEL and its mutants in native forms with more than 95% purity and in good yields. This system also allows us to produce HEL with engineered cysteines so that we can use these cysteines for orientation-specific conjugation onto liposomes, which is a key feature of viral surface proteins. We describe our approach to purify these proteins and report the production of well-behaved liposomal particles that have both HEL conjugation on the surface in an orientation-specific manner and encapsulation of nucleic acids in the interior of the liposomes. These particles therefore serve as synthetic mimicries of biological viruses for the study of B-cell responses to particulate antigens.
Purification of Wild-Type HEL and Mutants from E. coli with >95% Purity.
The pET system for expression of recombinant proteins in E. coli has been widely used over two decades. In this system, the strong T7 RNA polymerase activity can be inhibited by the T7 lysozyme expressed in trans,37 which forms the basis for inclusion of pLysS or pLysE plasmids in this commercialized system. On the basis of this phenomenon, we reasoned that it might be possible for E. coli to harbor a high concentration of active lysozyme in the cytosol without concern on bacterial lysis, provided that the enzyme was expressed in the cytosol and had no access to the periplasmic space. Follow this reasoning, we constructed a plasmid in which a HEL open reading frame carrying a C-terminal hexahistidine tag is directly placed under the control of the bacteriophage T7 promoter and used Shuffle T7 express (NEB) as the host for protein expression. As shown in Figure 2a, we engineered a free accessible cysteine close to the C-terminus of the protein for maleimide chemistry. We have developed a protocol and purified five recombinant HEL proteins in native forms from this system: wild-type HEL (wtHEL), a double mutant of HEL (R73E and D101R, named HELD herein), a tripple mutant of HEL (R21Q, R73E, and D101R, named HELT herein), HELD that carries an engineered free cysteine close to the C-terminus of the protein (named HELDC herein), and HELT that carries an engineered free cysteine close to the C-terminus of the protein (named HELTC herein). These mutations are shown in spacing-filling models in Figure 2a and are important for the binding between HEL and the transgenic B-cell antigen receptor, IgHEL.36
Figure 2.
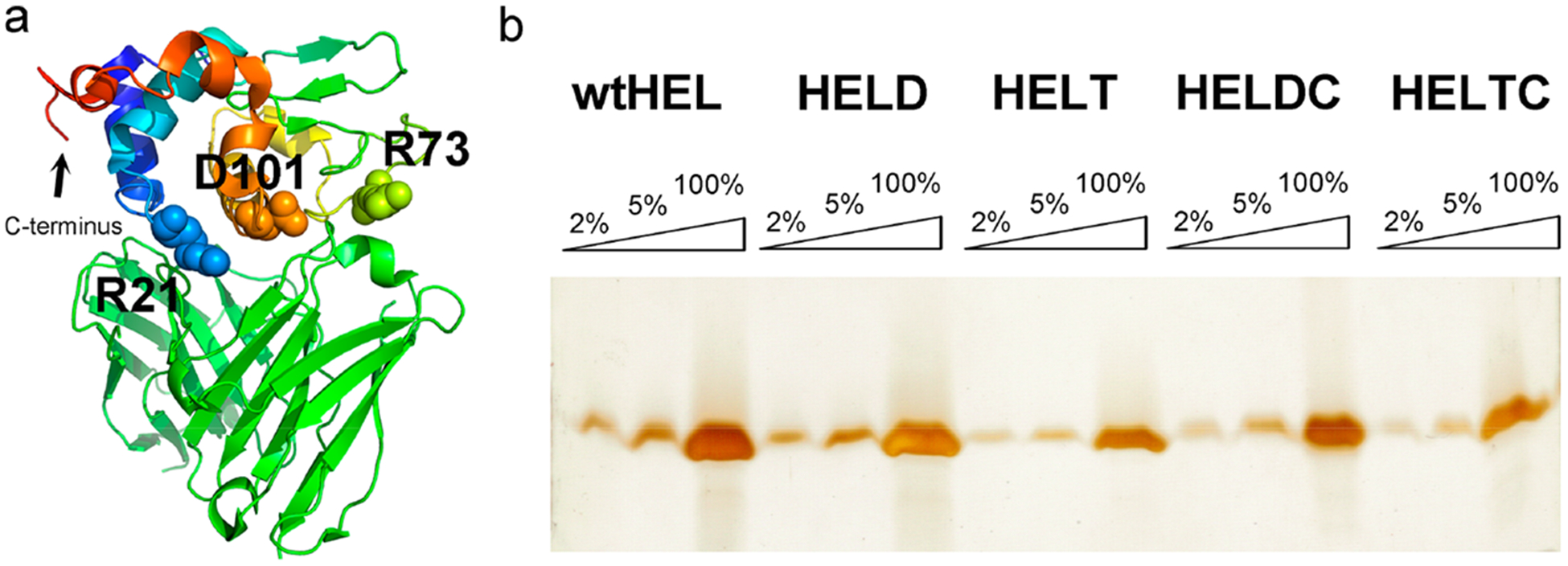
HEL protein and mutant purification. (a) Crystal structure of HEL-Fv complex showing three residues, R21, R73, and D101 in HEL that are important for binding to the transgenic BCR, IgHEL (PDB 1c08). These three residues are highlighted in spacing-filling model. The C-terminus of the HEL protein is also denoted. (b) Silver staining of a 15% (29:1 acrylamide/Bis) reducing SDS–polyacrylamide gel to assess the purity of the purified proteins wtHEL, HELD, HELT, HELDC, and HELTC. For each protein, three lanes were loaded at 2%, 5%, and 100% relative to the quantity of each purified protein, respectively.
The purification procedure involves three major steps: (1) using a Ni–NTA column to purify the protein to 70–80% purity, (2) using a Heparin column to clean up nucleic acids and further purify the protein to ~90% purity, and (3) using Enrich S (Bio-Rad), a cation exchanger to purify the protein to >95% purity. As shown in Figure 2b for the final purified proteins on a denaturing polyacrylamide gel, we obtained these recombinant proteins that were more than 95% pure based on quantitation of band intensity. Two of the proteins were even more than 98% pure. Wild-type HEL has an isoelectric point of 11.35 at ionic strength 0.1.38 The five recombinant proteins above have theoretical pI values of 8.7. At pH 7.4, these proteins will carry positive charges. Because these proteins will be used for immunology studies, we have paid extra attention throughout our purification process to remove any nucleic acids that may bind nonspecifically to HEL via electrostatic interactions. We have two specific steps in our purification protocol to wash away nucleic acids. (1) We used an alkaline buffer with high ionic strength (0.1 M Tris, 2 M KCl, pH 10 at 4 °C) to wash the Ni–NTA column extensively (≥20 column volumes) before elution of HEL proteins from the column. (2) We added a Heparin column after Ni–NTA elution. Heparin is a polyanion that can effectively compete with DNA for binding to target proteins.39 We have established a condition to bind HEL on this column and elute HEL protein off the column using a buffer of high ionic strength (Materials and Methods). These steps ensure that the recombinant HEL proteins were not only highly pure in protein compositions but also free of nucleic acids contamination, as we will see from immunological studies below. The typical yield for these recombinant proteins from the current system is ~1 mg of protein from 2 L of culture.
Activity of HEL and Mutants in Stimulating Antigen-Specific B Cells.
To examine the activity of purified HEL in stimulating antigen-specific B cells in vitro, we incubated various HEL proteins at serially diluted concentrations with lymphocytes freshly harvested from IgHEL-Nur77-EGFP transgenic mice.40 The B cells in these transgenic mice carry a monoclonal B-cell antigen receptor, IgHEL, that is specific for HEL. These B cells also carry an enhanced green fluorescent protein (EGFP) marker that is under the control of the Nur77/Nr4a1 regulatory region. Nr4a1 is a primary response gene whose transcription is rapidly and robustly triggered in response to BCR signaling.41 As established previously, these B cells express EGFP upon BCR engagement of HEL proteins both in vitro and in vivo, and the amount of EGFP expression is quantitatively related to the strength of BCR signaling.40 As shown in Figure 3, three of the HEL proteins that we purified elicited dose-dependent responses of EGFP expression in these transgenic B cells after 6 h of incubation. Among these, wtHEL (squares) elicited responses that were quantitatively similar to that by HEL from a commercial source (sHEL, circles). Higher concentrations of HELD and HELT were required to observe the plateaus in EGFP expression from these reporter cells. These trends are consistent with expectation since both HELD and HELT have a lower affinity toward the BCR compared to wtHEL with an equilibrium association constant to the BCR Ka = 8.0 × 107 M−1 for HELD and Ka = 1.5 × 106 M−1 for HELT.36 In addition, we did not observe EGFP upregulation in Nur77-EGFP B cells that lack the HEL-specific BCR, IgHEL. Since EGFP upregulation in Nur77-EGFP B cells can be induced by CpG and LPS, this suggests that all responses were Ag specific and not due to contaminating TLR ligands.42
Figure 3.
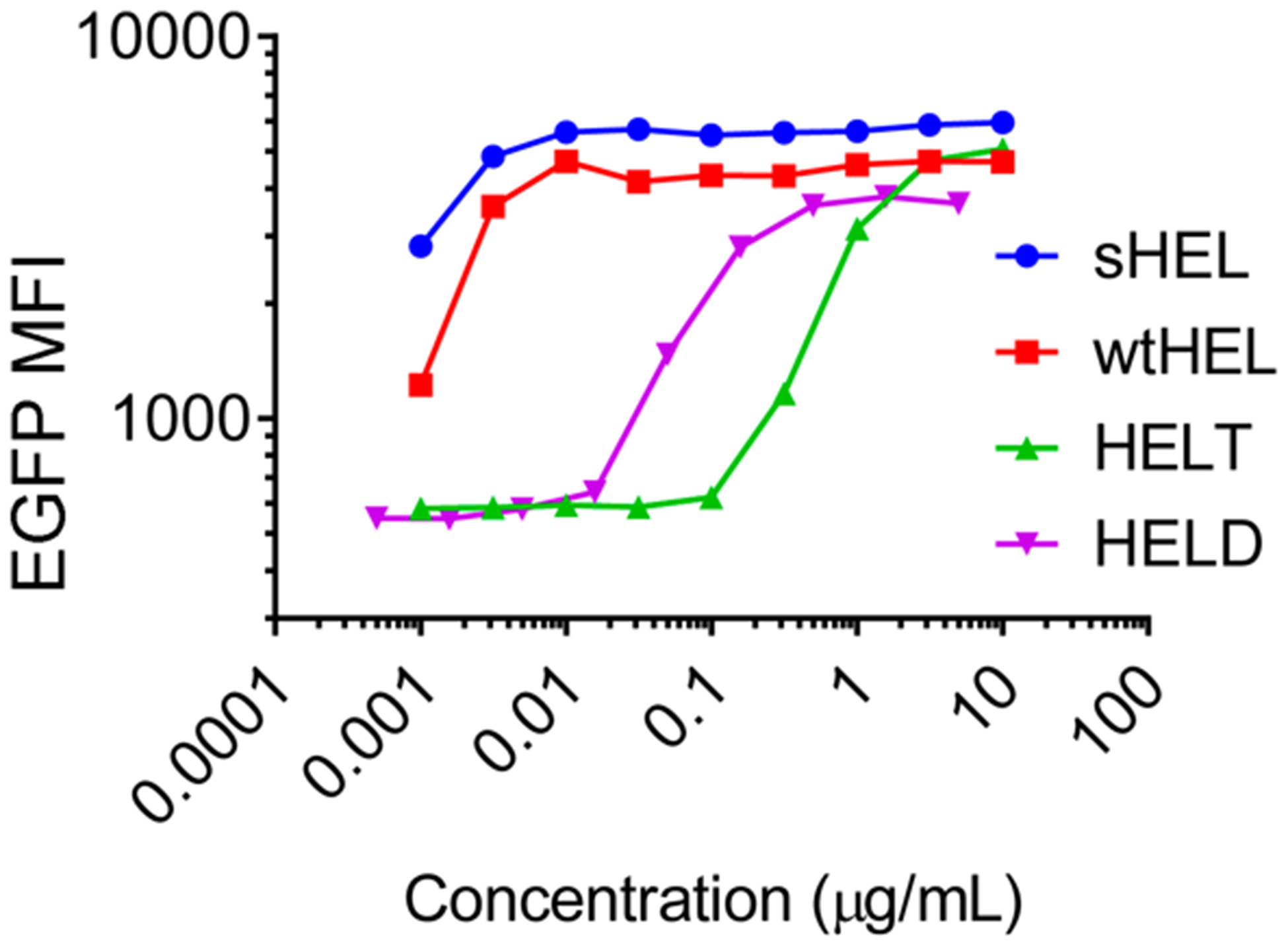
Activation of antigen-specific B cells in response to soluble HEL antigens. Lymphocytes freshly harvested from IgHEL-Nur77-EGFP transgenic (Tg) mice were incubated with soluble HEL at serially diluted concentrations for 6 h. After surface staining to detect B220 expression, the fluorescence intensity of EGFP per B cell was assessed by flow cytometry. EGFP mean fluorescence intensity (MFI) for each population was shown as a function of the concentration of soluble HEL antigen. sHEL is commercially produced by Sigma. Data are representative of four biological replicates.
Conjugation of HEL to Liposome Surface through Maleimide Chemistry.
To prepare unilamellar liposomes for conjugation of purified HEL proteins, we have followed the workflow that we developed previously for preparation of protein-conjugated liposomes14 but with important modifications: (1) we used maleimide-containing lipids instead of metal-chelating lipids; (2) we used 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) instead of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC); (3) the molar percentage of maleimide-containing lipids is limited to 5% or less to minimize undesired cross-linking between proteins and lipid molecules.
Previously we prepared liposomes using metal-chelating lipids, Ni–NTA-DGS (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt)).14 Histidine-tagged proteins were attached to these liposomes through Ni-chelating chemistry. Although the initial epitope density on these liposomes can be fine controlled, the density decreases with time in biologically relevant media due to the noncovalent nature of Ni-chelation chemistry. This time-dependent change of epitope density will confound the interpretation of the immunogenicity data. In this study, we have selected 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] ammonium salt (DSPE–PEG maleimide) for preparation of liposomes. The proteins of interest were conjugated to the surface of these liposomes through maleimide–thiol addition chemistry.43,44
This covalent linkage between the epitope and the liposome likely improves the stability of the epitope density with time. Second, we have selected 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) to pair with DSPE–PEG maleimide for preparation of these liposomes for two reasons: (1) DPSC has the same number of hydrocarbons as DSPE–PEG maleimide in their hydrophobic tails, which may afford better packing in the lipid bilayer than other lipid molecules with different numbers of hydrocarbons; (2) DSPC has a reported melting temperature of 55 °C.45 Thus, at physiological temperatures, these lipid molecules will remain closely packed in the ordered gel phase, which may afford better retention of molecules inside liposomes. This is an important issue for the current study because we plan to encapsulate nucleic acid molecules into these liposomes to qualitatively mimic the structural feature of biological viruses. Furthermore, the addition of cholesterol in the lipid formulation may further improve the cohesion of lipid bilayer46 and therefore enhance the stability of the liposomes in physiologically relevant media.47–49
To this end, we have developed a protocol (see Materials and Methods) to prepare unilamellar liposomes using a mixture of DSPC, DSPE–PEG maleimide, and cholesterol. After preparation of liposomes, we added purified HEL proteins that carried free engineered cysteines and incubated with liposomes for conjugation of HEL proteins onto the liposomal surface via maleimide–thiol chemistry. After this conjugation, the liposomes were purified away from free excess HEL proteins by running through a size exclusion column (SEC). As shown in Figure 4 for liposomes prepared with 1% DSPE–PEG maleimide, 1 h after HELDC incubation with liposomes at 22 °C, a protein band of higher molecular weight (MW) appeared (indicated by the downward arrow), the amount of which did not increase upon further incubation, consistent with the fast kinetics of maleimide reaction at this temperature. This higher MW species is consistent with HELDC conjugation with one molecule of DSPE–PEG maleimide, as indicated by the MW markers. Moreover, this species exclusively resulted from the conjugation between maleimide and the thiol from the free engineered cysteines. Omission of DSPE–PEG maleimide from lipid mixture did not produce this band. Incubation of the same maleimide-containing liposome with HELD without the engineered cysteine did not yield this band either, demonstrating the site-specific nature of this conjugate.
Figure 4.
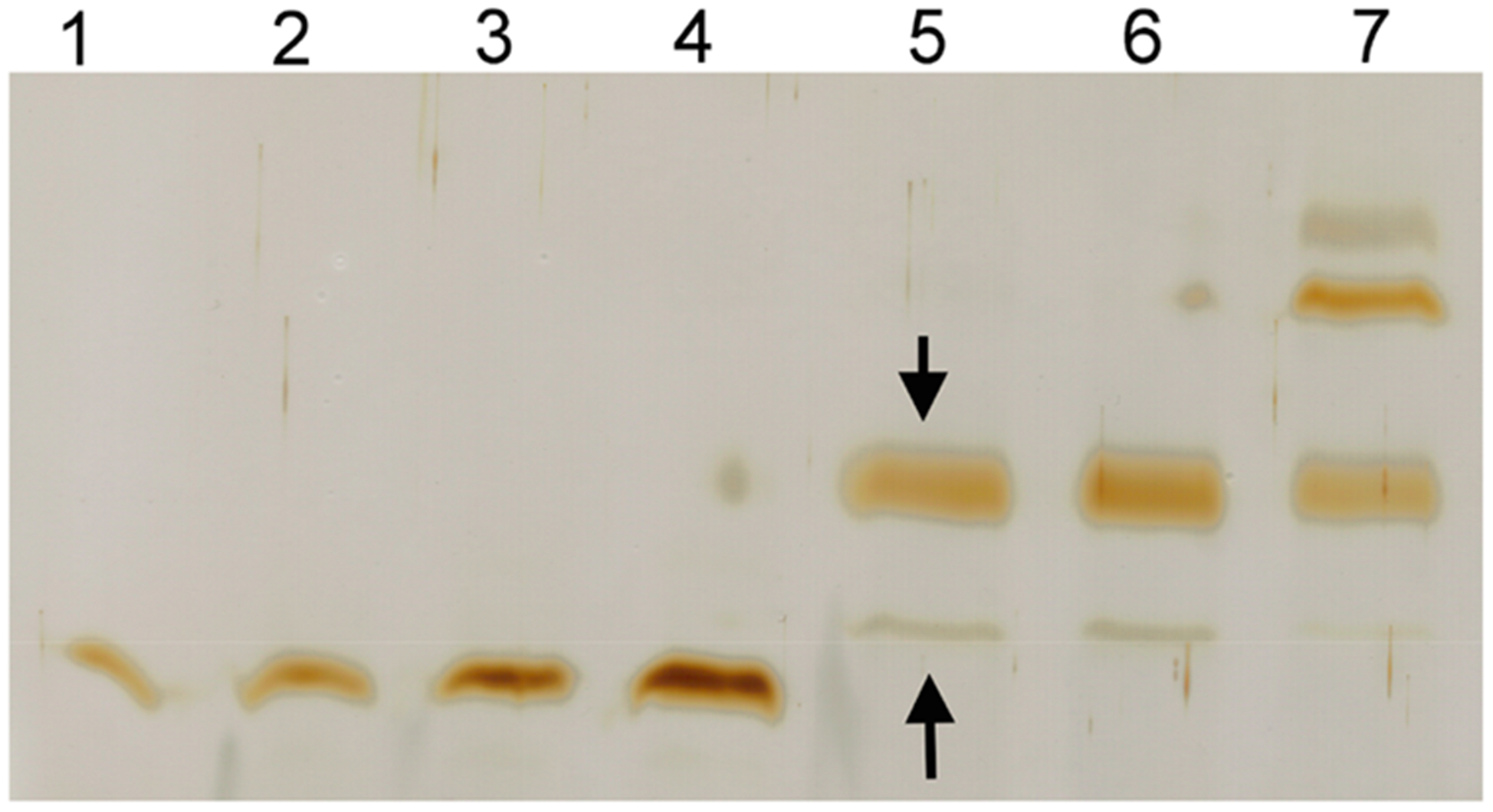
Conjugation of HEL proteins with maleimide-containing liposomes. Conjugation of purified HEL proteins with maleimide on the surface of liposomes was monitored using a 15% (29:1 acrylamide/Bis) reducing SDS–polyacrylamide gel followed by silver staining. Lanes 1–4: 20, 40, 80, and 160 ng of sHEL as part of reference for quantitation of HEL proteins on the gel. Lane 5: purified HELDC–liposome with 1% DSPE–PEG maleimide. Lane 6: purified HELTC–liposome with 1% DSPE–PEG maleimide. Lane 7: purified HELDC–liposome with 10% DSPE–PEG maleimide.
After SEC, most of the free HELDC proteins were removed but left with a tiny faint band on the gel (indicated by the upward arrow, Figure 4) whose intensity was 5 ± 3% of the cross-linked species. This band was consistent from run to run, in general <10% of the intensity compared to cross-linked species, and corresponded to noncovalent association between HEL and liposomes. This band did not appear at the elution time nor in the fractions expected for liposomes when the same quantity of free HEL proteins alone ran through the SEC, suggesting that the SEC could effectively separate liposome away from free proteins. However, it did appear on the gel when HEL proteins were incubated with liposomes followed by SEC, even when there was no maleimide in the lipids, suggesting that it was a noncovalent association between HEL and liposome. In the literature, noncovalent interactions between HEL and the polar headgroup of lipids at neutral pH have been noticed for a long time.50,51 We quantitated the number of HELDC molecules per liposome using methods that we established previously14 (Materials and Methods). There were on average 152 ± 18 (mean ± standard deviation unless otherwise noted) molecules of HELDC specifically conjugated on liposome and on average 8 ± 5 molecules of HELDC associated with the liposome in a noncovalent manner. By changing the molar percentage of DSPE–PEG maleimide in the lipid mixture from 0.1% to 5%, we were able to prepare a set of liposomes with epitope density ranging from 30 to 500 molecules per liposome (Table 1). Taking the size of these liposomes into account, they corresponded to a surface density ranging from 600 to 10000 molecules/μm2, which well covered the range of epitope density observed for naturally occurring biological viruses.1
Table 1.
Epitope Density of Liposomes That Display HEL Proteins on Their Surfaces
| molar percentage of DSPE-PEG maleimide | HEL protein conjugated | average number of HEL molecules per liposomea |
|---|---|---|
| 0.1 | HELDC | 30 ± 4 |
| 0.1 | HELTC | 44 ± 8 |
| 0.2 | HELDC | 62 ± 7 |
| 0.2 | HELTC | 62 ± 8 |
| 1 | HELDC | 152 ± 18 |
| 1 | HELTC | 137 ± 16 |
| 3 | HELDC | 254 ± 29 |
| 3 | HELTC | 288 ± 33 |
| 5 | HELDC | 413 ± 43 |
| 5 | HELTC | 503 ± 72 |
The average number of HEL molecules per liposome refers to HEL molecules specifically attached to the liposome surface through maleimide chemistry. The standard deviations were from three independent repeats of the same experiment.
When the percentage of DSPE–PEG maleimide is 10% or higher, additional cross-linking products appeared on the gel (Lane 7 Figure 4). On the basis of comparison with MW markers, they corresponded to HEL conjugated with two and three molecules of DSPE–PEG maleimide. This was surprising initially given the fact that all eight natural cysteines in HEL protein form four pairs of disulfide bonds that are important for HEL folding and stability27–30 and would not be accessible for cross-linking with maleimide. However, this observation was reproducible from time to time when the percentage of maleimide was high (≥10%). This suggest to us that after the first cross-linking reaction occurs between the engineered cysteine and the maleimide group on the liposome surface excess maleimide groups, when available, can attack the existing disulfide bonds in HEL protein, which results in HEL proteins cross-linked with two or more maleimide groups on the liposomal surface.
Among the four disulfide bonds in the native structure of HEL, three are deeply buried and one pair formed by C6 and C127 is relatively exposed to the solvent.52 This disulfide bond is also close to the free engineered cysteine in space. We have tried to overexpress and purify a HEL protein that carried a total of five mutations: R21Q, R73E, D101R, C6A, and C127I. The former three mutations are necessary to produce a version of HEL protein that has low affinity toward mouse IgHEL antigen-specific B cells. The latter two mutations were designed to remove this pair of disulfide bonds and replace it with van der Waals interactions. This mutant HEL was overexpressed in E. coli, but the amount of soluble proteins available for purification was very low. Several attempts to purify this protein in native states were not successful. This was consistent with the fact that the disulfide bond formed between C6 and C127 is critical for HEL stability.27 In order to preserve the stability of HEL protein on the liposomal surface after cross-linking, we have thus decided to add a large excess of free cysteines at the end of a 1 h cross-linking reaction to quench all of the available maleimide groups that remain on the liposomal surface (Materials and Methods). By limiting the percentage of DSPE–PEG maleimide and by using free cysteines to quench excess maleimide groups, we were able to produce HEL-conjugated liposomes that were sufficiently stable under physiological conditions (see below).
HEL Liposome Stability in 50% Serum.
To examine the stability of HEL-conjugated liposomes under physiologically relevant conditions, we took HEL liposomes prepared from the above procedures, mixed them with 100% freshly thawed fetal bovine serum (FBS) in a 1:1 volume ratio, and incubated the mixture at 37 °C. At the designated time, aliquots of the mixture were taken for measurement of both particle size and epitope density. As shown in Figure 5 for HELDC liposomes prepared with 3% DSPE–PEG maleimide, the size of liposomes did not change over a 2-week period (Figure 5a), with an average diameter of 120 ± 3 nm and no sign of aggregation. The average number of HELDC molecules per liposome was 254 ± 29 on Day 0 and slowly dropped to 208 ± 28 on Day 14 (Figure 5b). By the end of 2 weeks, more than 80% of the epitope remained covalently associated with liposomes. This trend was quantitatively similar to the stability of another sets of maleimide-based liposomes that we prepared recently8 and was much better than proteins conjugated onto the liposomal surface through Ni–NTA noncovalent chemistry as we reported previously.14 This slow dissociation of proteins off of the liposome surface is likely a result of the reverse or exchange reaction of thiol maleimide addition,53 the kinetics of which is slow under biologically relevant conditions54 and modulated by the local environment around the attachment site.55 Since antigen uptake well occurs within 1 week through typical routes of delivery, we can interpret the results in terms of epitope density with confidence.
Figure 5.
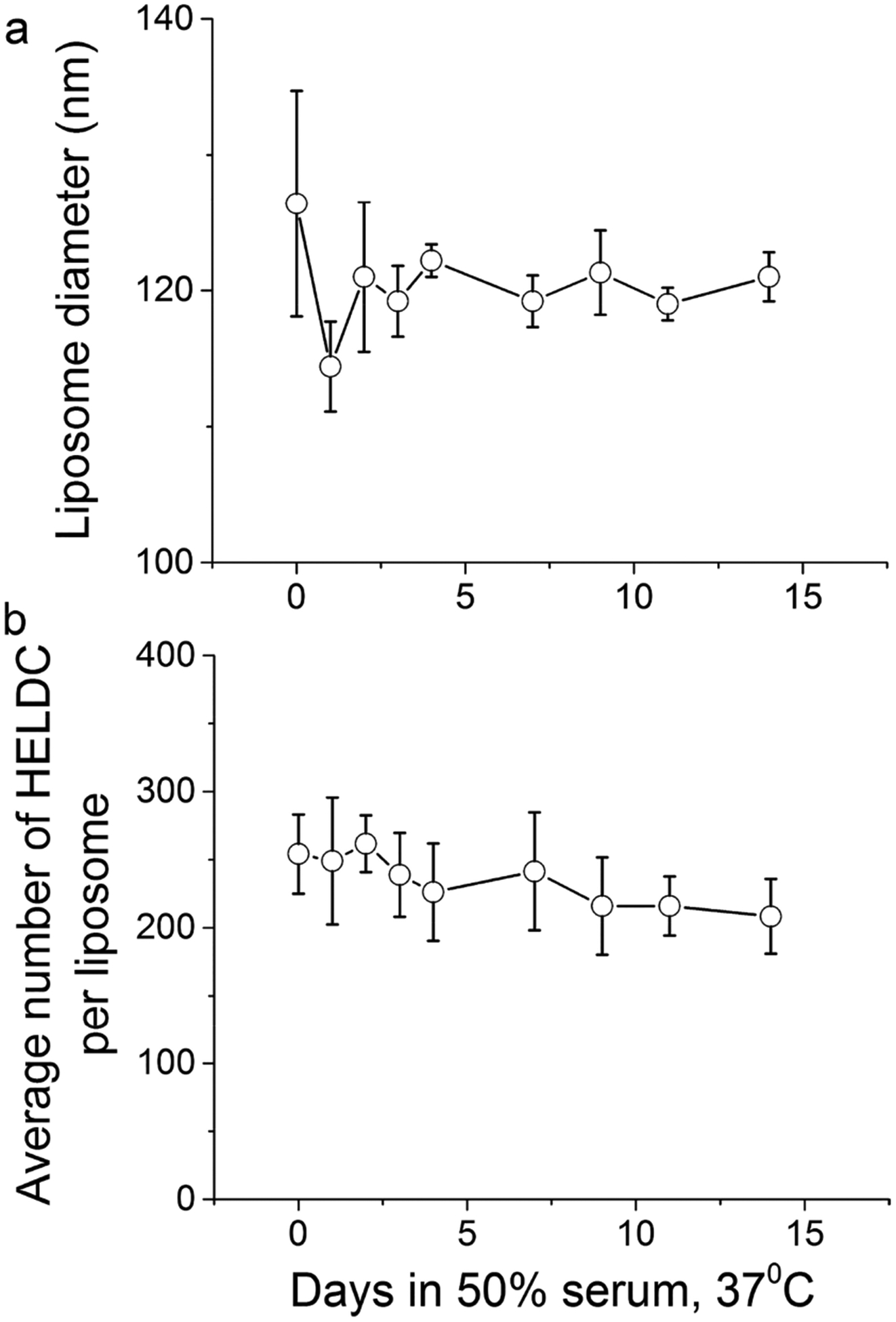
Stability of HEL–liposomes in biologically relevant conditions. (a) Size of HELDC-conjugated liposomes as a function of time upon incubation in 50% FBS at 37 °C. (b) Average number of HELDC molecules per liposome as a function of time upon incubation in 50% FBS at 37 °C. Error bars were standard deviations from three independent repeats of the same experiments.
Activity of HEL-Conjugated Liposomes in Stimulating Antigen-Specific B Cells.
To examine the activity of HEL-conjugated liposomes in stimulating antigen-specific B cells in vitro, we incubated HEL liposomes at serially diluted concentrations with freshly harvested lymphocytes from IgHEL-Nur77-EGFP Tg mice,40 similar to what we did for soluble HEL proteins. As shown in Figure 6a and 6b HELDC-conjugated liposomes (pHELDC) and HELTC-conjugated liposomes (pHELTC) each elicited dose-dependent responses of EGFP expression in these transgenic B cells 24 h upon incubation (black bars). To confirm that these responses were completely Ag specific, liposomes were incubated with lymphocytes from control transgenic mice. B cells from these control transgenic mice carry an EGFP reporter that is under the control of Nur77 regulatory region, but they harbor a wild-type and unrestricted BCR repertoire and lack monoclonal BCRs that are specific for HEL. As a result, we do not expect pHELDC or pHELTC to activate WT Nur77-EGFP B cells through their BCR, but these “control” B cells can nevertheless sensitively detect any non-Ag-specific stimulatory contaminants in liposome preps. Indeed, we can confirm the complete absence of such dose-dependent upregulation of EGFP in “control” B cells treated with HEL–liposomes (WT Nur77-EGFP, gray bars, Figure 6a and 6b) even for this longer time of incubation. This result thus demonstrates that the response we observed for pHELDC and pHELTC in IgHEL Nur77-EGFP B cells is a response specific to HELDC and HELTC proteins conjugated on the liposomal surface. Furthermore, as a control, we also prepared a liposome containing 5% DSPE–PEG maleimide but without any HEL proteins conjugated on them. As shown in Figure 6c, this liposome did not elicit any dose-dependent response in either IgHEL Nur77-EGFP B cells or the control WT Nur77-EGFP B cells. Therefore, the responses in Figure 6a and 6b were exclusively from HEL proteins conjugated on liposomes, not from liposome themselves.
Figure 6.
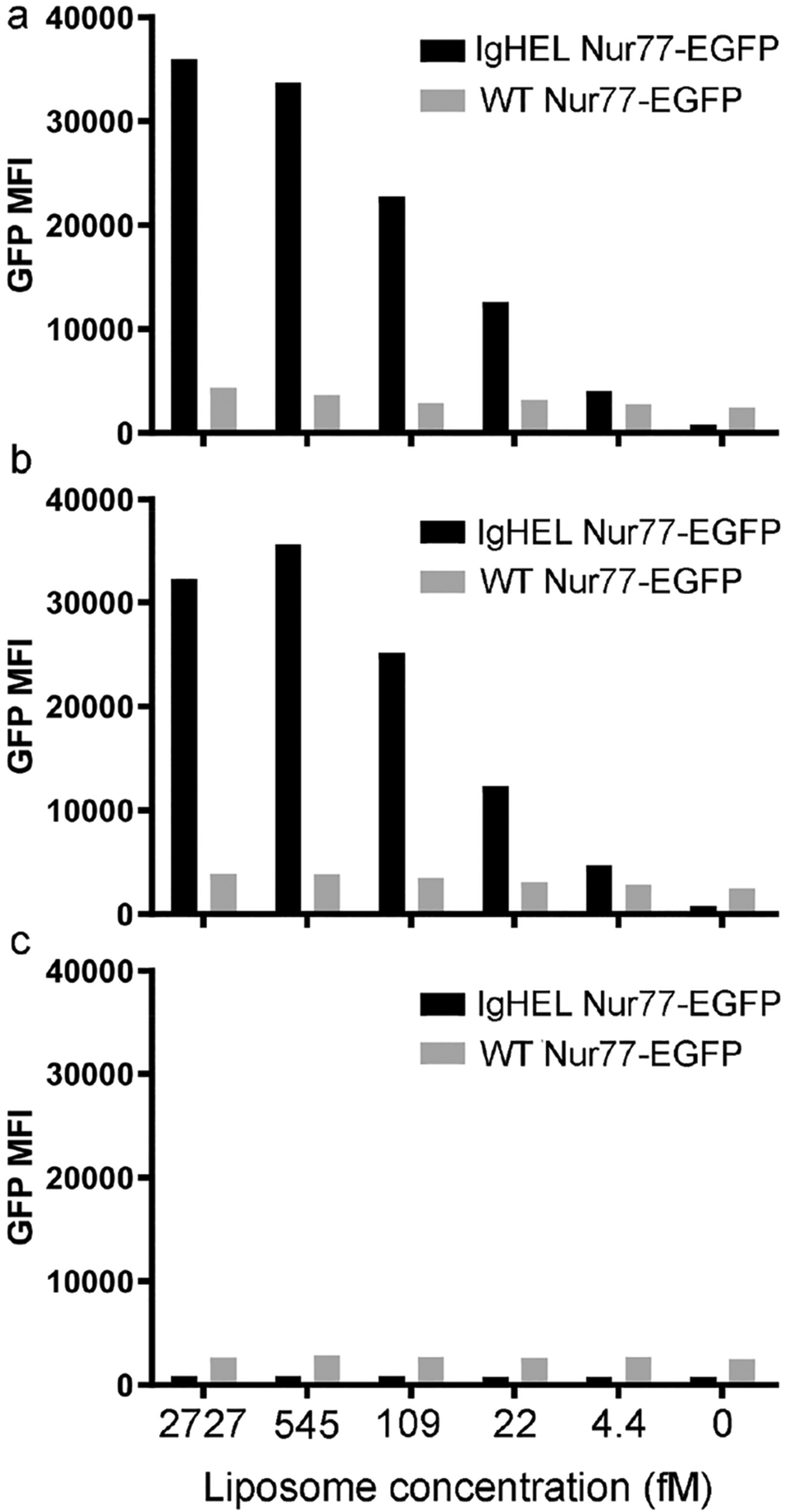
Activation of antigen-specific B cells by HEL-conjugated liposomes. Lymphocytes harvested from CD45.1/CD45.2 IgHEL Tg-Nur77-EGFP and WT-Nur77-EGFP were mixed in a 1:1 ratio and incubated with HEL-conjugated liposomes at serially diluted concentrations for 24 h. After surface staining to detect CD45.1/CD45.2 and B220 in order to distinguish B cells of each genotype, the fluorescence intensity of EGFP per cell was assessed by flow cytometry. EGFP mean fluorescence intensity (MFI) for each B-cell population is shown as a function of the molar concentration of pHELDC (a), pHELTC (b), and control liposome (c) for B cells of each genotype. Data are representative of at least three independent experiments.
Encapsulation of DNA Oligos into Liposomes and Liposomal Stability.
A very important aspect of biological viruses is the presence of nucleic acids genome inside the viral particles. To mimic this aspect qualitatively, we decided to encapsulate nucleic acids molecules into the liposomes. This was done by extrusion of lipid emulsions in the presence of an oligomeric single-stranded DNA followed by purification of the resulting liposomes through a SEC to remove excess free nucleic acid molecules.
Under the current conditions, there were 153 ± 8 molecules of DNA associated with each particle of liposomes on average. This was quantitated by denaturing the liposomes and running the samples in a polyacrylamide gel, followed by fluorescence staining of the single-stranded DNA oligos using SYBR Green II and imaging using a Typhoon scanner (Materials and Methods). As shown in Figure 7a, we could clearly observe the DNA from liposomal samples (last lane indicated by the arrow). By comparing the fluorescence intensity of the DNA with a standard curve of reference DNA molecules on the same gel (Figure 7b), we were able to quantitate the concentration of the DNA associated with liposomes. Control experiments in which the DNA at the same concentration was incubated with empty liposomes after extrusion did not reveal any presence of DNA in the liposomal sample after SEC, indicating that free DNA molecules could be efficiently separated from liposomes by SEC. Thus, the DNA we observed on the gel was exclusively associated with liposomes via the extrusion process and likely encapsulated inside the liposomes instead of nonspecific binding on the outer surface of the liposomes. At this step, the diameter of the liposomes was 110 ± 4 nm. The maximum number of DNA molecules that can be encapsulated in the internal volume of the liposome is 243 at the DNA concentration we used. Comparison with this theoretical estimation yielded an encapsulation efficiency of 63% under current conditions.
Figure 7.
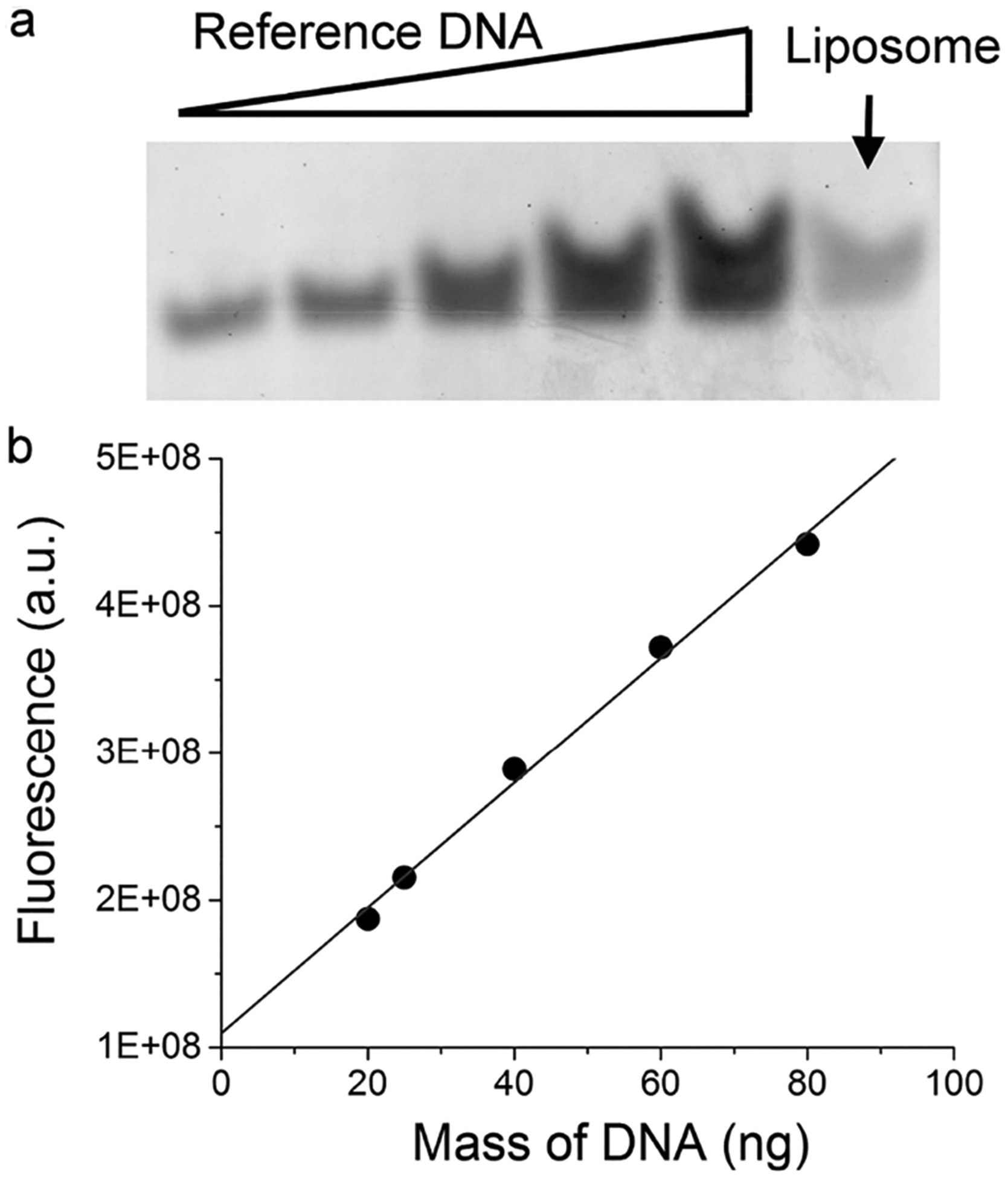
Quantitation of DNA encapsulated inside liposomes. (a) Polyacrylamide gel showing the band of DNA from liposomes, indicated by the arrow. Reference DNA was the same as the DNA encapsulated inside the liposomes except that 20, 25, 40, 60, and 80 ng of the free DNA was loaded in Lanes 1–5. (b) Standard curve that correlates the measured fluorescence of DNA in relation to the mass of the DNA in the gel. Fluorescence intensity for the DNA from liposomes was within the range of fluorescence from reference DNA molecules. Specifically, there was 24.8 ± 1.3 ng of DNA for the liposome sample in panel a. Standard deviation was from three independent repeats of the same experiments.
After this SEC purification to remove free excess DNA, HEL proteins that carry free accessible cysteines were then mixed with the purified liposomes for conjugation onto the liposomal surface, which was followed by a second SEC to remove free excess HEL proteins. The purified liposomes were then assayed for particle size, surface epitope density, and internal nucleic acids content upon incubation in 50% FBS for various amounts of time at 37 °C. As shown in Figure 8a for a liposome containing 3% DSPE–PEG maleimide and conjugated with HELTC, the size of these particles remains unchanged during a 2-week period, with an average diameter of 124 ± 3 nm. The average number of HELTC molecules per liposome dropped slowly from 272 ± 31 on Day 0 to 221 ± 46 on Day 14 (Figure 8b). By the end of 2 weeks, more than 80% of the epitope remained covalently associated with liposomes, which was quantitatively consistent with the trend observed in Figure 5b.
Figure 8.
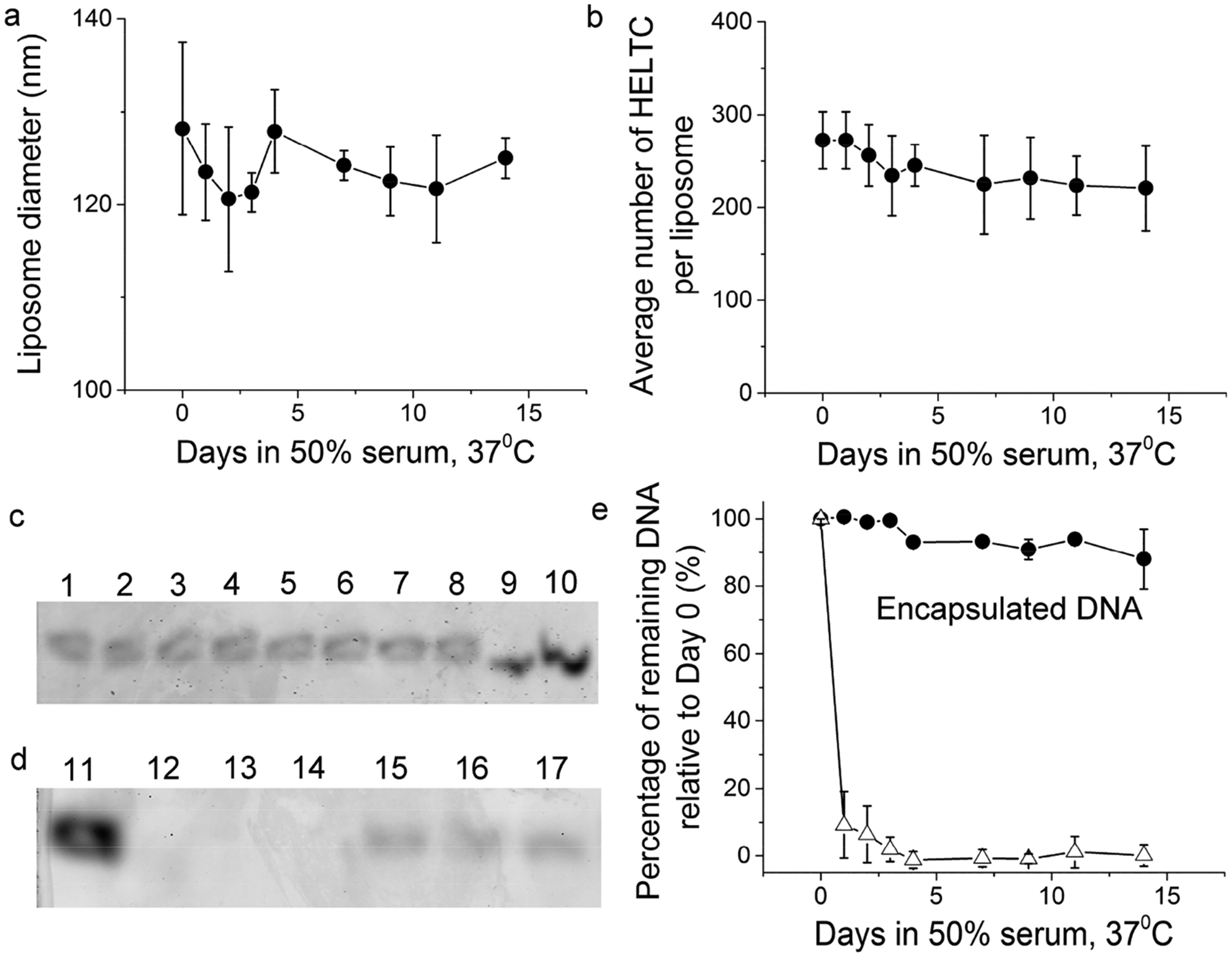
Stability of HEL-conjugated liposomes encapsulating nucleic acids. (a) Size of HELTC-conjugated liposomes as a function of time upon incubation in 50% FBS at 37 °C. (b) Average number of HELTC molecules per liposome as a function of time upon incubation in 50% FBS at 37 °C. (c) Polyacrylamide gels showing the bands of DNA from liposomes (top gel) and admixture of free DNA with liposomes (bottom gel) as a function of time upon incubation in 50% FBS at 37 °C. Lanes 1, 2, 3, 4, 5, 6, 7, and 8 were samples from Day 0, 1, 2, 3, 4, 7, 9, and 11, respectively. Lanes 9 and 10 were 10 and 20 ng of a free reference DNA that was the same as the DNA encapsulated inside the liposomes. Lanes 11, 12, 13, and 14 were samples from Day 0, 1, 2, and 3, respectively. Lanes 15, 16, and 17 were from liposomes encapsulating DNA on Day 0, 1, and 2 and loaded side-by-side for comparison. (e) Percentage of remaining DNA relative to Day 0 as a function of time upon incubation in 50% FBS at 37 °C, shown in black circles for the DNA encapsulated within liposomes and open triangles for the free DNA in an admixture with liposomes. Throughout, error bars were standard deviations from three independent repeats of the same experiments.
Most importantly, the nucleic acids content associated with liposomes only declined minimally during this time period, as shown by the fluorescence gel image of the DNA from various time points after this incubation (Lanes 1–8 in Figure 8c). In sharp contrast, when the same DNA molecules were mixed with empty control liposomes and incubated in 50% FBS at 37 °C, these nucleic acid molecules were quickly degraded within 24 h (Lanes 11–14 in Figure 8d). For the liposomes encapsulating DNA, quantitation of the DNA band intensity showed that more than 90% of the initial DNA content remained after 11 days in 50% FBS at 37 °C (black circles in Figure 8e). Comparison with the control (open triangles) thus demonstrates that a simple admixture of DNA and liposomes did not help protect the DNA from degradation by nucleases in FBS. It was encapsulation into liposomes that protected these DNA molecules from degradation by nucleases present in FBS.
DISCUSSION
The study of cellular and molecular immunology has long benefited from the use of model antigens. The availability of soluble antigens such as HEL, ovalbumin, and polymeric antigens such as NP-based polymers has fueled the deep mechanistic understanding of both humoral and cell-mediated arms of the adaptive immune system. In contrast to these soluble proteins or polymers, biological viruses are more complex supramolecular structures, the immune responses to which are also likely to be multilayered. Although various viral agents are available for in vitro and in vivo studies of immune responses to these antigens, it is often difficult to dissect and therefore understand the contributions of individual viral components and features to the overall integrated immune response. A potential alternative to the direct use of viral particles is man-made synthetic particles that mimic both the qualitative and the quantitative features of biological viruses. Because these particles are assembled from purified components, the molecular basis of immunogenicity is well defined and specific features can be independently modulated to isolate their contribution to B-cell responses. In this study, we explore this idea and report the development of viral mimics based on synthetic liposomal nanoparticles. Because these synthetic particles were built in a stepwise manner, this allows us to isolate the contribution of individual features of the particle to the overall immune response. For example, what is the contribution of epitope display at certain spatial density to B-cell activation? What is the role of nucleic acid genome in shaping B- and T-cell responses? It is our belief that only through this reductionist approach can one build a layer-by-layer integrated and mechanistic understanding of the immune responses toward a complex antigen such as a biological virus.
To afford both in vitro and in vivo studies, these particles need to be stable under physiological conditions. Our previous attempt using Ni-chelating chemistry to attach protein antigens onto liposomes did not fulfill this purpose.14 In this study, we have tested the maleimidE–thiol reaction as the alternative approach to conjugate epitopes onto the liposome surface in an orientation-specific manner. Under physiologically relevant conditions, this covalent chemistry yielded a spatial density of epitopes that was relatively stable over time (Figures 5b and 8b). The fact that these liposomes activated B cells in a highly antigen-specific manner (Figure 6) indicates that the HEL proteins were displayed on the surface of liposomes and accessible for binding to monoclonal BCRs on the surface of those B cells. Moreover, that the DNA encapsulated in these liposomes was protected from degradation by nucleases present in FBS (Figure 8e) indicates that the internal DNA molecules were shielded underneath the lipid bilayer of the liposomes as we expected. Thus, these particles can be applied for both in vitro and in vivo studies, and the result can be rigorously interpreted in molecular terms relevant to viral structures. This approach also has a better potential for clinical translation compared to Ni-chelation chemistry because the maleimidE–thiol conjugation has been used in several FDA-approved drug products.56–58 The wide interest in the pharmaceutical and biotech industry to further optimize the stability of this conjugation54,59 may offer even better and more stable maleimide-based cross-linkers in the future.
Our approach developed here is intrinsically modular in nature. First, it has the potential to be applied to many other proteins, provided that there are specific free cysteines available, either intrinsic in the protein structure or through protein engineering. The stability of the liposome under physiological conditions likely varies with each protein, which needs to be assessed on an individual basis. Second, the internal content of these particles can be replaced with RNA molecules to mimic that of RNA viruses, provided that a RNA solution of sufficiently high concentration is available for encapsulation.
The particles we prepared herein elicited antigen-specific B-cell responses in vitro. It will be of great future interest to study both in vitro and in vivo how the epitope density on these liposomal particles modulates B-cell responses, how epitope density together with internal content contributes to B-cell activation, and how they act independently or synergistically. B-cell activation is a very complex process. The availability of these modular and synthetic viral-like antigens will allow systematic study and dissection of this complex biological process to unravel the detailed mechanisms underneath. These studies will be extremely valuable in light of the cascade of events involved in B-cell antibody responses, from initial BCR engagement to antigen processing, and presentation of antigens on MHC class II for recruitment of T cell help. The availability of these designer particles will allow us to examine in detail how the features in a biological virus influence each step in B-cell activation, and this in turn will greatly facilitate rational vaccine design in the future.
MATERIALS AND METHODS
Purification of HEL Proteins.
We have purified five recombinant HEL proteins in this study: wild-type HEL (wtHEL), a double mutant of HEL (R73E and D101R, called HELD), a tripple mutant of HEL (R21Q, R73E, and D101R, called HELT), HELD that carries an engineered free cysteine close to the C-terminus of the protein (HELDC), and HELT that carries an engineered free cysteine close to the C-terminus of the protein (HELTC). The C-terminal sequence of HELDC or HELTC is as follows: GCRLGGGCHHHHHH, where the cysteine residue underlined is the engineered cysteine free for cross-linking with maleimide. All of these proteins carry a hexahistidine tag at the C-terminus of the respective proteins to facilitate their purification using nickeL–nitrilotriacetic acid (Ni–NTA) technology as the first step of the purification. All proteins were house expressed in E. coli Shuffle T7 express strain (NEB). We have developed a protocol to purify these recombinant HEL proteins to greater than 95% purity as judged by intensity comparisons on an acrylamide gel loaded with different amounts of final purified proteins (Figure 2b). All protein purification procedures were performed at 4 °C unless otherwise noted. Briefly, the E. coli cell paste was resuspended in Buffer A (50 mM Na2HPO4, 0.3 M NaCl, and 10 mM imidazole pH 8.0 at 22 °C). The cell slurry was incubated at 4 °C with constant stirring for 1 h, followed by sonication using a tip sonicator on ice for 15 min, with a 5 min on and 5 min off cycle. The resulting cell lysate was centrifuged at 18.5 kg for 1 h. The supernatant was then loaded onto a prepacked Ni–NTA agarose column at a flow rate of 0.5 mL/min. The column was first washed with Buffer A to baseline and then sequentially washed with Buffer A containing 20 and 35 mM imidazole for 30 min, which was followed by a wash using an alkaline buffer containing 0.1 M Tris and 2 M KCl pH 10 at 4 °C. The bound HEL proteins were then eluted in a linear gradient from 35 to 250 mM increasing concentrations of imidazole in Buffer A over 15 column volumes at a flow rate of 1 mL/min. The fractions containing HEL proteins were pooled, diluted with Buffer C (10 mM Na2HPO4, pH 7.0 at 22 °C) to 100 mM NaCl and loaded onto a HiTrap heparin column (GE) at a flow rate of 1.0 mL/min. The column was washed with Buffer C to baseline and then sequentially washed with Buffer C containing 200 and 300 mM NaCl for 30 min. The bound HEL proteins were then eluted in a linear gradient from 300 to 1000 mM increasing concentrations of NaCl in Buffer C over 15 column volumes at a flow rate of 1 mL/min. The fractions containing HEL proteins were pooled, diluted with Buffer C to 100 mM NaCl, and loaded onto an Enrich S column (Bio-Rad) at a flow rate of 1.0 mL/min. The column was washed with Buffer C to baseline and then sequentially washed with Buffer C containing 100 and 200 mM NaCl for 30 min. The bound HEL proteins were then eluted in a linear gradient from 200 to 1000 mM increasing concentrations of NaCl in Buffer C over 15 column volumes at a flow rate of 1 mL/min. At this stage, the eluted HEL proteins were >95% pure. For HEL proteins that did not have free engineered cysteines, the purified proteins were dialyzed using a 3.5 kDa molecular weight cutoff membrane against 1 × PBS at 4 °C with 3 changes over a course of 48 h. The protein was then filtered through a 0.1 μm sterile syringe filter, the concentration was determined by absorbance at 280 nm, and it was flash frozen in liquid N2 and stored in a −80 °C freezer. For HEL proteins that carried free engineered cysteines, the purified proteins were directly filtered through a 0.1 μm sterile syringe filter, the concentration was determined by absorbance at 280 nm, and they were flash frozen in liquid N2 and stored in −80 °C freezer. For all HEL proteins, introduction of mutations did not change the extinction coefficient of the protein at 280 nm under denaturing conditions, and an extinction coefficient of 3.85 × 104 M−1 cm−1 was used for calculation of protein concentrations.
Preparation of Maleimide-Containing Liposomes.
All liposomes used in this study were prepared using oil-in-water emulsion precursor followed by membrane extrusion as originally described in the literature.60–62 Three different lipids of designated molar ratios were used in the synthesis of liposomes: 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] ammonium salt (DSPE–PEG (2000) maleimide), and cholesterol (Avanti lipids). Briefly, a lipid mixture (2.25 μmol in total) in chloroform was added to a round-bottom flask, blown dry with purified argon, and desiccated by vacuum to form a thin film at the bottom of the flask. For formation of empty liposomes, 300 μL of 1 × PBS buffer was added to hydrate the lipid film through vortex and short bursts of sonication in a water bath. After hydration, the lipid film resuspension was extruded using polycarbonate membrane with a pore size of 100 nm 19 times at 70 °C. The resulting liposomes were then stored at 4 °C for all experiments. For encapsulation of nucleic acid molecules inside the liposomes, a DNA oligo of the following sequence TCCATGACGTTCCTGACGTT (IDT, Coralville, IA) was dissolved in 1 × PBS buffer at a concentration of 0.83 mM, and 300 μL of this solution was used to hydrate the lipid film as described above, followed by extrusion using polycarbonate membrane with a pore size of 100 nm 35 times at 70 °C. The DNA sequence harbors two unmethylated CpG dinucleotides and serves as an effective ligand for Toll-like receptor 9.63 The resulting liposomes were then applied to a Sepharose CL-4B (GE Life Sciences) gel filtration column, as we described previously,14 to separate liposomes away from free excess nucleic acids. The purified liposomes were then stored at 4 °C for all experiments.
Conjugation of HEL Proteins to Liposomes and Purification.
For attachment of HEL proteins onto the liposome surface, the purified proteins were incubated with synthesized liposomes at 22 °C for 1 h at a designated molar ratio between the proteins and the maleimide group. At the end of 1 h, 5 μL of 1 M cysteine solution was added to the reaction mixture and further incubated at 22 °C for 20 min. The mixture was then directly applied to the top of a 20 mL Sepharose CL-4B gel filtration column that was already equilibrated in PBS in order to remove excess free proteins not bound to liposomes. The sample was allowed to enter the column by gravity and then eluted with 1 × PBS at a flow rate of 0.35 mL/min at 4 °C. Absorbance at 280 nm was used to indicate fractions containing liposomes, which were further verified offline using Stewart assay (see below). The fractions with most of the liposomes were stored at 4 °C.
Stewart Assay To Measure Phospholipid Content.
We have used the established Stewart assay64 to determine the phospholipid content in the purified liposomes, based on which the molarity of the liposomes can be determined. It is worth noting, however, that under Stewart assay conditions DSPE–PEG (2000) maleimide alone can also give rise to nonnegligible absorbance at 470 nm that is in linear proportion to the molarity of this specific lipid. Thus, throughout our studies, the phospholipid content measured has been corrected based on the percentage of DSPE–PEG (2000) maleimide in the liposome sample, although this correction was minor for most cases. Purified liposomes conjugated with HEL proteins were examined using Stewart assay for phospholipid quantitation. Briefly, 20 μL of liposome samples was added to 0.5 mL of chloroform in a clean test tube, and then 0.5 mL of ferrothiocynate solution containing 0.1 M ferric chloride hexahydrate and 0.4 M ammonium thiocyanate was added. The mixture was vigorously vortexed for 20 s and centrifuged at 1 kg for 5 min to separate chloroform from the aqueous layers. The lower chloroform layer was then taken for absorption measurement at 470 nm. The phospholipid content was calculated based on comparison with a standard curve of DSPC lipids measured under identical conditions.
Estimation of Liposome Molarity Based on Geometric Considerations.
To determine the molarity of the liposome based on the phospholipid content, we used geometric considerations of liposomes as described previously.14 Briefly, we calculated the number of lipid molecules N per liposome using the following equation
| (1) |
where D is the diameter of the liposome, h is the thickness of the bilayer, and a is the area of the lipid molecule headgroup. In our calculation for liposomes prepared using DSPC as the major lipids, the bilayer thickness h was taken as 6.2 nm, the value for DSPC in the gel state based on small-angle neutron scattering measurements;65 and the area of DSPC lipid headgroup was taken as 0.56 nm2 at 293 K.66 The average diameters of the liposomes were measured using dynamic light scattering for each liposomal sample using a Malvern Zetasizer Nano ZSP at 20 °C. The molarity of the liposomes was then calculated based on the measured phospholipid content and the value of N determined from eq 1.
Quantitation of Epitope Density on Liposomes.
Quantitation of epitope density follows the ensemble method that we described previously.14 This ensemble method was validated previously by single-molecule measurements.14 Briefly, the epitope density, or the average number of protein molecules per liposomal particle, was determined by measuring both the protein concentration and the liposome concentration of the sample in molarity. To determine both concentrations, the liposome sample was first purified through SEC to remove free proteins. The purified liposome was then loaded onto a 15% (29:1 acrylamide/Bis) reducing SDS polyacrylamide gel, together with a set of known quantities of HEL proteins, ranging from 2 to 200 ng, for construction of a standard curve. A minimum of eight reference points were included in each standard curve. Most of the time the gel was silver stained for quantitation. When FBS was included with liposomes, the gel was electrotransferred onto a supported nitrocellulose membrane and then further probed by Western blotting. Primary antibody used for detection of HEL was rabbit anti-HEL polyclonal antibodies (Rockland ImmunoChemicals), which was diluted at 1:1000. The secondary antibody was anti-rabbit alkaline phosphatase-conjugated secondary antibody (Santa Cruz Biotech), which was diluted at 1:2000. Protein bands were developed with the Nitroblue tetrazolium chloride/5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt substrates (NBT/BCIP, Roche) in a buffer containing 0.1 M Tris·HCl, 0.1 M NaCl, and 0.05 M MgCl2 at pH 9.5. The silver-stained gel or the Western blot was scanned using a Dell V305 all-in-one scanner, and the band intensities were quantitated using ImageJ (NIH, Bethesda, MD, USA, http://imagej.nih.gov/ij/). The concentration of proteins in the liposome sample was quantitated by comparison with the standard curve. All intensities to be measured were within the range of intensities shown by known reference samples. The average number of protein molecules per liposome was then taken as the ratio between the molar concentration of proteins and the molar concentration of liposomes.
Quantitation of Nucleic Acids Content within Liposomes.
The nucleic acids content, or the average number of DNA molecules per liposomal particle, was determined by measuring both the DNA concentration and the liposome concentration of the sample in molarity. To determine DNA concentrations, the liposome sample that was purified through SEC was loaded onto a 15% polyacrylamide gel (19:1 acrylamide/Bis), together with a set of known quantities of the same DNA molecules, ranging from 5 to 100 ng, for construction of the standard curve. A minimum of five reference points were included in each standard curve. The gel was stained with SYBR Green II (ThermoFisher), and the fluorescence was imaged using a multimode Typhoon scanner. The band intensities were quantitated using ImageQuant 5.0 (Molecular Dynamics). The concentration of DNA in the liposome sample was quantitated by comparison with the standard curve as shown in Figure 7b. All intensities to be measured were within the range of intensities shown by known reference samples. The average number of DNA molecules per liposome was then taken as the ratio between the molar concentration of DNA and the molar concentration of liposomes.
Stability Assay for Liposomes.
To measure the stability of liposomes under physiologically relevant conditions, we mixed liposome samples with an equal volume of freshly thawed FBS on ice. An aliquot was taken immediately for both particle size and epitope density measurements as time zero. The mixture in a polypropylene test tube was then placed in an air-circulated incubator that was set at 37 °C. At the designated time, aliquots were taken out of the tube for both particle size and epitope density measurements. The original mixture in the test tube was immediately put back to the incubator for continued incubation and the next time points.
Mice.
Nur77-EGFP mice, IgHEL Tg (MD4) mice were previously described.40,67 CD45.1+ BoyJ mice were obtained from The Jackson Laboratory. All strains were fully back-crossed to C57BL/6 genetic background. All mice were housed in a specific pathogen-free facility at UCSF according to University and National Institutes of Health guidelines.
In Vitro B Cell Culture and Stimulation.
Lymphocytes were harvested into single-cell suspension and plated at a concentration of 5 × 105 cells/200 μL in round-bottom 96-well plates in complete RPMI media with stimuli for 6 or 24 h with stimuli (soluble HEL protein, HEL–liposomes, or blank liposomes). The antigen-specific B cells can stay alive 3 days or longer in the presence of HEL-conjugated liposomes. The complete RPMI media contained the following components: RPMI 1640 with ʟ-glutamine (MediaTech), PenStrepGlut (Life Technologies), Non-Essential Amino Acids (Life Technologies), 0.88 mM sodium pyruvate (Life Technologies), 8.8 mM HEPES Buffer (Life Technologies), 48 μM β-mercaptoethanol and 10% FCS, heat inactivated (Omega Scientific). Commercial soluble HEL was purchased from Sigma. In vitro cultured cells were then stained with fluorophore-conjugated antibodies against B220, CD45.1, and CD45.2 in FACS buffer on ice for 15 min and subsequently washed twice. Samples were then stained with fixable near IR live/dead stain (Invitrogen) per the manufacturer’s instructions and subsequently washed and fixed with BD cytofix per the manufacturer’s instructions. Stained samples were analyzed on a Fortessa flow cytometer (Becton Dickson). Data analysis was performed using FlowJo (v9.7.6) software (Treestar Incorporated, Ashland, OR). Statistical analysis and graphs were generated using Prism v6 (GraphPad Software, Inc.).
ACKNOWLEDGMENTS
This work was supported by an NIH/NIAID grant (1R21AI135559-01A1) to W.C., MCubed project #8290 and the Team Science Award to W.C. by the University of Michigan, and NIH/NIAMS grant (R01AR069520) to J.Z. We thank the Cheng and Zikherman Lab members for helpful discussions.
ABBREVIATIONS
- HEL
hen egg lysozyme
- HELD
HEL mutant R73E and D101R
- HELT
HEL mutant R21Q, R73E, and D101R
- HELDC
HELD that carries an engineered free cysteine close to the C-terminus of the protein
- HELTC
HELT that carries an engineered free cysteine close to the C-terminus of the protein
- sHEL
soluble HEL from commercial sources
- pHELDC
liposomes that display HELDC
- pHELTC
liposomes that display HELTC
- EGFP
enhanced green fluorescent protein
- DSPC
1,2-distearoyl-sn-glycero-3-phosphocholine
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocho-line
- DSPE–PEG maleimide
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] ammonium salt
- Tg
transgenic
- BCR
B-cell antigen receptors
- IgHEL
monoclonal B-cell antigen receptor specific for HEL
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.9b00825
The authors declare no competing financial interest.
Contributor Information
Wei-Yun Wholey, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
James L. Mueller, Division of Rheumatology, Rosalind Russell and Ephraim P. Engleman Arthritis Research Center, Department of Medicine, University of California, San Francisco, California 94143, United States
Corey Tan, Division of Rheumatology, Rosalind Russell and Ephraim P. Engleman Arthritis Research Center, Department of Medicine, University of California, San Francisco, California 94143, United States.
Jeremy F. Brooks, Division of Rheumatology, Rosalind Russell and Ephraim P. Engleman Arthritis Research Center, Department of Medicine, University of California, San Francisco, California 94143, United States
Julie Zikherman, Division of Rheumatology, Rosalind Russell and Ephraim P. Engleman Arthritis Research Center, Department of Medicine, University of California, San Francisco, California 94143, United States.
Wei Cheng, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, Michigan 48109, United States;.
REFERENCES
- (1).Cheng W (2016) The Density Code for the Development of a Vaccine? J. Pharm. Sci 105 (11), 3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cyster JG, and Allen CDC (2019) B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 177 (3), 524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, and Zinkernagel RM (1993) The influence of antigen organization on B cell responsiveness. Science 262 (5138), 1448–51. [DOI] [PubMed] [Google Scholar]
- (4).Dintzis HM, Dintzis RZ, and Vogelstein B (1976) Molecular determinants of immunogenicity: the immunon model of immune response. Proc. Natl. Acad. Sci. U. S. A 73 (10), 3671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chackerian B, Lowy DR, and Schiller JT (1999) Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. U. S. A 96 (5), 2373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Feldmann M, and Basten A (1971) The relationship between antigenic structure and the requirement for thymus-derived cells in the immune response. J. Exp. Med 134 (1), 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chackerian B, Lowy DR, and Schiller JT (2001) Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J. Clin. Invest 108 (3), 415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen Z, Wholey WY, Hassani Najafabadi A, Moon JJ, Grigorova I, Chackerian B, and Cheng W (2020) Self-Antigens Displayed on Liposomal Nanoparticles above a Threshold of Epitope Density Elicit Class-Switched Autoreactive Antibodies Independent of T Cell Help. J. Immunol 204 (2), 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Takahashi Y, Dutta PR, Cerasoli DM, and Kelsoe G (1998) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med 187 (6), 885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Han S, Zheng B, Dal Porto J, and Kelsoe G (1995) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J. Exp. Med 182 (6), 1635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jacob J, Kassir R, and Kelsoe G (1991) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med 173 (5), 1165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jacob J, and Kelsoe G (1992) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. II. A common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J. Exp. Med 176 (3), 679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jacob J, Przylepa J, Miller C, and Kelsoe G (1993) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J. Exp. Med 178 (4), 1293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen Z, Moon JJ, and Cheng W (2018) Quantitation and Stability of Protein Conjugation on Liposomes for Controlled Density of Surface Epitopes. Bioconjugate Chem. 29 (4), 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, and DeFranco AL (2011) Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity 34 (3), 375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eckl-Dorna J, and Batista FD (2009) BCR-mediated uptake of antigen linked to TLR9 ligand stimulates B-cell proliferation and antigen-specific plasma cell formation. Blood 113 (17), 3969–77. [DOI] [PubMed] [Google Scholar]
- (17).Wigton EJ, DeFranco AL, and Ansel KM (2019) Antigen Complexed with a TLR9 Agonist Bolsters c-Myc and mTORC1 Activity in Germinal Center B Lymphocytes. Immunohorizons 3 (8), 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Marques-Gallego P, and de Kroon AI (2014) Ligation strategies for targeting liposomal nanocarriers. BioMed Res. Int 2014, 129458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, and Moudgil K (1993) Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol 11, 729–66. [DOI] [PubMed] [Google Scholar]
- (20).Unanue ER, Turk V, and Neefjes J (2016) Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu. Rev. Immunol 34, 265–97. [DOI] [PubMed] [Google Scholar]
- (21).Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, Bourne K, Reed JH, Patterson K, Porebski BT, Brink R, Christ D, and Goodnow CC (2018) Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science 360 (6385), 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chan TD, Wood K, Hermes JR, Butt D, Jolly CJ, Basten A, and Brink R (2012) Elimination of germinal-center-derived self-reactive B cells is governed by the location and concentration of self-antigen. Immunity 37 (5), 893–904. [DOI] [PubMed] [Google Scholar]
- (23).Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. (1988) Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334 (6184), 676–82. [DOI] [PubMed] [Google Scholar]
- (24).Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, and Goodnow CC (1991) Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 353 (6346), 765–9. [DOI] [PubMed] [Google Scholar]
- (25).Blake CC, Koenig DF, Mair GA, North AC, Phillips DC, and Sarma VR (1965) Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature 206 (4986), 757–61. [DOI] [PubMed] [Google Scholar]
- (26).Matagne A, and Dobson CM (1998) The folding process of hen lysozyme: a perspective from the ‘new view’. Cell. Mol. Life Sci 54 (4), 363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cooper A, Eyles SJ, Radford SE, and Dobson CM (1992) Thermodynamic consequences of the removal of a disulphide bridge from hen lysozyme. J. Mol. Biol 225 (4), 939–43. [DOI] [PubMed] [Google Scholar]
- (28).Tachibana H, Oka T, and Akasaka K (2001) Native-like tertiary structure formation in the alpha-domain of a hen lysozyme two-disulfide variant. J. Mol. Biol 314 (2), 311–20. [DOI] [PubMed] [Google Scholar]
- (29).Shioi S, Imoto T, and Ueda T (2004) Analysis of the early stage of the folding process of reduced lysozyme using all lysozyme variants containing a pair of cysteines. Biochemistry 43 (18), 5488–93. [DOI] [PubMed] [Google Scholar]
- (30).Silvers R, Sziegat F, Tachibana H, Segawa S, Whittaker S, Gunther UL, Gabel F, Huang JR, Blackledge M, Wirmer-Bartoschek J, and Schwalbe H (2012) Modulation of structure and dynamics by disulfide bond formation in unfolded states. J. Am. Chem. Soc 134 (15), 6846–54. [DOI] [PubMed] [Google Scholar]
- (31).DeSantis MC, Kim JH, Song H, Klasse PJ, and Cheng W (2016) Quantitative Correlation between Infectivity and Gp120 Density on HIV-1 Virions Revealed by Optical Trapping Virometry. J. Biol. Chem 291 (25), 13088–13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, and Schwalbe H (2002) Long-range interactions within a nonnative protein. Science 295 (5560), 1719–22. [DOI] [PubMed] [Google Scholar]
- (33).Yokota A, Izutani K, Takai M, Kubo Y, Noda Y, Koumoto Y, Tachibana H, and Segawa S (2000) The transition state in the folding-unfolding reaction of four species of three-disulfide variant of hen lysozyme: The role of each disulfide bridge. J. Mol. Biol 295 (5), 1275–1288. [DOI] [PubMed] [Google Scholar]
- (34).Fischer B, Perry B, Phillips G, Sumner I, and Goodenough P (1993) Physiological consequence of expression of soluble and active hen egg white lysozyme in Escherichia coli. Appl. Microbiol. Biotechnol 39 (4–5), 537–40. [DOI] [PubMed] [Google Scholar]
- (35).Lamppa JW, Tanyos SA, and Griswold KE (2013) Engineering Escherichia coli for soluble expression and single step purification of active human lysozyme. J. Biotechnol 164 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Paus D, Phan TG, Chan TD, Gardam S, Basten A, and Brink R (2006) Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med 203 (4), 1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Studier FW (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol 219 (1), 37–44. [DOI] [PubMed] [Google Scholar]
- (38).Wetter LR, and Deutsch HF (1951) Immunological studies on egg white proteins. IV. Immunochemical and physical studies of lysozyme. J. Biol. Chem 192 (1), 237–242. [PubMed] [Google Scholar]
- (39).Gadgil H, and Jarrett HW (1999) Heparin elution of transcription factors from DNA-Sepharose columns. J. Chromatogr A 848 (1–2), 131–8. [DOI] [PubMed] [Google Scholar]
- (40).Zikherman J, Parameswaran R, and Weiss A (2012) Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature 489 (7414), 160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mittelstadt PR, and DeFranco AL (1993) Induction of early response genes by cross-linking membrane Ig on B lymphocytes. J. Immunol 150 (11), 4822–4832. [PubMed] [Google Scholar]
- (42).Noviski M, Mueller JL, Satterthwaite A, Garrett-Sinha LA, Brombacher F, and Zikherman J IgM and IgD B cell receptors differentially respond to endogenous antigens and control B cell fate. eLife 2018, 7 DOI: 10.7554/eLife.35074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fleiner M, Benzinger P, Fichert T, and Massing U (2001) Studies on protein-liposome coupling using novel thiol-reactive coupling lipids: influence of spacer length and polarity. Bioconjugate Chem. 12 (4), 470–5. [DOI] [PubMed] [Google Scholar]
- (44).Harasym TO, Tardi P, Longman SA, Ansell SM, Bally MB, Cullis PR, and Choi LS (1995) Poly(ethylene glycol)-modified phospholipids prevent aggregation during covalent conjugation of proteins to liposomes. Bioconjugate Chem. 6 (2), 187–94. [DOI] [PubMed] [Google Scholar]
- (45).Mabrey S, and Sturtevant JM (1976) Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proc. Natl. Acad. Sci. U. S. A 73 (11), 3862–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Needham D, and Nunn RS (1990) Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J 58 (4), 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Watson DS, Endsley AN, and Huang L (2012) Design considerations for liposomal vaccines: influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine 30 (13), 2256–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gregoriadis G, and Davis C (1979) Stability of liposomes in vivo and in vitro is promoted by their cholesterol content and the presence of blood cells. Biochem. Biophys. Res. Commun 89 (4), 1287–93. [DOI] [PubMed] [Google Scholar]
- (49).Kirby C, Clarke J, and Gregoriadis G (1980) Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem. J 186 (2), 591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zschornig O, Paasche G, Thieme C, Korb N, and Arnold K (2005) Modulation of lysozyme charge influences interaction with phospholipid vesicles. Colloids Surf., B 42 (1), 69–78. [DOI] [PubMed] [Google Scholar]
- (51).Posse E, De Arcuri BF, and Morero RD (1994) Lysozyme interactions with phospholipid vesicles: relationships with fusion and release of aqueous content. Biochim. Biophys. Acta, Biomembr 1193 (1), 101–6. [DOI] [PubMed] [Google Scholar]
- (52).David C, Foley S, and Enescu M (2009) Protein S-S bridge reduction: a Raman and computational study of lysozyme interaction with TCEP. Phys. Chem. Chem. Phys 11 (14), 2532–42. [DOI] [PubMed] [Google Scholar]
- (53).Baldwin AD, and Kiick KL (2011) Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjugate Chem. 22 (10), 1946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lyon RP, Setter JR, Bovee TD, Doronina SO, Hunter JH, Anderson ME, Balasubramanian CL, Duniho SM, Leiske CI, Li F, and Senter PD (2014) Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol 32 (10), 1059–62. [DOI] [PubMed] [Google Scholar]
- (55).Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al. (2012) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol 30 (2), 184–9. [DOI] [PubMed] [Google Scholar]
- (56).Senter PD, and Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol 30 (7), 631–7. [DOI] [PubMed] [Google Scholar]
- (57).Peddi PF, and Hurvitz SA (2013) Trastuzumab emtansine: the first targeted chemotherapy for treatment of breast cancer. Future Oncol. 9 (3), 319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Lang L (2008) FDA approves Cimzia to treat Crohn’s disease. Gastroenterology 134 (7), 1819. [DOI] [PubMed] [Google Scholar]
- (59).Fontaine SD, Reid R, Robinson L, Ashley GW, and Santi DV (2015) Long-term stabilization of maleimide-thiol conjugates. Bioconjugate Chem. 26 (1), 145–52. [DOI] [PubMed] [Google Scholar]
- (60).Hope MJ, Bally MB, Webb G, and Cullis PR (1985) Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta, Biomembr 812 (1), 55–65. [DOI] [PubMed] [Google Scholar]
- (61).Mayer LD, Hope MJ, and Cullis PR (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta, Biomembr 858 (1), 161–8. [DOI] [PubMed] [Google Scholar]
- (62).Olson F, Hunt CA, Szoka FC, Vail WJ, and Papahadjopoulos D (1979) Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta, Biomembr 557 (1), 9–23. [DOI] [PubMed] [Google Scholar]
- (63).Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, and Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408 (6813), 740–5. [DOI] [PubMed] [Google Scholar]
- (64).Stewart JC (1980) Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem 104 (1), 10–4. [DOI] [PubMed] [Google Scholar]
- (65).Ashkar R, Nagao M, Butler PD, Woodka AC, Sen MK, and Koga T (2015) Tuning membrane thickness fluctuations in model lipid bilayers. Biophys. J 109 (1), 106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Qin SS, Yu ZW, and Yu YX (2009) Structural characterization on the gel to liquid-crystal phase transition of fully hydrated DSPC and DSPE bilayers. J. Phys. Chem. B 113 (23), 8114–23. [DOI] [PubMed] [Google Scholar]
- (67).Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink R. a., Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, and Raphael K (1988) Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682. [DOI] [PubMed] [Google Scholar]