

Abstract
Advances in genomics over the past two decades have allowed for the elucidation of the genetic alterations leading to the development adrenocortical tumors and/or hyperplasias. These molecular changes were initially discovered through the study of rare familial tumor syndromes such as McCune-Albright Syndrome, Carney complex, Li-Fraumeni syndrome, and Beckwith-Wiedemann syndrome with identification of alterations in genes and molecular pathways that subsequently lead to the discovery of aberrations in these or related genes and pathways in sporadic tumors. Genetic alterations in GNAS, PRKAR1A, PRKACA, PRKACB, PDE11A, and PDE8B, leading to aberrant cyclic adenosine monophosphate-protein kinase A signaling were identified as playing a major role in the development of benign cortisol-producing adrenocortical tumors and/or hyperplasias, whereas genetic defects in KCNJ5, ATP1A1, ATP2B3, CACNA1D, CACNA1H, and CLCN2 were implicated in the development of benign aldosterone-producing tumors and/or hyperplasias through modification of intracellular calcium signaling. Germline ARMC5 defects were found to cause the development of primary bilateral macronodular adrenal hyperplasia with cortisol and/or aldosterone oversecretion. Adrenocortical carcinoma was linked primarily to aberrant Wnt-β-catenin signaling, p53 signaling, and/or IGF2 overexpression, with frequent genetic alterations in TP53, ZNRF3, CTNNB1, and 11p15. This review focuses on the genetic underpinnings of benign cortisol- and aldosterone-producing adrenocortical tumors and adrenocortical carcinoma.
Keywords: Adrenocortical adenoma, adrenocortical hyperplasia, adrenocortical carcinoma, Cushing syndrome, primary aldosteronism
Introduction
Primary adrenocortical tumors and hyperplasias are predominantly due to benign adenomas and/or hyperplasias and less frequently carcinomas, with these tumors being further classified as unilateral or bilateral, secreting or non-secreting, and sporadic or familial. Advances in genomics over recent years has allowed for the elucidation of the genetic underpinnings of adrenocortical tumorigenesis, initially through the study of rare familial tumor syndromes. Aberrant cAMP-protein kinase A signaling was initially implicated in the development of Cushing syndrome in McCune-Albright syndrome through early embryonic postzygotic somatic activating defects in the GNAS gene, with subsequent discovery of inactivating germline PRKAR1A defects as the causative genetic alteration in Carney complex and primary pigmented nodular adrenocortical disease. Activating somatic PRKACA defects were later found to be a major cause of cortisol-producing adrenocortical adenomas. Additionally, germline defects in ARMC5, a tumor suppressor gene, were identified as the primary underlying genetic alteration in primary bilateral macronodular adrenal hyperplasia (PBMAH). The pathogenesis of primary aldosteronism was linked to aberrant intracellular calcium signaling with genes encoding ion channels such as KCNJ5, CLCN2, CACNA1H, CACNA1D, and ATPases such as ATP1A1 and ATP2B3, implicated in adrenocortical tumorigenesis. Lastly, germline defects in TP53 were identified in Li-Fraumeni syndrome, whereas molecular alterations affecting IGF2 expression were implicated in the pathogenesis of Beckwith-Wiedemann syndrome, two familial tumor syndromes, amongst others, associated with adrenocortical carcinomas. Further studies have identified somatic molecular alterations affecting p53 and Wnt-β-catenin signaling, and IGF2 expression as major drivers of adrenocortical carcinoma development. This review aims to describe the reported genomic alterations in secreting adrenocortical tumors with a focus on cortisol- and aldosterone-producing tumors and adrenocortical carcinoma.
Benign Cortisol-Producing Adrenocortical Adenomas and Hyperplasias
Cushing’s syndrome (CS) is a compilation of signs and symptoms that develop as a result of prolonged tissue exposure to excess glucocorticoids, with an incidence in European-based population studies of 2 to 3 cases per 1 million inhabitants per year [1, 2]. Approximately 20-30% of cases of endogenous CS are the result of primary adrenocortical hyperfunction which is predominantly due to benign-cortisol producing adrenocortical tumors (ACTs) such as cortisol-producing adenomas (CPAs) (10-15%) or adrenocortical hyperplasia which is almost always bilateral (10%), and less commonly due to adrenocortical carcinoma (ACC) (less than 5%) [3]. Bilateral adrenocortical hyperplasia (BAH) is characterized by multiple adrenocortical nodules and can be divided into two main groups according to the histopathological size of the majority of the nodules, with micronodular adrenocortical hyperplasia describing BAH with nodules less than 1 centimeter in diameter, and macronodular adrenocortical hyperplasia, such as PBMAH, describing BAH with nodules greater than 1 centimeter in diameter (Table 1). BAH can be further subdivided according to the presence of atrophy or hyperplasia of the internodular cortex, or the presence of pigment (lipofuscin) within the nodules or in the surrounding adrenal cortex [4]. The bilateral nature of the adrenocortical tumors in BAH suggests an underlying genetic predisposition. Recently a gene-based classification for BAH in which patients are grouped according to the presence of disease-causing germline genetic alterations or other genetic changes was proposed (Figure 1) [5]. This classification focuses on the causative genes for each form of BAH, listing the implicated gene name before the type of hyperplasia with the goal of guiding genetic testing, counseling, and treatment as well as decreasing the risk of misclassification of familial cases as sporadic.
Table 1.
Classification and characteristics of primary cortisol-producing adrenocortical hyperplasia.*
| Adrenocortical lesions |
Genes (locus) | Histolopathology | Characteristics |
|---|---|---|---|
| PBMAH |
ARMC5 (16p 11.2) MEN1 (11 q13) FH (1q42.3-43) APC (5q22.2) PDE11A (2q31.2) PDE8B (5q13.3) GNAS (20q13) PRKACA duplication (19p13.1) |
-Nodules or macroadenomatous, > 1cm, with (type 1) or without (type 2) internodular atrophy. -Hyperplasia with dominant nodule. |
-Middle age, mild hypercortisolism and/or mineralocorticoid excess -Associated with MEN-1, FAP, MAS, HLRCC, isolated (AD) -Most lesions have aberrant GPCRs (vasopressin, serotonin, catecholamines, GIP, luteinizing hormone) -PMAH carry the ability of intra-adrenal production of ACTH with an autocrine/paracrine effect on glucocorticoid or mineralocorticoid production |
| PBAD | GNAS (20q13; mosaic) | -Distinct adenomas (>1cm), with occasional microadenomas and internodular atrophy | -Infants and very young children -MAS |
| FDCS (GIP-depedent) | GIPR gene (19q13.32) duplication | -Large adenomas and/or macronodules | -Isolated or familial -Aberrant GPCRs (GIPR) -Low fasting cortisol, hypercortisolism post-meals |
| i-PPNAD |
PRKAR1A (17q22-24) PRKACA duplications (19p13.1) |
-Microadenomatous (<1cm) hyperplasia with pigmentation | -Children and young Adults -Lentiginosis in few cases |
| c-PPNAD | PRKAR1A (17q22-24, CNC1 locus) 2p16 (CNC2 locus, unkown gene) | -Microadenomatous (<1cm) hyperplasia with (mostly) internodular atrophy and pigmentation | -Children, young and middle aged adults -Disease at a younger age and a higher frequency of myxomas, schwannomas, and thyroid and gonadal tumors than patients without PRKAR1A variants. -In-frame deletion of exon 3 and the c.708 +1G>T appears to confer a more severe CNC phenotype, while the splice variant c.709(−7-2)del6 and the initiation alternating substitution c.1A>G/p.M1Vp has been associated with incomplete penetrance of CNC, as seen in i-PPNAD -CNC1: The hot spot c.491-492delTG is most closely associated with lentigines, cardiac myxoma, and thyroid tumors when opposed to all other PRKAR1A variants -Expressed RIα mutant protein present with more severe and aggressive CNC-phenotype -CNC2: Sporadic disease later in life with a lower frequency of myxomas, schwannomas, thyroid and LCCSCT |
| i-MAD |
PDE11A (2q31.2) PDE8B (5q13) PRKACA (19p13.1) 2p16 (unkown gene) |
-Microadenomatous (<1cm) hyperplasia with internodular hyperplasia and limited or absent pigmentation | -Mostly children and young adults -Cyclical hypercortisolism -May be associated with a paradoxical rise of glucocorticoid excretion during the Liddle's test -Isolated or AD |
Abbreviations: APC, adenomatous polyposis coligene; c-PPNAD, CNC-associated primary pigmented nodular adrenocortical disease; CNC, Carney complex; FAP, familial adenomatous polyposis; FDCS, food-dependent Cushing syndrome; GNAS, gene coding for the stimulatory subunit α of the G-protein (Gsα); GPCR, G-protein-coupled receptor; HLRCS, hereditary leiomyomatosis and renal cancer syndrome; i-MAD, isolated micronodular adrenocortical disease; i-PPNAD, isolated PPNAD; LCCSCT, large cell calcifying Sertoli cell tumor; MAS, McCune–Albright syndrome; MEN1, multiple endocrine neoplasia type 1; PBAD, primary bimorphic adrenocortical disease; PBNMAH, primary bilateral macronodular adrenocortical hyperplasia; PDE8B, phosphodiesterase 8B gene; PDE11A, phosphodiesterase 11A gene; PRKAR1A, protein kinase, cAMP-dependent, regulatory, type I, α gene
Adapted from Hannah-Shmouni F, Stratakis CA: A Gene-Based Classification of Primary Adrenocortical Hyperplasias. Hormone and Metabolic Research 2020, 52(3): 133-141.
Figure 1. Gene-based diagnostic algorithm for primary cortisol-producing adrenocortical hyperplasias.
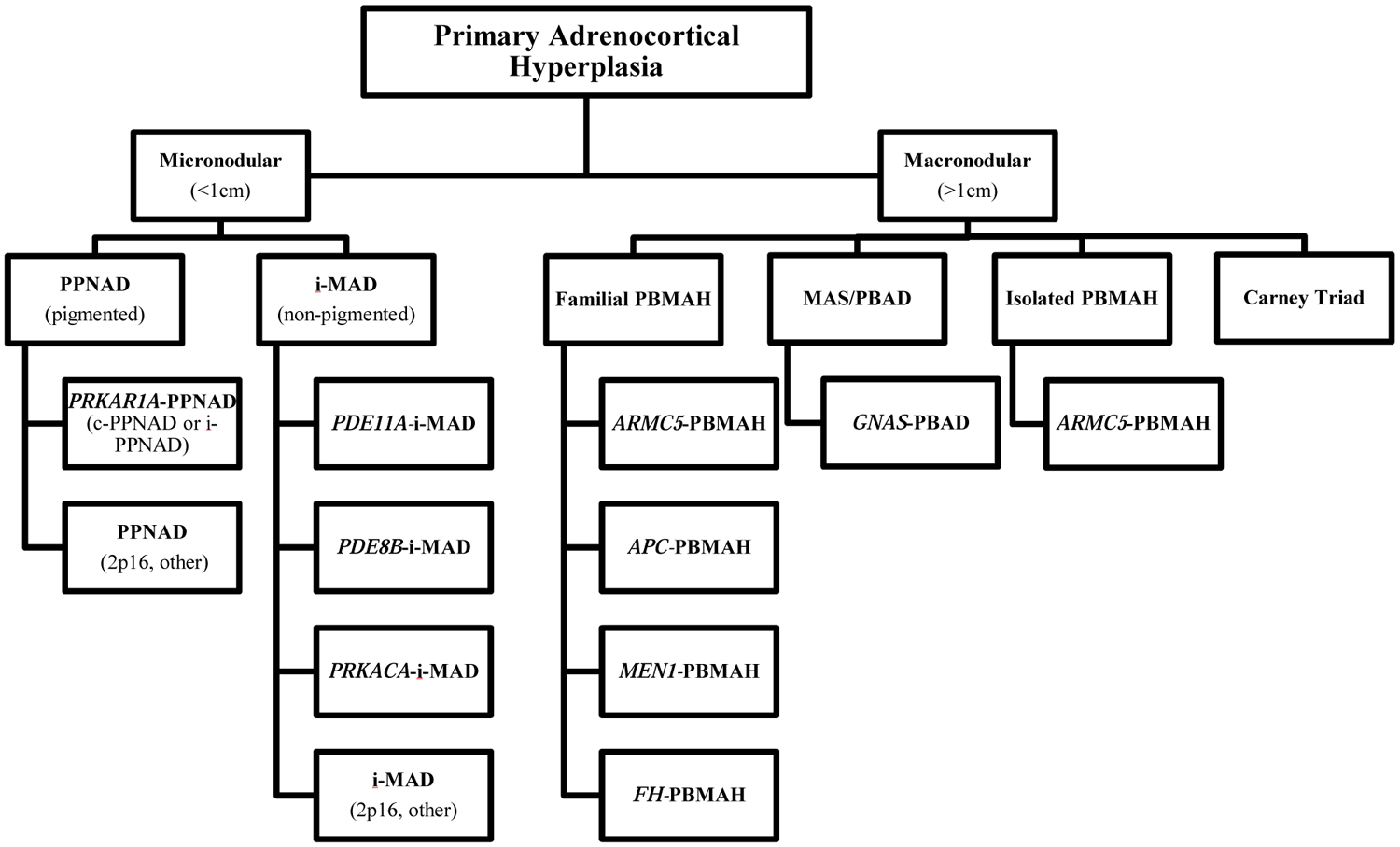
Abbreviations: APC, adenomatous polyposis coligene; ARMC5, armadillo repeat-containing protein 5; c-PPNAD, CNC-associated primary pigmented nodular adrenocortical disease; FH, fumarate hydratase; GNAS, gene coding for the stimulatory subunit α of the G-protein (Gsα); i-MAD, isolated micronodular adrenocortical disease; i-PPNAD, isolated PPNAD; MAS, McCune–Albright syndrome; MEN1, multiple endocrine neoplasia type 1; PBAD, primary bimorphic adrenocortical disease; PBMAH, primary bilateral macronodular adrenocortical hyperplasia; PDE8B, phosphodiesterase 8B gene; PDE11A, phosphodiesterase 11A gene PPNAD, primary pigmented nodular adrenocortical disease; PRKACA, protein kinase, cAMP-dependent, catalytic, alpha; PRKAR1A, protein kinase, cAMP-dependent, regulatory, type I, α gene.
Aberrant cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling has been shown to play a key role in the development of most benign cortisol-producing adrenocortical tumors. The cAMP-PKA pathway is fundamental in regulating adrenocortical cell development, proliferation, and function. This pathway is activated by the binding of ACTH to the ACTH receptor, a G-protein coupled receptor (GPCR) on the adrenocortical cell membrane that is encoded by the MC2R gene. This catalyzes exchange of guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on the α subunit of the associated heterotrimeric G protein, with dissociation of the α subunit (encoded by GNAS) from the βγ dimer. The α subunit then binds to adenylyl cyclase (AC) and activates its enzymatic activity, with activated AC producing cAMP from adenosine triphosphate (ATP). Subsequently, cAMP binds to the regulatory subunits (R) of PKA which allows for release of its catalytic subunits (C), that then mediate serine-threonine phosphorylation of target molecules, including the transcription factor CREB (cAMP response element-binding protein). PKA is a ubiquitous cAMP-dependent serine-threonine kinase that is involved in the regulation of a wide range of cellular processes including transcription, metabolism, cell cycle progression, and apoptosis. Four isoforms exist for both the regulatory subunits (R1α, R1β, R2α, and R2β) and catalytic subunits (Cα, Cβ, Cγ, and PRKX) of PKA, with each isoform having individual localization and specificity [6]. Phosphodiesterases (PDEs), which are the only known enzymes that degrade cyclic nucleotides, play a crucial role in the regulation of the cAMP-PKA pathway through regulating cAMP levels. The involvement of aberrant cAMP-PKA signaling in adrenocortical tumorigenesis was first described in 1991 when the genetic cause of McCune-Albright syndrome (MAS) was discovered to be early embryonic postzygotic somatic activating defects of the GNAS gene. This gene encodes the α-subunit of the stimulatory G protein (Gsα), with mosaic gain-of-function mutations leading to constitutive activation of the cAMP-PKA pathway [7]. MAS is comprised of café-au-lait skin macules, skeletal fibrous dysplasia, and multiple endocrinopathies, including precocious puberty, testicular and thyroid lesions, phosphate wasting, growth hormone excess, and, rarely, neonatal hypercortisolism [8]. CS in MAS results primarily from bilateral adrenocortical hyperplasia that develops from adrenocortical cells with fetal features and is termed primary bimorphic adrenocortical disease (PBAD) [9].
MICRONODOULAR ADRENOCORTICAL HYPERPLASIA
Micronodular BAH may be familial or sporadic and can be divided into at least two subgroups: primary pigmented nodular adrenocortical disease (PPNAD) and isolated micronodular adrenocortical disease (i-MAD). In PPNAD adrenocortical nodules are pigmented with surrounding atrophic adrenocortical tissue, whereas in i-MAD pigment is limited or absent and hyperplasia is present the surrounding zona fasciculata [10]. PPNAD is more common than i-MAD, and is associated with Carney complex (CNC) in 90% of cases (c-PPNAD) though it may be isolated (i-PPNAD) [11, 12]. Both PPNAD and i-MAD can be inherited in an autosomal dominant manner, however, PPNAD is more frequently familial.
CNC was the first disease identified to be caused by a defect in a gene encoding a component of the PKA enzyme. It is a rare, autosomal dominant, multiple endocrine neoplasia and lentiginosis syndrome that is familial in 70% of cases, and is comprised of abnormal cutaneous and mucosal pigmentation, myxomas predominantly of the heart, skin, and breast, endocrine neoplasms, psammomatous melanotic schwannomas, breast ductal adenomas, osteochondromyxomas, and other non-endocrine tumors. The most common endocrine neoplasm in CNC is c-PPNAD, which occurs in 25-60% of patients and has been found in almost all patients on autopsy. Other endocrine tumors include pituitary adenomas or hyperplasia, thyroid tumors, and gonadal tumors [13-15]. Germline inactivating defects in PRKAR1A gene are found in 37% of patients with sporadic CNC and more than 70% of patients with familial CNC, with almost 100% penetrance. The PRKAR1A gene is situated at the 17q24.2-24.3 locus (CNC1 locus) of the long arm of chromosome 17 and encodes the regulatory subunit type 1α (R1α) of PKA [11, 16, 17]. Inactivating defects of PRKAR1A lead to constitutive activation of the cAMP-PKA pathway through loss of regulation of the catalytic subunits of PKA. Most pathogenic PRKAR1A variants are subject to mRNA nonsense-mediated decay of the mutant sequence, leading to predicted absence of mutant protein products in affected cells and PRKAR1A haploinsufficiency [11, 18]. Initial data suggested that PRKAR1A functions as a “classic” tumor-suppressor gene, with tumors from CNC patients exhibiting germline mutations and subsequent loss of heterozygosity at the PRKAR1A locus. However, some data demonstrate that haploinsufficiency of PRKAR1A may be sufficient for increased PKA activity and the early development of certain tumors [19, 20]. In the majority of the remaining cases of CNC that do not harbor a germline PRKAR1A defect, genetic linkage analysis of tumors has revealed a second affected locus on chromosome 2p16 (CNC2 locus) [21, 22]. The responsible gene at this locus has not yet been identified. Copy number gains on chromosome 1 of the PRKACB gene locus, encoding the catalytic subunit β (Cβ) of PKA, were also found in a single patient with CNC that presented with abnormal skin pigmentation, myxomas, and acromegaly, though defects in PRKACB have not been linked to c-PPNAD. This patient had increased levels of Cβ in her lymphocytes, fibroblasts, and in a breast myxoma with cAMP-induced increased kinase activity in her lymphocytes similar to that noted in CNC due to PRKAR1A defects [23]. PRKAR1A mutations also cause i-PPNAD, where there is a genotype-phenotype correlation, as demonstrated in a study that included 353 patients with germline PRKAR1A defects or a diagnosis of CNC and/or PPNAD, where among patients with i-PPNAD and PRKAR1A mutations, most had a germline c.709-7del6 mutation whereas the remainder of these patients carried the p.Met1Val mutation. Patients that were younger than 8 years old were only rarely found to have PRKAR1A defects [11]. In addition to PPNAD, somatic mutations in PRKAR1A have less frequently been described in other cortisol-producing adrenal tumors, including CPAs and ACCs [24]. ACC has also been reported in two patients with CNC and c-PPNAD due to PRKAR1A defects [24, 25]. Hypercortisolism can be rarely ACTH-dependent in CNC, from a corticotropinoma [26].
Defects in genes encoding cyclic nucleotide PDEs have also been implicated in the pathogenesis of micronodular BAH and other cortisol producing ACTs. This was demonstrated in patients with i-MAD or i-PPNAD not caused by known genetic defects (mutations in GNAS or PRKAR1A) through a single-nucleotide polymorphism-based genome-wide association study that included both leukocyte and tumor DNA. In this study mutations in genetic loci harboring PDE genes were most likely to be associated with the disease, with inactivating mutations of the PDE11A gene, encoding phosphodiesterase type 11A, being the most frequently linked, followed by the PDE8B gene, which encodes phosphodiesterase type 8B. Tumor specimens most often demonstrated loss of heterozygosity in the 2q31–2q35 region (the location of PDE11A) with decreased protein expression as well as evidence of increased cAMP-PKA signaling with high cyclic nucleotide levels and cAMP-responsive element binding protein (CREB) phosphorylation. [27]. A higher frequency of PDE11A variants has also been noted in patients with CNC. Among patients with CNC and PRKAR1A defects, those with PPNAD and/or testicular large-cell calcifying Sertoli cell tumors (LCCSCT) were more commonly carriers of PDE11A germline variants compared to those without PPNAD and/or LCCSCT, suggesting that PDE11A may be a genetic modifying factor for the development of adrenal and testicular tumors in this population [28]. PDE11A heterozygous germline defects have also been described in one non-secreting adrenocortical adenoma and were found to be more frequent in patients with other ACTs as compared to age and/or sex-matched controls including ACC, CPAs, and PBMAH [29]. PDE11A- inactivating mutations may additionally play a role in the development of prostate cancer and testicular germ cell tumors [28, 30, 31]. A single germline PDE8B missense substitution was initially reported in a pediatric patient with i-MAD and CS who inherited the mutation from her father [32]. Her father was not diagnosed with CS but did have elevated midnight cortisol. In a subsequent case-control study of 216 unrelated patients with adrenocortical tumors (including PPNAD, PBMAH, CPAs, non-secreting adrenocortical tumors, and ACC) and 192 controls, nine different PDE8B sequence changes were identified in the patients and controls, with two variations that were seen only in the patient group, demonstrating significant potential to impair protein function in vitro and in silico [33].
Genetic alterations involving genes encoding the catalytic subunits of PKA have also been linked to micronodular BAH. In addition to the aforementioned patient with CNC due to germline PRKACB copy number gains, germline copy number gains resulting in amplification of PRKACA, which encodes the catalytic subunit (Cα) of PKA, have been implicated in the development of i-MAD. Germline copy number gains of the genomic region on chromosome 19p that includes the entire PRKACA gene were initially described in three patients with sporadic i-MAD, as well as 2 patients with familial PBMAH. Tumor tissues from patients with PRKACA copy number gains had higher PKA Cα mRNA and protein levels with associated higher basal and cAMP stimulated PKA activity [34, 35].
Finally, the wingless-type (Wnt)–β-catenin pathway has also been implicated in the development of micronodular BAH as a possible genetic modifier. The key player in this pathway is the cytoplasmic protein β-catenin, the stability of which is regulated by the Axin complex. In the absence of Wnt, β-catenin is continually degraded by the action of the Axin complex. This complex is comprised of the scaffolding protein Axin and the tumor suppressor adenomatous polyposis coli (APC) gene product, as well as two kinases, casein kinase 1 (CK1) and glycogen synthase kinase 3 (GSK3). Axin and APC bind newly synthesized β-catenin while CKI and GSK3 sequentially phosphorylate the amino terminal region of this protein resulting in β-catenin recognition by β-Trcp, a E3 ubiquitin ligase subunit, with resulting β-catenin ubiquitination and proteasomal degradation. This prevents β-catenin from translocating to the nucleus, with subsequent repression of Wnt target genes by the DNA-bound T cell factor/lymphoid enhancer factor (TCF/LEF) family of proteins. When the Wnt-β-catenin pathway is activated through binding of Wnt ligands to a receptor complex including a member of the frizzled family of seven-transmembrane receptors and a member of the LDL receptor family (LRP 5 or 6), the Axin complex is inhibited. This leads to stabilization of β-catenin which accumulates and translocates to the nucleus to activate TCF/LEF transcription factors thereby activating Wnt target gene expression [36]. Somatic beta-catenin gene (CTNNB1) defects were identified in 2 of 18 (11%) of patients with PPNAD in one study (a germline PRKAR1A mutation was identified in 1 of the 2 patients with somatic CTNNB1 mutations). These defects occurred in relatively large adrenocortical adenomas that had formed in the background of PPNAD and were absent in the surrounding hyperplastic adrenocortical tissue [37]. In another study, involving tissue from 9 subjects with PPNAD (with 8 of the 9 harboring PRKAR1A defects), including 5 with macronodules, β-catenin accumulation was found in all PPNAD tissues including within the macronodules, micronodules, and internodular tissue, whereas activating somatic CTNNB1 mutations were found in 2 of the 5 macronodules but not in the micronodules or in the contralateral adrenal gland [38].
MACRONODULAR ADRENOCORTICAL HYPERPLASIA
PBMAH, which has also been called bilateral macronodular adrenal hyperplasia (BMAH), ACTH-independent macronodular adrenal hyperplasia (AIMAH), primary macronodular adrenal hyperplasia (PMAH), massive macronodular adrenocortical disease (MMAD), autonomous macronodular adrenal hyperplasia (AMAH), ACTH-independent massive bilateral adrenal disease (AIMBAD), and "giant" or "huge" macronodular adrenal disease, is a rare cause of adrenal CS (<2%) that is most commonly isolated or sporadic and inherited in an autosomal dominant manner when hereditary. Macronodular BAH was previously deemed to be an ACTH-independent process and thus called AIMAH; however, studies have shown that adrenal cortisol secretion in PBMAH may be partially regulated through ACTH secretion from clusters of adrenocortical cells by paracrine action [39]. PBMAH can be sub-grouped into two histologic categories: type I PBMAH and, more commonly, type II PBMAH, with type I PBMAH having internodular atrophic tissue, and type II PBMAH showing diffuse hyperplasia with no residual normal or atrophic surrounding tissue [40]. In 77-87% of cases of PBMAH, adrenocortical cells express aberrant (ectopic or excessive) hormone receptors that are primarily members of the GPCR family and are linked to steroidogenesis, leading to significant elevation in plasma cortisol after stimulation of these receptors. They include receptors for glucose-dependent insulinotropic peptide (GIP) leading to food-dependent CS, vasopressin, β-adrenergic agonists, luteinizing hormone/choriogonadotropin (LH/hCG), serotonin, angiotensin II, and glucagon [41-51]. Aberrant receptors have been identified less frequently in adrenocortical adenomas and ACC [40]. The genetic alterations causing this ectopic receptor expression have not yet been well elucidated. A study investigating the molecular pathogenesis of ectopic GIP receptor (GIPR) expression in food dependent CS, a rare subtype of macronodular BAH, that included adrenal tissue from CPAs and macronodular BAH demonstrated that GIPR expression occurred through transcriptional activation of a single allele of the GIPR gene, with 3 of 15 tumor samples demonstrating somatic duplications in the chromosome region 19q13.32 containing the GIPR locus. The duplicated 19q13.32 region was rearranged with other chromosome regions in two of the samples that were consistent with CPAs. These chromosome rearrangements did not result in gene fusion, but rather placed the GIPR gene in a genomic environment near cis-acting regulatory regions favoring transcriptional activation. In the third sample that was consistent with macronodular BAH, 19q duplication without chromosome rearrangement was identified [52].
As in micronodular BAH, increased cAMP-PKA signaling also plays a role in the development of PBMAH. This is evident in cases of PBMAH with ectopic receptor expression as most of these receptors are GPCRs that stimulate AC, with resulting increased cAMP-PKA signaling. As aforementioned, germline PDE11A variants have also been implicated in PBMAH with a prevalence of 24-28%, as have germline PDE8B mutations, somatic GNAS mutations without MAS, and PRKACA copy number gains [29, 33, 40, 53]. In MAS, CS due to PBAD, another subtype of macronodular BAH has been described [9, 54]. Although PRKAR1A mutations have not been described in PBMAH, somatic losses of the 17q22–24 region in PBMAH lead to PKA subunit and enzymatic activity changes, which is indicative that PKA signaling is altered in PBMAH similarly to that of other adrenal tumors with PRKAR1A defects or 17q losses [55]. A single case of PBMAH has been described due to 2 mutations in the same allele of the MC2R gene encoding the ACTH or melanocortin 2 receptor, which resulted in clinical hypersensitivity to ACTH through constitutive activation of the cAMP-PKA pathway. Either mutation alone (p.C21R and p.S247G mutation) would have produced an inactive receptor with loss of ligand binding and responsiveness; however, it appears that the presence of both mutations in the same molecule resulted in a receptor with constitutive activity, and the co-expression of the normal MC2R allele lead to retention of a normal response to ACTH [56].
Rarely, PBMAH is a component of autosomal dominant multiple tumor syndromes, such as familial adenomatous polyposis (FAP), multiple endocrine neoplasia type 1 (MEN1), or hereditary leiomyomatosis and renal cell carcinoma (HLRCC) [57-59]. These predominantly were the only genetic defects linked to PBMAH until inactivating mutations of the ARMC5 gene were discovered in this disease [60]. FAP is caused by germline inactivating defects in the adenomatous polyposis coli (APC) tumor suppressor gene, which encodes the APC protein that comprises the β-catenin degradation complex and acts as a negative regulator of the Wnt-β-catenin signaling pathway. Mutations of both APC alleles in a single cell result in the absence of functional APC protein, aberrant accumulation of beta-catenin, and transcriptional activation of the Wnt-signaling pathway. Classic FAP is characterized by multiple colorectal adenomas (>100) that predispose to colorectal cancer as well as fundic gland polyps and duodenal adenomas with increased risk of duodenal cancer. Extraintestinal manifestations may include but are not limited to follicular or papillary thyroid cancer, childhood hepatoblastoma, central nervous system tumors, desmoid tumors, sebaceous or epidermoid cysts, lipomas, osteomas, fibromas, supernumerary teeth, and juvenile nasopharyngeal angiofibromas. Adrenal masses, including PBMAH, adrenocortical adenomas, and ACC, have also been described in FAP, with 16% of patients in one cohort having adrenal masses of which 97% were benign and 80% were adenomas, with 23% of the adrenal masses being bilateral. The median diameter of these adrenal masses at diagnosis was 1.7 cm (interquartile range (IQR) 1.4–3.0) with median maximal diameter of 2.5 cm (IQR 1.7—4.1) [61].
MEN1 is characterized by a predisposition to a multitude of endocrine neoplasms primarily of parathyroid, enteropancreatic, and anterior pituitary origin, as well as nonendocrine neoplasms. Other endocrine tumors in MEN1 include foregut carcinoid tumors, adrenocortical tumors, and rarely pheochromocytoma. Nonendocrine manifestations include meningiomas and ependymomas, lipomas, angiofibromas, collagenomas, and leiomyomas [62]. This syndrome is caused by inactivating defects of the tumor suppressor gene MEN1 located at the 11q13 locus, that encodes the protein menin [63, 64]. Menin, is implicated in the regulation of transcription, genome stability, cell division, and cell proliferation, though it’s exact role in tumorigenesis is yet to be elucidated [63-66]. In a retrospective cohort study of 715 patients with MEN1, 146 patients (20.4%) were found to have adrenal enlargement, with adrenal tumors exceeding 1 centimeter in size accounting for 58.1% of these cases (10.1% of the whole patient cohort) and bilateral tumors occurring in 12.5% of cases. Hormonal hypersecretion occurred in 15.3% and only in patients with ACTs. MEN1-related adrenal tumors exhibited more cases of primary aldosteronism and ACC when compared to controls with adrenal incidentalomas, with 13.8% of adrenal tumors being ACC in MEN1 compared to 1.3% of controls, and a prevalence of ACC in MEN1 of 1.3% [57].
HLRCC is a syndrome characterized by the development of cutaneous and uterine leiomyomas (rarely leiomyosarcomas) and renal cell carcinoma that has been linked to germline genetic defects in the fumarate hydratase (FH) gene, a possible tumor suppressor gene [67]. Fumarate hydratase is part of the mitochondrial Krebs or tricarboxylic acid cycle. Though the mechanism through which FH defects lead to HLRCC has not been completely elucidated, it may involve increased cellular dependence on glycolysis and pseudohypoxia [68, 69]. This condition also predisposes to adrenocortical tumors including PBMAH and adrenocortical adenomas that may produce cortisol or may be non-functional. This was highlighted in a study including 255 patients with HLRCC where 20 (7.8%) had primary adrenal lesions, with 4 having bilateral adrenal lesions and 4 with multiple nodules. Two patients were found to have hypercortisolism [59, 70].
A genetic origin for PBMAH was suggested by the bilateral nature of the disease, the cases of familial PBMAH, and the association of PBMAH with familial tumor syndromes. Genotyping (both blood and tumor) of 33 patients with PBMAH, led to detection of inactivating mutations in the ARMC5 gene in 55% (18/33) of tumors [60]. The prevalence of ARMC5 defects has subsequently estimated to be around 21-26% [71, 72]. Patients with PBMAH due to ARMC5 defects are more likely to have overt CS, larger adrenal glands, and more adrenal nodules, when compared to those with PBMAH that do not have ARMC5 mutations [71]. ARMC5 is a cytosolic protein without enzymatic activity with little known about its function or mechanism of action. It encodes a protein that contains an armadillo repeat domain, which is similar to the gene for β-catenin that also contains armadillo repeats [73, 74]. Of the 18 cases of PBMAH with ARMC5 defects that were initially described, biallelic defects of ARMC5 were identified at the tumor level with four cases harboring nodule-specific secondary ARMC5 mutations, whereas leukocyte DNA only carried one of the two genetic alterations, suggesting that ARMC5 is a tumor suppressor gene [60]. Studies in Armc5 knockout mice have demonstrated that this gene may be important in fetal development, T-cell function, and adrenal gland growth homeostasis, with Armc5 haploinsufficiency causing CS in mice later in life, with the involvement of the cAMP-PKA and the Wnt-β–catenin pathways [73, 75]. Initial functional investigations demonstrated that inactivation of ARMC5 was associated with reduced expression of steroidogenic enzymes and MC2R with abnormal cortisol production and that the large size of the adrenal glands may be related to loss of the ability to induce apoptosis in adrenocortical cells with ARMC5 mutations [60, 71]. A gradual process of adrenocortical cell dedifferentiation and growth of bilateral masses was apparent. Hypercortisolemia in these patients is more likely related to increased adrenocortical mass than to cortisol overproduction per se. In addition, ARMC5 mutations have been associated with primary aldosteronism, as was first described in 2015 [76]. ARMC5 defects may also play a role in the development of meningiomas, which was described in one family with adrenal hyperplasia and meningioma, with ARMC5 loss of heterozygosity in the meningioma DNA [77].
Other possible genetic alterations associated with PBMAH have been reported in a small number of patients including somatic mutations the genes DOTIL, which encodes a histone H3 lysine methyl-transferase, and HDAC9, which encodes a histone deacetylase, both of which are involved in histone modification, chromatin organization and modification of gene transcription. A single study demonstrated a mutation in the Endothelin receptor type A EDNRA gene, which encodes a G-coupled protein, in adrenal tissue from two siblings from a family with familial PBMAH [78].
CORTISOL PRODUCING ADRENOCORTICAL ADENOMAS
The cAMP-PKA pathway also plays a crucial role in the development of CPAs, however in contrast to BAH, in CPAs, somatic genetic alterations affecting this pathway predominate [79]. Whole exome sequencing of tumor-tissue specimens from 10 patients with unilateral CPAs revealed somatic mutations in PRKACA in 8 out of 10 adenomas. Additional sequencing of another 129 adenomas revealed a p.Leu206Arg variant in 14 of these 129 adrenocortical adenomas. PRKACA gene mutations were found only in patients with overt CS and were associated with a more severe phenotype [34]. Following the publication of these data in 2013, another 3 studies from China, Japan, and the United States demonstrated similar findings, with PRKACA mutations identified in 86 of 206 (42%) CPAs with overt CS [80]. Activating mutations of PRCACA abolish the interaction between the regulatory and catalytic subunits of PKA, leading to constitutive activation of PKA, and may also alter substrate specificity with hyperphosphorylation of certain PKA substrates [81]. Recently, an activating somatic mutation in PRKACB was found in a patient with a CPA, with in vitro studies indicating increased sensitivity to cAMP [82]. As aforementioned, somatic inactivating defects in PRKAR1A have also been identified in CPAs with an estimated prevalence of 5%, as have somatic activating defects in GNAS with a prevalence 4.5-11% [18, 24]. Though both PRKAR1A and GNAS genetic alterations may lead to increased cAMP-PKA signaling, a whole genome expression profile study revealed that not all cAMP activation is the same, with adrenal lesions harboring PRKAR1A or GNAS defects both showing overexpression of the MAPK and p53 signaling pathways but GNAS-mutant tissues showing increased expression of genes involved in extracellular matrix receptor interaction and focal adhesion pathways (NFKB, NFKBIA, and TNFRSF1A) whereas PRKARlA-mutant tissues overexpressed genes related to the Wnt-signaling pathway (CCND1, CTNNB1, LEF1, LRP5, WISP1, and WNT3). Furthermore, in a study of samples from 27 patients with CPAs without mutations in GNAS, PRKAR1A, PDE11A, or PDE8B, abnormalities of the cAMP-signaling pathway were found, with mutation-negative CPAs having significantly decreased PDE activity [83].
Benign Aldosterone-Producing Adrenocortical Adenomas and Hyperplasias
Primary aldosteronism (PA) describes a group of disorders characterized by autonomous aldosterone secretion that is inappropriately high for sodium status and is not suppressed by sodium loading. This condition leads to hypertension, sodium retention, suppression of plasma renin, and increased potassium excretion [84]. The two most important stimuli of aldosterone synthesis are intravascular volume depletion and hyperkalemia which both exert their effects through generating a cytoplasmic calcium signal. Intravascular volume depletion causes release of angiotensin II (AT-II) through the renin-angiotensin system, which binds to a GPCR on the zona glomerulosa cells. Hyperkalemia directly raises aldosterone production in zona glomerulosa cells, whose resting potential is set by potassium channel activity [85]. These physiologic stimuli can cause membrane depolarization with activation of voltage-gated calcium channels and increased intracellular calcium, that results in increased aldosterone synthase expression (CYP11B2) and aldosterone production as well as glomerulosa cell proliferation. Secreted aldosterone acts on the kidneys at the level of the distal convoluted tubule, connecting tubule, and cortical collecting duct through the mineralocorticoid receptor, with increased sodium reabsorption and potassium excretion. PA is the most frequent form of secondary hypertension and is estimated to account for approximately 8% of cases of hypertension [86, 87]. The two most common causes of PA are BAH (65%) and aldosterone-producing adenomas (APAs) (35%), with less common causes being unilateral hyperplasia, aldosterone-producing ACC, and familial hyperaldosteronism [87].
FAMILIAL HYPERALDOSTERONISM
Familial hyperaldosteronism (FH) accounts for 1-5% of cases of PA. Four types of FH, type I through IV (FH-I through FH-IV), have been described that are inherited in an autosomal dominant fashion. FH-I or glucocorticoid remediable hyperaldosteronism (GRA) was first described in a single family in 1966, with discovery of the causative chimeric gene 26 years later [88, 89]. GRA occurs as the result of unequal crossing over between 2 highly homologous genes on chromosome 8 that encode isozymes of 11-beta-hydroxylase, to form a chimeric gene: CYP11B1, encoding 11β-hydroxylase that catalyzes conversion of 11-deoxycortisol to cortisol, and CYP11B2, encoding aldosterone synthase which converts deoxycorticosterone to corticosterone and 18-hydroxycorticosterone to aldosterone. The fusion of the promoter region of CYP11B1 with CYP11B2, results in ectopic expression of CYP11B2 in the zona fasciculata and ACTH-dependent activation of the aldosterone synthase with aldosterone overproduction. In GRA, the hypersecretion of aldosterone can be reversed with physiologic doses of glucocorticoid. FH-II was initially described as familial PA due to APA and/or BAH without response to glucocorticoid administration. Though FH-II was initially described in 1992, it did not have a known genetic cause until the recent discovery of mutations in the CLCN2 gene in a study of a family with FH-II and 80 additional probands with unsolved early-onset PA. This study identified eight probands with heterozygous variants in CLCN2, including two de novo mutations, with all relatives with early-onset PA carrying the CLCN2 variant found in the probands [90]. CLCN2 encodes the chloride channel ClC-2, with gain of function mutations causing increased chloride permeability and depolarization that results in voltage-gated calcium influx [90, 91]. FH-III was first described in 2008 in a father and his two daughters who presented with a new form of glucocorticoid-refractory PA due to a germline mutation in KCNJ5. The KCNJ5 gene encodes the GIRK4 (G-protein-activated inward rectifier potassium channel 4), an inwardly rectifying potassium channel, with mutations causing altered channel selectivity that leads to increased sodium conductance and cell depolarization, ultimately resulting in increased intracellular calcium and calcium signaling [92]. Recently a case of early-onset PA due to BAH was described that was caused by mosaicism for KCNJ5 [93]. FH-IV was first described in 2015, when germline mutations in CACNA1H, which encodes a T-type calcium channel, were found to be the cause of early-onset PA in five unrelated individuals with family analysis suggesting incomplete penetrance and demonstrating de novo occurrence in two kindreds. These mutations cause impaired channel inactivation and activation at more hyperpolarized potentials, leading to increased intracellular calcium [94]. Germline mutations in CACNA1D, encoding an L-type calcium channel, can also cause PA, however these mutations exclusively occur de novo due to the severity of the associated phenotype. Such germline defects lead to early onset PA, seizures, and neurologic abnormalities, referred to as PA with seizures and neurological abnormalities or PASNA syndrome. Mutant channels show activation at less depolarized membrane potentials and impaired inactivation, leading to increased calcium influx [95].
ALDOSTERONE-PRODUCING ADRENOCORTICAL ADENOMAS
Somatic mutations in genes encoding ion channels or transporters are found in more than half of APAs. Such mutations include those in KCNJ5 and CACNA1D, as well as in ATPases, ATP1A1 and ATP2B3 [95-98]. Somatic mutations in KCNJ5 are associated with 40% of APAs. In a study of 22 patients with APAs, two recurrent somatic mutations (p.G151R and p.L168R), in and near the selectivity filter of KCNJ5, were present in 36% of APA samples. These two hotspot mutations were later shown to be accountable for the majority of KCNJ5 mutations in APAs. With some exceptions, APA-causing somatic KCNJ5 mutations lead to a more severe phenotype when found in the germline, whereas KCNJ5 mutations found solely in the germline are typically associated with a milder phenotype. KCNJ5 mutations appear to be more common in females than in males (53-63% vs. 22-31%), with higher frequency in some Asian cohorts (60-70% APAs) compared to European cohorts [98-102]. Additionally, APAs due to KCNJ5 mutations are larger and are associated with more pronounced PA at a younger age [101, 103]. CACNA1D mutations are the second most common somatic driver mutations in APAs with a prevalence of 21-42% when using panel sequencing of the entire coding sequence to detect these genetic alterations [104]. Somatic CACNA1D mutations were the most common genetic defects in APAs from blacks, accounting for 42%, followed by KCNJ5 defects which accounted for 34%, then ATP1A1 defects which comprised 8%, and ATP2B3 mutations which were found in 4%. These CACNA1D variants were more common in APAs from black males than those from black females, whereas KCNJ5 genetic alterations were more frequent in APAs from black females compared with those from black males [105]. Gain of function somatic mutations in the ATPases ATP1A1 and ATP2B3 are responsible for 3-17% of APAs [97, 104]. These mutations cause abnormal Na+ or H+ permeability and enhanced aldosterone production. In addition to genetic alterations that affect ion channels and ATPases, activating somatic mutations in CTNNB1 have been described in 2-5% of APAs, with a high proportion of APAs found to have constitutive activation of the Wnt-β-catenin pathway [95, 106-109]. Somatic CTNNB1 mutations have also been described in two women with APAs that presented in pregnancy that had highly upregulated adrenocortical expression of the LH/hCG receptor and gonadotropin releasing hormone (GnRH) receptor [110].
Adrenocortical Carcinoma
Adrenocortical carcinoma (ACC) is a rare and aggressive disease, with an incidence of 1 to 2 per million population per year [111-113]. Most tumors are secretory leading to clinically apparent hormone excess in approximately 60%, most often CS and/or hyperandrogenemia and less frequently PA or feminization. Most ACC cases are sporadic, though 5-10% of ACCs have been associated with germline mutations. ACC has been described in several hereditary cancer syndromes including Li-Fraumeni syndrome, Lynch syndrome, Beckwith-Wiedemann syndrome, MEN-1, FAP, neurofibromatosis type 1, and CNC [114-116]. Investigation of the genetic underpinnings and molecular characteristics of these ACC-predisposing familial syndromes has allowed for the elucidation of signaling pathways involved in tumorigenesis that can also lead to the development of sporadic ACC.
Li-Fraumeni syndrome (LFS) is a rare, autosomal dominant, cancer predisposition syndrome associated with germline genetic alterations in the tumor protein p53 gene (TP53), a tumor suppressor gene located on chromosome 17p13.1 [117]. TP53 encodes a multi-domain homo-tetrameric transcription factor with more than 100 direct transcriptional target genes, that plays key roles in mediating the cellular response to genotoxic stress and oncogene activation, by activating pathways involved in cell-cycle arrest and DNA-damage repair, as well as apoptosis pathways when catastrophic genomic compromise has occurred. [118]. LFS is characterized by a wide range of malignancies that appear at a young age, primarily osteosarcoma, ACC, central nervous system tumors, breast cancer, and soft tissue sarcoma, among others [119]. Germline defects in TP53 are found in 70% of families with classical LFS and 40% of families with Li-Fraumeni-like syndrome, while de novo TP53 mutations account for 7-20% of cases with LFS [118, 119]. The frequency of TP53 germline mutations in ACC decreases with age with approximately 50% of children diagnosed with ACC harboring germline TP53 mutations, as opposed to less than 10% of adults diagnosed with ACC [120, 121]. A TP53 p.R337H mutation is highly prevalent among children with ACC from Southern Brazil, with this founder mutation accounting for 90% of cases in this region [122].
Lynch syndrome (LS) or hereditary nonpolyposis colorectal cancer (HNPCC) is another autosomal dominant cancer predisposition syndrome, that is attributed to germline mutations in one of several DNA-mismatch repair (MMR) genes including MLH1, MSH2, MSH6, and PMS2 or loss of expression of MSH2 due to deletion in the EPCAM gene [123]. Biallelic inactivation of MMR genes in a cell lead to failure to repair DNA mismatches that occur during normal DNA synthesis and resulting genomic instability, with tumors demonstrating microsatellite instability. LS is characterized by an increased risk for colorectal cancer and cancers of the endometrium, stomach, ovary, small bowel, hepatobiliary tract, urinary tract, brain, and skin. An increased prevalence of ACC has also been associated with LS, with a prevalence of 3.2% in a cohort of 94 patients, which is higher than the ACC prevalence in the general population and comparable to the prevalence of LS in colorectal and endometrial cancer. Immunohistochemistry to evaluate for MMR deficiency in these three cases and another 5 previously reported showed lack of expression of the mutated MMR gene in 6 of the 8 cases though all tumors were microsatellite stable [124].
Beckwith-Wiedemann syndrome (BWS) is a rare overgrowth disorder characterized by a spectrum of clinical features including neonatal hypoglycemia, macrosomia, macroglossia, hemihyperplasia, omphalocele, visceromegaly, renal abnormalities, and ear creases/pits. Neoplasms associated BWS include embryonal tumors (Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma), adrenocortical cytomegaly, and ACC, with 5-10% of patients developing tumors, of which 7% are ACC [125]. Tumors occur primarily in the first 8 years of life. Most cases are sporadic with autosomal dominant familial transmission in 15%. In greater than 80% of patients, molecular/cytogenetic testing detects one of the following alterations on chromosome 11p15.5 affecting imprinted genes in this region: loss of methylation of maternal imprinting center (IC) 2 (50%), gain of methylation of maternal IC1 (5%) , paternal uniparental disomy (20%), mutation of the CDKN1C (5% in sporadic cases), or rarely duplication, inversion or translocation of 11p15.5 [126]. The critical BWS genes in the 11p15.5 region include insulin-like growth factor 2 (IGF2) and H19, regulated by IC1, and CDKN1C, KCNQ1, and KCNQ1OT1, regulated by IC2. H19 is a possible tumor suppressor gene that is maternally expressed and codes for a long 2.3kb-noncoding RNA, whereas IGF2 is paternally expressed and encodes for a potent fetal growth factor that is involved in the differentiation and growth of the adrenal cortex. Physiologically, IC1 is unmethylated on the maternal allele and methylated on the paternal allele. Molecular defects in 11p15.5 can lead to overexpression of IGF2, decreased expression of H19, and/or decreased expression of CDKN1C, with gain of methylation at IC1 or paternal uniparental disomy being associated with a higher risk of tumor development in BWS. This suggests a potential role of overexpression of IGF2 and/or decreased expression of H19 in tumorigenesis in this syndrome [127].
As aforementioned, ACC has a prevalence of 1-2% in patients with MEN1, and cases of ACC have also been associated with FAP and CNC [24, 25, 57, 61, 116]. Cases of patients with neurofibromatosis type 1 (NF1) and ACC have also described. NF1 is an autosomal dominant genetic disorder that is familial in half of the cases, and is due to inactivating defects in the tumor suppressor gene NF1, located at chromosome 17q11.2, that encodes the protein neurofibromin [128, 129]. Neurofibromin is part of a family of guanosine triphosphate hydrolase (GTPase)-activating proteins (GAPs). These GAPs stimulate intrinsic GTPase activity in the ras p21 family (21 kD rat sarcoma viral oncogene homologs), with ras being an activator of several signaling pathways including the mechanistic target of rapamycin (mTOR), stem cell factor (SCF)/c-kit signaling, and mitogen-activated protein kinase (MAPK) pathways [130-132]. Clinical manifestations that have a high penetrance include neurofibromas, cafe-au-lait spots, axillary freckling, Lisch nodules, and skeletal malformations. Other neoplasms associated with NF1 include optic pathway gliomas and other central nervous system tumors, soft tissue sarcomas, gastrointestinal stromal tumors, glomus tumors, juvenile myelomonocytic leukemia, pheochromocytoma, and breast cancer. There have been cases reported of ACC in patients with NF1, primarily affecting young children, however it is unclear if this is a true association [116]. Recently, nine germline inactivating variants in PDEs and related genes (PDE3B, PDE5A, PDE6B, PDE8A, and PDE11A, and the phosphodiesterase interacting protein PDE4DIP) were identified in a cohort of 37 pediatric patients with ACT (24%), with tumor DNA loss of heterozygosity, suggesting that these germline variants may play a role in the development of ACC in children [133].
As in familial ACC, somatic genetic alterations leading to changes in p53 signaling, Wnt-β-catenin signaling, IGF2 overexpression, and less so cAMP-PKA signaling, amongst others, have been implicated in the development of sporadic ACC. A study published in 2014 based on the molecular analysis of 122 ACCs identified nine pathogenic genetic alterations (mutations, homozygous deletions or high-level amplifications) that were present in at least 5% of ACCs including in the ZNRF3, CTNNB1, TP53, CDKN2A, RB1, MEN1, DAXX, MED12 and TERT genes. The most frequently altered pathway was the Wnt-β-catenin pathway with 39% of the genetic defects affecting this pathway. The most common genetic abnormality identified in 21% of tumors was that of the ZNRF3 (zing finger and ring finger protein 3) gene, a potential tumor suppressor gene related to the β-catenin pathway which encodes a cell surface E3 ubiquitin ligase that plays a role in the negative feedback of Wnt-β-catenin signaling through promoting degradation of the frizzled and LRP6 receptors. CTNNB1 mutations were present in 16% of ACCs and were mutually exclusive with ZNRF3 alterations, whereas APC mutations were identified in 2%. The MED12 gene was found to be mutated in 5% of tumors with mutations affecting the C-terminal region of the protein that was found to physically interact with β-catenin, leading to the recruitment of mediator to Wnt-responsive genes. The p53-Rb pathway was the second most frequently affected pathway, with alterations identified in 33% of ACCs. Genetic changes affecting this pathway included defects in the tumor suppressor genes TP53 (16%), CDKN2A (11%) and RB1 (7%) and amplifications in the oncogenes CDK4 (2%) and MDM2 (2%). Mutations in genes associated with chromatin remodeling or telomere maintenance were also identified including MEN1 (7%), DAXX (6%), ATRX (2 tumors), and TERT (6%). Chromosomal instability was found to be very high in most samples, with significant gains in chromosomes 4, 5, 7, 8, 12, 16, 19, and 20, and significant deletion in chromosome arm 1p and chromosome 22, with loss of heterozygosity in more than 30% of cases. Loss of heterozygosity including the 11p15 IGF2 locus was found in 82% and was mostly copy neutral [134]. A subsequent study published in 2016, based on the comprehensive genomic characterization of 91 ACCs, expanded the list of known ACC driver genes to include PRKAR1A, RPL22, TERF2, CCNE1, and NF1. In this study, somatic genetic alterations were noted most frequently in the following genes: TP53 (21%), ZNRF3 (19%), CDKN2A (15%), CTNNB1 (16%), TERT(14%), and PRKAR1A (11%). Genetic alterations in genes affecting Wnt-β-catenin pathway activity (ZNRF3, CTNNB1, APC and MEN1) were identified in 41% of cases, whereas somatic alterations in genes affecting p53-Rb signaling (TP53, CDKN2A, RB1, CDK4 and CCNE1) were found in 44.9% of the cases. Collectively, 22% of cases demonstrated genetic changes in histone modification genes (MLL, MLL2, and MLL4) and chromatin remodeling genes (ATRX and DAXX). IGF2 expression was relatively high in 86% of ACCs with no association to either promoter DNA methylation or genome rearrangement. As aforementioned, high chromosomal aneuploidy has been found to be a characteristic of ACC which distinguishes it from benign adrenocortical tumors. In this cohort, three groups were identified among ACCs, the “chromosomal” group (61%) with gains or deletions of chromosomes or chromosomal arms, the “noisy” group (30%) with a high number of chromosomal breaks, and the “quiet” group (9%) with few somatic copy number alterations, with the “noisy” group having a poorer prognosis. Additionally, genome-wide DNA copy number analysis demonstrated increased frequency of DNA loss, followed by whole genome doubling, which was associated with more aggressive tumors [135].
Tumorigenesis in sporadic ACC has been hypothesized to be a multistep process with possible progression from a benign to a malignant adrenocortical tumor. Such an example is that of a 43-year-old patient incidental adrenal mass, who underwent unilateral adrenalectomy. Pathology demonstrated two different tumor components: a central tumor component that harbored ACC and a surrounding (peripheral) tumor component that was consistent with benign tumor. Molecular analysis of the tumor demonstrated 17p13 loss of heterozygosity, 11p15 uniparental disomy, and IGF2 gene overexpression, as well as chromosomal gains and losses in the malignant tissue, which were not present in the benign tissue [136].
Conclusion
In recent years, with the study of adrenocortical tumors using genomic tools and next generation sequencing, significant strides have been made in understanding the genomic underpinnings of adrenocortical tumorigenesis highlighting the roles of the cAMP-PKA pathway and ARMC5 in the development of benign cortisol-producing tumors, aberrant intracellular calcium signaling in PA, and molecular alterations in the Wnt-β-catenin and p53 pathways and IGF2 expression in ACC. These discoveries may pave the way for future tumor classifications as well as targeted new diagnostic, therapeutic, and prognostic tools that may lead to less invasive diagnostic and therapeutic methods, improved genetic counseling and screening for familial cases, and better prognosis.
Practice Points and Research Agenda.
Advances in genomics over the past two decades have allowed for the identification of the genetic alterations causing the development adrenocortical tumors (ACTs). This was achieved initially through the study of rare familial tumor syndromes such as McCune-Albright syndrome (MAS), Carney complex (CNC), Li-Fraumeni syndrome (LFS), and Beckwith-Wiedemann syndrome (BWS), with identification of disease-causing alterations in genes and molecular pathways that subsequently led to the discovery of aberrations in these or related genes and pathways in sporadic ACTs.
ACTs are predominantly comprised of benign adrenocortical tumors and/or hyperplasias and less frequently ACC. These tumors can be unilateral or bilateral, secreting or non-secreting, and familial or sporadic. Bilateral adrenocortical hyperplasia (BAH) is divided into micronodular BAH (nodules less than 1 centimeter in diameter) and macronodular BAH (nodules greater than 1 centimeter in diameter). Micronodular adrenocortical hyperplasia can be divided into primary pigmented nodular adrenocortical disease (PPNAD), characterized by pigmented nodules with atrophic surrounding adrenocortical tissue, and isolated micronodular adrenocortical disease (i-MAD) that has absent or limited pigment and hyperplasia of surrounding adrenocortical tissue. PPNAD is associated with CNC (c-PPNAD) in the majority of cases, and is less frequently an isolated occurrence (i-PPNAD).
The pathogenesis of most benign cortisol-producing ACTs can be linked to aberrant cAMP-protein kinase A (PKA) signaling. c-PPNAD is associated with inactivating germline genetic defects in the PRKAR1A gene encoding the regulatory subunit type 1α (R1α) of PKA, that lead to constitutive activation of the cAMP-PKA pathway, and less frequently is associated with germline alterations in chromosome 2p16. i-PPNAD and i-MAD have been linked to germline defects in PDE11A and PDE8B, encoding phosphodiesterases that play a regulatory role in the cAMP-PKA pathway and i-MAD has been associated with germline copy number gains in PRKACA, encoding the catalytic subunit (Cα) of PKA, that lead to increased cAMP-PKA signaling. Activating mutations in PRKACA account for up to 42% of cortisol-producing adrenocortical adenomas presenting with Cushing syndrome.
Macronodular BAH is predominantly due to primary bilateral macronodular adrenal hyperplasia (PBMAH). In PBMAH adrenocortical cells may secrete ACTH in addition to glucocorticoids and/or mineralocorticoids and may express aberrant (ectopic or excessive) receptors such as LH/hCG, that lead to cortisol secretion when stimulated. Germline defects in the tumor suppressor gene ARMC5 account for 21-26% of cases of PBMAH, whereas PBMAH is rarely associated with familial tumor syndromes such as familial adenomatous polyposis (FAP), multiple endocrine neoplasia type 1 (MEN1), or hereditary leiomyomatosis and renal cell carcinoma. A subtype of macronodular BAH called primary bimorphic adrenocortical disease (PBAD) has been found in MAS.
Primary aldosteronism (PA) is predominantly due to BAH and less commonly aldosterone-producing adenomas (APAs), with rare causes including unilateral adrenal hyperplasia, ACC, and familial hyperaldosteronism (FH). Somatic and germline genetic defects affecting intracellular calcium signaling have been found to cause PA, with more than half of APAs demonstrating somatic genetic defects affecting ion channels (KCNJ5 and CACNA1D) or ATPases (ATP1A1 and ATP2B3).
FH is caused by germline mutations that are inherited in an autosomal dominant manner. FH-I is caused by fusion of the promoter region of the gene for CYP11B1 and the coding sequences of CYP11B2, resulting in ACTH-dependent activation of aldosterone synthase. FH-II is caused by defects in CLCN2, whereas FH-III is caused by KCNJ5 defects, and FH-IV is caused by defects in CACNA1H. CACNA1D germline defects cause PA with seizures and neurological abnormalities or PASNA syndrome, which is not familial due to the severity of the disease.
The development of adrenocortical carcinoma (ACC) has been linked with abnormal p53 and Wnt-β-catenin signaling, as well as IGF2 overexpression, amongst others. Most cases of ACC are sporadic with 5-10% being familial. Hereditary cancer syndromes associated with ACC include LFS, Lynch syndrome, BWS, MEN-1, FAP, neurofibromatosis type 1, and CNC. 39-41% of sporadic ACC cases harbor genetic alterations affecting Wnt-β-catenin signaling, whereas 33-44.9% harbor molecular changes that affect p53-Rb signaling. These genetic alterations most often involve the ZNRF3, CTNNB1, TP53, CDKN2A, and RB1 genes, amongst others. Mutations in genes associated with chromatin remodeling or telomere maintenance have also been identified in ACC including MEN1, DAXX, ATRX, and TERT. High chromosomal aneuploidy is a characteristic of ACC with a high number of chromosomal breaks being associated with a poorer prognosis.
Acknowledgments
Funding statement:
This work was supported by the research project Z01-HD008920 (Principal Investigator: Dr. Constantine A Stratakis) of the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA.
Footnotes
Conflict statement:
Dr. Stratakis holds patents on the PRKAR1A, PDE11A and GPR101 genes and/or their function and has received research funding from Pfizer Inc. on the genetics and treatment of abnormalities of growth hormone secretion.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Etxabe J, Vazquez JA: Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin Endocrinol (Oxf) 1994, 40(4):479–484. [DOI] [PubMed] [Google Scholar]
- 2.Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, Jorgensen J, Kosteljanetz M, Kristensen L et al. : Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab 2001, 86(1): 117–123. [DOI] [PubMed] [Google Scholar]
- 3.Lacroix A, Feelders RA, Stratakis CA, Nieman LK: Cushing's syndrome. Lancet 2015, 386(9996):913–927. [DOI] [PubMed] [Google Scholar]
- 4.Stratakis CA, Boikos SA: Genetics of adrenal tumors associated with Cushing's syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab 2007, 3(11):748–757. [DOI] [PubMed] [Google Scholar]
- *5.Hannah-Shmouni F, Stratakis CA: A Gene-Based Classification of Primary Adrenocortical Hyperplasias. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme 2020, 52(3): 133–141. [DOI] [PubMed] [Google Scholar]
- 6.Taylor SS, Ilouz R, Zhang P, Kornev AP: Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol 2012, 13(10):646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM: Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991, 325(24):1688–1695. [DOI] [PubMed] [Google Scholar]
- 8.Boyce AM, Florenzano P, de Castro LF, Collins MT: Fibrous Dysplasia/McCune-Albright Syndrome In: GeneReviews((R)). Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Seattle (WA); 1993. [Google Scholar]
- *9.Carney JA, Young WF, Stratakis CA: Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-Albright syndrome. Am J Surg Pathol 2011, 35(9): 1311–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tirosh A, Valdes N, Stratakis CA: Genetics of micronodular adrenal hyperplasia and Carney complex. Presse Med 2018, 47(7-8 Pt 2):e127–e137. [DOI] [PubMed] [Google Scholar]
- 11.Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, Rene-Corail F, Stergiopoulos S, Bourdeau I et al. : Mutations in regulatory subunit type 1A of cyclic adenosine 5'-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009, 94(6): 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL: The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985, 64(4):270–283. [DOI] [PubMed] [Google Scholar]
- 13.Espiard S, Bertherat J: Carney Complex. Front Horm Res 2013, 41:50–62. [DOI] [PubMed] [Google Scholar]
- 14.Kamilaris CDC, Faucz FR, Voutetakis A, Stratakis CA: Carney Complex. Exp Clin Endocrinol Diabetes 2019, 127(2-03):156–164. [DOI] [PubMed] [Google Scholar]
- 15.Stratakis CA, Kirschner LS, Carney JA: Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 2001, 86(9):4041–4046. [DOI] [PubMed] [Google Scholar]
- 16.Cazabat L, Ragazzon B, Groussin L, Bertherat J: PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary 2006, 9(3):211–219. [DOI] [PubMed] [Google Scholar]
- *17.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA: Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000, 26(1):89–92. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet-Serrano F, Bertherat J: Genetics of tumors of the adrenal cortex. Endocr Relat Cancer 2018, 25(3):R131–R152. [DOI] [PubMed] [Google Scholar]
- 19.Tsilou ET, Chan CC, Sandrini F, Rubin BI, Shen DF, Carney JA, Kaiser-Kupfer M, Stratakis CA: Eyelid myxoma in Carney complex without PRKAR1A allelic loss. Am J Med Genet A 2004, 130A(4):395–397. [DOI] [PubMed] [Google Scholar]
- 20.Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA: Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet 2003, 12(13): 1475–1484. [DOI] [PubMed] [Google Scholar]
- 21.Matyakhina L, Pack S, Kirschner LS, Pak E, Mannan P, Jaikumar J, Taymans SE, Sandrini F, Carney JA, Stratakis CA: Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet 2003, 40(4):268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stratakis CA, Carney JA, Lin JP, Papanicolaou DA, Karl M, Kastner DL, Pras E, Chrousos GP: Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest 1996, 97(3):699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, Zuffardi O: PRKACB and Carney complex. N Engl J Med 2014, 370(11):1065–1067. [DOI] [PubMed] [Google Scholar]
- 24.Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent-Dejean C et al. : Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res 2003, 63(17):5308–5319. [PubMed] [Google Scholar]
- 25.Bertherat J: Adrenocortical cancer in Carney complex: a paradigm of endocrine tumor progression or an association of genetic predisposing factors? J Clin Endocrinol Metab 2012, 97(2):387–390. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Ramirez LC, Tatsi C, Lodish MB, Faucz FR, Pankratz N, Chittiboina P, Lane J, Kay DM, Valdes N, Dimopoulos A et al. : Corticotropinoma as a Component of Carney Complex. J Endocr Soc 2017, 1(7):918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *27.Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP et al. : A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 2006, 38(7):794–800. [DOI] [PubMed] [Google Scholar]
- 28.Libe R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, Ragazzon B, Guillaud-Bataille M, Groussin L, Clauser E et al. : Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab 2011, 96(1):E208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libe R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, Rene-Corail F, Groussin L, Bertagna X, Raffin-Sanson ML et al. : Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res 2008, 14(12):4016–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libe R, Raffin-Sanson ML, Bertherat J, Carraro DM, Soares FA, Molina Gde C et al. : Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J Clin Endocrinol Metab 2011, 96(1):E135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pathak A, Stewart DR, Faucz FR, Xekouki P, Bass S, Vogt A, Zhang X, Boland J, Yeager M, Loud JT et al. : Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr Relat Cancer 2015, 22(6):909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath A, Mericq V, Stratakis CA: Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med 2008, 358(7):750–752. [DOI] [PubMed] [Google Scholar]
- 33.Rothenbuhler A, Horvath A, Libe R, Faucz FR, Fratticci A, Raffin Sanson ML, Vezzosi D, Azevedo M, Levy I, Almeida MQ et al. : Identification of novel genetic variants in phosphodiesterase 8B (PDE8B), a cAMP-specific phosphodiesterase highly expressed in the adrenal cortex, in a cohort of patients with adrenal tumours. Clin Endocrinol (Oxf) 2012, 77(2):195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *34.Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR et al. : Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome. N Engl J Med 2014, 370(11): 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lodish MB, Yuan B, Levy I, Braunstein GD, Lyssikatos C, Salpea P, Szarek E, Karageorgiadis AS, Belyavskaya E, Raygada M et al. : Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: molecular mechanisms and clinical presentations. Eur J Endocrinol 2015, 172(6):803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald BT, Tamai K, He X: Wnt/beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev Cell 2009, 17(1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I: Detection of somatic beta-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf) 2008, 69(3):367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaujoux S, Tissier F, Groussin L, Libe R, Ragazzon B, Launay P, Audebourg A, Dousset B, Bertagna X, Bertherat J: Wnt/beta-catenin and 3',5'-cyclic adenosine 5'-monophosphate/protein kinase A signaling pathways alterations and somatic beta-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab 2008, 93(10):4135–4140. [DOI] [PubMed] [Google Scholar]
- 39.Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, Libe R, Bram Z, Groussin L, Caron P et al. : Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 2013, 369(22):2115–2125. [DOI] [PubMed] [Google Scholar]
- 40.Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, Robinson-White AJ, Nesterova M, Lacroix A, Stratakis CA: Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab 2009, 94(8):2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gagliardi L, Hotu C, Casey G, Braund WJ, Ling KH, Dodd T, Manavis J, Devitt PG, Cutfield R, Rudzki Z et al. : Familial vasopressin-sensitive ACTH-independent macronodular adrenal hyperplasia (VPs-AIMAH): clinical studies of three kindreds. Clin Endocrinol 2009, 70(6):883–891. [DOI] [PubMed] [Google Scholar]
- 42.Hofland J, Hofland LJ, van Koetsveld PM, Steenbergen J, de Herder WW, van Eijck CH, de Krijger RR, van Nederveen FH, van Aken MO, de Groot JW et al. : ACTH-independent macronodular adrenocortical hyperplasia reveals prevalent aberrant in vivo and in vitro responses to hormonal stimuli and coupling of arginine-vasopressin type 1a receptor to 11 beta-hydroxylase. Orphanet J Rare Dis 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imohl M, Koditz R, Stachon A, Muller KM, Nicolas V, Pfeilschifter J, Krieg M: Catecholamine-dependent hereditary Cushing's syndrome - Follow-up after unilateral adrenalectomy. Med Klin 2002, 97(12):747–753. [DOI] [PubMed] [Google Scholar]
- 44.Lacroix A, Bolte E, Tremblay J, Dupre J, Poitras P, Fournier H, Garon J, Garrel D, Bayard F, Taillefer R et al. : Gastric inhibitory polypeptide-dependent cortisol hypersecretion--a new cause of Cushing's syndrome. N Engl J Med 1992, 327(14):974–980. [DOI] [PubMed] [Google Scholar]
- 45.Lacroix A, Hamet P, Boutin JM: Leuprolide acetate therapy in luteinizing hormone--dependent Cushing's syndrome. N Engl J Med 1999, 341(21):1577–1581. [DOI] [PubMed] [Google Scholar]
- 46.Lee S, Hwang R, Lee J, Rhee Y, Kim DJ, Chung UI, Lim SK: Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin Endocrinol 2005, 63(6):625–630. [DOI] [PubMed] [Google Scholar]
- 47.Libe R, Coste J, Guignat L, Tissier F, Lefebvre H, Barrande G, Ajzenberg C, Tauveron I, Clauser E, Dousset B et al. : Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: a frequent finding in a prospective study of 32 patients with overt or subclinical Cushing's syndrome. European Journal of Endocrinology 2010, 163(1):129–138. [DOI] [PubMed] [Google Scholar]
- 48.Mircescu H, Jilwan J, N'Diaye N, Bourdeau I, Tremblay J, Hamet P, Lacroix A: Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing's syndrome? J Clin Endocrinol Metab 2000, 85(10):3531–3536. [DOI] [PubMed] [Google Scholar]
- 49.Miyamura N, Taguchi T, Murata Y, Taketa K, Iwashita S, Matsumoto K, Nishikawa T, Toyonaga T, Sakakida M, Araki E: Inherited adrenocorticotropin-independent macronodular adrenal hyperplasia with abnormal cortisol secretion by vasopressin and catecholamines - Detection of the aberrant hormone receptors on adrenal gland. Endocrine 2002, 19(3):319–325. [DOI] [PubMed] [Google Scholar]
- 50.Reznik Y, Allali-Zerah V, Chayvialle JA, Leroyer R, Leymarie P, Travert G, Lebrethon MC, Budi I, Balliere AM, Mahoudeau J: Food-dependent Cushing's syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med 1992, 327(14):981–986. [DOI] [PubMed] [Google Scholar]
- 51.Vezzosi D, Cartier D, Regnier C, Otal P, Bennet A, Parmentier F, Plantavid M, Lacroix A, Lefebvre H, Caron P: Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. European Journal of Endocrinology 2007, 156(1):21–31. [DOI] [PubMed] [Google Scholar]
- 52.Lecoq AL, Stratakis CA, Viengchareun S, Chaligne R, Tosca L, Demeocq V, Hage M, Berthon A, Faucz FR, Hanna P et al. : Adrenal GIPR expression and chromosome 19q13 microduplications in GIP-dependent Cushing's syndrome. JCI Insight 2017, 2(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vezzosi D, Libe R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, Rene-Corail F, Ragazzon B, Stratakis CA, Vandecasteele G et al. : Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab 2012, 97(11):E2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown RJ, Kelly MH, Collins MT: Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab 2010, 95(4): 1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis CA: 17q22-24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab 2006, 91(9):3626–3632. [DOI] [PubMed] [Google Scholar]
- 56.Swords FM, Noon LA, King PJ, Clark AJ: Constitutive activation of the human ACTH receptor resulting from a synergistic interaction between two naturally occurring missense mutations in the MC2R gene. Mol Cell Endocrinol 2004, 213(2): 149–154. [DOI] [PubMed] [Google Scholar]
- 57.Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, Young J, Delemer B, Du Boullay H, Verger MF et al. : Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol 2012, 166(2):269–279. [DOI] [PubMed] [Google Scholar]
- 58.Gaujoux S, Pinson S, Gimenez-Roqueplo AP, Amar L, Ragazzon B, Launay P, Meatchi T, Libe R, Bertagna X, Audebourg A et al. : Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res 2010, 16(21):5133–5141. [DOI] [PubMed] [Google Scholar]
- 59.Shuch B, Ricketts CJ, Vocke CD, Valera VA, Chen CC, Gautam R, Gupta GN, Gomez Macias GS, Merino MJ, Bratslavsky G et al. : Adrenal nodular hyperplasia in hereditary leiomyomatosis and renal cell cancer. J Urol 2013, 189(2):430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *60.Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, Barreau O, Lefevre L, Sibony M, Guignat L et al. : ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med 2013, 369(22):2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shiroky JS, Lerner-Ellis JP, Govindarajan A, Urbach DR, Devon KM: Characteristics of Adrenal Masses in Familial Adenomatous Polyposis. Dis Colon Rectum 2018, 61(6):679–685. [DOI] [PubMed] [Google Scholar]
- 62.Kamilaris CDC, Stratakis CA: Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front Endocrinol (Lausanne) 2019, 10:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA et al. : Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276(5311):404–407. [DOI] [PubMed] [Google Scholar]
- 64.Lemmens I, VandeVen WJM, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, DeWitte K, Salandre J, Lenoir G et al. : Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum Mol Genet 1997, 6(7): 1177–1183. [DOI] [PubMed] [Google Scholar]
- 65.Lemos MC, Thakker RV: Multiple endocrine neoplaslia type 1 (MEN 1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008, 29(1):22–32. [DOI] [PubMed] [Google Scholar]
- 66.Matkar S, Thiel A, Hua XX: Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci 2013, 38(8):394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alam NA, Bevan S, Churchman M, Barclay E, Barker K, Jaeger EE, Nelson HM, Healy E, Pembroke AC, Friedmann PS et al. : Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3-q43. Am J Hum Genet 2001, 68(5):1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pollard P, Wortham N, Barclay E, Alam A, Elia G, Manek S, Poulsom R, Tomlinson I: Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J Pathol 2005, 205(1):41–49. [DOI] [PubMed] [Google Scholar]
- 69.Sudarshan S, Pinto PA, Neckers L, Linehan WM: Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer--a distinct form of hereditary kidney cancer. Nat Clin Pract Urol 2007, 4(2):104–110. [DOI] [PubMed] [Google Scholar]
- 70.Matyakhina L, Freedman RJ, Bourdeau I, Wei MH, Stergiopoulos SG, Chidakel A, Walther M, Abu-Asab M, Tsokos M, Keil M et al. : Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 2005, 90(6):3773–3779. [DOI] [PubMed] [Google Scholar]
- 71.Espiard S, Drougat L, Libe R, Assie G, Perlemoine K, Guignat L, Barrande G, Brucker-Davis F, Doullay F, Lopez S et al. : ARMC5 Mutations in a Large Cohort of Primary Macronodular Adrenal Hyperplasia: Clinical and Functional Consequences. J Clin Endocrinol Metab 2015, 100(6):E926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, Berthon A, Libe R, Assie G, Espiard S et al. : Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab 2014, 99(6):E1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu Y, Lao L, Mao J, Jin W, Luo H, Charpentier T, Qi S, Peng J, Hu B, Marcinkiewicz MM et al. : Armc5 deletion causes developmental defects and compromises T-cell immune responses. Nat Commun 2017, 8:13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, Rene-Corail F, Jullian E, Gicquel C, Bertagna X et al. : Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res 2005, 65(17):7622–7627. [DOI] [PubMed] [Google Scholar]
- 75.Berthon A, Faucz FR, Espiard S, Drougat L, Bertherat J, Stratakis CA: Age-dependent effects of Armc5 haploinsufficiency on adrenocortical function. Hum Mol Genet 2017, 26(18):3495–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, Batsis M, Sinaii N, Quezado MM, Merino M et al. : Primary Aldosteronism and ARMC5 Variants. J Clin Endocrinol Metab 2015, 100(6):E900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elbelt U, Trovato A, Kloth M, Gentz E, Finke R, Spranger J, Galas D, Weber S, Wolf C, Konig K et al. : Molecular and clinical evidence for an ARMC5 tumor syndrome: concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab 2015, 100(1):E119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu J, Cui L, Wang W, Hang XY, Xu AX, Yang SX, Dou JT, Mu YM, Zhang X, Gao JP: Whole exome sequencing identifies mutation of EDNRA involved in ACTH-independent macronodular adrenal hyperplasia. Fam Cancer 2013, 12(4):657–667. [DOI] [PubMed] [Google Scholar]
- 79.Stratakis CA: Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): the expected, the unexpected, and what might be next. Journal of Pathology 2018, 244(3):257–259. [DOI] [PubMed] [Google Scholar]
- 80.Lodish M, Stratakis CA: A genetic and molecular update on adrenocortical causes of Cushing syndrome. Nat Rev Endocrinol 2016, 12(5):255–262. [DOI] [PubMed] [Google Scholar]
- 81.Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A, Fassnacht M, Calebiro D: Alterations in Protein Kinase A Substrate Specificity as a Potential Cause of Cushing Syndrome. Endocrinology 2019, 160(2):447–459. [DOI] [PubMed] [Google Scholar]
- 82.Espiard S, Knape MJ, Bathon K, Assie G, Rizk-Rabin M, Faillot S, Luscap-Rondof W, Abid D, Guignat L, Calebiro D et al. : Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 2018, 3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bimpaki EI, Nesterova M, Stratakis CA: Abnormalities of cAMP signaling are present in adrenocortical lesions associated with ACTH-independent Cushing syndrome despite the absence of mutations in known genes. Eur J Endocrinol 2009, 161(1): 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF Jr.: The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101(5): 1889–1916. [DOI] [PubMed] [Google Scholar]
- 85.Spat A, Hunyady L: Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 2004, 84(2):489–539. [DOI] [PubMed] [Google Scholar]
- 86.Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M et al. : A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006, 48(11):2293–2300. [DOI] [PubMed] [Google Scholar]
- 87.Young WF: Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007, 66(5):607–618. [DOI] [PubMed] [Google Scholar]
- 88.Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM: A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355(6357):262–265. [DOI] [PubMed] [Google Scholar]
- 89.Sutherland DJ, Ruse JL, Laidlaw JC: Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J 1966, 95(22): 1109–1119. [PMC free article] [PubMed] [Google Scholar]
- 90.Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V et al. : CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018, 50(3):349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, Boulkroun S, Jeunemaitre X, Amar L, Lefebvre H et al. : A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 2018, 50(3):355–361. [DOI] [PubMed] [Google Scholar]
- *92.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ et al. : K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331(6018):768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maria AG, Suzuki M, Berthon A, Kamilaris C, Demidowich A, Lack J, Zilbermint M, Hannah-Shmouni F, Faucz FR, Stratakis CA: Mosaicism for KCNJ5 Causing Early-Onset Primary Aldosteronism due to Bilateral Adrenocortical Hyperplasia. Am J Hypertens 2020, 33(2): 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC et al. : Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015, 4:e06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW et al. : Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013, 45(9):1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W et al. : Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 2013, 45(9): 1055–1060. [DOI] [PubMed] [Google Scholar]
- 97.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E et al. : Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013, 45(4):440–444, 444e441-442. [DOI] [PubMed] [Google Scholar]
- 98.Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, Strom TM, Monticone S, Amar L, Meatchi T et al. : Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (Dallas, Tex : 1979) 2014, 64(2):354–361. [DOI] [PubMed] [Google Scholar]
- 99.Akerstrom T, Crona J, Verdugo AD, Starker LF, Cupisti K, Willenberg HS, Knoefel WT, Saeger W, Feller A, Ip J et al. : Comprehensive Re-Sequencing of Adrenal Aldosterone Producing Lesions Reveal Three Somatic Mutations near the KCNJ5 Potassium Channel Selectivity Filter. Plos One 2012, 7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hong AR, Kim JH, Song YS, Lee KE, Seo SH, Seong MW, Shin CS, Kim SW, Kim SY: Genetics of Aldosterone-Producing Adenoma in Korean Patients. Plos One 2016, 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP: A Meta-Analysis of Somatic KCNJ5 K+ Channel Mutations In 1636 Patients With an Aldosterone-Producing Adenoma. J Clin Endocr Metab 2015, 100(8):E1089–E1095. [DOI] [PubMed] [Google Scholar]
- 102.Wu VC, Huang KH, Peng KY, Tsai YC, Wu CH, Wang SM, Yang SY, Lin LY, Chang CC, Lin YH et al. : Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci Rep-Uk 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, Mulatero P, Samson-Couterie B, Hahner S, Quinkler M et al. : Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension (Dallas, Tex : 1979) 2012, 59(3):592–598. [DOI] [PubMed] [Google Scholar]
- 104.Seidel E, Schewe J, Scholl UI: Genetic causes of primary aldosteronism. Exp Mol Med 2019, 51(11): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, Thompson LDR, Cohen DL, Luther JM, Gellert L et al. : Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension (Dallas, Tex : 1979) 2019, 73(4):885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, Stalberg P, Robinson B, Alexander Iwen K, Dralle H et al. : Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 2016, 6:19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro MC, Plouin PF, Skah S et al. : WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet 2014, 23(4):889–905. [DOI] [PubMed] [Google Scholar]
- 108.Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, Horne MJ, Dietrich D, Riemer J, Kucukkoylu S et al. : Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin Endocrinol (Oxf) 2015, 83(6):779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tadjine M, Lampron A, Ouadi L, Bourdeau I: Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf) 2008, 68(2):264–270. [DOI] [PubMed] [Google Scholar]
- 110.Teo AE, Garg S, Shaikh LH, Zhou J, Karet Frankl FE, Gurnell M, Happerfield L, Marker A, Bienz M, Azizan EA et al. : Pregnancy, Primary Aldosteronism, and Adrenal CTNNB1 Mutations. N Engl J Med 2015, 373(15):1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Allolio B, Fassnacht M: Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006, 91(6):2027–2037. [DOI] [PubMed] [Google Scholar]
- 112.Hsing AW, Nam JM, Co Chien HT, McLaughlin JK, Fraumeni JF Jr.: Risk factors for adrenal cancer: an exploratory study. Int J Cancer 1996, 65(4):432–436. [DOI] [PubMed] [Google Scholar]
- 113.Ng L, Libertino JM: Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol 2003, 169(1): 5–11. [DOI] [PubMed] [Google Scholar]
- 114.Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, Lyssikatos C, Horvath A, Carney JA, Stratakis CA: A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab 2012, 97(2):351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ, Hammer GD: Adrenocortical carcinoma. Endocrine reviews 2014, 35(2):282–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Petr EJ, Else T: Adrenocortical carcinoma (ACC): When and why should we consider germline testing? Presse Med 2018, 47(7-8 Pt 2):e119–e125. [DOI] [PubMed] [Google Scholar]
- 117.Malkin D, Li FP, Strong LC, Fraumeni JF Jr., Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA et al. : Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250(4985):1233–1238. [DOI] [PubMed] [Google Scholar]
- 118.Wasserman JD, Zambetti GP, Malkin D: Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Molecular and Cellular Endocrinology 2012, 351(1): 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, Bremer RC, Rosenberg PS, Savage SA: Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122(23):3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GD: Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab 2013, 98(1):E119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wasserman JD, Novokmet A, Eichler-Jonsson C, Ribeiro RC, Rodriguez-Galindo C, Zambetti GP, Malkin D: Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children's oncology group study. J Clin Oncol 2015, 33(6):602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Custodio G, Parise GA, Kiesel Filho N, Komechen H, Sabbaga CC, Rosati R, Grisa L, Parise IZ, Pianovski MA, Fiori CM et al. : Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol 2013, 31(20):2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moreira L, Balaguer F, Lindor N, de la Chapelle A, Hampel H, Aaltonen LA, Hopper JL, Le Marchand L, Gallinger S, Newcomb PA et al. : Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308(15): 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ et al. : Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 2013, 31(24):3012–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lapunzina P: Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 2005, 137C(1):53–71. [DOI] [PubMed] [Google Scholar]
- 126.Shuman C, Beckwith JB, Weksberg R: Beckwith-Wiedemann Syndrome In: GeneReviews((R)). Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Seattle (WA); 1993. [PubMed] [Google Scholar]
- 127.Weksberg R, Shuman C, Beckwith JB: Beckwith-Wiedemann syndrome. Eur J Hum Genet 2010, 18(1): 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Feldkamp MM, Gutmann DH, Guha A: Neurofibromatosis type 1: piecing the puzzle together. Can J Neurol Sci 1998, 25(3): 181–191. [DOI] [PubMed] [Google Scholar]
- 129.Ledbetter DH, Rich DC, O'Connell P, Leppert M, Carey JC: Precise localization of NF1 to 17q11.2 by balanced translocation. Am J Hum Genet 1989, 44(1):20–24. [PMC free article] [PubMed] [Google Scholar]
- 130.Gutmann DH, Blakeley JO, Korf BR, Packer RJ: Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs 2013, 22(4):443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O'Connell P, Cawthon RM et al. : The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63(4):843–849. [DOI] [PubMed] [Google Scholar]
- 132.Weiss B, Bollag G, Shannon K: Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet 1999, 89(1): 14–22. [PubMed] [Google Scholar]
- 133.Pinto EM, Faucz FR, Paza LZ, Wu G, Fernandes ES, Bertherat J, Stratakis CA, Lalli E, Ribeiro RC, Rodriguez-Galindo C et al. : Germline Variants in Phosphodiesterase Genes and Genetic Predisposition to Pediatric Adrenocortical Tumors. Cancers (Basel) 2020, 12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- *134.Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F et al. : Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 2014, 46(6):607–612. [DOI] [PubMed] [Google Scholar]
- *135.Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G et al. : Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29(5):723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *136.Bernard MH, Sidhu S, Berger N, Peix JL, Marsh DJ, Robinson BG, Gaston V, Le Bouc Y, Gicquel C: A case report in favor of a multistep adrenocortical tumorigenesis. J Clin Endocrinol Metab 2003, 88(3):998–1001. [DOI] [PubMed] [Google Scholar]