

Abstract
Energy metabolism is the process of generating energy (i.e. ATP) from nutrients. This process is indispensable for cell homeostasis maintenance and responses to varying conditions. Cells require energy for growth and maintenance and have evolved to have multiple pathways to produce energy. Both genetic and functional studies have demonstrated that energy metabolism, such as glucose, fatty acid, and amino acid metabolism, plays important roles in the formation and function of bone cells including osteoblasts, osteocytes, and osteoclasts. Dysregulation of energy metabolism in bone cells consequently disturbs the balance between bone formation and bone resorption. Metabolic diseases have also been reported to affect bone homeostasis. Bone morphogenic protein (BMP) signaling plays critical roles in regulating the formation and function of bone cells, thus affecting bone development and homeostasis. Mutations of BMP signaling-related genes in mice have been reported to show abnormalities in energy metabolism in many tissues, including bone. In addition, BMP signaling correlates with critical signaling pathways such as mTOR, HIF, Wnt, and self-degradative process autophagy to coordinate energy metabolism and bone homeostasis. These findings will provide a newly emerging target of BMP signaling and potential therapeutic strategies and the improved management of bone diseases. This review summarizes the recent advances in our understanding of (1) energy metabolism in regulating the formation and function of bone cells, (2) function of BMP signaling in whole body energy metabolism, and (3) mechanistic interaction of BMP signaling with other signaling pathways and biological processes critical for energy metabolism and bone homeostasis.
Keywords: BMP signaling, Energy metabolism, Bone homeostasis, Diabetes, Autophagy
1. Introduction
The prevalence of metabolic disorders, such as diabetes, has increased drastically worldwide in recent years [1]. This phenomenon prompts a renewed interest in the study of energy metabolism and cellular bioenergetics. Recent advances in mouse genetics, together with biochemical methods for analyzing energy metabolism, have greatly improved our understanding of the mechanisms that integrate energy metabolism in the whole organism. Cellular metabolism has become a focus for basic investigations into the pathophysiology of chronic diseases.
Bone remodeling occurs throughout life and involves the coordination between bone resorption and new bone formation [2,3]. It has been known that bone resorption and formation are regulated by systemic and local release of cytokines and growth factors, including the bone morphogenetic proteins (BMPs) [4]. The bone remodeling unit is composed of multiple cell types including osteoblasts, osteocytes, and osteoclasts [5]. The remodeling cycle is initiated by bone resorbing osteoclasts via releasing protons and selected proteases. Then, stromal-derived mesenchymal cells are recruited to the freshly excavated site and differentiate into bone forming osteoblasts [6]. Mature osteoblasts produce and secrete type I collagen and mineralize bone, and in turn, give rise to bone lining cells or osteocytes [7]. Osteocytes secrete os-teokines such as sclerostin and function as mechanical sensors [8]. The process of remodeling takes ~100 days in humans from resorption to formation, and the entire skeleton is remodeled every 10 years [9]. It is therefore plausible that a high energetic cost is associated with these diverse skeletal functions [9]. Proper regulation and coordination of differentiation and function of each cell type is essential to maintain the adult skeletal homeostasis by coupling bone resorption to bone formation. Imbalances between osteoclast-mediated bone resorption and osteoblast-mediated bone formation will cause bone diseases, such as osteoporosis [10]. Osteoporosis is a major public health condition leading to an increased susceptibility to fractures. Recently, studies both in mouse models and in clinical cases showed that osteoporosis can be attributed to many pathological conditions that impact energy metabolism, such as diabetes mellitus and anorexia nervosa. Therefore, changes in bone cell energy metabolism have significant roles in regulating the differentiation and function of bone cells.
BMPs belong to the transforming growth factor-β (TGF-β) super-family [11,12]. BMPs bind to serine/threonine kinase receptors and initiate an intracellular transduction cascade through the canonical signaling mediated through SMADs and/or non-canonical BMP signaling through small GTPases, PI3K/AKT, LIM kinase-1 (LIMK1), and various types of MAPKs. BMP signaling has diverse functions in skeletogenesis and bone homeostasis [11]. BMPs have been demonstrated to play important roles in regulating the differentiation and function of osteoblasts and osteoclasts [11–15]. Genetic mutations of BMP signaling cause a wide variety of inheritable bone diseases in humans [11,12]. Recently, BMP signaling has been identified to have novel functions in regulating energy metabolism in many tissues, including adipose. This occurs directly or indirectly through dynamic cross-talk with other signals or cell processes such as mTOR, hypoxia, Wnt, autophagy, parathyroid hormone (PTH), and reactive oxygen species (ROS) [16,17]. These findings suggest that machinery for energy metabolism in bone cells is an unexplored target of BMP signaling in regulating bone homeostasis. Until recently, information about the energy metabolism of bone cells, especially the role of BMP signaling in regulating energy metabolism for bone formation and resorption, has been largely absent.
In this article, we provide a review of the experiment evidence of (1) the metabolic properties of bone cells and the mechanisms that control energy substrate utilization and bioenergetics, (2) clinical correlations of metabolic diseases with bone homeostasis, (3) the roles of BMP signaling in controlling energy metabolism, and (4) interaction of BMP signaling with intrinsic mechanisms of energy metabolism in bone. Special attention is devoted to identifying gaps in our current understanding regarding the involvement of BMP signaling in this new area of skeletal biology. Additional studies are necessary to fill these gaps and better define the significance of BMP signaling function in bioenergetics of bone cells. These studies have opened new prospects for identifying novel targets for better prognostics and therapies against bone diseases.
2. Energy metabolism of bone cells
2.1. Overview of energy metabolism
Cells harvest the energy to generate adenosine 5′-triphosphate (ATP) from ingested or stored carbohydrates, fats, and proteins by processing them through a series of enzyme-catalyzed reactions [18]. Glucose derived from either dietary carbohydrates or glycogen break-down is the preferred fuel for most cells in the body. Glucose is processed to generate ATP through glycolysis in the cytoplasm and oxidative phosphorylation in the mitochondria [19]. During the nine sequential reactions of glycolysis, ATP is generated from glucose by dismantling and reshuffling chemical bonds in a series of linked enzymatic reactions that produce pyruvate. The terminal product, pyruvate, can be converted to acetyl coenzyme A (acetyl CoA) by pyruvate dehydrogenase in the presence of oxygen for entry into the tricarboxylic acid (TCA) cycle or, alternately, can be converted to lactate by lactate dehydrogenase in hypoxia conditions. In some situations, glucose is converted to lactate in the presence of oxygen, referred to as aerobic glycolysis (aka the “Warburg Effect”) [20]. In addition to glucose, fatty acids and amino acids can also be used as substrates for generating energy through oxidative phosphorylation [21–23]. Free fatty acids, from either dietary fats or generated through lipolysis of adipocyte triglycerides, are another potent substrate for energy production and can also be stored in tissues. Oxidative metabolism of fats yields more than twice the energy of an equal weight of dry carbohydrates or proteins [21]. Certain amino acids can deaminate into “glucogenic” carbon-based molecules, and thus be used as substrates for glycolysis and/or further progression through the TCA cycle to generate ATP [23]. In this chapter, we will review the current understanding of the exogenous energy sources and bioenergetic pathways that are utilized by bone cells to meet their ATP need.
2.2. Energy metabolism in osteoblasts
2.2.1. Glucose metabolism in osteoblasts
Glucose has been long known as a major energy source for osteoblasts. Previous studies using radiolabeled glucose analogs have confirmed a significant uptake of glucose by bone [24,25]. Recent evidence suggests that osteoblast lineage cells generate ATP from glucose through both glycolysis and the TCA cycle (Fig. 1A). It has been shown that progenitors of osteoblasts, including mesenchymal stem cells, primarily use glycolysis as their energy source, but switch to use both glycolysis and the TCA cycle after osteogenic induction [26–28]. Of note, in fully differentiated osteoblasts, glycolysis becomes predominant again to maintain ATP production (Fig. 1A) [28]. Increases in the oxygen consumption and extracellular acidification rates have been identified during osteogenic differentiation in both preosteoblastic cell line MC3T3-E1C4 and primary calvarial osteoblasts [28]. C3H/HeJ mice have a higher bone formation rate in vivo than C57BL6 mice. It has been identified that the oxygen consumption rates are higher in calvarial osteoblasts from C3H mice than those from C57BL6 mice [28]. These findings suggest that the activity of the TCA cycle positively correlates with bone formation. More carefully designed in vitro studies have confirmed that glycolysis is predominant in primary calvarial osteoblasts even in the presence of oxygen, regarded as “aerobic glycolysis” [29,30]. The necessity of aerobic glycolysis is speculated as need for (1) generating ATP at a faster rate, (2) providing metabolic intermediates (e.g. glucose carbons) to support a synthesis of matrix proteins, (3) generating citrate that is structurally important for the formation of apatite nanocrystals in bones. However, it is not clear yet which of these are relevant for osteoblasts. Altogether, these findings suggest that osteoblast differentiation and function are coupled with bioenergetic programs. Osteoblasts can adjust energy mechanisms to meet changing functional demands during their lifespan (Fig. 1A).
Fig. 1.
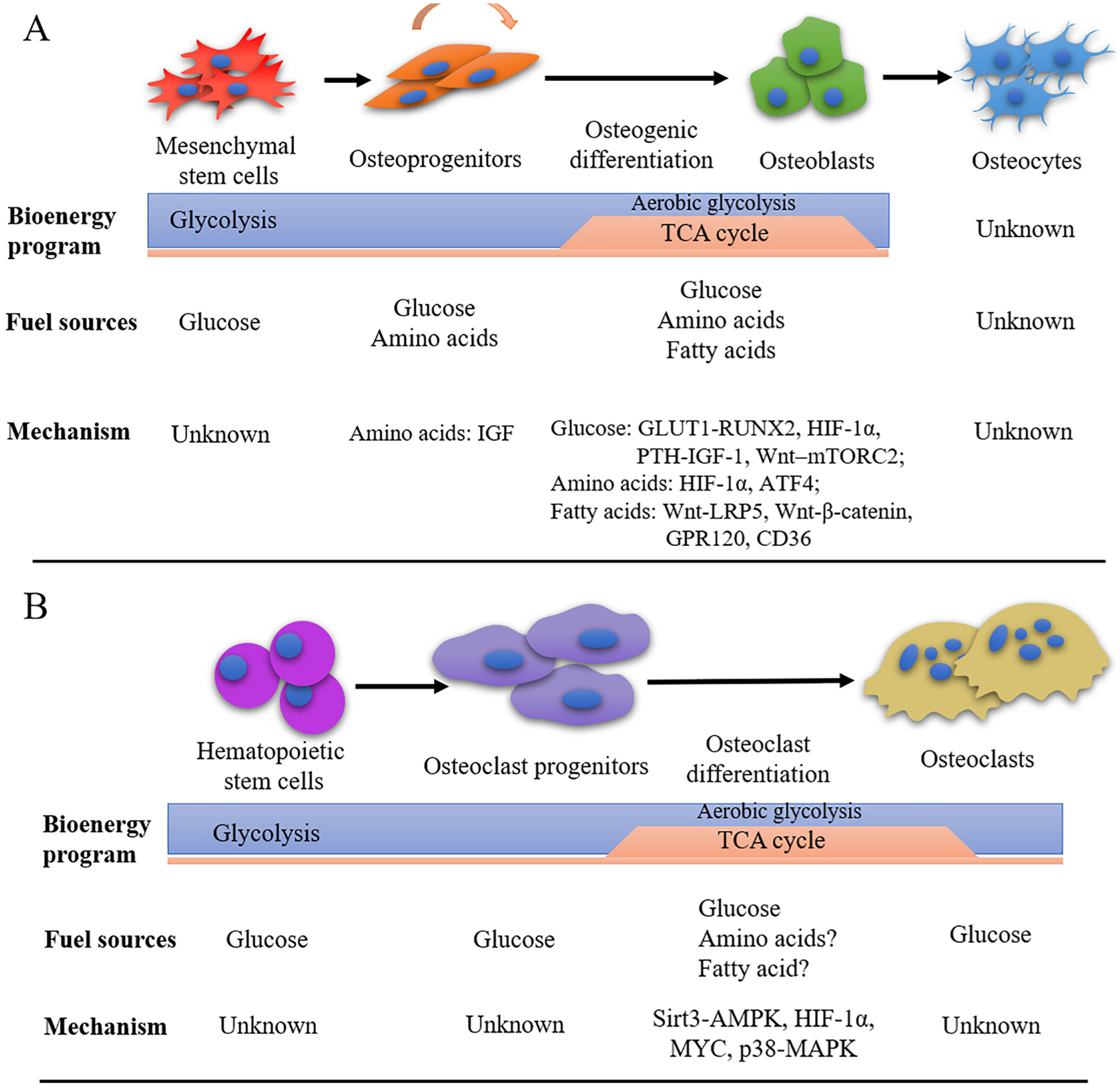
Summary of cell type and differentiation stage specific bioenergy program in osteoblasts and osteoclasts. Different energy programs are required for distinct stages of the osteoblasts and osteoclasts. (A) Proliferative mesenchymal stem cells (MSCs) and osteoprogenitors primarily use glycolysis as their energy source. While the mechanism involved glucose metabolism at this stage is unknown yet, the effects of essential amino acids on the proliferation and differentiation of osteoprogenitors are dependent on IGF signaling. After osteogenic induction, osteoblasts switch to use both glycolysis (including aerobic glycolysis) and the TCA cycle. During differentiation, glucose metabolism is controlled by GLUT1-RUNX2, HIF-1α, PTH-IGF-1, or Wnt-mTORC2 signaling. Amino acids metabolism is controlled by HIF-1α and ATF4. Fatty acid metabolism is controlled by Wnt-LRP5, Wnt-β-catenin, GPR120, CD36. In fully differentiated osteoblasts, glycolysis becomes predominant again with unknown mechanism. The preferred fuel sources and bioenergetic programs of osteocytes remains unknown. (B) Glucose is an important energy source for the differentiation and function of osteoclasts. TCA cycle is dominant over glycolysis during osteoclast differentiation, while glycolysis is suggested to be critical for bone resorption. Glucose metabolism is dependent on Sirt3-AMPK, HIF-1α, MYC, and p38-MAPK signaling. The possible involvement of amino acids and fatty acids in osteoclast differentiation and function are unknown.
How glucose metabolism is involved in osteoblast differentiation and function is largely unknown. Glucose transporters are a group of membrane proteins that are critical to transport glucose into the cell [31]. Osteoblasts express specific members of the glucose transports such as Glut1, Glut3, and Glut4 [32]. Deletion of Glut1 diminishes osteoblast differentiation in vitro and essentially abolishes an increase of bone formation in Wnt7b overexpressing mice [33]. Mechanistic studies have identified that GLUT1 promotes osteoblast differentiation via suppression of the degradation of runt-related transcription factor 2 (RUNX2), which is a critical transcription factor for osteoblast differentiation [25]. RUNX2 also promotes Glut1 expression [25]. This feedforward regulation between RUNX2 and GLUT1 determines the onset and extent of bone formation. GLUT4 is increased during osteoblast differentiation in direct association with up-regulation of glucose uptake [32]. However, genetic deletion of Glut4 does not cause an obvious skeletal phenotype [32]. These facts suggest potential functional redundancy of glucose transporters in osteoblasts differentiation.
Until now, only limited factors have been shown to regulate glucose metabolism in the differentiation and function of osteoblasts such as HIF-1α, PTH and Wnt (Fig. 1A). For example, stimulated aerobic glycolysis through HIF-1α stabilization in preosteoblasts significantly increases osteogenic differentiation and bone formation [34]. This phenotype is independent of the increase in angiogenesis but susceptible to glycolytic suppression. PTH induces glucose uptake and lactate production via aerobic glycolysis in osteoblasts through induction of insulin-like growth factor 1 (IGF-1) and up-regulation of glycolytic genes [30]. In addition, increased glycolysis is associated with osteoblast differentiation in response to Wnt signaling. Wnt3a, a known osteogenic factor, induces glycolysis through an mTORC2-dependent pathway [35]. GLUT1 and glucose consumption are involved in Wnt7b-mediated osteoblast differentiation in vitro and bone formation in vivo [33]. Further studies are required to clarify the mechanisms regulating glucose metabolism in osteoblasts. Details will be discussed in Section 5.
2.2.2. Amino acid metabolism of osteoblasts
Amino acids, the basic building blocks of protein biosynthesis, are divided into essential and nonessential amino acids. Amino acid metabolism has been shown to be important for osteoblast differentiation through protein intake studies. In primary rat calvarial osteoblasts, essential amino acids (e.g. Arg, Lys, Met, Thr, and Trp) have been shown to stimulate cell proliferation and differentiation [36]. Such effects are likely influenced both directly and/or indirectly by the insulin/insulin-like growth factor (IGF) signaling pathway (Fig. 1A) [36]. Of note, oxidized metabolites of certain amino acids are detrimental to osteoblasts such as dityrosine and kynurenine, metabolites from tyrosine and tryptophan respectively [37]. Therefore, different amino acids may have opposing effects in regulating the differentiation and function of osteoblasts.
Glutamine is the most abundant amino acid in circulation [38]. In addition to its direct contribution to protein synthesis, glutamine functions as an important energy source and an essential carbon and nitrogen donor for the synthesis of amino acids, nucleotides, glutathione, and hexosamine [38]. Recent studies have revealed an important role of glutamine metabolism in osteoblasts as an alternate fuel source. The active uptake and metabolism of glutamine is first identified in bone explants, suggesting the involvement of glutamine in bones [39]. The direct evidence of the involvement of glutamine in osteoblast precursors has been identified by using stable isotope tracing experiments [40]. Evidence suggests that glutamine is converted to citrate through oxidation in the TCA cycle, thus contributing to energy production in the mitochondria of the cells. Pharmacological inhibitors of glutamine metabolism suppress excessive bone formation caused by hyperactivation of Wnt signaling in vivo. In addition, glutamine may be a good alternative energy source for osteoblasts, as glutamine supplementation increases osteoblast viability in a low-glucose environment [40,41]. These findings indicate that glutamine is essential for osteoblast differentiation and function. Intriguingly, consumption of glutamine by bone marrow stromal cells, a population containing osteoblast precursors, declines with aging suggesting a link between glutamate usage and levels of osteoblast differentiation [42]. In addition, glutamine has also been reported to play an important role in maintaining redox homeostasis, thus enhancing the viability of osteoblasts [42]. This effect is stimulated by HIF-1α in instances of hypoxia and other cellular stress. Thus, glutamine is critical for producing energy and maintains redox homeostasis in osteoblast lineage cells. Further mechanisms and functions for glutamine in these cells remain to be clarified.
Correlations between amino acid transport and osteoblast-dependent collagen synthesis have been suggested by studies regarding the role of ATF4 in collagen synthesis [43]. ATF4 has been shown to regulate amino acid import in some cell types, such as CD4+ T cells [44]. In osteoblasts, deletion of Atf4 suppresses type I collagen synthesis without affecting the expressions of α1(I) Collagen, Runx2 and Osterix. Interestingly, addition of nonessential amino acids in culture medium restores the defect in collagen synthesis in osteoblasts lacking ATF4 [43]. These results indicate that the decrease in type I collagen synthesis observed in Atf4-deficient osteoblasts is secondary to a decrease in amino acid import.
2.2.3. Fatty acid metabolism of osteoblasts
Intracellular fatty acids have also been identified as energy sources for osteoblast differentiation in bone marrow stromal cells (BMSCs) (Fig. 1A). Studies have demonstrated that oxidation of fatty acids by osteoblasts for energy generation is controlled by both Wnt-Lrp5 signaling and β-catenin, and that a loss in this process results in decreased bone mass along with increased whole body fat mass [45–49]. Fatty acid oxidation in osteoblasts is shown to be required for normal bone acquisition in a sex and diet-dependent manner [50]. These effects of fatty acids on osteoblast function could be mediated by fatty acid translocase (e.g. CD36) or specific receptors (e.g. GPR120) [51,52]. Recent findings showed that osteoblasts express the fatty acid translocase CD36. Mice lacking CD36 exhibit a low bone mass phenotype secondary to reduced osteoblast numbers and activity [51]. GPR120 transcripts are present in primary bone marrow-derived MSCs and expression levels gradually increase during osteogenic induction [52]. In addition to serving as fuel substrates for osteoblasts, certain lipids exert detrimental effects on osteoblasts. As reported recently, conditioned medium from adipocytes inhibits proliferation of human MSCs and reduces expressions of several osteoblast differentiation marker genes. Of note, these inhibitory effects are ameliorated by pretreatment of adipocytes with inhibitors of fatty acid synthesis [53]. Thus, fatty acids function as (1) energy source to facilitate bone formation, and (2) detrimental effectors to suppress osteogenic differentiation possibly via specific receptors. Future studies are likely to reveal additional mechanisms and functions of fatty acids in osteoblast lineage cells.
2.3. Energy metabolism in osteocytes
Osteocytes are terminally differentiated osteoblasts and embedded in bone matrix. Although osteocytes make up close to 95% of the cells in the adult skeleton [54], there are virtually no data on osteocyte bioenergetics (Fig. 1A). Osteocytes are exposed to low oxygen tensions and highly express inducible factor-1α (HIF-1α) and related genes [54,55]. The absolute local oxygen tension (pO2) is quite low in the bone marrow of live mice (< 32 mmHg, or < 4.2%), especially in deeper peri-sinusoidal regions (pO2 9.9 mmHg, or 1.3%) [55]. In addition, recent finding identified that oxygen sensing in osteocytes negatively regulates bone mass. Enhanced HIF-1α signaling caused by a conditional deletion of Phd2 or Vhl (HIF-1α-degrading machinery) in osteocytes results in a high bone mass phenotype due to increased bone formation and decreased resorption [56,57]. There is an interesting contrast since Hif1α deletion in osteocytes has no overt effects on bone [57]. There is also suggestive evidence showing a highly correlation between glycolysis related proteins and mitochondria content with the expression of oxygen-regulated protein 150 (ORP150) in osteocytes [58,59]. Functions of HIF signaling on bone homeostasis will be further discussed in Section 5.2. These facts lead to the hypothesis that they would be highly glycolytic in their energy production. Osteocytes have been shown to acquire the capacity to resorb bone via generating protons and acidifying their microenvironment during extreme calcium deficiency [60]. Bone-lining cells can become osteoblasts with PTH treatment, but they can also remain quiescent or retract to expose bone surfaces prior to osteoclast-mediated bone resorption [61]. These finding suggest that osteocytes should be metabolically active during bone remodeling. Immortalized cell models and new isolation techniques of primary osteocytes should provide greater insights into their energy status or their ultimate fate during remodeling.
2.4. Energy metabolism in osteoclasts
Osteoclasts would be expected to need a substantial amount of energy due to their motility and specialized functions related to bone resorption. Glucose is an important energy source for osteoclasts (Fig. 1B). Depletion of glucose suppresses osteoclast function, as replacement of glucose with galactose reduced collagen degradation in an in vitro culture method used to model bone resorption [62]. Both glycolysis and the TCA cycle are important for osteoclast formation and function [62,63]. Studies with osteoclasts differentiated from mouse marrow cells on plastic (i.e. non-resorbing cells) show that RANKL treatment results in increased lactate production, along with large increases in oxygen consumption, and the expression of mitochondrial respiratory chain enzymes. These findings suggest that both aerobic and anaerobic respiration occur during osteoclast differentiation and function. Glycolytic enzymes PKM2 and GAPDH have been found to localize at the sealing zone, close to the actin ring. The specific location of these glycolytic enzymes suggests that glycolysis may function at the bone resorption site, allowing for rapid, effective generation and delivery of ATP for motility and activity for the vacuolar H+ pump during resorption [62]. During osteoclast differentiation, mitochondrial respiration is dominant to produce high level of ATP (Fig. 1B) [63]. Inhibition of mitochondria respiration using rotenone, an inhibitor of mitochondrial complex 1, strongly decreases osteoclastogenesis and blocks resorption pit formation in vitro [63,64]. Rotenone also reduces lipopolysaccharide (LPS) -induced bone loss in vivo [64]. Deletion of the peroxisome proliferator-activated receptor-gamma coactivator 1 beta (Ppargc1β, aka Pgc1β) gene in mice impairs mitochondria biogenesis and suppresses osteoclast differentiation and function [65]. Recent study using macrophage-specific mutant mice with LysM-Cre found that Ppargc1b-deficient macrophages differentiate normally into osteoclasts, but albeit with impaired resorptive function due to cytoskeletal disorganization [66]. Of note, suppression of mitochondria function has also been shown to up-regulate osteoclast functions. For example, deletion of mitochondrial transcription factor A (Tfam) leads to severe ATP depletion but elevates bone-resorbing activity [67]. Sirt3, a mitochondrial (NAD)+-dependent protein deacetylase, is identified as a negative regulator of osteoclastogenesis [68,69]. Mice lacking Sirt3 have enhanced production of osteoclasts, due to the loss of normal osteoclast inhibitory activity of Sirt3 to inhibit RANKL-mediated osteoclastogenesis through stabilization of AMPK protein [69]. Taken together, osteoclast formation uses oxidative phosphorylation as a main bioenergetic source, while bone resorption function mainly relies on glycolysis.
The molecular mechanisms involved in regulating energy metabolism during osteoclast differentiation and function have been revealed recently (Fig. 1B). MYC is a key upstream regulator for metabolic reprogramming to oxidative phosphorylation in osteoclasts [70]. Genetic studies identified that osteoclast-specific Myc-deficient mice exhibit defective in osteoclast development, and protect mice from osteoporosis-induced bone loss [70]. Besides MYC, researchers found that hypoxia-inducible factors (HIFs) also regulate metabolic pathways in osteoclasts. The study found that hypoxia increases glutamine consumption, glycolysis activity, ATP production, and mitochondrial electron transport chain activity in osteoclasts [71]. These effects permit rapid bone resorption in a short term, while compromising long-term cell survival. In cultured chicken osteoclasts, bone-resorbing activity is increased in a medium supplemented with glucose, mechanistically associated with signaling through p38 MAPK [72,73]. Besides glucose, whether and to what extent other substrates such as fatty acids and amino acids contribute to oxidative phosphorylation during osteoclastogenesis are yet to be clarified.
Various metabolic products or by-products, including ATP, pyruvate, H+ ions, and reactive oxygen species (ROS), are known to exert direct actions on osteoclast function. ATP is released in small amounts into the extracellular environment by cells. It acts on P2 receptors in an autocrine or paracrine fashion to modulate cell function. At concentrations within the physiological range (2–5 mM inside cells), ATP is a potent stimulator of both the formation and activity of osteoclasts. However, higher concentration of ATP inhibits the formation and activity of osteoclasts [74]. Pyruvate plays important roles in boosting oxygen consumption of non-resorbing osteoclasts derived from mouse marrow cells [75]. It is noteworthy that a moderate increase in pyruvate levels significantly enhances osteoclast-like cell formation from RAW264.7 cells [75]. H+ ions are universal by-products of cellular metabolism to make extracellular environment acidic. Acidosis is demonstrated to play significant roles in the differentiation and activation of osteoclasts via up-regulating various genes responsible for cell adhesion, migration, survival and bone matrix degradation [76,77]. ROS, including superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) are produced at the electron transport chain during aerobic respiration. ROS has been shown to be important components that stimulate both the formation and resorptive activity of osteoclasts [78,79]. Functions of other metabolic products or by-products in regulating the formation and function of osteoclasts remain to be clarified.
3. Correlation of metabolic diseases with bone homeostasis
In addition to the local cell energy metabolism, many clinical conditions that impact whole body energy metabolism, such as diabetes mellitus (DM), anorexia nervosa (AN), and anabolic treatments for osteoporosis, have been reported to affect the bone homeostasis.
3.1. Diabetes mellitus
Diabetes mellitus (DM) is a chronic disease characterized by impaired glucose metabolism in liver, fat, and skeletal muscle, leading to varying degrees of fuel starvation in those tissues [80]. Clinically, systemic metabolic disruptions associated with DM are known to decrease bone turnover and alter the bone material properties and microstructure (Fig. 2A) [81,82]. The relationship between DM and osteoporosis is complex. One possible mechanism is the compromised properties of collagen (the most abundant of the bone proteins) due to complications with hyperglycemia, oxidative stress, and glycation end-product accumulation [83]. In addition, DM is associated with declining renal function and microvascular complications, which limit blood flow to the bone [84]. Consequently, the circulating bioactive hormones, including osteocalcin (OCN) are decreased in bone, which also contribute to skeletal fragility. Moreover, both type 1 and late-stage type 2 DM impair insulin dependent actions of the liver. Defects in liver function suppress production and release of IGF-1, thus affecting the bioenergetics and function of osteoblasts [85]. However, how exactly fuel metabolism in bone cells is altered in DM is not clear at present. The linkage between DM and bone creates possibilities that certain antidiabetic therapies may affect bone homeostasis.
Fig. 2.
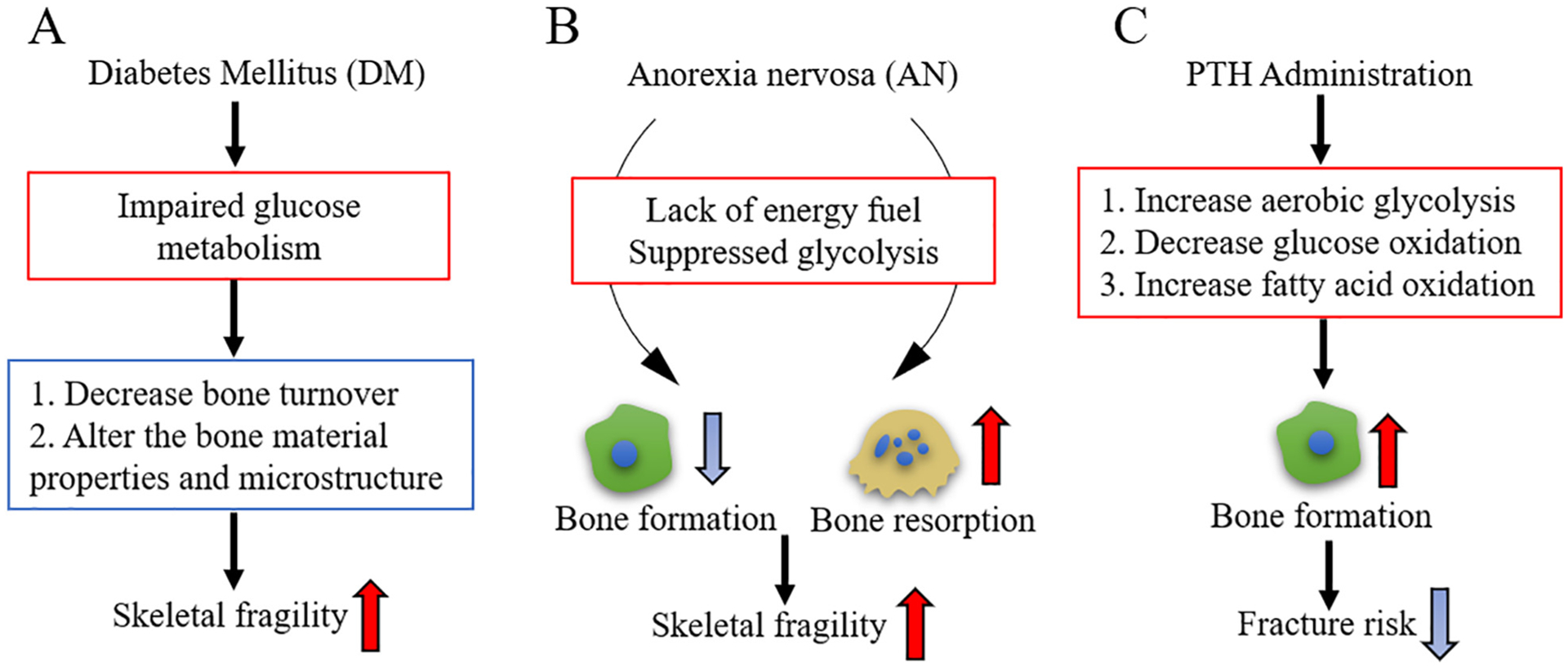
Examples of metabolic diseases that affect bone homeostasis. (A) Diabetes mellitus (DM) impairs glucose metabolism leading to varied degrees of fuel starvation in the whole body. Systemic metabolic disruptions associated with DM decrease bone turnover and alter the bone material properties and microstructure, thus increasing skeletal fragility. (B) Patients with Anorexia nervosa (AN) have low nutrient substrates, such as glucose, fatty acids, and amino acids, thus lack fuel source for energy metabolism. Glycolysis is possibly suppressed in AN patients. AN have a profound impact on increasing skeletal fragility with suppressed bone formation and increased bone resorption. (C) Parathyroid hormone (PTH) administration is known to stimulate new bone formation, increase bone mass, and reduce fracture risk. Glycolysis is increased by PTH treatment.
3.2. Anorexia nervosa
Anorexia nervosa (AN) is a psychiatric disease characterized by a voluntary food restriction [86,87]. Malnutrition in the form of calorie deficit occurs in AN, as energy is utilized to maintain tissue homeostasis rather than anabolic activity. This causes significant changes in whole body energy metabolism. AN has a profound impact on skeletal remodeling. Bone formation is suppressed while bone resorption is increased in AN in both mice and humans (Fig. 2B). Anorexic patients have very low bone mass and markedly enhanced skeletal fragility, as well as persistent difficulties with hypoglycemia and hypothermia. It is not clear how bioenergetic profiles in bone cells change during severe nutritional stress. Patients with AN also have low nutrient substrates, such as glucose, fatty acids, and amino acids, thus lack fuel source for energy metabolism [88] (Fig. 2B). In addition, serum IGF-1 level is reduced in AN [89]. These facts suggest that glycolysis is suppressed (Fig. 2B). Further studies are needed to determine how AN affect energy metabolism, and its role in regulating bone homeostasis.
3.3. Anabolic treatments for osteoporosis
Osteoporosis is a disease that causes bone fragility and an increased risk of fractures due to low bone mass and structural deterioration of bone tissue. Parathyroid hormone (PTH) 1–34 and abaloparatide, a PTH-related protein analog, are currently the only FDA-approved anabolic drugs to treat osteoporosis. Mechanistically, intermittent administration of PTH1–34 or abaloparatide, stimulates new bone formation acting via PTH1 receptor (PTH1R), thus increasing bone mass and reducing fracture risk (Fig. 2C) [90]. This effect is probably due to the function of PTH1R on the induction of osteoblastogenesis from marrow stromal cells and bone-lining cells [91]. As previously noted, several studies have shown that PTH stimulates aerobic glycolysis in osteoblast lineage cells [30]. The bone anabolic effect of intermittent PTH was shown to be diminished when reducing glycolysis with the PDK1 inhibitor dichloroacetate in mice [30]. These findings indicate glycolysis is critical for PTH induced bone formation. Of note, PTH treatment also increases the oxygen consumption rate, suggesting the possible use of other fuel sources through the TCA cycle [30]. However, the alternative fuel source is not clear yet. One possible source is fatty acids from the bone marrow adipocytes. Marrow adipose tissue occupies > 70% of the bone marrow in rodents and humans [92]. PTH could use endogenous marrow to fuel osteoblasts by lipolyzing triglycerides to produce free fatty acids and glycerol [46]. Therefore, energy metabolism, especially glycolysis, is a critical mechanism of PTH-mediated bone formation. Further studies are remained to clarify the alternative sources needed to fuel mitochondrial oxidative phosphorylation and the mechanism involved. The molecular mechanisms regarding how PTH regulates glycolysis and its possible interaction with BMP signaling will be discussed in Section 5.4.
4. Role of BMP signaling in controlling whole body energy metabolism
In addition to its critical functions in bone development and homeostasis, BMP signaling plays a pivotal role in glucose and fatty acid metabolism. It was recently reported that glucose metabolism induced by BMP signaling is essential for murine skeletal development [93]. The authors found that deletion of Bmpr1a in chondrocytes markedly reduces GLUT1 levels in vivo, whereas recombinant BMP-2 administration increases Glut1 mRNA and protein levels in vitro, boosting glucose metabolism in primary chondrocytes. These findings suggest that energy metabolism may be a novel mechanism downstream of BMP signaling in regulating bone homeostasis. In this chapter, we will review recent publications reporting the function of BMP signaling in glucose metabolism and fatty acid metabolism.
4.1. BMP signaling in glucose metabolism
BMPs have recently been implicated in pancreas development as well as controlling adult glucose homeostasis (Fig. 3). The entire BMP signaling machinery, including BMP ligands (BMP-2, 3, 4, 6, 7, and 9), receptors (BMPR1A, BMPR1B, BMPR2), and downstream signaling components (SMADs, IDs), are present in pancreatic islets [16]. Systemically, BMP expression and secretion may arise from several tissues in response to the inflammatory diabetogenic environment. For example, cytokine exposure of primary islets induces up-regulation and secretion of BMP-2. The released BMP-2 increases expression of Id1, 2, 3, 4, direct targets of BMP signaling, indicating autocrine effects BMP signaling [94]. These observations are also reflected in increased expression of Bmp2, 4 and Id-1 in islets from type 2 DM mice [95]. An age-dependent increase in serum BMP-4 and decrease in serum BMP-7 levels are observed in animal models of type 2 DM [96]. Moreover, there has been reported a negative correlation of BMPR1A level, while a positive correlation of BMPR2 level, in visceral and subcutaneous adipose tissues with parameters of glucose metabolism, insulin sensitivity and obesity [97]. In addition, insulin secretion is stimulated in mice expressing Bmp4 under the control of the beta-cell specific promoter, and in mice treated by systemic administration of BMP-4 [98]. These changes in diabetes development suggest that BMP signaling pathway has important functions in regulating glucose metabolism.
Fig. 3.
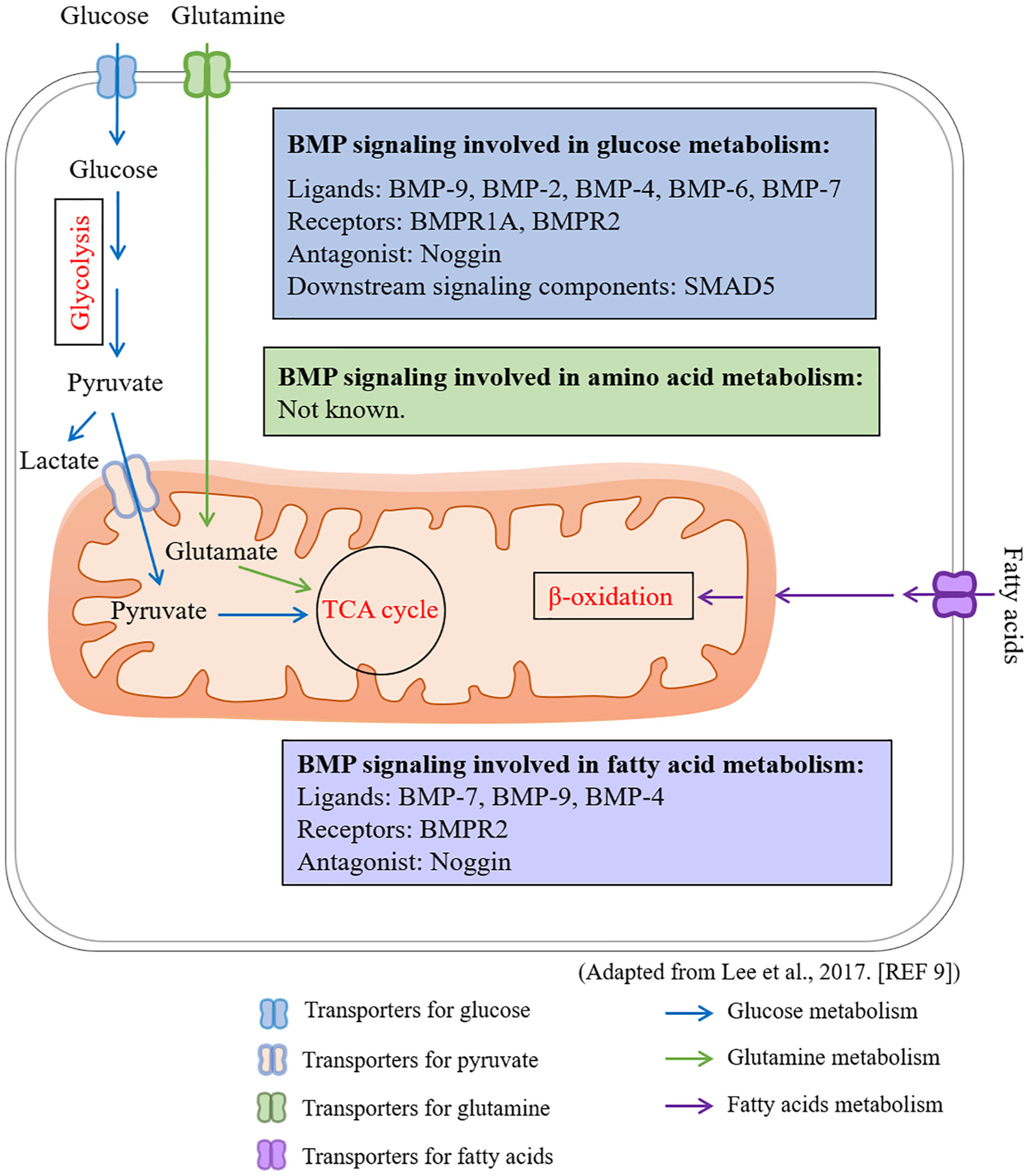
BMP signaling regulates glucose metabolism and fatty acid oxidation. Many BMP signaling components have been shown to regulate glucose metabolism, including BMP ligands BMP-9, BMP-2, BMP-4, BMP-6, and BMP-7; BMP receptors BMPR1A and BMPR2; BMP signaling antagonist Noggin; and downstream signaling components SMAD5. Several BMP signaling components have been shown to regulate fatty acid oxidation, including BMP-9, BMP-4, BMPR2, and Noggin. The possible involvement of BMP signaling in amino acid metabolism is unknown yet. Adapted from Lee et al. (ref [9]).
BMP signaling has been shown to play a pivotal role in regulating glucose metabolism via recent genetic studies (Fig. 3). Mice heterozygous for Bmpr1a null allele show attenuated glucose homeostasis [99]. Beta-cell specific homozygous deletion of Bmpr1a shows a similar phenotype on glucose homeostasis [98,100]. This phenomenon is associated with abnormal expression of metabolic genes, glucose sensing, and reduced expression of several key regulators of β-cell function. Mutation or deficiency of BMPR2 is associated with increased glycolysis in pulmonary arterial endothelial cells [101]. These findings demonstrate a critical role of BMP signaling in glucose homeostasis. Among BMP ligands, BMP-9 has been identified as a factor that regulates blood glucose homeostasis via inhibition of hepatic glucose production [102]. Overexpression of BMP-9 causes a downregulation of gluconeogenic genes, increase of phosphorylation of insulin signaling molecules, amelioration of triglyceride accumulation and inhibition of lipogenic gene expressions in the liver [103].
BMP-2, 4, 6, and 7, in addition to BMP-9, also have been reported to regulate glucose metabolism (Fig. 3). BMP-2 enhances mitochondrial metabolism and oxidative phosphorylation with a minimal effect on key glycolytic regulators in articular chondrocytes [104]. BMP-2 boosts glucose metabolism via increasing Glut1 mRNA and protein levels in primary chondrocytes [93]. BMP-6, which signals through BMPR1A, BMPR1B and ACVR2a, reduces circulating glucose in obese mice, and suppresses gluconeogenesis and glucose output in hepatoma cells [105]. BMP-2 and BMP-6 enhances insulin-mediated glucose uptake through increasing GLUT4 protein levels in both insulin-sensitive and -insensitive adipocytes [106]. It is noted, however, that subsets of genes, which are involved in glucose and fatty acid metabolism, and adipokine expression, are regulated exclusively by BMP-2 or BMP-6. These results indicate that the effects of BMP-2 and BMP-6 are not completely redundant [106]. BMP-4 has a dual role in regulating glucose metabolism. A transgenic mouse line with forced expression of BMP-4 in adipose tissues shows decreased levels of fasting serum glucose and insulin. In addition, BMP-4 significantly reduces levels of fasting glucose and insulin that are elevated by high fat diet [107]. These results indicate that BMP-4 increases insulin sensitivity. Contrarily, BMP-4 has been shown to decrease insulin sensitivity. In the liver, muscle, and adipose cell lines, BMP-4 inhibits insulin signaling through activation of PKC-θ isoform followed by serine phosphorylation of IRS-1, thus resulting in insulin resistance in diabetic subjects [96]. In addition to functions of BMP-2, 4, and 6, BMP-7 treatment enhances glucose uptake in the insulin sensitive tissues, such as the adipose tissues and muscles, by increasing GLUT4 translocation to the plasma membrane through phosphorylation and activation of PDK1 and Akt [96]. Restoration of BMP-7 levels in serum decreases blood glucose levels in diabetic animals. BMP-7 enhances glucose utilization in the basal ganglia ipsilateral to stroke [108]. Therefore, BMP ligands play important roles in regulating glucose metabolism.
Enhanced BMP signaling via inhibition of its antagonist Noggin also shows defects in glucose metabolism [109]. It has been shown that SMAD5, a BMP signaling molecule, physically interacts with hexokinase 1 (HK1) in the cytoplasm to enhance glycolysis [110]. Loss of function of SMAD5 causes glycolysis defects and irreversible dysregulation of cellular bioenergetic homeostasis. Similarly, promotion of phospho-SMAD1/5/8 translocation into the nucleus impaired glycolytic metabolism, thus causing chondrocyte hypertrophy-like changes. Altogether, these finding suggest BMP signaling plays important roles in regulating glucose metabolism in a highly context dependent manner.
4.2. BMP signaling in fatty acid metabolism
BMP signaling also regulates fatty acid metabolism (Fig. 3). High-throughput screens using C. elegans reveal that importance of BMP signaling in mitochondrial lipid oxidation. Mutant worms defective in each level of BMP signaling pathways, including ligands, receptors, signaling molecules and co-transcription factors, exhibit increased rates of lipid mobilization in association with the induction of mitochondrial β-oxidation and mitochondrial fusion [111]. BMP signaling is also critical in Drosophila fat body where it positively regulates nutrient storage and energy homeostasis [112]. In vertebrates, genetic involvement of ligands and receptors in regulating fatty acid metabolism are also reported. BMP-4 has a dual role in either promoting or repressing fatty acid oxidation in a cell context-dependent manner. BMP-4 treatment suppresses expression of β-oxidation genes (e.g. Hadha, Hadhb, Acadm, and Acadvl) and mitochondrial genes involved in fatty acid uptake (e.g. Cpt1-β, Cpt2, and Crat) in brown pre-adipocytes. Moreover, administration of exogenous free fatty acids causes a blunted increase in oxygen consumption rate in BMP-4-treated brown pre-adipocytes [113]. Similarly, forced expression of BMP-4 in white adipose tissues (WATs) increases mitochondrial biogenesis (e.g. UCP-1, PGC1α, and mtTFA), and the expression of fatty acid-oxidizing genes (e.g. Cycs, CPT1b, and MCAD) [107]. These findings indicate that BMP-4 reduces fatty acids oxidation in adipocytes. However, BMP-4 depletion restores fatty acids and possibly enhances fatty acid oxidation in EGFR-mutant non-small-cell lung cancer cells [114]. In addition to BMP-4, BMP-7 increases mitochondrial activity and fatty acid uptake of mature brown adipocytes [115]. Overexpression of Bmp9 ameliorates lipid accumulation both in vivo and in vitro primarily through the suppression of lipogenic genes [116]. BMP-9 has also been shown to regulate key enzymes of fatty acid synthesis in the liver, promote insulin release from the pancreas, suppress hepatic glucose production and increase brown adipogenesis in adipose tissues [102]. Patients with BMPR2-associated heritable pulmonary arterial hypertension also show fatty acid oxidation defects and cardiac steatosis. Impairment in fatty acid oxidation and increased glycolysis in endothelial cells from the patients are direct consequences of the mutation in BMPR2 [117]. Similarly, abrogation of BMP signaling via expressing a dominant-negative form of Bmpr2 causes an increase in long-chain fatty acids coupled with increased lipid import and impaired fatty acid oxidation in the cardiomyocytes [118]. In addition, enhanced BMP signaling in an adipocyte-specific deletion of Noggin promotes age-related obesity along with changes in expressions of adipogenesis and lipid metabolism related genes [109]. Altogether, these data highly support the idea that BMP signaling regulates fatty acid metabolism. However, it remains to be clarified how BMP signaling regulates fatty acid metabolism in bone homeostasis.
Adipose tissues are metabolically active tissues with endocrine effects, which include white adipose tissues (WATs), brown adipose tissues (BATs), and bone marrow adipose tissues (MATs) [119,120]. Adipose tissues function to both take up fatty acids via the action of lipoprotein lipase and release fatty acids by the hydrolysis (lipolysis) of stored intracellular triacylglycerol [121]. White adipocytes in WATs store energy and brown adipocytes in BATs are thermogenic through lipolysis. Cold exposure induces brown adipose-like tissues within WATs capable for thermogenesis, which are classified as beige (or “brite” as brown-in-white) adipocytes [13,122,123]. Adipose tissues, especially MATs, regulate bone remodeling via extracellular vesicles, adipokines (i.e. leptin and adiponectin), or cytokines (e.g. IL-1β, IL-6 and TNF-α) [124–126]. Osteoblasts and bone marrow adipocytes are derived from same progenitor cells, such as bone marrow mesenchymal stem cells. Marrow adipocytes inhibit the osteogenic differentiation of mesenchymal stem cells via suppressing BMP signaling [53]. Hypoxia also plays critical roles in regulating the osteogenesis and adipogenesis of bone marrow mesenchymal stem cells in a highly context dependent manner [127].
There is accumulating evidence that BMP signaling plays a critical role in adipogenesis, including the commitment of undifferentiated progenitor cells, maturation of pre-adipocytes, and browning of white adipocytes [13]. BMP-4 and BMP-7 are the most studied BMP ligands in adipocytes formation, with major roles in white adipogenesis and brown adipogenesis, respectively [16,128,129]. Genetic mouse models reveal that type 1 receptors ACVR1 and BMPR1A, but not BMPR1B, are essential for brown adipogenesis [130,131]. In lower temperature condition, brown adipocytes generate heat by dissipating energy. Brown adipocytes also generate hypoxic microenvironment induced by BMP ligands, which drives the early steps of heterotopic endochondral ossification [132]. Taken together, these facts suggest that BMP signaling differentially influences the bone metabolism via regulating adipose tissues at regular, hypoxia, or lower temperature conditions.
5. Crosstalk of BMP signaling with intrinsic mechanism of energy metabolism in bone
To achieve the metabolic flexibility of bone cells at different stages in their life span, signaling molecules and transcription factors are coupled with extracellular queues to downstream bioenergetics pathways. Several key metabolic signaling pathways have been reported to program the bioenergetics of bone cells, such as mTOR, HIF, Wnt, PTH, autophagy, and ROS (Figs. 4–8). BMP signaling has been reported to regulate these signaling pathways, thus may regulate energy metabolism of bone cells secondarily through these pathways.
Fig. 4.
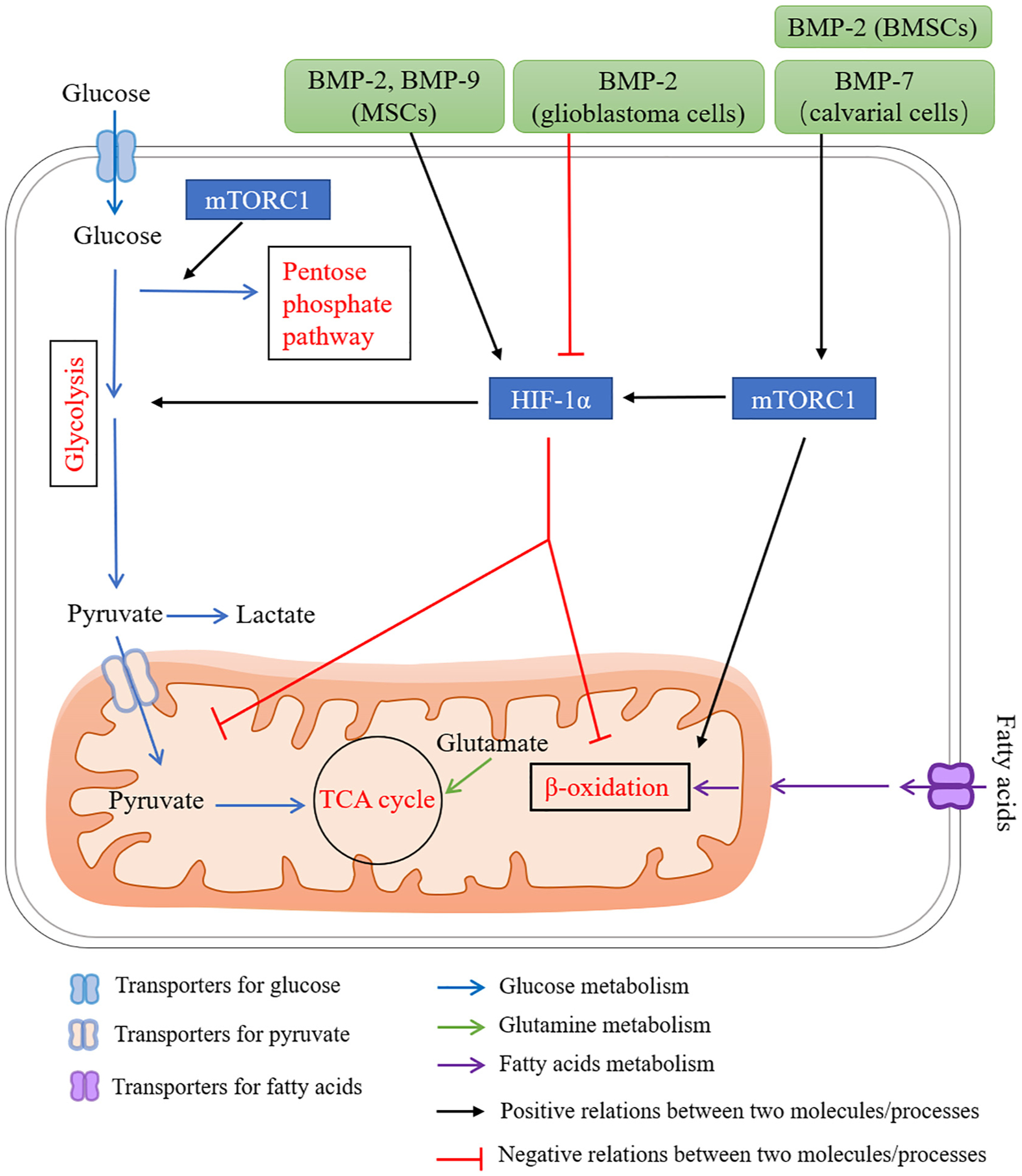
A proposed network for BMP signaling in regulating energy metabolism via mTOR and HIF-1. mTORC1 plays important roles in regulating energy metabolism, including (1) promoting a shift from TCA cycle to glycolysis via HIF-1α, (2) increasing flux through the oxidative pentose phosphate pathway and fatty acid synthesis. BMP signaling induces glucose metabolism via mTORC1. BMP-2 and BMP-7 activate mTORC1 in bone marrow stromal cells (BMSCs) and fetal rat calvaria, respectively. BMPs have dual functions in regulating HIF-1α signaling dependent on cell types. For examples, BMP-2 and BMP-9 induce Hif1α expression in mesenchymal stromal cells (MSCs), while BMP-2 downregulates HIF-1α in glioblastoma cells. BMP-mTORC1-HIF-1α signaling increases glycolysis activity in chondrocytes during skeletal development.
Fig. 8.
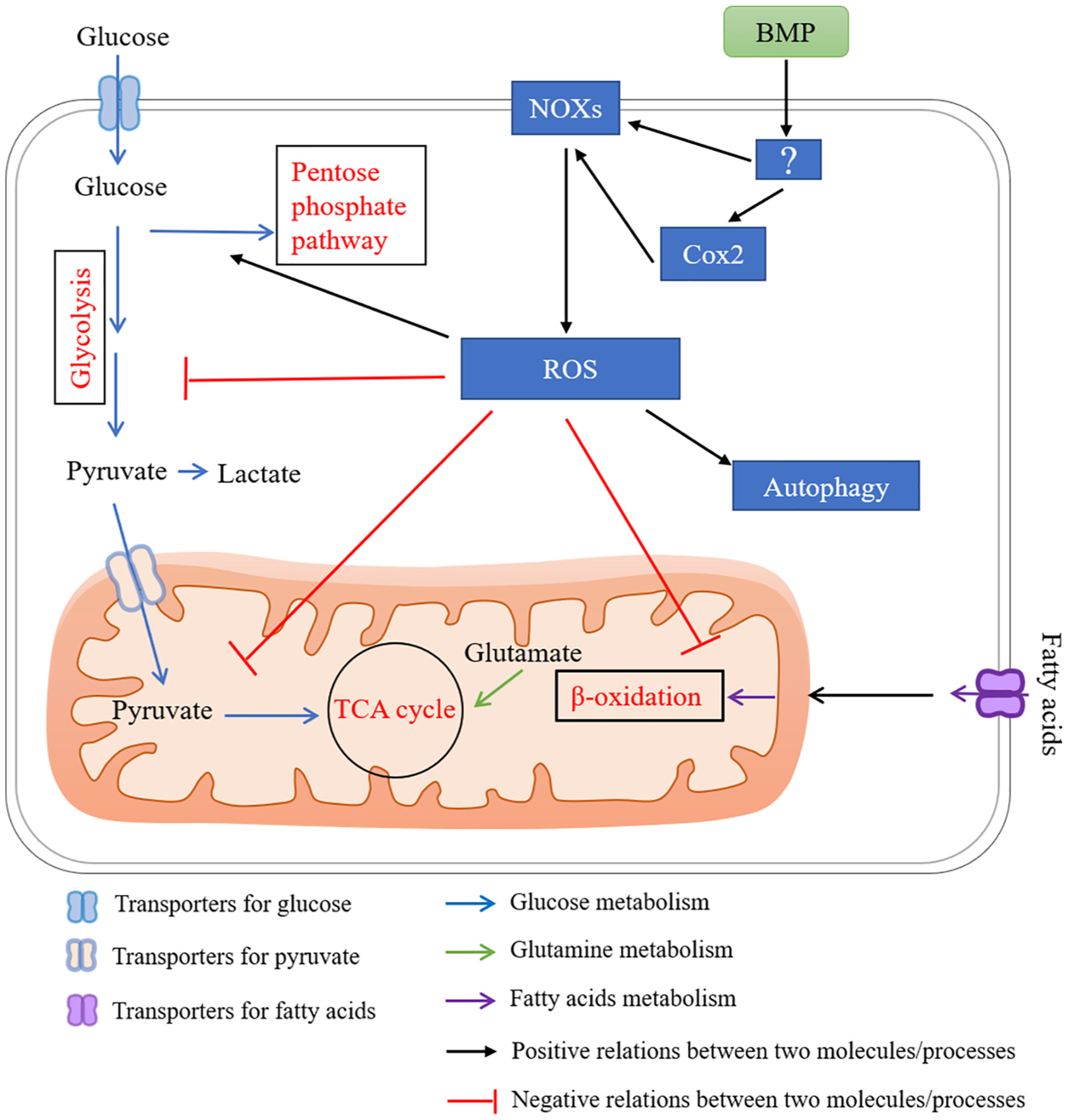
A proposed network for BMP signaling in regulating energy metabolism via ROS. Cytosolic reactive oxygen species (ROS) inhibit the glycolytic enzymes causing accumulation of upper glycolytic intermediates, and increase flux through the pentose phosphate pathway. ROS inhibit reactions in the mitochondria, including the TCA cycle and β-oxidation. ROS also activate autophagy. BMPs stimulate ROS production by inducing the nicotinamide adenine dinucleotide phosphate [NADPH] oxidase (NOX) family. BMPs also stimulate ROS production through cyclooxygenase (COX)-2.
5.1. mTOR
The mammalian target of rapamycin (mTOR) represents an important energy sensor, and integrates intracellular and extracellular signals that affect cell growth, energy metabolism, and autophagy [133]. mTOR is a serine/threonine kinase that functions via two functionally distinct complexes, the rapamycin-sensitive mTOR complex 1 (mTORC1; mTOR-Raptor) and the rapamycin insensitive mTORC2 (mTOR-Rictor). mTORC1 plays a central role in regulating cell size and proliferation through regulating the balance between anabolism and catabolism in response to environmental conditions [134]. To facilitate cell growth, mTORC1 has been shown to play important roles in regulating energy metabolism (Fig. 4), including (1) promoting a shift in glucose metabolism from oxidative phosphorylation to glycolysis via the transcription factor HIF-1α, which drives the expression of several glycolytic enzymes such as phospho-fructo kinase (PFK), (2) activating sterol responsive element binding protein (SREBP), resulting to increased flux through the oxidative pentose phosphate pathway and fatty acid synthesis [135]. Consistent with in vitro studies, recently developed transgenic mouse models have revealed an essential role of mTOR signaling in regulating energy metabolism at the organismal level, such as glucose and lipid homeostasis. The dysregulation of mTOR signaling causes a number of metabolic pathological conditions, including type 2 DM [134]. Genetics studies have shown that mTOR signaling plays important roles in regulating the differentiation and functions of both osteoblasts and osteoclasts [136]. Recent work suggests that the osteoblast differentiation program is linked to whole body metabolism through mTOR signaling [137]. Constitutively activate mTOR signaling via deletion of Tsc2 in osteoblasts results in excessive proliferation and the production of disorganized bone. These skeletal changes are accompanied by metabolic disturbances, including hypoglycemia, hyperinsulinemia, and increased undercarboxylated osteocalcin, a metabolic profile in osteoblasts [137]. Therefore, mTOR plays an important role in regulating glucose and lipid metabolism to facilitate cell growth and function, and links osteoblast differentiation with whole body metabolism.
As an energy sensor, mTORC1 can respond to growth factors, intracellular signaling, and environmental stress such as low ATP levels and hypoxia [138]. Recent reports showed that mTORC1 senses glucose through phosphorylation and activation of TSC2 indirectly and phosphorylation of Raptor directly by AMPK signaling, or inhibition of the Rag GTPases in cells lacking AMPK [139]. Similarly, hypoxia inhibits mTORC1 in part through AMPK activation, but also through the induction of REDD1-TSC [140]. It is reported that BMP signaling regulates mTOR signaling in several cell types (Fig. 4). In the osteogenic differentiation of bone marrow stromal cells (BMSCs), BMP-2 activates mTOR signaling and its downstream target genes regulating protein anabolism, such as Atf4 and Ddit3 [141]. Similarly, BMP-7 has been shown to stimulate osteogenesis and adipogenesis in a primary culture of fetal rat calvaria through mTOR signaling [142]. Recent genetic studies show the most direct evidence that BMP signaling induces glucose metabolism via mTORC1 during murine skeletal development [93]. These findings suggest that mTOR functions downstream of BMP signaling in osteoblast differentiation. However, further studies are required to obtain a more detailed understanding of BMP-mTOR signaling in the differentiation and function of other bone cells, as well as the involvement of energy metabolism downstream of this signaling.
5.2. HIF
The bone cells exist and operate in a low-oxygen (hypoxic) environment of bone marrow (refer to Section 2.3 for more supporting evidence). HIFs mediate the cellular response to hypoxia by activating genes controlling bioenergetics, oxygen supply, and consumption to allow cells to survive, proliferate, and differentiate in a hypoxic environment. Three human HIF family members have been identified, HIF-1, HIF-2, and HIF-3. HIFs function as heterodimers comprised of α subunit and β subunit, which dissociate in normal oxygen (normoxic) conditions. The most extensive characterization has been provided for HIF-1α. HIF-1α is degraded by the proteasome in a normoxic condition, while stabilized by hypoxia. Thus, HIF-1α activity increases in hypoxic cells [143]. HIFs have previously been linked to a promotion of angiogenesis and coupling between osteogenic and angiogenic processes [144]. In many cells, HIF-1 serves as a molecular switch diverting energy production from oxidative to glycolytic metabolism under hypoxic conditions [145] (Fig. 4). It has been reported that in human mesenchymal stromal cells (MSCs), hypoxia induces the expression of HIF-1α protein and its target genes (GLUT1, LDHA, PGK1), indicating up-regulation of glycolysis [144]. Reduction of HIF-1α expression suppresses the hypoxia-induced osteogenic differentiation of human MSCs. Studies using in vivo postnatal mouse models have shown that stabilization of HIF-1α in osteoblast precursors increases osteoblast population and stimulates cancellous bone formation [34]. Pharmacological inhibition of glycolytic enzyme pyruvate dehydrogenase kinase 1 (PDK1) reverses HIF-1α-driven bone formation in vivo [34]. These findings indicate that glycolysis is an important downstream target of HIF-1 in osteoblast differentiation and bone formation.
BMPs have dual functions in regulating HIF-1 signaling dependent on cell type (Fig. 4). BMP-2 and BMP-9/SMAD-1/5/9 mediated signaling induces expression of HIF-1α in MSCs, while BMP-2 downregulates HIF-1α in glioblastoma cells [103,146,147]. HIF1α gene silencing inhibits the effect of hBMP-2 on endothelial tube formation [148]. Dorsomorphin, an inhibitor for AMP-activated kinase (AMPK) and BMP type 1 receptor kinases, blocks hypoxic activation of HIF-1α in vitro [149]. HIF-1α is a critical downstream intermediate in the mTOR signaling pathway and rapamycin inhibits HIF-1α through mTOR. A recent report has identified a BMP-mTORC1-HIF-1α signaling cascade resulting in up-regulation of Glut1 in chondrocytes during development [93]. These findings indicate HIF-1α, which functions downstream of BMP signaling, regulates glycolysis activity in skeletal formation. Conversely, hypoxia also activates BMP signaling. Hypoxia and ischemic reperfusion in intestinal epithelium upregulate the expression levels of Bmp2, Bmp4, Bmpr1a, and Bmpr2 [150]. In cartilage and osteoblasts, hypoxia induces Bmp2 expression partially through HIF-1α induced ILK-mTOR-AKT pathways [151]. However, it remains to be clarified how BMP signaling regulates energy metabolism through its correlation with HIF-1 in bone cells.
5.3. Wnt signaling cascades
Wnt signaling is a major mechanism for stimulating bone accrual in both mice and humans by activating genes required for osteoblast differentiation and function [152]. In the developing skeletal system, Wnt signaling regulates the differentiation of progenitor cells into osteoblasts. Wnt signaling also helps to increase bone mass through stimulating pre-osteoblast replication, inducing osteoblastogenesis, or inhibiting apoptosis of osteoblasts and osteocytes [152].
Recent work has linked the bone anabolic function of Wnt signaling with increased energy metabolism in osteoblast-lineage cells, including glycolysis, fatty acid, and glutamine metabolism [153] (Fig. 5). For example, Wnt3a, Wnt7b and Wnt10b, which promote osteoblast differentiation of a bone marrow stromal cell line ST2, also stimulate aerobic glycolysis with increased glucose consumption, and lactate production [35]. Further studies have identified that Wnt signaling regulates glucose metabolism through its coreceptor LRP5. The authors found that Wnt signaling-regulated aerobic glycolysis is independent of β-catenin and GSK3β, but relies on LRP5 to activate mTORC2-AKT signaling during osteoblast differentiation [35]. Consistent with in vitro studies, mice lacking Lrp5, which have defects in bone formation, contain low levels of serum lactate and glycolysis enzymes HK2, LDHA and PDK1 in bones. In comparison, high-bone-mass mice harboring the point mutation of A214V in LRP5 show higher expression levels of HK2, PDK1 and LDHA in bones [35]. These findings indicate that Wnt-LRP5 induced metabolic reprogramming contributes to osteoblast differentiation. Mice lacking Lrp5 also exhibit an increased body fat due to reductions in the fatty acid oxidative potential of Lrp5-deficient osteoblasts [48]. In comparison, activation of Wnt signaling through either Lrp5 hyperactivation or Wnt10b stimulation increases the expression genes involved in fatty acid metabolism in murine calvarial osteoblasts [48]. Therefore, Wnt/Lrp5 also regulates fatty acid oxidation in osteoblasts (Fig. 5). In addition to glycolysis and fatty acid oxidation, Wnt signaling (Wnt3a) also stimulates glutamine oxidation in the TCA cycle via mTORC1 [40]. Importantly, pharmacological inhibition of glutaminase normalizes the high bone mass caused by enhanced Wnt signaling in mice. However, the involvement of energy metabolism in Wnt signaling-induced osteoclast formation and function remain to be clarified.
Fig. 5.
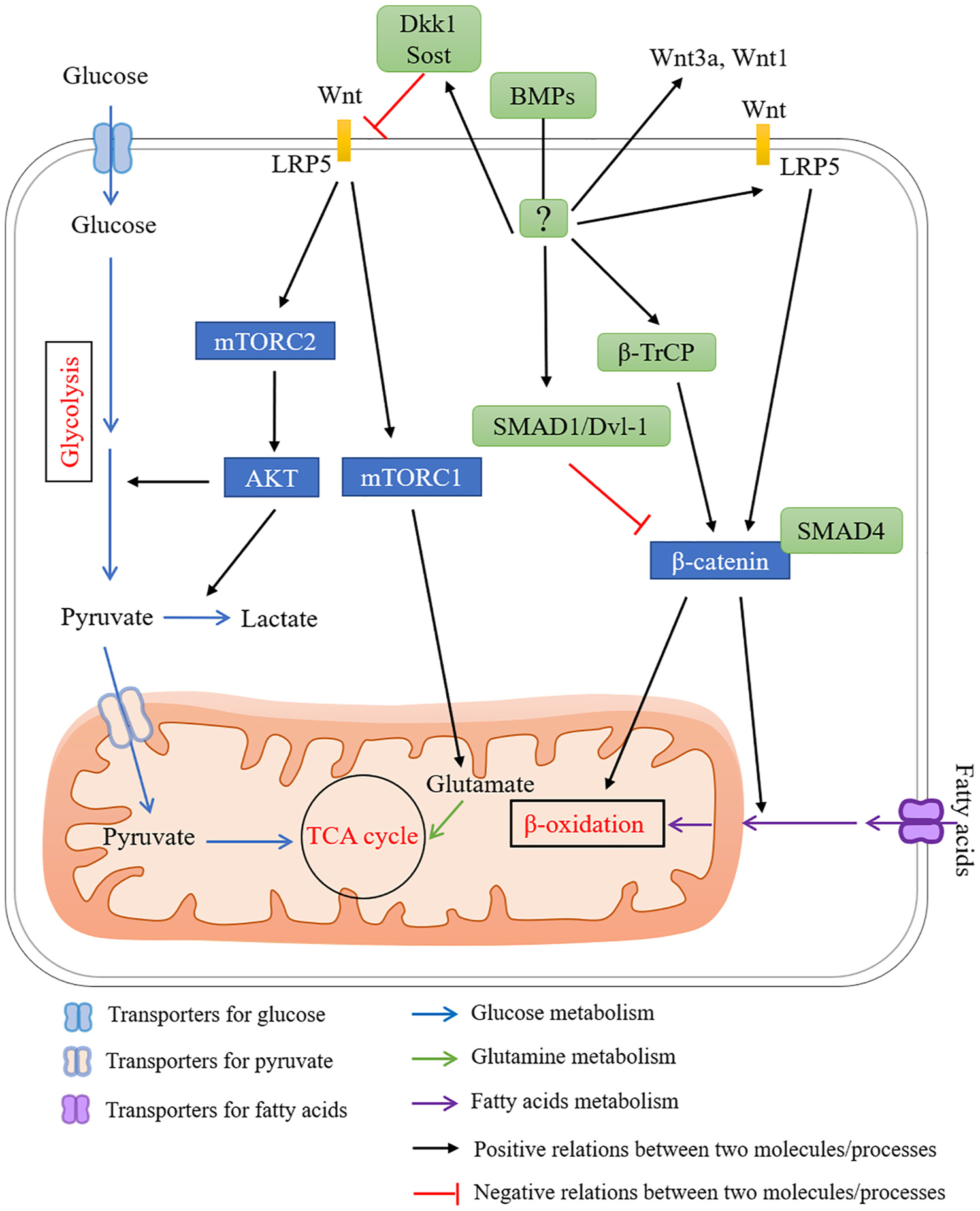
A proposed network for BMP signaling in regulating energy metabolism via Wnt signaling. Wnt signaling regulates energy metabolism in bones, which is highly corelated with bone homeostasis. Wnt signaling has been shown to regulate energy metabolisms by (1) increasing glycolysis activity via activation of mTORC2-AKT signaling through LPR5, a receptor for Wnt ligands (yellow boxes), (2) regulating fatty acid oxidation through LRP5, (3) stimulating glutamine oxidation in the TCA cycle via mTORC1. BMP signaling have dual functions in regulating Wnt signaling. BMP signaling inhibits Wnt signaling by (1) inducing Dkk1 and Sost expression, (2) inhibiting the activity of β-catenin via inducing the formation of SMAD1-Dvl-1 complex, (3) facilitating the formation of SMAD4-β-catenin complex thus preventing the binding of β-catenin with a TCF/LEF transcription complex. In contrast, BMP signaling has been also shown to increase Wnt signaling by (1) inducing the expression of Wnt3a, Wnt1 and Lrp5, (2) inhibiting β-catenin degradation via β-TrCP. Blue rectangles indicate signaling components downstream of Wnt signaling in regulating energy metabolism. Green rounded rectangles indicate components downstream of BMP signaling in regulating Wnt signaling. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
BMPs have dual functions in regulating Wnt signaling (Fig. 5). BMP induces Dkk1 and Sost expression through MAPK and SMAD signaling, respectively, leading to suppressed Wnt signaling in osteoblasts and reduced bone mass [154]. BMP-2 inhibits the activity of β-catenin via inducing the formation of SMAD1-Dvl-1 complex [155]. SMAD4 competitively binds β-catenin and prevents its binding with a TCF/LEF transcription complex, thus suppressing Wnt signaling in osteoblasts [156]. On the other hand, osteoblastogenesis is maximized in the presence of both BMP and Wnt signaling. One mechanism is a cooperative action of downstream transcription co-factors. It is reported that SMAD and TCF/LEF/β-catenin form a complex to achieve the highest expression of osteoblast genes (Dlx5, Msx2, and Runx2) [157]. Alternatively, BMP-2 also promotes canonical Wnt signaling by inducing Wnt3a, Wnt1, Lrp5 expression and inhibiting β-catenin degradation via β-TrCP (F-box E3 ligase) in osteoblasts [158] (Fig. 5). Ablation of SMAD4 in preosteoblasts significantly downregulates Lrp5 expression and impairs both BMP and Wnt signaling [156]. Further studies are required to clarify the possible involvement of energy metabolism in BMP-Wnt signaling mediated bone formation and resorption.
5.4. Parathyroid hormone (PTH)
Parathyroid hormone (PTH) is a major endocrine regulator of extracellular calcium and phosphate levels [159,160]. Continuous PTH administration (cPTH) causes loss of bone mass, as found in patients with chronic hyperparathyroidism. The bone loss caused by cPTH is due to excessive stimulation of bone resorption by osteoclasts, despite increased osteoblasts number and bone formation [160,161]. In contrast, intermittent PTH administration (iPTH), either full-length (amino acid 1–84) or a fragment (amino acid 1–34, known as teriparatide), results in increased bone mass and bone formation, and reduced incidence of fracture in osteoporotic patients [160,161]. Extensive studies have indicated that iPTH has no significant effect on osteoclasts, but stimulates bone formation via regulation of osteoblast-lineage cells at multiple levels, such as increasing osteoblast activity, stimulating osteoblast differentiation, suppressing osteoblast apoptosis, and activating quiescent bone-lining cells [161].
PTH plays important roles in regulating energy metabolism (Fig. 6). PTH promotes bone formation by stimulating aerobic glycolysis via IGF signaling [30]. In osteoblast lineage cells, PTH stimulates glucose consumption and lactate production in the presence of oxygen, indicating the presence of aerobic glycolysis. Experiments with radio-actively labeled glucose demonstrate that PTH suppresses glucose entry into the TCA cycle [30]. In addition, suppression of glucose consumption using dichloroacetate, which inhibits the activity of PDK1, greatly attenuates the teriparatide-induced increase in trabecular bone parameters [30]. Another report has shown that iPTH influences glucose metabolism partially through its effect on bone turnover, without influence on insulin secretion, resistance, pancreatic β cell function, and fat mass [162]. PTH also has been reported to significantly reduce mitochondrial oxygen consumption in skeletal muscles [163]. In addition to glucose metabolism, PTH is indicated to be involved in fatty acids metabolism. PTH induces adipocyte lipolysis via phosphorylation of hormone-sensitive lipase [164]. PTH may also promote fatty acid utilization though its major effector cAMP signaling [165,166]. In addition, PTH induces a browning program in human white adipocytes for thermogenesis when exposed to cold [167]. The role of PTH-mediated energy metabolism in regulating bone homeostasis remains to be determined.
Fig. 6.
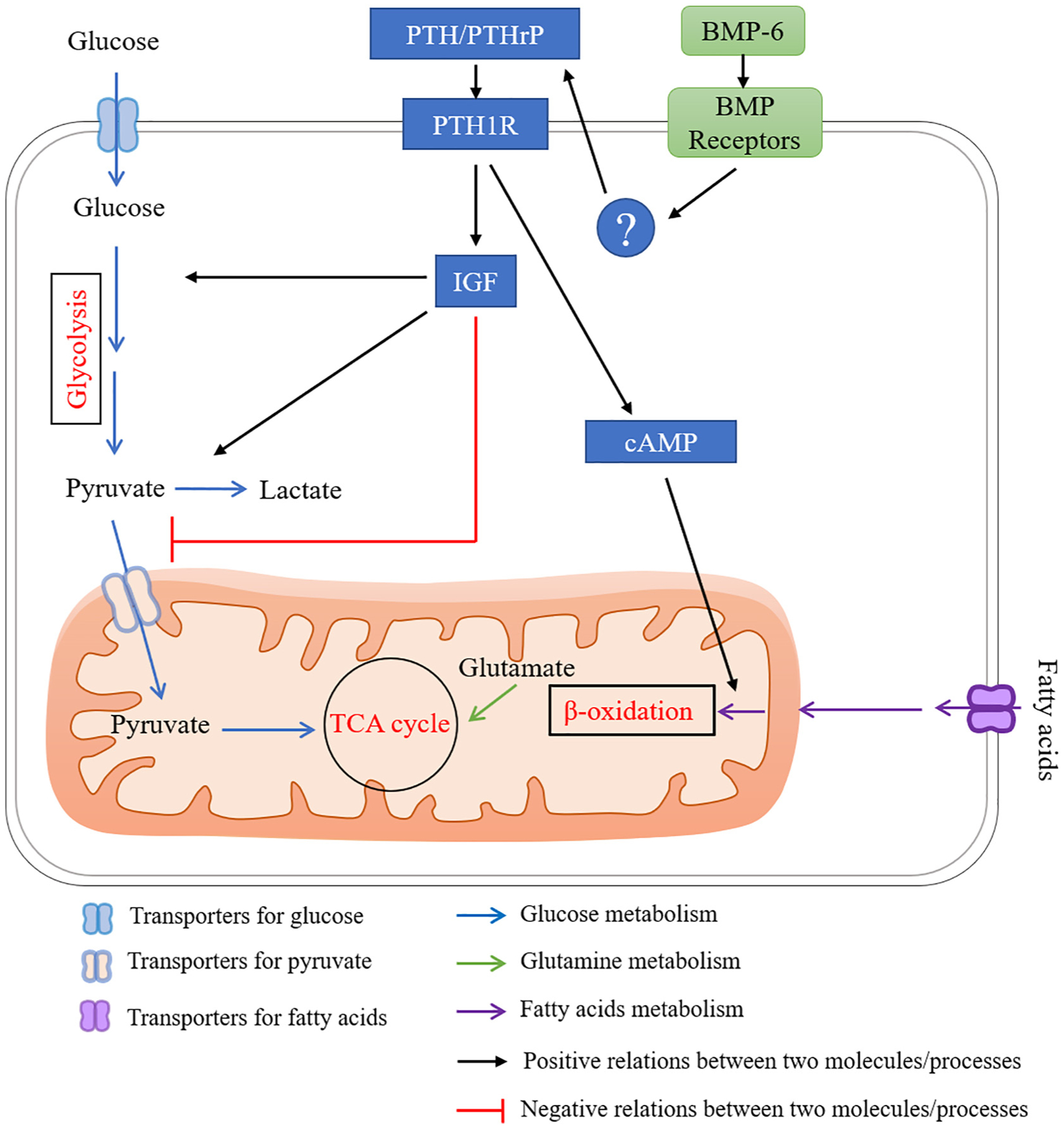
A proposed network for BMP signaling in regulating energy metabolism via PTH signaling. PTH stimulates glycolysis, while suppresses TCA cycle. PTH promotes fatty acid utilization though cAMP signaling. PTHrP expression is increased with BMP-6 stimulation or activating of type 1 BMP receptors with unknown mechanism in chondrocytes.
Although PTH has been shown to promote bone formation via activating BMP signaling, recent findings have shown that BMP signaling also functions upstream of PTH signaling (Fig. 6). BMP signaling positively regulates the levels of Parathyroid hormone-related peptide (PTHrP), as demonstrated in chick limbs expressing a constitutively active form of BMP receptors [168]. In addition, gene silencing of Bmp6 decreases expression levels of PTH, PTHrP and protein levels of PTHrP in chicken cartilage cells [169]. Therefore, BMP signaling may regulate energy metabolism via PTH signaling in osteoblast lineage cells.
5.5. Autophagy
Autophagy is a self-digesting process by which cell components, including proteins, lipids, and whole organelles, are degraded by lysosomes and recycled inside the cell. Extensive studies have identified that multiple autophagy related proteins play important roles in regulating the survival, differentiation, and function of bone cells, including osteoblasts, osteocytes, and osteoclasts [170]. Dysregulation of autophagic activity disturbs the balance between bone formation and bone resorption, thus mediating the onset and progression of multiple bone diseases, including osteoporosis [170].
Autophagy is also a critical and fine-tuned process in maintaining energy homeostasis (Fig. 7) [171]. As the degradation products, such as monosaccharides, fatty acids and amino acids, are transported back to the cytoplasm, and then used to sustain cell homeostasis. These molecules can be further used to produce ATP through catabolic reactions and/or provide building blocks for the synthesis of essential proteins. Up-regulation of autophagy is important for generating amino acids and free fatty acids, and maintaining cellular ATP levels in both human breast cancer cells and normal breast epithelial cells [172]. Autophagy-deficient neutrophil precursors fail to shift toward mitochondrial respiration and display excessive glycolysis, lipid droplet accumulation, and ATP depletion [173]. Autophagy also increases glucose production and blood sugar levels via degradation of CRY1 [174]. In addition, autophagic degradation of mitochondria (mitophagy) contributes to metabolic adaptation during differentiation as has been shown in innate NK lymphocytes [175,176]. Autophagy also functions to clear lipids (lipophagy), thus playing an essential regulatory role in lipid metabolism, lipoprotein assembly and lipid metabolic homeostasis [177]. Both animal experiments and human clinical evidence have shown that suppression of autophagy disturbs cellular energy homeostasis, thus contributing to the onset and development of many metabolic diseases, such as diabetes [178].
Fig. 7.
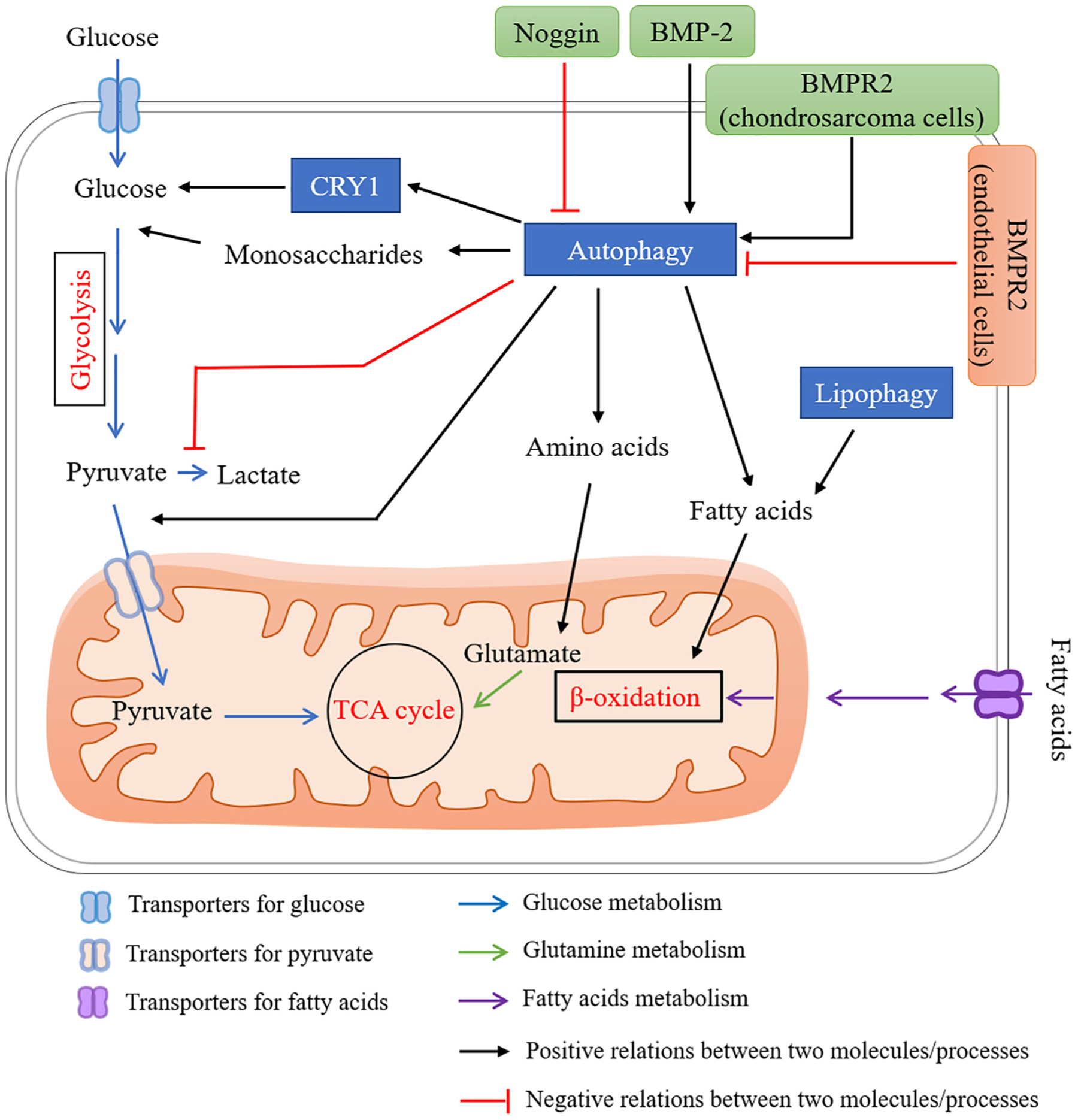
A proposed network for BMP signaling in regulating energy metabolism via autophagy. Autophagy has been shown to regulate energy metabolism by (1) providing fuel sources such as monosaccharides, amino acids and free fatty acids, (2) increasing glucose production and blood sugar levels via degradation of CRY1, (3) regulating shift from glycolysis to TCA cycle, (4) lipophagy to regulate lipid metabolism. BMP signaling plays distinct roles in regulating autophagic activity depending on cell types. Several studies using BMP-2, BMPR2, or Noggin have shown BMP signaling activates autophagy in several different cell types (black arrows). Contrary to above, reduced BMP signaling in Bmpr2 heterozygous mutation increases autophagy activity in endothelial cells.
BMP signaling plays distinct roles in regulating autophagic activity depending on cell types (Fig. 7). BMP-2 up-regulates autophagy-related proteins ATG5, ATG7, ATG12, BECLIN-1 and LC3-II/LC3-I in hair follicle stem cells [179]. Gene silencing of Bmp2 reduced accumulation of LC3-II (an autophagosome marker) in colon cancer stem cells [180]. Gene silencing of Bmpr2 inhibits autophagy in chondrosarcoma cells [181]. BMP-2 also increases autophagy activity in osteoblast lineage cells. In α7 integrin-positive human skeletal muscle-derived stem cells (α7 + hSMSCs), BMP-2 induces ATG7 expression and causes accumulation of autophagosome during their osteogenic differentiation [182]. Furthermore, hVPS34, a factor that is believed to function in autophagosome biogenesis, strongly binds to BMP-2 at the late endosome stage, suggesting a possible involvement of BMP signaling in autophagy [180]. Similarly, Noggin, an endogenous BMP antagonist, has been shown to reduce autophagic vacuole formation in acute pancreatitis [183]. These facts demonstrate a positive relation between BMP signaling activity and autophagy (Fig. 7). In contrast to these facts, there are some reports showing that BMP signaling suppresses autophagic activity. For example, BMPR2 heterozygosity causes an increase in autophagy in human endothelial cells [184]. Given these preliminary studies, the possible involvement of BMP-autophagy in bone homeostasis remains to be clarified.
5.6. Reactive oxygen species (ROS)
Reactive oxygen species (ROS) are highly reactive molecules derived from the reduction of molecular oxygen as a consequence of cellular metabolism or the activity of specific enzymes, such as the nicotinamide adenine dinucleotide phosphate [NADPH] oxidase (NOX) complexes [185]. Since ROS are highly unstable, they react with proteins, nucleic acids, lipids and other cellular components and often disrupt their cellular functions. Even though oxidative stress occurs in several pathologies, numerous studies demonstrate that ROS signaling is also important in normal physiology and in the generation of proper redox biological responses. Cytosolic ROS influence metabolic processes including glycolysis, the TCA cycle, pentose phosphate pathway activity, and autophagy (Fig. 8). Cytosolic ROS inhibit the glycolytic enzymes (e.g. GAPDH and PKM2) causing accumulation of upper glycolytic intermediates. Thus, ROS increase flux through the pentose phosphate pathway. ROS also inhibit reactions in the mitochondria, including the TCA cycle and β-oxidation of fatty acids. Regardless of the sources of ROS, it has been shown that osteogenic differentiation, osteoclast differentiation and function are blunted by elevated ROS [78,186,187]. The possible involvement of energy metabolism in ROS-mediated differentiation and function of bone cells remain to be clarified.
BMPs stimulate ROS production by up-regulating expression and activity of NOX in several cell types including osteoblasts (Fig. 8) [188–190]. It is also reported that BMP signaling induces cyclooxygenase (COX)-2 expression in osteoblasts, which produces prostanoides, known to modulate NOX activity and ROS production (Fig. 8) [191]. Therefore, BMP signaling may regulate energy metabolism via ROS in bone cells.
6. Summary and perspective
Proper metabolic programming is essential for the normal formation and function of each cell in the bone remodeling unit. Substrate utilization and ATP generation are both context and cell specific. To date, the best characterized metabolic profile in bone cells is the pattern of glucose utilization by osteoblasts or osteoclasts during differentiation of their progenitors in response to signaling peptides such as the Wnts and the IGFs. It is less clear how bone cells use fatty acids, citrate, and intracellular proteins and lipids during their differentiation and function. In part, this is due to the challenges of recapitulating the marrow niche ex vivo. Currently, there are some 3D models of the bone marrow in development, which may be helpful research tools [192,193]. Intriguingly, metabolic diseases, such as diabetes mellitus, might offer better clues into the importance of specific metabolic programs for bone cells. BMP signaling plays important roles in regulating whole body energy metabolism and is highly correlated with major signaling pathways or biological process regulating energy metabolism of bone cells. The evidence presented here highlights a potential role of BMP signaling in regulating energy metabolism of bone cells to maintain bone homeostasis. Further understanding of the complex role of BMP signaling in energy metabolism to maintain bone homeostasis may help to improve the treatment of abnormal skeletal status during aging and in certain bone metabolic disorders, and ultimately, open new therapeutic targets for bone regenerative medicine.
Acknowledgments
The authors thank Drs. Benjamin Levi, Vesa Kaartinen and Fei Liu for long-term collaboration. We thank Drs. Ernestina Schipani, Benton W. Swanson, Ke’ale Louie, Honghao Zhang and Maiko Omi for critical reading of the manuscript. We apologize to colleagues whose work we could not discuss due to the space limitations. This study was supported by the National Institutes of Health (R01DE020843 to YM).
References
- 1.Gurka MJ, Filipp SL, DeBoer MD, Geographical variation in the prevalence of obesity, metabolic syndrome, and diabetes among US adults, Nutr Diabetes 8 (1) (2018) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakahama K, Cellular communications in bone homeostasis and repair, Cell. Mol. Life Sci 67 (23) (2010) 4001–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eriksen EF, Cellular mechanisms of bone remodeling, Rev. Endocr. Metab. Disord 11 (4) (2010) 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez-Duffhues G, et al. , Bone morphogenetic protein signaling in bone homeostasis, Bone 80 (2015) 43–59. [DOI] [PubMed] [Google Scholar]
- 5.Zaidi M, Skeletal remodeling in health and disease, Nat. Med 13 (7) (2007) 791–801. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, et al. , TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation, Nat. Med 15 (7) (2009) 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rey C, et al. , Bone mineral: update on chemical composition and structure, Osteoporos. Int 20 (6) (2009) 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonewald LF, Kneissel M, Johnson M, Preface: the osteocyte, Bone 54 (2) (2013) 181. [DOI] [PubMed] [Google Scholar]
- 9.Lee WC, et al. , Energy metabolism of the osteoblast: implications for osteoporosis, Endocr. Rev 38 (3) (2017) 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanis JA, et al. , European guidance for the diagnosis and management of osteoporosis in postmenopausal women, Osteoporos. Int 30 (1) (2019) 3–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salazar VS, Gamer LW, Rosen V, BMP signalling in skeletal development, disease and repair, Nat Rev Endocrinol 12 (4) (2016) 203–221. [DOI] [PubMed] [Google Scholar]
- 12.Katagiri T, Watabe T, Bone morphogenetic proteins, Cold Spring Harb. Perspect. Biol 8 (6) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grafe I, et al. , TGF-beta family signaling in mesenchymal differentiation, Cold Spring Harb. Perspect. Biol (5) (2018) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Mishina Y, Phenotypic analyses of genetically modified mice for BMP receptors, Methods Mol. Biol 1891 (2019) 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omi M, Mishina, Bone morphogenetic protein signaling in chondrogenic differentiation during skeletogenesis: new perspective from recent mouse genetic studies, in: Caplan M (Ed.), Reference Module in Biomedical Sciences, Elsevier, 2020. [Google Scholar]
- 16.Grgurevic L, et al. , Bone morphogenetic proteins in inflammation, glucose homeostasis and adipose tissue energy metabolism, Cytokine Growth Factor Rev. 27 (2016) 105–118. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-de-Diego C, et al. , Interplay between BMPs and reactive oxygen species in cell signaling and pathology, Biomolecules 9 (10) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rolfe DF, Brown GC, Cellular energy utilization and molecular origin of standard metabolic rate in mammals, Physiol. Rev 77 (3) (1997) 731–758. [DOI] [PubMed] [Google Scholar]
- 19.Chen LQ, et al. , Transport of sugars, Annu. Rev. Biochem 84 (2015) 865–894. [DOI] [PubMed] [Google Scholar]
- 20.Karner CM, Long F, Glucose metabolism in bone, Bone 115 (2018) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glatz JF, Luiken JJ, Bonen A, Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease, Physiol. Rev 90 (1) (2010) 367–417. [DOI] [PubMed] [Google Scholar]
- 22.Kushwaha P, Wolfgang MJ, Riddle RC, Fatty acid metabolism by the osteoblast, Bone 115 (2018) 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schweikhard ES, Ziegler CM, Amino acid secondary transporters: toward a common transport mechanism, Curr. Top. Membr 70 (2012) 1–28. [DOI] [PubMed] [Google Scholar]
- 24.Zoch ML, et al. , In vivo radiometric analysis of glucose uptake and distribution in mouse bone, Bone Res 4 (2016) 16004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei J, et al. , Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation, Cell 161 (7) (2015) 1576–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shum LC, et al. , Energy metabolism in mesenchymal stem cells during osteogenic differentiation, Stem Cells Dev 25 (2) (2016) 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komarova SV, Ataullakhanov FI, Globus RK, Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts, Am J Physiol Cell Physiol 279 (4) (2000) C1220–C1229. [DOI] [PubMed] [Google Scholar]
- 28.Guntur AR, et al. , Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass, Endocrinology 155 (5) (2014) 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esen E, Long F, Aerobic glycolysis in osteoblasts, Curr Osteoporos Rep 12 (4) (2014) 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esen E, et al. , PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling, J. Bone Miner. Res 30 (11) (2015) 1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan N, A glimpse of membrane transport through structures-advances in the structural biology of the GLUT glucose transporters, J. Mol. Biol 429 (17) (2017) 2710–2725. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, et al. , Glucose transporter-4 facilitates insulin-stimulated glucose uptake in osteoblasts, Endocrinology 157 (11) (2016) 4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, et al. , Increased glycolysis mediates Wnt7b-induced bone formation, FASEB J. 33 (7) (2019) 7810–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regan JN, et al. , Up-regulation of glycolytic metabolism is required for HIF1alpha-driven bone formation, Proc. Natl. Acad. Sci. U. S. A 111 (23) (2014) 8673–8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esen E, et al. , WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation, Cell Metab. 17 (5) (2013) 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fini M, et al. , Effect of L-lysine and L-arginine on primary osteoblast cultures from normal and osteopenic rats, Biomed. Pharmacother 55 (4) (2001) 213–220. [DOI] [PubMed] [Google Scholar]
- 37.El Refaey M, et al. , Oxidation of the aromatic amino acids tryptophan and tyrosine disrupts their anabolic effects on bone marrow mesenchymal stem cells, Mol. Cell. Endocrinol 410 (2015) 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cruzat V, et al. , Glutamine: metabolism and immune function, supplementation and clinical translation, Nutrients 10 (11) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biltz RM, et al. , Glutamine metabolism in bone, Miner. Electrolyte Metab 9 (3) (1983) 125–131. [PubMed] [Google Scholar]
- 40.Karner CM, et al. , Increased glutamine catabolism mediates bone anabolism in response to WNT signaling, J. Clin. Invest 125 (2) (2015) 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown PM, Hutchison JD, Crockett JC, Absence of glutamine supplementation prevents differentiation of murine calvarial osteoblasts to a mineralizing phenotype, Calcif. Tissue Int 89 (6) (2011) 472–482. [DOI] [PubMed] [Google Scholar]
- 42.Zhou T, et al. , Glutamine metabolism is essential for stemness of bone marrow mesenchymal stem cells and bone homeostasis, Stem Cells Int. 2019 (2019) 8928934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elefteriou F, et al. , ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae, Cell Metab. 4 (6) (2006) 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, et al. , ATF4 regulates CD4(+) T cell immune responses through metabolic reprogramming, Cell Rep. 23 (6) (2018) 1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rendina-Ruedy E, Guntur AR, Rosen CJ, Intracellular lipid droplets support osteoblast function, Adipocyte 6 (3) (2017) 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maridas DE, et al. , Progenitor recruitment and adipogenic lipolysis contribute to the anabolic actions of parathyroid hormone on the skeleton, FASEB J. 33 (2) (2019) 2885–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rendina-Ruedy E, Rosen CJ, Lipids in the bone marrow: an evolving perspective, Cell Metab. 31 (2) (2020) 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frey JL, et al. , Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast, Mol. Cell. Biol 35 (11) (2015) 1979–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frey JL, et al. , Beta-catenin directs long-chain fatty acid catabolism in the osteoblasts of male mice, Endocrinology 159 (1) (2018) 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SP, et al. , Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner, JCI Insight 2 (16) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kevorkova O, et al. , Low-bone-mass phenotype of deficient mice for the cluster of differentiation 36 (CD36), PLoS One 8 (10) (2013) e77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao B, et al. , GPR120: a bi-potential mediator to modulate the osteogenic and adipogenic differentiation of BMMSCs, Sci. Rep 5 (2015) 14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abdallah BM, Marrow adipocytes inhibit the differentiation of mesenchymal stem cells into osteoblasts via suppressing BMP-signaling, J. Biomed. Sci 24 (1) (2017) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robling AG, Bonewald LF, The osteocyte: new insights, Annu. Rev. Physiol 82 (2020) 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spencer JA, et al. , Direct measurement of local oxygen concentration in the bone marrow of live animals, Nature 508 (7495) (2014) 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stegen S, et al. , Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin, Nat. Commun 9 (1) (2018) 2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loots GG, et al. , Vhl deficiency in osteocytes produces high bone mass and hematopoietic defects, Bone 116 (2018) 307–314. [DOI] [PubMed] [Google Scholar]
- 58.Guo D, et al. , Identification of osteocyte-selective proteins, Proteomics 10 (20) (2010) 3688–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frikha-Benayed D, et al. , Regional differences in oxidative metabolism and mitochondrial activity among cortical bone osteocytes, Bone 90 (2016) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jahn K, et al. , Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo, J. Bone Miner. Res 32 (8) (2017) 1761–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim SW, et al. , Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts, J. Bone Miner. Res 27 (10) (2012) 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lemma S, et al. , Energy metabolism in osteoclast formation and activity, Int. J. Biochem. Cell Biol 79 (2016) 168–180. [DOI] [PubMed] [Google Scholar]
- 63.Kim JM, et al. , Osteoclast precursors display dynamic metabolic shifts toward accelerated glucose metabolism at an early stage of RANKL-stimulated osteoclast differentiation, Cell. Physiol. Biochem 20 (6) (2007) 935–946. [DOI] [PubMed] [Google Scholar]
- 64.Kwak HB, et al. , Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression, Bone 46 (3) (2010) 724–731. [DOI] [PubMed] [Google Scholar]
- 65.Ishii KA, et al. , Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation, Nat. Med 15 (3) (2009) 259–266. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, et al. , PGC1beta organizes the osteoclast cytoskeleton by mitochondrial biogenesis and activation, J. Bone Miner. Res 33 (6) (2018) 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyazaki T, et al. , Intracellular and extracellular ATP coordinately regulate the inverse correlation between osteoclast survival and bone resorption, J. Biol. Chem 287 (45) (2012) 37808–37823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim H, et al. , SOD2 and Sirt3 control osteoclastogenesis by regulating mitochondrial ROS, J. Bone Miner. Res 32 (2) (2017) 397–406. [DOI] [PubMed] [Google Scholar]
- 69.Huh JE, et al. , Sirtuin 3 (SIRT3) maintains bone homeostasis by regulating AMPK-PGC-1beta axis in mice, Sci. Rep 6 (2016) 22511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bae S, et al. , MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha, J. Clin. Invest 127 (7) (2017) 2555–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morten KJ, Badder L, Knowles HJ, Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts, J. Pathol 229 (5) (2013) 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams JP, et al. , Regulation of osteoclastic bone resorption by glucose, Biochem. Biophys. Res. Commun 235 (3) (1997) 646–651. [DOI] [PubMed] [Google Scholar]
- 73.Larsen KI, et al. , Glucose-dependent regulation of osteoclast H(+)-ATPase expression: potential role of p38 MAP-kinase, J. Cell. Biochem 87 (1) (2002) 75–84. [DOI] [PubMed] [Google Scholar]
- 74.Morrison MS, et al. , ATP is a potent stimulator of the activation and formation of rodent osteoclasts, J. Physiol 511 (Pt 2) (1998) 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fong JE, et al. , Moderate excess of pyruvate augments osteoclastogenesis, Biol Open 2 (4) (2013) 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pereverzev A, et al. , Extracellular acidification enhances osteoclast survival through an NFAT-independent, protein kinase C-dependent pathway, Bone 42 (1) (2008) 150–161. [DOI] [PubMed] [Google Scholar]
- 77.Yuan FL, et al. , The roles of acidosis in osteoclast biology, Front. Physiol 7 (2016) 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Agidigbi TS, Kim C, Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated osteoclast diseases, Int. J. Mol. Sci 20 (14) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Callaway DA, Jiang JX, Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases, J. Bone Miner. Metab 33 (4) (2015) 359–370. [DOI] [PubMed] [Google Scholar]
- 80.American Diabetes A, Diagnosis and classification of diabetes mellitus, Diabetes Care 32 (Suppl. 1) (2009) S62–S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Napoli N, et al. , Mechanisms of diabetes mellitus-induced bone fragility, Nat Rev Endocrinol 13 (4) (2017) 208–219. [DOI] [PubMed] [Google Scholar]
- 82.Valderrabano RJ, Linares MI, Diabetes mellitus and bone health: epidemiology, etiology and implications for fracture risk stratification, Clin Diabetes Endocrinol 4 (2018) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh VP, et al. , Advanced glycation end products and diabetic complications, Korean J Physiol Pharmacol 18 (1) (2014) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rask-Madsen C, King GL, Vascular complications of diabetes: mechanisms of injury and protective factors, Cell Metab. 17 (1) (2013) 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guntur AR, Rosen CJ, IGF-1 regulation of key signaling pathways in bone, Bonekey Rep (2) (2013) 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fazeli PK, Klibanski A, Effects of anorexia nervosa on bone metabolism, Endocr. Rev 39 (6) (2018) 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fazeli PK, Klibanski A, Anorexia nervosa and bone metabolism, Bone 66 (2014) 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marzola E, et al. , Nutritional rehabilitation in anorexia nervosa: review of the literature and implications for treatment, BMC Psychiatry 13 (2013) 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Misra M, Klibanski A, Endocrine consequences of anorexia nervosa, Lancet Diabetes Endocrinol. 2 (7) (2014) 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neer RM, et al. , Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis, N. Engl. J. Med 344 (19) (2001) 1434–1441. [DOI] [PubMed] [Google Scholar]
- 91.Datta NS, Abou-Samra AB, PTH and PTHrP signaling in osteoblasts, Cell. Signal 21 (8) (2009) 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suchacki KJ, Cawthorn WP, Molecular interaction of bone marrow adipose tissue with energy metabolism, Curr Mol Biol Rep 4 (2) (2018) 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee SY, Abel ED, Long F, Glucose metabolism induced by Bmp signaling is essential for murine skeletal development, Nat. Commun 9 (1) (2018) 4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bruun C, et al. , Inhibition of beta cell growth and function by bone morphogenetic proteins, Diabetologia 57 (12) (2014) 2546–2554. [DOI] [PubMed] [Google Scholar]
- 95.Nielsen SS, et al. , Regulation of pancreatic alpha-cell function and proliferation by bone morphogenetic protein 4 (BMP4) in vitro, Endocrinology 157 (10) (2016) 3809–3820. [DOI] [PubMed] [Google Scholar]
- 96.Chattopadhyay T, et al. , Bone morphogenetic protein-7 (BMP-7) augments insulin sensitivity in mice with type II diabetes mellitus by potentiating PI3K/AKT pathway, Biofactors 43 (2) (2017) 195–209. [DOI] [PubMed] [Google Scholar]
- 97.Schleinitz D, et al. , Genetic and evolutionary analyses of the human bone morphogenetic protein receptor 2 (BMPR2) in the pathophysiology of obesity, PLoS One 6 (2) (2011) e16155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goulley J, et al. , BMP4-BMPR1A signaling in beta cells is required for and augments glucose-stimulated insulin secretion, Cell Metab. 5 (3) (2007) 207–219. [DOI] [PubMed] [Google Scholar]
- 99.Scott GJ, et al. , Abnormal glucose metabolism in heterozygous mutant mice for a type I receptor required for BMP signaling, Genesis 47 (6) (2009) 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang FX, et al. , Transcriptome of pancreas-specific Bmpr1a-deleted islets links to TPH1–5-HT axis, Biol Open 4 (8) (2015) 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frump A, Prewitt A, de Caestecker MP, BMPR2 mutations and endothelial dysfunction in pulmonary arterial hypertension (2017 Grover Conference Series), Pulm Circ 8 (2) (2018) (2045894018765840). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen C, et al. , An integrated functional genomics screening program reveals a role for BMP-9 in glucose homeostasis, Nat. Biotechnol 21 (3) (2003) 294–301. [DOI] [PubMed] [Google Scholar]
- 103.Hu N, et al. , BMP9-regulated angiogenic signaling plays an important role in the osteogenic differentiation of mesenchymal progenitor cells, J. Cell Sci 126 (Pt 2) (2013) 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang C, et al. , Distinct metabolic programs induced by TGF-beta1 and BMP2 in human articular chondrocytes with osteoarthritis, J Orthop Translat 12 (2018) 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pauk M, et al. , A novel role of bone morphogenetic protein 6 (BMP6) in glucose homeostasis, Acta Diabetol. 56 (3) (2019) 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schreiber I, et al. , BMPs as new insulin sensitizers: enhanced glucose uptake in mature 3T3-L1 adipocytes via PPARgamma and GLUT4 upregulation, Sci. Rep 7 (1) (2017) 17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qian SW, et al. , BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis, Proc. Natl. Acad. Sci. U. S. A 110 (9) (2013) E798–E807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Y, et al. , The effect of bone morphogenetic protein-7 (BMP-7) on functional recovery, local cerebral glucose utilization and blood flow after transient focal cerebral ischemia in rats, Brain Res. 905 (1–2) (2001) 81–90. [DOI] [PubMed] [Google Scholar]
- 109.Blazquez-Medela AM, et al. , Noggin depletion in adipocytes promotes obesity in mice, Mol Metab 25 (2019) 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fang Y, et al. , Smad5 acts as an intracellular pH messenger and maintains bioenergetic homeostasis, Cell Res. 27 (9) (2017) 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu Y, et al. , High-throughput screens using photo-highlighting discover BMP signaling in mitochondrial lipid oxidation, Nat. Commun 8 (1) (2017) 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ballard SL, Jarolimova J, Wharton KA, Gbb/BMP signaling is required to maintain energy homeostasis in Drosophila, Dev. Biol 337 (2) (2010) 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Modica S, et al. , Bmp4 promotes a brown to white-like adipocyte shift, Cell Rep. 16 (8) (2016) 2243–2258. [DOI] [PubMed] [Google Scholar]
- 114.Bach DH, et al. , BMP4 upregulation is associated with acquired drug resistance and fatty acid metabolism in EGFR-mutant non-small-cell lung cancer cells, Mol Ther Nucleic Acids 12 (2018) 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Townsend KL, et al. , Increased mitochondrial activity in BMP7-treated brown adipocytes, due to increased CPT1- and CD36-mediated fatty acid uptake, Antioxid. Redox Signal 19 (3) (2013) 243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang M, et al. , Role of bone morphogenetic protein-9 in the regulation of glucose and lipid metabolism, FASEB J. 33 (9) (2019) 10077–10088. [DOI] [PubMed] [Google Scholar]
- 117.Andruska A, Spiekerkoetter E, Consequences of BMPR2 deficiency in the pulmonary vasculature and beyond: contributions to pulmonary arterial hypertension, Int. J. Mol. Sci 19 (9) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Talati MH, et al. , Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle, Am. J. Respir. Crit. Care Med 194 (6) (2016) 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Choe SS, et al. , Adipose tissue remodeling: its role in energy metabolism and metabolic disorders, Front Endocrinol (Lausanne) 7 (2016) 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Luo L, Liu M, Adipose tissue in control of metabolism, J. Endocrinol 231 (3) (2016) R77–R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frayn KN, Arner P, Yki-Jarvinen H, Fatty acid metabolism in adipose tissue, muscle and liver in health and disease, Essays Biochem. 42 (2006) 89–103. [DOI] [PubMed] [Google Scholar]
- 122.Wu J, et al. , Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human, Cell 150 (2) (2012) 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harms M, Seale P, Brown and beige fat: development, function and therapeutic potential, Nat. Med 19 (10) (2013) 1252–1263. [DOI] [PubMed] [Google Scholar]
- 124.Muruganandan S, Govindarajan R, Sinal CJ, Bone marrow adipose tissue and skeletal health, Curr Osteoporos Rep 16 (4) (2018) 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cornish J, Wang T, Lin JM, Role of marrow adipocytes in regulation of energy metabolism and bone homeostasis, Curr Osteoporos Rep 16 (2) (2018) 116–122. [DOI] [PubMed] [Google Scholar]
- 126.Hawkes CP, Mostoufi-Moab S, Fat-bone interaction within the bone marrow milieu: impact on hematopoiesis and systemic energy metabolism, Bone 119 (2019) 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen Q, et al. , Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 23 (7) (2016) 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tseng YH, et al. , New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure, Nature 454 (7207) (2008) 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blazquez-Medela AM, Jumabay M, Bostrom KI, Beyond the bone: bone morphogenetic protein signaling in adipose tissue, Obes. Rev 20 (5) (2019) 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schulz TJ, et al. , Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat, Nature 495 (7441) (2013) 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schulz TJ, et al. , Loss of BMP receptor type 1A in murine adipose tissue attenuates age-related onset of insulin resistance, Diabetologia 59 (8) (2016) 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Olmsted-Davis E, et al. , Hypoxic adipocytes pattern early heterotopic bone formation, Am. J. Pathol 170 (2) (2007) 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Laplante M, Sabatini DM, mTOR signaling in growth control and disease, Cell 149 (2) (2012) 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Saxton RA, Sabatini DM, mTOR signaling in growth, metabolism, and disease, Cell 168 (6) (2017) 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Duvel K, et al. , Activation of a metabolic gene regulatory network downstream of mTOR complex 1, Mol. Cell 39 (2) (2010) 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen J, Long F, mTOR signaling in skeletal development and disease, Bone Res 6 (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Riddle RC, et al. , Tsc2 is a molecular checkpoint controlling osteoblast development and glucose homeostasis, Mol. Cell. Biol 34 (10) (2014) 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sengupta S, Peterson TR, Sabatini DM, Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress, Mol. Cell 40 (2) (2010) 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Agarwal S, et al. , AMP-activated protein kinase (AMPK) control of mTORC1 is p53- and TSC2-independent in pemetrexed-treated carcinoma cells, J. Biol. Chem 290 (46) (2015) 27473–27486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brugarolas J, et al. , Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex, Genes Dev. 18 (23) (2004) 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Karner CM, Lee SY, Long F, Bmp induces osteoblast differentiation through both Smad4 and mTORC1 signaling, Mol. Cell. Biol 37 (4) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yeh LC, et al. , Rapamycin inhibits BMP-7-induced osteogenic and lipogenic marker expressions in fetal rat calvarial cells, J. Cell. Biochem 114 (8) (2013) 1760–1771. [DOI] [PubMed] [Google Scholar]
- 143.Kaelin WG Jr., P.J. Ratcliffe, G.L. Semenza, Pathways for oxygen regulation and homeostasis: the 2016 Albert Lasker Basic Medical Research Award, JAMA 316 (12) (2016) 1252–1253. [DOI] [PubMed] [Google Scholar]
- 144.Wagegg M, et al. , Hypoxia promotes osteogenesis but suppresses adipogenesis of human mesenchymal stromal cells in a hypoxia-inducible factor-1 dependent manner, PLoS One 7 (9) (2012) e46483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Thomas LW, Ashcroft M, Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria, Cell. Mol. Life Sci 76 (9) (2019) 1759–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Francois S, et al. , Synergistic effect of human bone morphogenic protein-2 and mesenchymal stromal cells on chronic wounds through hypoxia-inducible factor-1 alpha induction, Sci. Rep 7 (1) (2017) 4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Persano L, et al. , BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1alpha stability and MGMT expression, Cell Death Dis. 3 (2012) e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Belmokhtar K, et al. , Regeneration of three layers vascular wall by using BMP2-treated MSC involving HIF-1alpha and Id1 expressions through JAK/STAT pathways, Stem Cell Rev. Rep 7 (4) (2011) 847–859. [DOI] [PubMed] [Google Scholar]
- 149.Emerling BM, et al. , Compound C inhibits hypoxic activation of HIF-1 independent of AMPK, FEBS Lett. 581 (29) (2007) 5727–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang J, et al. , Hypoxia inducible factor-1-dependent up-regulation of BMP4 mediates hypoxia-induced increase of TRPC expression in PASMCs, Cardiovasc. Res 107 (1) (2015) 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tseng WP, et al. , Hypoxia induces BMP-2 expression via ILK, Akt, mTOR, and HIF-1 pathways in osteoblasts, J. Cell. Physiol 223 (3) (2010) 810–818. [DOI] [PubMed] [Google Scholar]
- 152.Regard JB, et al. , Wnt signaling in bone development and disease: making stronger bone with Wnts, Cold Spring Harb. Perspect. Biol 4 (12) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Karner CM, Long F, Wnt signaling and cellular metabolism in osteoblasts, Cell. Mol. Life Sci 74 (9) (2017) 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kamiya N, et al. , Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts, J. Bone Miner. Res 25 (2) (2010) 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu Z, et al. , A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells, J. Biol. Chem 281 (25) (2006) 17156–17163. [DOI] [PubMed] [Google Scholar]
- 156.Salazar VS, et al. , Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with beta-catenin, J. Cell Sci 126 (Pt 24) (2013) 5598–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mbalaviele G, et al. , Beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation, J. Cell. Biochem 94 (2) (2005) 403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang M, et al. , BMP-2 modulates beta-catenin signaling through stimulation of Lrp5 expression and inhibition of beta-TrCP expression in osteoblasts, J. Cell. Biochem 108 (4) (2009) 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gensure RC, Gardella TJ, Juppner H, Parathyroid hormone and parathyroid hormone-related peptide, and their receptors, Biochem. Biophys. Res. Commun 328 (3) (2005) 666–678. [DOI] [PubMed] [Google Scholar]
- 160.Wein MN, Kronenberg HM, Regulation of bone remodeling by parathyroid hormone, Cold Spring Harb Perspect Med 8 (8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Silva BC, Bilezikian JP, Parathyroid hormone: anabolic and catabolic actions on the skeleton, Curr. Opin. Pharmacol 22 (2015) 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.D’Amelio P, et al. , Effect of intermittent PTH treatment on plasma glucose in osteoporosis: a randomized trial, Bone 76 (2015) 177–184. [DOI] [PubMed] [Google Scholar]
- 163.Baczynski R, et al. , Effect of parathyroid hormone on energy metabolism of skeletal muscle, Kidney Int 28 (5) (1985) 722–727. [DOI] [PubMed] [Google Scholar]
- 164.Larsson S, et al. , Parathyroid hormone induces adipocyte lipolysis via PKA-mediated phosphorylation of hormone-sensitive lipase, Cell. Signal 28 (3) (2016) 204–213. [DOI] [PubMed] [Google Scholar]
- 165.Louet JF, et al. , The coactivator PGC-1 is involved in the regulation of the liver carnitine palmitoyltransferase I gene expression by cAMP in combination with HNF4 alpha and cAMP-response element-binding protein (CREB), J. Biol. Chem 277 (41) (2002) 37991–38000. [DOI] [PubMed] [Google Scholar]
- 166.Luiken JJ, et al. , Effects of cAMP modulators on long-chain fatty-acid uptake and utilization by electrically stimulated rat cardiac myocytes, Biochem. J 367 (Pt 3) (2002) 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hedesan OC, et al. , Parathyroid hormone induces a browning program in human white adipocytes, Int. J. Obes 43 (6) (2019) 1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang D, et al. , ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development, J. Bone Miner. Res 18 (9) (2003) 1593–1604. [DOI] [PubMed] [Google Scholar]
- 169.Ye F, et al. , The role of BMP6 in the proliferation and differentiation of chicken cartilage cells, PLoS One 14 (7) (2019) e0204384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yin X, et al. , Autophagy in bone homeostasis and the onset of osteoporosis, Bone Res 7 (2019) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang J, Zhou R, Ma Z, Autophagy and energy metabolism, Adv. Exp. Med. Biol 1206 (2019) 329–357. [DOI] [PubMed] [Google Scholar]
- 172.Thomas M, et al. , Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells, Cell Biochem. Funct 36 (2) (2018) 65–79. [DOI] [PubMed] [Google Scholar]
- 173.Riffelmacher T, et al. , Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation, Immunity 47 (3) (2017) 466–480 (e5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Toledo M, et al. , Autophagy regulates the liver clock and glucose metabolism by degrading CRY1, Cell Metab. 28 (2) (2018) 268–281 (e4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O’Sullivan TE, et al. , BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory, Immunity 43 (2) (2015) 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Salio M, et al. , Essential role for autophagy during invariant NKT cell development, Proc. Natl. Acad. Sci. U. S. A 111 (52) (2014) E5678–E5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yang M, Zhang Y, Ren J, Autophagic regulation of lipid homeostasis in cardio-metabolic syndrome, Front Cardiovasc Med 5 (2018) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Choi AM, Ryter SW, Levine B, Autophagy in human health and disease, N. Engl. J. Med 368 (7) (2013) 651–662. [DOI] [PubMed] [Google Scholar]
- 179.Cai B, et al. , BMP2-mediated PTEN enhancement promotes differentiation of hair follicle stem cells by inducing autophagy, Exp. Cell Res 385 (2) (2019) 111647. [DOI] [PubMed] [Google Scholar]
- 180.Panda PK, et al. , Abrus agglutinin stimulates BMP-2-dependent differentiation through autophagic degradation of beta-catenin in colon cancer stem cells, Mol. Carcinog 57 (5) (2018) 664–677. [DOI] [PubMed] [Google Scholar]
- 181.Jiao G, et al. , BMPR2 inhibition induced apoptosis and autophagy via destabilization of XIAP in human chondrosarcoma cells, Cell Death Dis. 5 (2014) e1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ozeki N, et al. , Bone morphogenetic protein-induced cell differentiation involves Atg7 and Wnt16 sequentially in human stem cell-derived osteoblastic cells, Exp. Cell Res 347 (1) (2016) 24–41. [DOI] [PubMed] [Google Scholar]
- 183.Cao Y, et al. , Noggin attenuates cerulein-induced acute pancreatitis and impaired autophagy, Pancreas 42 (2) (2013) 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gomez-Puerto MC, et al. , Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension, J. Pathol 249 (3) (2019) 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Forrester SJ, et al. , Reactive oxygen species in metabolic and inflammatory signaling, Circ. Res 122 (6) (2018) 877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Atashi F, Modarressi A, Pepper MS, The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review, Stem Cells Dev. 24 (10) (2015) 1150–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li M, et al. , Inhibition of osteoclastogenesis by stem cell-derived extracellular matrix through modulation of intracellular reactive oxygen species, Acta Biomater. 71 (2018) 118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Simone S, et al. , BMP-2 induces a profibrotic phenotype in adult renal progenitor cells through Nox4 activation, Am J Physiol Renal Physiol 303 (1) (2012) F23–F34. [DOI] [PubMed] [Google Scholar]
- 189.Chandrasekaran V, et al. , Reactive oxygen species are involved in BMP-induced dendritic growth in cultured rat sympathetic neurons, Mol. Cell. Neurosci 67 (2015) 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sorescu GP, et al. , Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase, Circ. Res 95 (8) (2004) 773–779. [DOI] [PubMed] [Google Scholar]
- 191.Chikazu D, et al. , Bone morphogenetic protein 2 induces cyclo-oxygenase 2 in osteoblasts via a Cbfal binding site: role in effects of bone morphogenetic protein 2 in vitro and in vivo, J. Bone Miner. Res 17 (8) (2002) 1430–1440. [DOI] [PubMed] [Google Scholar]
- 192.Raic A, et al. , 3D models of the bone marrow in health and disease: yesterday, today and tomorrow, MRS Commun 9 (1) (2019) 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ham J, et al. , In vitro 3D cultures to reproduce the bone marrow niche, JBMR Plus 3 (10) (2019) e10228. [DOI] [PMC free article] [PubMed] [Google Scholar]