

Abstract
Background
Aortic aneurysms and dissections are highly lethal diseases for which an effective treatment strategy is critically needed to prevent disease progression. The nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)–caspase‐1 inflammasome cascade was recently shown to play an important role in aortic destruction and disease development. In this study, we tested the effects of MCC950, a potent, selective NLRP3 inhibitor, on preventing aortic destruction and aortic aneurysm and dissection formation.
Methods and Results
In a model of sporadic aortic aneurysm and dissection induced by challenging wild‐type mice with a high‐fat, high‐cholesterol diet and angiotensin II infusion, MCC950 treatment significantly inhibited challenge‐induced aortic dilatation, dissection, and rupture in different thoracic and abdominal aortic segments in both male and female mice. Aortic disease reduction by MCC950 was associated with the prevention of NLRP3–caspase‐1 upregulation, smooth muscle cell contractile protein degradation, aortic cell death, and extracellular matrix destruction. Further investigation revealed that preventing matrix metallopeptidase 9 (MMP‐9) expression and activation in macrophages is an important mechanism underlying MCC950's protective effect. We found that caspase‐1 directly activated MMP‐9 by cleaving its N‐terminal inhibitory domain. Moreover, the genetic knockdown of Nlrp3 or Casp‐1 in mice or treatment of mice with MCC950 diminished the challenge‐induced N‐terminal cleavage of MMP‐9, MMP‐9 activation, and aortic destruction.
Conclusions
Our findings suggest that the NLRP3–caspase‐1 inflammasome directly activates MMP‐9. Targeting the inflammasome with MCC950 is a promising approach for preventing aortic destruction and aortic aneurysm and dissection development.
Keywords: aortic destruction, MCC950, MMP‐9 activation, NLRP3 inflammasome inhibitor
Subject Categories: Aneurysm, Aortic Dissection, Vascular Disease, Vascular Biology, Basic Science Research
Clinical Perspective
What Is New?
The nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3–caspase‐1 inflammasome activates matrix metallopeptidase 9.
Caspase‐1 directly activates matrix metallopeptidase 9 by cleaving its N‐terminal inhibitory domain.
MCC950 prevents challenge‐induced nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3–caspase‐1 inflammasome activation, aortic destruction, and aortic aneurysm and dissection formation.
What Are the Clinical Implications?
Targeting the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3–caspase‐1 inflammasome with MCC950 is a promising approach for preventing aortic destruction and aortic aneurysm and dissection development.
Introduction
Aortic aneurysms and dissections (AADs) are life‐threatening cardiovascular diseases for which there is no effective pharmacologic therapy.1, 2, 3 Therefore, developing an effective treatment strategy that prevents disease progression is critical. An increasing amount of evidence has indicated that the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)–caspase‐1 inflammasome cascade, an important regulator of the inflammatory response,4, 5 plays an important role in vascular inflammation and aortic aneurysm formation in mice that were challenged with angiotensin II (AngII).6, 7, 8 The NLRP3–caspase‐1 inflammasome has been shown to activate macrophages and promote aortic inflammation and destruction.6, 7 Furthermore, the NLRP3–caspase‐1 inflammasome also degrades contractile proteins in vascular smooth muscle cells (SMCs), leading to aortic dysfunction.8
MCC950 is a novel, potent selective small‐molecule NLRP3‐inflammasome inhibitor9 that was recently shown to reduce myocardial infarct size in a porcine model of myocardial infarction10 and to inhibit atherosclerotic lesion development in apolipoprotein E–deficient mice.11 Moreover, MCCC950 has superior pharmacologic characteristics including high potency (7.5 nM), oral bioavailability (68%), and temporal application.12 Given the critical role of the NLRP3–caspase‐1 inflammasome in AAD formation, we sought to determine the therapeutic potential of preventing AAD progression by targeting this cascade with MCC950. We found that the NLRP3–caspase‐1 inflammasome directly activates matrix metallopeptidase (MMP) 9 (MMP‐9) by cleaving its N‐terminal inhibitory domain. Our findings indicated that MCC950 prevents the NLRP3–caspase‐1 inflammasome from activating MMP‐9, thus protecting the aorta from destruction and AAD formation.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Patient Enrollment
Patients with sporadic ascending thoracic AAD (TAAD) who were undergoing elective open repair operations were prospectively enrolled in this study approved by the institutional review board at Baylor College of Medicine after written informed consent was obtained from patients or family members. In addition, control ascending aortic tissues were collected from age‐matched organ donors without aortic aneurysm, dissection, coarctation, or previous aortic repair (International Institute for the Advancement of Medicine). Characteristics of patients and controls are shown in the Table. The aortic tissues were rinsed with 0.9% normal saline and fixed in 10% formalin. Samples were then embedded in paraffin for histology analysis, embedded in optimal cutting temperature compound for immunofluorescence staining, or snap‐frozen in liquid nitrogen for protein extraction.
Table 1.
Characteristics of Patients and Organ Donors (Controls)
| Characteristics | Control (n=8) | ATAA (n=10) | ATAD (n=10) |
|---|---|---|---|
| Age, y | 66.0±2.2 | 65.1±3.1 | 62.6±2.9 |
| Male | 4 (50) | 5 (50) | 5 (50) |
| Hypertension | 7 (88) | 9 (90) | 10 (100) |
| COPD | 2 (25) | 0 (0) | 2 (20) |
| Diabetes mellitus | 1 (13) | 1 (10) | 1 (10) |
| History of smoking | 4 (50) | 3 (30) | 3 (30) |
| Use of antilipid medication | 3 (38) | 0 (0) | 1 (10) |
| Use of cyclooxygenase inhibitor | 3 (8) | 2 (20) | 4 (40) |
| Aortic diameter, cm | NA | 5.5±0.3 | 6.2±0.4 |
Data are expressed as the number (percentage) or as the mean±SD. ATAA indicates ascending thoracic aortic tissue from patients with ascending aortic aneurysm without dissection; ATAD, ascending thoracic aortic tissue from patients with chronic ascending aortic dissection; COPD, chronic obstructive pulmonary disease; NA, not available.
Animal Studies
The wild‐type (WT) (C57BL/6J) mice used in this study were purchased from the Jackson Laboratory. Both male and female mice were studied. Sporadic AAD was induced as previously described.8, 13 Four‐week‐old male and female WT mice (n=95) were challenged with a high‐fat, high‐cholesterol diet for 8 weeks and infused with 2000 ng/min per kg AngII (Sigma‐Aldrich Co) during the last 4 weeks through an osmotic minipump (Model 2004; ALZA Scientific Products). During the AngII infusion period, the challenged mice were also given daily intraperitoneal injections of either MCC950 (20 mg/kg, from Dr. Matthew A. Cooper's laboratory at the University of Queensland, Australia) (n=45) or PBS (n=50). The high‐fat, high‐cholesterol diet (Research Diets, Inc, D12108C) contained 20% protein, 40% carbohydrate, 40% fat, and 1.25% cholesterol. Unchallenged 4‐week‐old male and female control mice (n=20) were fed a chow diet and infused with saline during the last 4 weeks. Systolic blood pressure was measured weekly in conscious mice using a noninvasive tail cuff BP‐2000 system (Visitech Systems, Inc). Body weight and glucose levels were measured weekly. At the end of the 8‐week study period, mice were euthanized. Blood serum was collected and sent to the Center for Comparative Medicine at Baylor College of Medicine for lipid profile testing. The aortas were exposed and rinsed with cold PBS, and the periaortic tissues were removed. The aorta was then excised, further cleaned, and rinsed with cold PBS to remove any residual blood in the lumen. Next, aortas were imaged for diameter measurement and disease evaluation. The aortic segments were then embedded in optimal cutting temperature compound for histology and immunofluorescence staining or were snap‐frozen for protein analysis. All animal experiments were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine in accordance with the guidelines of the National Institutes of Health.
Aortic Diameter Measurement
For each extracted aorta, we evaluated the ascending, arch, descending thoracic, suprarenal abdominal, and infrarenal aortic segments. Images of the aorta were obtained using an Olympus SZX7 microscope at a magnification of ×0.4 (scale bar=2 mm), and the diameter of each aortic segment was measured with DP2‐BSW software (Olympus Life Science Solutions) by 2 independent observers who were blinded to the experimental group. The mean diameter of the different regions was calculated and compared among the groups.
Classification of Aortic Disease
The severity of disease was classified using our previously reported13 modification of the classification system described by Daugherty and colleagues.14 Dilatation of an aortic segment was defined as an aortic diameter ≥1.25 but <1.5 times the mean aortic diameter of the corresponding segment in unchallenged mice. Aneurysm was defined as an aortic diameter ≥1.5 times the mean aortic diameter of the corresponding segment in unchallenged mice. Aortic dissection was defined as the presence of hematoma within the aortic wall detected on gross examination, or as the presence of layer separation within the aortic media or medial‐adventitial boundary (with a false lumen hematoma) detected on aortic histology. Aortic rupture was defined as hemorrhage into the adjacent body cavity resulting in premature death. We defined the category AAD as the presence of aneurysm, dissection, or rupture. Severe AAD was defined as the presence of dissection or rupture. Aneurysmal aortas were evaluated by 4 independent observers who were blinded to the experimental groups. In the case of a discrepancy, the observers discussed the case and agreed on the classification.
Hematoxylin and Eosin Staining and Elastic Fiber Staining
Aortic sections were stained with hematoxylin and eosin (Sigma‐Aldrich Co) and Verhoeff–van Gieson elastin stain (Sigma‐Aldrich Co) according to the manufacturer's instructions. Aortic sections were examined by 2 independent observers who were blinded to the experimental groups. The extent of elastic fiber fragmentation was scored on a scale of 0 to 3 (grade 0=none, grade 1=minimal, grade 2=moderate, and grade 3=severe).
In Situ Zymography
MMP activity in the aortic wall was determined using an EnzCheck Gelatinase Assay Kit (Life Technologies Corp) according to the manufacturer's instructions, with modifications. Briefly, frozen aortic sections were overlaid with 0.1 mg/mL DQ gelatin that was dissolved in 1% low‐melting multipurpose agarose (Roche) containing reaction buffer. The slides were incubated at room temperature overnight. Green fluorescence indicative of gelatinase activity was examined using fluorescence microscopy.
In‐Gel Zymography
Cell culture supernatants were collected, and 10 μL of each were mixed in Novex Tris‐Glycine SDS Sample Buffer (Thermo Fisher Scientific) and then loaded on Novex 10% Zymogram Plus (Gelatin) Protein Gels. Samples were electrophoresed and separated at 125 V for 2 hours. After electrophoresis, gels were renatured in Zymogram Renaturing Buffer (Thermo Fisher Scientific) at room temperature for 30 minutes and incubated overnight at 37°C in Zymogram Developing Buffer (Thermo Fisher Scientific). Gels were then stained with Coomassie blue staining solution (0.1% Coomassie R250 in 40% ethanol, 10% acetic acid; Thermo Fisher Scientific) for 2 hours and destained twice for 30 minutes in destaining solution (10% ethanol and 7.5% acetic acid). MMP activity appeared as a clear band on a dark background. Images of stained gels were captured using a CanoScan 9950F Flatbed Scanner (Canon).
Cell Culture and Transfection
Human monocytic THP‐1 cells (American Type Culture Collection) were cultured in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% heat‐inactivated fetal bovine serum (Thermo Fisher Scientific) under standard conditions (humidified atmosphere of 5% CO2 at 37°C). Cells were seeded into a 6‐well plate at a density of 3×105 cells/mL and treated with 100 ng/mL phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich Co) for 48 to 72 hours to induce differentiation into macrophages. Cells were starved overnight and then treated with lipopolysaccharide (100 ng/mL, Sigma‐Aldrich Co) for an additional 16 hours. Culture media was replaced with fresh RPMI 1640 medium, and cells were exposed to hydrogen peroxide (H2O2; 1000 mM, Sigma‐Aldrich Co) for 24 hours.
In a subset of experiments, macrophages were transfected with 50 nM NLRP3 siRNA, caspase‐1 siRNA (Dharmacon), or scrambled siRNA using Lipofectamine RNAiMax (Life Technologies) according to the manufacturer's instructions. After 24 hours, the transfected cells were treated with lipopolysaccharide (100 ng/mL, Sigma‐Aldrich Co) for 16 hours overnight and then stimulated with H2O2 (1000 mM, Sigma‐Aldrich Co) for 24 hours. Transfection efficiency was confirmed by means of Western blot analysis.
Overexpressing NLRP3 (WT and mutant) and caspase‐1 was achieved using NLRP3 (Addgene) and caspase‐1 plasmids (Addgene).15 Mutant caspase‐1 plasmid Casp‐1 C285A was generated by GenScript. Plasmid (1–2 μg) was transiently transfected into macrophages using Lipofectamine 2000 (Thermo Fisher Scientific) and Opti‐MEM medium (Thermo Fisher Scientific) for 24 hours. Then, cells were supplied with fresh growth media overnight (14 hours).
Western Blot Analysis
Protein lysates from treated cells or aortic tissues were prepared as previously described.16 Protein samples (15 μg per lane) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and were transferred to polyvinylidene fluoride membranes. The membranes were blocked for 1 hour in blocking solution comprising Tris‐buffered saline containing 5% nonfat dried milk and 0.5% Tween 20 and were incubated overnight with primary antibodies against the following proteins: NLRP3 (AdipoGen Life Sciences), caspase‐1 (Santa Cruz Biotechnology), IL‐1β (Santa Cruz), and MMP‐9 (Santa Cruz). The membranes were washed and incubated with horseradish peroxidase–conjugated anti‐rabbit or anti‐mouse secondary antibody. Protein bands were visualized using Clarity Enhanced Chemiluminescence (Bio‐Rad Laboratories, Inc) and were exposed with HyBlot ES autoradiography film (Denville Scientific Inc). The blots were quantified with ImageJ software (National Institutes of Health). We confirmed equal protein loading by immunoblotting with horseradish peroxidase–conjugated β‐actin antibody (Santa Cruz).
Immunofluorescence Staining and Imaging
Optimal cutting temperature–embedded human or mouse aortic sections were fixed with Cytofix (BD Biosciences) and permeabilized with Perm/Wash (BD Biosciences). Nonspecific staining was reduced by blocking with 5% normal blocking serum. The sections were then incubated with primary antibody against CD68 (Abcam), NLRP3 (AdipoGen), caspase‐1 (Santa Cruz), and MMP‐9 (Santa Cruz) at 4°C overnight; washed with Perm/Wash; and incubated with secondary antibody conjugated to an Alexa Fluor dye such as Alexa Fluor‐488, ‐568, or ‐647 (Thermo Fisher Scientific). Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (Thermo Fisher Scientific). The slides were mounted with Dako Fluorescence Mounting Medium (Dako North America, Inc). Slides incubated with secondary antibody alone were used as negative controls. Tissue sections were examined using a Leica SP5 confocal microscope (Leica Microsystems Inc) or an Olympus immunofluorescence microscope.
TUNEL Assay and Immunofluorescence Staining
Apoptosis was studied using an in situ cell death detection kit (Roche Applied Science). We performed terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) staining according to the manufacturer's instructions. In some instances, we performed TUNEL and immunofluorescence costaining. First, frozen sections of aorta or cells were fixed with Cytofix (BD Biosciences), permeabilized with Perm/Wash (BD Biosciences), and subjected to TUNEL staining. After TUNEL staining, sections or cells were blocked with 10% donkey serum at room temperature for 1 hour and stained with anti‐SM22‐α antibody at 4°C overnight. Sections or cells were then stained with secondary antibody at room temperature for 1 hour, as previously described.13 Tissue sections or cells were observed using a Leica SP5 confocal microscope (Leica). For each aorta or cell treatment condition, images were captured from 3 randomly selected views. For each image, the number of positive cells and the number of total cell nuclei were quantified, and the percentage of positive cells was calculated.
Coimmunoprecipitation Analysis
Protein lysates from treated cells or aortic tissues were prepared as previously described.13 Coimmunoprecipitation experiments were performed using the Dynabeads Protein G Coimmunoprecipitation Kit (Invitrogen Corp) according to the manufacturer's instructions but with several modifications. Briefly, aortic tissue lysates or cell lysates were precleared with IgG‐conjugated Dynabeads Protein G at 4°C for 1 hour. Lysates were then incubated at 4°C overnight with the antibody‐conjugated beads, and beads were washed extensively with cold cell lysis buffer. The beads were then mixed with sample buffer, heated at 95°C for 10 minutes, and subjected to Western blot analysis to detect protein‐protein interactions.
Caspase‐1 Cleavage Assay
Recombinant MMP‐9 (EMD Millipore) was mixed with recombinant caspase‐1 (EMD Millipore) with or without caspase‐1 inhibitor Z‐VAD‐FMK (Santa Cruz) at 37°C for 60 to 120 minutes. Samples were denatured with 1X sample buffer at 95°C for 10 minutes, and proteins were separated by performing sodium dodecyl sulfate polyacrylamide gel electrophoresis (4–15% sodium dodecyl sulfate) followed by incubation with the appropriate antibody.
Statistical Analysis
All quantitative data are presented as the mean±SD or as the mean±SEM. Data were analyzed using SPSS software, version 11.0 (SPSS Inc). Normality of the data was examined using the Shapiro–Wilk test. For normally distributed data, the equality of variances was examined using the Levene test. Two‐group comparisons were performed using the 2‐sample t test. Multigroup comparisons were performed using 1‐way ANOVA with the post hoc Tukey test for equal variance or Dunnett T3 test for unequal variance. For data that were not normally distributed, 2‐group comparisons were performed using the Mann–Whitney test, and multigroup comparisons were performed using the Kruskal–Wallis test. P values were adjusted with the Bonferroni method for pairwise comparisons when indicated. The incidence of disease categories was analyzed using the Fisher exact test. Kaplan–Meier survival curves were plotted to examine mouse survival rates, and the differences were analyzed with the log‐rank (Mantel‐Cox) test. For all statistical analyses, 2‐tailed probability values were used. A probability value of P<0.05 was considered significant.
Study Approval
Informed consent was obtained from patients before aortic surgery, and aortic tissues were collected according to our institutional review board–approved protocol (Baylor College of Medicine and Affiliated Hospitals Protocol H‐12515). Mice were maintained in the Center for Comparative Medicine at Baylor College of Medicine, and procedures were performed according to a protocol approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. All animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publications No. 8023, revised 1978).
Results
MCC950 Treatment Alleviates Challenge‐Induced AAD Formation in Mice
We first examined whether inhibiting the NLRP3–caspase‐1 inflammasome with MCC950 affected disease development in a mouse model of sporadic AAD. WT male and female mice were challenged with a high‐fat, high‐cholesterol diet and AngII infusion. During the AngII infusion period, mice were given a daily intraperitoneal injection of either MCC950 or vehicle (PBS). AngII infusion significantly increased blood pressure in challenged WT mice, whereas MCC950 treatment had no effect on blood pressure (Figure S1A). Furthermore, no difference was observed in body weight, circulating lipid level, or blood glucose level between challenged vehicle‐treated WT mice and challenged MCC950‐treated WT mice (Figure S1B through S1D). Compared with aortas of unchallenged WT mice, aortas of challenged vehicle‐treated WT mice showed significantly increased levels of procaspase‐1, active caspase‐1, pro–interleukin (IL)‐1β, and IL‐1β after 1 week and 4 weeks of AngII infusion (Figure 1A). However, the challenge‐induced expression of these proteins was reduced in aortas of challenged WT mice treated with MCC950 after 1 week and 4 weeks of AngII infusion (Figure 1A). Importantly, treating challenged WT mice with MCC950 preserved aortic structure (Figure 1B) and reduced challenge‐induced aortic enlargement in thoracic and abdominal aortic segments (Figure 1C) in both male and female mice. MCC950 treatment also reduced challenge‐induced aortic dilatation, AAD, severe AAD, aortic rupture (Figure 1D), and premature death (Figure 1E). The reduction of challenge‐induced AAD and severe AAD was observed in different thoracic and abdominal regions in both male and female MCC950‐treated mice (Figure 1F through 1H). These observations suggest that MCC950 reduces AAD development in mice by inhibiting NLRP3 inflammasome activation.
Figure 1.
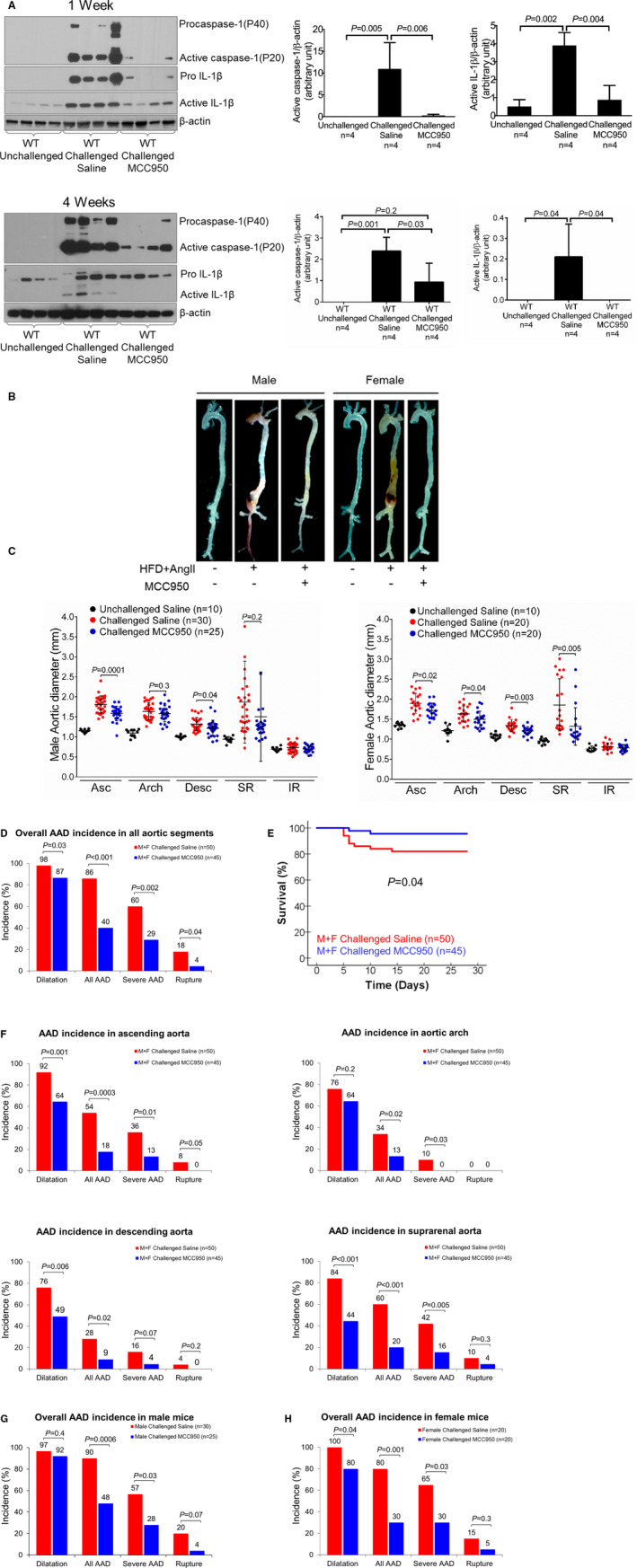
Attenuation of challenge‐induced aortic degeneration and aneurysm and dissection (AAD) development in wild‐type (WT) mice treated with the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3) inflammasome inhibitor MCC950. A, Representative Western blot images and quantification of protein lysates of ascending aortas after 1 week and after 4 weeks of angiotensin II (AngII) infusion during the aortic challenge period, showing lower levels of procaspase‐1, active caspase‐1, pro–interleukin (IL)‐1β, and active IL‐1β in aortic tissues from challenged WT MCC950‐treated mice than in those from challenged WT mice treated with vehicle. B, Excised aortas showing that treatment with MCC950 attenuated challenged‐induced AAD formation in WT mice. C, Aortic diameters in several segments in male and female challenged WT mice were reduced with MCC950 treatment. D, The overall incidence of AAD in challenged male and female WT mice was reduced with MCC950 treatment. E, Kaplan–Meier survival curve analysis showing that survival was increased at 28 days in challenged male and female WT mice that received MCC950 compared with those that received vehicle. F, In male and female challenged WT mice, the incidence of AAD in different aortic segments was significantly lower when mice were treated with MCC950 than when mice were treated with vehicle. G and H, The overall incidence of challenge‐induced AAD in male and female mice treated with MCC950 was similar. Data are presented as mean±SEM. One‐way ANOVA with the post hoc Dunnett T3 test was used for pairwise comparisons in (A and C). The Fisher exact test was used for (D through H). Kaplan–Meier survival curves in (E) were compared using the log‐rank (Mantel‐Cox) test. Asc indicates ascending; Desc, descending thoracic; F, female; HFD, high‐cholesterol, high‐fat diet; IR, infrarenal; M, male; SR, suprarenal abdominal.
MCC950 Treatment Preserves Aortic Structure and Reduces SMC Apoptosis and Contractile Protein Degradation
Consistent with the effect of MCC950 on AAD reduction, we observed better preserved elastic fiber architecture and less elastic fiber fragmentation (Figure 2A) in the aortic wall of challenged WT mice treated with MCC950 than in that of challenged WT mice treated with vehicle.
Figure 2.
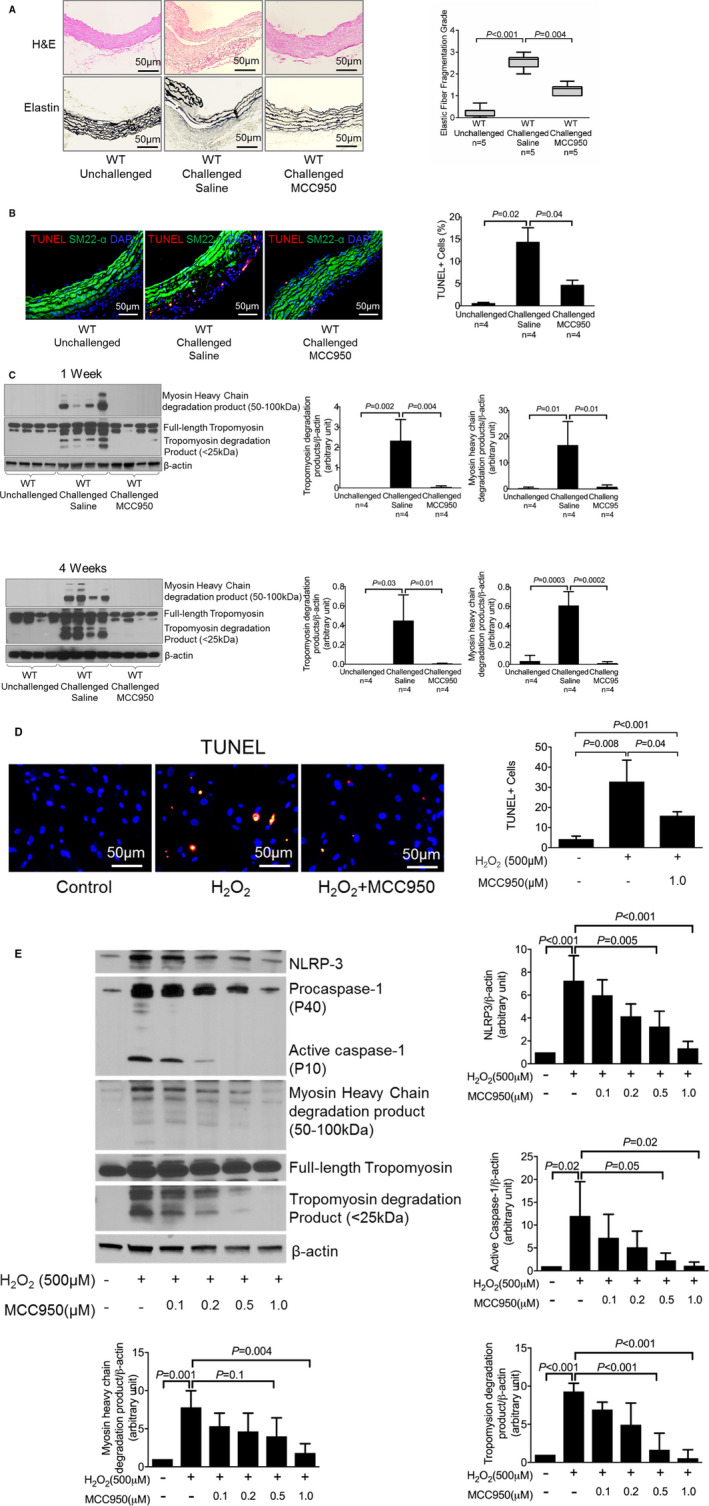
Attenuation of challenge‐induced aortic degeneration, elastic fiber fragmentation, and reduced smooth muscle cell (SMC) apoptosis and SMC contractile protein degradation in vitro and in vivo after MCC950 treatment. A, Hematoxylin and eosin (H&E) staining and Verhoeff–van Gieson elastin staining showing that challenge‐induced aortic degeneration and elastic fiber fragmentation were reduced in mice treated with MCC950 after 4 weeks of angiotensin II (AngII) infusion. Elastic fiber fragmentation grades are also shown. B, Representative terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) staining showing that MCC950 prevented SMC apoptosis in challenged wild‐type (WT) mice. C, Representative Western blot images and quantification showing that MCC950 decreased levels of myosin heavy chain and tropomyosin degradation in challenged WT mice after 1 week and 4 weeks of AngII infusion. SMCs were treated with increasing doses of MCC950 as indicated. D, Representative TUNEL staining showing that MCC950 prevented SMC apoptosis induced by hydrogen peroxide (H2O2). E, Representative Western blot images and quantification showing that MCC950 dose‐dependently decreased the levels of nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3), caspase‐1, myosin heavy chain, and tropomyosin degradation. Data are presented as mean±SEM. One‐way ANOVA with the post hoc Dunnett T3 test was used for pairwise comparisons in (A, B, and D). The post hoc Tukey test was used for pairwise comparisons in (C and E). DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
We previously showed that the NLRP3–caspase‐1 inflammasome cascade induces the degradation of contractile proteins, leading to SMC contractile dysfunction.8 As shown in Figure 2B, we observed reduced levels of SMC apoptosis and degradation of tropomyosin and myosin heavy chain (MHC) in the aortas of MCC950‐treated mice (Figure 2B and 2C). Therefore, we further examined whether the NLRP3 inhibitor MCC950 affects SMC apoptosis and contractile protein degradation in vitro. SMCs were treated with H2O2 in the presence or absence of increasing concentrations of MCC950. In the absence of MCC950, H2O2 induced SMC apoptosis and degradation of tropomyosin and MHC. However, treatment with MCC950 reduced H2O2‐induced SMC apoptosis and degradation of tropomyosin and MHC (Figure 2D and 2E). Thus, our findings suggest that MCC950 inhibits SMC apoptosis and contractile protein degradation both in vitro and in vivo.
MCC950 Treatment Prevents MMP Upregulation in Macrophages
We further investigated the potential mechanisms by which MCC950 protects the aorta. We observed less inflammatory cell infiltration in the aortic wall of challenged WT mice treated with MCC950 than in that of challenged WT mice treated with vehicle (Figure 3A). We then examined the effects of MCC950 on the activity of MMPs, which are important proteases in aortic destruction and AAD formation. As shown in Figure 3B, we found that the challenge‐induced MMP‐9 activity observed in the aortas of WT mice treated with vehicle was significantly reduced in the aortas of challenged WT mice treated with MCC950. Further analysis showed that MCC950 also reduced the challenge‐induced expression of MMP‐9 in the aortas of WT mice. Interestingly, using an antibody against the C‐terminus of MMP‐9, we detected a fragment of MMP‐9 in which the N‐terminal inhibitory domain was removed or truncated (Figure 3C), indicating MMP‐9 activation. Importantly, after both 1 week and 4 weeks of AngII infusion, the level of the truncated MMP‐9 fragment was lower in the aortas of WT challenged mice treated with MCC950 than in the aortas of WT challenged mice treated with vehicle, indicating that MCC950 prevents MMP‐9 cleavage and activation. We further examined whether MCC950 directly inhibited MMP‐9 activity in macrophages. Macrophages derived from the human monocytic THP‐1 cell line were first primed with lipopolysaccharide and then treated with H2O2 in the presence or absence of increasing concentrations of MCC950. Treating macrophages with MCC950 dose‐dependently inhibited caspase‐1 and IL‐1β activation (Figure 3D), as well as MMP‐9 cleavage/activation and activity (Figure 3E and 3F). Thus, our findings suggest that MCC950 may directly block MMP‐9 activation by targeting the NLRP3–caspase‐1 cascade.
Figure 3.
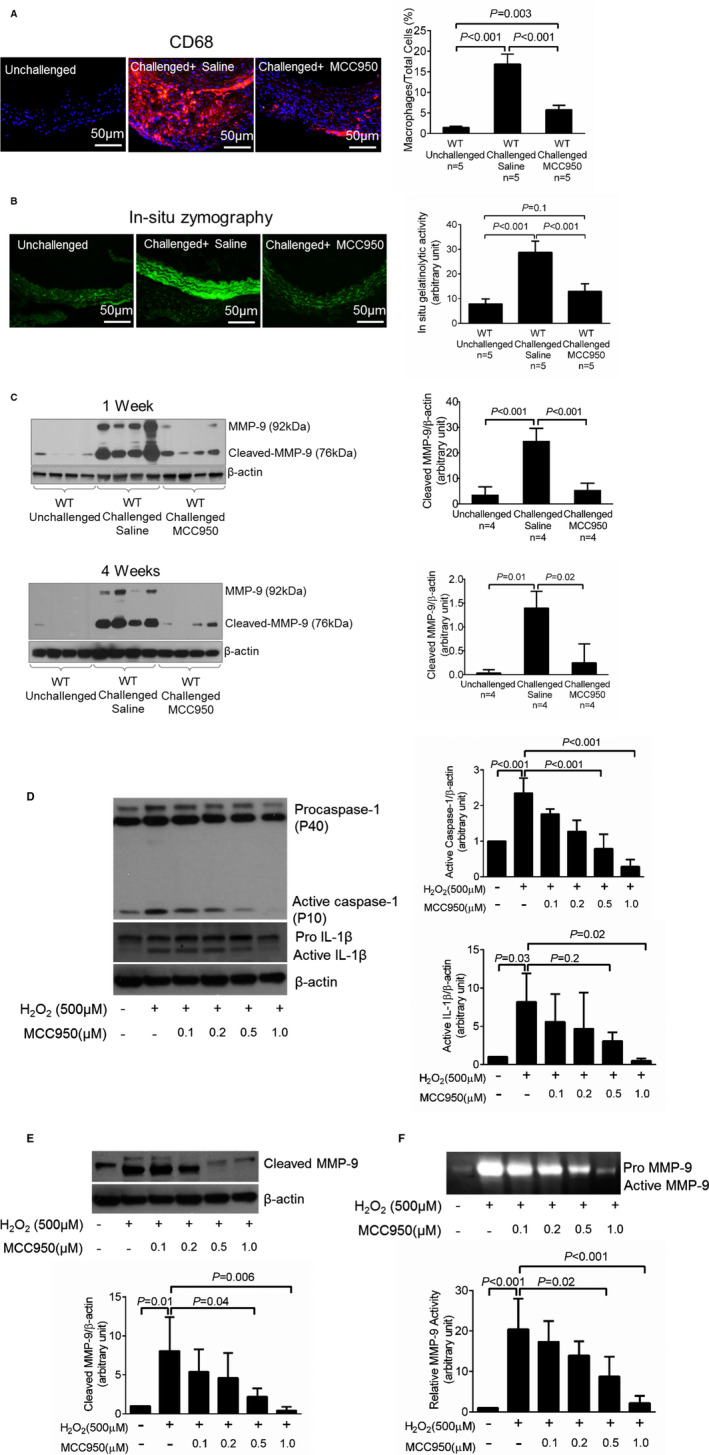
Attenuation of challenge‐induced macrophage infiltration, matrix metallopeptidase 9 (MMP‐9) expression level, and MMP‐9 cleavage in challenged wild‐type (WT) mice treated with MCC950. A, Representative immunofluorescence staining showing that challenge‐induced macrophage infiltration was reduced in the aortic wall of mice treated with MCC950. B, In situ zymography images and the corresponding quantification of data showing that challenge‐induced MMP‐9 activity was decreased in aortas of mice treated with MCC950. C, Representative Western blot images and quantification showing that the challenge‐induced expression and cleavage/activation of MMP‐9 was attenuated in the aortas of mice treated with MCC950 both after 1 week and after 4 weeks of angiotensin II infusion. Macrophages were treated with increasing doses of MCC950 as indicated. D, Representative Western blot images and quantification showing that MCC950 dose‐dependently decreased the levels of active caspase‐1 and active interleukin (IL)‐1β. E and F, Representative Western blot images, in‐gel zymography images, and the corresponding quantification of data showing that MCC950 treatment decreased MMP‐9 protein level and activity. Data are presented as mean±SEM. One‐way ANOVA with the post hoc Tukey test was used for pairwise comparisons.
NLRP3–Caspase‐1 Cascade Directly Activates MMP‐9 in Macrophages
We further explored the possible role of the NLRP3–caspase‐1 inflammasome cascade in MMP‐9 cleavage and activation in human macrophages. Treating macrophages with H2O2 (500 μmol/L) increased levels of NLRP3, procaspase‐1, active caspase‐1, and cleaved/activated MMP‐9 (Figure 4A). Importantly, the knockdown of NLRP3 or caspase‐1 prevented H2O2‐induced MMP‐9 cleavage and activation (Figure 4A and 4B). Conversely, overexpressing WT caspase‐1 but not a dominant‐negative mutant of caspase‐1 (C285A) increased MMP‐9 cleavage and activation (Figure 4C and 4D). Moreover, coimmunoprecipitation analysis showed that caspase‐1 bound to MMP‐9 (Figure 4E). Finally, we found that recombinant caspase‐1 directly cleaved (Figure 4F) and activated (Figure 4G) recombinant MMP‐9, and this activation was reduced by the caspase‐1 inhibitor Z‐VAD‐FMK.
Figure 4.
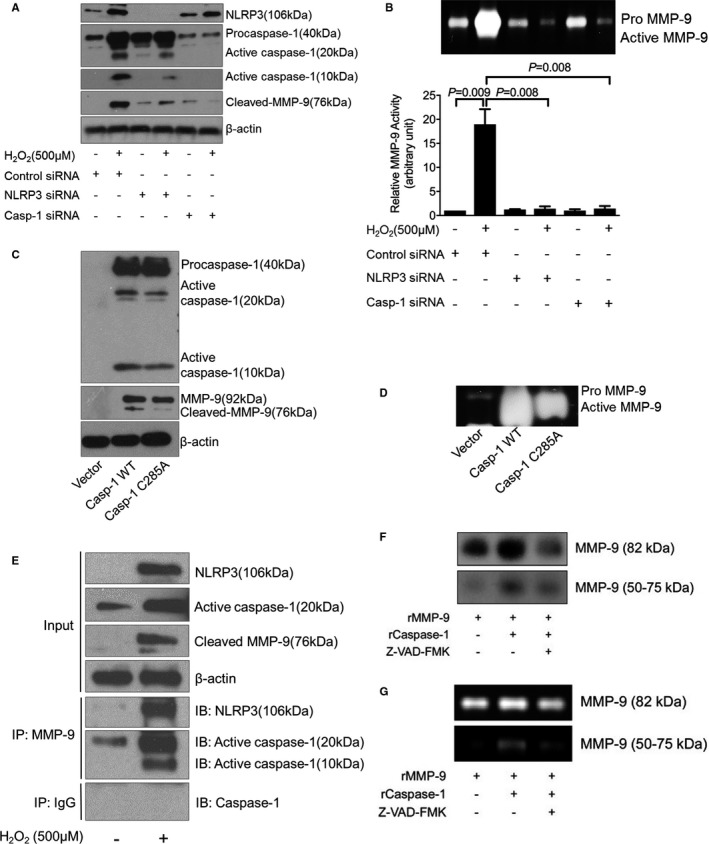
Activation of matrix metallopeptidase 9 (MMP‐9) by the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)–caspase‐1 cascade in human THP‐1 macrophages. A, Representative Western blot results showing that silencing NLRP3 or caspase‐1 with siRNA prevented H2O2‐induced MMP‐9 activation. B, In‐gel zymography results showing that silencing NLRP3 or caspase‐1 with siRNA reduced MMP‐9 activity. C, Representative Western blot results showing that the overexpression of capase‐1 increased the cleavage/activation of MMP‐9 but that the overexpression of a dominant‐negative caspase‐1 mutant (Casp‐1 S285A) failed to induce MMP‐9 cleavage/activation. D, In‐gel zymography results showing that the overexpression of capase‐1 increased the activity of MMP‐9 but that the overexpression of a dominant‐negative caspase‐1 mutant (Casp‐1 S285A) reduced MMP‐9 activity. E, Results of coimmunoprecipitation analysis showing evidence of an interaction between NLRP3/capase‐1 with MMP‐9. F, Representative cleavage assay results showing that recombinant caspase‐1 (rCaspase‐1) directly cleaves recombinant MMP‐9 (rMMP‐9). Z‐VAD‐FMK, caspase‐1 inhibitor. G, The cleaved MMP‐9 fragment (50‐75 kDa) exhibiting gelatin digestive ability. Data are presented as mean±SEM. One‐way ANOVA with the post hoc Dunnett T3 test was used for pairwise comparisons. WT indicates wild‐type.
NLRP3–Caspase‐1 Cascade is Involved in Aortic Challenge–Induced MMP‐9 Activation and Aortic Destruction
We also examined MMP‐9 cleavage in aortic tissues of Nlrp3‐/‐ and Casp1‐/‐ mice. Compared with aortic tissues of unchallenged WT mice, those of challenged WT mice showed increased MMP‐9 levels, MMP‐9 cleavage (Figure 5A), and MMP activity (Figure 5B). Importantly, the challenge‐induced increases in MMP‐9 levels, MMP‐9 cleavage, and MMP activity observed in WT mice were partially prevented in Nlrp3‐/‐ and Casp1‐/‐ mice (Figure 5A and 5B). These findings suggest that the NLRP3–caspase‐1 inflammasome cascade is involved in MMP‐9 activation, which may contribute to aortic destruction and AAD formation.
Figure 5.
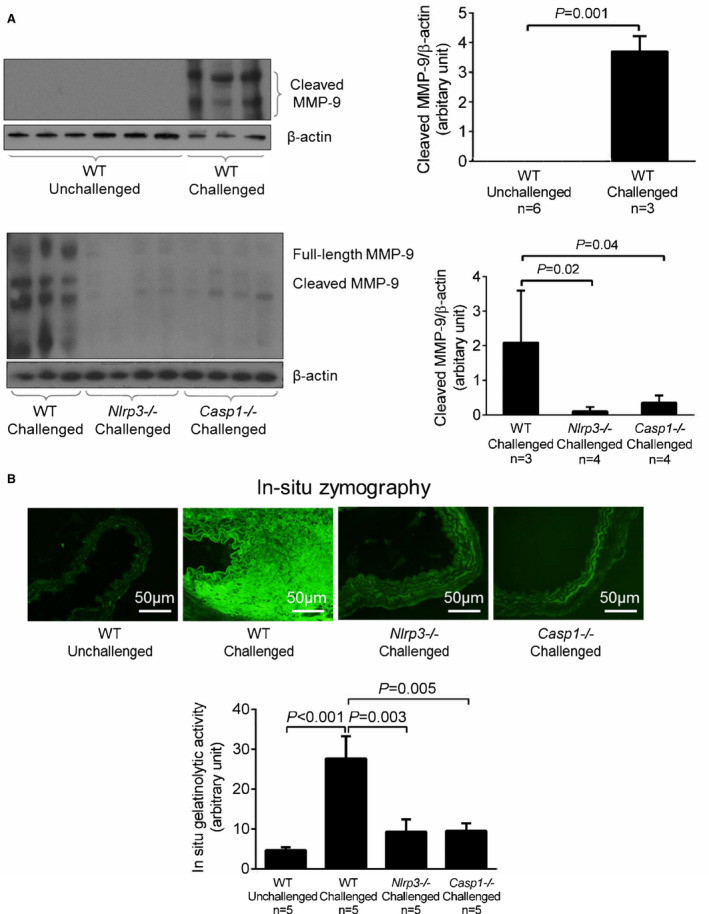
Involvement of the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)–caspase‐1 cascade in aortic challenge–induced matrix metallopeptidase 9 (MMP‐9) activation. A, Representative Western blot analysis of aortic protein lysates, (B) in situ zymography images of aortas, and the corresponding quantification of data showing that the challenge‐induced increase in MMP‐9 levels and activity observed in wild‐type (WT) mice was reduced in challenged Nlrp3‐/‐ and Casp1‐/‐ mice. Data are presented as mean±SEM. A 2‐sample t test was used for 2‐group comparisons in (A). One‐way ANOVA with the post hoc Dunnett T3 test was used for pairwise comparisons in (B).
MMP‐9 Activation Correlates With NLRP3–Caspase‐1 Inflammasome Cascade Activation in Human Sporadic TAAD Tissues
Finally, we examined the association between the NLRP3–caspase‐1 inflammasome cascade and MMP‐9 activation in human sporadic thoracic aortic aneurysm and thoracic aortic dissection tissues, as well as in the aortic tissues of organ donors (controls) (Table). Compared with control aortic tissues, thoracic aortic aneurysm and thoracic aortic dissection tissues showed increased levels of MMP‐9, cleaved/activated MMP‐9, NLRP3, procaspase‐1, and active caspase‐1 (p10 and p20) (Figure 6A). Immunofluorescence staining showed that NLRP3, caspase‐1, and MMP‐9 colocalized in macrophages of human thoracic aortic aneurysm and thoracic aortic dissection tissues (Figure 6B). Moreover, the results of coimmunoprecipitation experiments showed that caspase‐1 was bound to MMP‐9 in diseased aortic tissues from patients with thoracic aortic aneurysm and thoracic aortic dissection (Figure 6C). These findings suggest a potential association between the NLRP3−caspase‐1 inflammasome cascade and MMP‐9 activation in human sporadic TAAD tissues.
Figure 6.
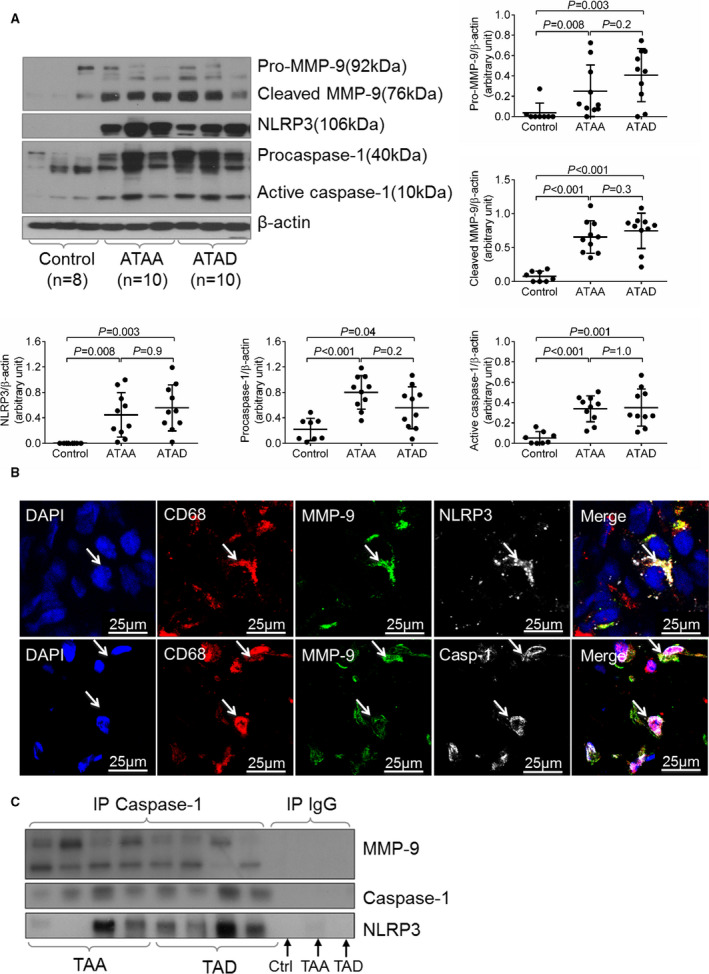
Association between matrix metallopeptidase 9 (MMP‐9) activation and the nucleotide‐binding oligomerization domain–like receptor pyrin domain containing 3 (NLRP3)–caspase‐1 inflammasome in human sporadic ascending aortic aneurysm and dissection tissues. Aortic tissues from patients with ascending thoracic aortic aneurysm (ATAA) without dissection, patients with chronic ascending thoracic aortic dissection (ATAD), and organ donors (controls) were analyzed. A, Representative Western blot data showing that the NLRP3–caspase‐1 inflammasome and MMP‐9 were activated in the aortic wall of patients with ATAA and those with ATAD. B, Immunostaining showing the colocalization of NLRP3 and caspase‐1 with MMP‐9 in the macrophages (CD68) of ATAD tissues. C, Results of coimmunoprecipitation experiments showing that caspase‐1 was bound to MMP‐9 in ATAA and ATAD tissues. Data are presented as mean±SEM. One‐way ANOVA with the post hoc Tukey test was used for pairwise comparisons. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; IP, immunoprecipitation; TAA, thoracic aortic aneurysm; TAD, thoracic aortic dissection.
Discussion
In this study, we tested the effects of MCC950, a potent, selective NLRP3 inhibitor, on preventing aortic destruction and AAD formation in a mouse model of sporadic AAD. We observed that MCC950 treatment significantly inhibited challenge‐induced inflammasome activation, aortic destruction, SMC contractile protein degradation and death, and the development of aortic aneurysm, dissection, and rupture. Disease reduction was observed in different thoracic and abdominal aortic segments, in both male and female mice. Interestingly, the aortic wall of MCC950‐treated mice showed significantly diminished levels of activated MMP‐9 with reduced cleavage of the N‐terminal inhibitory domain of MMP‐9, a phenomenon that was also observed in Nlrp3 −/− or Casp‐1 −/− mice. Further mechanistic studies revealed that caspase‐1 directly bound and cleaved the N‐terminal inhibitory domain of MMP‐9, leading to its activation. Finally, in human sporadic TAAD tissues, we observed the colocalization of NLRP3 and caspase‐1 with MMP‐9 in macrophages, and caspase‐1 directly interacted with MMP‐9 in patient tissues. Our findings suggest that the NLRP3–caspase‐1 inflammasome directly activates MMP‐9 by removing its N‐terminal inhibitory domain and that targeting the inflammasome with MCC950 is a promising approach for preventing AAD development.
Currently, no drugs are available that effectively prevent sporadic TAAD development or progression; surgical repair is the only effective treatment. Therefore, developing new treatment strategies to prevent disease progression is crucial. For the first time to our knowledge, we have described the protective effects of MCC950, a small‐molecule inhibitor of the NLRP3 inflammasome, in an animal model of sporadic AAD. MCC950 has previously been implicated as a potential treatment in an increasing number of inflammatory diseases, including atherosclerosis,11 nonalcoholic fatty liver disease,17 Alzheimer's disease,18, 19 and myocardial infarction.10 The common underlying mechanism of MCC950 in these pathologic conditions is blocking NLRP3 inflammasome activation to reduce inflammation by decreasing inflammatory cell infiltration.10, 11, 17, 18, 19 Recently, MCC950 was shown to prevent the secretion of IL‐1β induced by NLRP3 stimulation, both in vitro and in vivo.3 In addition, the efficacy of MCC950 under hyperlipidemic conditions has been previously established; treating apolipoprotein E–deficient mice with MCC950 reduces the development of atherosclerotic lesions by reducing intraplaque macrophage content.11 Furthermore, in a porcine model of myocardial infarction, the administration of MCC950 results in reduced infarct size and preserved cardiac function.10 Moreover, MCC950 was recently shown to selectively inhibit the NLRP3 inflammasome, subsequently leading to the suppression of MMP‐9 expression in human umbilical vein endothelial cells.20 These findings are in line with our results reported here. We showed that MCC950 reduces vascular inflammation by inhibiting MMP‐9 activation, suggesting a novel target for treating inflammatory disease. We further showed that MCC950 can dose‐dependently inhibit the NLRP3−caspase‐1 inflammasome cascade to prevent MMP‐9 activation in macrophages in vitro. Moreover, consistent with these effects of MCC950 in macrophages in vitro, administering MCC950 in challenged WT mice inhibited the activation of the NLRP3−caspase‐1 inflammasome and MMP‐9 in the aorta and prevented the development of aortic aneurysm, dissection, and rupture. Together, these observations suggest that MCC950 can reduce AAD development by inhibiting NLRP3 inflammasome activation.
In our study, we did not observe a significant difference in the incidence of all AAD or severe AAD between male and female mice. Interestingly, treating mice with MCC950 produced a slightly better reduction in all AAD in female mice (from 85% to 30%) than in male mice (from 90% to 48%). Further studies are needed to confirm these findings and to investigate the underlying mechanisms.
MMPs,21 particularly MMP‐9,22, 23, 24, 25, 26 play critical roles in aortic destruction and AAD progression.27 MMPs can be regulated by the alteration of their expression level or activation. Although the regulation of MMP expression during disease development has been extensively studied, the molecular mechanisms underlying MMP activation are poorly understood. The structure of the MMP‐9 protein consists of an N‐terminal inhibitory prodomain, a catalytic domain, a hinge region, and a C‐terminal hemopexin domain.28 Pro‐MMP‐9 (92kD) is cleaved into an active 82‐kD isoform (amino acids 107‐707) via the removal of an N‐terminal inhibitory prodomain (amino acids 20‐93) by other MMPs or proteases at the plasma membrane or in the extracellular space. Identifying and blocking the pathways that regulate MMP‐9 activation may prevent its activity and the subsequent induction of aortic damage.
Study Strengths and Limitations
Here, we identified caspase‐1 as a novel protease that directly cleaves the N‐terminal inhibitory domain of MMP‐9, leading to its activation. Caspase‐1 is a protease that has been shown to have a role in the inflammatory response by cleaving the precursors of IL‐1β, IL‐18, and IL‐33.28 Research has extensively focused on caspase‐1 activation and the release of active IL‐1β, whereas less is known about the role of caspase‐1 in the activation of other inflammatory factors. In addition to its role in inflammation, caspase‐1 has a broad range of substrates related to the ontologies of cell death, cytoskeleton, and metabolism.30 Accordingly, we found that the NLRP3–caspase‐1 inflammasome cascade is involved in MMP‐9 activation that contributes to aortic inflammation in the development of AAD. However, a limitation of our study is that the mechanism by which MMP‐9 is activated by the NLRP3–caspase‐1 inflammasome cascade in vivo remains unclear. The changes in MMP‐9 expression and activity that we observed may be a consequence of the reduced expression and secretion of IL‐1β, IL‐18, or downstream cytokines such as IL‐6 that are known to play important roles in the regulation of MMP expression and activity. Further studies are needed to demonstrate the direct contribution of the NLRP3–caspase‐1 inflammasome cascade to MMP‐9 activation in vivo.
Conclusions
Our findings suggest a novel mechanism by which the NLRP3–caspase‐1 inflammasome activates MMP‐9 and disrupts aortic wall homeostasis. The activation of MMP‐9 by the NLRP3–caspase‐1 inflammasome cascade in macrophages accelerated the destruction of the aortic wall in a mouse model of sporadic AAD. Furthermore, MCC950 reduced the development of aortic disease in mice by inhibiting NLRP3–caspase‐1 inflammasome activation and MMP‐9 activation. These findings have important clinical implications and suggest that the blockade of the NLRP3–caspase‐1 inflammasome by MCC950 may have therapeutic potential for preventing or treating aortic disease.
Sources of Funding
LeMaire is supported in part by the Jimmy and Roberta Howell Professorship in Cardiovascular Surgery at Baylor College of Medicine. This study was supported by grants from the National Institutes of Health (NIH; R01 HL127111) and the Roderick D. MacDonald Research Fund at Baylor St. Luke's Medical Center (13RDM006). The Thoracic Aortic Disease Tissue Bank at Baylor College of Medicine was supported in part through the Tissue Banking Core of the Specialized Center of Clinically Oriented Research in Thoracic Aortic Aneurysms and Dissections (NIH P50 HL083794). Wu was supported by a training grant (NIH T32 HL007676) through the Department of Molecular Physiology and Biophysics at Baylor College of Medicine.
Conflict of Interest
AABR is a co‐inventor on granted patents (US 10,538,487, EP 3259253) and patent applications (WO2018215818, WO2017140778, WO2016131098) for NLRP3 inhibitors, which are licensed to Inflazome Ltd, a company headquartered in Dublin, Ireland. Inflazome is developing drugs that target the NLRP3 inflammasome to address unmet clinical needs in inflammatory disease.
Disclosures
LeMaire serves as an advisory panel member for Biom'up and Acer Therapeutics, as a consultant and principal investigator for clinical studies sponsored by Terumo Aortic and CytoSorbants, and as a coinvestigator for clinical studies sponsored by W.L. Gore & Associates. The remaining authors have no disclosures to report.
Supporting information
Figure S1. Effect of MCC950 on blood pressure, body weight, blood glucose level, and lipid profile in unchallenged and challenged mice treated with saline or MCC950. A, Systolic blood pressure in mice before and after 4 weeks of angiotensin II (AngII) infusion (n=5). B and C, Body weight and blood glucose level before and after 4 weeks of AngII infusion (n=5). D, Blood serum total cholesterol, triglyceride, very low‐density lipoprotein (VLDL), low‐density lipoprotein cholesterol (LDL‐C), and high‐density lipoprotein cholesterol (HDL‐C) levels after 4 weeks of AngII infusion (n=5). Data are presented as mean±SEM. One‐way ANOVA with post hoc Tukey test was used for pairwise comparisons.
Acknowledgments
We gratefully acknowledge Nicole Stancel, PhD, ELS(D), of Scientific Publications at the Texas Heart Institute, for editorial support, and Scott Weldon, MA, of the Division of Cardiothoracic Surgery, Baylor College of Medicine, for creating the illustration.
Author contributions: The study was designed by Shen and LeMaire. The patient samples were prepared by Wu and Ren. MCC950 was prepared by Robertson and Cooper. The experiments were conducted by Ren, Wu, Appel, L. Zhang, C. Zhang, and Luo. The data were analyzed by Ren with consultation from Shen, LeMaire, and Milewicz. All authors discussed the results. The article was written by Ren, edited by Shen and LeMaire, and approved by all authors.
(J Am Heart Assoc. 2020;9:e014044 DOI: 10.1161/JAHA.119.014044.)
Contributor Information
Ying H. Shen, Email: slemaire@bcm.edu.
Scott A. LeMaire, Email: hyshen@bcm.edu.
References
- 1. Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2014;64:1725–1739. [DOI] [PubMed] [Google Scholar]
- 2. Pape LA, Awais M, Woznicki EM, Suzuki T, Trimarchi S, Evangelista A, Myrmel T, Larsen M, Harris KM, Greason K, Di Eusanio M, Bossone E, Montgomery DG, Eagle KA, Nienaber CA, Isselbacher EM, O'Gara P. Presentation, diagnosis, and outcomes of acute aortic dissection: 17‐year trends from the International Registry of Acute Aortic Dissection. J Am Coll Cardiol. 2015;66:350–358. [DOI] [PubMed] [Google Scholar]
- 3. Aggarwal B, Raymond CE. Therapeutic goals in patients with acute aortic dissection: management before surgery. J Am Coll Cardiol. 2015;65:1599–1600. [DOI] [PubMed] [Google Scholar]
- 4. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 5. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II‐induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:127–136. [DOI] [PubMed] [Google Scholar]
- 7. Sun W, Pang Y, Liu Z, Sun L, Liu B, Xu M, Dong Y, Feng J, Jiang C, Kong W, Wang X. Macrophage inflammasome mediates hyperhomocysteinemia‐aggravated abdominal aortic aneurysm. J Mol Cell Cardiol. 2015;81:96–106. [DOI] [PubMed] [Google Scholar]
- 8. Wu D, Ren P, Zheng Y, Zhang L, Xu G, Xie W, Lloyd EE, Zhang S, Zhang Q, Curci JA, Coselli JS, Milewicz DM, Shen YH, LeMaire SA. NLRP3 (nucleotide oligomerization domain‐like receptor family, pyrin domain containing 3)‐caspase‐1 inflammasome degrades contractile proteins: implications for aortic biomechanical dysfunction and aneurysm and dissection formation. Arterioscler Thromb Vasc Biol. 2017;37:694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O'Neill LA. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pasterkamp G, Hoefer IE. The selective NLRP3‐inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. 2017;38:828–836. [DOI] [PubMed] [Google Scholar]
- 11. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E‐deficient mice‐brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461. [DOI] [PubMed] [Google Scholar]
- 12. Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ren P, Hughes M, Krishnamoorthy S, Zou S, Zhang L, Wu D, Zhang C, Curci JA, Coselli JS, Milewicz DM, LeMaire SA, Shen YH. Critical role of ADAMTS‐4 in the development of sporadic aortic aneurysm and dissection in mice. Sci Rep. 2017;7:12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E‐deficient mice. J Clin Invest. 2000;105:1605‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature. 2009;458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen YH, Zhang L, Ren P, Nguyen MT, Zou S, Wu D, Wang XL, Coselli JS, LeMaire SA. AKT2 confers protection against aortic aneurysms and dissections. Circ Res. 2013;112:618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, Masters SL, Schroder K, Cooper MA, Feldstein AE, Farrell GC. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qi Y, Klyubin I, Cuello AC, Rowan MJ. NLRP3‐dependent synaptic plasticity deficit in an Alzheimer's disease amyloidosis model in vivo. Neurobiol Dis. 2018;114:24–30. [DOI] [PubMed] [Google Scholar]
- 19. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O'Neill LAJ, Lynch MA. Inhibiting the NLRP3 inflammasome with MCC950 promotes non‐phlogistic clearance of amyloid‐beta and cognitive function in APP/PS1 mice. Brain Behav Immun. 2017;61:306–316. [DOI] [PubMed] [Google Scholar]
- 20. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine‐N‐Oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3‐SOD2‐mtROS signaling pathway. J Am Heart Assoc. 2017;6:e006347. 10.1161/JAHA.117.002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papazafiropoulou A, Tentolouris N. Matrix metalloproteinases and cardiovascular diseases. Hippokratia. 2009;13:76–82. [PMC free article] [PubMed] [Google Scholar]
- 22. Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase‐9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang X, Wu D, Choi JC, Minard CG, Hou X, Coselli JS, Shen YH, LeMaire SA. Matrix metalloproteinase levels in chronic thoracic aortic dissection. J Surg Res. 2014;189:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson WR, Anderton M, Choke EC, Dawson J, Loftus IM, Thompson MM. Elevated plasma MMP1 and MMP9 are associated with abdominal aortic aneurysm rupture. Eur J Vasc Endovasc Surg. 2008;35:580–584. [DOI] [PubMed] [Google Scholar]
- 25. Wilson WR, Anderton M, Schwalbe EC, Jones JL, Furness PN, Bell PR, Thompson MM. Matrix metalloproteinase‐8 and ‐9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. 2006;113:438–445. [DOI] [PubMed] [Google Scholar]
- 26. McMillan WD, Tamarina NA, Cipollone M, Johnson DA, Parker MA, Pearce WH. Size matters: the relationship between MMP‐9 expression and aortic diameter. Circulation. 1997;96:2228–2232. [DOI] [PubMed] [Google Scholar]
- 27. Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013;184:907–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Page‐McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arend WP, Palmer G, Gabay C. IL‐1, IL‐18, and IL‐33 families of cytokines. Immunol Rev. 2008;223:20–38. [DOI] [PubMed] [Google Scholar]
- 30. Denes A, Lopez‐Castejon G, Brough D. Caspase‐1: is IL‐1 just the tip of the ICEberg? Cell Death Dis. 2012;3:e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of MCC950 on blood pressure, body weight, blood glucose level, and lipid profile in unchallenged and challenged mice treated with saline or MCC950. A, Systolic blood pressure in mice before and after 4 weeks of angiotensin II (AngII) infusion (n=5). B and C, Body weight and blood glucose level before and after 4 weeks of AngII infusion (n=5). D, Blood serum total cholesterol, triglyceride, very low‐density lipoprotein (VLDL), low‐density lipoprotein cholesterol (LDL‐C), and high‐density lipoprotein cholesterol (HDL‐C) levels after 4 weeks of AngII infusion (n=5). Data are presented as mean±SEM. One‐way ANOVA with post hoc Tukey test was used for pairwise comparisons.