

Abstract
Interleukin 7 (IL-7) and its receptor, formed by IL-7Rα (encoded by IL7R) and γc, are essential for normal T-cell development and homeostasis. Here we show that IL7R is an oncogene mutated in T-cell acute lymphoblastic leukemia (T-ALL). We find that 9% of individuals with T-ALL have somatic gain-of-function IL7R exon 6 mutations. In most cases, these IL7R mutations introduce an unpaired cysteine in the extracellular juxtamembrane-transmembrane region and promote de novo formation of intermolecular disulfide bonds between mutant IL-7Rα subunits, thereby driving constitutive signaling via JAK1 and Independently of IL-7, γc or JAK3. IL7R mutations induce a gene expression profile partially resembling that provoked by IL-7 and are enriched in the T-ALL subgroup comprising TLX3 rearranged and HOXA deregulated cases. Notably, IL7R mutations promote cell transformation and tumor formation. Overall, our findings indicate that IL7R mutational activation is involved in human T-cell leukemogenesis, paving the way for therapeutic targeting of IL-7R–mediated signaling in T-ALL.
Signaling mediated by IL-7 and IL-7R is essential for normal T-cell development and homeostasis1,2. Mice with IL-7 or IL-7R deficiency show an early block in thymocyte development and reduced numbers of non-functional peripheral T cells3,4. In humans, IL7R-inactivating mutations result in severe combined immunodeficiency5,6, whereas IL7R polymorphisms have been shown to confer susceptibility to multiple sclerosis7,8. There is also evidence that IL-7 and IL-7R may contribute to T-cell leukemia progression. For example, IL-7 transgenic mice develop lymphomas9,10, and AKR/J mice, which develop spontaneous thymic lymphomas, have high IL-7R levels11. In addition, T-ALL cells respond to IL-7 in vitro in a majority of cases12–14. Notably, IL-7Rα is transcriptionally upregulated by NOTCH1 (ref. 15), one of the most commonly mutated genes in T-ALL16, and appears to be involved in Notch-mediated leukemia cell maintenance15. The possibility that IL-7R–mediated signaling may play a role in T-cell leukemia is further supported by the observation that 18% of adult and 2% of pediatric T-ALL cases have activating mutations in JAK1, which encodes a tyrosine kinase that directly binds IL-7Rα17, among other receptors. Here we provide direct evidence that IL-7R–mediated signaling plays an active role in the T-cell leukemogenic process in humans.
RESULTS
Somatic IL7R mutations in diagnostic pediatric T-ALL samples
Based on the evidence that IL-7– and IL-7R–mediated signaling contribute to T-cell leukemia survival and proliferation in vitro and in vivo and given the existence of JAK1 activating mutations in some T-ALL cases, we hypothesized that gain-of-function mutations in IL7R could be present in some T-ALL cases. Analysis of the complete coding sequence of IL7R in 68 pediatric diagnostic T-ALL samples treated in Centro Infantil Boldrini, Campinas, Brazil showed that five (7%) of the cases had heterozygous mutations in IL7R that exclusively affected exon 6. All cases with these mutations had in-frame insertions or deletions-insertions (Table 1. Fig. 1a and Supplementary Fig. 1) in the juxtamembrane-transmembrane domain at the interface with the extracellular region (Fig. 1a and Supplementary Fig. 2). The mutations were somatic, as they were detected at diagnosis but not in samples from the same individuals in remission (n = 5) (Fig. 1b and Supplementary Fig. 1). Subsequent analysis of IL7R exon 6 in the Dutch Childhood Oncology Group (DCOG) and the Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia (COALL) case series confirmed these results and showed the presence of heterozygous mutations in 12 out of 133 cases, with the majority of mutations targeting the same hotspot (Table 1 and Fig. 1a). In total, 17 of 201 (9%) T-ALL samples from three independent cohorts had IL7R exon 6 mutations (Fig. 1c). This frequency was independently confirmed by a parallel study describing IL7R mutations in 10.5% of T-ALL cases18.
Table 1.
Mutational and immunophenotypical characteristics of T-All cases with mutated IL7R
| Subject | Cohort | IL7R mutationa | IL-7Rα predicted amino acid alterationsa |
NOTCH1 and FBXW7 mutational status |
PTEN mutational status | Oncogenetic group | EGIL maturation stage | CD3, CD4 or CD8 stage |
|---|---|---|---|---|---|---|---|---|
| P1 | Boldrini | c.726_727insAACCCATGC | p.Leu242_Leu243insAsn ProCys | Wild typeb | Wild type | Unknown | Cortical | TP |
| P2 | Boldrini | c.731_732insTTGTCCCAC | p.Thr244_Ile245 insCysProThr | Unknown | Wild type | Unknown | Pre-T | DP |
| P3 | Boldrini | c.722_730delTCTTACTAA insGCGCAAACTGTGGGG | p.Ile241_Thr244delins SerAlaAsnCysGlyAla | HDb | Wild type | Unknown | Cortical | TP |
| P4 | Boldrini | c.728_729insGGTATCTT GTCC | p.Leu243_Thr244insVal SerCysPro | Wild typeb | Wild type | Unknown | Cortical | TP |
| P5 | Boldrini | c.731C>T; 741delTinsCC AATGG | p.Thr244I; Ile247_Leu248 insGlnTrp | Wild typeb | Exon 7 mutation | Unknown | Pre-T | ISP4 |
| P6 | DCOG | c.717_727delTCCTATCTTAC insCCAGTCCCCCTCCTGCT | p.Pro240_Leu242 delinsGlnSerProSerCys | HD | Wild type | Unknown | Pre-T | ISP4 |
| P7 | DCOG | c.721_722delATinsTG; 726_727insGAAGGC | p.Ile241Cys; Leu242_ Leu243insGluGly | HD/PEST | Wild type | TLX3 | n.d. | DN |
| P8 | DCOG | c.755_761del CTGTCGCinsGGAA | p.Ser252_254delinsTrpAsn | Wild type | Wild type | HOXA/MLL | Pre-T | DP |
| P9 | COALL | c.719_731delCTATCTTACTA ACinsGGTTTTGTCCCCA | p.Pro240_Thr244delins ArgPheCysProHis | HD | Wild type | TLX3 | Pre-T | ISP4 |
| P10 | COALL | c.719_736delCTATCTTACT AACCATCAinsTTAAGT | p.Pro240_Ser246delins LeuLysCys | Wild type | Wild type | TLX3 | Pre-T | DN |
| P11 | COALL | c.726_730delACTAAinsTCA CCCTTTTAACTGTGGAC | p.Leu242_Thr244delins PheHisProPheAsnCysGlyPro | HD | Wild type | TLX3 | Mature | ISP4 |
| P12 | COALL | c.730_731insTGTGCCCAA | p.Leu243_The244isn MetCysPro | JM | Wild type | HOXA | Mature | DP |
| P13 | COALL | c.757_758insGCCCATCCC | p.Val253delinsGlyPro SerLeu | PEST | Wild type | HOXA | Pre-T | DN |
| P14 | COALL | c.727_728insGACTTGA GTGCG | p.Leu243delinsArgLeu GluCysVal | PEST | Wild type | HOXA/inv-7 | Mature | DP |
| P15 | COALL | c.724_736delTTACTAACC ATCAinsCCCCAGGGCGGGT | p.Leu242_Ser246delins ProGlnGlyGlyCys | HD/FBXW7 | Wild type | HOXA/SET-NUP214 | Mature | DP |
| P16 | COALL | c.719_736delCTATCTTACT AACCATCAinsTCCAATCAT | p.Pro240_Ser246delins LeuGlnSerCys | Wild type | Wild type | TAL1/LMO2-like | Cortical | DP |
| P17 | COALL | c.726_729delACTA insTCCCCATCAGCATTGT | p.Leu242_Leu243delins PheProHisGlnHisCys | FBXW7 | Wild type | Unknown | Mature | ISP4 |
All mutations were heterozygous.
FBXW7 mutational status not analyzed. n.d., not determined or inconclusive; HD, Notch heterodimerization domain; JM, Notch juxtamembrane region; PEST, Notch Pro-Glu-Ser-Thr–rich domain; pre-T, pre–T-ALL; TP, triple positive; DP, double positive; ISP4, immature CD4 single positive; DN, double negative.
Figure 1.

IL7R exon 6 somatic mutations in pediatric T-ALL. (a) Scheme of IL-7Rα protein (top) and predicted amino acid alterations (bottom). Indicated are the two extracellular fibronectin type III–like domains containing four paired cysteines and a WS×WS motif, the transmembrane domain and the cytoplasmic tail with the Box 1 motif and the tyrosine residues involved in signal transduction. The region where the mutations occur is denoted by an empty box. Amino acid changes involving de novo introduction of a cysteine are indicated in orange; filled boxes denote deletions-insertions and are aligned with the respective deleted amino acid sequence; arrows point to where simple insertions occur. (b) Representative homoduplex and heteroduplex analysis of PCR products (left) and sequencing chromatograms (right) of paired diagnosis and remission samples indicating the somatic, tumor-associated origin of exon 6 mutations. (c) Frequency of T-ALL mutations in the three different case cohorts analyzed.
Biological and clinical features associated with IL7R mutations
To identify possible transcriptional patterns associated with IL7R mutations in T-ALL, we analyzed microarray data from 8 IL7R mutated and 109 non-mutated diagnostic case samples. We tested differential gene expression by regression analysis using the LIMMA package (see URLs). IL7R mutations were associated with upregulation of 39 and downregulation of 41 probe sets (false discovery rate P < 0.05) (Fig. 2a and Supplementary Table 1). Notably, gene set enrichment analysis (GSEA) of these T-ALL samples showed significant enrichment of a set of genes activated upon IL-7 stimulation in normal lymphocytes (enrichment score = 0.67, P = 0.045)19. These genes include SOCS1, SOCS2, PIM1, BCL2, DPP4 (CD26) and CCND2 (Fig. 2b), all of which have been reported as transcriptional targets of the JAK-STAT pathway.
Figure 2.
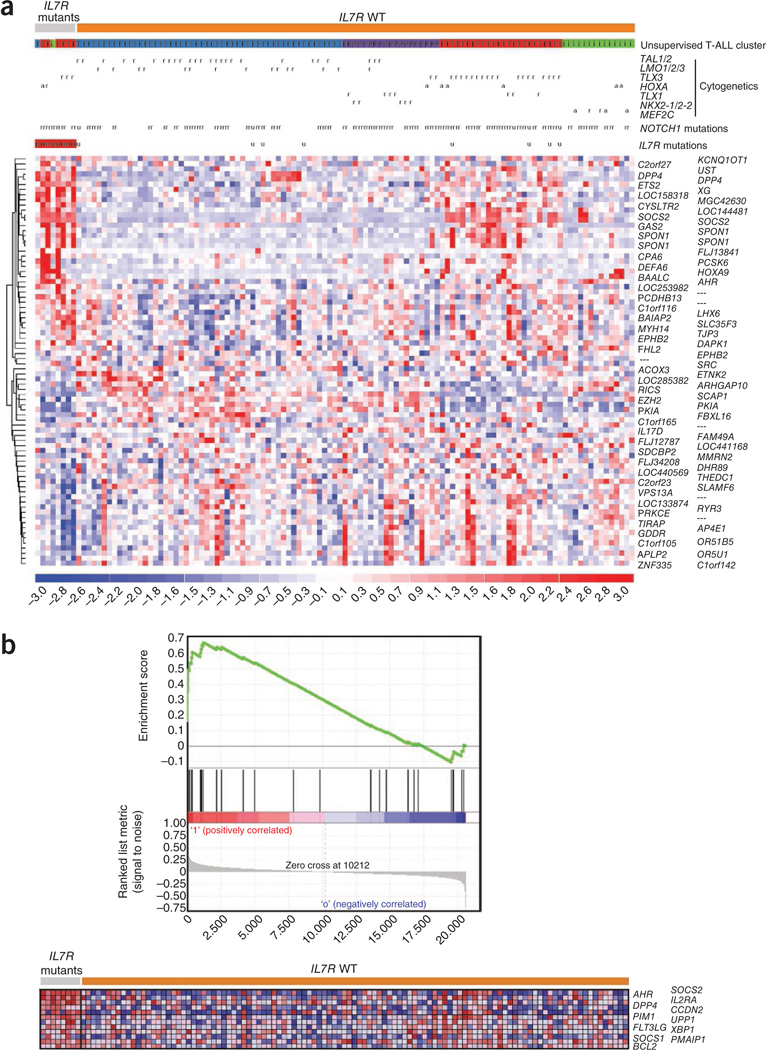
Molecular signatures associated with IL7R mutations in T-ALL. (a) Heat-map diagram of the 80 top ranking differentially expressed genes (supplementary table 1) in IL7R mutant (n = 8) compared to wild-type (n = 109) T-ALLs as determined by empirical Bayes linear models (LIMMA package; cutoff false discovery rate P = 0.05). Genes are shown in rows; each individual sample is shown in one column The scale bar shows color-coded differential expression from the mean in s.d. units, with red indicating higher expression and blue indicating lower expression. Unsupervised gene expression T-ALL clusters were defined as previously described20 and are indicated as: T (blue), TAL/LMO; T (red), TLX; i (green), immature; and P (violet), proliferative. Cytogenetic defects are denoted as: r, rearranged or mutated; a, aberrant expression; and u, unavailable data. (b) Gene set enrichment analysis (GSEA) plot (top) showing that genes overexpressed in human normal lymphocytes following IL-7 exposure19 were significantly enriched in IL7R mutant T-ALL cases (enrichment score = 0.67, P = 0.045). Heat-map diagram (bottom) of the 12 top-ranking genes in the leading edge.
Individuals with T-ALL are categorized into several oncogenetic subgroups that are characterized by rearrangements and aberrant expression of transcription factors such as TAL1 and LMO1 or LMO2, TLX1 (HOX11), TLX3 (HOX11L2), HOXA, NKX2–1 or MEF2C (ref. 20). We found IL7R mutations predominantly in cases belonging to the HOXA subgroup (Table 2). Recently, we identified unsupervised T-ALL gene expression clusters that closely recapitulate oncogenetic T-ALL subgroups, namely the TAL/LMO subgroup (enriched for TAL1 or TAL2 and/or LMO1, LMO2 or LMO3 rearranged cases), the proliferative subgroup (enriched for TLX1, or NKX2–1 or NKX2–2 rearranged cases), the TLX subgroup (enriched for TLX3 rearranged and HOXA deregulated cases) and the immature/ETP-ALL cases (enriched for MEF2C deregulated cases)20. Our current analyses showed that IL7R mutations were especially associated with the TLX subgroup (Fig. 2a and Table 2), which is in agreement with the fact that this unsupervised gene expression T-ALL subset is enriched in HOXA deregulated cases.
Table 2.
Association of IL7R mutations with genetic features of T-All cases
|
IL7R |
||||
|---|---|---|---|---|
| n | Mutant (%) | Wild type (%) | P | |
| Gene expression clustersa | 8 (7) | 101 (93) | ||
| TAL-LMO | 49 | 1 (2) | 48 (98) | 0.284 |
| Proliferative | 19 | 0 (0) | 19 (100) | 1.0 |
| TLX | 26 | 6 (23) | 20 (77) | 0.008 |
| Immature | 15 | 1 (7) | 14 (93) | 1.0 |
| Genetics (oncogenetic subgroups) | 12 (9) | 123 (91) | ||
| TAL1 or TAL2 b | 28 | 0 (0) | 28 (100) | 0.568 |
| LMO1, LMO2 or LMO3 b | 19 | 0 (0) | 19 (100) | 1.0 |
| TLX3 | 25 | 4 (16) | 21 (84) | 1.0 |
| TLX1 | 8 | 0 (0) | 8 (100) | 1.0 |
| HOXA | 13 | 5 (38) | 8 (62) | 0.016 |
| NKX2–1 or NKX2–2 | 6 | 0 (0) | 6 (100) | 1.0 |
| MEF2C | 6 | 0 (0) | 6 (100) | 1.0 |
| Unknown | 32 | 3 (9) | 29 (91) | 1.0 |
| NOTCH1 or FBXW7 | 12 (9) | 122 (91) | ||
| Mutant | 86 | 9 (10) | 77 (90) | 1.0 |
| Wild type | 48 | 3 (6) | 45 (94) | |
Unsupervised gene expression cluster analysis (109 T-ALL cases had known IL7R mutational status). The subgroups used are as previously defined20.
Two T-ALL cases have both TAL1 and TAL2 and LMO1 and LMO2 aberrations.
As some oncogenic rearrangements in T-ALL are associated with specific immunophenotypic development stages21,22, we evaluated whether IL7R mutations predominated in particular immunophenotypes. IL7R gene alterations did not associate with any specific T-ALL maturation stage based on European Group for the Classification of Acute Leukemia (EGIL) criteria23. Although IL7R mutations were negatively and positively associated with CD2 and CD10 expression, respectively (Supplementary Fig. 3), they did not associate with CD34, CD33, CD5, CD1, CD4, CD8, cytoplasmic CD3, surface CD3, TCRαβ or TCRγδ expression.
JAK1 and JAK3 are essential for physiologic IL-7–mediated signaling1. None of the IL7R mutants analyzed (n = 5) had gene alterations in the JH2 pseudokinase domain of JAK1 or JAK3, reported to be mutated in pediatric T-ALL17, and in breast cancer24 and acute megakaryoblastic leukemia25, respectively. The PI3K-Akt signaling pathway is activated by IL-7 in T-ALL cells26, and PTEN, the major negative regulator of the PI3K-Akt signaling pathway, is mutated in up to 20% of T-ALL cases27–31. Only 1 of the 17 mutated IL7R samples showed PTEN gene alterations (Table 1). NOTCH1 is a major oncogene in T-ALL, with more than 60% of T-ALL cases having gene alterations in NOTCH1 or FBXW7, which encodes the E3 ubiquitin ligase that targets NOTCH1 for degradation16,32–34. We observed no significant difference in the distribution of IL7R mutations in NOTCH1- and/or FBXW7-mutated versus non-mutated cases (Table 2).
We also evaluated whether IL7R mutations could predict treatment response and clinical outcome. We did not find any association of these mutations to initial in vivo prednisone response. Moreover, there was no difference in survival between cases with wild-type and mutant IL7R. Disease-free (P = 0.82, log-rank test), event-free (P = 0.84) and overall survival (P = 0.51; Supplementary Fig. 4) were similar for both groups.
IL7R mutations induce constitutive signaling
The high-affinity IL-7R complex is formed by IL-7Rα and γc. Triggering of IL-7R by IL-7 involves recruitment of both subunits and consequent activation of the tyrosine kinases JAK1 (associated with IL-7Rα) and JAK3 (associated with γc), leading to the downstream activation of different pathways, most prominently PI3K-Akt and STAT5 (refs. 1,2). We hypothesized that T-ALL– associated IL7R mutations should promote either constitutive signaling or increased responsiveness to IL-7. We first compared two primary leukemia samples collected at diagnosis that differed in their IL7R mutational status. In contrast to the T-ALL case with wild-type IL7R, the case sample harboring an IL7R mutation (P1, p.Leu242-Leu243insAsnProCys; Table 1) showed constitutive JAK1 and STAT5 phosphorylation (Fig. 3a). To exclude the possibility that this difference resulted from lesions other than IL7R mutation, we transduced the IL-7–dependent thymocyte cell line D1 (ref. 35) with retroviral vectors driving the expression of the human wild-type IL-7Rα chain or two of the mutants (P1; and P2, p.Thr244-Ile245insCysProThr). Analysis of the JAK-STAT and PI3K-Akt pathways showed that the IL7R mutations are gain of function, inducing ligand-independent constitutive hyperactivation of IL-7R–mediated signal transduction. IL7R mutations induced phosphorylation of JAK1 and STAT5 (Fig. 3b), STAT1 and STAT3 (Supplementary Fig. 5), as well as Akt and its direct target Bad (Fig. 3c). Notably, the mutant proteins did not promote JAK3 phosphorylation, which is a hallmark of physiological IL-7– mediated signaling (Supplementary Fig. 5). We obtained similar results with Ba/F3 cells (Supplementary Fig. 6). Strikingly, reconstitution of the IL-7R machinery in 293T cells (which express endogenously only JAK1 and lack IL-7Rα, γc and JAK3) further showed that the IL-7Rα mutant proteins signal constitutively in a manner that is independent of γc (Fig. 3d,e) and JAK3 (Fig. 3e). In contrast, knockdown of JAK1 resulted in abrogation of mutant IL7R-dependent constitutive STAT5 phosphorylation (Fig. 3f and Supplementary Fig. 7). Because, similar to JAK3, JAK2 and TYK2 are not activated by the IL7R mutants (Supplementary Fig. 5), our results indicate that JAK1 is the only Janus kinase that is mandatory for signaling triggered by mutant IL-7Rα.
Figure 3.

IL7R mutations induce constitutive signaling in a manner that is independent of IL-7, γc and JAK3 and relies on disulfide bond promotion of homodimer formation. (a) We analyzed primary T-ALL cells collected at diagnosis from cases with mutant (P1) and wild-type IL7R by immunoblot for JAK1 and STAT5 phosphorylation. (b,c) We cultured D1 cells expressing human wild-type or mutant (P1 and P2) IL-7Rα without IL-7 for 4 h, stimulated them or not with IL-7 for 20 min and evaluated them for activation of JAK-STAT (b) and PI3K-Akt (c) pathway activation by immunoblot. (d) We analyzed 293T cells reconstituted with JAK3, STAT5 and wild-type or mutant IL-7Rα, and expressing or not expressing γc, for constitutive and IL-7–induced (15 min stimulation) STAT5 phosphorylation. (e) We transfected 293T cells with IL-7Rα P2 and the remaining components of the IL-7R signaling machinery as indicated and evaluated them for STAT5 phosphorylation. (f) We transfected 293T cells with IL-7Rα P1 or P2 and small interfering RNA (siRNA) against JAK1 (+) or control non-targeting siRNA (−) and evaluated them after 36 h for JAK1 expression and STAT5 phosphorylation. (g) Lysates from D1 cells expressing wild-type or mutant IL-7Rα were treated or untreated with the reducing agent DTT and analyzed for IL-7Rα expression by immunoblot. The monomeric and dimeric forms of the receptor are denoted by black and white arrowheads, respectively. (h) We pretreated 293T cells expressing IL-7Rα P1 and P2 and the remaining components of the IL-7R signaling machinery with β-mercaptoethanol (β-ME), stimulated or unstimulated them with IL-7 for 15 min and subsequently evaluated them for STAT5 phosphorylation by immunoblot. (i) We analyzed the D1 cells expressing each of the indicated IL-7R constructs for IL-7Rα expression by immunoblot. (j) We assessed the signaling elicited by each indicated mutant form expressed in D1 (top) or 293T (bottom) cells by detection of STAT5 phosphorylation.
IL7R mutant proteins form homodimers via disulfide bonds
Most IL7R mutations (14/17; 82%) created an unpaired cysteine residue in the extracellular juxtamembrane-transmembrane interface region (Fig. 1a and Supplementary Fig. 2). Mutations that introduce cysteines in this region in receptors such as EpoR36, RET37 and Her2/Neu38 have been implicated in intermolecular disulfide bond formation with consequent homodimerization and signaling activation. A similar mechanism was suggested to account for the oncogenic activity of the p.Phe232Cys alteration in the TSLP receptor (encoded by CRLF2) recently found in B-ALL39. Expression of human IL-7Rα in γc-expressing D1 cells or in 293T cells, which do not express γc, followed by immunoblot analysis under non-reducing conditions showed that the mutants are detected mostly as dimers and oligomers, whereas wild-type IL-7Rα is found mainly in a monomeric form. In contrast, we detected both wild-type and mutant IL-7Rα essentially in the monomeric form when we resolved the protein lysates under reducing conditions (Fig. 3g and Supplementary Fig. 8). We obtained similar results by transducing Il7r−/− bone marrow cells (Supplementary Fig. 9). Accordingly, constitutive, ligand-independent phosphorylation of STAT5 was markedly downregulated by pretreatment of mutant IL-7Rα–expressing cells with β-mercaptoethanol (Fig. 3h). Furthermore, receptor dimerization and constitutive signaling were abrogated upon substitution of the mutated cysteine to alanine or serine (Fig. 3i,j). These data indicate that constitutive hyperactivation of IL-7R–mediated signaling in T-ALL cells results, in most cases, from intermolecular disulfide bond formation arising from the introduction of an unpaired cysteine in the extracellular juxtamembrane-transmembrane region of IL-7Rα that leads to homotypic dimerization and/or oligomerization.
IL7R mutations promote transformation and tumor formation
We then investigated the cellular consequences of constitutive signaling emanating from IL-7Rα mutants. Expression of mutant, but not wild-type, IL-7Rα into IL-7–dependent D1 cells and IL-3–dependent Ba/F3 cells promoted both cell cycle progression (Fig. 4a and Supplementary Figs. 10 and 11) and viability (Fig. 4b and Supplementary Figs. 10 and 11) independently of IL-7. Accordingly, mutation of IL-7Rα conferred growth factor independence to Ba/F3 cells (Fig. 4c), indicating that the IL7R mutants have a transforming capacity. In agreement with the signaling data (Fig. 3d–f), the functional effect of the mutants was also independent of γc and JAK3, as shown by increased survival of bone marrow cells from Il2rg−/− (Supplementary Fig. 12) and Jak3−/− (Supplementary Fig. 13) mice transduced with two of the mutants, and was dependent on JAK1, as determined by inhibition of mutant IL7R-mediated survival in Ba/F3 and D1 cells upon JAK1, but not γc or JAK3, knockdown (Fig. 4d and Supplementary Fig. 14). Furthermore, substitution of the de novo inserted cysteine residue to serine or alanine resulted in reversal of the transforming capacity of the IL-7Rα mutants (Fig. 4e and Supplementary Fig. 15), suggesting that intermolecular disulfide-bond–dependent homodimerization is mandatory not only for signaling but also for the functional effects of IL-7Rα mutants.
Figure 4.

IL7R mutations induce cell-cycle progression, increase cell viability and promote growth factor independence. (a,b) We cultured Ba/F3 cells stably expressing wild-type or mutant IL-7Rα for 96 h in medium and analyzed them for (a) cell cycle distribution (percentage of cells in cycle (S+G2/M) is indicated for each condition) and (b) viability (percentage of viable, early apoptotic and late apoptotic or necrotic cells is indicated in the respective quadrant). (c) We cultured Ba/F3 cells stably expressing IL-7Rα in the absence of growth factors or with IL-3 or IL-7 and measured expansion at the indicated time points. (d) We transfected Ba/F3 cells stably expressing P1 or P2 mutant IL-7Rα with siRNA against JAK1, JAK3, γc (IL-2Rγ) or with non-targeting (NT) control and evaluated them for cell viability after 48 h. (e) We cultured Ba/F3 cells transduced with IL-7Rα P2 or with the indicated introduced mutations in the absence of growth factors and measured expansion at the indicated time points. Results in c–e represent the average of triplicates ± s.e.m.
Although IL7R mutations induced cell transformation, growth factor independence or immortalization in vitro does not necessarily implicate the acquisition of a malignant phenotype in vivo. Therefore, we next evaluated the in vivo tumorigenic potential of IL7R mutations. In contrast to D1 cells transduced with empty vector or wild-type IL-7Rα, subcutaneous injection of mutant IL-7Rα–expressing D1 cells in Rag1−/− mice resulted in tumor formation (Fig. 5a). Notably, ill mice showed a phenotype typical of T-ALL, with substantial homing of mutant IL-7Rα–expressing cells into their bone marrow and with infiltration into various organs that are normally affected in advanced-stage disease, such as the lymph nodes, liver and spleen (Fig. 5b–e, Supplementary Fig. 16 and data not shown). The tumors were transplantable into secondary recipient animals (data not shown) and were not dependent on the presence of IL-7, as injection of mutant IL-7Rα–expressing cells led to tumor development in IL-7–deficient mice (Fig. 5f). Taken together, our results indicate that IL7R mutational activation is an oncogenic event involved in T-ALL.
Figure 5.

In vivo tumorigenic effect of IL7R mutations. We subcutaneously injected D1 cells expressing wild-type or mutant IL-7Rα into Rag1−/− mice and evaluated them for tumor progression and organ infiltration. (a) Subcutaneous tumor volume growth curves. (b) Phase contrast and fluorescence imaging of D1 cells (GFP-positive) infiltrated into liver, spleen and bone marrow. (c) Representative images of spleens from mice culled at day 20 and (d) respective spleen cellularity. (e) Histological analysis (hematoxylin and eosin staining) of |indicated organs from a representative mouse transplanted with cells expressing mutant IL-7Rα P2; the right panel shows a 20× magnification of the area denoted by a square on the left panel. (f) We subcutaneously injected D1 cells expressing wild-type or mutant IL-7Rα into Il7−/− Rag2−/− mice and evaluated them for tumor size at day 20. Results in a, d and f represent the average of triplicates ± s.e.m.
Targeting IL7R mutant cells with JAK-STAT pathway inhibitors
To test the potential therapeutic application of our findings, we reasoned that mutant IL-7Rα–expressing cells should rely on constitutive signaling downstream from the receptor. We first evaluated the efficacy of several JAK inhibitors, including Pyridone 6 (JAK inhibitor I), CP-690550 and INCB018424. The latter two inhibitors are of particular relevance because they are currently being used in clinical trials for rheumatoid arthritis and several cancers, including hematological malignancies. Notably, all three drugs significantly downregulated JAK1 phosphorylation and consequent downstream activation of STAT5 and Akt (Fig. 6a) and induced cell death in a dose- and time-dependent manner (Fig. 6b,c and Supplementary Fig. 17) in Ba/F3 cells expressing mutant IL-7Rα. Likewise, CP-690550, INCB018424 and another clinically relevant JAK inhibitor, CYT387, inhibited the proliferation of mutant IL-7Rα–expressing D1 cells (Supplementary Fig. 18). Furthermore, a STAT5-specific small-molecule inhibitor40 promoted significant killing of Ba/F3 cells expressing mutant IL-7Rα (Fig. 6d and Supplementary Fig. 19). Finally, we found that primary T-ALL cells harboring IL7R mutations are also sensitive to JAK-STAT pathway inhibition. With the exception of CP-690550, the remaining drugs had differential but always significant cytotoxic effects on diagnostic leukemia cells (Fig. 6e). These results illustrate the potential therapeutic value of JAK-STAT pathway small-molecule inhibitors in the context of IL7R mutant T-ALL.
Figure 6.

Targeting IL7R mutants using JAK-STAT pathway inhibitors. We cultured Ba/F3 cells expressing mutant IL-7Rα P1 in medium alone in the presence or absence of the indicated doses of different JAK and STAT5 pharmacological inhibitors. (a) We analyzed the cells at 48 h for effective JAK-STAT pathway inhibition by immunoblot. (b,c) We analyzed cell viability (b) at 48 h (INCB018424) and 72 h (CP-690550 and JAK inhibitor 1) after increasing doses of each drug and (c) at different time points with a single dose of each inhibitor. (d) We analyzed cell viability at 72 h with increasing doses or at different culture time points with 200 ~M of STAT5-specific inhibitor. (e) We cultured primary T-ALL cells from subject P1 in the presence of the indicated JAK-STAT pathway inhibitors and evaluated them for cell viability at 24 h. Ns, P ≥ 0.05; *P < 0.05; **P < 0.01; ***P < 0.001. Viability results in b–e represent the average of triplicates ± s.e.m.
DISCUSSION
T-ALL is an aggressive hematological cancer resulting from leukemic transformation of thymocytes. Although there has been a remarkable increase in our knowledge of T-ALL molecular pathogenesis, the identification and characterization of the players and mechanisms driving proliferation and survival of leukemia T cells remains relatively poor. IL-7 and its receptor are essential for normal T-cell development and have been suggested to play a role in T-ALL. In the present study, we showed that 9% of pediatric T-ALL cases have IL7R exon 6 mutations that are gain of function and have oncogenic ability. Thus, our findings expand the spectrum of disease-associated IL7R genetic alterations to cancer. Moreover, this is the first example of an oncogene in the γc family of cytokine receptors, which is critically involved in numerous lymphoid cell functions41.
Notably, IL7R mutations do not occur in the cytoplasmic tail, which recruits signaling effectors, but do occur at the extracellular juxtamembrane-transmembrane interface. The vast majority of IL7R mutations we identified create an unpaired cysteine residue, which is necessary for disulfide-bond–dependent IL-7Rα homodimerization and bypasses the requirement for ligand binding and γc heterodimerization to trigger downstream signaling. Moreover, all IL7R mutations insert additional amino acids rather than involving a single amino acid change to cysteine. This may indicate that these additional amino acids are required for the optimal conformation leading to maximal signaling, perhaps by allowing for the most adequate alignment and exposure of the unpaired cysteine and/or by maximizing the interactions between downstream effectors at the cytoplasmic tail of the receptor. The three remaining T-ALL cases resulted in the inclusion of either a tryptophan or an SxxxG motif in the transmembrane domain. Although we did not analyze the mechanisms by which these mutations might contribute to T-cell leukemia, tryptophan residues and SxxxG motifs have both been reported to promote association of transmembrane helices42,43 that could result in homodimer or heterodimer formation with possibly similar outcomes to cysteine mutations. However, preliminary analyses of mutant P5, which has the insertion of a tryptophan in the transmembrane domain (Table 1), suggest that it does not form dimers (data not shown) and suggest that the pro-survival effect of this mutation is relatively minor: P5 expression in D1 cells deprived of IL-7 for 48 h resulted in a 2.8-fold increase in viability relative to wild-type IL-7R, as compared to 7.4-, 9.1- and 6.0-fold increases for P1, P2 and P4, respectively. In accordance, P5 appears to be relatively inefficient at inducing constitutive signaling as compared to the other IL7R mutations (Supplementary Fig. 7). These results suggest that IL7R mutations not involving cysteine insertion are not as potent and probably require additional cooperating oncogenic events compared to those that result in the introduction of an unpaired cysteine, which constitute the vast majority of the cases we identified in childhood T-ALL and characterized in our study.
IL7R gene alterations appear to be highly predominant in T-cell compared to B-cell leukemia. We did not detect exon 6 mutations in any of the 50 childhood pre–B-ALL cases we analyzed, and a recent report indicated that IL7R mutations occur in only 0.6% pre–B-ALL cases. In contrast to T-ALL, half of the B-cell–associated mutations affect exon 5, rather than exclusively exon 6, and require cooperation with TSLPR (CRLF2) overexpression18. TSLPR expression is rare in T-ALL and is not necessary for signaling driven by the IL7R mutations, as we showed here in 293T cells, which do not express TSLPR yet display constitutive signaling after expression of mutant IL7R. Notably, the fact that IL-7Rα is apparently expressed in various carcinoma cell lines and breast cancer tissue44 raises the intriguing question of whether mutations in IL7R might also occur in solid tumors.
We found IL7R mutations in different T-ALL oncogenetic subgroups, but they tended to associate predominantly with HOXA aberrant expression. Although the exact biological importance of this link remains to be fully understood, it is noteworthy that Hoxa9−/− mice show impaired early T-cell development with reduced Bcl-2 and IL-7Rα expression45. Curiously, IL7R gene alterations did not associate with T-ALL maturation stage or with most T-cell differentiation markers. These observations are reminiscent of the fact that primary T-ALL cells, in contrast to normal developing thymocytes, respond to IL-7 independent of their maturation stage14.
We showed that pharmacological inhibition of the JAK-STAT pathway induces cell death of mutant IL-7Rα–expressing cells. The preliminary data on the effect of these inhibitors in one primary T-ALL case sample was evident but not as striking as in cell lines. This may relate to the early time point at which we assessed viability (which may have prevented the inhibitors from having the maximal effect), to the importance of other alternative downstream signaling pathways in the regulation of cell survival in primary leukemia and/or to higher dependence on other oncogenic defects in the leukemia cells of the case analyzed. Irrespective of these considerations, our results suggest that JAK-STAT pathway inhibitors are cytotoxic to mutated IL-7Rα–expressing T-ALL cells. Whether inhibitors of other signaling components activated by gain-of-function IL7R mutations, such as Akt, can be exploited on their own or in combination with JAK-STAT antagonists to target IL7R mutant T-ALL cells requires further investigation.
The extraordinary improvement in T-ALL treatment outcome in recent years is mitigated by the long-term side effects associated with current regimens and by the dismal prognosis of relapsed cases. Further improvement requires an in-depth understanding of T-ALL molecular genetics and leukemogenic pathways, which will ultimately lead to the identification of new molecular players and to the development of effective targeted therapies. This line of reasoning has led, for instance, to the identification of CREBBP (CBP) mutations that are associated with ALL relapse46 or CRLF2 rearrangements, which are particularly frequent in Down syndrome ALL47. PTPN2 and PHF6 mutational loss48,49 are among the most recently characterized genetic lesions involved in T-ALL. Our present work, and that of a parallel study18, indicates that IL7R mutational activation takes part in human T-cell leukemogenesis, thereby expanding the spectrum of genetic alterations in T-ALL to a long recognized major regulator of lymphoid biology. Notably, our findings provide a strong rationale for specific targeting of IL-7R–mediated signaling as a treatment option for T-ALL.
URLs.
LIMMA, http://bioconductor.org/packages/release/bioc/html/limma.html.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
ONLINE METHODS
Cells.
Primary leukemia cells were obtained from the bone marrow and/or peripheral blood of diagnostic pediatric T-ALL and pre–B-ALL cases. Samples were enriched by density centrifugation over Ficoll-Paque (GE Healthcare), washed twice in culture medium (RPMI-1640 supplemented with 10% FBS, 2 mM L-glutamine and penicillin streptomycin), subjected to immunophenotypic analysis by flow cytometry and classified according to their maturation stage (Table 1). Informed consent and institutional review board approval were obtained for all primary leukemia collections from Centro Infantil Boldrini, Campinas, São Paulo, Brazil (Boldrini); the Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia, Germany (COALL); and the Dutch Childhood Oncology Group, The Hague, The Netherlands (DCOG). Primary leukemia cells from subject P1 were cultured in culture medium as 2 × 106 cells/ml. Growth-factor–dependent D1 and Ba/F3 cells were maintained in culture medium plus 50 ng/ml rmIL-7 (PeproTech) or 1% (v/v) WEHI-3B–conditioned medium as source of mIL-3, respectively. The phoenix-Eco packaging cell line and 293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Mediatech or Gibco) supplemented with 10% FBS and penicillin streptomycin.
IL7R sequencing and mutational analysis.
Total RNA was extracted and RNA integrity was confirmed by agarose gel electrophoresis. We reverse transcribed 1 ~g of total RNA to complementary DNA (cDNA) using the ImProm-II Reverse Transcriptase (Promega). The complete coding sequence of IL7R and the JH2 domain of JAK1 and JAK3 were amplified by RT-PCR and sequenced on both strands for a total of 68, 52 and 52 T-ALL samples, respectively, from Centro Infantil Boldrini. The same primers were used for amplification and sequencing (Supplementary Table 2). Mutations found in the IL7R were confirmed in the corresponding genomic DNA by PCR amplification of exon 6 coding and flanking intronic sequences followed by homoheteroduplex formation analysis32 and/or sequencing. Mutations in exon 6 coding and flanking intronic sequences were further investigated in 119 T-ALL cases from the DCOG and COALL case series by sequencing and in 50 precursor B-ALL cases from Centro Infantil Boldrini by homoduplex and heteroduplex formation analysis.
Gene expression microarray analysis and unsupervised cluster analysis.
RNA isolation for 117 pediatric T-ALL case samples, integrity analyses of RNA, copy-DNA and cRNA syntheses and hybridizations to Human Genome U133 plus 2.0 oligonucleotide microarrays have been described20. Differentially expressed genes associated with IL7R mutations were obtained by regression analysis using the LIMMA package. Unsupervised cluster analyses were performed in dChip as described20.
Geneset enrichment analysis (GSEA).
GSEA was performed on our Affymetrix U133 plus 2.0 microarray expression dataset for 117 T-ALL cases20 using 100 random permutations. The microarray dataset is available at http://www.ncbi.nlm.nih.gov/geo/ under accession number GSE26713. An enrichment score and nominal P value were obtained for genes that are upregulated in human lymphocytes following exposure to IL-7 as described19, for which probesets were present on the U133 plus 2.0 expression array (SOCS2, CCL4, CCL3, TNF, PMAIP1, LRP1, PIM1, AHR, UPP1, GARS, CCND2, DUSP5, FLT3LG, IL2RA, LIF, CEACAM1, MX1, TNFSF10, CSF2, CD69, CXCR4, CSF1, SOCS1, IL18R1, DPP4, CASP3, XBP1 and BCL2).
Construction of IL7R expression vectors.
The coding sequence of IL7R was PCR amplified from cDNA of blood mononuclear cells of a healthy donor using primers IL7R 3U32 and IL7R 1434L39 (Supplementary Table 2). The reverse primer did not incorporate the stop codon. The undigested PCR product was cloned into pGEM-T Easy (Promega) and verified by sequencing. The cloned fragment was subsequently digested with XmaI, treated with the Klenow fragment of DNA polymerase I and then digested with KpnI and cloned into the XbaI (blunted with Klenow) and KpnI sites of the pUC19 vector, resulting in the clone pUC19/IL7R. By doing so, a stop codon was re-inserted, but the last C-terminal amino acids QN of the normal IL7Rα were changed to QNPG. A lentiviral expression vector of IL7R, #304/IL7R, was obtained by subcloning the IL7R EcoRI(Klenow)-SalI fragment of pUC19/IL7R in place of the ΔLNGFR SmaI-SalI fragment of a pCCL.sin.cPPT.minCMV.eGFP.PGK.ΔNGFR.WPRE lentivirus vector50 (kindly provided by L. Naldini). To obtain a retroviral expression vector, the IL7R fragment was amplified from pUC19/IL7R using primers hIL7R5′BglII and hIL7R3′EcoRI. The PCR product was digested with BglII and EcoRI and cloned into pMIG (Addgene 9044, contributed by W. Hahn). Equal procedures were used to obtain the expression vectors for the mutant IL7R cDNAs. Site-directed mutagenesis of the new cysteine was obtained by PCR amplification of a BamHI-BbsI fragment spanning positions 803–934 of the IL7R sequence (NM_002185.2) using the pUC19/IL7R clone as a template, one of the following forward primers (hIL7R_cP1s, hIL7R_cP2s, hIL7R_cP2a) and the reverse primer hIL7R_BbsI. The amplified fragments were digested with BamHI and BbsI and inserted into pUC19/IL7R, thus replacing the IL7R fragment containing the cysteine codon. Subsequently, the mutants of the IL7R coding sequences were cloned into the lentiviral and retroviral vectors, as described above. All of the above clones were verified by sequencing.
Retroviral infection of D1, Ba/F3 and mouse bone marrow cells.
Wild-type or mutant full-length human IL7R was cloned into pCCL.sin.cPPT.minCMV. eGFP.PGK.ΔNGFR.WPRE lentiviral50 or pMIG retroviral vectors, both of which also drive the expression of enhanced GFP. Where indicated, C>A or C>S mutations were introduced into the mutant IL7R using PCR strategies. All subcloned genes and constructs were verified by DNA sequencing. D1 cells were infected in RetroNectin (TaKaRa)-coated plates with pMIG supernatant produced using the phoenix-Eco packaging cell line. Ba/F3 cells were infected with either pMIG or lentiviral supernatants produced in 293T cells. Equivalent levels of expression of GFP and IL-7Rα were confirmed for all established D1 and Ba/F3 cell lines. Bone marrow cells were harvested from tibia and femur of Il7r−/− or Il2rg−/− mice, and progenitors were enriched by lineage cell depletion kit (Miltenyi Biotec) and cultured in X-vivo 10 medium (Bio Whittaker) supplemented with 5% FBS, mouse SCF (100 ng/ml), mouse IL-6 (50 ng/ml) and flt-3 ligand (100 ng/ml) (PeproTech). After 48 h, cells were infected on RetroNectin (TaKaRa)-coated plates overnight with different retroviral supernatant from the packaging line, and the infection was repeated after 72 h. On the fourth day, cells were harvested, washed and cultured with or without IL-7.
Transfection of 293T cells.
pCDNA3.1 vectors (Invitrogen) bearing human JAK3, human γC and mouse Stat5a, and pMIG-IL7R constructs were used, in the indicated combinations, to transfect 293T cells by calcium phosphate precipitation. Transfected cells were stimulated or not with IL-7 (100 ng/ml) for 15 min at 37 °C. Where indicated, cells were pretreated with 1 mM 2β-mercaptoethanol or vehicle (PBS) for 2 h at 37 °C. Reactions were stopped by placing samples on ice.
Small interfering RNA (siRNA) transfection of 293T and Ba/F3 cells.
For 293T cells, 50 pmol of ON-TARGETplus Non-Targeting pool or ON-TARGETplus SMARTpool JAK1 siRNA (Dharmacon) were co-transfected with the indicated plasmid DNA constructs (600 ng) using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. Cells were harvested 36 h after transfection, and whole-cell lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Ba/F3 cells were electroporated (300 V, 1,500 microfarads) in a Gene Pulser II (Bio-Rad) with 200 pmol of ON-TARGETplus Non-Targeting pool, ON-TARGETplus SMARTpool Jak1 or Jak3 (Dharmacon) or Silencer siRNA Il2rg (Ambion) siRNAs. At the indicated time points, cells were harvested for viability assay and cell counts.
Short hairpin RNA (shRNA) transduction of D1 cells.
The retroviral vector containing mouse Jak1-specific 29-mer shRNA expressed under U6 promoter and the puromycin selection marker was bought from OriGene. Retroviral supernatant from the packaging line was used to infect mutant IL-7Rα–expressing D1 cells on RetroNectin-coated plates overnight. At 24 h after infection, cells were put in fresh culture medium containing 50 ng/ml of mIL-7 and 5 ~g/ml of puromycin (Invitrogen) for another 48h. mIL-7 and puromycin were washed away, and cells were placed in culture without mIL-7 for 48 h. Cell viability and proliferation were measured by MTT assay.
Treatment with pharmacological inhibitors.
Ba/F3 cells stably expressing mutant IL7R or primary T-ALL cells bearing IL7R mutations were cultured in medium alone or with the indicated concentrations of Pyridone 6 (JAK Inhibitor I), STAT5 inhibitor N′-((4-Oxo-4H-chromen-3-yl)methylene)n icotinohydrazide (both purchased from Calbiochem), Ruxolitinib (INCB 018424) or Tasocitinib (CP-690550) (both purchased from Axon Medchem), and viability was determined at the indicated time by flow cytometry analysis. D1 cells stably expressing mutant IL7R were plated in 96-well plate at a density of 1 × 105 cells per well in IL-7–free medium and incubated for 48 h with or without JAK inhibitors at the indicated concentrations. Cell viability and proliferation were determined by MTT assay.
Immunoblotting.
Cell lysates were resolved by 10% or 12% SDS-PAGE, and equal amounts of protein were transferred onto nitrocellulose membranes and immunoblotted with antibodies against p-JAK3 (Y980), JAK3, JAK1, STAT5, γC, actin, (Santa Cruz Biotechnology), p-STAT5a/b (Y694/Y699) (Upstate Biotechnology), p-TYK2 (Y1054/1055), p-JAK1 (Y1022/1023), p-JAK2 (Y1007/1008), JAK1, p-STAT5 (Y694), p-STAT3 (Y705), p-STAT1 (Y701), p-Akt (S473), Akt, p-Bad (S112), Bad (Cell Signaling Technology) and IL-7Rα (R&D Systems). Immunodetection was performed by incubation with horseradish-peroxidase–conjugated appropriate secondary antibodies and developed by chemiluminescence. For the analysis of IL-7Rα dimer formation, whole-cell lysates were resolved in denaturing, non-reducing SDS-PAGE, transferred onto nitrocellulose membranes and immunoblotted. When indicated, lysates were incubated with 100 mM dithiothreitol (DTT) (Sigma-Aldrich) for 5 min at room temperature (20–25 °C) before non-reducing SDS-PAGE.
Cell-cycle analysis.
Cells were either permeabilized in 0.1% BSA, 0.01 M HEPES, 0.1% saponin in PBS at a concentration of 1 × 106 cells/ml and an equal volume of detergent buffer containing 50 ~g/ml of propidium iodide (Sigma) and 50 ~g/ml of RNase (Puregene), or treated as described13, and analyzed by flow cytometry. Cell cycle distribution was determined using ModFit LT software (Verity).
Cell viability assay.
Quantitative determination of cell viability was performed using Annexin V–based apoptosis detection kits and the manufacturer’s instructions (R&D Systems or eBioscience). Briefly, cells were resuspended in the appropriate binding buffer, stained with APC-conjugated Annexin V and propidium iodide or 7-AAD at room temperature for 15 min and subsequently analyzed by flow cytometry.
Cell counts.
Ba/F3 cells were cultured as 2 × 105/ml in medium deprived of growth factors or in the presence of IL-3–conditioned medium (1%; v/v) or IL-7 (10 ng/ml). Total cell counts were calculated by trypan blue exclusion using a hemocytometer at the indicated time points.
MTT assay.
We added 8 ~l of MTT (3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide; 5 mg/ml; Sigma) to each well and kept cells at 37 °C for 4 h, after which 100 ~l of solubilization solution (Promega) was added, and cells were incubated overnight at 37 °C. Absorbance was measured by spectrophotometry at wavelengths of 590 and 630 nm.
Mice.
Rag1−/− mice were originally purchased from The Jackson Laboratory, and Il7−/− Rag2−/− mice were obtained from R. Murray (DNAX Research Institute). Mice were maintained by homozygous breeding at the National Cancer Institute (NCI)-Frederick, Maryland. Animal care was provided in accordance with US NIH Animal Use and Care guidelines. Experiments were performed following protocols approved by NCI-Frederick Animal Care and Use Committee. All mice used were 8–12 weeks old.
Tumor model.
Mice were treated with 0.64 mg/ml of sulfamethoxazole in drinking water 2 days before the injection, and treatment continued for up to a week after the injection. Mice received 3 Gy of whole-body γ irradiation 4 h before the injection. D1 cells harboring the empty vector or human IL7R (2 × 106 cells in 100 ~l of PBS) were injected subcutaneously into the right flank. On day 20, mice were killed, and tumor size was measured by caliper. Tumor volume was calculated by the modified ellipsoidal formula51: tumor volume = ½ (length × width2).
Statistical analysis.
A Fisher’s exact test with Bonferroni correction was used to compare the frequency of IL7R mutations between T-ALL subgroups. Differences between populations were calculated using an unpaired two-tailed Student’s t-test. Differences were considered significant at P < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the subjects and their families for providing the specimens for this study. We thank S. Walsh (University of Maryland) for helpful discussions on the IL7R transmembrane domain; K. Czarra and M. Karwan for animal technical assistance; A. Silva, I. Antunes, A. Melão and J. Buijs-Gladdines for experimental support; P. Vandenabeele for kindly providing the WEHI3B cell line; and J. O’Shea for providing Jak3−/− bone marrow and CP-690550. This work was supported by grants from Fundação para a Ciência e a Tecnologia (FCT; PTDC/SAU-OBD/104816/2008, J.T.B.), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 08/10034–1, J.A.Y.) and the intramural program of the National Cancer Institute, US National Institutes of Health (NIH) (S.K.D.). P.P.Z. and A.B.S. have Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) PhD scholarships. L.M.S. has a postdoctoral fellowship; D.R., B.A.C. and N.C. have PhD scholarships, and M.C.S. had a Bolsa de Investigação (BI) fellowship, all from the FCT. L.Z. was supported by a grant (2007–012) from the foundation Children Cancer-Free (Stichting Kinderen Kankervrij; KiKa).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Jiang Q et al. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. 16, 513–533 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Fry TJ & Mackall CL The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J. Immunol. 174, 6571–6576 (2005). [DOI] [PubMed] [Google Scholar]
- 3.von Freeden-Jeffry U et al. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 181, 1519–1526 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peschon JJ et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor–deficient mice. J. Exp. Med. 180, 1955–1960 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puel A, Ziegler SF, Buckley RH & Leonard WJ Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat. Genet. 20, 394–397 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Roifman CM, Zhang J, Chitayat D & Sharfe N A partial deficiency of interleukin-7Rα is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood 96, 2803–2807 (2000). [PubMed] [Google Scholar]
- 7.Lundmark F et al. Variation in interleukin 7 receptor α chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 39, 1108–1113 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Hafler DA et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 357, 851–862 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Rich BE, Campos-Torres J, Tepper RI, Moreadith RW & Leder P Cutaneous lymphoproliferation and lymphomas in interleukin 7 transgenic mice. J. Exp. Med. 177, 305–316 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham N et al. Haploinsufficiency identifies STAT5 as a modifier of IL-7–induced lymphomas. Oncogene 24, 5252–5257 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Laouar Y, Crispe IN & Flavell RA Overexpression of IL-7Rα provides a competitive advantage during early T-cell development. Blood 103, 1985–1994 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Touw I et al. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood 75, 2097–2101 (1990). [PubMed] [Google Scholar]
- 13.Barata JT, Cardoso AA, Nadler LM & Boussiotis VA Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 98, 1524–1531 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Barata JT, Keenan TD, Silva A, Boussiotis VA & Cardoso AA Common γ chain-signaling cytokines promote proliferation of T-cell acute lymphoblastic leukemia. Haematologica 89, 1459–1467 (2004). [PubMed] [Google Scholar]
- 15.González-García S et al. CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7Rα gene expression in early human thymopoiesis and leukemia. J. Exp. Med. 206, 779–791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weng AP et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Flex E et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 205, 751–758 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shochat C et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 208, 901–908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovanen PE et al. Analysis of γ c-family cytokine target genes. Identification of dual-specificity phosphatase 5 (DUSP5) as a regulator of mitogen-activated protein kinase activity in interleukin-2 signaling. J. Biol. Chem. 278, 5205–5213 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Homminga I et al. Integrated transcript and genome analyses reveal NKX2–1 and MEF2C as potential oncogenes in T-cell acute lymphoblastic leukemia. Cancer Cell 19, 484–497 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Ferrando AA et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1, 75–87 (2002). [DOI] [PubMed] [Google Scholar]
- 22.van Grotel M et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia 22, 124–131 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Bene MC et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9, 1783–1786 (1995). [PubMed] [Google Scholar]
- 24.Jeong EG et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin. Cancer Res. 14, 3716–3721 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Walters DK et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 10, 65–75 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Barata JT et al. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J. Exp. Med. 200, 659–669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maser RS et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 447, 966–971 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palomero T et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 13, 1203–1210 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva A et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Invest. 118, 3762–3774 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutierrez A et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jotta PY et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 24, 239–242 (2010). [DOI] [PubMed] [Google Scholar]
- 32.O’Neil J et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J. Exp. Med. 204, 1813–1824 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson BJ et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 204, 1825–1835 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuurbier L et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia 24, 2014–2022 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Kim K et al. Characterization of an interleukin-7–dependent thymic cell line derived from a p53(−/−) mouse. J. Immunol. Methods 274, 177–184 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Gross AW & Lodish HF Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J. Biol. Chem. 281, 7002–7011 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Kjaer S, Kurokawa K, Perrinjaquet M, Abrescia C & Ibanez CF Self-association of the transmembrane domain of RET underlies oncogenic activation by MEN2A mutations. Oncogene 25, 7086–7095 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Burke CL & Stern DF Activation of Neu (ErbB-2) mediated by disulfide bond-induced dimerization reveals a receptor tyrosine kinase dimer interface. Mol. Cell. Biol. 18, 5371–5379 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoda A et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 107, 252–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X et al. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nat. Med. 16, 191–197 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Kovanen PE & Leonard WJ Cytokines and immunodeficiency diseases: critical roles of the γ(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 202, 67–83 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Ridder A, Skupjen P, Unterreitmeier S & Langosch D Tryptophan supports interaction of transmembrane helices. J. Mol. Biol. 354, 894–902 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Russ WP & Engelman DM The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 296, 911–919 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Al-Rawi MA, Mansel RE & Jiang WG Interleukin-7 (IL-7) and IL-7 receptor (IL-7R) signalling complex in human solid tumours. Histol. Histopathol. 18, 911–923 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Izon DJ et al. Loss of function of the homeobox gene Hoxa-9 perturbs early T-cell development and induces apoptosis in primitive thymocytes. Blood 92, 383–393 (1998). [PubMed] [Google Scholar]
- 46.Mullighan CG et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–239 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullighan CG et al. Rearrangement of CRLF2 in B-progenitor– and Down syndrome–associated acute lymphoblastic leukemia. Nat. Genet. 41, 1243–1246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleppe M et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 42, 530–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Vlierberghe P et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 42, 338–342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amendola M, Venneri MA, Biffi A, Vigna E & Naldini L Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat. Biotechnol. 23, 108–116 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Jensen MM, Jorgensen JT, Binderup T & Kjaer A Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med. Imaging 8, 16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.