

Abstract
Purpose
We sought to determine the role of glycogen synthase kinase-3β (GSK-3β) in the pathogenesis of Graves’ orbitopathy(GO).
Methods
Expression of the GSK-3β gene in whole orbital tissue explants was compared between GO and non-GO donors using quantitative real-time PCR (RT-PCR). The expression of proinflammatory molecules in the presence of the GSK-3β inhibitor CHIR 99021 was analyzed using RT-PCR, western blot, and ELISA. Adipogenic differentiation was identified using Oil Red O staining, and the levels of peroxisome proliferator activator gamma (PPARγ) and CCAAT-enhancer-binding proteins (C/EBPs) α and β were determined by western blot.
Results
The expression of GSK-3β was significantly higher in GO tissues than in control tissues. The addition of CHIR 99021 led to a decrease in the active form of the kinase in which the Y216 residue is phosphorylated. When GO and non-GO fibroblasts were stimulated with IL-1β or TNF-α, IL-6, IL-8, intercellular adhesion molecule-1 (ICAM-1), cyclooxygenase-1 (COX-1), and monocyte chemoattractant protein 1 (MCP-1) showed increased production, which was blunted when CHIR 99021 was added. The activation of Akt, PI3K, nuclear factor (NF)-κB, Erk, Jnk, and p38 kinase by IL-1β and TNF-α was diminished with CHIR 99021 in GO cells. A decrease in lipid droplets and expression of PPARγ and c/EBPα and -β was noted in fibroblasts treated with CHIR 99021 during adipocyte differentiation. The inhibition of Wnt and β-catenin in adipogenesis was reversed by CHIR 99021.
Conclusions
GSK-3β plays a significant role in GO pathogenesis. The inhibition of the kinase attenuated the proinflammatory cytokines production and fibroblast differentiation into adipocytes. GSK-3β may be a potential target for anti-inflammatory and anti-adipogenic treatment of GO.
Keywords: Graves’ orbitopathy, inflammation, adipogenesis, glycogen synthase kinase-3β, orbital fibroblast
Graves’ orbitopathy (GO) is characterized by extensive remodeling of soft tissues in the orbit.1 Many studies have attempted to elucidate its pathogenesis. Although the importance of different cell types has been debated, orbital fibroblasts in particular have recently gained considerable attention as the main agent of the disease process.2,3 In response to the inflammatory cells that infiltrate the space, fibroblasts proliferate and secrete proinflammatory cytokines, actively inciting and perpetuating inflammation.4 During the process, they may also differentiate into adipocytes and myofibroblasts and secrete extracellular matrix components, including hydrophilic glycosaminoglycans.5 Ultimately, the resultant increase in soft tissue volume within the confined bony orbit clinically leads to proptosis, compressive optic neuropathy, and periorbital edema.
Recent advances in genomic studies have allowed discoveries of molecular mechanisms driving the pathogenesis of GO. The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, Wnt/β-catenin pathway, and phosphoinositide 3-kinase (PI3K)/Akt pathways are among a number of signaling pathways that have been found to play cardinal roles.2,6,7 At the intersection of these pathways lies one molecule: glycogen synthase kinase-3beta (GSK-3β). However, other than a small number of studies that have examined its role in inflammation in adipose tissues,8,9 no studies to date have conducted an extensive analysis of its function with respect to the pathogenesis of GO. In this regard, the present study was designed to characterize the role of GSK-3β in GO fibroblasts in vitro. We investigated the expression of GSK-3β in GO fibroblasts and identified its role in inflammation and adipocyte differentiation of fibroblasts obtained from the orbits of patients with GO by employing the GSK-3β-specific inhibitor CHIR 99021.
Methods
Reagents
The antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA), BD Biosciences (San Jose, CA, USA), Santa Cruz Biotechnology (Santa Cruz, CA, USA), Novus Biologicals (Centennial, CO, USA), and Abcam (Cambridge, UK). The antibodies used are listed in detail in Supplementary Table S1. CHIR 99021 was a product of Tocris Bioscience (Minneapolis, MN, USA). Recombinant human IL-1β was a product from R&D Systems (Minneapolis, MN, USA). The enzyme-linked immunosorbent assay (ELISA) kits for IL-6 and IL-8 were purchased from R&D Systems (Minneapolis); for MCP-1, from R&D Systems (Abingdon, UK); and for ICAM-1, from Abcam. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTT) assay and Oil Red O were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, and gentamicin were purchased from Hyclone Laboratories, Inc. (Logan, UT, USA).
Subjects and Preparation of Tissues and Cells
The GO patient cohort was comprised of six men and nine women (mean age, 43.9 ± 5.7 years). Their orbital adipose/connective tissues were obtained during orbital decompression from tissues normally discarded. All GO patients were in a stable euthyroid state for a minimum of 3 months with clinical activity scores less than 4 and were free of steroid or radiotherapy treatment for at least 3 months prior to surgery. The control patient population was comprised of three men and six women (mean age, 42.2 ± 6.7 years) undergoing either evisceration or orbital wall fracture repair. The control patients had no known history or clinical evidence of any thyroid disease. All patients provided written informed consent for the surgical procedure and study participation. The study protocol was reviewed and approved by the Institutional Review Board of Severance Hospital, and the study adhered to the tenets of Declaration of Helsinki. The clinical characteristics of the patient population of this study are detailed in Supplementary Table S2.
The isolation of the orbital fibroblasts followed a standard protocol in published literature.10 In short, the harvested tissues were minced and placed in DMEM/F12 (1:1 ratio) medium containing 20% FBS, penicillin (100 U/mL), and gentamycin (20 µg/mL). Following incubation, tissues were maintained in solution containing DMEM, antibiotics, and 10% FBS. When the growth of the fibroblasts had been confirmed, the cells were treated with trypsin/EDTA and passaged in monolayers. The cell strains were stored in liquid nitrogen and only those between the third and fifth passages were used for experiments.
Cell Viability Assay
The MTT assay was employed to test cell viability. Following the manufacturer's protocol (Sigma-Aldrich), orbital fibroblasts isolated from GO patients and healthy non-GO donors were seeded into 24-well culture plates (1 × 105 cells/well) and treated with dimethylsulfoxide (DMSO; vehicle control) and different concentrations of CHIR 99021 (1, 5, 10, and 20 µM) for 18, 24, 48, and 72 hours. Thereafter, cells were washed and incubated in 5-mg/mL MTT solution for 3 hours at 37°C. Ice-cold isopropanol was applied for solubilization, and the absorbance of the converted dye was measured with a microplate reader (EL 340 Microplate Bio Kinetics Reader; Bio-Tek Instruments, Winooski, VT, USA) at 560 nm, with background subtraction at 630 nm.
Quantitative Real-Time PCR
The RNA was extracted from cells with Invitrogen TriZol (Thermo Fisher Scientific, Waltham, MA, USA). Out of the extract, 1 µg was reverse-transcribed into cDNA (Qiagen, Hilden, Germany) and amplified with SYBR Green Real-Time PCR Master Mix in a StepOne Plus real-time PCR thermocycler (Applied Biosystems, Carlsbad, CA, USA). The sequence of primers is listed in detail in Supplementary Table S3. The PCR results for each type of mRNA were normalized to the level of glyceraldehyde-3-phosphate dehydrogenase and expressed as fold change in the Ct value relative to the control group in accordance with the 2–ΔΔCt method.11
Western Blot Assay
Orbital fibroblasts subjected to different study conditions were washed with PBS and then lysed with cell lysis buffer on ice.12 The lysates were centrifuged, and the supernatants were resolved in 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer onto polyvinylidene fluoride membranes (Immobilon; Millipore Corp., Billerica, MA, USA). The membranes were then probed with primary antibodies in tris-buffered saline and Tween 20 along with 1% BSA and 0.01% sodium azide. Then, the membranes were probed with horseradish peroxidase-conjugated secondary antibody with chemiluminescence (Cell Signaling Technology), using 5% skim milk as the blocking solution. The bands were detected on x-ray films (Agfa, Mortsel, Belgium), and their intensities were quantified and normalized to that of the β-actin in the same sample.
ELISA
According to the manufacturer's instructions, supernatants were collected from cell cultures and diluted, and assays were performed to quantify the levels of IL-6, IL-8, ICAM-1, and MCP-1. The absorbance was measured at 405 nm to determine the percentage of binding for each sample, and a standard binding curve was then drawn to determine the concentrations. The average value of three repeated assays was used for statistical analyses.
Adipogenesis
Fibroblasts were induced to differentiate into adipocytes following a previously published protocol.13,14 Briefly, cells were cultured in serum-free DMEM supplemented with T3, insulin (Boehringer-Mannheim, Mannheim, Germany), carbaprostaglandin (cPGI2; Calbiochem, La Jolla, CA, USA), and dexamethasone, along with a peroxisome proliferator activator gamma (PPARγ) agonist, rosiglitazone (10 µM; Cayman Chemical, Ann Arbor, MI, USA) for 10 days.
Oil Red O Staining
A working solution was prepared by diluting 6 mL of a stock solution (0.5% Oil Red O in isopropanol) with 4 mL of distilled water. The cells were fixed with 3.7% formalin at 4°C for 15 minutes before being washed with PBS and mixed with the solution for 1 hour at room temperature. The cell–solution mixture was visualized under a light microscope (Olympus BX60; Olympus Corp., Melville, NY, USA). The Oil Red O staining protocol has been described in detail by Green and Kehinde.15
Statistical Analysis
All experiments were performed at least three times with samples from each patient, and the results were expressed as mean ± SD. Independent t-tests and ANOVA were employed for comparisons between GO and non-GO tissue explants and also among DMSO-treated, stimulant-treated, and stimulant- and inhibitor-treated cell groups. The Bonferroni test was performed as a post hoc test. SPSS Statistics 20 (IBM, Armonk, NY, USA) was used. P < 0.05 was considered statistically significant.
Results
GO Tissues Show Increased Expression of GSK-3β
RNA was extracted from whole orbital tissue explants of 15 GO and nine non-GO subjects, and GSK-3β transcript levels were compared between GO and non-GO using real-time PCR (RT-PCR (Fig. 1). The RT-PCR results showed that the expression of GSK-3β mRNA was greater in GO tissues (n = 15) than in the control orbital tissues (n = 9).
Figure 1.
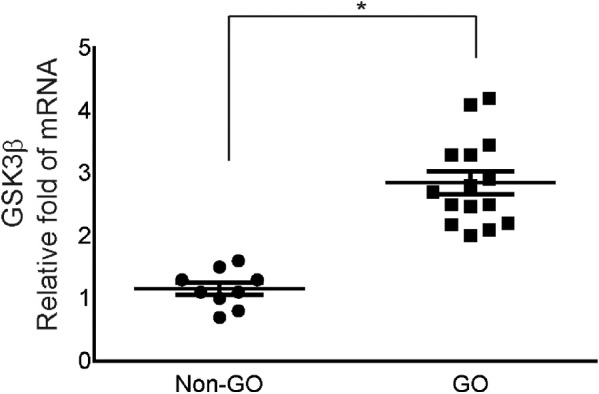
Expression of GSK-3β mRNA in GO tissues in comparison to non-GO tissues. RNA was extracted from GO (n = 15) and non-GO (n = 9) orbital tissues, and the GSK-3β transcript levels were compared between GO and non-GO tissues using real-time PCR. A single dot represents the value obtained from the experiment on a single donor (*P < 0.05 vs. non-GO tissues).
CHIR 99021 Inhibits GSK-3β in GO Fibroblasts
An MTT assay was conducted in order to identify the non-toxic concentrations of the GSK-3β inhibitor CHIR 99021 for orbital fibroblasts (Fig. 2). The results revealed that more than 95% of both GO and non-GO fibroblasts were viable when CHIR 99021 was applied in concentrations up to 20 µM for at least 72 hours. Based on these results, when analyzing the effect of CHIR 99021, CHIR 99021 was applied in concentrations below 20 µM for less than 72 hours for the remainder of the study.
Figure 2.

Effect of CHIR 99021 on the viability of orbital fibroblasts. Orbital fibroblasts of GO and non-GO patients were seeded in 24-well culture plates, 1 × 105 cells per well. Various concentrations (0–20 µM) of CHIR 99021 were applied to wells for 18, 24, 48, and 72 hours, and an MTT assay was conducted to test cell viability. Results are expressed as a percentage of untreated control and are presented as mean ± SD. Assays were performed at least three times in triplicate. Differences between treated and untreated cells are indicated (*P < 0.05).
When GO fibroblasts were challenged with IL-1β and TNF-α, increased production of both active (phosphorylation of Y216 residue) and inactive (phosphorylation of S9 residue)16,17 forms of GSK-3β were observed in western blot (Fig. 3A, Supplementary Fig. S2). The addition of CHIR 99021 to the medium significantly inhibited the expression of both active and inactive forms of the kinase while the total amount of GSK-3β remained unchanged. When GO fibroblasts were treated with CHIR 99021, the cell lysates of fibroblasts collected at different time points showed a gradual decrease in production of both the active and inactive forms GSK-3β (Fig. 3B). The decrease was especially prominent for the active form (Y216).
Figure 3.

CHIR 99021 suppresses GSK-3β. (A) Confluent orbital fibroblasts obtained from GO patients (n = 3) were treated with DMSO and 10 ng/mL of IL-1β or TNF-α for 15 minutes with or without pretreatment with 10-µM CHIR 99021 for 48 hours. Western blot analyses were performed to investigate the levels of inactive GSK-3β (p-GSK-3β [S9]), active GSK-3β (p-GSK-3β [Y216]), and total GSK-3β. Although the treatment with proinflammatory cytokines led to a significant increase in the levels of inactive GSK-3β (p-GSK-3β [S9]) and active GSK-3β (p-GSK-3β [Y216]), pretreatment with CHIR 99021 significantly suppressed the increase. (B) Confluent orbital fibroblasts obtained from GO patients (n = 3) were treated with CHIR 99021 for increasing lengths of time (0–48 hours) without stimulant. Increasing durations of CHIR 99021 treatment resulted in decreased p-GSK-3β (Y216) levels. Data in the columns indicate the mean density ratio ± SD, normalized to the level of β-actin in the same sample. Representative gel images are also shown. Differences between treated and untreated cells are indicated (*P < 0.05). For full-length gel images, see Supplementary Figure S2.
GSK-3β Mediates Elevated Expression of Proinflammatory Cytokines in GO
The GO and non-GO fibroblasts were treated with DMSO for 3 hours, 10 ng/mL of IL-1β for 3 hours, or IL-1β for 3 hours following pretreatment with the GSK-3β inhibitor CHIR 99021 for 1 hour (Fig. 4A). In comparison to the DMSO-treated cells, IL-1β-challenged fibroblasts showed an increased expression of mRNA of the proinflammatory cytokines IL-6, IL-8, ICAM-1, cyclooxygenase-2 (COX-2), and MCP-1. When CHIR 99021 was added to the medium, the elevated mRNA expression of all the cytokines was blunted in GO and control fibroblasts. The investigation was repeated with the stimulant switched from IL-1β to TNF-α (Fig. 4B). Likewise, the RT-PCR results showed that the stimulation with TNF-α led the GO and control fibroblasts to increase mRNA expression of all of the proinflammatory cytokines, which was then suppressed when CHIR 99021 was added. The western blot analyses with the same condition echoed the results of RT-PCR (Fig. 5). The increased production of the five cytokines by GO and control fibroblasts in response to challenge with IL-1β or with 10-ng/mL TNF-α for 16 hours was blunted with the addition of CHIR 99021 (Fig. 5, Supplementary Fig. S3). The effect of CHIR 99021 on the release of proinflammatory cytokines was analyzed with ELISA in both GO and non-GO cells (Supplementary Fig. S1). The increased level of secretory proinflammatory cytokines in response to IL-1β and TNF-α stimulation was significantly suppressed by the addition of CHIR 99021 in both GO and non-GO fibroblasts.
Figure 4.

The effect of GSK-3β inhibitor on the transcription of proinflammatory cytokines in GO fibroblasts in comparison to control fibroblasts. (A) Cells obtained from GO patients (n = 3) and control patients (n = 3) were treated with DMSO for 3 hours, as well as 10 ng/mL of IL-1β for 3 hours and IL-1β for 3 hours after pretreatment with GSK-3β inhibitor (10-µM CHIR 99021) for 1 hour. The RNA extracted from each sample was reverse-transcribed by real-time PCR and quantified. The results showed elevated levels of IL-6, IL-8, ICAM-1, COX-2, and MCP-1 in response to IL-1β treatment, which was significantly suppressed by the addition of CHIR 99021. (B) The same experiment was repeated with TNF-α. The results are shown as mean ± SD fold expression of three separate experiments (*P < 0.05 vs. IL-1β- or TNF-α-stimulated cells).
Figure 5.

The effect of GSK-3β inhibitor on the protein levels of proinflammatory cytokines in GO and control fibroblasts. (A) Orbital fibroblasts obtained from GO patients (n = 3) and control non-GO patients (n = 3) were treated with 10-ng/mL IL-1β and 10-ng/mL TNF-α for 16 hours. The cells were also subjected to pretreatment with 10-µM CHIR 99021 for 48 hours before the challenge with either stimulant. Western blot analyses were conducted to compare levels of the proinflammatory cytokines IL-6, IL-8, ICAM-1, COX-2, and MCP-1. The representative gel images are also shown. The molecular weights of the detected bands are marked to the left of the bands (kDa). (B) The results from GO fibroblasts are presented as the mean density ratio ± SD, normalized to the level of β-actin in the same sample. (C) The same experiment was repeated in the control fibroblasts, and results are shown as the mean density ratio ± SD fold expression of three separate experiments, normalized to the level of β-actin in the same sample (*P < 0.05 vs. IL-1β- or TNF-α-stimulated cells). For full-length gel images, see Supplementary Figure S3.
Adipocyte Differentiation in GO is Dependent on the Activity of GSK-3β
When confluent orbital fibroblasts taken from GO patients were treated with the adipogenic medium for 10 days to induce their differentiation into adipocytes, the fibroblasts lost their stellate appearance and morphologically conformed into a spherical shape with intracellular lipid droplets (Fig. 6A). When the GSK-3β inhibitor CHIR 99021 was added to the medium in increasing concentrations, adipogenesis identified with Oil Red O staining decreased in a dose-dependent manner. Quantification of the optical density of Oil Red O staining at 490 nm confirmed the histochemical results (Fig. 6B). Increasing concentrations of CHIR 99021 led to dose-dependent decreases in the adipogenic transcription factors PPARγ and CCAAT-enhancer-binding protein (C/EBP) α and β (Fig. 6C, Supplementary Fig. S4). Markers of mature adipocytes, fatty acid-binding protein 4 (FABP4) and Acrp30, which were detected on day 10 of adipogenic differentiation in control-treated fibroblasts, were significantly suppressed upon CHIR 99021 treatment. Further, the Wnt/β-catenin signaling pathway was assessed. The inactive and active forms of GSK-3β that showed an increase during adipogenesis decreased following the treatment with CHIR 99021. The β-catenin levels that decreased during adipogenesis significantly increased in a dose-dependent manner with CHIR 99021 treatment, and cyclin D1 that increased during adipogenesis significantly decreased in a dose-dependent manner with CHIR treatment. The results suggest that GSK-3β plays a role in inducing adipogenic differentiation of fibroblasts in GO through the canonical Wnt/β-catenin signaling pathway.
Figure 6.

GSK-3β mediates adipogenic differentiation of GO fibroblasts. Orbital fibroblasts from GO (n = 3) patients were cultured in adipogenic medium to induce differentiation into adipocytes. (A) Oil Red O staining showed that increasing concentrations of CHIR 99021 inhibited adipogenesis in a dose-dependent manner. (B) Measurements of the optical density of cell lysates at 490 nm showed the same results. Experiments were conducted on three cell strains, and the results are expressed as the mean optical density (%) ± SD (*P < 0.05 vs. untreated controls). (C) Western blot analyses of adipogenic transcription factors PPARγ, C/EBPα, and C/EBPβ showed decreased levels in response to increasing concentrations of CHIR. The levels of mature adipocyte markers, FABP4 and Acrp30, were significantly reduced in fibroblasts treated with higher concentrations of CHIR 99021. Increasing concentrations of CHIR led to decreased levels of inactive and active GSK-3β and increased levels of inactive (np-β-catenin) and total β-catenin. The molecular weights of the detected bands are marked to the left of the bands (kDa). Data in the columns indicate the mean density ratio ± SD, normalized to the level of β-actin in the same sample (n = 3), and representative gel images are shown (*P < 0.05 vs. control fibroblasts on day 10 of adipogenesis). For full-length gel images, see Supplementary Figure S4.
Inhibition of GSK-3β Decreases Activation of Proinflammatory Signaling Molecules
In order to identify signaling pathways affected by the regulation of the kinase, multiple transcription factors were assayed. Figure 7 illustrates the results of the western blot analyses. In comparison to the DMSO-treated fibroblasts, IL-1β-treated GO fibroblasts (10 ng/mL of IL-1β for 15 minutes) showed increased levels of phosphorylated forms of proinflammatory signaling molecules, including Akt, PI3K, NF-κB, extracellular signal-regulated kinase (ERK), p38 and c-Jun NH(2)-terminal kinase (JNK). When CHIR 99021 was applied for 48 hours prior to the challenge with IL-1β, the level of phosphorylated forms relative to the total amount of the same protein significantly decreased for all eight of the transcription factors tested. The response of the GO fibroblasts to incubation with 10-ng/mL TNF-α and CHIR 99021 demonstrated a similar trend (Fig. 7A). In control fibroblasts, treatment with CHIR 99021 led to significant reduction in only Akt, ERK, and JNK (Fig. 7B). In response to 10-ng/mL TNF-α, the reduction by CHIR 99021 was significant only for Akt, PI3K, p38, and JNK (Supplementary Fig. S5).
Figure 7.

The role of GSK-3β in the activation of signaling molecules in response to proinflammatory cytokines treatment. (A) Confluent orbital fibroblasts obtained from GO patients (n = 3) were treated with DMSO (D), and 10-ng/mL of IL-1β (IL) or TNF-α (TN) for 15 minutes with or without pretreatment with 10-µM CHIR 99021 (C) for 48 hours. Representative gel images are shown. The molecular weights of the detected bands are marked to the right of the bands (kDa). Data in the columns indicate the mean density ratio ± SD of the bands obtained from the GO patients, normalized to the level of β-actin in the same sample. Treatment with IL-1β or TNF-α resulted in a significant increase in the levels of phosphorylated forms of Akt, PI3K, NF-κB, Erk, p38, and Jnk. Pretreatment with the GSK-3β inhibitor CHIR 99021 blunted the increases in the transcription factors. (B) The same experiment was repeated with fibroblasts obtained from non-GO patients (n = 3). Representative gel images are shown, where the molecular weight of each band is marked on the right. The mean density ratios ± SD from the control fibroblasts were normalized to the level of β-actin in the same sample (*P < 0.05 vs. IL-1β- or TNF-α-stimulated cells). For full-length gel images, see Supplementary Figure S5.
Discussion
In this study, we examined the role of GSK-3β in the pathogenesis of GO. The challenge with proinflammatory cytokines led to an increase in both active and inactive forms of GSK-3β. The GSK-3β inhibitor CHIR 99021 significantly decreased proinflammatory cytokine production, adipogenic transcription factors, and adipocyte formation. In response to inflammatory and adipogenic stimulants applied to primary culture cells to mimic in vivo conditions, the rise in secretory cytokines and transcription factors was more prominent in GO fibroblasts in comparison to the non-GO fibroblasts. Together with the higher mRNA level of GSK-3β in GO orbital tissues, which most closely resembles the in vivo conditions, our study demonstrates that GSK-3β plays a central role in inflammatory reactions and adipocyte differentiation of GO fibroblasts (Fig. 8).
Figure 8.

Schematic diagram of the GSK-3β signaling in orbital fibroblasts of Graves’ orbitopathy. GSK-3β mediates proinflammatory reactions by activating PI3K, MAP kinases (p38, JNK, and ERK), and NF-κB. By inducing nuclear translocation of β-catenin and activating C/EBPα and β, the kinase also plays a critical role in inducing adipocyte differentiation.
A number of studies have made attempts at clarifying the importance of GSK-3β in thyroid diseases. For instance, Rao et al.18 suggested a possible role for GSK-3β in the regulation of thyrocyte proliferation; inhibition of the kinase with lithium allowed the Wnt/β-catenin signaling pathway to stimulate the proliferation of thyrocytes. Indeed, the kinase has been included as an important tumor suppressor in thyroid carcinogenesis models in several studies.19,20 In the context of GO, however, studies on the kinase are scarce. In 2018, Yang et al.21 confirmed that single nucleopeptide polymorphisms within the genetic sequence of GSK-3β were associated with 1.4 to 2.7 times greater susceptibility to GO. In 2015, Shen et al.22 postulated that downregulation of GSK-3β was the mechanism by which microRNA (miR-224-5p), when over-expressed, increased the sensitivity of GO to glucocorticoid therapy. To our knowledge, this study is the first of its kind to confirm the role of GSK-3β in the pathogenesis of GO. Our results showed that GSK-3β is expressed at higher levels in GO tissues compared to the control. More importantly, the inhibition of this enzyme ameliorated proinflammatory and adipogenic reactions of orbital fibroblasts of GO.
Previous reports have partially discovered the mechanism of GO at a molecular level. From what has been identified, orbital fibroblasts are believed to actively interact with immune cells, secrete proinflammatory cytokines, and maintain orbital inflammation in GO.23,24 Evidence shows activation of the p38, Erk, JNK, and NF-κB pathways for expression of proinflammatory genes, including ICAM-1, IL-6, IL-8, and MCP-1.25,26 In accordance with previous studies,27,28 our study showed increased production of proinflammatory cytokines, as well as NF-κB protein, in the IL-1β- and TNF-α-stimulated GO fibroblasts, which was then curtailed with the addition of the GSK-3β inhibitor. According to the literature, GSK-3β is believed to activate NF-κB and produce a proinflammatory effect.29,30 Some studies suggest that IκB kinase, IκB, and p65 are substrates of GSK-3β.31 Other studies have associated GSK-3β with cAMP response element-binding protein (CREB); the suppression of GSK-3β activity is believed to allow CREB to remove the co-activator CREB-binding protein from NF-κB.31–33 The decreased activation of NF-κB, PI3K/Akt, and MAPK signaling pathways as presented in our study may be associated with the ability of GSK-3β inhibitor to contain inflammation in the confluent GO fibroblasts.
Current literature on adipogenesis indicates that the process largely takes place in two phases: the early phase, in which C/EBPα is expressed immediately following the exposure to an adipogenic medium to mediate cell proliferation, and the late phase, in which PPARγ and C/EBPβ are responsible for terminal differentiation of fibroblasts into adipocytes.34 Recently, a study on murine 3T3-L1 preadipocytes found that both stages are regulated by GSK-3β.35 The inhibition of GSK-3β decreased the expression of the transcription factors responsible for the two stages. These results are in accordance with those of our study. Increasing concentrations of CHIR 99021, a highly selective adenosine triphosphate (ATP)-competitive synthetic inhibitor of GSK-3β,36 led to the loss of adipocyte morphology under the microscope. Inhibition of GSK-3β also led to corresponding decreases in PPARγ, C/EBPα, and C/EBPβ in our cell populations. GSK-3β appears to achieve this effect, at least in part, by regulating the expression of Wnt genes. Among the Wnt subtypes, Wnt4 and Wnt5b are promoters of adipogenesis and are downregulated upon inhibition of the enzyme,37 whereas Wnt3a and Wnt10a hinder adipogenesis and are upregulated by CHIR 99021 treatment.38,39 Although in our study we did not investigate the expression of Wnts in detail, GSK-3β inhibition caused upregulation of Wnt3 and increased nuclear translocation of β-catenin that culminated in suppression of adipogenesis. In addition to the canonical Wnt pathway that was examined in our analyses, GSK-3β may also regulate adipogenesis by affecting non-canonical Wnt signaling pathways.35 Studies have shown in the past that activation of non-canonical Wnt pathways by suppression of GSK-3β caused JNK to be phosphorylated and curb adipogenesis.40,41 As such, evidence regarding the importance of Wnt signaling pathways in the pathogenesis of GO is mounting; recently, we found that suppression of the adipogenesis of GO fibroblasts by curcumin involved activation of the canonical Wnt signaling pathway, as well.42
The changes in the phosphorylation of multiple transcription factors upon the addition of GSK-3β inhibitor in our GO fibroblasts indicate that a more complex network of signaling pathways may exist than once believed. Kumar et al.28 discovered that an auto-antibody against thyroid-stimulating hormone receptor stimulated the PI3K/Akt pathway downstream and induced adipogenesis of orbital preadipocytes in GO. They argued in their study that this pathway triggered the terminal stages of adipogenesis. Similarly, the present study showed that GSK-3β inhibition decreased the activation of PI3K and Akt and attenuated adipogenesis in orbital fibroblasts also taken from GO patients. Based on the results of our study, we suspect that GSK-3β may be involved in the feedback regulation of PI3K/Akt signaling. A number of studies have presented results that support our view. In breast cancer cell lines and murine embryonic fibroblasts, the loss of GSK-3β by genetic deletion has been shown to cause reduced activation of Akt.43 Additionally, studies in the past have shown that active GSK-3β regulates the functional antagonist of PI3K, PTEN; it phosphorylates PTEN at Thr262 at the regulatory C-terminal tail, reducing the function of the antagonist.44,45 As the mechanism by which GSK-3β is regulated remains elusive, further studies are necessary to identify the interaction of GSK-3β with other signaling molecules.
Despite the admittedly perplexing role of GSK-3β in a cell system, multiple studies have repeatedly proven the therapeutic benefits of GSK-3β inhibition in a variety of disorders, including neurodegenerative, cognitive, and psychological diseases.46,47 Lithium reversibly binds the enzyme at the magnesium binding site or reduces the activity of the phosphatase that removes a phosphate from the serine 9 residue of the enzyme.48 Another inhibitor, thiadiazolidinone 8, is a non-ATP competitive inhibitor of GSK-3β.49 Apart from psychiatric diseases, for which the two inhibitors are already widely in use,50 they have been tried in Alzheimer's diseases, bone diseases, and inflammatory diseases with limited success.51,52 Even though a number of other GSK-3β inhibitors do exist and have shown promise in many studies, in the context of thyroid diseases no attempts at targeting GSK-3β for treatment have been made, to our knowledge. The results of this study may stimulate future development of clinical trials aimed at testing the effect of GSK-3β inhibition on Graves’ orbitopathy in vivo.
In conclusion, the inhibition of GSK-3β blunted the elevated expression of proinflammatory cytokines in GO fibroblasts and inhibited activation of proinflammatory transcription factors. The GSK-3β inhibitor CHIR 99021 also attenuated differentiation of fibroblasts into adipocyte by regulating both the PPARγ–C/EBPα/β and β-catenin pathways. Future studies are needed to establish the response to GSK-3β inhibitors in vivo and to explore the use of the inhibitor as a therapeutic agent.
Supplementary Material
Acknowledgments
Supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science and ICT (NRF-2016M3A9E9941746).
Disclosure: J.S. Lee, None; M.K. Chae, None; D.O. Kikkawa, Horizon Therapeutics (C); E.J. Lee, None; J.S. Yoon, None
References
- 1. Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010; 362: 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dik WA, Virakul S, van Steensel L. Current perspectives on the role of orbital fibroblasts in the pathogenesis of Graves’ ophthalmopathy. Exp Eye Res. 2016; 142: 83–91. [DOI] [PubMed] [Google Scholar]
- 3. Kuriyan AE, Phipps RP, Feldon SE. The eye and thyroid disease. Curr Opin Ophthalmol. 2008; 19: 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lehmann GM, Feldon SE, Smith TJ, Phipps RP. Immune mechanisms in thyroid eye disease. Thyroid. 2008; 18: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McLachlan SM, Prummel MF, Rapoport B. Cell-mediated or humoral immunity in Graves’ ophthalmopathy? Profiles of T-cell cytokines amplified by polymerase chain reaction from orbital tissue. J Clin Endocrinol Metab. 1994; 78: 1070–1074. [DOI] [PubMed] [Google Scholar]
- 6. Xu J, Liao K. Protein kinase B/AKT 1 plays a pivotal role in insulin-like growth factor-1 receptor signaling induced 3T3-L1 adipocyte differentiation. J Biol Chem. 2004; 279: 35914–35922. [DOI] [PubMed] [Google Scholar]
- 7. Ko J, Kim JY, Lee EJ, Yoon JS. Inhibitory effect of idelalisib, a selective phosphatidylinositol 3-kinase delta inhibitor, on adipogenesis in an in vitro model of Graves’ orbitopathy. Invest Ophthalmol Vis Sci. 2018; 59: 4477–4485. [DOI] [PubMed] [Google Scholar]
- 8. Wang L, Wang Y, Zhang C, et al.. Inhibiting glycogen synthase kinase 3 reverses obesity-induced white adipose tissue inflammation by regulating apoptosis inhibitor of macrophage/CD5L-mediated macrophage migration. Arterioscler Thromb Vasc Biol. 2018; 38: 2103–2116. [DOI] [PubMed] [Google Scholar]
- 9. Lappas M. GSK3β is increased in adipose tissue and skeletal muscle from women with gestational diabetes where it regulates the inflammatory response. PLoS One. 2014; 9: e115854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoon JS, Lee HJ, Choi SH, Chang EJ, Lee SY, Lee EJ. Quercetin inhibits IL-1β-induced inflammation, hyaluronan production and adipogenesis in orbital fibroblasts from Graves’ orbitopathy. PLoS One. 2011; 6: e26261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 12. Kim CY, Lee HJ, Chae MK, Byun JW, Lee EJ, Yoon JS. Therapeutic effect of resveratrol on oxidative stress in Graves’ orbitopathy orbital fibroblasts. Invest Ophthalmol Vis Sci. 2015; 56: 6352–6361. [DOI] [PubMed] [Google Scholar]
- 13. Kim SE, Lee JH, Chae MK, Lee EJ, Yoon JS. The role of sphingosine-1-phosphate in adipogenesis of Graves’ orbitopathy. Invest Ophthalmol Vis Sci. 2016; 57: 301–311. [DOI] [PubMed] [Google Scholar]
- 14. Valyasevi RW, Erickson DZ, Harteneck DA, et al.. Differentiation of human orbital preadipocyte fibroblasts induces expression of functional thyrotropin receptor. J Clin Endocrinol Metab. 1999; 84: 2557–2562. [DOI] [PubMed] [Google Scholar]
- 15. Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975; 5: 19–27. [DOI] [PubMed] [Google Scholar]
- 16. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001; 65: 391–426. [DOI] [PubMed] [Google Scholar]
- 17. Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992; 1114: 147–162. [DOI] [PubMed] [Google Scholar]
- 18. Rao AS, Kremenevskaja N, Resch J, Brabant G. Lithium stimulates proliferation in cultured thyrocytes by activating Wnt/beta-catenin signalling. Eur J Endocrinol. 2005; 153: 929–938. [DOI] [PubMed] [Google Scholar]
- 19. Ago K, Saegusa Y, Nishimura J, et al.. Involvement of glycogen synthase kinase-3beta signaling and aberrant nucleocytoplasmic localization of retinoblastoma protein in tumor promotion in a rat two-stage thyroid carcinogenesis model. Exp Toxicol Pathol. 2010; 62: 269–280. [DOI] [PubMed] [Google Scholar]
- 20. Zhang J, Gill AJ, Issacs JD, et al.. The Wnt/beta-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum Pathol. 2012; 43: 1044–1050. [DOI] [PubMed] [Google Scholar]
- 21. Yang G, Fu Y, Lu X, Wang M, Dong H, Li Q. The interactive effects of genetic polymorphisms within LFA-1/ICAM-1/GSK-3β pathway and environmental hazards on the development of Graves’ opthalmopathy. Exp Eye Res. 2018; 174: 161–172. [DOI] [PubMed] [Google Scholar]
- 22. Shen L, Huang F, Ye L, et al.. Circulating microRNA predicts insensitivity to glucocorticoid therapy in Graves’ ophthalmopathy. Endocrine. 2015; 49: 445–456. [DOI] [PubMed] [Google Scholar]
- 23. Weetman AP, Cohen S, Gatter KC, Fells P, Shine B. Immunohistochemical analysis of the retrobulbar tissues in Graves’ ophthalmopathy. Clin Exp Immunol. 1989; 75: 222–227. [PMC free article] [PubMed] [Google Scholar]
- 24. Yang D, Hiromatsu Y, Hoshino T, Inoue Y, Itoh K, Nonaka K. Dominant infiltration of T(H)1-type CD4+ T cells at the retrobulbar space of patients with thyroid-associated ophthalmopathy. Thyroid. 1999; 9: 305–310. [DOI] [PubMed] [Google Scholar]
- 25. Zhao LQ, Wei RL, Cheng JW, Cai JP, Li Y. The expression of intercellular adhesion molecule-1 induced by CD40-CD40L ligand signaling in orbital fibroblasts in patients with Graves’ ophthalmopathy. Invest Ophthalmol Vis Sci. 2010; 51: 4652–4660. [DOI] [PubMed] [Google Scholar]
- 26. Hwang CJ, Afifiyan N, Sand D, et al.. Orbital fibroblasts from patients with thyroid-associated ophthalmopathy overexpress CD40: CD154 hyperinduces IL-6, IL-8, and MCP-1. Invest Ophthalmol Vis Sci. 2009; 50: 2262–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hiromatsu Y, Yang D, Bednarczuk T, Miyake I, Nonaka K, Inoue Y. Cytokine profiles in eye muscle tissue and orbital fat tissue from patients with thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2000; 85: 1194–1199. [DOI] [PubMed] [Google Scholar]
- 28. Kumar S, Bahn RS. Relative overexpression of macrophage-derived cytokines in orbital adipose tissue from patients with Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2003; 88: 4246–4250. [DOI] [PubMed] [Google Scholar]
- 29. Klamer G, Song E, Ko KH, O'Brien TA, Dolnikov A. Using small molecule GSK3beta inhibitors to treat inflammation. Curr Med Chem. 2010; 17: 2873–2881. [DOI] [PubMed] [Google Scholar]
- 30. Cheng YL, Wang CY, Huang WC, et al.. Staphylococcus aureus induces microglial inflammation via a glycogen synthase kinase 3β-regulated pathway. Infect Immun. 2009; 77: 4002–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock. 2007; 27: 113–123. [DOI] [PubMed] [Google Scholar]
- 32. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005; 6: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nat Immunol. 2005; 6: 751–752. [DOI] [PubMed] [Google Scholar]
- 34. Kim AR, Yoon BK, Park H, et al.. Caffeine inhibits adipogenesis through modulation of mitotic clonal expansion and the AKT/GSK3 pathway in 3T3-L1 adipocytes. BMB Rep. 2016; 49: 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang L, Wang Y, Meng Y, Zhang C, Di L. GSK3-activated STAT5 regulates expression of SFRPs to modulate adipogenesis. FASEB J. 2018; 32: 4714–4726. [DOI] [PubMed] [Google Scholar]
- 36. Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011; 4: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishizuka M, Koyanagi A, Osada S, Imagawa M. Wnt4 and Wnt5a promote adipocyte differentiation. FEBS Lett. 2008; 582: 3201–3205. [DOI] [PubMed] [Google Scholar]
- 38. Bennett CN, Ross SE, Longo KA, et al.. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002; 277: 30998–31004. [DOI] [PubMed] [Google Scholar]
- 39. Kennell JA, MacDougald OA. Wnt signaling inhibits adipogenesis through beta-catenin-dependent and -independent mechanisms. J Biol Chem. 2005; 280: 24004–24010. [DOI] [PubMed] [Google Scholar]
- 40. Camp HS, Tafuri SR, Leff T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-gamma1 and negatively regulates its transcriptional activity. Endocrinology. 1999; 140: 392–397. [DOI] [PubMed] [Google Scholar]
- 41. Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007; 282: 22910–22920. [DOI] [PubMed] [Google Scholar]
- 42. Lee JS, Kim J, Lee EJ, Yoon JS. Therapeutic effect of curcumin, a plant polyphenol extracted from Curcuma longae, in fibroblasts from patients with Graves’ orbitopathy. Invest Ophthalmol Vis Sci. 2019; 60: 4129–4140. [DOI] [PubMed] [Google Scholar]
- 43. Lu Y, Muller M, Smith D, et al.. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011; 30: 4567–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005; 280: 35195–35202. [DOI] [PubMed] [Google Scholar]
- 45. Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phosphorylation on Thr366. Biochem J. 2007; 405: 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013; 288: 1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beurel E, Kaidanovich-Beilin O, Yeh WI, et al.. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J Immunol. 2013; 190: 5000–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003; 24: 441–443. [DOI] [PubMed] [Google Scholar]
- 49. Castro A, Encinas A, Gil C, et al.. Non-ATP competitive glycogen synthase kinase 3β (GSK-3β) inhibitors: study of structural requirements for thiadiazolidinone derivatives. Bioorg Med Chem. 2008; 16: 495–510. [DOI] [PubMed] [Google Scholar]
- 50. Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry. 2004; 55: 781–784. [DOI] [PubMed] [Google Scholar]
- 51. Martin R, Yu K. Assessing performance of prediction rules in machine learning. Pharmacogenomics. 2006; 7: 543–550. [DOI] [PubMed] [Google Scholar]
- 52. Wang H, Brown J, Gu Z, et al.. Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-β-signaling pathways regulates the innate inflammatory response. J Immunol. 2011; 186: 5217–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.