

Abstract
Introduction
Mounting evidence supports an association between antihypertensive medication use and reduced risk of Alzheimer's disease (AD). Consensus on possible pathological mechanisms remains elusive.
Methods
Human brain tissue from a cohort followed to autopsy that included 96 cases of AD (46 medicated for hypertension) and 53 pathological controls (33 also medicated) matched for cerebrovascular disease was available from the New South Wales Brain Banks. Quantified frontal cortex amyloid beta (Aβ) and tau proteins plus Alzheimer's neuropathologic change scores were analyzed.
Results
Univariate analyses found no difference in amounts of AD proteins in the frontal cortex between medication users, but multivariate analyses showed that antihypertensive medication use was associated with a less extensive spread of AD proteins throughout the brain.
Discussion
The heterogeneous nature of the antihypertensive medications is consistent with downstream beneficial effects of blood pressure lowering and/or management being associated with the reduced spreading of AD pathology observed.
Keywords: Alzheimer's disease, amyloid, antihypertensive medication, neuropathology, tau
1. BACKGROUND
Alzheimer's disease (AD) is pathologically diagnosed by the excessive accumulation of amyloid beta (Aβ) peptides in extracellular plaques and the fibrillization of hyperphosphorylated tau protein in neurons. 1 These AD proteins concentrate in predilection sites, increasing in their accumulation to progressively involve multiple brain regions causing clinical dementia. 1 The National Institue on Aging‐Alzheimer's Association (NIA‐AA) consensus on the neuropathologic assessment of AD requires the evaluation of the spreading of AD pathology through the brain. 1 The increased spread of AD pathology in the brain is then used to give a probability assessment of AD neuropathologic change as not sufficient for AD, a low probability of AD, or the diagnosis of AD when intermediate or high levels of AD pathology in the brain are achieved. 1
Although AD is common clinically, only a minority of cases have pure AD neuropathology post mortem (3% to 24%), while the majority has a mixture of AD and vascular pathologies as well as other neurodegenerative pathologies. 2 , 3 The additional pathologies observed in patients with clinical AD impact the pathological progression of AD. 2 , 3 One significant harbinger for the vascular pathologies observed is midlife hypertension, 4 which has been associated with an increased risk of AD. 5 Of interest is the finding that treating hypertension with any type of antihypertensive medication prevents cognitive decline 6 , 7 and progression to AD. 8 The precise tissue mechanism of such protective effects is not yet established; however, the most plausible explanation points to these medications working to minimize damaging cerebrovascular events in the brain. Alternatively, and possibly more contentious, is that the protective effects seen may be due to the medications working directly on cellular targets in the brain. The third explanation is that it is a combination of the above two. Regardless, the types of brain pathologies that antihypertensive medication(s) have an impact on is still contentious and in need of further investigation to understand this clinical effect and its underlying mechanism.
Most studies of the impact of antihypertensive medications on AD pathology have assessed regional pathological densities in cohorts not matched for concomitant pathologies. The Aβ plaque load in the frontal cortex assessed by immunohistochemistry is marginally lower in normotensive AD cases (by ≈1%) compared to hypertensive AD cases, although this was not a treatment effect as there were no significant differences between those on or not on antihypertensive medications. 9 Similarly, counts of silver‐stained neurofibrillary tangles (NFTs) and neuritic plaques (NPs) in neocortical regions are higher in hypertensive compared to normotensive AD cases 10 and these silver‐stained neuritic pathologies are also increased in hypertensive compared to normotensive non‐demented cases. 11 With respect to antihypertensive treatment, there are lower densities of silver‐stained NFT and NP in treated AD or mild cognitive impairment cases compared to untreated hypertensive or normotensive cases. 8 , 12 These immunohistochemical and classic histological studies suggest that antihypertensive medications may mainly impact the densities of silver‐positive neuritic and not immunohistochemically assessed Aβ pathologies.
Two studies have assessed the association of medications targeting the renin‐angiotensin system (RAS) with the staging of silver‐stained NFT pathology throughout the brain, again in cohorts not matched for concomitant pathologies. 8 , 13 RAS‐treated AD cases had more limited NFT formation throughout the brain compared to those untreated, 13 although there were no differences in the spread of NFT in those progressing from mild cognitive impairment to AD despite lower densities of NFT in most regions in those medicated. 8 Whether all antihypertensive medications impact disease progression remains to be determined. Hence, this study assessed the relationship in an elderly population at death between antihypertensive medication use and the extent of regional pathological thresholds as well as the spread of AD pathologies in cohorts with equivalent cerebrovascular disease but not other neurodegenerative disorders. As additional pathologies have been shown to impact the pathological progression of AD, 2 , 3 these exclusion criteria ensured that the relationship between antihypertensive medication use and AD pathology amount and spread could be determined in cohorts matched for concomitant pathologies.
Research in Context
Systematic review: Pubmed and Embase searches using the search terms “hypertension” and/or “hypertensive,” “dementia” and/or “Alzheimer,” “pathology” and/or “amyloid,” as well as citation lists from landmark studies and reviews were used to evaluate the accumulated knowledge in this area.
Interpretation: The data show no difference in the early deposition of Alzheimer's disease (AD)‐specific proteins in the brain between antihypertensive medication users but those on antihypertensives had less extensive anatomical spread of AD pathology.
Future directions: Identifying how antihypertensive medications potentially reduce the progression of AD pathologies may suggest novel strategies for their prevention and/or amelioration.
Highlights
Amyloid beta and tau levels did not differ in frontal cortex with antihypertensive medication use.
There was a reduced spread of Alzheimer's disease pathology with antihypertensive medication usage.
Antihypertensive medications’ heterogeneity points to downstream vascular benefit.
2. METHODS
2.1. Cohort inclusion and exclusion criteria
Cases were selected from the New South Wales Brain Banks database (n = 1009) of longitudinally followed participants in ethically approved brain donor programs specializing in neurodegenerative diseases and/or aging (without neurological or neuropsychiatric diseases). Consent for brain donation was obtained before death with brain retrieval occurring within 24 to 48 hours after death. Upon retrieval, brains were either hemisected with half frozen at –80°C and the other half fixed in 10% formalin or the whole brain fixed in 10% formalin. Fixed tissue was processed for histological characterization using current neuropathological consensus criteria. Study inclusion was based on the dominant type of diagnostic neuropathology present; being either no significant neuropathology (NSN, incidental, non‐diagnostic pathologies allowed) or AD neuropathologic change (ADNC) 1 or cerebrovascular disease 1 , 14 , 15 ± ADNC (n = 149, see Table 1). Exclusion included those reaching pathological diagnoses of tauopathies, synucleinopathies, neoplasms, or other rarer neuropathologies either alone or in association with ADNC. 1 Those aged at death <75 years old (median age at death in Australia for both men and women is 85–89 years old, see https://www.aihw.gov.au/reports/life-expectancy-death/deaths-in-australia/contents/age-at-death) were also excluded (see also Brayne et al. 16 ) to further reduce potential for neuropathologies other than AD or cerebrovascular disease (dementia and cerebrovascular disease are the second and third most prevalent causes of death in Australians >75 years of age at death, see https://www.aihw.gov.au/reports/life-expectancy-death/deaths-in-australia/contents/age-at-death) as well as those with incomplete clinical data (see Figure S1 in supporting information). Cerebrovascular disease included all cases except those with lobar infarction equating to >50 mL of tissue loss 15 as current pathological criteria for AD require the systematized sampling of multiple lobar regions. 1
TABLE 1.
Demographic, clinical, and pathological profile of cases
| Clinical Dementia Rating (n) | 0‐0.5 (60) | 1‐3 (89) |
|---|---|---|
| AD intermediate/high level change, n (%) | 26 (43%) | 70 (79%) ‡ |
| Age at death, mean years (SD) | 89 (6) | 87 (5) * |
| Male, n (%) | 29 (48%) | 31 (35%) |
| PMD, mean hours (SD) | 23 (18) | 23 (19) |
| Hypertensive, n (%) | 33 (55%) | 40 (45%) |
| Hypertension duration, mean years (SD) a | 16 (10) | 11 (9) * |
| Hypertensive and medicated, n (% of hypertensive) | 28 (85%) | 29 (73%) |
| Normotensive, n (%) | 27 (45%) | 49 (55%) |
| Normotensive and medicated, n (% of normotensive) | 14 (52%) | 8 (16%) † |
| Cerebrovascular disease and vascular brain injury, n (%) | 29 (48%) | 48 (54%) |
| Single infarct (non‐lobar), n (% of CVD) | 12 (41%) | 21 (44%) |
| Multiple infarcts (non‐lobar), n (% of CVD) | 17 (59%) | 27 (56%) |
Hypertensive cases based on clinical assessment.
Abbreviations: AD, Alzheimer's disease: CVD, cerebrovascular disease and vascular brain injury cases; CDR, Clinical Dementia Rating; n, number of cases; PMD, post mortem delay; SD, standard deviation.
Average duration in years of hypertensive cases, incomplete data for some cases; five from 0 to 0.5 CDR group and two from 1 to 3 CDR group due to incomplete clinical record (confirmation of hypertensive status but no information on date of diagnosis). Statistically significant results (P < .05) are in bold.
P < .05.
P < .01.
P < .001. For further breakdown of this data with all CDR categories see Table S1. For further medication data see Table S2. For further breakdown of this data with the experimental subsets used see Table S3.
2.2. Clinical data extraction
Clinical data were extracted from brain donor program databases plus review of original clinical case files. These files included general practitioner health summaries, specialist reports, and annual self‐report survey responses. From these sources, details on hypertension diagnosis, antihypertensive medication use, antihypertensive medication dose (where available), hypertension status, and clinical dementia rating (CDR) were obtained (see Table 1 and Table S1 in supporting information). Prescribed antihypertensive medications were cross‐referenced with the Monthly Index of Medical Specialties (MIMS) online database to confirm the antihypertensive medication class, which included diuretics, calcium channel blockers (CCBs), angiotensin receptor blockers (ARBs), angiotensin converting enzyme (ACE) inhibitors, and beta blockers. For more information on antihypertensive medications see Table S2 in supporting information.
2.3. Analysis of the amount of AD pathology
The middle frontal cortex was selected to quantify AD protein load using western blotting on protein extracts from frozen tissue and immunohistochemistry on formalin‐fixed, paraffin‐embedded tissue sections, as this is an initial site for Aβ deposition 17 and is also prone to tau pathology in the aged 18 with all AD subtypes accumulating tau in this region. 19 Due to the more limited availability in the cohort with frozen tissue, only a subset of cases could be assessed. This provided 44 cases with available frozen cortical tissue for this analysis (14 NSN). This group was more than doubled to 34 for the immunohistochemistry analysis to give 58 of the original 149 cases for the areal fraction validation analysis. This was to ensure adequate representation. For a further breakdown of the cohorts see Table S3 in supporting information.
For western blotting, insoluble pathological proteins were extracted from 200 mg of frozen brain tissue. Tissue was homogenized using an IKA T‐10 hand‐held homogenizer in 2 mL (10 µl/mg) of cold reassembly (RAB) homogenization buffer (0.75 M NaCl, 100 mM MES, 1 mM EGTA, 0.5 mM MgSO4, 1 mM DTT, pH 6.8 with a Roche complete EDTA‐free protease inhibitor cocktail tablet added prior to use). Samples were centrifuged at 100,000 × g for 60 minutes at 4°C using a Beckman Optima L‐90K ultracentrifuge with a 70.1 Ti rotor, and the pellet repeatedly resuspended in 2 mL of cold paired helical filament (PHF) extraction buffer (10 mM Tris, 10% sucrose, 0.85 M NaCl, 1 mM EGTA, pH 7.4) then centrifuged at 15,000 × g for 2 × 20 minutes at 4°C. The supernatants were combined and 10% sarkosyl added prior to incubation on a shaking platform for 60 minutes at room temperature. The sarkosyl resuspensions were centrifuged at 100,000 × g for 30 minutes at 4°C and the remaining pellet resuspended in Tris‐HCl (0.2 µl/mg; starting volume) and collected as the sarkosyl insoluble fraction which was used to investigate the most toxic forms of the pathological proteins. Protein concentrations were determined by bicinchoninic protein assay (BCA) as per the manufacturer's instructions (Pierce Biotechnology, Rockford, IL). Six grams of protein lysate from the sarkosyl insoluble fraction was combined with 1x LDS sample buffer and 2.5% β‐mercaptoethanol, separated at a constant 225 V for ≈40 minutes using 10% to 20% Novex tris‐glycine gels (Life Technologies, Carlsbad, CA) and transferred at a constant 10 V for 60 minutes to nitrocellulose membranes (Biorad, Hercules, CA) using the Mini Gel Tank and Mini Blot Module (Life Technologies, Carlsbad, CA).
For insoluble Aβ protein amount, membranes were blocked for 1 hour in 5% skim milk powder in Tris‐buffered saline with 0.1% (v/v) Tween 20 (TBST) after antigen retrieval (microwave incubation in boiling citrate buffer for 2 minutes on each side of the membrane). Membranes were probed for monoclonal mouse anti‐human Aβ (Dako, Cat M0872 [Clone 6F/3D], diluted 1:500 in 1% skim milk TBST) overnight at 4°C on a rocker, washed in TBST (3 × 10 minutes) the next morning, and then probed for goat anti‐mouse horseradish peroxidase secondary antibody (Thermo Scientific, Cat 31430, diluted 1:5000 in 1% skim milk TBST) for 1 to 2 hours at room temperature on rocker. Clarity Max western enhanced chemiluminescence (ECL) substrate (Biorad, Hercules, CA) was used to detect protein bands that were visualized using a Chemidoc MP digital imaging system (Biorad). Each membrane was visualized using the same imaging protocol and exposure times or all membranes were imaged together.
For insoluble phospho‐tau protein amount, membranes were then incubated (2 × 10 minutes) in mild stripping buffer (0.2 M glycine, 0.1% SDS, 1% Tween‐20, pH 2.2) to remove residual primary and secondary antibody complexes. Membrane stripping efficacy was tested by further incubation in ECL substrate and Chemidoc visualization. Membranes were then re‐probed for Alz50 (kind gift from Prof. Peter Davies, diluted 1:250) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (Sigma, Cat G9545, diluted 1:10,000) as a loading control. Quantification of relative protein band intensity were analyzed using the gels plugin on Fiji (Image J, National Institutes of Health, Bethesda, MD) and expressed as arbitrary intensity units normalized to GAPDH as a protein loading control and also standardized to an internal control to compare between membranes. All western blots were replicated, and arbitrary intensity units were averaged, except for rare data points that exceeded two standard deviations from the mean. These were considered extreme outliers and were excluded from the analysis.
For an orthogonal analysis, the amount of the cortex containing pathology was evaluated using immunostaining for Aβ (mouse monoclonal anti‐human, Dako, Cat M0872 [Clone 6F/3D], 1:500) with hematoxylin counterstaining on a Ventana Discovery XT autostainer as per the manufacturer's instructions. Slides were viewed on a Zeiss Axio Lab.A1 microscope and imaged using a Zeiss AxioCam ICc 5 camera and AxioVision software v4.9.1. Four representative images of the densest area of Aβ immunopositivity were acquired at 5x magnification making sure that the cortex filled the entire area of the image taken. Images were then analyzed using Fiji (Image J, National Institutes of Health, Bethesda, MD) for pixel area of Aβ immunopositivity in each image using color threshold and particle analysis operations to obtain area percentage of cortex with pathology. Areas were averaged across the four representative images taken from each case to obtain an average Aβ load for the middle superior frontal cortex.
2.4. Analysis of the regional spread of AD pathology using AD pathological stages
To investigate the spread of AD pathology, the A and B scores for each case as set out in the NIA‐AA guidelines 1 were used. This involves the assessment of the presence of any Aβ plaques (used method detailed above) or NFTs (used phosphorylated tau clone AT8; mouse; 1:1000; Cat. No. MN1012; Thermo Scientific, Pittsburgh, USA, with hematoxylin counterstaining on a Ventana Discovery XT autostainer as per the manufacturer's instructions) in tissue sections prepared from hierarchically affected brain regions (cortical association regions in all lobes, hippocampus and entorhinal cortex, basal ganglia and nucleus basalis of Meynert at the level of the anterior commissure, midbrain with substantia nigra, the cerebellar cortex, and dentate nucleus). 1 Briefly, an A0 score corresponds to no Aβ plaques in any region examined; an A1 score corresponds to Aβ plaques in any association cortices; an A2 score corresponds to cortical, hippocampal, and basal ganglia Aβ plaques; and an A3 score corresponds to cortical, basal ganglia, cerebellar, and brainstem Aβ plaques (see Figure 3B for schematic). A B0 score corresponds to no NFTs in any brain region examined; a B1 score corresponds to medial temporal lobe NFT involvement; a B2 score corresponds to additional neocortical temporal lobe NFT involvement; and a B3 score corresponds to additional frontal, parietal, and occipital neocortical involvement (see Figure 4B for schematic).
FIGURE 3.
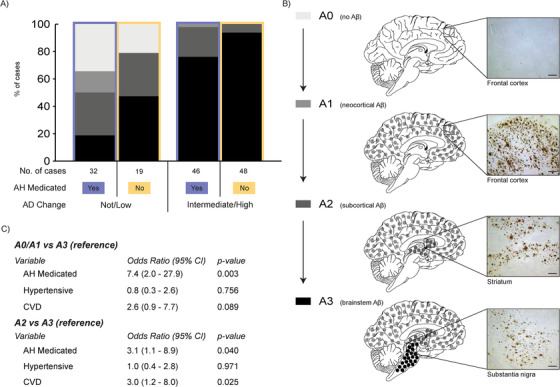
A, Stacked bar chart representing the percentage distribution of A scores of cases with a not/low or intermediate (int)/high level of Alzheimer's disease (AD) neuropathologic change that is further dichotomized into antihypertensive medication (AH Medicated) use (yes = orchid color, no = cantaloupe color). Case types and number of cases illustrated in key underneath the stacked bar chart. B, Schematic illustrating the progression of amyloid beta (Aβ) through the four A score stages (A0 to A3). The left panel of (B) shows the key used for the percentage distributions depicted in (A) with descriptors of Aβ brain regional involvement. The middle panel of (B) presents a schematic of the brain at each A score with colored round circles representing the involvement of Aβ seen at each A score level. The right panel of (B) show representative immunomicrographs of Aβ normally seen at each A score level with the brain region noted under each. Scale bars = 200 µm. C, Pertinent results of the multinomial logistic regression carried out to assess the effect of variables of interest on likelihood of A score level membership using the most severe A score level (A3) as the reference group. For full details of these results please see Table S4
FIGURE 4.

A, Stacked bar chart representing the percentage distribution of B scores of cases with a not/low or intermediate (int)/high level of Alzheimer's disease (AD) neuropathologic change that is further dichotomized into antihypertensive medication (AH Medicated) use (yes = orchid color, no = cantaloupe color). Case types and number of cases illustrated in key underneath the stacked bar chart. B, Schematic illustrating the progression of neurofibrillary tangles (NFT) through the four B score stages (B0 to B3). The left panel of (B) shows the key used for the percentage distributions depicted in (A) with descriptors of NFT brain regional involvement. The middle panel of (B) presents a schematic of the brain at each B score with colored NFT icons representing the involvement of NFTs seen at each B score level. The right panel of (B) shows representative micrographs of NFTs normally seen at each B score level with the brain region noted under each. The main micrograph is of modified Bielschowsky silver stain while the insert in the top right hand corner is of the same region immunostained for tau (AT8). Scale bars = 200 μm. C, Pertinent results of the multinomial logistic regression carried out to assess the effect of variables of interest on likelihood of B score level membership using the most severe B score level (B3) as the reference group. For full details of these results please see Table S4
2.5. Statistical analyses
Statistical analyses were carried out using SPSS software (IBM Corp. Released 2016. IBM SPSS Statistics for Macintosh, Version 24.0. Armonk, NY: IBM Corp.). T‐tests and chi‐square analyses were used to investigate differences in demographic data. Univariate analysis of variance was conducted to examine main group effects (disease group, medication group, or interaction of the two) on AD pathology (Aβ and tau) covarying for age, sex, and post mortem delay. Multinomial logistic regression was carried out to determine whether medication group predicted the spread of the AD neuropathologies through the brain assessed via A and B scores covarying for age, sex, post mortem delay, the presence of cerebrovascular disease and hypertensive status. Probability (P) values < .05 were judged as statistically significant.
3. RESULTS
3.1. Demographics, clinical, and pathological characteristics
The overall average age at death was 88 years, which is typical for the Australian population (see https://www.aihw.gov.au/reports/life-expectancy-death/deaths-in-australia/contents/age-at-death). When dichotomizing with respect to dementia (CDR 0 and 0.5 vs 1, 2, and 3), the groups were well matched for post mortem delay, sex, and presence of cerebrovascular disease, although those with a CDR of 0 or 0.5, now referred to as non‐demented group, lived longer by 2 years on average (Table 1). As expected, there was a significant association between CDR and AD level of neuropathologic change.
Assessment of hypertension parameters between these two clinicopathological groups found that they were well matched for hypertension status, antihypertensive medication usage and cerebrovascular disease (see Figure S2), but those with limited cognitive impairment had an average 5‐year increase in their duration of hypertension (note this group did live longer, see above and Table 1). The most commonly used antihypertensive medication class was diuretics (63%) followed by beta blockers (43%), ACE inhibitors (41%), CCBs (34%), and ARBs (14%). The use of multiple classes of antihypertensive medications was common, with 73% of antihypertensive‐medicated individuals taking multiple classes either concurrently or sequentially. There was a significant association between CDR and medication usage in normotensive subjects, with a larger proportion of the non‐demented group taking antihypertensive medications. Chi‐square analysis revealed that this association held for either of the major classes of antihypertensive medications used except for ARBs.
3.2. Regional amounts of Aβ and tau do not differ with antihypertensive medication usage
As expected, there was a significant disease effect in the average amount of insoluble Aβ in the middle frontal cortex with approximately four‐ to six‐fold more Aβ on average in the AD group compared to pathological controls (Figure 1A). No significant medication effect or medication by disease interaction was identified in relation to the amount of Aβ in frontal cortex as measured using either method (Figure 1B and C). No effect on histological frontal Aβ deposition was confirmed with either of the major classes of antihypertensive medications (sample size too small for additional western statistical analyses). Similarly, as expected, there was a significant disease effect in the average amount of insoluble phospho‐tau (Alz50) in the middle frontal cortex with on average a nine‐fold increase in the AD group compared to pathological controls (Figure 2A). Again, no medication or medication by disease interaction effect was observed in the whole cohort as measured using either method (Figure 2B and C). These data indicate that antihypertensive medications do not impact significantly the amount of AD pathological proteins in affected cortical regions.
FIGURE 1.
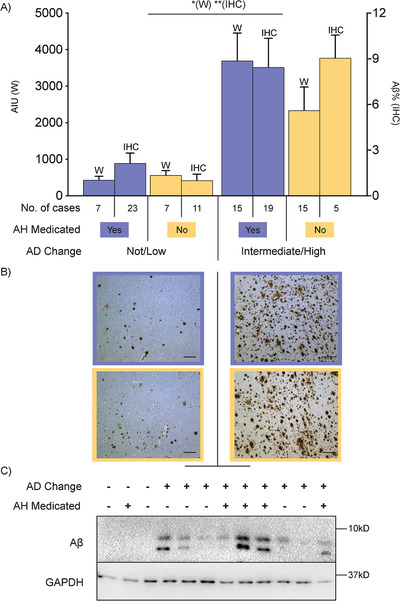
A, Bar graph showing amounts of amyloid beta (Aβ) in the frontal cortex of cases with a not/low or intermediate (int)/high level of Alzheimer's disease (AD) neuropathologic change that is further dichotomized into antihypertensive medication (AH Medicated) use (yes = “+” and orchid color, no = “−” and cantaloupe color) as measured in arbitrary intensity units (AIU, left axis) by western immunoblotting (w) or average percentage area of Aβ positivity by immunohistochemistry (IHC, right axis). Case types and number of cases illustrated in key underneath bar graph. Error bars = standard error of the mean. *P < .01 Western and **P < .001 IHC for difference between not/low AD and int/high AD. B, Representative micrographs of Aβ immunohistochemistry in the frontal cortex. Those on the left‐hand side are cases that have a not/low level of AD change while cases on the right‐hand side are those that have an int/high level of AD change. Micrographs with an orchid‐colored border denote cases that had taken antihypertensive medications while micrographs with a cantaloupe‐colored border denote cases that had not taken antihypertensive medications. Scale bars = 200 µm. C, Representative western immunoblots of the data presented in (A). Lanes denoted as AD Change “‐” indicate not/low level of AD change while those denoted as AD Change “+” indicate int/high level of AD change. Lanes denoted as AH Medicated “−” indicate cases that had not taken antihypertensive medications while those denoted as AH Medicated “+” indicate cases that had taken antihypertensive medications
FIGURE 2.
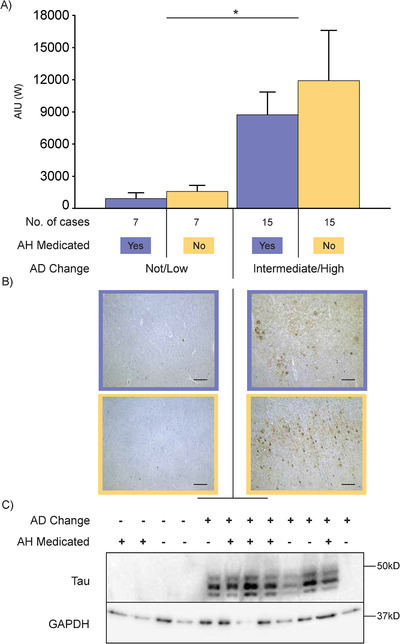
A, Bar graph showing amounts of tau (Alz50) in the frontal cortex of cases with a not/low or intermediate (int)/high level of Alzheimer's disease (AD) neuropathologic change that is further dichotomized into antihypertensive medication (AH Medicated) use (yes = “+” and orchid color, no = “−” and cantaloupe color) as measured in arbitrary intensity units (AIU) by western immunoblotting. Case types and number of cases illustrated in key underneath the bar graph. Error bars = standard error of the mean. *P < .05 Western for not/low AD versus int/high AD. B, Representative immunomicrographs of tau (AT8) in the frontal cortex. Those on the left‐hand side are cases that have a not/low level of AD change while cases on the right‐hand side are those that have an int/high level of AD change. Micrographs with an orchid‐colored border denote cases that had taken antihypertensive medications while micrographs with a cantaloupe‐colored border denote cases that had not taken antihypertensive medications. Scale bars = 200 µm. C, Representative immunoblots of the western data presented in (A). Lanes denoted as AD Change “−” indicate not/low level of AD change while those denoted as AD Change “+” indicate int/high level of AD change. Lanes denoted as AH Medicated “‐” indicate cases that had not taken antihypertensive medications while those denoted as AH Medicated “+” indicate cases that had taken antihypertensive medications
3.3. There is an association between less progression of AD pathology through the brain and antihypertensive medication usage
Multinomial logistic regression was used to explore the relationship between the progression of AD pathology throughout the brain and medication use, specifically whether being medicated for hypertension predicted a higher or lower A score (progression of Aβ throughout the brain, Figure 3B) or a higher or lower B score (progression of NFTs throughout the brain, Figure 4B). Age, post mortem delay, sex, the presence of cerebrovascular disease and hypertension were covaried in the model. For the progression of Aβ deposits throughout the brain, a main medication effect was detected (Figure 3C), indicating that the odds of someone who is medicated being in either the less severe (A0 or A1 groups) compared to the most severe (A3, see Figure 3A for proportion of cases involved) group was 7.4 times greater than someone who was not medicated for hypertension (Figure 3C and Table S4 in supporting information for statistics). There was no medication effect comparing A2 to A3. This effect was confirmed when analyzing the major classes of antihypertensive medications individually (ie, diuretics, beta blockers, and ACE inhibitors). For the progression of NFT throughout the brain, similar to the progression of Aβ deposits, there was a significant medication effect on NFT regional progression (Figure 4C). Those taking medication for hypertension were 4.3 and 3.8 times more likely to have limited NFTs (B0 or B2 respectively) compared to the most severe NFT stage (B3, see Figure 4A for proportion of cases involved) than someone who was not medicated (Figure 4C and Table S4 for statistics). There was no medication effect comparing B1 to B3. When analyzing the major classes individually, this effect was confirmed for all B score comparisons for diuretics.
4. DISCUSSION
While it is hoped that the use of antihypertensive medications will adequately control hypertension, this is not guaranteed, as observed in this study (Table 1). The recent SPRINT (Systolic Blood Pressure Intervention Trial) results confirmed the positive impact on cognitive decline and dementia of aggressively controlling hypertension, 20 a finding independent of the type of antihypertensive medications used. 6 How such an effect impacts brain tissue is poorly understood and was the focus of our clinicopathological study of AD pathological proteins. In dementia and non‐dementia cohorts matched for cerebrovascular disease, we found that antihypertensive medication use did not associate with the amount of either insoluble Aβ or phospho‐tau in a predilection site for such protein depositions when measured using two different methods. These robust results are consistent with those of others (no difference in frontal Aβ plaque load 9 or overall amyloid load 8 ), but differs from the studies of the scaled density measures of neuritic pathologies where treated AD 12 , 13 or MCI 8 cases had reduced pathologies compared to untreated hypertensive, normotensive AD cases, or non RAS medicated MCI cases, respectively. Of course, these scaled density ratings are performed using traditional histopathological silver stains rather than the measurement of the amount of insoluble protein in a sample, as performed in the present study. Additionally, in these studies 8 , 13 only RAS medications had this effect, and in one study 13 those taking this antihypertensive class had a higher incidence of stroke with histopathologic large vessel infarction and hemorrhage. In our cohort, the presence of large vessel cerebrovascular disease was low and well matched in the clinicopathological groups assessed, negating the potential to weight any group for large vessel disease over AD. Overall the data suggest that, independent of cerebrovascular disease presence, antihypertensive medications do not reduce the amounts of AD pathologic proteins in a predilection site for Aβ pathology.
The demographics of our cohort with AD pathology and matched cerebrovascular disease assessed for cognition (Table 1) show a slight 2‐year difference in the age at death and the expected high proportion of those with dementia also with intermediate to high levels of AD neuropathologic change, although this was not universal. Approximately 20% of those with dementia had insufficient pathology for a diagnosis of AD, but had cerebrovascular disease and vascular brain injury instead. People with cerebrovascular pathologies are two times more likely to have dementia than those without cerebrovascular pathologies. 2 When we excluded lobar infarctions in the present study of the elderly, there was no difference in the relative prevalence of cerebrovascular disease between those with and without dementia. Additionally, 43% of those without dementia also fulfilled pathologic criteria for AD (Table 1). Such cases have been previously shown to have a suppressed neuroinflammatory response in the brain. 21 , 22 Apart from dementia, the main clinical differences between the groups related to the duration of hypertension and the adequate control of hypertension by antihypertensive medications (Table 1). Those with dementia were less likely to be normotensive and medicated (16% compared to 52% without dementia) and had a shorter duration of hypertension (11 vs 16 years). This implies a potential medication effect at the tissue level on the progressive toxicity of the AD pathologies.
It has become more obvious that after the initial regional deposition of AD pathologic proteins, the pathology spreads throughout the brain. The impact of antihypertensive medication use on the spread of AD pathologies has only been assessed in two studies 8 , 13 with only one of these studies 13 suggesting that only ARBs associated with the spread of NFT, but again in a cohort with a high incidence of stroke. In the present study assessing the progression of Aβ and tau through the brain controlling for cerebrovascular disease, we show that antihypertensive medication use associated with less progression of pathology compared to those not medicated. This suggests that the effect of antihypertensive medications is more on progression from initiating predilection sites. Recent observational studies report similar findings, that antihypertensive medications slow the conversion of MCI to AD 8 , 23 and the rapidity of cognitive decline 24 but perhaps are less effective once AD has taken hold and pathology has plateaued. 25 The mechanism of pathological progression of AD is under active research, but if antihypertensive medications impact this mechanism, it may involve vascular tissues. There is some evidence for this with many recent studies showing relationships between increasing white matter hyperintensities and amyloid using positron emission tomography (PET) imaging. 26 , 27 Also, white matter hyperintensities are associated independently with hypertension and lower cerebrospinal fluid Aβ (indicative of higher brain Aβ loads). 28 A previous community autopsy sample showed more cerebrovascular damage in non‐medicated older adults 29 supporting the concept that adequate hypertension treatment may reduce dementia by minimizing microvascular injury to the cerebrum. Our results suggest that this may impact the progression of AD pathological proteins infiltrating the brain.
There are several limitations to our study. While the heterogeneity in the cohort is reflective of the aged population, the diversity and numbers of antihypertensive medications used differs from those reported in other studies (mainly acting on RAS). In our study of elderly cases without lobar infarctions and other neurodegenerative disorders at death, the majority (63%, see Table S2) were taking the diuretic class of antihypertensive medication with 41% taking ACE inhibitors and a mere 14% taking ARBs. While the comparative effects of different antihypertensive classes cannot be singled out in our study, our data support the concept that multiple types of antihypertensive medications impact AD, 6 , 7 including diuretics. 30 In terms of other medications identified as having a potential effect on AD, statins have been associated with an increased rather than decreased AD neuropathology 31 while antidiabetics are associated with cerebrovascular but not AD neuropathology. 32 , 33 In our cohort, statins were taken by 14%, and only in those on antihypertensive medications (27%), while antidiabetics were taken by only 4% of the cohort, making it unlikely that either type of medication contributed significantly to the association of antihypertensive medication use with reduced progression of AD neuropathology. Apolipoprotein E (APOE) ε4 genotype has also been associated with increased rather than decreased AD neuropathology, 34 , 35 , 36 and this was also the case in our cohort (89% of those with ε4 genotype had AD neuropathology). The small cohort of not AD APOE ε4 cases that were not medicated (<3) does not allow for statistical modelling of the influence of this factor. As with many post mortem human studies in the aged, the exclusion of many confounding variables to give confidence in the interpretation of the data reduces the cohort that can be studied. On the other hand, diagnostic accuracy for pathological AD or not is ensured. Further validation in additional well‐documented pathologically confirmed cohorts is necessary.
Our results show antihypertensive medication use does not alter the amount of AD pathology seen in a brain region known to be involved early in AD. Instead the data show that antihypertensive medication use is associated with a reduction in the progression of AD pathologies throughout the brain. This is consistent with a recent amyloid PET imaging study that found that angiotensin receptor blockers and diuretic use predicted less accumulation of Aβ, 31 supporting a role in reducing the spread of Aβ in the brain. The precise mechanism by which this occurs remains to be elucidated; however, due to the heterogeneous nature of the medications taken, the associations detected here are likely due to the impact these medications have on blood pressure lowering and the cerebral vessels themselves, a topic of current debate. 37
FUNDING INFORMATION
This study was funded by the National Health and Medical Research Council of Australia (NHMRC dementia team grant 1095127). GMH is an NHMRC Senior Principal Research Fellow (1079679).
CONFLICTS OF INTEREST
AA is a recipient of UNSW Brain Sciences Collaborative PhD Grant. PS is supported by research grants from the National Health & Medical Research Council, the Australian Research Council, the National Institute of Aging, the NHMRC National Institute for Dementia Research, and several philanthropic foundations. He reports no conflicts of interest in relation to this work. JS is funded by a research grant from the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health (AA012725). GH consults for the National Health and Medical Research Council of Australia (NHMRC); received travel funds from AAIC, Int Soc Neurochemistry, Int DLB Conference, AAN, Int MSA Conference, NHMRC National Institute for Dementia Research, Japanese Neurology Soc; is on the editorial boards of Acta Neuropathol, J Neural Transm, J Parkinson Dis, Transl Neurodegen, Neuropathol Appl Neurobiol, receives royalties from Academic press, Elsevier & Oxford University Press; receives research grant funding from NHMRC, MJ Fox Foundation, Shake‐it‐up Australia, Parkinson's NSW, University of Sydney (infrastructure & equipment); and holds stock in Cochlear (2004 on) & NIB Holdings (2007 on).
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We wish to thank donors and their families who made this study possible. Tissues were received from the New South Wales Brain Tissue Resource Centre at the University of Sydney and the Sydney Brain Bank at Neuroscience Research Australia, which are supported by The University of New South Wales, Neuroscience Research Australia, and by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under Award Number R28AA012725. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We also wish to thank and acknowledge The Sydney Memory and Ageing Study (MAS) and the Sydney Centenarian Study (SCS). Both studies being funded by National Health & Medical Research Council (NHMRC) Program Grants; MAS‐ ID No. ID350833, ID568969, and APP1093083 and SCS‐ ID No. 568969, 1093083 and 630593. We thank the participants and their informants from both of these studies for their time and generosity in contributing to this research. We also acknowledge the MAS and SCS research teams: https://cheba.unsw.edu.au/research‐projects.
Affleck AJ, Sachdev PS, Stevens J, Halliday GM. Antihypertensive medications ameliorate Alzheimer's disease pathology by slowing its propagation. Alzheimer's Dement. 2020;6:e12060 10.1002/trc2.12060
REFERENCES
- 1. Hyman B, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Azarpazhooh MR, Avan A, Cipriano LE, Munoz DG, Sposato LA, Hachinski V. Concomitant vascular and neurodegenerative pathologies double the risk of dementia. Alzheimers Dement. 2018;14:148‐156. [DOI] [PubMed] [Google Scholar]
- 3. Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lennon MJ, Makkar SR, Crawford JD, Sachdev PS. Midlife hypertension and alzheimer's disease: a systematic review and meta‐analysis. J Alzheimers Dis. 2019;71(1):307‐316. [DOI] [PubMed] [Google Scholar]
- 6. Peters R, Yasar S, Anderson CS, et al. An investigation of antihypertensive class, dementia, and cognitive decline: a meta‐analysis. Neurology. 2020;94(3):e267‐e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rouch L, Cestac P, Hanon O, et al. Blood pressure and cognitive performances in middle‐aged adults: the aging, health and work longitudinal study. J Hypertens. 2019;37(6): 1244‐1253. [DOI] [PubMed] [Google Scholar]
- 8. Wharton W, Zhao L, Steenland K, et al. Neurofibrillary tangles and conversion to mild cognitive impairment with certain antihypertensives. J Alzheimers Dis. 2019;70:153‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ashby EL, Miners JS, Kehoe PG, Love S. Effects of hypertension and anti‐hypertensive treatment on amyloid‐β (Aβ) plaque load and Aβ‐synthesizing and Aβ‐degrading enzymes in frontal cortex. J Alzheimers Dis. 2016;50:1191‐1203. [DOI] [PubMed] [Google Scholar]
- 10. Petrovitch H, White LR, Izmirilian G, et al. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu‐Asia Aging Study. Neurobiol Aging. 2000;21:57‐62. [DOI] [PubMed] [Google Scholar]
- 11. Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC. 3rd. Increased incidence of neurofibrillary tangles (NFT) in non‐demented individuals with hypertension. J Neurol Sci. 1995;131:162‐169. [DOI] [PubMed] [Google Scholar]
- 12. Hoffman LB, Schmeidler J, Lesser GT, et al. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology. 2009;72:1720‐1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hajjar I, Brown L, Mack WJ, Chui H. Impact of angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol. 2012;69:1632‐1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry. 1997;63:749‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci. 2004;226:75‐80. [DOI] [PubMed] [Google Scholar]
- 16. Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of dementia in over‐80‐year‐old brain donors from the population‐based Cambridge city over‐75s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645‐658. [DOI] [PubMed] [Google Scholar]
- 17. Grothe MJ, Barthel H, Sepulcre J, et al. In vivo staging of regional amyloid deposition. Neurology. 2017;89:2031‐2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 19. Petersen C, Nolan AL, de Paula Franca Resende E, et al. Alzheimer's disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol. 2019;138:597‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. SMIftSR Group, JD Williamson, Pajewski NM, et al. Effect of intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA. 2019;321:553‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barroeta‐Espar I, Weinstock LD, Perez‐Nievas BG, et al. Distinct cytokine profiles in human brains resilient to Alzheimer's pathology. Neurobiol Dis. 2019;121:327‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez‐Nievas BG, Stein TD, Tai HC, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. 2013;136:2510‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wharton W, Goldstein FC, Zhao L, Steenland K, Levey AI, Hajjar I. Modulation of renin‐angiotensin system may slow conversion from mild cognitive impairment to Alzheimer's disease. J Am Geriatr Soc. 2015;63:1749‐1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mielke MM, Rosenberg PB, Tschanz J, et al. Vascular factors predict rate of progression in Alzheimer disease. Neurology. 2007;69:1850‐1858. [DOI] [PubMed] [Google Scholar]
- 25. Eldholm RS, Persson K, Barca ML, et al. Association between vascular comorbidity and progression of Alzheimer's disease: a two‐year observational study in Norwegian memory clinics. BMC Geriatr. 2018;18:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Graff‐Radford J, Arenaza‐Urquijo EM, Knopman DS, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142:2483‐9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kao Y‐H, Chou M‐C, Chen C‐H, Yang Y‐H. White matter changes in patients with Alzheimer's disease and associated factors. J Clin Med Res. 2019;8(2):167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al‐Janabi OM, Brown CA, Bahrani AA, et al. Distinct white matter changes associated with cerebrospinal fluid amyloid‐β1‐42 and hypertension. J Alzheimers Dis. 2018;66:1095‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang LY, Larson EB, Sonnen JA, et al. Blood pressure and brain injury in older adults: findings from a community‐based autopsy study. J Am Geriatr Soc. 2009;57:1975‐1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta‐analysis and meta‐regression of prospective studies. J Hypertens. 2016;34:1027‐1035. [DOI] [PubMed] [Google Scholar]
- 31. Glodzik L, Rusinek H, Kamer A, et al. Effects of vascular risk factors, statins, and antihypertensive drugs on PiB deposition in cognitively normal subjects. Alzheimers Dement. 2016;2:95‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. dos Santos Matioli MNP, Suemoto CK, Rodriguez RD, et al. Diabetes is not associated with Alzheimer's disease neuropathology. J Alzheimers Dis. 2017;60:1035‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abner EL, Nelson PT, Kryscio RJ, et al. Diabetes is associated with cerebrovascular but not Alzheimer's disease neuropathology. Alzheimers Dement. 2016;12:882‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Skillback T, Lautner R, Mattsson N, et al. Apolipoprotein E genotypes and longevity across dementia disorders. Alzheimers Dement. 2018;14:895‐901. [DOI] [PubMed] [Google Scholar]
- 35. Iacono D, Feltis GC. Impact of apolipoprotein E gene polymorphism during normal and pathological conditions of the brain across the lifespan. Aging. 2019;11:787‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall A, Pekkala T, Polvikoski T, et al. Prediction models for dementia and neuropathology in the oldest old: the Vantaa 85+ cohort study. Alzheimers Res Ther. 2019;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peters R, Warwick J, Anstey KJ, Anderson CS. Blood pressure and dementia: what the SPRINT‐MIND trial adds and what we still need to know. Neurology. 2019;92:1017‐1018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information