

Abstract
Hematopoietic stem cell (HSC) gene therapy has the potential to cure many genetic, malignant, and infectious diseases. We have shown in a nonhuman primate gene therapy and transplantation model that the CD34+CD90+ cell fraction was exclusively responsible for multilineage engraftment and hematopoietic reconstitution. In this study, we show the translational potential of this HSC-enriched CD34 subset for lentivirus-mediated gene therapy. Alternative HSC enrichment strategies include the purification of CD133+ cells or CD38low/– subsets of CD34+ cells from human blood products. We directly compared these strategies to the isolation of CD90+ cells using a good manufacturing practice (GMP) grade flow-sorting protocol with clinical applicability. We show that CD90+ cell selection results in about 30-fold fewer target cells in comparison to CD133+ or CD38low/– CD34+ hematopoietic stem and progenitor cell (HSPC) subsets without compromising the engraftment potential in vivo. Single-cell RNA sequencing confirmed nearly complete depletion of lineage-committed progenitor cells in CD90+ fractions compared to alternative selections. Importantly, lentiviral transduction efficiency in purified CD90+ cells resulted in up to 3-fold higher levels of engrafted gene-modified blood cells. These studies should have important implications for the manufacturing of patient-specific HSC gene therapy and gene-engineered cell products.
Keywords: Hematopoietic Stem Cells, Gene Therapy, CD90 CD34, Lentivirus Transduction, Stem Cell Enrichment / Cell Sorting, single cell RNA sequencing, Mouse xenograft transplants, Closed System Application
Graphical Abstract
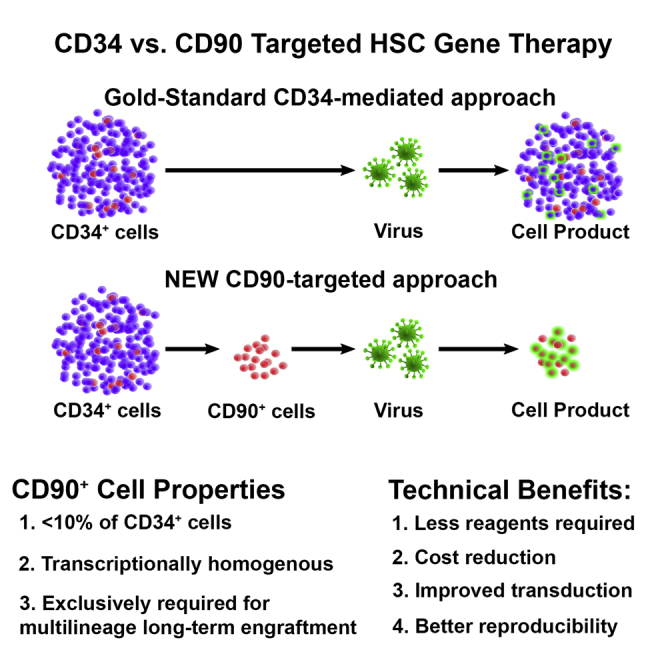
Current CD34-mediated hematopoietic stem cell (HSC) gene therapy approaches are untargeted and expensive. Purification and targeting of the HSC-enriched CD34+CD90+ subset is less expensive and more efficient with predictable results ex vivo and in vivo. Modifying CD34+CD90+ cells can overcome current roadblocks for the routine clinical use of HSC gene therapy.
Introduction
Hematopoietic stem cell (HSC)-mediated gene therapy for the treatment of hematologic diseases is commonly performed with single-marker purified CD34+ cells. However, it is well established that CD34+ cells are a heterogeneous mix of predominantly lineage-committed progenitor cells containing only very few true HSCs with long-term multilineage engraftment potential.1,2 Moreover, the proportion of true HSCs is variable across individuals, on average requiring 2–5 × 106 viable CD34+ cells per kg of body weight to reach consistent engraftment.3,4 This results in excessive use of costly reagents (e.g., lentiviral vectors or nucleases) to reliably target HSCs.5, 6, 7, 8, 9, 10, 11 Moreover, the efficiency of gene modification seen ex vivo does not correlate with the in vivo engraftment of modified cells often stabilizing at a significantly lower frequency in pre-clinical and clinical studies.6,8,12, 13, 14 These major hurdles significantly limit the broad availability of this very promising technology as a potential cure for many patients.
HSC gene therapy and transplantation would benefit from the ability to isolate, target, and modify a more HSC-enriched subset that provides short-term reconstitution as well as long-term multilineage engraftment. Availability of a defined target could overcome all currently existing limitations simultaneously, which would (1) greatly reduce the amount of modifying reagents needed for manufacturing, (2) result in more reliable genetic modification of HSCs, and (3) increase the predictability of transplant success in vivo.
Within the last four decades, a plethora of cell surface markers have been described for the identification and purification of human HSCs.1,15, 16, 17, 18, 19 Similarly, a great variety of HSC enrichment strategies have been proposed for gene therapy approaches. Some of the most promising candidates include the purification of the CD38low/–,20,21 CD133+,22 or CD90+23 CD34+ hematopoietic stem and progenitor cell (HSPC) subsets. While each strategy claims to improve individual aspects of gene therapy, a side-by-side analysis of these phenotypes has not been reported.
In this study, we applied up-to-date readouts (in vitro, in silico, and in vivo) for a systematic and objective side-by-side comparison of candidate HSC enrichment strategies with the goal to identify the most applicable HSC-enriched target for gene therapy and transplantation.
Results
CD34+CD90+ Cells Are Phenotypically the Most Defined Target for HSC Gene Therapy
Multilineage long-term engraftment and bone marrow (BM) reconstitution are driven by CD34+CD90+ HSPCs in the pre-clinical nonhuman primate (NHP) stem cell transplantation and gene therapy model.13,23,24 To evaluate the translational potential of the CD34+CD90+ phenotype, we compared this subset to alternative HSC enrichment strategies, including CD34+CD133+22 and CD34+CD38low/–20,21 on human HSPCs.
To compare the fold reduction of target cells for each strategy, we initially analyzed the frequency of CD133+, CD38low/−, and CD90+ subsets in steady-state BM (ssBM)-derived CD34+ cells across nine different healthy donors (Figures 1A and 1B). On average 42% (±2.8%, SD) of CD34+ cells co-expressed CD133 on the cell surface, reducing the target cell count for HSC gene therapy approximately 2.4-fold. Lack of CD38 expression (CD38low/−) was observed on 16.5% (±5.2%, SD) of CD34+ cells or a 5.8-fold enrichment of more primitive HSCs. A 12.5-fold reduction of target cells was reached for CD90+ cells comprising on average 7.5% (±4.7%, SD) of bulk CD34+ HSPCs.
Figure 1.

Quantitative Comparison of Phenotype-Defined Target CD34 Subsets
(A) Average frequency of CD133+, CD38low/–, and CD90+ HSPCs within bulk ssBM-derived CD34+ cells (mean ± SD, n ≥ 9 independent healthy donors). (B) Schematic of the quantitative and hierarchical relationship of phenotypic CD34 subsets. (C) Frequency of phenotype-defined subsets in bulk CD34+ cells before FACS (top row) and cross-contamination of phenotypic subsets within FACS-purified CD133+ (first column), CD38low/– (second column), and CD90+ (third column) HSPCs from a representative ssBM donor used for scRNA-seq analysis. (See also Figure S1.)
Since the purification of CD90+ HSPCs is currently limited to fluorescence-activated cell sorting (FACS), we next evaluated the feasibility of flow cytometric cell sorting for the enrichment of human HSC-enriched CD34 subsets for CD133+, CD38low/–, and CD90+ HSPCs in a side-by-side comparison. All three phenotypes were FACS-purified from ssBM on the FACSAria platform (BD Biosciences) and the phenotypic composition of purified subsets analyzed (Figures 1C and S1). The CD133+ subpopulation showed approximately 50% enrichment of CD38low/– but only 27% of the CD90+ subset. Purified CD38low/– cells were markedly enriched for CD133+ (78%) and CD90+ (49%) cells but still contained a significant proportion of CD90– HSPCs (51%). CD90+ cells demonstrated more than 86% enrichment of CD133+ as well as CD38low/– HSPCs.
Based on this phenotypic assessment, CD90+ HSPCs demonstrate the most defined HSC-enriched CD34+ subset. Purification of this phenotype yields the greatest reduction of target cells for HSC gene therapy in comparison to CD133+ or CD38low/– CD34 subsets.
CD90+ HSPCs Are Depleted for Transcriptionally Lineage-Committed Progenitors
Single-cell transcriptome analysis has proven useful in determining the heterogeneity of complex cell populations.25 Here, we used single-cell RNA sequencing (scRNA-seq) on the 10x Genomics Chromium platform to assess the transcriptional heterogeneity and depletion of lineage-committed progenitor cells within FACS-purified CD133+, CD38low/–, and CD90+ CD34+ subsets.
To identify transcriptionally distinct CD34 subsets of primitive HSPCs and lineage-committed progenitor cells, we initially generated a scRNA-seq reference map. CD34+ cells from ssBM of two healthy human individuals were sequenced (Table S1), and the data were analyzed using t-Distributed Stochastic Neighbor Embedding (tSNE) and principal-component analysis (PCA) for each donor individually.
Seven transcriptionally distinct groups of single cells were identified within CD34+ cell populations using the Seurat unsupervised graph-based clustering tool26 (Table S2). Clusters were mapped onto both the tSNE (Figures 2A and S2A) and PCA (Figures 2B and S2B) visualizations. Comparison of clusters demonstrated expression of genes associated with primitive HSPCs predominantly localized in cluster 1 (Figure 2A, S3, and S4). Increased expression of genes for lymphoid, myeloid, and erythroid differentiation were observed in clusters 2 and 3, clusters 4 and 5, and clusters 6 and 7, respectively. CD34 expression was detectable in all clusters, with a gradual increase toward more primitive HSPCs (Figures 2C and S5A). In good agreement with previous publications,1,27, 28, 29, 30 CD133 mRNA expression was restricted to transcriptionally lympho-myeloid-committed progenitors as well as immature HSPCs (clusters 1–5). CD38 expression was scattered throughout the reference map and showed no obvious association or absence in distinct clusters. Surprisingly, CD90 expression was undetectable in both donors.
Figure 2.

scRNA-Seq of ssBM-Derived CD34 HSPCs and FACS-Purified CD34 Subsets
(A) Dimensional reduction (tSNE) and graph-based clustering (PCA) of scRNA-seq data from ssBM-derived CD34+ cells and representative feature plots showing the expression of genes associated with lymphoid-primed, myeloid-primed, erythroid-primed, and immature HSPCs. Level of expression is color coded as shown in the legend in (C). (B) PCA-based transformation with clusters defined in (A) projected onto the PCA analysis and expression of representative genes as shown in (A). (C) Expression of CD34, CD133, CD38, and CD90 in ssBM-derived CD34+ cells. Level of expression is color coded as shown in the legend. (D) Overlay of scRNA-seq data from CD34+ cells (black, first plot) with FACS-purified CD133+ (green, second plot), CD38low/– (purple, third plot), and CD90+ (pink, fourth plot) HSPCs. (See also Figures S2, S3, S4 and S5 as well as Tables S1 and S2.)
To now compare phenotypic CD34+ HSPC subsets, we FACS-purified CD133+, CD38low/–, and CD90+ cell fractions from ssBM, performed scRNA-seq, and overlaid the resulting data onto the CD34+ reference map (Figures 2D and S5B; Table S1). Similar to the observed CD133 mRNA expression, FACS-purified CD133+ HSPCs exclusively colocalized with lympho-myeloid and more primitive HSPCs. FACS-purified CD38low/– cells predominantly overlaid with cluster 1, with individual cells spreading into both the lympho-myeloid as well as the erythroid branch. Surprisingly, mapping of FACS-purified CD90+ cells was almost indistinguishable from CD38low/– cells, demonstrating a significant enrichment of immature HSPCs (cluster 1), as well as depletion of lympho-myeloid and erythroid primed progenitors.
Since CD90+ and CD38low/– cells from ssBM show significant phenotypic differences but are indistinguishable by scRNA-seq, we hypothesized that the mRNA capture efficiency of scRNA-seq may be insufficient31, 32, 33, 34 to detect subtle transcriptional differences between both phenotypes. Consequently, we performed bulk RNA-seq on CD90+ (population a) and CD38low/– (population e) cells from ssBM and mapped the data onto our scRNA-seq reference map (Figure 3A). To validate this “bulk onto scRNAseq” mapping strategy, additional CD34+ subsets from ssBM enriched for lympho-myeloid and erythro-myeloid progenitor cells were added. Briefly, HSPCs were sorted into CD34+CD90+ (population a), CD34+CD90–CD45RA–CD133+ (population b), CD34+CD90–CD45RA+CD133+ (population c), and CD34+CD90–CD45RA–CD133low/– (population d) subsets29,35,36 as well as CD34+CD38low/– (population e), CD34+CD38low/–CD90+ (population f), and CD34+CD38low/–CD90– (population g) populations20,21,30 (Figures 3A and S6).
Figure 3.

Bulk RNA-Seq of ssBM-Derived CD34 Subsets
(A) Gating strategy for phenotypic human CD34 subpopulations. (B) Heatmap of the top 200 differentially expressed genes in phenotypic CD34 subsets listed in (A) from two independent human donors. (C) Overlay of bulk RNA-seq data from FACS-purified ssBM subsets (populations a–g are color coded as defined in A) on the ssBM scRNA-seq PCA reference map (black dots). (D) Overlay of bulk RNA-seq data from FACS-purified G-CSF-mobilized CD34 subsets (populations a–d are color coded as defined in A) on the ssBM scRNA-seq PCA reference map (black dots). (See also Figures S6 and S7 as well as Tables S3 and S4.)
Consistent with the reported lineage potentials of the FACS-purified CD34+ subsets,20,21,23,29,30,35,36 we observed upregulation of lymphoid genes (JCHAIN, DNTT, CD2, IGLL1) in population c (CD34+CD90–CD45RA+CD133+), erythro-myelo-megakaryocytic genes (HBB, HDC, GATA1, CD36) enriched in population d (CD34+CD90–CD45RA–CD133low/–), and expression of more immature marker genes (AVP, HES1, DLK1) in populations a (CD34+CD90+), e (CD34+CD38low/–), f (CD34+CD38low/–CD90+), and g (CD34+CD38low/–CD90–) (Figure 3B; Table S3). Individual myeloid genes (ELANE, MPO, CEBPD) were simultaneously upregulated in populations c (CD34+CD90–CD45RA+CD133+) and d (CD34+CD90–CD45RA–CD133low/–). Interestingly, population b (CD34+CD90–CD45RA–CD133+) did not show a unique cluster of differentially expressed genes but shared features of genes associated with lympho-myeloid and myeloid-primed as well as immature HSPCs (Figure 3B; Table S3).
To confirm this manual assessment, we mapped the bulk RNA-seq data from ssBM CD34+ HSPC subsets onto the CD34 scRNA-seq reference map (Figure 3C). As expected, lympho-myeloid primed cells (population c, CD34+CD90–CD45RA+CD133+) mapped within clusters 4 and 5 and erythro-myelo-megakaryocytic primed HSPCs (population d, CD34+CD90–CD45RA–CD133low/–) plotted within cluster 7. Population b (CD34+CD90–CD45RA–CD133+) showed greater heterogeneity (distance between dots) and localized within clusters 2 and 3 of the lympho-myeloid arm. Populations a (CD34+CD90+), e (CD34+CD38low/–), f (CD34+CD38low/–CD90+), and g (CD34+CD38low/–CD90–) were closely co-localized within cluster 1 at the top of the reference map. More detailed comparison of populations a, e, f, and g revealed that CD90+ HSPCs (population a) demonstrated the lowest donor-to-donor variability. In contrast, CD38low/– (population e) HSPCs showed the greatest heterogeneity despite nearly identical proportions of CD38low/–CD90+ (donor 1, 36%; donor 2, 33%) and CD38low/–CD90– (donor 1, 64%; donor 2, 67%) subsets in both donors. CD38low/–CD90– (population g) subsets were similarly heterogeneous and shifted toward the erythroid-primed clusters matching the reported enrichment of erythro-myeloid progenitors in CD133low/–CD38–CD90– HSPCs.36,37 Most importantly, sorting of the CD38low/–CD90+ subset (population f) significantly reduced the donor-to-donor variability and led to closer co-localization with CD90+ HSPCs (population a).
Finally, we FACS-purified populations a–d (a, CD34+CD90+; b, CD34+CD90–CD45RA–CD133+; c, CD34+CD90–CD45RA+CD133+; d, CD34+CD90–CD45RA–CD133low/–) HSPCs from two granulocyte colony-stimulating factor (G-CSF)-mobilized CD34+ donors, the most frequently used stem cell source for HSC gene therapy approaches, and bulk RNA-seq data were mapped onto the reference map (Figure 3D). No meaningful differences in the transcriptional signature were observed between CD90+ (population a) HSPCs from both sources (Tables S3 and S4). However, CD90–CD45RA–CD133+ (population b) HSPCs reported to contain multipotent progenitors30,37 co-localized much closer with CD90+ HSPCs (population a). Pairwise comparison of CD90+ (population a) with CD90–CD45RA–CD133+ (population b) HSPCs confirmed that G-CSF mobilization significantly reduced the transcriptional heterogeneity between both CD34+ HSPC subsets (ssBM, 615 genes; G-CSF, 131 genes; Figure S7; Tables S5, S6, S7, and S8).
In conclusion, combining single-cell and bulk RNA-seq we herein show that CD90+ HSPCs are almost entirely depleted for transcriptionally lineage-committed progenitor cells and significantly enriched for transcriptionally primitive HSPCs. Most importantly for the clinical application, the transcriptional signatures within the CD90+ subset are not impacted by G-CSF-mediated mobilization.
BM Reconstitution Potential Is Restricted to CD34+CD90+ Cell Fractions
Pre-clinical experiments in the NHP and mouse showed that G-CSF-primed CD34+CD90+ HSPCs were solely responsible for the reconstitution of the BM stem cell compartment.23,24 To ensure that the depletion of CD90− HSPCs in humans does not impact the in vivo multilineage engraftment potential, bulk CD34+ and FACS-purified population a–d cells from human G-CSF-mobilized CD34+ HSPCs were transplanted into sub-lethally irradiated adult NSG (non-obese diabetic [NOD].Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice.
The highest engraftment in the peripheral blood (PB), BM, and thymus was observed in cohorts transplanted with CD90+ (population a) cells (Figures 4A, 4B, S8A–S8D, S9A, and S9B). Lower level multilineage engraftment of human cells was observed upon transplantation of population b (CD90–CD45RA–CD133+). Representing a mix of CD90+ (population a) and CD90–CD45RA–CD133+ (population b) cells, the average human chimerism in CD34 transplanted mice fell in between the observed engraftment for population a and b observed in almost all tissues. Mice receiving population c showed locally restricted human chimerism in the thymus, whereas population d did not show any human engraftment in the analyzed tissues. Engraftment and reconstitution of the entire BM stem cell compartment, including the recovery of phenotypic primitive human CD34+CD90+ HSPCs, was exclusively observed after transplantation of CD90+ cells as well as CD90-containg bulk CD34+ HSPCs. (Figures 4C, S8E, S8F, and S9C). Similarly, erythroid, myeloid, and erythro-myeloid colony-forming cell (CFC) potentials were only detected in mice transplanted with CD90+ or CD34+ cells (Figures 4D and S9D).
Figure 4.

Multilineage Engraftment Potential of Human CD34 Subpopulations
(A and B) Flow cytometric assessment of the frequency of human chimerism in the (A) PB and (B) BM, spleen, and thymus after transplantation of bulk CD34+ HSPCs as well as FACS-purified CD34 subpopulations (1 × 105 cells per mouse) from a single G-CSF-mobilized human donor. Engraftment data from a second donor can be found in Figure S9. (C) Frequency of engrafted human CD34+ and CD90+ HSPCs. CD34+ frequency, left y axis; CD90+ frequency, right y axis. (D) Human CD34+ cells from the murine BM were flow-sorted into CFC assays and erythroid, myeloid, and erythro-myeloid CFC potentials were quantified after 12–14 days. Horizontal line at 0.1% in A and B indicates threshold for the detection of human chimerism. Horizontal bars in (B) and (C) indicate the mean for each population. CFU, colony-forming unit; CFU-M, CFU macrophages; CFU-G, CFU granulocytes; CFU-GM, CFU, granulocytes/macrophages; BFU-E, burst forming unit erythroids; CFU-MIX, CFU erythro-myeloid colonies. (See also Figure S8, S9, and S10.)
To confirm that human CD90–CD45RA–CD133+ HSPCs (population b) from G-CSF-mobilized CD34+ HSPCs do not contain primitive HSCs with multilineage long-term engraftment potential, we performed limiting dilution experiments (Figure S10). Gradually increasing the number of transplanted cells from population b led to greater multilineage engraftment of human cells in all tissues, including CD34+ cells in the BM stem cell compartment (Figure S10A–S10E). However, none of the mice demonstrated human CD34+CD90+ HSPCs in vivo after transplant with this population, and engrafted CD34+ cells were restricted to erythroid and myeloid colony types lacking mixed colony-forming unit (CFU) potentials (Figure S10F). The number of SRCs (severe combined immunodeficiency [SCID]-repopulating cells) in population b was calculated to be 1 in 4.6 × 105 transplanted cells (Figure S10G).
In summary, mouse xenograft experiments confirm enrichment of primitive human HSPCs with multilineage engraftment and BM reconstitution potential in the CD34+CD90+ phenotype (population a). Furthermore, CD34+CD90–CD133+CD45RA– HSPCs (population b) contain low-level multilineage engraftment potential but lack the ability to recover the entire stem cell compartment.
FACS Purification Increases the Transduction Efficiency in HSC-Enriched CD90+ Cells
After validating that the human CD90+CD34+ cell subset is similarly enriched for phenotypically, transcriptionally, and functionally primitive HSPCs, we next evaluated the feasibility of a flow cytometry-based, GMP-compatible, large-scale FACS purification strategy for the enrichment and direct targeting of CD90+ cells from G-CSF-mobilized CD34+ HSPCs in a clinical setting.
CD34+ cells from six healthy G-CSF-mobilized donors were enriched on the Miltenyi CliniMACS Prodigy according to our previously established protocol (Figure 5A; Table S9).38 Leukapheresis products yielded on average 2.56 × 108 CD34+ cells with a purity greater than 93.5% ± 1.9%. Donor #4 insufficiently mobilized CD34+ cells and was excluded from the study.
Figure 5.

FACS Purification and Quality Control of CD90+ HSPCs
(A) Schematic of the experimental design. (B) Flow cytometric assessment of cells before FACS (CD34+, first plot) and purified CD90+ cells after sorting on the Sony (second plot) and Tyto (plot). (C) Comparison of the purity, yield, and fold enrichment of CD90+ HSPCs on the Sony and Tyto sorters. (D) CFC potential of CD90+ cells within bulk CD34+ HSPCs and FACS-purified CD90+ subsets. CD90+ cells from all three conditions were sorted into CFC assays to exclude contaminating CD90– cells. (E) Flow cytometric quantification of GFP-expressing CD90+ cells within bulk CD34+ cell and FACS-purified CD90+ subsets (left) and the delta-MFI of GFP expression in gene-modified cells (right). (F) Erythroid, myeloid, and erythro-myeloid CFC potential of gene-modified CD90+ cells from bulk CD34+ cells and FACS-purified CD90+ subsets. CD90+ cells from all three conditions were sorted into CFC assays. (G and H) Individual colonies from all three conditions in (F) were picked, and (G) the gene-modification efficiency in CFCs was determined by PCR as well as (H) the VCN in modified CFCs quantified by qPCR. (See also Table S9.) Statistics, means ± SEM; in C, third graph, median and range; significance values, two-tailed paired t test.
FACS purification of CD90+ HSPCs was performed comparing the jet-in-air sorter FX500 from Sony with the cartridge-based closed-system sorter MACSQuant Tyto from Miltenyi Biotec (Figure 5A). Sorted CD90+ cell fractions on both machines showed no significant phenotypic or quantitative differences. They both reached an average of 79.5% ± 6.9% (Sony) and 77.1% ± 5.5% (Tyto) purity with a yield of 52.3% ± 9.6% and 49.9% ± 11.1%, respectively (Figures 5B and 5C). Of special interest for HSC gene therapy, the median fold enrichment of target cells relative to the available target cell number in the bulk CD34+ product was 29.2-fold (range, 15.7- to 47.17-fold) on the Sony sorter and 33.1-fold (range, 23.32- to 65.36-fold) on the Tyto (Figure 5C). To determine whether the FACS purification impacted the differentiation potential of HSC-enriched CD34+ HSPC subsets, cells were introduced into CFC assays. FACS-purified cell fractions did not demonstrate any differences in the total or compositional CFC potential compared to unprocessed cells (Figure 5D).
Next, sorted CD90+ as well as bulk CD34+ cells were transduced with a lentiviral vector encoding for green fluorescent protein (GFP) according to our clinically approved CD34-mediated gene therapy protocol.39,40 Five days post-transduction, the gene modification efficiency in the CD90+ subset of bulk-transduced CD34+ cells reached 16.5% ± 3.4% (SEM), whereas 2.3-fold (Tyto, 38.1% ± 5.5%, SEM, p = 0.012) and 3.1-fold (Sony, 51.9% ± 5.8%, SEM, p = 0.006) higher frequencies of GFP were seen in the FACS-purified CD90+ cell fractions (Figure 5E). Despite significant differences in the gene modification efficiency, no significant increase in the overall mean fluorescence intensity (MFI) of GFP was observed transducing purified CD90+ cells (Figure 5E). Cells were further introduced into CFC assays which demonstrated no significant quantitative and qualitative differences (Figure 5F). Finally, individual colonies were extracted to determine the gene modification efficiency within erythro-myeloid colonies as well as to precisely quantify the vector copy number (VCN). No obvious differences were found in the expression of GFP within CFCs from all three conditions (Figure 5G). Similar to the MFI of GFP, almost identical VCNs were observed in all colonies (Figure 5H).
In summary, the FACS purification of CD90+ cells is technically feasible and does not impact the cells phenotypic and functional properties. Most importantly, purification of CD90+ cells reduces the number of target cells and simultaneously improves the efficiency of lentivirus-mediated gene transfer without increasing the MFI as well as VCN and the need for additional transduction enhancers.
Improved In Vivo Engraftment of FACS-Purified and Gene-Modified CD90+ HSPCs
Regardless of high gene modification efficiency ex vivo, the frequency of long-term in vivo engraftment of modified cells is often both unpredictable and, in many studies, significantly lower compared to values determined during quality control of the infusion product.6,12,13 To compare the multilineage long-term engraftment potential of gene-modified bulk CD34+ with FACS-purified CD90+ HSPCs from G-CSF-mobilized donors, cells were transplanted into sublethally irradiated adult NSG mice. Of note, in order to mimic the average percentage of CD90+ cells within bulk CD34+ cell fractions mice transplanted with FACS-purified CD90+ cells received one-tenth the cell number compared to mice transplanted with bulk CD34+ cells.
Multilineage engraftment of human cells in the PB of transplanted mice was followed longitudinally by flow cytometry (Figure 6A). Mice transplanted with bulk CD34+ as well as Tyto-sorted CD90+ cells showed persisting levels of human engraftment, whereas human chimerism in the vast majority of mice receiving Sony-sorted CD90+ HSPCs gradually declined toward the end of study. Similar trends in the overall frequency of human multilineage engraftment were seen at the day of necropsy in the BM, spleen, and thymus (Figures 6B and S11). Overall, the greatest frequency of human chimerism was found in mice transplanted with bulk CD34+ and Tyto-sorted CD90+ HSPCs followed by Sony-sorted CD90+ cells.
Figure 6.

Transplantation of FACS-Purified and Gene-Modified CD90+ Cells into NSG Mice
(A) Frequency of human chimerism in the PB over time. (B) Human chimerism in the BM, spleen, and thymus at 20 weeks post-transplant. (C and D) Frequency of GFP+ human cells in (C) the PB over time and (D) tissues at 20 weeks post-transplant. (E) Engraftment of human CD34+, CD90+, and CD90+GFP+ HSPCs in the BM at necropsy. (F) Human CD34+ cells from the murine BM were flow-sorted into CFC assays and erythroid, myeloid, and erythro-myeloid CFC potentials were determined. Horizontal line at 0.1% in A and B indicates the threshold for the detection of human chimerism. Shapes indicate different human donors (n = 3). (See also Figure S11.) Statistics, means ± SEM.
Lentiviral-mediated delivery of GFP enabled us to flow cytometrically follow the frequency of gene modification longitudinally in the PB as well as at the day of necropsy in tissues. Consistent with the efficiency of gene modification we saw ex vivo (Figure 5E), a 2- to 3-fold higher frequency of GFP+ in human cells was seen in the PB and tissues of animals receiving FACS-purified CD90+ cells compared to bulk CD34+ cells (Figures 6C and 6D). The frequency of gene modification in the PB of animals transplanted with FACS-purified CD90+ cells remained consistent throughout the follow-up, whereas in animals receiving gene-modified bulk CD34+ cells the frequency of GFP+ cells showed great variance toward the end of study. In some mice receiving transduced human CD34+ cells, mature human cells in the PB either (1) gradually lost the gene modification, leveling out at about 10%–15%; or (2) started to show a rapid increase in GFP+ human cells up to almost 80%. This trend was less often observed in mice receiving Sony- or Tyto-sorted CD90+ cells, suggesting an overall more stable engraftment of gene-modified cells over time.
Next, we analyzed the BM stem cell compartment of engrafted mice. Mice transplanted with bulk CD34+ cells demonstrated significantly greater engraftment of human CD34+ and CD34+CD90+ cells compared to Sony- and Tyto-sorted CD90+ HSPCs (Figure 6E, graph 1 and 2). However, on average more GFP+CD90+ cells were found in mice transplanted with Tyto-sorted CD90+ cells in comparison to mice receiving CD34+ or Sony-sorted CD90+ cells, respectively (Figure 6E, graph 3). Most importantly, the frequency of gene modification in CD90+ cells was on average 2- to 3-fold higher in mice engrafted with FACS-purified and gene-modified CD90+ cells compared to bulk CD34+ HSPCs (Figure 6E, graph 4). Finally, we introduced BM-resident human CD34+ cells into CFC assays to test the maintenance of erythro-myeloid differentiation potentials (Figure 6F). Engrafted human HSPCs in only two mice transplanted with either bulk CD34+ or Sony-sorted CD90+ gave rise to colonies, whereas in eight out of nine cases erythro-myeloid colonies were seen in mice receiving Tyto-sorted CD90+ cells.
In summary, gene modification of FACS-purified CD34+CD90+ cells does not alter the multilineage long-term engraftment potential of human HSPCs in the mouse xenograft model. Most importantly, gene modification efficiencies observed ex vivo successfully translated into the in vivo transplant and remained stable throughout the long-term follow-up.
Discussion
In this study, we show that the enrichment of the human CD34+CD90+ population has the potential to overcome current limitations of HSC gene therapy. FACS purification of this HSC-enriched CD34 subset resulted in about 30-fold reduction of target cells and consequently significant savings of costly reagents (here lentiviral vectors). Most importantly, purification of this phenotype improved the gene modification efficiency of HSCs with long-term in vivo engraftment potential up to 3-fold, reflecting the ex vivo assessment, enhancing the predictability of HSC gene therapy, and improving the long-term stability of modified cells at high levels. Thus, CD34+CD90+ HSCs demonstrate a potential target population for HSC gene therapy and transplantation.
In order to systematically and objectively compare human candidate HSC-enriched target cell populations for HSC gene therapy, in this study, we combined phenotypic, transcriptional, and functional readouts. We initially analyzed the phenotypic heterogeneity, the relationship between subsets, as well as the quantitative reduction of target cells for each CD34+ HSPC subset using flow cytometry and cell sorting. Next, we purified previously reported HSC-enriched cell populations as well as our recently defined CD34+CD90+ subset and performed high-throughput scRNA-seq analysis in combination with bulk RNA-seq. In addition, mouse xenograft assays were performed to evaluate the multilineage engraftment and BM reconstitution potential of human CD34+ subsets. In this comprehensive side-by-side comparison, we observed the greatest reduction of target cells in the CD34+CD90+ cell fraction without compromising the multilineage engraftment and BM reconstitution potential in mouse xenograft transplants. Furthermore, scRNA-seq data showed that CD34+CD90+ cells are almost entirely depleted for lineage-committed progenitor cells, while phenotypically, transcriptionally, and functionally primitive HSPCs are markedly enriched. Most importantly for the clinical translation, we compared two GMP-compatible FACS platforms for the purification and gene modification of HSC-enriched CD34 subsets. Purity and yield of sorting, gene-modification efficiency, as well as maintenance of stem cell features were confirmed by flow cytometry, in vitro assays, and mouse xenograft transplants. We demonstrate that direct transduction of FACS-purified CD34+CD90+ cells significantly reduced gene-modifying reagents and, at the same time, enhanced the gene modification efficiency without compromising the long-term multilineage engraftment potential.
These findings are all in line with our recently performed competitive reconstitution studies in NHPs to identify a defined target cell population for HSC gene therapy and transplantation.23 In our previous study, we reported the CD34+CD90+ phenotype to be exclusively responsible for both rapid short-term as well as robust long-term hematopoietic reconstitution. In this study, we show that human multilineage engraftment potential with full recovery of the BM stem cell compartment is similarly restricted to CD34+CD90+ HSPCs, whereas CD34+ cells that lost CD90 expression lack full multilineage xenograft reconstitution potential. This finding is also consistent with previous publications describing the enrichment of human HSCs within CD90-expressing CD34 subsets. The first CD90-mediated isolation of human HSCs from fetal BM, adult BM, and cytokine-mobilized stem cells was reported in the 1990s.15,41 Thereafter, lineage (Lin)–CD34+CD90+ subsets were shown to be enriched for long-term culture-initiating cells (LTC-ICs) with thymus engraftment potential in the SCID mouse model.15,41 However, alternative CD90/Thy-1 antibody clones (F15 421-5 and GM201) were used at this time that recognized a significantly higher expressed CD90 variant on up to 60% of cytokine-mobilized CD34+ cells.15 Refining the phenotype with additional cell surface markers, switching to the currently used CD90 clone 5E10, and using an improved mouse model, Majeti et al.30 later confirmed the enrichment of human HSCs in CD34+CD38–CD90+CD45RA– cell fractions when using umbilical cord blood (UCB)- and BM-derived CD34 subsets. With the highest level of purification, Notta et al.1 demonstrated engraftment and reconstitution potential of single human CD34+CD38–CD90+/–CD45RA–CD49f+ cells from UCB after intrafemoral injection into NSG mice. Despite significant differences in the phenotype, cell source, level of HSC purification, mouse model (NOD/SCID or NSG), and mode of transplantation (intravenous, intrafemoral), all groups, including our studies, associate CD90 expression with human HSCs, whereas HSPCs lacking CD90 demonstrate limited engraftment potential and do not re-express or recover the HSC-enriched CD90+ subset.
Studies in the 1990s used CD90-enriched cells in patients undergoing autologous transplantation for solid tumors, but the absence of a gene marker did not permit the discrimination of remaining endogenous stem cells competing with sort-purified and transplanted CD34+CD90+ cells. In contrast, using subleathal myeloablative conditioning and transplanting lentivirus gene-modified cells, we were able to show in both the large animal and the mouse models that NHP cells as well as human CD90+ cells are the primary CD34+ HSPC subset required for reconstitution after transplantation.23,24
Recent studies aiming to refine the target for HSC gene therapy and transplantation combined flow cytometry with functional in vitro and/or in vivo readouts.20,21 In the present study, we additionally applied single-cell as well as bulk RNA sequencing to compare the CD34+CD90+ phenotype with alternative HSC enrichment strategies. In contrast to most approaches performing transcriptomics,42, 43, 44 we initially performed scRNA-seq on CD34+ cells to build a reference map rather than using historically defined human CD34 subsets for comparison. This strategy allowed us to define transcriptionally distinct clusters and perform an unbiased assessment of the expression profile in these clusters. Furthermore, we were able to determine the reproducibility of scRNA-seq for CD34+ HSPCs and ultimately establish a baseline for the comparison of phenotype-defined HSC-enriched cell fractions. Having this baseline further enabled us to objectively compare scRNA-seq data from different donors as well as reliably map FACS-purified CD34+ HSPC subsets. Most importantly, this strategy provided the foundation to overlay scRNA-seq with bulk RNA-seq data from multiple donors without any computational correction or manipulation of the dataset for donor-, batch-, source-, or preparation-dependent variability.
Interestingly, we were not able to reliably determine transcriptional differences between CD34+CD90+ and CD34+CD38low/– HSPCs by scRNA-seq. Using the 10x Genomics scRNA-seq v2 platform, we were unable to determine transcriptionally distinct and meaningful clusters or define more detailed hierarchical structures within either subset. Similar to previous reports on Lin–CD34+CD38low/– cells describing a continuum of low-primed undifferentiated (CLOUD) cells without hierarchical structures,42, 43, 44, 45 CD34+CD90+ and CD34+CD38low/– HSPCs were indistinguishable even though CD38low/– cells were displaying clear differences in their phenotypic composition. Platform-dependent limitations in the mRNA capturing efficiency,34 technical noise,46 and low-level expression of key genes currently limit the ability to reliably distinguish differences in RNA expression43 required to identify cell subsets with short- and long-term engraftment potential within the CD34+CD90+ phenotype.
Despite the current limitation in scRNA-seq, our approach clearly shows that purified CD34+CD90+ as well as CD34+CD38low/– HSPCs are similarly depleted for transcriptionally lineage-committed progenitor cells. However, combining phenotypic, quantitative, and transcriptional data, the CD34+CD90+ subset was determined to have the greatest overall reduction of target cells. With an average 12.5-fold reduction in total cells compared to 5.8-fold for the CD34+CD38low/– subset, isolation and targeting of CD34+CD90+ HSPCs significantly increases the feasibility of currently existing HSC gene therapy approaches. Most importantly, transplantation of purified CD34+CD90+ HSPCs did not compromise the multilineage engraftment potential and BM reconstitution capacity of CD34+ HSPC subsets from G-CSF-mobilized leukapheresis products.
Particularly important for the application of HSC gene therapy as a routine treatment option, we have successfully demonstrated that the large-scale FACS purification of the HSC-enriched CD90+ subset for human stem cell sources is technically feasible and significantly reduces the target cell number in contrast to reported small-molecule-mediated approaches where bulk CD34+ cells are commonly targeted. CD34+CD90+ cells can be efficiently purified using either the Sony droplet cell sorter, which is clean room-dependent, or the fully closed, portable MACSQuant Tyto from Miltenyi without any significant differences in the purity and yield of cells. However, more comprehensive follow-up studies are required to further investigate the underlying biological mechanism impacted by both cell-sorting strategies causing the shown differences in multilineage engraftment potential as well as the efficiency of gene modification. Specific analysis of stress-mediated responses, as well as activation of cell cycle pathways, may help to explain the observed variances in the frequency of gene modification and long-term engraftment seen comparing both systems.
Most surprisingly, we found that the FACS purification CD34+CD90+ cells significantly increased the transduction efficiency of HSCs with multilineage long-term engraftment potential compared to gold standard CD34+ cells. In particular, the observation that the gene modification efficiencies determined in culture directly translate into the frequency of gene modification in vivo will help to increase consistency of quality in stem cell products and ultimately make HSC gene therapy applications more predictable. These findings should have important implications for currently available as well as future HSC gene therapy protocols; hence, the presented FACS purification of CD34+CD90+ HSPCs can be performed in a clinical-grade large scale and embedded into previously established CD34-mediated approaches. Isolation of this phenotype will allow more targeted and more efficient gene modification of multipotent human HSCs without the application of a small molecule transduction enhancer.
Application of this HSC-enriched subset will require the implementation of additional processing steps into existing clinical protocols. While the FACS purification will generate further expenses, costs performing these steps will be easily compensated by the about 30--fold savings in expensive modifying reagents. Gene therapy vectors are currently estimated to be the biggest hurdle in terms of large-scale production and by far the dominating cost factor limiting the routine application of HSC gene therapy.9, 10, 11 Our HSC-targeted gene therapy strategy will help to overcome both bottlenecks at the same time: a single large-scale virus production that was previously needed for a single patient could be used for multiple patients, overcoming the current shortage of GMP grade vectors; at the same time, modifying reagents will no longer be the major factor for the price determination in HSC gene therapy. Availability of new sorting technology for GMP grade, closed-system cell sorting of human HSCs47 in combination with our HSC-enriched target population will bring HSC gene therapy a step closer toward a technically and financially feasible routing application.
Of special interest for the clinical routine, we have seen a remarkable consistency in gene marking of CD90+ cells before transplantation, in ex vivo culture, and long-term in the mouse xenograft model. Commonly observed donor-to-donor variability in the CD34+ cell population leading to unpredictable levels of transduction in vivo was not seen for FACS-purified and gene-modified CD90+ cells. Especially the drop of ex vivo gene marking onto a significantly lower level in vivo previously observed in humans and NHPs6,8,12, 13, 14 was not evident in our experiments. These findings not only provide additional evidence that CD90+ HSPCs are uniquely required for robust long-term engraftment of gene-modified cells at high levels, but they further provide a rapid quality control before transplantation to predict the long-term success for gene-modification strategies.
In summary, this study describes a human HSC-enriched cell population with unique phenotypic, transcriptional, and functional features. Isolation of these HSC-enriched CD34+CD90+ HSPCs has the potential to improve the targeting efficiency of current clinical HSC gene therapy applications. Conservation of this HSC-enriched phenotype among different human stem cell sources and in the pre-clinical NHP model23 further highlights its biological relevance. Species-independent similarities of engraftment patterns in mouse xenograft (human) and autologous NHP transplants,23,30 as well as robust long-term multilineage engraftment with more than 2 years of follow-up in the NHP,23 are promising indicators for a successful clinical translation of this HSC enrichment strategy.
Materials and Methods
Cell Sources and CD34+ Enrichment
Fresh, whole BM in sodium heparin for scRNA-seq was purchased from StemExpress (Folsom, CA, USA). G-CSF-mobilized leukapheresis collections were purchased from the Co-operative Center for Excellence in Hematology (CCEH) at the Fred Hutchinson Cancer Research Center. All human samples were obtained after informed consent according to the Declaration of Helsinki, stating that the protocols used in this study have been approved by a local Ethics Committee/Institutional Review Board of the Fred Hutchinson Cancer Research Center. Human CD34+ cells from steady-state BM and G-CSF-mobilized leukapheresis collections were harvested and enriched as previously described.23,38 Enrichment of CD34+ cell fractions was performed according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany).
Flow Cytometry Analysis and FACS
Fluorochrome-conjugated antibodies used for flow cytometric analysis and FACS of human cells are listed in Table S10. Dead cells and debris were excluded via forward light scatter (FSC)/side light scatter (SSC) gating. Flow cytometric analysis was performed on an LSR IIu (BD Biosciences, Franklin Lakes, NJ, USA), LSRFortessa X50 (BD Biosciences), and FACSAria IIu (BD Biosciences). Cells for scRNA-seq, bulk RNA-seq, and in vitro assays were sorted using a FACSAria IIu cell sorter (BD Biosciences). Large-scale clinical-grade purification of CD34+CD90+ cells was performed on the FX500 (Sony Biotechnology, San Jose, CA, USA) and the MACSQuant Tyto (Miltenyi Biotec). Post-FACSpurity was assessed on the FACSAria IIu reanalyzing at least 500 cells for each sample. Data were acquired using FACSDiva version 6.1.3 and newer (BD Biosciences). Data analysis was performed using FlowJo version 8 and higher (BD Biosciences).
RNA Isolation for Bulk RNA-Seq
Total RNA from FACS-purified CD34 subsets of G-CSF-mobilized leukapheresis products was extracted with the Arcturus PicoPure RNA isolation kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Total RNA from FACS-purified CD34 subsets of steady-state BM was extracted with the RNeasy micro kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. Detailed methods on the data analysis can be found in Supplemental Materials and Methods.
RNA Quality Control for Bulk RNA-Seq
Total RNA integrity was analyzed using an Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, CA, USA) and quantified using a Trinean DropSense96 spectrophotometer (Caliper Life Sciences, Hopkinton, MA, USA).
scRNA-Seq
Steady-state BM-derived CD34+ cells and CD34 subsets for single-cell RNA sequencing were FACS-purified and processed using the Chromium Single Cell 3ʹ (v2) platform from 10x Genomics (Pleasanton, CA, USA). Separation of single cells, RNA extraction, and library preparation were performed in accordance with the 10x Chromium Single Cell Gene Expression Solution protocol. Detailed methods on the data analysis can be found in Supplemental Materials and Methods.50,51
Next-Generation Sequencing
Sequencing of G-CSF-mobilized and steady-state BM bulk RNA-seq samples was performed using Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) in rapid mode using a paired-end, 50-base read length (PE50) sequencing strategy. Sequencing of ssRNA-seq samples was performed using an Illumina HiSeq 2500 in rapid mode using 26-base read length for read 1 (10x barcode and 10-bp unique molecular index [UMI]) and 98-base read length for read 2 (cDNA sequence). Image analysis and base calling were performed using Illumina’s real-time analysis (v1.18) software, followed by “demultiplexing” of indexed reads and generation of fastq files, using Illumina’s bcl2fastq conversion software (v1.8.4).
Lentiviral Vectors
The vector used in this study (pRSCSFFV.P140K.PGK.eGFP-sW) is a self-inactivating (SIN) lentiviral vector produced with a third-generation split packaging system and pseudo-typed by the vesicular stomatitis virus G protein (VSV-G). The vector for these studies was produced by our institutional Vector Production Core (principal investigator [PI] H.-P.K.). Infectious titer was determined by flow cytometry evaluating EGFP protein expression following titrated transduction of HT1080 human fibrosarcoma-derived cells with research grade lentiviral vector preparations.
CFC Assay
For CFC assays, 1,000–1,200 FACS-purified CD34 subpopulations were seeded into MethoCult H4435 (STEMCELL Technologies) or H4230 supplemented with human interleukin (hIL)-3, IL-5, G-CSF, stem cell factor (SCF), thyroperoxidase (TPO), and granulocyte-macrophage colony-stimulating factor (GM-SCF) (all PeproTech), each at 100 μg/mL, as well as erythropoietin (EPO) (Amgen, Thousand Oaks, CA, USA) at 4 U/mL for the large-scale clinical grade experiments according to our established clinical protocols.39,48 Colonies were scored after 12–14 days, discriminating CFU-granulocyte (CFU-G), CFU-macrophage (CFU-M), granulocyte-macrophage (CFU-GM), and burst forming unit-erythrocyte (BFU-E). Colonies consisting of erythroid and myeloid cells were scored as CFU-MIX.
Transduction of Cell, Measurement of Transduction Efficiency, and VCN
For lentiviral transduction, CD34+ cells and FACS-purified CD90+ subsets were seeded in culture media (StemSpan ACF [STEMCELL Technologies, Vancouver, BC, Canada] supplemented with 1% penicillin streptomycin [Pen/Strep] [Thermo Fisher Scientific], as well as SCF, TPO, and fms-related tyrosine kinase 3 [FLT3] at 100 μg/mL [all three from PeproTech, Rocky Hill, NJ, USA]). Cells were seeded at a density of 1 × 106 per mL and cultured overnight for pre-stimulation. After 12–16 h, cells were harvested, washed, and transferred to RetroNectin-coated (Takara Bio, Kusatsu, Japan) tissue culture plastic in culture medium supplemented with 4 μg/mL protamine sulfate (Sigma-Aldrich, St. Louis, MO, USA) and a density of approximately 250,000 cells per cm2. Two doses of lentiviral vector at an MOI of 10 were added 6–8 h apart, and cells were kept at 37°C, 5% CO2 for 12–16 hours. Transduced cells were harvested, residual virus was removed, and cells were introduced into ex vivo and in vivo readouts.
To measure the transduction efficiency and VCN, at least 80 colonies were picked for each condition. Genomic DNA (gDNA) was isolated by incubating tubes at 95°C for 2 h on a thermal cycler. Crude DNA preparations were then subjected to PCR using lentiviral-specific primers (forward [Fwd], 5′-AGAGATGGGTGCGAGAGCGTCA-3′ and reverse [Rev], 5′-TGCCTTGGTGGGTGCTACTCCTAA-3′ [Integrated DNA Technologies (IDT), Coralville, IA, USA]) and, in a separate reaction, actin-specific primers were used (human Fwd, 5′-TCCTGTGGCACTCACGAAACT-3′ and Rev, 5′-GAAGCATTTGCGGTGGACGAT-3′ [IDT]). Colonies containing expected bands for both lentivirus and actin were scored as transduced. Reactions that did not yield actin products were considered non-evaluable.
VCN per genome equivalent was determined as previously described.38 Briefly, VCNs were assessed by a multiplex TaqMan 5′ nuclease quantitative real-time PCR assay in triplicate reactions. Colony gDNA samples were subjected to a lentivirus-specific primer/probe combination (Fwd, 5′-TGAAAGCGAAAGGGAAACCA-3′; Rev, 5′-CCGTGCGCGCTTCAG-3′; probe, 5′- AGCTCTCTCGACGCAGGACTCGGC [IDT]) as well as an endogenous control (TaqMan copy number reference assay RNaseP, Thermo Fisher Scientific, Pittsburgh, PA, USA) using TaqMan GTXpress master mix (Applied Biosystems, Foster City, CA, USA). Samples with an average VCN ≥0.5 were considered transduced.
Mouse Xenograft Transplantation
Adult (8- to 12-week-old) NSG mice received a radiation dose of 275 cGy followed 4 h later by a 200-μL intravenous injection of FACS-purified human CD34+ cells , CD34 subpopulations, gene-modified bulk CD34+ cells, or gene-modified CD34 subpopulations. Beginning at 8 weeks post-injection, blood samples were collected every 2–4 weeks and analyzed by flow cytometry. After 16–20 weeks, animals were sacrificed, and tissues were harvested and analyzed. All animal studies were carried out at the Fred Hutchinson Cancer Research Center in compliance with the approved Institutional Animal Care and Use Committee (IACUC) protocol #1483.
Statistical Analysis
Data analysis of limiting dilution experiments was performed as previously described.49 Statistical analysis of data was performed using GraphPad Prism version 5. Significance analyses were performed with the unpaired, two-sided Student’s t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Data and Materials Availability
All original RNA-seq data were uploaded to the NCBI database. The BioProject accession code is available upon request.
Author Contributions
S.R. and H.-P.K. designed the study. S.R., Y.-Y.C., and A.M.P. performed RNA-seq. D.P. and M.E. analyzed RNA-seq data. S.R., Y.-Y.C., A.M.P., and M.C. performed longitudinal mouse follow-up, necropsy, and data analysis. S.R., A.M.P., Y.-Y.C., M.C., S.S., A.B., and T.E. performed clinical-grade sorting experiments, follow-up experiments, and data analysis. S.R., D.P., and M.E. generated the figures. H.-P.K. and J.E.A. funded the study. S.R. and H.-P.K. wrote the manuscript. All authors reviewed and edited the final manuscript.
Conflicts of Interest
S.R. is a consultant to Forty Seven Inc. (Gilead Sciences). H.P.K. is a consultant to and has ownership interests with Rocket Pharma and Homology Medicines. H.P.K. is a consultant to CSL Behring and Magenta Therapeutics. S.R., J.E.A., and H.-P.K. are inventors on patent applications (#62/351,761, #62/428,994, and #PCT/US2017/037967) submitted by the Fred Hutchinson Cancer Research Center that cover the selection and use of cell populations for research and therapeutic purposes as well as strategies to assess and/or produce cell populations with predictive engraftment potential. The remaining authors declare no competing interests.
Acknowledgments
Human PB stem cells and steady-state BM were kindly provided by Shelly Heimfeld. We thank Helen Crawford for help in preparing this manuscript and figures. This work was supported in part by grants to H.-P.K. from the Evergreen Fund from the Fred Hutchinson Cancer Research Center, the National Institutes of Health (R01 AI135953-01), and the Immunotherapy Integrated Research Center, as well as by funds to J.E.A. from the Fred Hutchinson Cancer Research Center, the Cuyamaca Foundation, the Evergreen Fund, and from the NIH/NCI Cancer Center Support Grant P30 CA015704. H.P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. JEA holds the Fleischauer Family Endowed Chair in Gene Therapy Translation.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.07.010.
Contributor Information
Stefan Radtke, Email: sradtke@fredhutch.org.
Hans-Peter Kiem, Email: hkiem@fredhutch.org.
Supplemental Information
References
- 1.Notta F., Doulatov S., Laurenti E., Poeppl A., Jurisica I., Dick J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 2.Doulatov S., Notta F., Eppert K., Nguyen L.T., Ohashi P.S., Dick J.E. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat. Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 3.Barnett D., Janossy G., Lubenko A., Matutes E., Newland A., Reilly J.T., General Haematology Task Force of the British Committee for Standards in Haematology Guideline for the flow cytometric enumeration of CD34+ haematopoietic stem cells. Prepared by the CD34+ haematopoietic stem cell working party. Clin. Lab. Haematol. 1999;21:301–308. doi: 10.1046/j.1365-2257.1999.00253.x. [DOI] [PubMed] [Google Scholar]
- 4.Trébéden-Negre H., Rosenzwajg M., Tanguy M.L., Lefrere F., Azar N., Heshmati F., Belhocine R., Vernant J.P., Klatzmann D., Norol F. Delayed recovery after autologous peripheral hematopoietic cell transplantation: potential effect of a high number of total nucleated cells in the graft. Transfusion. 2010;50:2649–2659. doi: 10.1111/j.1537-2995.2010.02746.x. [DOI] [PubMed] [Google Scholar]
- 5.Peterson C.W., Haworth K.G., Burke B.P., Polacino P., Norman K.K., Adair J.E., Hu S.L., Bartlett J.S., Symonds G.P., Kiem H.P. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol. Ther. Methods Clin. Dev. 2016;3:16007. doi: 10.1038/mtm.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterson C.W., Wang J., Norman K.K., Norgaard Z.K., Humbert O., Tse C.K., Yan J.J., Trimble R.G., Shivak D.A., Rebar E.J. Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood. 2016;127:2416–2426. doi: 10.1182/blood-2015-09-672337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J., Exline C.M., DeClercq J.J., Llewellyn G.N., Hayward S.B., Li P.W., Shivak D.A., Surosky R.T., Gregory P.D., Holmes M.C., Cannon P.M. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 10.Morrison C. $1-Million price tag set for Glybera gene therapy. Nat. Biotechnol. 2015;33:217–218. doi: 10.1038/nbt0315-217. [DOI] [PubMed] [Google Scholar]
- 11.Melchiorri D., Pani L., Gasparini P., Cossu G., Ancans J., Borg J.J., Drai C., Fiedor P., Flory E., Hudson I. Regulatory evaluation of Glybera in Europe—two committees, one mission. Nat. Rev. Drug Discov. 2013;12:719. doi: 10.1038/nrd3835-c1. [DOI] [PubMed] [Google Scholar]
- 12.Hoban M.D., Cost G.J., Mendel M.C., Romero Z., Kaufman M.L., Joglekar A.V., Ho M., Lumaquin D., Gray D., Lill G.R. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray L., Chen B., Galy A., Chen S., Tushinski R., Uchida N., Negrin R., Tricot G., Jagannath S., Vesole D. Enrichment of human hematopoietic stem cell activity in the CD34+Thy-1+Lin− subpopulation from mobilized peripheral blood. Blood. 1995;85:368–378. [PubMed] [Google Scholar]
- 16.Yin A.H., Miraglia S., Zanjani E.D., Almeida-Porada G., Ogawa M., Leary A.G., Olweus J., Kearney J., Buck D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 17.Civin C.I., Strauss L.C., Brovall C., Fackler M.J., Schwartz J.F., Shaper J.H. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J. Immunol. 1984;133:157–165. [PubMed] [Google Scholar]
- 18.Lansdorp P.M., Sutherland H.J., Eaves C.J. Selective expression of CD45 isoforms on functional subpopulations of CD34+ hemopoietic cells from human bone marrow. J. Exp. Med. 1990;172:363–366. doi: 10.1084/jem.172.1.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terstappen L.W., Huang S., Safford M., Lansdorp P.M., Loken M.R. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38− progenitor cells. Blood. 1991;77:1218–1227. [PubMed] [Google Scholar]
- 20.Zonari E., Desantis G., Petrillo C., Boccalatte F.E., Lidonnici M.R., Kajaste-Rudnitski A., Aiuti A., Ferrari G., Naldini L., Gentner B. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports. 2017;8:977–990. doi: 10.1016/j.stemcr.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masiuk K.E., Brown D., Laborada J., Hollis R.P., Urbinati F., Kohn D.B. Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Mol. Ther. 2017;25:2163–2175. doi: 10.1016/j.ymthe.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon P.R., Leimig T., Babarin-Dorner A., Houston J., Holladay M., Mueller I., Geiger T., Handgretinger R. Large-scale isolation of CD133+ progenitor cells from G-CSF mobilized peripheral blood stem cells. Bone Marrow Transplant. 2003;31:17–22. doi: 10.1038/sj.bmt.1703792. [DOI] [PubMed] [Google Scholar]
- 23.Radtke S., Adair J.E., Giese M.A., Chan Y.Y., Norgaard Z.K., Enstrom M., Haworth K.G., Schefter L.E., Kiem H.P. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl. Med. 2017;9:eaan1145. doi: 10.1126/scitranslmed.aan1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radtke S., Chan Y.Y., Sippel T.R., Kiem H.P., Rongvaux A. MISTRG mice support engraftment and assessment of nonhuman primate hematopoietic stem and progenitor cells. Exp. Hematol. 2019;70:31–41.e1. doi: 10.1016/j.exphem.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Povinelli B.J., Rodriguez-Meira A., Mead A.J. Single cell analysis of normal and leukemic hematopoiesis. Mol. Aspects Med. 2018;59:85–94. doi: 10.1016/j.mam.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Görgens A., Radtke S., Möllmann M., Cross M., Dürig J., Horn P.A., Giebel B. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 2013;3:1539–1552. doi: 10.1016/j.celrep.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 28.Radtke S., Görgens A., Kordelas L., Schmidt M., Kimmig K.R., Köninger A., Horn P.A., Giebel B. CD133 allows elaborated discrimination and quantification of haematopoietic progenitor subsets in human haematopoietic stem cell transplants. Br. J. Haematol. 2015;169:868–878. doi: 10.1111/bjh.13362. [DOI] [PubMed] [Google Scholar]
- 29.Radtke S., Haworth K.G., Kiem H.P. The frequency of multipotent CD133+CD45RA−CD34+ hematopoietic stem cells is not increased in fetal liver compared with adult stem cell sources. Exp. Hematol. 2016;44:502–507. doi: 10.1016/j.exphem.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majeti R., Park C.Y., Weissman I.L. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saliba A.E., Westermann A.J., Gorski S.A., Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Islam S., Zeisel A., Joost S., La Manno G., Zajac P., Kasper M., Lönnerberg P., Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 34.Svensson V., Natarajan K.N., Ly L.H., Miragaia R.J., Labalette C., Macaulay I.C., Cvejic A., Teichmann S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods. 2017;14:381–387. doi: 10.1038/nmeth.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dmytrus J., Matthes-Martin S., Pichler H., Worel N., Geyeregger R., Frank N., Frech C., Fritsch G. Multi-color immune-phenotyping of CD34 subsets reveals unexpected differences between various stem cell sources. Bone Marrow Transplant. 2016;51:1093–1100. doi: 10.1038/bmt.2016.88. [DOI] [PubMed] [Google Scholar]
- 36.Görgens A., Ludwig A.K., Möllmann M., Krawczyk A., Dürig J., Hanenberg H., Horn P.A., Giebel B. Multipotent hematopoietic progenitors divide asymmetrically to create progenitors of the lymphomyeloid and erythromyeloid lineages. Stem Cell Reports. 2014;3:1058–1072. doi: 10.1016/j.stemcr.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Görgens A., Radtke S., Horn P.A., Giebel B. New relationships of human hematopoietic lineages facilitate detection of multipotent hematopoietic stem and progenitor cells. Cell Cycle. 2013;12:3478–3482. doi: 10.4161/cc.26900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adair J.E., Waters T., Haworth K.G., Kubek S.P., Trobridge G.D., Hocum J.D., Heimfeld S., Kiem H.-P. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat. Commun. 2016;7:13173. doi: 10.1038/ncomms13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adair J.E., Beard B.C., Trobridge G.D., Neff T., Rockhill J.K., Silbergeld D.L., Mrugala M.M., Kiem H.P. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci. Transl. Med. 2012;4:133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adair J.E., Chandrasekaran D., Sghia-Hughes G., Haworth K.G., Woolfrey A.E., Burroughs L.M., Choi G.Y., Becker P.S., Kiem H.P. Novel lineage depletion preserves autologous blood stem cells for gene therapy of Fanconi anemia complementation group A. Haematologica. 2018;103:1806–1814. doi: 10.3324/haematol.2018.194571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baum C.M., Weissman I.L., Tsukamoto A.S., Buckle A.M., Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc. Natl. Acad. Sci. USA. 1992;89:2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karamitros D., Stoilova B., Aboukhalil Z., Hamey F., Reinisch A., Samitsch M., Quek L., Otto G., Repapi E., Doondeea J. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat. Immunol. 2018;19:85–97. doi: 10.1038/s41590-017-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velten L., Haas S.F., Raffel S., Blaszkiewicz S., Islam S., Hennig B.P., Hirche C., Lutz C., Buss E.C., Nowak D. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017;19:271–281. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Notta F., Zandi S., Takayama N., Dobson S., Gan O.I., Wilson G., Kaufmann K.B., McLeod J., Laurenti E., Dunant C.F. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson N.K., Kent D.G., Buettner F., Shehata M., Macaulay I.C., Calero-Nieto F.J., Sánchez Castillo M., Oedekoven C.A., Diamanti E., Schulte R. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell. 2015;16:712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brennecke P., Anders S., Kim J.K., Kołodziejczyk A.A., Zhang X., Proserpio V., Baying B., Benes V., Teichmann S.A., Marioni J.C., Heisler M.G. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 47.Czechowska K., Lannigan J., Wang L., Arcidiacono J., Ashhurst T.M., Barnard R.M., Bauer S., Bispo C., Bonilla D.L., Brinkman R.R. Cyt-Geist: current and future challenges in cytometry: reports of the CYTO 2018 Conference Workshops. Cytometry A. 2019;95:598–644. doi: 10.1002/cyto.a.23777. [DOI] [PubMed] [Google Scholar]
- 48.Adair J.E., Johnston S.K., Mrugala M.M., Beard B.C., Guyman L.A., Baldock A.L., Bridge C.A., Hawkins-Daarud A., Gori J.L., Born D.E. Gene therapy enhances chemotherapy tolerance and efficacy in glioblastoma patients. J. Clin. Invest. 2014;124:4082–4092. doi: 10.1172/JCI76739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 51.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.