

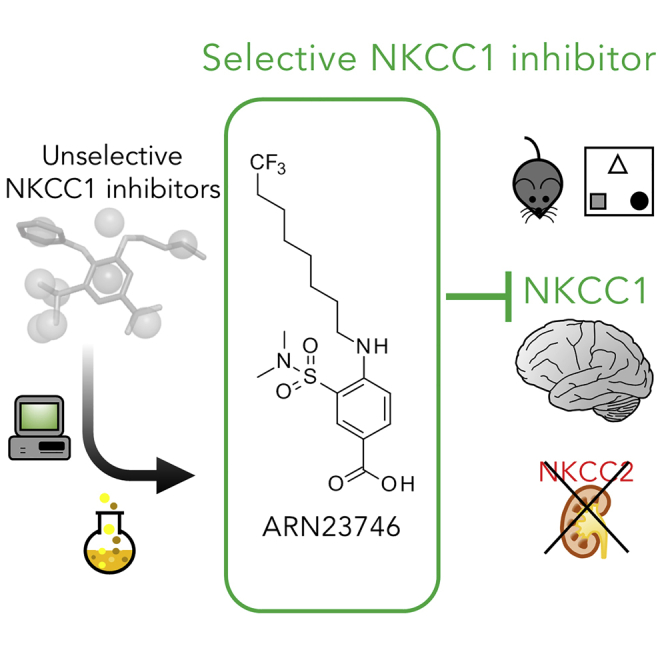
Summary
Aberrant expression ratio of Cl− transporters, NKCC1 and KCC2, is implicated in several brain conditions. NKCC1 inhibition by the FDA-approved diuretic drug, bumetanide, rescues core symptoms in rodent models and/or clinical trials with patients. However, bumetanide has a strong diuretic effect due to inhibition of the kidney Cl− transporter NKCC2, creating critical drug compliance issues and health concerns. Here, we report the discovery of a new chemical class of selective NKCC1 inhibitors and the lead drug candidate ARN23746. ARN23746 restores the physiological intracellular Cl− in murine Down syndrome neuronal cultures, has excellent solubility and metabolic stability, and displays no issues with off-target activity in vitro. ARN23746 recovers core symptoms in mouse models of Down syndrome and autism, with no diuretic effect, nor overt toxicity upon chronic treatment in adulthood. ARN23746 is ready for advanced preclinical/manufacturing studies toward the first sustainable therapeutics for the neurological conditions characterized by impaired Cl− homeostasis.
Keywords: NKCC1, NKCC2, KCC2, small molecule, brain disorders, chloride homeostasis, GABAergic transmission, Down syndrome, autism, drug discovery
Graphical Abstract
Highlights
-
•
NKCC1 is a promising target for the treatment of brain disorders
-
•
The newly discovered ARN23746 presents selective NKCC1 versus NKCC2 and KCC2 inhibition
-
•
ARN23746 restores altered neuronal chloride homeostasis in vitro
-
•
ARN23746 rescues core behaviors in DS and ASD mice with no diuretic effect or toxicity
The Bigger Picture
In the last few decades, drug development for brain disorders has struggled to deliver effective small molecules as novel breakthrough classes of drugs. Discovery of effective chemical compounds for brain disorders has been greatly hampered by the fact that the few currently clinically used drugs were identified by serendipity, and these drugs’ mechanism of action is often poorly understood. Here, by leveraging drug repurposing as a means to quickly and safely evaluate the new pharmacological target NKCC1 and its implications in brain disorders in animal models and patients, we report an integrated strategy for the rational design and discovery of a novel, selective, and safe NKCC1 inhibitor, active in vivo. This compound has the potential to become a clinical drug candidate to treat several neurological conditions in patients. Eventually, this integrated drug-discovery strategy has the prospective to revive the appeal of drug-discovery programs in the challenging field of neuroscience.
Currently, therapeutic options for several neurological disorders are scant or not highly effective. This is possibly due to the poor understanding of the mechanisms underlying these conditions. Here, starting from former validation of the new pharmacological target NKCC1 in brain disorders, we developed a novel, potent, and safe NKCC1 inhibitor, able to restore core behaviors in Down syndrome and autistic mouse models. This compound has the potential to become a solid drug candidate for the treatment of several neurological conditions.
Introduction
In recent years, a large and growing body of literature has indicated that intracellular chloride concentration ([Cl−]i) is defective in diverse neurological conditions,1, 2, 3 including Down syndrome (DS)4 and autism spectrum disorders (ASD).5 In neurons, [Cl−]i is mainly regulated by the opposing action of the sodium (Na+)- potassium (K+)-Cl− importer NKCC1 and the K+-Cl− exporter KCC2. In agreement with aberrant Cl− homeostasis, the NKCC1/KCC2 expression ratio is altered in animal models of DS,6 ASD,7 and several other brain disorders.1, 2, 3 In mature neurons, this leads to a depolarizing (versus hyperpolarizing and inhibitory) GABA neurotransmitter action through Cl−-permeable GABAA receptors. Accordingly, NKCC1 inhibition by the FDA-approved diuretic, bumetanide, rescues GABA transmission and behavioral deficits in the animal models characterized by aberrant Cl− homeostasis.2,8 Notably, the NKCC1/KCC2 expression ratio is also defective in individuals with DS,6 and bumetanide reduced some of the diagnostic symptoms of ASD in seven independent clinical studies,9, 10, 11, 12, 13, 14, 15 including a phase I and a phase II clinical trial. Moreover, bumetanide treatment has shown positive outcomes in clinical case studies of patients with other neurological/psychiatric conditions.16, 17, 18, 19, 20, 21, 22, 23 However, bumetanide has a strong diuretic effect due to its inhibition of the kidney Cl− transporter NKCC2. This creates critical issues with drug compliance and health concerns for chronic treatment,11, 12, 13 strongly jeopardizing bumetanide from becoming a solid therapy for patients. Selective NKCC1 inhibition would be devoid of the diuretic effect, thus solving these problems.
Here, we present a new chemical class that selectively inhibits NKCC1 over renal NKCC2. We designed, synthesized, and tested these new molecular entities in vitro, and optimized the initial hit compounds into a potent lead inhibitor (ARN23746). ARN23746 is able to selectively block NKCC1 in a human cell line and restore the physiological [Cl−]i in murine DS neurons in culture. ARN23746 has excellent drug-like properties, with great solubility, metabolic stability, and target specificity, while also presenting no overt organ toxicity following in vivo chronic treatment. Moreover, ARN23746 is characterized by an improved in vivo pharmacokinetic profile when compared with bumetanide. Importantly, ARN23746 treatment recovers cognitive deficits in a DS mouse model and rescues behaviors related to ASD core symptoms in an ASD mouse model, with no diuretic effect. These results show that selective NKCC1 inhibition devoid of diuretic effect is able to rescue core diagnostic behaviors in DS and ASD mice. Thus, our study reports the discovery of ARN23746 as a solid drug candidate, which can be developed into a sustainable therapeutic strategy for DS, ASD, and the other brain disorders characterized by increased [Cl−]i and depolarizing GABAergic transmission.
Results
Design and Synthesis of NKCC1 Novel Inhibitors
To discover and develop selective NKCC1 inhibitors, we first sought to identify and isolate the structural features of bumetanide that could generate selective inhibition of NKCC1 over NKCC2. NKCC1 inhibition is responsible for bumetanide’s beneficial central nervous system (CNS) effect. We wanted to distinguish the structural features responsible for NKCC1 inhibition from the ones responsible for peripheral NKCC2 inhibition in the kidney, which causes the undesirable diuretic effect. We thus designed, synthesized (Schemes S1 and S2; Supplemental Experimental Procedures), and tested novel bumetanide analogs with diverse substituents in positions R1, R3, and R5 in bumetanide’s core structure (Figures 1A and S1A).
Figure 1.

In Vitro Selection of the Selective NKCC1 Inhibitor ARN23746 as a Lead Compound
(A) Schematic representation of the intervention point in bumetanide’s structure for synthesizing novel bumetanide analogs.
(B) Quantification of the inhibitory activity of bumetanide (Bume) and bumetanide analogs (10, 100 μM) in NKCC1-(left) or NKCC2-(right) transfected HEK293 cells in the Cl− influx assay. Data are presented as a percentage of the respective control DMSO. Data represent mean ± standard error of the mean (SEM) from 3–4 independent experiments (Kruskal-Wallis one way ANOVA on Ranks, NKCC1 10 μM: H = 84.898, DF = 6, p < 0.001; NKCC1 100 μM: H = 86.799, DF=6, p < 0.001; NKCC2 10 μM: H = 40.700, DF = 6, p < 0.001; NKCC2 100 μM: H = 70.569, DF = 6, p < 0.001, Dunn’s post hoc test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(C) Representation of the ligand-based computational strategy to discover novel molecular scaffolds that inhibit NKCC1. The obtained bumetanide pharmacophore (1) consists of three H-bond acceptor (HBA) features (red spheres), three H-bond donor (HBD) interactions (blue spheres), one lipophilic feature (green sphere), and one stacking feature (brown sphere) anchored around the central aromatic core. Ligand disposition was then implemented by superimposing other known unspecific NKCC1 inhibitors (2), revealing shared features and different dihedral dispositions of substituent around the central aromatic core. This model was used as a search filter for the virtual screening (3) of our internal chemical collection (∼15,000 compounds). Results generated from in vitro testing of the 165 initial hits (4) were then used to retrain the model (5) and perform a second more specific screening of our chemical library and commercial chemical libraries (∼135,000 compounds). This iterative computational cycle led to hit compounds (6) ARN22393 and ARN22394. (D) Quantification of the inhibitory activity of the indicated compounds (10, 100 μM) in NKCC1-transfected HEK293 cells (Cl− influx assay). Data are presented as a percentage of the respective control DMSO. Data represent mean ± SEM from 3–4 independent experiments (Kruskal-Wallis one way ANOVA on Ranks, 10 μM: H = 37.119, DF = 3, p < 0.001; 100 μM: H = 33.724, DF = 3, p < 0.001, Dunn’s post hoc test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(E) Chemical structures of NKCC1 inhibitors with novel scaffold. (F) Left, example traces obtained in the Cl− influx assay on NKCC1-transfected HEK293 cells for each compound (100 μM). The arrow indicates the addition of NaCl (74 mM) to initiate the NKCC1-mediated Cl− influx. Right, quantification of the NKCC1 inhibitory activity of the indicated compounds (10, 100 μM) in experiments such as those on the right. Data are presented as a percentage of the respective control DMSO. Data represent mean ± SEM from 3–4 independent experiments (10 μM: one way ANOVA, F(4,84) = 33.048, p < 0.001, Dunnett's post hoc test, ∗p < 0.05, ∗∗∗p < 0.001; 100 μM: Kruskal-Wallis one way ANOVA on Ranks, H = 50.796, DF = 4, p < 0.001, Dunn’s post hoc test, ∗∗∗p < 0.001.
(G) Left, example traces obtained in the Ca2+ influx assay on 3DIV hippocampal mouse neurons for each tested compound (100 μM). The arrows indicates the addition of GABA (100 μM) and KCl (90 mM). Right, quantification of the effect of the indicated compounds (10, 100 μM) in the Ca2+ influx assay on 3DIV neurons. Data are presented as a percentage of the control DMSO. Data represent mean ± SEM from three independent experiments. 10 μM: one-way ANOVA, F(5,161)= 77.184, p < 0.001, Dunnett's post hoc test ∗∗p < 0.01, ∗∗∗p < 0.001; 100 μM: Kruskal-Wallis One ANOVA on Ranks, H = 134.681, DF = 5, p < 0.001, Dunn’s post hoc test, ∗∗∗p < 0.001).
(H) Amplitude change average and single cell data points of GABA-evoked currents obtained by voltage-clamp recordings of 12–20 DIV hippocampal mouse neurons before (gray example recordings in the inset above) and after (black example traces) bath application of the indicated drugs (10 μM). Data are presented as mean ± SEM (Paired t test, ∗p < 0.05, ∗∗p < 0.01). Scale bars: 250 pA, 250 ms. See also Figures S1A–S1D; Tables S1 and S2.
To test our newly synthesized bumetanide analogs, we developed a functional NKCC transporter assay (Cl− influx assay), based on the detection of variations of [Cl−]i by a Cl−-sensitive, membrane-tagged yellow fluorescent protein (mbYFPQS).24 We performed the Cl− influx assay in human HEK293 kidney cells transfected with NKCC1, NKCC2 (or the corresponding empty plasmid as control-mock; (Figure S1B), together with the mbYFPQS plasmid. mbYFPQS fluorescence is inversely dependent on [Cl−]i. In this assay, cells are kept in a Cl−-free-hypotonic solution and, upon application of NaCl to the assay well, Cl− ions enter into cells by NKCC1 or NKCC2 transport, bind mbYFPQS, and determine a decrease in fluorescence.
First, we validated the assay by assessing NKCC1 or NKCC2 functionality upon application of 74 mM NaCl in the well (Figures S1C and S1E). Then, we assessed the inhibitory activity (upon NaCl application) of two known unselective inhibitors of both NKCC1 and NKCC2 (i.e., bumetanide and furosemide; Figures S1D and S1F), which we used as positive controls. Then, on the newly developed Cl− influx assay, we tested all our 15 synthesized bumetanide analogs. We found that the carboxylic acid group, although suboptimal in CNS drug design, in position R1 was essential for the inhibition of NKCC1 (e.g., ARN21902, Figure S1A). Moreover, small modifications of the linear (six to eight carbon atoms) alkyl chain in position R3 (e.g., ARN21878, ARN22351, and ARN22381, Figure S1A) and dimethylation of the sulfonamide in R5 (e.g., ARN23837, Figure S1A) decreased activity against NKCC2, while retaining significant activity against NKCC1 (Figure 1B). Nevertheless, all the modifications that reduced NKCC2 inhibition also led to a significant loss of potency for NKCC1 inhibition (Figure 1B). Altogether, these data indicated a major difficulty to develop new and potent bumetanide derivatives with significant selectivity for NKCC1 over NKCC2. This is in line with the literature on other existing bumetanide analogs25 and prodrugs.26 We therefore sought for new molecular entities, structurally unrelated to bumetanide, as selective NKCC1 inhibitors.
Because of the importance of the carboxylic group and the contribution to the selectivity of a linear alkyl chain attached to an aromatic core, we applied a ligand-based computational strategy to build a pharmacophore model based on bumetanide and other known NKCC1 inhibitors (Figure 1C). We first performed a force field-based conformational search on bumetanide’s structure. This approach returned the preferred spatial arrangement of the pharmacophoric features of bumetanide, such as H-bond donor and acceptor, lipophilic, and aromatic groups (Figure 1C). We then implemented ligand disposition by superimposing this template pharmacophore with other structures of unselective NKCC1 inhibitors (e.g., diuretics furosemide, asozemide, piretanide, and chlorothiazide) to identify shared chemicophysical properties and three-dimensional (3D) localization (Figure 1C). We used this pharmacophore model as a filter for the virtual screening of our institution’s diverse and non-redundant internal library of ∼20,000 molecules (Figure 1C). In a first round of experiments, 165 new compounds from the library emerged as initial hits and were individually tested for their ability to inhibit NKCC1 in the Cl− influx assay (not shown). Of these compounds, 20% showed NKCC1 inhibition between 5%–10% at 10 μM. The other 80% showed no activity (not shown). Using the structural data acquired in the first round, we refined the pharmacophore model (Figure 1C). We then performed a second and more specific screening of the internal library and other chemical libraries from commercial vendors for a total of ∼135,000 compounds (Figure 1C). In this second round, we selected the best 90 compounds (structures have been deposited to Mendeley Data: https://doi.org/10.17632/x9ttg84pzt.2) and tested them in the Cl− influx assay. Two 2-amino-5-nitro-benzenesulfonamide derivatives showed significant (although moderate) inhibitory activity against NKCC1 (ARN22393 29.4 ± 2.8% at 100 μM; ARN22394, 17.7 ± 3.9% at 10 μM and of 28.6 ± 4.3% at 100 μM; Figure 1D). Notably, both compounds had the sulfonamide moiety and a nitro group (i.e., an isostere of a carboxylic acid), linked to an aromatic core, although in a different disposition in respect to the other substituents, as compared with bumetanide.
To improve the potency of the NKCC1 inhibition of the two 2-amino-5-nitro-benzenesulfonamide derivatives, we synthesized twelve 2-amino-5-nitro-benzene-sulfonamide and thirty-one 4-amino-3-sulfamoyl-benzoic acid new compounds (Scheme S3; Supplemental Experimental Procedures), which shared the core chemical structure of the two 2-amino-5-nitro-benzenesulfonamide derivatives. The 2-amino-5-nitro-benzene-sulfonamides did not exhibit enhanced potency. However, the 4-amino-3-sulfamoyl-benzoic acids showed promising activity against NKCC1 (not shown). In particular, potent NKCC1 inhibition was displayed by compounds bearing the carboxylic group in R1, a (methylated) sulfonamide in R3, and an extended (eight carbon atom) alkyl chain in R4 (i.e., ARN22642 35.4 ± 17.3% at 100 μM; ARN22430 70.6 ± 9.1% at 100 μM; Figures 1E and 1F). Nevertheless, ARN22642 and ARN22430 (Figure 1E) showed poor solubility and metabolic stability (Table S1). To overcome these poor drug-like properties, we manipulated the most eligible point of metabolism, i.e., the terminal methyl group on the n-octyl carbon chain. We added a bioisosteric trifluoromethyl group (Schemes S3 and S4; Supplemental Experimental Procedures), which is often used to improve the drug-likeness of promising compounds. The resulting trifluoromethylated analog (ARN23746; Figure 1E) showed not only a substantially improved solubility and metabolic stability (Table S1), but also an increased potency (NKCC1 inhibition 31.8 ± 5.4% at 10 μM, and 95.2 ± 7.6% at 100 μM, Figure 1F).
Encouraged by these results, we next investigated the three most promising compounds (ARN22642, ARN22430, and ARN23746; Figure 1E) for their NKCC1 inhibition in primary hippocampal mouse neurons. First, we used an indirect assay (the Ca2+ influx assay). The Ca2+ influx assay takes advantage of the endogenous high expression of NKCC1 in immature neurons, which causes depolarizing actions of GABA to activate voltage-gated Ca2+ channels.27 In immature neurons, a compound that blocks NKCC1 is, therefore, predicted to inhibit Ca2+ responses upon GABA application. We thus used a fluorescent, Ca2+-sensitive dye (Fluo4) to monitor intracellular Ca2+ variations upon application of GABA (100 μM) in the presence of ARN22642, ARN22430, ARN2376, bumetanide, furosemide (as positive controls), or vehicle, in immature neurons (Figure 1G). The tested compounds significantly reduced the Ca2+ influx upon GABA application in comparison to DMSO-treated controls, without affecting the cell viability (measured as fluorescence level upon KCl [90 mM] application and consequent neuronal depolarization) (Figure 1G, left). Both bumetanide and ARN23746 displayed a level of alleged NKCC1 inhibition that corresponded to the levels obtained for the Cl− influx assay. Conversely, furosemide, ARN22642, and ARN22430 showed higher potency than in the Cl− influx assay, at 10 and 100 μM (compare Figure 1G, right with Figures 1F, right and S1D, right). As described for furosemide,28 this was possibly due to direct off-target inhibition of Cl−-permeable GABAA receptors. Indeed, voltage-clamp patch-cell recordings in primary hippocampal mouse neurons showed that, while furosemide, ARN22642, and ARN22430 exhibited variable inhibitory activity on GABAA receptors, this was not the case for bumetanide or ARN23746 (Figure 1H). Based on NKCC1-inhibition potency, metabolic stability, solubility, and lack of GABAA inhibition, we thus selected ARN23746 as a lead compound for further characterization.
First, to directly measure the effect of ARN23746 on [Cl−]i, we used primary hippocampal cultures obtained from the Ts65Dn mouse model of DS, which is characterized by increased [Cl−]i due to NKCC1 upregulation.6 Chloride imaging with the Cl−-sensitive fluorescent dye MQAE6 showed that bumetanide and ARN23746 (10 μM) both restored aberrant [Cl−]i to physiological levels in mature Ts65Dn neurons,6 without significantly affecting the [Cl−]i in wild-type (WT) neurons (Figure 2A). Next, we used the Cl− influx assay to test ARN23746’s NKCC2 inhibition potency in HEK cells transfected with NKCC2. Notably, ARN23746 (10 μM) did not show significant NKCC2 inhibition (−6.9 ± 7.8%, Figure 2B), in contrast to the potent NKCC2 inhibition of bumetanide (82.7 ± 20.4%, Figure 2B). Finally, we tested ARN23746’s potency against KCC2 (Figure 2C). To this aim, we used a thallium (Tl) influx assay based on a Tl-sensitive fluorescent dye29 in HEK293 cells transfected with KCC2 or empty plasmid (as control-mock; Figure S1B, right). Cells maintained in Cl−-free-hypotonic solution were monitored upon sequential application of TlSO4 (2 mM) followed by NaCl (74 mM). Upon activation of KCC2 by Cl− ions, Tl also enters the cells where it binds a Tl-sensitive dye, thus determining a fluorescence increase (Figure 2C, left). We validated the assay by assessing KCC2 functionality (Figure S1G) by application of the known KCC2 inhibitor R-(+)-[(2-n-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]acetic acid (DIOA). DIOA (10 μM) showed significant KCC2 inhibition (37.6 ± 6.7%, Figure 2C). Notably, ARN23746 (10 μM) did not show KCC2 inhibition (4.6 ± 6.9%, Figure 2C).
Figure 2.

ARN23746 Restores [Cl−]i in DS Neurons, Does Not Inhibit NKCC2 in HEK Cells and Shows Improved Brain Penetration In Vivo in Comparison to Bumetanide
(A) Left, representative pseudo-color images (colored scale below) of the [Cl−]i measured with the MQAE Cl−-sensitive dye, in WT and Ts65Dn hippocampal neurons at 15 DIV, after treatment with control DMSO (0.1%) and the indicated compounds (10 μM). Scale bar: 70 μm. Right, quantification of the indicated compounds (10 μM) in modulating [Cl−]i in experiments such as those on the left. Data represent mean ± SEM from three independent experiments (two-way ANOVA, Finteraction (2,30)= 3.815, p = 0.033, Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(B) Left, example traces obtained in the Cl− influx assay on NKCC2-transfected HEK293 cells for each compound (10 μM). The arrow indicates the addition of NaCl (74 mM) to initiate the NKCC2-mediated Cl− influx. Right, quantification of the NKCC2 inhibitory activity in experiments such as those on the right. Data are presented as a percentage of the respective control DMSO. Data represent mean ± SEM from four independent experiments (Kruskal-Wallis one way ANOVA on Ranks, H = 16,962, DF = 2, p < 0.001, Dunn’s post hoc test, ∗∗∗p < 0.001).
(C) Left, example traces obtained in the Tl influx assay on KCC2-transfected HEK293 cells for each compound (10 μM). The arrows indicate the additions of TlSO4 and NaCl. Right, quantification of the KCC2 inhibitory activity in experiments such as those on the left. Data are presented as a percentage of the respective control DMSO. Data represent mean ± SEM from 4 independent experiments (one way ANOVA, F (2,51) = 10.676, p < 0.001, Dunnett's post hoc test ∗∗∗p < 0.001).
(D) Quantification of the level of bumetanide and ARN23746 in the brain at diverse time points (5, 15, 30, 60, 120 min) after injection in C57BL/6N mice (BioLASCO Taiwan). (Two-way ANOVA, Ftreatment (1,50) = 6.510, p = 0.014, Tukey’s post hoc test, ∗p < 0.05.).
(E) Quantification of the ratio between brain and plasma concentration of bumetanide and ARN2346 5 min after the injection (two-tailed t test, t = 7.915, p < 0.001). See also Figures S1E–S1G.
Efficacy of the Lead Compound ARN23746
Since ARN23746 displayed an optimal absorption-distribution-metabolism-excretion (ADME) profile in vitro (Table S2), no significant off-target activity as agonist or antagonist for a panel of 47 classical pharmacological targets (Table S3), and an improved in vivo pharmacokinetic profile in comparison to bumetanide (Figures 2D and 2E), we proceeded to in vivo evaluations of our lead. First, we evaluated the diuretic effect of ARN23746 and bumetanide (as positive control) in adult (postnatal day P60–120) Ts65Dn mice and their WT littermates. We measured the urine volume for 2 h following treatment (0.2 mg kg−1 IP6; Figure 3A). As expected, bumetanide administration significantly increased the urine volume in WT and Ts65Dn mice relative to vehicle-treated mice. Conversely, ARN23746 treatment had no significant diuretic effect in WT or Ts65Dn mice (Figure 3B). This is in agreement with the lack of significant NKCC2 inhibition in vitro. Next, we investigated ARN23746’s efficacy in rescuing cognitive impairment in Ts65Dn mice, as previously tested with bumetanide.6 We evaluated the short-term working memory and the long-term hippocampus-dependent explicit memory after chronic (7–28 days) systemic treatment with ARN23746 (0.2 mg kg−1 IP, daily; Figure 3C). ARN23746 administration completely rescued the poor short-term working memory of Ts65Dn mice,30 which we assessed in the T-maze task by analyzing the correct choice of the previously unexplored arm (Figure 3D). Moreover, in the object-location (OL) test, ARN23746 treatment completely rescued the poor spatial memory of Ts65Dn mice to the level of WT littermates (Figure 3E). Similarly, ARN23746 administration completely rescued the poor novel-discrimination ability of Ts65Dn mice in the novel-object recognition (NOR) test (Figure 3F). ARN23746’s effect in the OL and NOR tests was not due to object preference (Table S4) or to alterations in total object exploration (Table S4). Finally, ARN23746 treatment fully restored associative memory in Ts65Dn mice in the contextual fear-conditioning (CFC) test, as demonstrated by the rescue of the poor freezing response induced after re-exposure to the training context 24 h after conditioning (Figure 3G). The rescue of the freezing response was not induced by altered sensitivity to shock or by changes in non-associative freezing (Table S4). Notably, 4 weeks of daily treatment with ARN23746 did not affect the general health of the mice, as evaluated by daily eye assessment, body weight measures (Figure S2A), and locomotor activity measures (Table S4). Moreover, there was no change in the weight of internal organs (kidneys, liver, spleen, lungs, and brain; Figure S2B) or in the histopathological profile of liver, kidney, and spleen (Figure S2C). This indicates that the compound was well-tolerated during chronic administration.
Figure 3.

ARN23746 Rescues Cognitive Impairment in the Ts65Dn Mouse Model of Down Syndrome with no Diuretic Effect
(A) Schematic cartoon of the experimental protocol for the treatment of adult WT and Ts65Dn mice to assess the diuretic effect.
(B) Quantification of the mean ± SEM and single animal cases of the urine volume collected for 120 min after mice were treated with the indicated drugs (two-way ANOVA, Ftreatment (2,69) = 10.516, p < 0.001,Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01).
(C) Schematic cartoon of the experimental protocol for the treatment of adult WT and Ts65Dn mice with ARN23746 for in vivo efficacy assessment of improved cognitive impairment in DS mice.
(D) Top, schematic representation of the T-maze test. Bottom, quantification of the mean ± SEM and single animal cases of correct choices in mice treated with the indicated drugs (two-way ANOVA, Finteraction (1,46) = 5.475, p = 0.024, Tukey’s post hoc test, ∗∗p < 0.01, ∗∗∗p < 0.001).
(E) Top, schematic representation of the OL test. Bottom, quantification of the mean ± SEM and single animal cases of discrimination index in mice treated with the indicated drugs (two-way ANOVA, Finteraction (1,45) = 15.523, p < 0.001, Tukey’s post hoc test; ∗∗∗p < 0.001).
(F) Top, schematic representation of the NOR task. Bottom, quantification of the mean ± SEM and single animal cases of the discrimination index in mice treated with the indicated drugs (two-way ANOVA, Ftreatment (1,46)= 7.154, p = 0.010, Tukey’s post hoc test, ∗ p < 0.05, two-way).
(G) Top, schematic representation of the CFC test. Bottom, quantification of the mean ± SEM and single animal cases of the freezing response in mice treated with the indicated drugs (two-way ANOVA on Ranks, Ftreatment (1,45)= 4.425, p = 0.041, Tukey’s post hoc test, ∗ p < 0.05, ∗∗p < 0.01). See also Figure S2; Table S4.
Finally, to further corroborate ARN23746 as a therapeutic for neurological disorders with impaired Cl− homeostasis and depolarizing GABA transmission,1, 2, 3 we assessed its efficacy compared with bumetanide in reducing behaviors related to ASD core symptoms in the valproic acid (VPA) mouse model of ASD.7 To the best of our knowledge, the literature contains no preclinical data on the efficacy of bumetanide treatment in postnatal mouse models of ASD, although bumetanide has showed positive outcomes in seven different clinical trials on children and young adults.9, 10, 11, 12, 13, 14, 15 As with the DS mice, we first evaluated the diuretic effect of ARN23746 and bumetanide (as positive control) in young adult (P40–60) VPA mice and controls (CTR) by measuring the urine volume for 2 h following treatment (0.2 mg kg−1 IP6; Figure 4A). Similarly to DS mice, bumetanide significantly increased the urine volume in control and VPA mice relative to vehicle-treated mice, whereas ARN23746 treatment had no significant diuretic effect in control or VPA mice (Figure 4B).
Figure 4.

ARN23746 Rescues Social Deficits and Repetitive Behaviors in the Valproic Acid Mouse Model of Autism, with No Diuretic Effect
(A) Schematic cartoon of the experimental protocol for the treatment of control (CTR) and VPA mice with bumetanide or ARN23746 to the assess the diuretic effect.
(B) Quantification of mean ± SEM and single animal cases of the urine volume collection for 120 min after mice were treated with the indicated drugs (two-way ANOVA on Ranks, Ftreatment (2,63) = 11.635, p < 0.001, Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01)
(C) Schematic cartoon of the experimental protocol for the treatment of young adult WT and VPA mice with ARN23746 for in vivo efficacy assessment of improved sociability and repetitive behaviors in ASD mice.
(D) Top, schematic representation of the three-chamber test. Bottom left, quantification of the mean ± SEM and single animal cases of sociability index in mice treated with the indicated drugs (two-way ANOVA, Fcondition (1,42) = 9.575, p = 0.003, Tukey’s post hoc test, ∗∗p < 0.01; ∗∗∗p < 0.001). Bottom right, quantification of the mean ± SEM and single animal cases of social novelty index in mice treated with the indicated drugs (two-way ANOVA, Fcondition (1,42)= 8.195, p = 0.007, Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01).
(E) Top, schematic representation of the male-female interaction test. Bottom quantification of the mean ± SEM and single animal cases of the interaction time in mice treated with the indicated drugs (two-way ANOVA, Ftreatment (1,42) = 6.351, p = 0.016, Tukey’s post hoc test, ∗∗p < 0.01).
(F) Top, schematic representation of the marble burying test. Bottom, quantification of the mean ± SEM and single animal cases of the number of marbles buried by mice treated with the indicated drugs (two-way ANOVA, Fcondition (1,56) = 6.727, p = 0.012, Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01).
(G) Top, schematic representation of the grooming test. Bottom quantification of the mean ± SEM and single animal cases of the grooming time for mice treated with the indicated drugs (two-way ANOVA on Ranks, Finteraction (1,59)= 5.019, p = 0.029, Tukey’s post hoc test, ∗p < 0.05, ∗∗p < 0.01).
(H) Top, representative immunoblots for NKCC1 and KCC2 on extracts of membrane-enriched protein fractions from prefrontal cortices of individuals diagnosed with ASD and controls. Bottom left, quantification of KCC2 in samples from individuals with ASD in comparison to age- and sex-matched controls (two-tailed t test, t = 2.128, p = 0.0492). Bottom right, quantification of the NKCC1/KCC2 expression ratio in samples from individuals with ASD in comparison to age- and sex-matched controls (Mann-Whitney rank sum test, p = <0,001). See also Figures S3 and S4 and Tables S4–S6.
Then, we evaluated the social interactions upon exposure to a stranger mouse and the repetitive behaviors (grooming and marble burying) after a chronic (7–21 day) systemic treatment with ARN23746 or bumetanide (0.2 mg kg−1 IP, daily; Figure 4C; Figure S3A) in young adult (P40 to P60) VPA mice. ARN23746 completely rescued the poor social interaction of VPA mice. This occurred for interaction with a never-met intruder versus an object (expressed as sociability index) and for a novel mouse versus an already-met mouse (expressed as social novelty index) in the three-chamber test (Figure 4D). Bumetanide also fully rescued the poor social behavior of VPA mice (Figure S3B). Moreover, ARN23746 rescued poor sociability during male-female interaction (Figure 4E), as did bumetanide (Figure S3C). Finally, ARN23746 reduced the repetitive behaviors in the marble burying test (Figure 4F) and in the grooming task (Figure 4G), while bumetanide significantly reduced repetitive behaviors in the marble burying test (Figure S3D) but showed only a non-significant trend toward WT levels in the grooming task (Figure S3E).
Finally, to increase our study’s translational value, we conducted a parallel investigation of the expression levels of NKCC1 and KCC2 in the cortex of adult VPA versus control mice and adult ASD individuals versus age/sex-matched controls (Table S5). In particular, for human samples, we focused on the prefrontal cortex (PFC), a brain region possibly involved in social and repetitive behaviors.31 Compared to controls, VPA mice had significantly lower expression of KCC2 protein in the membrane-enriched fraction (Figure S3F). This agrees with studies on hippocampi from VPA rats.7 Moreover, we found no significant differences in NKCC1 expression between VPA and control mice (WT: 100 ± 21.87; VPA: 104.8 ± 27.44). The NKCC1/KCC2 expression ratio was thus significantly increased in VPA versus control mice (Figure S3F). In agreement with these mice data, KCC2 expression in the membrane-enriched fraction of the PFC was significantly lower in samples from ASD individuals than from controls (Figure 4H). There were no significant differences in NKCC1 expression between ASD or control samples (control: 100 ± 28.97; ASD: 109.6 ± 43.39). Again, the NKCC1/KCC2 expression ratio was thus significantly increased in ASD PFC samples versus controls (Figure 4H).
Discussion
We discovered and developed a first selective NKCC1 inhibitor prompted by recent reports of the neurological activity of the diuretic bumetanide.2 Bumetanide inhibits NKCC1 and reduces core symptoms in several brain disorders characterized by aberrant Cl− homeostasis in animal models and in humans.2 Nevertheless, bumetanide also inhibits kidney NKCC2, exerting a strong diuretic effect that endangers drug compliance and may lead to hypokalemia and general ionic imbalance.12,13 In particular, the chronic administration of bumetanide in autistic subjects during a phase II clinical trial (3 months treatment)12 led to a high percentage of subjects presenting several treatment emergent adverse events (TEAEs; e.g., hypokalemia in 30% of the subjects at 0.5 mg bis in die (b.i.d.) bumetanide dosage and in 76% of the subjects at 2 mg b.i.d. bumetanide dosage). This caused a high percentage of drop out from the study (33% at the 2 mg b.i.d. dosage). These drawbacks prevent bumetanide from becoming a viable clinical option compatible with chronic treatment over possibly several years.6,12 Moreover, in the younger population, bumetanide can be dangerous because it can lead to dehydration and asthenia, loss of appetite,13 enuresis,12 encephalopathy in children (due to the general ionic imbalance),32 and potential ototoxicity in neonates (due to the inhibition of both NKCC1 and NKCC2).33, 34, 35 To overcome bumetanide’s limitations as a neurological drug, we report here on the discovery of a new molecular entity, ARN23746, with an NKCC1/NKCC2 inhibition ratio that is markedly higher than that of bumetanide. ARN23746 shows no significant NKCC2 inhibition in vitro and diuresis in vivo. It is, therefore, predicted to avoid the general ionic imbalance and to reduce, at least in part,34 the dangerous side effects and ototoxicity, possibly decreasing the dangers of early treatment. Given that most brain defects occurring in DS, ASD, and other neurodevelopmental disorders originate during development, an early therapeutic strategy could be indeed even more beneficial than in adults. Although we found here that cognitive impairment in DS, social and repetitive behavior in ASD can be rescued by NKCC1 inhibition in adult animals, chronic treatments are required.6 Earlier treatments with ARN23746 during development may result in ameliorated brain wiring and possibly in the requirement of no (or lower dosage) treatment in adulthood. Moreover, other disease symptoms, such as increased susceptibility to seizures and hyperactivity in DS, are not rescued by NKCC1 inhibition in adult animals.6 Earlier NKCC1 inhibition by ARN23746 during development may thus ameliorate brain wiring and possibly rescue these other symptoms too. However, since depolarizing/excitatory GABA-mediated responses are essential during early physiological brain development,1,3 it will be imperative to accurately determine when blockage of NKCC1 becomes detrimental and ensure that normal development is not hindered by premature exposure to ARN23476. In this overall context, ARN23746 may thus become a sustainable therapeutic for chronic treatment in adults and possibly also in the younger population.
Another drawback of bumetanide is its suboptimal blood-brain barrier (BBB) penetration. To enhance BBB penetration, some studies have evaluated lipophilic analogs of bumetanide.36,37 However, these studies have no insights about the analogs’ diuretic effect relative to bumetanide or about the establishment of a potential association between their activity and factual targeting of NKCC1.36 This has suggested the possibility that bumetanide may influence brain-related behaviors not by NKCC1 inhibition but by being a diuretic (i.e., through its well-known osmotic regulation and/or by altering ionic balance).38 Here, our study shows that core symptoms in DS and ASD mice are rescued by selective pharmacological NKCC1 inhibition with a compound with a better brain/plasma ratio than bumetanide. This further strengthens the idea that NKCC1 plays a major role in brain-related behaviors. Moreover, our pharmacological study rules out the possibility that bumetanide’s brain-related effects are due to osmotic regulation and/or ionic imbalance through excessive NKCC2-mediated diuresis.
In addition to DS and ASD, bumetanide rescues core symptoms in mouse models of several other brain disorders. Most importantly, bumetanide has shown positive outcomes in patients during clinical trials and case studies of neurodevelopmental (autism, fragile X, Asperger syndrome, schizophrenia), neurodegenerative (Parkinson disease), and neurological disorders (epilepsy).2 In particular, in seven independent clinical studies, including a phase II clinical trial, bumetanide reduced some of the diagnostic signs of ASD in children and young adults.9, 10, 11, 12, 13, 14, 15 Nevertheless, NKCC1 and KCC2 expression had not been investigated in ASD human brain samples. Here, we found a lower KCC2 expression in prefrontal cortices of adult human donors diagnosed with non-genetic forms of ASD. Moreover, bumetanide treatment in rodent models of ASD (i.e., rats exposed in utero to VPA and a mouse model of fragile X) had only been investigated at the end of gestation, which is not indicative of the current clinical protocols in patients.7 Our data show that ARN23746 and bumetanide both rescued the social deficits and repetitive behaviors in young adult animals of a mouse model of ASD. These findings offer preclinical evidence in support of the protocol performed in the several clinical trials with ADS individuals treated with bumetanide.9, 10, 11, 12, 13, 14, 15 We chose the VPA mouse model of ASD with the purpose of trying to mimic, at least in part, the non-genetic condition of ASD human samples that we had analyzed with biochemical approaches. In agreement with our findings in humans, our biochemistry data in VPA mice showed dysregulation of KCC2 not NKCC1. This indicates that, no matter which of the two transporters is dysregulated, it is the NKCC1/KCC2 ratio imbalance that must be targeted to restore the physiological Cl− homeostasis. This highlights the potential of ARN23746 not only to treat pathologies or disorders characterized by increased NKCC1 expression (e.g., DS,6 stroke, cerebral edema,39 glioma40) but also all the neurological disorders characterized by KCC2 downregulation (e.g., autism,7 Rett syndrome,41, 42, 43, 44 Huntington's disease45). In this respect, a combination therapy of ARN23746 with the newly discovered KCC2 activator CLP25746,47 (but see 48) or KCC2 expression-enhancing compounds49 could synergize their beneficial effects in improving the core symptoms of pathologies or disorders characterized by impaired Cl− homeostasis.
Conclusions
This is a time where neuroscience drug discovery is often dismissed in the pharma industry. This is due to repeated failures in developing small molecules that typically act on targets validated in animal models only, since target validation in humans is extremely challenging. In this context, drug repurposing is often proposed as an alternative strategy. However, this strategy often results in suboptimal care of the patient, since the repurposed drug still exerts the effects associated with its initial clinical indication. Here, we propose a new rational strategy for neuroscience drug discovery that leverages drug repurposing as a tool to safely validate drug targets shared between patients and animal models, and links it to the computer-aided drug design of new molecules based on the structure and mechanism of action of the repurposed drug. In doing so, we have discovered and developed a new compound ARN23746. ARN23746 is a selective NKCC1 inhibitor devoid of diuretic effects in vivo, displays no issues with off-target activity, has excellent solubility and metabolic stability in vitro, and does not induce toxicity following chronic treatment in vivo. This makes it a solid compound candidate ready for advanced preclinical and manufacturing studies toward development into a clinically relevant drug for unprecedented sustainable therapeutics in DS, ASD, and possibly the several other neurological conditions characterized by impaired Cl− homeostasis.
Experimental Procedures
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laura Cancedda (laura.cancedda@iit.it).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The original data generated during this study have been deposited to Mendeley Data (https://doi.org/10.17632/x9ttg84pzt.2).
Pharmacophore Generation and Ligand-Based Virtual Screening (LBVS)
The first pharmacophore model in this study was built starting from bumetanide and other NKCC1 inhibitors (i.e., furosemide, azosemide, piretanide, bendroflumethiazide, benzthiazide, chlorothiazide, metolazone, quinethazone). The nine NKCC1 inhibitors were designed with Maestro (Schrödinger suite version 2015-4) and prepared with LigPrep to retrieve the most probable protonation and tautomerization states at physiological pH. Since the active conformation of NKCC1 inhibitors is unknown, we decided to use the most energetically favored conformation (i.e., the energetic minimum) computed via MacroModel. This conformation was used for the subsequent pharmacophore modeling. For each of the nine compounds, phase software was used to obtain the pharmacophore features, such as H-bond donor and acceptor, lipophilic, and aromatic groups. The first pharmacophore model was obtained by removing from bumetanide's pharmacophore those features that were shared by the other eight compounds. The ‘generate phase database’ utility in Phase was used to prepare the internal and commercial molecular libraries for the LBVS. Subsequently, the libraries were filtered to retain only the molecules that obey Lipinsky’s rule and that do not bear reactive functional groups. The LBVS was conducted using Phase’s ‘find matches to hypothesis’ utility. In the first run (i.e., screening of the internal database), we used default parameters. In the second run (i.e., screening of the commercial library), we customized our settings by applying a strict tolerance value (i.e., < 1 Å) for the features that strongly impacted activity toward NKCC1 (e.g., sulfonamide moiety) and by increasing the tolerance values (i.e., >1 Å) for the remaining features. After visually inspecting the resulting matches, we selected the structures with the best scores for the in vitro tests.
Compound Synthesis
The detailed synthetic procedures of all compounds are available in Supplemental Information.
Compound Preparation for In Vitro Testing
All the compounds tested in vitro were dissolved in the assay buffer to a final concentration of 10 or 100 μM from a DMSO stock solution of 10 mM. As control, we used assay buffer supplemented with 0.1% DMSO or 1% DMSO.
HEK Cell Culture and Transfection
HEK293 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 1% L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and maintained at 37°C in a 5% CO2 humidified atmosphere. To assess NKCC1 and NKCC2 activity (Cl− influx assay), 3 million HEK cells were plated in a 10 cm cell-culture dish and transfected with a transfection mixture comprising 5 mL of DMEM, 4 mL Opti-MEM, 8 μg of DNA plasmid coding for NKCC1 (PRK-NKCC1 obtained from Medical Research Council and the University of Dundee), NKCC2 (OriGene plasmid #RC216145) subcloned in PRK5 plasmid, or mock control (empty vector), together with 8 μg of a plasmid coding for the Cl−-sensitive variant of the mbYFPQS (Addgene plasmid #80742),24 and 32 μL of Lipofectamin 2000. To assess KCC2 activity (Tl influx assay), cells were transfected with 16 μg of KCC250 subcloned in PRK5 plasmid or mock control (empty vector) and 32 μL of Lipofectamin 2000. After 4 h, the cells were collected and plated in 96-well black-walled, clear-bottomed plates at a density of 2.5 × 104. After 48 h, cells were used for the Cl− or Tl influx assays. All reagents were purchased from Life Technologies, unless otherwise specified.
Cl− Influx Assay in HEK Cells
Transfected cells were treated with 10 or 100 μM bumetanide and furosemide (as positive controls), DMSO (see negative control), or with each of our compounds in 100 μL/well of a Cl−-free-hypotonic solution (67.5 mM Na+ Gluconate, 2.5mM K+ Gluconate, 15mM HEPES pH 7.4, 50 mM Glucose, 1mM Na2HPO4, 1 mM NaH2PO4, 1 mM MgSO4, 1 mM CaSO4). After 30 min of incubation, plates were loaded into a Victor 3V (Perkin Elmer) multi-plate reader equipped with an automatic liquid injector system, and fluorescence of Cl−-sensitive mbYFPQS was recorded with excitation at 485 nm and emission at 535 nm. For each well, fluorescence was first recorded for 20 s of baseline and for 60 s after delivery of a NaCl concentrated solution (74 mM final concentration in assay well). Florescence of Cl−-sensitive mbYFPQS is inversely correlated to the intracellular Cl− concentration,24 therefore, chloride influx into the cells determined a decrease of mbYFPQS fluorescence. To represent the fluorescent traces in time, we normalized the fluorescence value for each time point to the average of the fluorescence value of the first 20 s of baseline (ΔF/F0). To quantify the average effects as represented by the bar plots, we expressed the decrease in fluorescence upon NaCl application as the average of the last 10 s of ΔF/F0 normalized traces. Moreover, for each experiment, to account for the contribution of Cl− changes that were dependent on transporters/exchangers other than NKCC1 or NKCC2, we subtracted the value of the last 10 s of ΔF/F0 normalized traces obtained from mock-transfected cells (either control or treated) from the respective ΔF/F0 value obtained from the cells transfected with NKCC1 or NKCC2. We then presented in the figures all the data as a percentage of the fluorescence decrease versus the value of the control DMSO.
Tl Influx Assay in HEK Cells
The Tl influx assay (FluxOR Potassium ion channel assay, Life Technologies) was modified from previously published protocols.29 In this assay, the amount of Tl+ ions (as a substitute for K+) entering the cells by KCC2 transport is detected with a Tl-sensitive fluorogenic dye that increases fluorescence upon Tl+ binding. Cell were loaded with a 1:1,000 dilution of Tl-sensitive fluorogenic dye (Component A) and 1:100 Probenecid (Component D) in 100 μL/well Cl− free-hypotonic solution (as above). After 1 h, cells were washed twice with 100 μL/well of hypotonic solution and treated with DIOA (Sigma, positive control), DMSO (as negative control), and ARN23746 diluted in 200 μL/well hypotonic solution and in the presence of 100 μM Ouabain to inhibit transport by the Na+/K+-ATPase pump. The plates were loaded onto a Spark (Tecan) multi-plate reader equipped with a double automatic liquid injector system. Fluorescence of Cl−-sensitive dye was recorded with excitation at 485 nm and emission at 535 nm. For each well, fluorescence was first recorded for 20 s of baseline, for another 20 s after TlSO4 delivery (2 mM final concentration in assay well), and for 60 s after delivery of a NaCl concentrated solution (74 mM final concentration in assay well). To represent the fluorescent traces in time, we normalized the fluorescence value for each time point to the average of the fluorescence value of the first 20 s of baseline (ΔF/F0). To quantify the average effects as represented by the bar plots, we expressed the increase in fluorescence upon NaCl application as the average of the last 10 s of ΔF/F0 normalized traces after subtracting the average of the last 10 s of ΔF/F0 normalized traces after TsSO4 injection. Moreover, for each experiment, to account for the contribution of Tl+ influx that was dependent on transporters/exchangers other than KCC2, we subtracted the value of the last 10 s of ΔF/F0 normalized traces obtained from mock-transfected cells (either control or treated) from the respective ΔF/F0 value obtained from the cells transfected with KCC2. We then presented in the figures all the data as a percentage of the fluorescence increase versus the value of the control DMSO.
Neuron Cultures
To perform the Ca2+ influx assay, primary neuronal cultures of dissociated hippocampal neurons were prepared from E18 C57BL/6J mice (Charles River), as previously described,51 and plated in 96-well black-walled, clear-bottomed plates coated with poly-L-lysine (Sigma; 0.1 mg/mL in 100 mM borate buffer, pH 8.5) at a density of 30,000 cells per well. Neurons were maintained in Neurobasal medium supplemented with 2% B-27 supplement, 0.5 mM glutamine, 50 U/mL of penicillin, and 50 μg/mL of streptomycin (all from Gibco). The cells were incubated at 37°C and 5% CO2 until DIV 3 for the Ca2+ influx assay. To perform the MQAE experiment and electrophysiology, primary neuronal cultures of dissociated hippocampal neurons were prepared from WT and Ts65Dn pups at postnatal day 2 (P2). Cells were plated on glass coverslips coated with poly-L-lysine at a density of 250–500 cells/mm2. Neurons were maintained in a culture medium consisting of Neurobasal-A supplemented with 2% B-27, 1% GlutaMax, and 5 μg/mL gentamycin (all from Gibco) at 37°C in humidified atmosphere (95% air, 5% CO2) until DIV 15 for the MQAE experiment and electrophysiology.
Ca2+ Influx Assay in Neuronal Cultures
At 3 DIVs, neurons were loaded with 2.5 μM of the calcium-sensitive day Fluo4-AM (Invitrogen) in extracellular solution (145 mM NaCl, 5 mM KCl, 10 mM HEPES, 5.55 mM Glucose, 1 mM MgCl2, 2 mM CaCl2, pH 7.4). After 15 min, cells were washed twice with extracellular solutions and treated with bumetanide or furosemide (as a positive control), DMSO (as negative control), or each of our compounds at 10 or 100 μM, in extracellular solution for 15 min. Plates were then loaded into a Victor 3V (Perkin Elmer) multi-plate reader equipped with an automatic liquid injector system and Fluo4 fluorescence was recorded with excitation at 485 nm and emission at 535 nm. For each well, fluorescence was first recorded for 20 s of baseline and for 20 s after delivery of GABA (100 μM). To evaluate neuronal viability, fluorescence was recorded for an additional 20 s after delivery of a depolarizing KCl stimulus (90 mM final concentration in wells). To represent the fluorescent traces in time, we normalized the fluorescence value for each time point to the average of the fluorescence value for the first 20 s of baseline (ΔF/F0). To quantify the average effects as represented by the bar plots, we measured the maximum peak increase in ΔF/F0 fluorescence upon GABA application normalized over the maximum peak increase in ΔF/F0 fluorescence upon KCl application. We then presented in the figures all the data as a percentage of the fluorescence increase versus the value of the control DMSO.
Electrophysiological Recordings in Neuronal Cultures
Hippocampal mouse neurons were recorded at DIV12-20 at room temperature (22°C–24°C) in an extracellular solution containing in mM: 145 NaCl, 5 KCl, 10 HEPES, 5.55 Glucose, 1 MgCl2, 2 CaCl2. Cells were visualized with an upright microscope (Olympus BX51WI) with infrared differential interference contrast optics. Patch pipettes (3–5 MΩ) were filled with an intracellular solution containing in mM: 130 K-Gluconate, 7 KCl, 10 HEPES, 4 MgATP, 0.6 EGTA, 0.3 NaGTP, 10 phospho-creatine; GABA currents were evoked by keeping the cell’s membrane potential at −65 mV and puffing 20 μM GABA (10 psi, 20–50 ms) using a Picospritzer III (Parker Instrumentation). Access resistance was monitored during voltage-clamp recordings, and cells with more than 25 MΩ were excluded. GABA-evoked currents were obtained by averaging five sweeps for each experimental condition; we discarded neurons with variations in GABA-current peaks greater than 20% compared with the first sweep recorded. Signals were sampled at 20 kHz, and low pass filtered at 10 kHz with an Axon Multiclamp 700B (Molecular Devices).
MQAE Cl− Imaging in Neuronal Cultures
Cl− imaging was performed with the fluorescent Cl−-sensitive indicator MQAE (N-(Ethoxycarbonylmethyl)-6-Methoxyquinolinium-Bromide; Molecular Probes) similarly to what was previously described.6 Neurons at 15 DIV were loaded with 5 mM MQAE in the presence of bumetanide (as positive control), DMSO (as negative control), or ARN23746 at 10 μM for 30 min at 37°C. Coverslips were then transferred to a holding chamber and perfused (2 mL∗min−1) with extracellular solution (NaCl 145 mM, KCl 5 mM, CaCl2 2 mM, MgCl2 1 mM, Hepes 10 mM, D-glucose 5.5 mM, pH 7.4) in the presence of the tested compound at 25°C for 5 min before imaging. Images were acquired with a Nikon A1 scanning confocal microscope equipped with a 20X air-objective (NA 0.75). MQAE was excited with a 405 nm diode laser, and fluorescence collected with a 525/50 nm band-pass emission filter. All excitation and acquisition parameters (laser intensity, PMT offset and gain) were kept constant throughout the experiments. Image analysis was performed with NIS-Elements software (Nikon) by measuring the mean fluorescent intensity of regions of interest (ROIs) centered on the cell body of single neurons from six randomly selected fields for each coverslip. For each experiment, the average fluorescent intensity of all ROIs from a coverslip was normalized to the average fluorescent intensity of control samples (WT neurons treated with DMSO) in the same experiment. Pseudo-color images were generated by ImageJ software (http://rsbweb.nih.gov/ij/).
Animals
All animal procedures were approved by IIT licensing in compliance with the Italian Ministry of Health (D.Lgs 26/2014) and EU guidelines (Directive 2010/63/EU). A veterinarian was employed to maintain the health and comfort of the animals. Mice were housed in filtered cages in a temperature-controlled room with a 12:12 h dark/light cycle and with ad libitum access to water and food. All efforts were made to minimize animal suffering and use the lowest possible number of animals required to produce statistical relevant results, according to the “3Rs concept.” In this study, we used Ts65Dn mice maintained in their original genetic background52 by crossing (more than 40 times) Ts65Dn female to C57BL/6JEi x C3SnHeSnJ (B6EiC3) F1 males (Jackson Laboratories). Ts65Dn mice were genotyped by PCR as previously described.53 We obtained the VPA mouse model of ASD by treating pregnant C57BL/6J (Charles River) dams at 12.5 days of pregnancy with 600 mg/kg (i.p.) of VPA dissolved in PBS. VPA-treated dams give birth to offspring that exhibit behaviors related to core signs of autism.7 As control, we used the offspring of C57BL/6J dams treated at E12.5 with PBS. Offspring aged between 4 and 16 weeks were used for experiments. For Ts65Dn and VPA mice and their respective controls, both males (for Ts65Dn and controls n = 48; for VPA and controls n = 41) and females (for Ts65Dn and controls n = 27; for VPA and controls n = 28) were used for the analysis of diuresis; only males were used for behavioral experiments. Both Ts65Dn and VPA mice with their respective controls, were randomly assigned to vehicle groups (2% DMSO in saline), bumetanide (Sigma, 0.2 mg∗Kg−1 body weight), or ARN23746 (0.2 mg∗Kg−1 body weight), and treated daily for 21–28 days by intraperitoneal injection (i.p.). On the day of behavioral testing, injection was performed 1 h before the task began.
Compound Preparation for In Vivo Experiments
For the in vivo experiments, bumetanide and ARN23746 were dissolved in DMSO in a stock solution of 1 mg/mL. On the day of the injection, the stock solution was dissolved in PBS at a concentration of 0.02 mg∗mL−1, and injected in a volume of 10 μL∗g−1 to have a final concentration of 0.2 mg∗kg−1.
Pharmacokinetic Analysis
An external contractor performed the study. Additional information on the study conditions can be obtained from the contractor’s website (https://www.pharmacologydiscoveryservices.com/). ARN23746 and bumetanide were administered i.v. at 2 mg/kg in phosphate-buffered saline (PBS), pH 7.4, with 2% DMSO to adult C57BL/6N mice (BioLASCO Taiwan). Blood collection was performed by cardiac puncture at 0, 5, 15, 30, 60, and 120 min. Blood aliquots (300–400 μL) were collected in tubes coated with lithium heparin, mixed gently, then kept on ice and centrifuged at 2,500 g for 15 min at 4°C, within 1 h of collection. The plasma was then harvested and stored frozen at (˗70°C) until further processing. Immediately after the blood sampling, mice were decapitated, and the whole brains were quickly removed, rinsed with cold saline (0.9% NaCl, g/mL), surface vasculature ruptured, blotted with dry gauze, weighed, and kept on ice until further processing, within 1 h of collection. Each brain was homogenized in 1.5 mL cold phosphate-buffered saline (PBS), pH 7.4, for 10 s on ice. The brain homogenate from each brain was then stored at ˗70°C until further processing. The plasma and brain samples were then processed using acetonitrile precipitation and analyzed by LC-MS/MS. Plots of plasma/brain concentration of the compound over time were constructed. The fundamental pharmacokinetic parameters of each compound after dosing (AUClast, AUCinf, T1/2, Cl, Vz, Vss, Tmax, and Cmax) were obtained from the non-compartmental analysis (NCA) of the plasma/brain data using WinNonlin.
Diuresis Analysis
Diuresis analysis was performed using mouse metabolic cages (Tecniplast,3600m021) equipped with a grid over a funnel and a plastic cone for the separate collection of urine and feces. Immediately after i.p. treatment with vehicle, bumetanide, or ARN23746 at 0.2 mg∗kg−1, animals were placed inside the metabolic cages (one animal per cage), where food and water were available ad libitum. After 2 h, mice were returned to their home cages, and the urine volume was measured.
Behavioral Testing
Ts65Dn male mice (8–16 weeks old) and VPA male mice (6–8 weeks old) were tested after 1 week of treatment with vehicle, ARN23746, or bumetanide (0.2 mg∗kg−1 i.p.). The battery of tests was run over a total period of 21–28 days (four behavioral tests for Ts65Dn and WT littermates in the following order: NOR, T-maze, NOL, CFC; four behavioral tests for VPA mice and their controls with the order and modalities described in Table S6). During the days of behavioral testing, animals were treated daily with the drug, with tests beginning 1 h after injection. The tasks were video-recorded and then analyzed manually by a blind operator. After each trial or experiment, the diverse apparatus and objects were cleaned with 70% ethanol.
T-Maze
The T-maze is a black opaque plastic apparatus with a starting arm and two perpendicular goal arms, each equipped with a sliding door and evenly illuminated by overhead red lighting (12–14 lux). The T-maze test (spontaneous alteration protocol, 11 trials) evaluates short-term memory by analyzing the correct choice of the unexplored arm. The test was performed in similar way to that previously conducted on Ts65Dn mice.30 In each trial, a mouse was first placed in the starting chamber for 20 s. Then, the sliding door was removed, and the animal was free to explore the apparatus. When the mouse entered (with all four limbs) one of the two goal arms, the opposite arm was closed with the sliding door. When the mouse (free to explore the remaining part of the apparatus) returned to the starting area, the previously closed goal arm was opened. The trial was repeated 11 times. Entry into a goal arm opposite the one previously chosen was considered a correct choice, while entry into the previously explored arm was considered an incorrect choice. Alternation score was calculated as the percentage of correct choices (i.e., left-right or right-left) over the total number of the ten possible alternations.
Object Location Test (OL)
The test evaluates the spatial memory by measuring the ability of mice to recognize the new location of a familiar object. The test was performed in a gray acrylic arena (44 × 44 cm), evenly illuminated by overhead red lighting (12–14 lux). Mice were first habituated to the chamber for 15 min on day 1. On day 2, during the acquisition phase, mice were exposed to two identical objects for 15 min. After 24 h, one of the two objects was moved during the test session to a novel location, and the mice were tested for 15 min for their ability to recognize the new location of the object. The time spent exploring each object was defined as the number of seconds during which mice showed investigative behavior (i.e., head orientation, sniffing occurring within < 1.0 cm) or clear contact between the nose and the object. A discrimination index was calculated as the percentage of time spent investigating the object in the new location minus the percentage of time spent investigating the object in the old location [discrimination index = (new object location exploration time/total exploration time × 100) – (old object location exploration time/total exploration time × 100]. As a control, we monitored object preference during the acquisition phase and the exploration time in the acquisition phase and trial phase. Of note, during the trial phase, Ts65Dn mice treated with ARN23746 had significantly longer exploration times (Table S4), possibly indicative of greater explorative interest for the objects. Nevertheless, given that the exploration of the single object was normalized against the total exploration time for each group, the increased exploration in Ts65Dn mice treated with ARN23746 did not affect the results of the discrimination index in the test.
NOR Test
The test evaluates long-term object recognition memory by measuring the ability of mice to recognize a new object with respect to familiar objects. The test was performed in a gray acrylic arena (44 × 44 cm), evenly illuminated by overhead red lighting (12–14 lux). On day 1, mice were habituated to the arena by freely exploring the chamber for 15 min. On day 2, during the acquisition phase, mice were free to explore three different objects (different in color, size, shape, material) for 15 min. After 24 h, one object from the acquisition phase was replaced with a novel object, and the mice were tested for 15 min for their ability to recognize the new object. The time spent exploring each object was defined as the number of seconds during which mice showed investigative behavior (i.e., head orientation, sniffing occurring within < 1.0 cm) or clear contact between the object and the nose. The time spent exploring each object, expressed as a percentage of the total exploration time, was measured for each trial. The discrimination index was calculated as the difference between the percentages of time spent investigating the novel object and investigating the familiar objects: discrimination index = (novel-object exploration time/total exploration time × 100) − (familiar object exploration time/total exploration time × 100). As a control, we monitored object preference during the acquisition phase and exploration time in the acquisition phase and trial phase. Of note, both during the acquisition phase and trial phase, Ts65Dn mice showed a significant increase in exploration time (Table S4), possibly indicative of their hyperactivity. Nevertheless, given that the exploration of the single object was normalized on the total exploration time for each group, the increased exploration in Ts65Dn did not affect the results on the discrimination index in the test. Moreover, although there was a preference of ∼10% for object B within the same group in all four groups, the preference was not significantly different among the four groups (Table S4). Moreover, although there was a significant general preference for object C in WT versus TS65Dn mice, the preference was not significantly different among the four single groups as assessed with Tukey post hoc test (Table S4).
CFC Test
The test evaluates the long-term associative memory by measuring the freezing time of the animals placed in a location where they had received an adverse stimulus (electric shock) 24 h earlier. The experiments were performed in a fear-conditioning system (TSE), which is a transparent acrylic conditioning chamber (23 × 23 cm) equipped with a stainless-steel grid floor. Mice were placed outside the experimental room in their home cages before the test and individually transported to the TSE apparatus in standard cages. Mice were placed in the conditioning chamber, and they received one electric shock (2 s, 0.75mA constant electric current) through the floor grid 3 min later. Mice were removed 15 s after the shock. After 24 h, mice were placed in the same chamber for 3 min. After 2 h, they were moved to a new context (black chamber with plastic gray floor and vanilla odor). The time spent frozen was scored and expressed as percentage of the total time analyzed.
Three-Chamber Test
The test evaluates the social approach of the tested mouse versus a never-met intruder in comparison to an object (sociability) or versus a novel never-met intruder in comparison to the already-met intruder (social novelty). It was performed in a similar way to that previously described for mouse models of ASD.54 The three-chamber apparatus comprises a rectangle, three-chambered box of gray acrylic, evenly illuminated by overhead red lighting (12–14 lux). The chambers are accessible by rectangle openings with sliding doors. In the first 10 min (habituation), the tested mouse was free to explore the apparatus of two inverted stainless-steel wire pencil cups (one in each of the two side chambers), with a weighted plastic cup on top to prevent the mouse climbing on the top. Then, the tested mouse was briefly confined in the center chamber, while a never-met intruder (previously habituated to the apparatus) was placed in one of the side chambers, under the pencil cup. For the following 10 min (sociability test), the tested mouse was allowed to explore all three chambers. Then, the tested mouse was again briefly confined in the center chamber, while a novel never-met intruder (previously habituated to the apparatus) was placed in the other side chamber under the pencil cup. For the following 10 min (social novelty test), the tested mouse was allowed to explore all the three chambers. The time spent exploring the object or the intruder was calculated by measuring the number of seconds when the mice showed investigative behavior (i.e., head orientation, sniffing occurring within < 1.0 cm). The sociability index was calculated as the difference between the time spent investigating the never-met intruder and the time spent investigating the familiar object divided by the total exploration time: sociability index = (never-met intruder exploration time − object exploration time) / (never-met intruder exploration time + object exploration time). The social novelty index was calculated as the difference between the time spent investigating the never-met intruder and the time spent investigating the already-met intruder divided by the total exploration time: social novelty index = (never-met intruder exploration time − already-met intruder exploration time) / (never-met intruder exploration time + already-met intruder exploration time).
Male-Female Interaction Test
The test evaluates the social approach of the tested mouse versus a never-met female mouse. It was performed in a similar way to that previously described for mouse models of ASD.55 Briefly, the tested mouse was placed alone in a cage (26 × 48 × 20 cm). After 5 min of habituation, an unfamiliar C57BL/6J female mouse was placed in the home cage of the isolated male mouse, and behavior was recorded for 5 min. The time spent interacting with the female intruder was calculated by measuring the seconds when the mice showed any of the following interacting behavior: anogenital sniffing, body sniffing, head sniffing, following, mounting.
Marble Burying Test
The test evaluates the repetitive behavior as the tendency to dig and bury marbles in a novel environment. It was performed as previously described in ASD mice.54 Briefly, mice were placed alone in a cage (26 × 48 × 20 cm) filled with 4 cm fresh mouse bedding material, evenly illuminated by overhead red lighting (12–14 lux). After 30 min habituation, the mouse was briefly removed, and bedding surface was leveled to place 15 glass marbles equidistantly distributed in a 3 × 5 arrangement. Then, the mouse was returned to the cage and allowed to explore for 30 min. Buried marbles were counted as those marbles that were covered by at least two-thirds.
Grooming
The test evaluates repetitive behavior in mouse grooming of all body parts. It was performed as previously described in ASD mice.56 Self-grooming was defined as licking or scratching the head or body parts with any of the forelimbs. Briefly, mice were placed individually into a clear Plexiglass cylinder (30 cm high, 10 cm wide). After 10 min habituation, the mouse was scored for 5 min for the time spent self-grooming in all body regions. For behavioral experiments, we adopted the following exclusion criteria independent of genotype or treatment (before blind code was broken). In the T-maze test, we excluded mice that did not conclude the 10 trials within 20 min of the test. In the CFC test, we excluded mice showing very high non-associative freezing in the new context. This was defined as more than 30 s freezing during the 3-min test. In the OL and NOR test, we excluded animals showing very low explorative behavior. This was defined as less than 10 s of direct object exploration during the 15-min test. Following these criteria, a total of 5 mice among the NOR, NOL, and T-maze tests were excluded. Original data used to calculate the discrimination index (for NOR and OL), sociability index, and social novelty index (for three-chamber test) have been deposited to Mendeley Data (https://doi.org/10.17632/x9ttg84pzt.2).
Human Brain Samples
PFC samples (Brodmann area 9) from adult humans with ASD and age- and sex-matched controls were obtained from the NeuroBioBank (NIH). Information on the samples is reported in Table S5. Samples were cryo-pulverized in dry ice (˗78°C) with a stainless-steel mortar. Aliquots of pulverized tissue were used for protein extraction.
Biochemistry
Cells were lysated in RIPA buffer (1% NP40, 0.5% Deoxycholic acid, 0.1% SDS, 150 mM NaCl 1 mM EDTA, 50 mM Tris, pH 7.4) containing 1% (v/v) protease and phosphatase inhibitor cocktails (Sigma). After 30 min incubation in ice, the samples were clarified by centrifugation at 20,000 × g for 20 min at 4°C. Brain samples from VPA and WT mice and from ASD people and controls were processed to obtain extracts of membrane-enriched fractions, as previously described.6 Samples were homogenized in ice-cold homogenization buffer (320 mM sucrose, 1 mM EDTA, 10 mM Tris, pH 7.4) supplemented with 1 mM PMSF, 10 mM NaF, 2 mM sodium orthovanadate, and 1% (v/v) protease and phosphatase inhibitor cocktail (Sigma). Homogenates were first centrifuged at 800 g to remove nuclei and debris. The resulting supernatant (S1) was further centrifuged at 9,200 g. The resulting pellet (P2), representing the membrane-enriched fraction, was extracted in RIPA buffer (as above) and used for western blot. The protein concentration of samples was determined with BCA kit (Pierce). To perform immunoblot, protein extracts were diluted in lithium-dodecyl-sulfate (LDS) sample buffer (ThermoFisher Scientific), with the addition of 50 mM DTT. To avoid NKCC1 and KCC2 protein precipitation or aggregation, all samples were warmed at 40°C for 5 min.6 Equivalent amounts of protein (20 μg) were loaded on the 4%–12% Bis-Tris NuPAGE pre-cast gels (Invitrogen), and electrophoresis were performed with MOPS buffer (Life Technologies). Next, gels were transferred overnight at 4°C onto nitrocellulose membranes (GE Healthcare) with Tris-Glycine transfer buffer (25 mM Tris-base, 192 mM glycine, 20% Methanol). Equal amounts of protein loading was verified by staining with 0.1% Ponceau. Membranes were blocked for 1 h in 5% milk in Tris-buffered saline (10 mM Tris, 150 mM NaCl, pH 8.0) plus 0.1% Tween-20 and incubated overnight at 4°C with primary antibodies: rabbit anti-actin (Sigma, catalog no: A2066; 1:10,000), mouse anti-NKCC1 (clone T4c, Developmental Studies Hybridoma Bank; 1:4,000), rabbit anti-KCC2 (Millipore catalog no: 07-432; 1:4000), sheep anti-NKCC2 (MRC-PPU, 1:2,000), and mouse anti-Na+/K+-ATPase (clone C464.6, Millipore catalog no. 05-369; 1:2,000). Next, membranes were washed and incubated for 2 h at room temperature with HRP-conjugated goat secondary antibodies (ThermoFisher Scientific; 1:10,000). Membranes were developed with SuperSignal West Pico chemiluminescent substrate (ThermoFisher Scientific). The chemiluminescent signals were acquired on the LAS 4000 Mini imaging system (GE Healthcare). Bands were later quantified by measuring the mean intensity of the band signal using ImageQuant software (GE Healthcare). Uncropped blots can be found in Figure S4. For KCC2 analysis, we considered the monomeric form, both in human and in mouse samples.
Statistical Analysis
The results are presented as the means ± SEM. The statistical analysis was performed using SigmaPlot 13.0 (Systat) software. Where appropriate, the statistical significance was assessed using the following parametric test: Student’s t test, one-way ANOVA followed by Dunnet post hoc test, two-way ANOVA followed by all pairwise Tukey post hoc test. Where normal distribution or equal variance assumptions were not valid, statistical significance was evaluated using Mann-Whitney Rank Sum Test, Kruskal-Wallis One Way ANOVA with Dunn’s post hoc test, or two-way ANOVA on ranks followed by all pairwise Dunn’s post hoc test. p values < 0.05 were considered significant. Outliers were excluded only from the final pool of data by a Grubb’s test run iteratively until no outliers were found.
Acknowledgments
This work was supported by Telethon (grant TCP15021 to L.C). This project received partial funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research And Innovation Program (grant agreement no 725563 to L.C.). Brain samples from individuals with ASD and controls were obtained from the NeuroBioBank (NIH). We thank Marina Nanni (IIT, NBT) for technical support, the staff of the IIT animal facility, Silvia Venzano, Giuliana Ottonello, and Sine Mandrup Bertozzi (IIT, D3) for technical assistance.
Author Contributions
A.S. designed and performed in vitro assays, diuresis experiment, behavioral experiments, western blot, and analyzed data; M.B. and J.A.O. designed and synthesized the compounds; R.N. collected and analyzed the electrophysiology data; G.L.S performed the virtual library screening; M.S. and R.B. performed the histopathological analysis; A.A. coordinated bioanalytical experiments; A.C. designed the in vitro assays and performed the MQAE assay; A.S., M.B., A.C., M.D.V., and L.C. designed the experiments and wrote the manuscript; all authors read and revised the manuscript.
Declaration of Interests
A.C. and L.C. are named as co-inventors on the following granted patent: US 9,822,368; EP 3083959; JP 6490077; A.C. and L.C. are named as co-inventors on the patent application WO 2018/189225. A.S., M.B., J.A.O., A.C., M.D.V., and L.C. are named as co-inventors on patent application IT 102019000004929.
Published: July 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chempr.2020.06.017.
Contributor Information
Marco De Vivo, Email: marco.devivo@iit.it.
Laura Cancedda, Email: laura.cancedda@iit.it.
Supplemental Information
References
- 1.Deidda G., Bozarth I.F., Cancedda L. Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci. 2014;8:119. doi: 10.3389/fncel.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Ari Y. NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. 2017;40:536–554. doi: 10.1016/j.tins.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Schulte J.T., Wierenga C.J., Bruining H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018;90:260–271. doi: 10.1016/j.neubiorev.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Contestabile A., Magara S., Cancedda L. The GABAergic hypothesis for cognitive disabilities in Down syndrome. Front. Cell. Neurosci. 2017;11:54. doi: 10.3389/fncel.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cellot G., Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2014;2:70. doi: 10.3389/fped.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deidda G., Parrini M., Naskar S., Bozarth I.F., Contestabile A., Cancedda L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015;21:318–326. doi: 10.1038/nm.3827. [DOI] [PubMed] [Google Scholar]
- 7.Tyzio R., Nardou R., Ferrari D.C., Tsintsadze T., Shahrokhi A., Eftekhari S., Khalilov I., Tsintsadze V., Brouchoud C., Chazal G. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 8.Kharod S.C., Kang S.K., Kadam S.D. Off-label use of bumetanide for brain disorders: an overview. Front. Neurosci. 2019;13:310. doi: 10.3389/fnins.2019.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadjikhani N., Åsberg Johnels J., Lassalle A., Zürcher N.R., Hippolyte L., Gillberg C., Lemonnier E., Ben-Ari Y. Bumetanide for autism: more eye contact, less amygdala activation. Sci. Rep. 2018;8:3602. doi: 10.1038/s41598-018-21958-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadjikhani N., Zürcher N.R., Rogier O., Ruest T., Hippolyte L., Ben-Ari Y., Lemonnier E. Improving emotional face perception in autism with diuretic bumetanide: a proof-of-concept behavioral and functional brain imaging pilot study. Autism. 2015;19:149–157. doi: 10.1177/1362361313514141. [DOI] [PubMed] [Google Scholar]
- 11.Lemonnier E., Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 2010;99:1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x. [DOI] [PubMed] [Google Scholar]
- 12.Lemonnier E., Degrez C., Phelep M., Tyzio R., Josse F., Grandgeorge M., Hadjikhani N., Ben-Ari Y. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry. 2012;2:e202. doi: 10.1038/tp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemonnier E., Villeneuve N., Sonie S., Serret S., Rosier A., Roue M., Brosset P., Viellard M., Bernoux D., Rondeau S. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry. 2017;7:e1056. doi: 10.1038/tp.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du L., Shan L., Wang B., Li H., Xu Z., Staal W.G., Jia F. A pilot study on the combination of applied behavior analysis and bumetanide treatment for children with autism. J. Child Adolesc. Psychopharmacol. 2015;25:585–588. doi: 10.1089/cap.2015.0045. [DOI] [PubMed] [Google Scholar]
- 15.Bruining H., Passtoors L., Goriounova N., Jansen F., Hakvoort B., de Jonge M., Poil S.S. Paradoxical benzodiazepine response: a rationale for bumetanide in neurodevelopmental disorders? Pediatrics. 2015;136:e539–e543. doi: 10.1542/peds.2014-4133. [DOI] [PubMed] [Google Scholar]
- 16.Grandgeorge M., Lemonnier E., Degrez C., Jallot N. The effect of bumetanide treatment on the sensory behaviours of a young girl with Asperger syndrome. BMJ Case Rep. 2014;2014 doi: 10.1136/bcr-2013-202092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemonnier E., Lazartigues A., Ben-Ari Y. Treating schizophrenia with the diuretic bumetanide: a case report. Clin. Neuropharmacol. 2016;39:115–117. doi: 10.1097/WNF.0000000000000136. [DOI] [PubMed] [Google Scholar]
- 18.Lemonnier E., Robin G., Degrez C., Tyzio R., Grandgeorge M., Ben-Ari Y. Treating fragile X syndrome with the diuretic bumetanide: a case report. Acta Paediatr. 2013;102:e288–e290. doi: 10.1111/apa.12235. [DOI] [PubMed] [Google Scholar]
- 19.Damier P., Hammond C., Ben-Ari Y. Bumetanide to treat Parkinson disease: a report of 4 cases. Clin. Neuropharmacol. 2016;39:57–59. doi: 10.1097/WNF.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 20.Eftekhari S., Mehvari Habibabadi J., Najafi Ziarani M., Hashemi Fesharaki S.S., Gharakhani M., Mostafavi H., Joghataei M.T., Beladimoghadam N., Rahimian E., Hadjighassem M.R. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia. 2013;54:e9–e12. doi: 10.1111/j.1528-1167.2012.03654.x. [DOI] [PubMed] [Google Scholar]
- 21.Rahmanzadeh R., Eftekhari S., Shahbazi A., Khodaei Ardakani M.R., Rahmanzade R., Mehrabi S., Barati M., Joghataei M.T. Effect of bumetanide, a selective NKCC1 inhibitor, on hallucinations of schizophrenic patients; a double-blind randomized clinical trial. Schizophr. Res. 2017;184:145–146. doi: 10.1016/j.schres.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Pressler R.M., Boylan G.B., Marlow N., Blennow M., Chiron C., Cross J.H., de Vries L.S., Hallberg B., Hellström-Westas L., Jullien V. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015;14:469–477. doi: 10.1016/S1474-4422(14)70303-5. [DOI] [PubMed] [Google Scholar]
- 23.Kahle K.T., Barnett S.M., Sassower K.C., Staley K.J. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(−) cotransporter NKCC1. J. Child Neurol. 2009;24:572–576. doi: 10.1177/0883073809333526. [DOI] [PubMed] [Google Scholar]
- 24.Watts S.D., Suchland K.L., Amara S.G., Ingram S.L. A sensitive membrane-targeted biosensor for monitoring changes in intracellular chloride in neuronal processes. PLoS One. 2012;7:e35373. doi: 10.1371/journal.pone.0035373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lykke K., Töllner K., Feit P.W., Erker T., MacAulay N., Löscher W. The search for NKCC1-selective drugs for the treatment of epilepsy: structure-function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav. 2016;59:42–49. doi: 10.1016/j.yebeh.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Töllner K., Brandt C., Töpfer M., Brunhofer G., Erker T., Gabriel M., Feit P.W., Lindfors J., Kaila K., Löscher W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 2014;75:550–562. doi: 10.1002/ana.24124. [DOI] [PubMed] [Google Scholar]
- 27.Yuste R., Katz L.C. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:333–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]
- 28.Korpi E.R., Lüddens H. Furosemide interactions with brain GABAA receptors. Br. J. Pharmacol. 1997;120:741–748. doi: 10.1038/sj.bjp.0700922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delpire E., Days E., Lewis L.M., Mi D., Kim K., Lindsley C.W., Weaver C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA. 2009;106:5383–5388. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleschevnikov A.M., Belichenko P.V., Faizi M., Jacobs L.F., Htun K., Shamloo M., Mobley W.C. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J. Neurosci. 2012;32:9217–9227. doi: 10.1523/JNEUROSCI.1673-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ha S., Sohn I.J., Kim N., Sim H.J., Cheon K.A. Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp. Neurobiol. 2015;24:273–284. doi: 10.5607/en.2015.24.4.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danziger J., Zeidel M.L. Osmotic homeostasis. Clin. J. Am. Soc. Nephrol. 2015;10:852–862. doi: 10.2215/CJN.10741013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delpire E., Lu J., England R., Dull C., Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 34.Kakigi A., Nishimura M., Takeda T., Taguchi D., Nishioka R. Expression of aquaporin1, 3, and 4, NKCC1, and NKCC2 in the human endolymphatic sac. Auris Nasus Larynx. 2009;36:135–139. doi: 10.1016/j.anl.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Ben-Ari Y., Damier P., Lemonnier E. Failure of the Nemo trial: bumetanide is a promising agent to treat many brain disorders but not newborn seizures. Front. Cell. Neurosci. 2016;10:90. doi: 10.3389/fncel.2016.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandt C., Seja P., Töllner K., Römermann K., Hampel P., Kalesse M., Kipper A., Feit P.W., Lykke K., Toft-Bertelsen T.L. Bumepamine, a brain-permeant benzylamine derivative of bumetanide, does not inhibit NKCC1 but is more potent to enhance phenobarbital's anti-seizure efficacy. Neuropharmacology. 2018;143:186–204. doi: 10.1016/j.neuropharm.2018.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Auer T., Schreppel P., Erker T., Schwarzer C. Functional characterization of novel bumetanide derivatives for epilepsy treatment. Neuropharmacology. 2020;162:107754. doi: 10.1016/j.neuropharm.2019.107754. [DOI] [PubMed] [Google Scholar]
- 38.Hochman D.W. The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia. 2012;53:18–25. doi: 10.1111/j.1528-1167.2012.03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan Y., Dempsey R.J., Flemmer A., Forbush B., Sun D. Inhibition of Na(+)-K(+)-Cl(−) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961:22–31. doi: 10.1016/s0006-8993(02)03832-5. [DOI] [PubMed] [Google Scholar]
- 40.Schiapparelli P., Guerrero-Cazares H., Magana-Maldonado R., Hamilla S.M., Ganaha S., Goulin Lippi Fernandes E., Huang C.H., Aranda-Espinoza H., Devreotes P., Quinones-Hinojosa A. NKCC1 regulates migration ability of glioblastoma cells by modulation of actin dynamics and interacting with cofilin. EBioMedicine. 2017;21:94–103. doi: 10.1016/j.ebiom.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duarte S.T., Armstrong J., Roche A., Ortez C., Pérez A., Mdel O'Callaghan, Mdel M., Pereira A., Sanmartí F., Ormazábal A., Artuch R. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One. 2013;8:e68851. doi: 10.1371/journal.pone.0068851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang X., Kim J., Zhou L., Wengert E., Zhang L., Wu Z., Carromeu C., Muotri A.R., Marchetto M.C., Gage F.H., Chen G. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA. 2016;113:751–756. doi: 10.1073/pnas.1524013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinz L., Torrella Barrufet J., Heine V.M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta neuropathol. Commun. 2019;7:196. doi: 10.1186/s40478-019-0852-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banerjee A., Rikhye R.V., Breton-Provencher V., Tang X., Li C., Li K., Runyan C.A., Fu Z., Jaenisch R., Sur M. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA. 2016;113:E7287–E7296. doi: 10.1073/pnas.1615330113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu Y.T., Chang Y.G., Chang C.P., Siew J.J., Chen H.M., Tsai C.H., Chern Y. Altered behavioral responses to gamma-aminobutyric acid pharmacological agents in a mouse model of Huntington's disease. Mov. Disord. 2017;32:1600–1609. doi: 10.1002/mds.27107. [DOI] [PubMed] [Google Scholar]
- 46.Gagnon M., Bergeron M.J., Lavertu G., Castonguay A., Tripathy S., Bonin R.P., Perez-Sanchez J., Boudreau D., Wang B., Dumas L. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gagnon M., Bergeron M.J., Perez-Sanchez J., Plasencia-Fernández I., Lorenzo L.E., Godin A.G., Castonguay A., Bonin R.P., De Koninck Y. Reply to the small molecule CLP257 does not modify activity of the K+-Cl- co-transporter KCC2 but does potentiate GABAA receptor activity. Nat. Med. 2017;23:1396–1398. doi: 10.1038/nm.4449. [DOI] [PubMed] [Google Scholar]
- 48.Cardarelli R.A., Jones K., Pisella L.I., Wobst H.J., McWilliams L.J., Sharpe P.M., Burnham M.P., Baker D.J., Chudotvorova I., Guyot J. The small molecule CLP257 does not modify activity of the K+-Cl- co-transporter KCC2 but does potentiate GABAA receptor activity. Nat. Med. 2017;23:1394–1396. doi: 10.1038/nm.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang X., Drotar J., Li K., Clairmont C.D., Brumm A.S., Sullins A.J., Wu H., Liu X.S., Wang J., Gray N.S. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cancedda L., Fiumelli H., Chen K., Poo M.M. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaech S., Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 52.Reeves R.H., Irving N.G., Moran T.H., Wohn A., Kitt C., Sisodia S.S., Schmidt C., Bronson R.T., Davisson M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 53.Reinholdt L.G., Ding Y., Gilbert G.J., Czechanski A., Solzak J.P., Roper R.J., Johnson M.T., Donahue L.R., Lutz C., Davisson M.T. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm. Genome. 2011;22:685–691. doi: 10.1007/s00335-011-9357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eissa N., Jayaprakash P., Azimullah S., Ojha S.K., Al-Houqani M., Jalal F.Y., Łażewska D., Kieć-Kononowicz K., Sadek B. The histamine H3R antagonist DL77 attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Sci. Rep. 2018;8:13077. doi: 10.1038/s41598-018-31385-7. 13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drapeau E., Riad M., Kajiwara Y., Buxbaum J.D. Behavioral phenotyping of an improved mouse model of Phelan-McDermid syndrome with a complete deletion of the Shank3 gene. E.Neuro. 2018;5 doi: 10.1523/ENEURO.0046-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campolongo M., Kazlauskas N., Falasco G., Urrutia L., Salgueiro N., Höcht C., Depino A.M. Sociability deficits after prenatal exposure to valproic acid are rescued by early social enrichment. Mol. Autism. 2018;9:36. doi: 10.1186/s13229-018-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data generated during this study have been deposited to Mendeley Data (https://doi.org/10.17632/x9ttg84pzt.2).