

Abstract
Background
Aberrant activation of the NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) inflammasome is thought to play a causative role in atherosclerosis. NLRP3 is kept in an inactive ubiquitinated state to avoid unwanted NLRP3 inflammasome activation. This study aimed to test the hypothesis that pharmacologic manipulating of NLRP3 ubiquitination blunts the assembly and activation of the NLRP3 inflammasome and protects against vascular inflammation and atherosclerosis. Since genetic studies yielded mixed results about the role for this inflammasome in atherosclerosis in low‐density lipoprotein receptor– or apolipoprotein E–deficient mice, this study attempted to clarify the discrepancy with the pharmacologic approach using both models.
Methods and Results
We provided the first evidence demonstrating that tranilast facilitates NLRP3 ubiquitination. We showed that tranilast restricted NLRP3 oligomerization and inhibited NLRP3 inflammasome assembly. Tranilast markedly suppressed NLRP3 inflammasome activation in low‐density lipoprotein receptor– and apolipoprotein E–deficient macrophages. Through reconstitution of the NLRP3 inflammasome in human embryonic kidney 293T cells, we found that tranilast directly limited NLRP3 inflammasome activation. By adopting different regimens for tranilast treatment of low‐density lipoprotein receptor– and apolipoprotein E–deficient mice, we demonstrated that tranilast blunted the initiation and progression of atherosclerosis. Mice receiving tranilast displayed a significant reduction in atherosclerotic lesion size, concomitant with a pronounced decline in macrophage content and expression of inflammatory molecules in the plaques compared with the control group. Moreover, tranilast treatment of mice substantially hindered the expression and activation of the NLRP3 inflammasome in the atherosclerotic lesions.
Conclusions
Tranilast potently enhances NLRP3 ubiquitination, blunts the assembly and activation of the NLRP3 inflammasome, and ameliorates vascular inflammation and atherosclerosis in both low‐density lipoprotein receptor– and apolipoprotein E–deficient mice.
Keywords: atherosclerosis, inflammasome, NLRP3 ubiquitination, tranilast, vascular inflammation
Subject Categories: Basic Science Research, Inflammation, Mechanisms, Vascular Biology
Nonstandard Abbreviations and Acronyms
- ApoE−/−
apolipoprotein E–deficient
- ASC
apoptosis‐associated speck‐like protein containing a caspase recruitment domain
- BMDMs
bone marrow–derived macrophages
- CANTOS
Canakinumab Antiinflammatory Thrombosis Outcome Study
- HEK293T
human embryonic kidney 293T
- ICAM‐1
intercellular cell adhesion molecule 1
- IL
interleukin
- K63
lysine 63
- Ldlr−/−
low‐density lipoprotein receptor–deficient
- MCP‐1
monocyte chemoattractant protein‐1
- HA‐NEK7
never in mitosis gene a‐related kinase 7
- NLRP3
nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3
- SMC
smooth muscle cells
- UbK63R
ubiquitin K63R
- WD
Western diet
- WT
wild‐type
Clinical Perspective
What Is New?
Tranilast enhances lysine 63‐linked ubiquitination of NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) and suppresses the activation of the NLRP3 inflammasome in low‐density lipoprotein receptor– and apolipoprotein E–deficient macrophages.
Tranilast represses the expression and activation of the NLRP3 inflammasome in the atherosclerotic lesions.
Tranilast blunts the initiation and progression of atherosclerosis in both low‐density lipoprotein receptor– and apolipoprotein E–deficient models.
What Are the Clinical Implications?
Pharmacologic manipulation of NLRP3 ubiquitination offers an opportunity for therapeutic targeting of vascular inflammation and atherosclerosis.
Application of tranilast combined with other lipid‐lowering therapies is beneficial in effective atherosclerosis therapy.
Atherosclerosis, a maladaptive chronic inflammatory disease, has consistently remained the leading cause of death in industrialized countries.1 To date, low‐density protein cholesterol–lowering statins remain the mainstay for the treatment of atherosclerosis.2 Nonetheless, atherosclerotic plaques still undergo progression to a great degree in a large proportion of patients receiving effective lipid‐lowering therapies.3 The “residual inflammatory risk” has increasingly become an attractive therapeutic target for atherosclerotic cardiovascular disease. Remarkably, CANTOS (Canakinumab Antiinflammatory Thrombosis Outcome Study) provides definitive evidence that inhibition in IL‐1β with canakinumab, an interleukin (IL)‐1β monoclonal antibody, robustly reduces inflammation risk for patients with atherosclerotic cardiovascular disease.4
IL‐1β is synthesized in cells, primarily macrophages, as an inactive precursor (designated pro–IL‐1β). As a consequence of the NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) inflammasome activation, pro–IL‐1β is processed by activated caspase‐1 to form the biologically active IL‐1β.5 The NLRP3 inflammasome is a multimeric protein complex consisting of 3 core components: NLRP3, apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC), and pro–caspase‐1.2 The activation of the NLRP3 inflammasome has been subjected to tight control.2 Thus, an important topic in this research field is to understand the regulation of NLRP3 inflammasome, which occurs at multiple levels.2 It is noteworthy that NLRP3 is kept in an inactive ubiquitinated state to avoid unwanted activation of the NLRP3 inflammasome.6 There are few, if any, reports concerning pharmacologic manipulation of NLRP3 ubiquitination. Because the NLRP3 inflammasome is fundamentally important for shaping immunity, complete inhibition of this inflammasome is detrimental.7 Accordingly, we sought to address whether pharmacologic targeting of NLRP3 ubiquitination can protect against the initiation and progression of atherosclerosis.
While accumulated evidence reveals the causal role for the NLRP3 inflammasome in dictating atherosclerosis, there is a long‐standing puzzle of why genetic studies have yielded mixed results. In brief, low‐density lipoprotein receptor–deficient (Ldlr−/−) mice transplanted with bone marrow lacking NLRP3, ASC, or IL‐1β exhibited substantially reduced atherosclerotic lesions when compared with those transplanted with wild‐type (WT) bone marrow.8 In sharp contrast, deletion of the NLRP3 inflammasome component (NLRP3, ASC, and pro–caspase‐1) in apolipoprotein E–deficient (ApoE−/−) mice had no significant impact on atherosclerosis.9 This raised the likelihood that the discrepancy is caused, at least in part, by employing different types of mouse models of atherosclerosis or conducting their experiments under distinct conditions in different studies.8, 9 Indeed, ApoE−/− and Ldlr−/− mice are the 2 most extensively used experimental models of atherosclerosis.10 The 2 models differ in important ways in the mechanisms whereby the absence of apolipoprotein E or low‐density lipoprotein receptor potentiates atherosclerosis.10 The majority of studies have employed only 1 model and investigators usually generalized their findings to fashion an overall picture of atherosclerosis. Although this might be legitimate under most circumstances, this is not always the case.11, 12, 13, 14, 15, 16 We therefore explored the effect of NLRP3 inflammasome inhibition on atherosclerosis in the 2 mouse models applying a pharmacologic approach.
Herein, we report the increase in lysine 63 (K63)‐linked ubiquitination of NLRP3 by tranilast, an analog of a tryptophan metabolite, which has been used as an anti‐inflammatory drug.17 Our study demonstrated that tranilast restricts NLRP3 oligomerization and thereby the assembly of the NLRP3 inflammasome. We showed that tranilast blunts the activation of the NLRP3 inflammasome in Ldlr−/− and ApoE−/− macrophages and in atherosclerotic lesions. This study provides the first evidence to indicate that tranilast protects against the initiation and progression of atherosclerosis in both Ldlr−/− and ApoE−/− models. Our findings suggest that pharmacologic manipulation of NLRP3 ubiquitination offers an opportunity for therapeutic targeting of vascular inflammation and atherosclerosis.
Data available on request.
Methods
Cell Culture and Transfection
Human embryonic kidney 293T (HEK293T) cells, L929, and mouse J774A.1 macrophages were obtained from ATCC. The cells were maintained in DMEM supplemented with 10% heat‐inactivated fetal bovine serum. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen) as previously described.18
Preparation and Treatment of Bone Marrow–Derived Macrophages
Mouse bone marrow–derived macrophages (BMDMs) were prepared by flushing the bone marrow from the tibiae and femorae obtained from 6‐ to 8‐week‐old mice. Cells were differentiated into macrophages by culturing in DMEM supplemented with 10% fetal bovine serum and 20% L929‐conditioned media for 7 days.
To induce NLRP3 inflammasome activation, macrophages were primed with lipopolysaccharides (100 ng/mL, 6 hours) before stimulation with ATP (5 mmol/L, 0.5–1.5 hours).
Immunoblotting
Immunoblotting was performed as previously described.19, 20 Primary antibodies used in this study include the following: anti–α‐tubulin (Yeasen), anti‐FLAG (Sigma), anti‐NLRP3 (AdipoGen), anti‐ASC (AdipoGen), anti–caspase‐1 (AdipoGen), anti–IL‐1β (R&D Systems), anti‐HA (BioLegend), anti‐ubiquitin (Santa Cruz Biotechnology), and anti‐GAPDH (Yeasen).
Detection of Proteins in Cultured Cell Medium
The cultured cell media were concentrated via chloroform and methanol precipitation. Macrophages were treated as detailed in the figure legends and the cultured media were collected. One fourth volume of chloroform and 1 V of methanol were added into the sample. The mixture was vortexed and spun down at 15 000g for 10 minutes at room temperature. After removing the water/methanol mix, an additional 1 V of methanol was added, followed by centrifugation at 15 000g for 10 minutes at room temperature. The protein pellet was air‐dried for 5 minutes at room temperature, then resuspended in Triton‐based lysis buffer21 and immunoblotted.
Reconstitution of the NLRP3 Inflammasome in HEK293T Cells
HEK293T cells were plated in 24‐well microplates at a density of 2×105 cells per well. The cells were transfected with the plasmids expressing Flag‐NLRP3 (200 ng), HA‐NEK7 (never in mitosis gene a‐related kinase 7, 200 ng), Flag‐ASC (20 ng), Flag‐pro–caspase‐1 (100 ng) and Flag‐pro–IL‐1β (200 ng). The cultured media were changed at 36 hours post‐transfection and the cells were cultured for an additional 12 hours. The concentrated media and cell lysates were assayed by immunoblotting.
Immunoprecipitation
Immunoprecipitation was performed as previously described.22 Cells were solubilized in lysis buffer.18 The precleared lysates were incubated with the corresponding antibody (about 1.5 μg each) in the presence of 20 μL of Protein A/G Agarose (Pierce) overnight with constant agitation. The immunoprecipitates were analyzed by immunoblotting. The in vivo ubiquitination assay was conducted as previously described.21
ASC Oligomerization Assay
Macrophages were harvested in lysis buffer (50 mmol/L Tris–HCl, pH 7.5, 150 mmol/L NaCl, 10% glycerol, 0.5% Triton X‐100, 1 mmol/L PMSF, and complete protease inhibitor cocktail) and incubated on ice for 30 minutes, followed by centrifugation at 6000g for 15 minutes at 4°C. The supernatants and pellets were used as the Triton‐soluble and ‐insoluble fractions, respectively. For detection of ASC oligomerization, the Triton‐insoluble fractions were washed with lysis buffer and the pellets were resuspended in 300 μL of lysis buffer. The pellets were crosslinked for 30 minutes at 37°C with 2 mmol/L disuccinimidyl suberate (Pierce) and then spun down for 15 minutes at 6000g. The pellets were eluted and analyzed by immunoblotting with anti‐ASC.
Semi‐Denaturing Detergent Agarose Gel Electrophoresis
To analyze NLRP3 oligomerization, semi‐denaturing detergent agarose gel electrophoresis was conducted as previously described23 with minor modifications. Cells were harvested in lysis buffer for 45 minutes at 4°C. The cleared lysates were resuspended in buffer containing 0.5× tris borate EDTA, 2% SDS, 10% glycerol, and 0.0025% bromophenol blue. The samples were resolved on a 1.5% vertical agarose gel in running buffer containing 1× tris borate EDTA (89 mmol/L Tris, pH 8.3, 89 mmol/L boric acid, and 2 mmol/L EDTA) and 0.1% SDS for 75 minutes with a voltage of 80 V. The proteins were blotted onto a polyvinylidenedifluoride membrane for 3 hours, followed by immunoblotting.
Immunohistochemistry and Double Immunofluorescence Staining
Immunohistochemistry was conducted as previously described24, 25 with modifications. Mouse tissues were fixed in 60% methanol and 10% acetic acid in H2O, and embedded in paraffin. Tissue sections were subjected to deparaffinization and rehydration, followed by treatment with 3% hydrogen peroxide solution for 20 minutes to block endogenous peroxides activity. Antigen retrieval was conducted by treatment of the slides with EDTA (pH 9.0) or 10 mmol/L sodium citrate buffer (pH 6.0) by microwaving for 10 minutes. The samples were blocked with 5% fetal bovine serum in 0.1% PBS/BSA and incubated with primary antibody overnight at 4°C. The standard streptavidin–biotin linked horseradish peroxidase technique was conducted with 3,3′‐diaminobenzidine tetrahydrochloride being used for the development of peroxidase activity. The sections were counterstained with hematoxylin. Double immunofluorescence staining was conducted as previously described.24, 25 Primary antibodies used include the following: anti–α‐smooth actin (Santa Cruz Biotechnology), anti‐MCP1 (Abcam), anti‐NLRP3 (AdipoGen), anti‐active caspase‐1 (Thermo Fisher Scientific), anti–IL‐1β (R&D systems), anti‐Mac3 (BD Pharmingen), anti‐CD68 (BD Pharmingen), and anti–intercellular cell adhesion molecule 1 (ICAM‐1) (Santa Cruz Biotechnology). Images were acquired through a Nikon upright microscope with an objective set to ×4 or ×20 magnification, quantified using cellSens Standard software (Olympus Corporation), and expressed as percent of plaque area (%)=(staining positive area/plaque area)×100%.
Serum Lipid Measurement
The mice were anesthetized with isoflurane (RWD, Life Science). Mouse blood was collected by heart puncture using a 1‐mL syringe treated with 0.9% saline supplemented with heparin (Wanbang Pharmaceuticals) immediately before application. Serum was separated by centrifugation at 845g for 5 minutes at room temperature. Serum lipid and lipoprotein profiles were measured according to the manufacturer's instructions (Leadman Biochemistry).
Animal Treatment and Characterization of Atherosclerotic Plaques
Animal studies were approved by the Animal Care and Use Committee from Renmin Hospital, the Hubei University of Medicine. ApoE−/− and Ldlr−/− mice on a C57BL/6 background (Jackson Laboratories) were maintained in specific pathogen free level, independent ventilation cage environment on a regular light‐dark cycle (12 hours light, 12 hours dark). To accelerate atherosclerotic lesion formation, 6‐ to 8‐week‐old male and female mice were fed a Western diet (WD; D12079B, Research Diets). As detailed in the figure legends, mice were treated daily by oral gavage with dimethyl sulfoxide (Sigma) or tranilast (Shelleck, dissolved in dimethyl sulfoxide) diluted in vehicle (0.5% Carboxymethylcellulose, Sigma) to a final volume of 500 μL for each mouse. At the end of the experiment, mice were anesthetized by isoflurane and blood was collected from the left ventricle by cardiac puncture. The mice were perfused via the left ventricle with 0.9% saline supplemented with heparin (50 U/mL), followed by another perfusion with 4% paraformaldehyde solution. The heart was harvested and embedded in paraffin or optimal cutting temperature compound (Tissue‐Tek, Sakura) and frozen in −80°C for cryostats tissue sectioning. The entire aorta from the heart outlet to the iliac bifurcation was dissected, cleaned of adventitial and fat tissues, opened longitudinally, and stained with Oil Red O as previously described,26 and pinned flat on a black wax surface. Aorta images were captured through a stereomicroscope (Olympus SZX10) with a digital camera (Olympus). Plaque area was quantified using cellSens Standard software and expressed as percent of stained area relative to total aortic area as suggested.27
For aortic sinus analysis, the optimal cutting temperature–embedded aortas were sectioned with 10‐μm thickness and sections were acquired sequentially beginning at the aortic valve. Sections were stained with Oil Red O. Mean lesion area (Oil Red O+ area) and the necrotic area was calculated by measuring cross‐sections. The fibrous cap of the lesions was visualized with Verhöeff staining reagent (Leagene, Beijing, China). Collagen was visualized with Sirius Red staining (Solarbio Life Sciences, Beijing, China). Atherosclerotic lesion area, necrotic area, collagen content, and fibrous cap were measured using cellSens Standard software. The content of the fibrous cap in plaque was calculated by a formula: (fibrous cap staining area/plaque area)×100%. All quantifications were conducted by 2 individuals in a blinded manner.
Statistical Analysis
Data analyses were performed using GraphPad Prism 7.0 (GraphPad Software). Student t test was used to assess statistical differences between the 2 groups. The data are presented as mean±SEM. Values of P<0.05 (*), P<0.01 (**), and P<0.001 (***) were considered statistically significant.
Results
Tranilast Increases K63‐Linked NLRP3 Ubiquitination
NLRP3 is kept in an inactive ubiquitinated state to avoid unexpected activation of the NLRP3 inflammasome.6 Furthermore, elimination of K63‐linked polyubiquitin chains from NLRP3 facilitates the assembly and activation of the NLRP3 inflammasome.28, 29 However, less is known regarding the pharmacologic regulation of NLRP3 ubiquitination. These findings and issues spurred us to identify an enhancer of NLRP3 ubiquitination to therapeutically target NLRP3 inflammasome–associated diseases including atherosclerosis.
We began to search a reagent able to increase NLRP3 ubiquitination by in vivo ubiquitination assay.18 Eventually, we identified tranilast as a strong candidate to elevate NLRP3 ubiquitination. As depicted in Figure 1A, HEK293T cells were transfected with NLRP3 together with or without ubiquitin in the presence or absence of tranilast. NLRP3 was ubiquitinated (Figure 1A), consistent with previous findings.6, 29 Tranilast treatment stimulated NLRP3 ubiquitination (Figure 1A). NLRP3 was reported to undergo K48‐ and K63‐linked polyubiquitination.6, 30 While K48‐linked ubiquitination marks protein for proteasomal decay, K63‐linked ubiquitination exerts nondegradative functions.31 To clarify the type of NLRP3 ubiquitination, NLRP3 was cotransfected into 293T cells with ubiquitin K63R (UbK63R; a ubiquitin mutant incapable of forming K63‐linked ubiquitin chain) or UbK63 (a ubiquitin mutant that can only assemble K63‐linked ubiquitin chain).18 As shown in Figure 1B, tranilast facilitated UbK63‐ but not UbK63R‐mediated NLRP3 ubiquitination, indicating that tranilast mainly promotes K63‐linked ubiquitination of NLRP3. In line with previous observations,6, 28 endogenous NLRP3 was ubiquitinated in resting macrophages (Figure 1C). This modification was substantially boosted by tranilast (Figure 1C). Tranilast had no significant effect on endogenous NLRP3 expression (Figure 1C), suggesting that tranilast‐furthered NLRP3 ubiquitination does not impact the abundance of the NLRP3 protein.
Figure 1. Tranilast (TR) enhances K63‐linked ubiquitination of NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3).
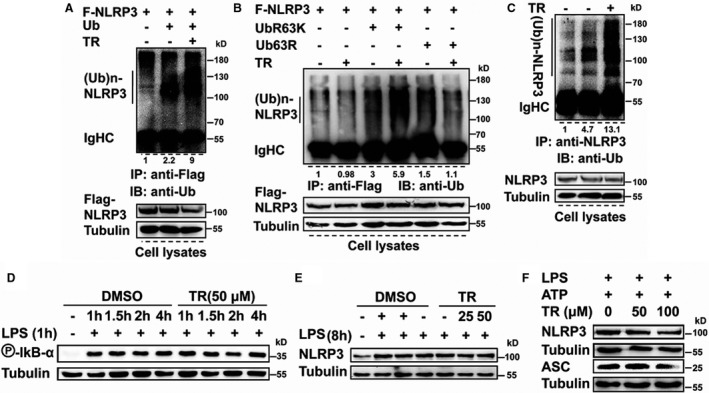
A and B, Human embryonic kidney 293T cells were transfected as indicated. Cell lysates were immunoprecipitated with anti‐Flag (F). The immunoprecipitates and lysates were immunoblotted with the indicated antibodies. C, J774A.1 cells were treated as indicated. Cell lysates were immunoprecipitated with anti‐NLRP3 and then immunoblotted with anti‐ubiquitin. Immunoblotting (IB) was conducted to examine the expression of indicated proteins. D through F, J774A.1 macrophages were treated as indicated. IB was conducted to examine the expression of proteins indicated. ASC indicates apoptosis‐associated speck‐like protein containing a caspase recruitment domain; DMSO, dimethyl sulfoxide; IgHC, heavy chain of immunoglobulin G; IP, immunoprecipitation; LPS, lipopolysaccharides; R, arginine; and Ub, ubiquitin.
We wanted to furnish more evidence for the effect of tranilast on the levels of the NLRP3 protein in macrophages. Nuclear factor κB‐dependent transcriptional upregulation of NLRP3 is essential for the assembly and activation of the NLRP3 inflammasome.2, 32 We addressed whether tranilast affects nuclear factor κB‐dependent upregulation of NLRP3 expression. As shown in Figure 1D, stimulation of macrophages with lipopolysaccharides efficiently elicited IкBα phosphorylation, a well‐recognized readout of nuclear factor кB activation.18 IкBα phosphorylation was kept intact upon tranilast treatment of macrophages (Figure 1D). Lipopolysaccharides promoted the expression of the NLRP3 protein (Figure 1E). The levels of the NLRP3 protein were unaffected by as high as 100 μmol/L of tranilast (Figure 1E and 1F). To the best of our knowledge, tranilast is the first chemical reagent that is able to regulate NLRP3 ubiquitination.
Tranilast Antagonizes the Assembly of the NLRP3 Inflammasome
NLRP3 inflammasome assembly and resultant activation require 2 signals: a priming signal (such as lipopolysaccharides) and an activation signal (as exemplified by ATP).2 The activation signal initiates the oligomerization of NLRP3. Polymerized NLRP3 recruits ASC, leading to the generation of ASC filaments, the latter of which nucleate pro–caspase‐1. The close proximity of pro–caspase‐1 then triggers the autoproteolytic maturation of pro–caspase‐1.7 Removal of K63‐linked polyubiquitin chains from NLRP3 precipitates the assembly and activation of the NLRP3 inflammasome.28, 29 Since tranilast improved NLRP3 ubiquitination with no effect on the abundance of the NLRP3 protein, we conjectured that tranilast may curb NLRP3 inflammasome assembly.
To examine whether tranilast interrupts NLRP3 inflammasome assembly, macrophages were primed by lipopolysaccharides, pretreated with tranilast, and then activated using ATP. Cellular lysates were immunoprecipitated with anti‐ASC and immunoblotted with anti‐NLRP3. NLRP3 was readily detected in the ASC immunoprecipitates (Figure 2A), indicating that NLRP3 interacts with ASC. However, this NLRP3‐ASC interaction was significantly impaired by tranilast (Figure 2A). As anticipated, the immunoprecipitation experiment showed that NLRP3 associated with pro–caspase‐1 (Figure 2B). This association was also dramatically counteracted by tranilast (Figure 2B). Consistent with a recent finding,33 NEK7 bound to NLRP3, which was efficiently dampened by tranilast (Figure 2C). In sum, tranilast antagonizes the assembly of the NLRP3 inflammasome.
Figure 2. Tranilast (TR) blunts NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) inflammasome assembly.
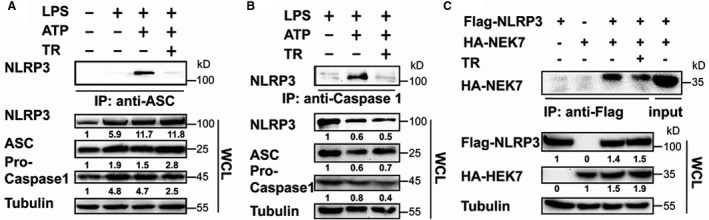
A and B, J774A.1 cells were treated as indicated. Cell lysates were immunoprecipitated with anti–apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC) (A) or anti–caspase‐1 (B). Immunoblotting was performed to assay for the expression of indicated proteins in the immunoprecipitates and whole cell lysates (WCLs). C, Human embryonic kidney (HEK) 293T cells were transfected as indicated. Cell lysates were immunoprecipitated with anti‐Flag. The immunoprecipitates and lysates were immunoblotted with the indicated antibodies. HA‐NEK7 indicates never in mitosis gene a‐related kinase 7; IP immunoprecipitation; and LPS, lipopolysaccharides.
Tranilast Blunts NLRP3 Oligomerization and IL‐1β Production in Atheroprone Macrophages
Since NLRP3 oligomerization is a prerequisite for NLRP3 inflammasome assembly,7 we assumed that tranilast could attenuate NLRP3 oligomerization. We first tested our hypothesis with BMDMs including those prepared from Ldlr−/− and ApoE−/− mice, respectively. In addition, IL‐1β, which has long been appreciated as a key atherogenic cytokine, is emerging as a therapeutic target of atherosclerotic cardiovascular disease,2 and we would measure the production of mature IL‐1β, a hallmark of NLRP3 inflammasome activation.2
We estimated NLRP3 oligomerization using semi‐denaturing detergent agarose gel electrophoresis in conjunction with immunoblotting.23 Ldlr−/− BMDMs were primed by lipopolysaccharides, pretreated with tranilast, and stimulated using ATP. We found that tranilast extremely restrained NLRP3 oligomerization (Figure 3A). NLRP3 polymerization ignites ASC oligomerization,34 a crucial event upstream of pro–caspase‐1 recruitment and activation.2 We assessed whether tranilast hinders ASC oligomerization by analyzing disuccinimidyl suberate cross‐linked Triton‐insoluble cell lysates. Exposure of Ldlr−/− BMDMs to ATP triggered ASC oligomerization (Figure 3B). Monomeric ASC was also seen in the Triton X‐100 insoluble fraction (Figure 3B). However, ASC oligomerization was impeded by tranilast pretreatment (Figure 3B).
Figure 3. Tranilast (TR) inhibits NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) oligomerization and activation.
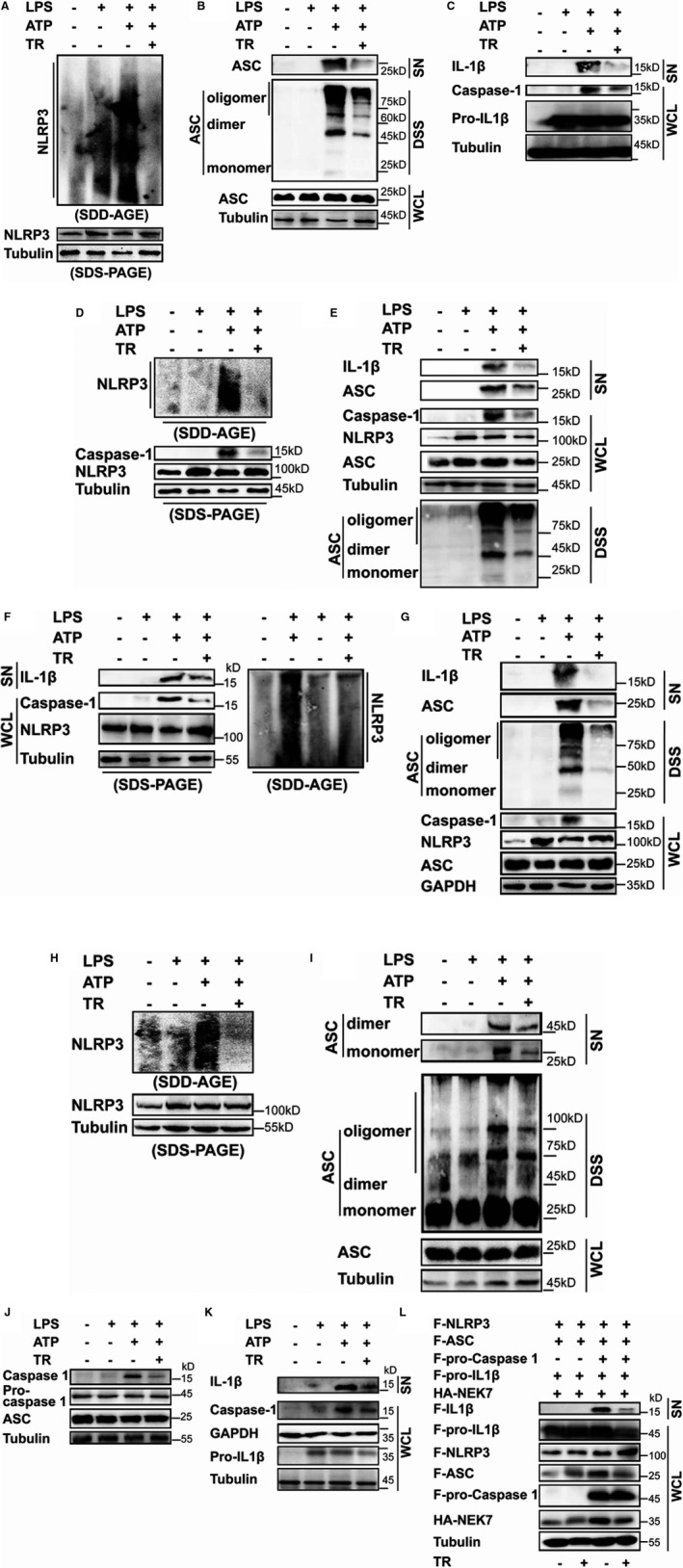
Low‐density lipoprotein receptor–deficient (Ldlr−/−) bone marrow–derived macrophages (BMDMs) (A through C), apolipoprotein E–deficient (ApoE−/−) BMDMs (D and E), wild‐type BMDMs (F and G), and J774A.1 macrophages (H through K) were treated as indicated. The supernatants (SNs), whole cell lysates (WCL) and the Triton‐insoluble fractions were analyzed by immunoblotting to determine the oligomerization of NLRP3 and apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC), caspase‐1 activation, and the release of interleukin (IL)‐1β and ASC. L, Human embryonic kidney 293T cells were transfected as indicated. At 8 hours before harvest, cells were treated without (–) or with TR (50 μmol/L). The SN and WCL were analyzed by immunoblotting as indicated. DSS indicates disuccinimidyl suberate; F, Flag; HA‐NEK7, never in mitosis gene a‐related kinase 7; LPS, lipopolysaccharides; SDD‐AGE, semi‐denaturing detergent agarose gel electrophoresis.
Figure 3C shows that exposure of Ldlr−/− BMDMs to tranilast led to a pronounced inhibition of caspase‐1 activation, revealing that tranilast suppressed NLRP3 inflammasome activation in Ldlr−/− BMDMs. Tranilast substantially retarded the production and secretion of IL‐1β (Figure 3C). ASC and its oligomers were also found in the extracellular space, serving as a danger signal to propagate inflammation.35 Interestingly, tranilast treatment significantly repressed the release of ASC from macrophages into the supernatants (Figure 3B).
We wondered whether tranilast exerts similar functions in ApoE−/− BMDMs. Treatment of ApoE−/− BMDMs with lipopolysaccharides plus ATP induced NLRP3 oligomerization, ASC oligomerization, caspase‐1 activation, and release of IL‐1β and ASC, all of which were dramatically inhibited by tranilast (Figure 3D and 3E). We conclude that tranilast suppresses the activation of the NLRP3 inflammasome in atheroprone macrophages.
We then tested whether tranilast also performs a similar function in WT (C57BL/6) BMDMs. Following stimulation with lipopolysaccharides plus ATP, NLRP3 inflammasome activation was markedly induced in WT BMDMs, as evidenced by NLRP3 oligomerization, ASC oligomerization (a strong activation of caspase‐1), and the release of IL‐1β and ASC (Figure 3F and 3G). Nonetheless, these changes were largely impeded by tranilast (Figure 3F and 3G). Likewise, similar findings were achieved with J774A.1 macrophage cell line (Figure 3H through 3K). Taken together, tranilast is a potent inhibitor of the NLRP3 oligomerization and IL‐1β production in macrophages, supporting the previous report indicating that tranilast blunted NLRP3 inflammasome activation in WT macrophages and macrophage cell line.36
Last, we reconstituted the NLRP3 inflammasome in HEK293T cells to ascertain whether tranilast directly modulates the activation of the NLRP3 inflammasome.33 In addition to the core members (NLRP3, ASC, and pro–caspase‐1), NEK7 is thought of as a new component.2 We transfected these molecules into HEK293T cells and measured IL‐1β production. Figure 3L shows that introduction of these molecules into HEK293T cells significantly induced the activation of caspase‐1 and the production of mature IL‐1β. In sharp contrast, exposure of the reconstituted cells to tranilast significantly repressed caspase‐1 activation and IL‐1β secretion (Figure 3L). This result demonstrated that tranilast directly inhibited the activation of the NLRP3 inflammasome.
Tranilast Inhibits the Initiation of Atherosclerosis in Ldlr−/− Mice
There is no evidence indicating whether increments in NLRP3 ubiquitination alleviate diet‐induced initiation and progression of atherosclerosis. Meanwhile, there is no report focusing on the role of tranilast in vascular inflammation and atherosclerosis in Ldlr−/− or ApoE−/− mice, the 2 best‐recognized experimental models of atherosclerosis.10
We set out to evaluate the role of tranilast in regulating the initiation of atherogenesis. We first treated Ldlr−/− mice with tranilast at the onset of WD feeding (Figure 4A). After 12 weeks of treatment, the mice were euthanized and the whole aortas were dissected for en face Oil Red O staining. The atherosclerotic lesions in mice treated with tranilast were much smaller than those in mice receiving vehicle (Figure 4B and 4C). The impact of tranilast on the initiation of atherosclerosis was also estimated in cross‐sectional lesions of the aortic sinus. As expected, treatment of mice with TR significantly reduced lesions in the aortic sinus (Figure 4D). Tranilast treatment led to an increase in size of the fibrous cap of the lesion compared with the vehicle (Figure 4E and 4F). In parallel, the necrotic core of the plaque in tranilast‐treated mice was much smaller than that in mice treated with vehicle (Figure 4F). Taken together, tranilast repressed the expression and activation of the NLRP3 inflammasome in the lesions and retarded the initiation of atherosclerosis in Ldlr−/− mice.
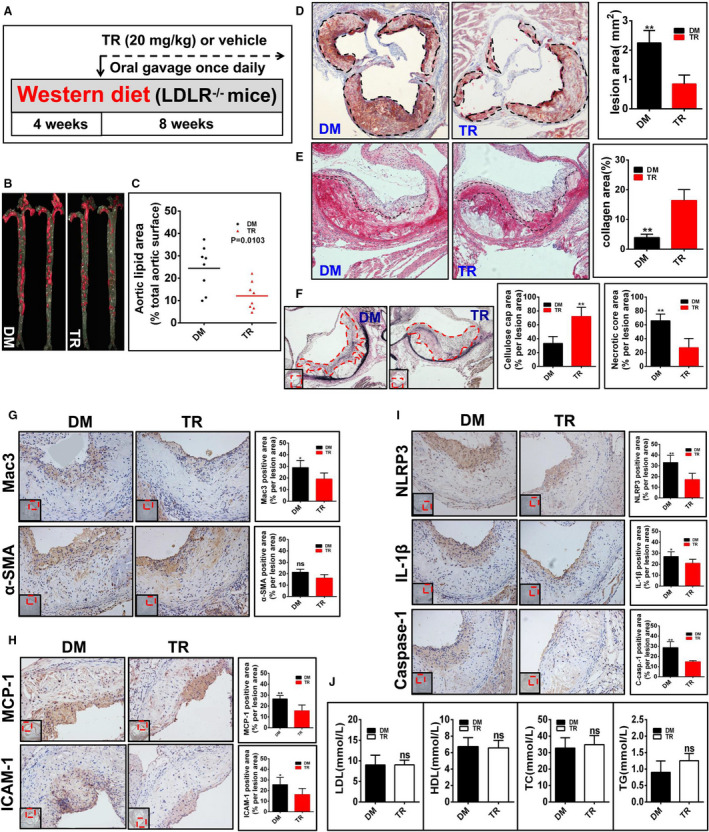
Figure 4. Tranilast (TR) inhibits the initiation of atherosclerosis in low‐density lipoprotein receptor–deficient (Ldlr−/−) mice (group 1).
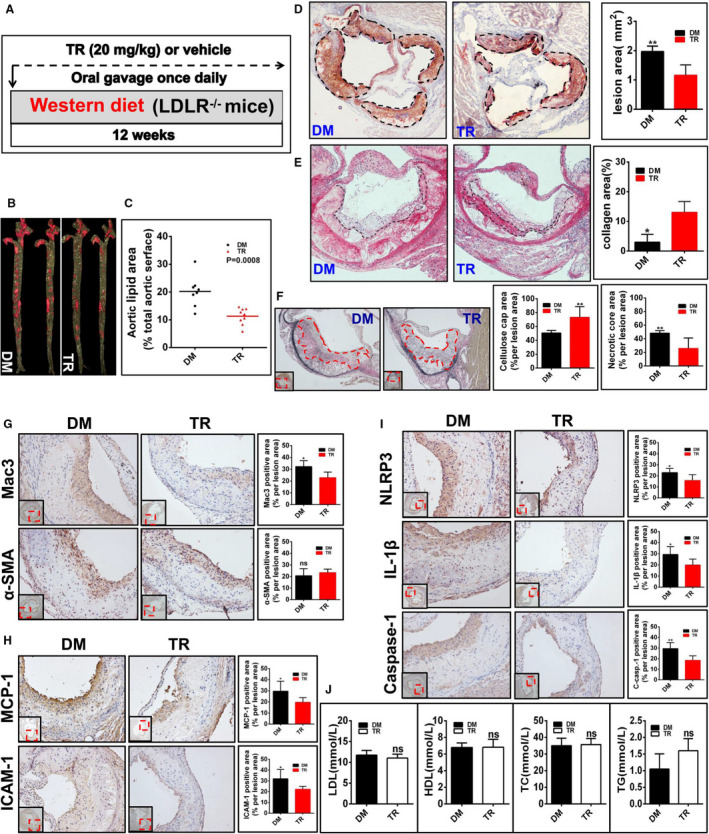
A, Shown is the regimen for treatment of Ldlr−/− mice (6 to 8 weeks old) with TR (n=9) or vehicle (n=8). The atherosclerotic lesions in the whole aortas (B) and in the aortic sinuses (D) were visualized by Oil Red O staining. The lesional size of the aortas (C) and aortic sinuses (D) were measured. E, The collagen was visualized by Sirius Red staining and the collagen content in the plaque was measured. F, The fibrous caps were stained by Verhöeff elastic fiber dye, outlined with red dashed line and quantified; the necrotic core areas of the plaques were calculated. G through I, The expression of the indicated proteins in the aortic sinuses was detected by immunohistochemical staining. The positive area of each protein was measured. J, The profile of the plasma lipids and lipoproteins. DM indicates vehicle containing dimethyl sulfoxide; NS, no significant difference; TC, total cholesterol; TG, triacylglycerol; and α‐SMA, α‐smooth muscle actin. *P<0.05; **P<0.01.
The development of atherosclerosis begins with the entry of monocytes into the arterial walls.2 Atherosclerosis is characterized by the accumulation of cholesterol‐laden macrophages,1 culminating in the activation of the NLRP3 inflammasome.2 Macrophages are prominent cellular components in the atherosclerotic lesions.37 We found that tranilast‐treated mice had dramatically reduced macrophage infiltration in the lesions in comparison to those given vehicle (Figure 4G). No significant difference in the content of smooth muscle cells (SMCs) between the 2 groups was seen (Figure 4G).
Blood‐borne monocytes differentiate into macrophages in the lesion.2 MCP‐1 (Monocyte chemoattractant protein‐1) and the adhesion molecules such as ICAM‐1 are essential for monocyte recruitment into the atherosclerotic lesions.2 As shown in Figure 4H, tranilast treatment caused a significant reduction in both ICAM‐1+ and MCP‐1+ areas in the lesions. IL‐1β is an apical inflammatory mediator that can induce the expression of various proinflammatory molecules such as MCP‐1 and ICAM‐1 in the vessel wall.2 We found that IL‐1β expression was markedly reduced in the lesion of mice treated with tranilast compared with that in control mice (Figure 4I). Tranilast treatment also inhibited NLRP3 expression in the lesions (Figure 4I). To delve into the role of tranilast in manipulating the activation of the NLRP3 inflammasome in the plaques, we performed immunohistochemical staining with an anti–caspase‐1 antibody that is a specifically recognized active caspase‐1 species. As expected, tranilast treatment pronouncedly impeded caspase‐1 activation in the atherosclerotic lesions (Figure 4I). Taken together, tranilast repressed the expression and activation of the NLRP3 inflammasome in the atherosclerotic lesions. However, tranilast had no impact on the levels of plasma lipids and lipoproteins (Figure 4J).
Tranilast Represses the Progression of Atherosclerosis in Ldlr−/− Mice
We next determined whether tranilast influences the progression of atherosclerosis in Ldlr−/− mice. Ldlr−/− mice were placed on a WD for 4 weeks, and then administrated with tranilast while feeding a WD for an additional 8 weeks (Figure 5A). The atherosclerotic lesions in the entire aortas (Figure 5B and 5C) and the aortic roots (Figure 5D) from tranilast‐treated mice were markedly declined when compared with those from the control group. The fibrous cap of the plaque in the tranilast‐treated mice was much larger than that in the control mice (Figure 5E and 5F). In parallel, tranilast restricted the formation of the necrotic core in the plaque (Figure 5F).
Figure 5. Tranilast (TR) suppresses the progression of atherosclerosis in low‐density lipoprotein receptor–deficient (Ldlr−/−) mice (group 2).
A, Diagramed is the regimen for treatment of Ldlr−/− mice (6 to 8 weeks old) with TR (n=8) or vehicle (n=8). The atherosclerotic lesions (B through D), the collagen content in the plaque (E), the fibrous caps and the necrotic core (F), the expression of the indicated proteins (G through I), and the profile of the plasma lipids and lipoproteins (J) in mice were analyzed and quantified as described in Figure 3.
The content of macrophages but not SMCs in the lesions was substantially reduced upon treatment with tranilast, as judged by the Mac3+ area and α‐smooth muscle actin+ area (Figure 5G). Of note, staining with normal IgG demonstrated the specificity of the immunohistochemistry results (Figure S1). The expression levels of ICAM‐1 and MCP‐1 were significantly decreased in the plaques of tranilast‐treated mice in comparison to those in the control mice (Figure 5H). Compared with the control mice, tranilast‐treated mice exhibited a significantly decreased expression and activation of the NLRP3 inflammasome in the lesions, as indicated by reduced levels of NLRP3, IL‐1β, and active caspase‐1 (Figure 5I). There was no significant change in the levels of lipids and lipoproteins between the 2 groups (Figure 5J). Taken together, tranilast represses the expression and activation of the NLRP3 inflammasome in the atherosclerotic lesions and impedes the progression of atherosclerosis in Ldlr−/− mice.
Tranilast Suppresses the Initiation of Atherosclerosis in ApoE−/− Mice
The ApoE−/− and Ldlr−/− models differ in significant ways in atherosclerosis‐facilitating mechanisms.10 A perusal of the literature showed that there are gene knockouts exhibiting different phenotypes in the 2 models.38 This is true for the NLRP3 inflammasome.2 Having investigated the relevance of tranilast in atherosclerosis in the Ldlr−/− model, we next gauged the role of tranilast in atherosclerosis employing the ApoE−/− model.
We evaluated the role for tranilast in the initiation of atherosclerosis in ApoE−/− mice (Figure 6A). The tranilast‐treated ApoE−/− mice exhibited a marked reduction in the atherosclerotic lesions in the whole aorta (Figure 6B and 6C) and in the aortic root (Figure 6D) compared with vehicle‐treated mice. It is worth noting that numerous studies reveal that sex is associated with the initiation and progression of atherosclerosis.27 It is important to determine whether the effect of tranilast on atherosclerosis is sex dependent or independent. Remarkably, tranilast blunted the initiation of atherosclerosis in both male (Figure S2A) and female (Figure S2B) ApoE−/− mice. The atherosclerotic plaque in the tranilast‐treated mice was characterized by a much larger fibrous cap and smaller necrotic core than vehicle‐treated ApoE−/− mice (Figure 6E and 6F). While the content of SMCs was comparable in both groups, fewer macrophages were present in the plaques of the tranilast‐treated mice (Figure 6G). Tranilast treatment gave rise to a marked decrease in the expression of MCP‐1 (Figure 6H), ICAM‐1 (Figure 6H), NLRP3 (Figure 6I), and IL‐1β (Figure 6I). NLRP3 inflammasome activation was retarded, as characterized by the reduction in the levels of active caspase‐1 in the lesions of the tranilast‐treated mice (Figure 6I). Tranilast administration did not significantly alter the plasma levels of lipids and lipoproteins (Figure 6J). Together, tranilast hinders the expression and activation of the NLRP3 inflammasome and dampens the initiation of atherosclerosis in ApoE−/− mice.
Figure 6. Tranilast (TR) represses the initiation of atherosclerosis in apolipoprotein E–deficient (ApoE−/−) mice (group 3).
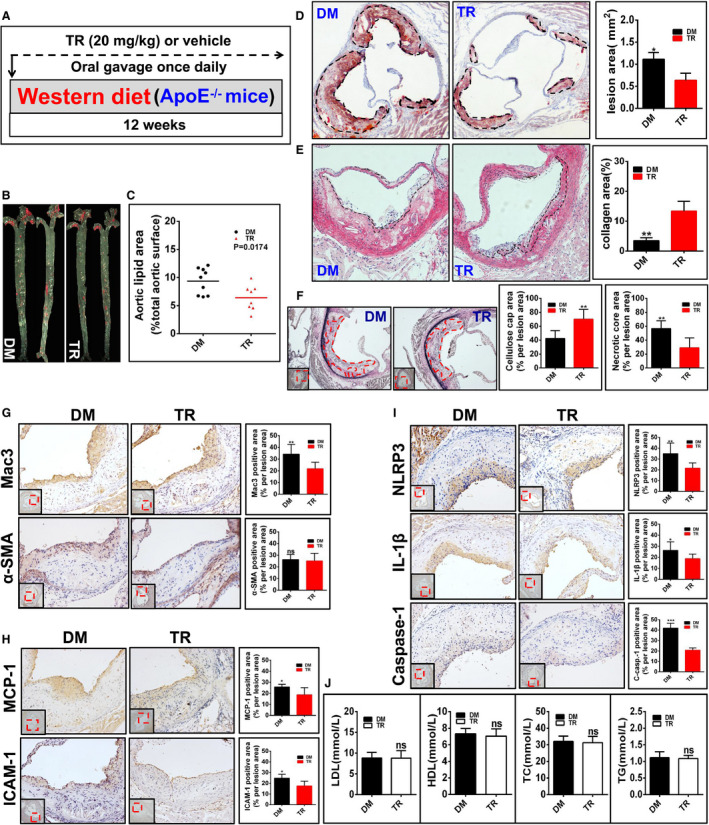
A, Illustrated is the regimen for treatment of ApoE−/− mice (6 to 8 weeks old) with TR (n=8) or vehicle (n=9). The atherosclerotic lesions (B through D), the collagen content in the plaque (E), the fibrous caps and the necrotic core (F), the expression of the indicated proteins (G through I), and the profile of the plasma lipids and lipoproteins (J) in mice were evaluated and quantified as described in Figure 3. *P<0.05; **P<0.01; ***P<0.001.
Tranilast Impedes the Progression of Atherosclerosis in ApoE−/− Mice
Finally, we assessed the impact of tranilast on the progression of atherosclerosis in ApoE−/− mice. ApoE−/− mice were first placed on a WD for 2 weeks. Tranilast was then administrated while continuing to feed the mice a WD for an additional 10 weeks (Figure 7A). As a consequence of tranilast treatment, ApoE−/− mice had a much smaller lesion in the whole aorta (Figure 7B and 7C) and in the aortic root (Figure 7D) than did the vehicle‐fed mice. Importantly, tranilast ameliorated the progression of atherosclerosis in both male (Figure S2C) and female (Figure S2D) ApoE−/− mice. The plaque in tranilast‐administrated mouse displayed a smaller necrotic core with a much thicker fibrous cap than did the control group (Figure 7E and 7F). Tranilast also curbed lesional infiltration of macrophages rather than SMCs (Figure 7G). In parallel, tranilast treatment caused a marked decline in the expression of MCP‐1 (Figure 7H), ICAM‐1 (Figure 7H), NLRP3 (Figure 7I), and IL‐1β (Figure 7I). Furthermore, NLRP3 inflammasome activation in tranilast‐treated ApoE−/− mice was pronouncedly inhibited, as demonstrated by decreased caspase‐1 abundance in the lesions (Figure 7I). The plasma levels of lipids and lipoproteins in the 2 groups were similar (Figure 7J). Together, tranilast repressed NLRP3 inflammasome expression and activation in the plaques and blunted the progression of atherosclerosis in ApoE−/− mice.
Figure 7. Tranilast (TR) curbs the progression of atherosclerosis in apolipoprotein E–deficient (ApoE−/−) mice (group 4).
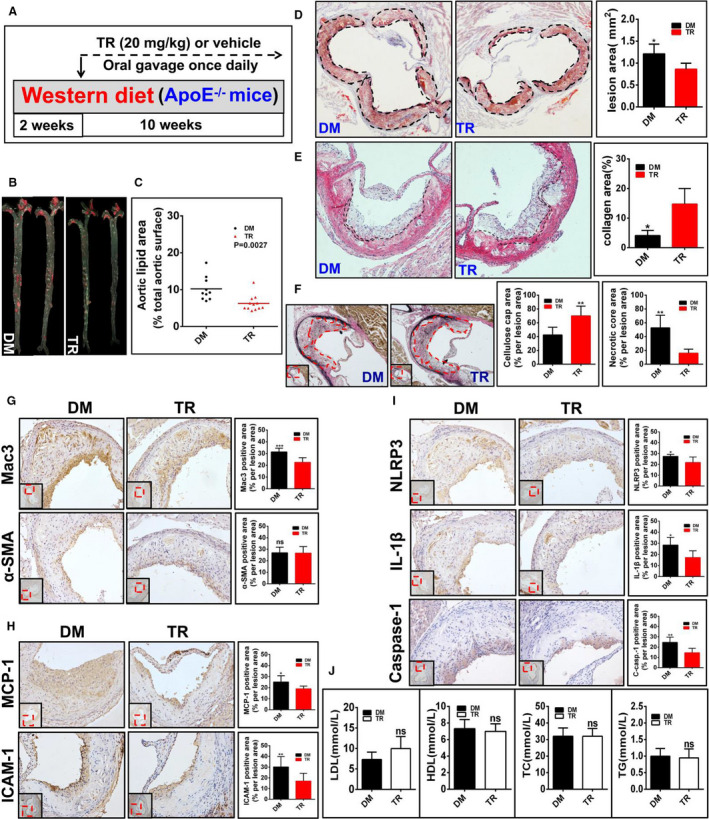
A, Depicted is the regimen for treatment of ApoE−/− mice (6 to 8 weeks old) with TR (n=12) or vehicle (n=10). The atherosclerotic lesions (B through D), the collagen content in the plaque (E), the fibrous caps and the necrotic core (F), the expression of the indicated proteins (G through I), as well as the profile of the plasma lipids and lipoproteins (J) in mice were examined and quantified as described in Figure 3.
Collectively, tranilast effectively suppresses the initiation and progression of atherosclerosis in both Ldlr−/− and ApoE−/− mice, at least partially, through targeting the NLRP3 inflammasome.
Tranilast Impeded the Activation of the NLRP3 Inflammasome Primarily in Macrophages
As described above, tranilast curbed NLRP3 inflammasome activation in cultured macrophages and atherosclerotic lesions. While numerous studies demonstrated that the activation of the NLRP3 inflammasome occurs predominantly in macrophages, NLRP3 inflammasome activation was also observed in SMCs.39 Given that SMCs are one of the most important cell types in atherosclerotic lesions, we performed double immunofluorescence to determine whether the activation of the NLRP3 inflammasome took place in SMCs and/or in macrophages in the atherosclerotic lesions and, if so, was subjected to regulation by tranilast. Dual immunostaining showed that the NLRP3 inflammasome was robustly activated in macrophages rather than in SMCs (Figure 8A). Tranilast treatment markedly suppressed the activation of macrophage NLRP3 inflammasome (Figure 8A). In addition, we also detected the expression of IL‐1β. Consistent with the above observation, the expression of IL‐1β was significantly upregulated in macrophages but not in SMCs (Figure 8B), which was pronouncedly repressed by tranilast (Figure 8B). Taken together, the activation of the NLRP3 inflammasome occurred primarily in macrophages, which was blunted by tranilast.
Figure 8. Tranilast (TR) predominantly targeted the NLRP3 (nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor family pyrin domain‐containing 3) inflammasome in macrophages.
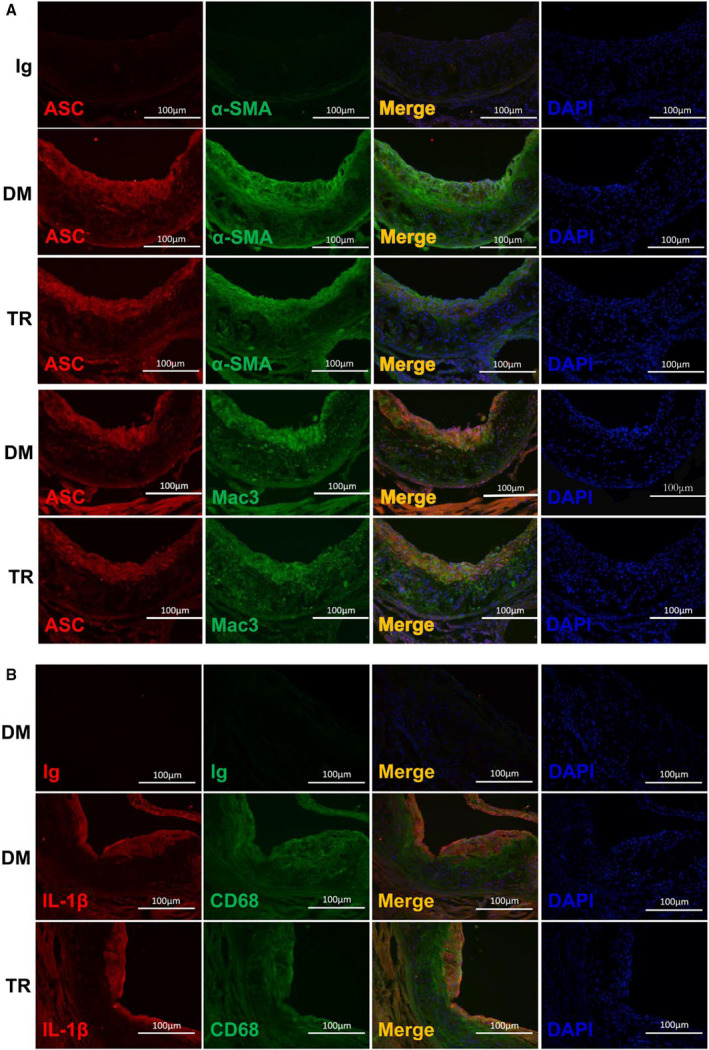
The frozen section of the aortic sinuses from mice treated with TR or vehicle containing dimethyl sulfoxide (DM) were analyzed by dual immunofluorescence staining. Double immunostaining was conducted with the antibodies against α–smooth muscle actin (α‐SMA) and apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC) (A), Mac3 and ASC (A), CD68 and interleukin (IL)‐1β (B).
Discussion
The novel findings of the current study involve the following: tranilast facilitates K63‐linked ubiquitination of NLRP3, blunts the activation of the NLRP3 inflammasome in atheroprone macrophages and in the atherosclerotic lesions, and hinders both the initiation and progression of atherosclerosis in both Ldlr−/− and ApoE−/− mice. To the best of our knowledge, this is the first study to identify a pharmacologic enhancer of NLRP3 ubiquitination, further revealing its inhibitory effect on the initiation and progression of atherosclerosis in both the Ldlr−/− and ApoE−/− models. Thus, pharmacologic manipulation of NLRP3 ubiquitination offers a new strategy for protection against atherosclerosis.
Ldlr−/− and ApoE−/− mice are the most widely accepted models of atherosclerosis although their atheroprone mechanisms differ in important ways.10 Most of investigators used one or other model in their studies to dissect the mechanism behind atherosclerosis and to discover novel therapeutics for this life‐threatening disorder.10 It remains an open question of whether the 2 models could yield comparable results to each other. Indeed, there are gene knockouts exhibiting different phenotypes in the 2 models. For instance, knocking out hepatic lipase led to an increase in lesion formation in Ldlr−/− mice11 but resulted in a decrease in lesional formation in ApoE−/− mice.12 Additionally, deletion of IL‐6 had no impact on atherosclerotic lesions in the aortic root in the Ldlr−/− model.13 In stark contrast, ablation of IL‐6 caused an increment in lesions in the entire aorta and the aortic arch in ApoE−/− mice.14 The same holds true for granulocyte‐macrophage colony‐stimulating factor.15, 16 The basis for the difference is likely ascribed to the differences in the genetic background and also in experimental design and conditions including the composition and feeding time of an atherogenic diet, the impact of the intestinal microbiome that may be various between the vivariums, the sampling for the evaluation of arterial lesions, and the sex of the mice used.10
Since genetic studies have also yielded mixed results about the role for the NLRP3 inflammasome in atherosclerosis,8, 9 we attempted to conduct our studies on tranilast in atherosclerosis using the 2 types of atheroprone mice in the same vivarium and on the same diet. Our work provided clear evidence showing that tranilast dramatically blocks the activation of the NLRP3 inflammasome in atherosclerotic lesions and blunts the initiation and progression of atherosclerosis in the 2 models. Our study demonstrates that tranilast is a potent suppressor of atherosclerosis. Of note, MCC950, a selective inhibitor of the NLRP3 inflammasome, was able to alleviate the development of atherosclerosis.40 Accordingly, our research highlights that a pharmacologic approach shows great advantage to clarify the significance of NLRP3 inflammasome activation in atherogenesis. There are 2 possible explanations for this. First, NLRP3 performs a variety of inflammasome‐independent functions in multiple types of cells.41, 42, 43, 44 Second, neither ASC nor pro–caspase‐1 is a specific component for the NLRP3 inflammasome.7 Absence of these molecules may cause NLRP3‐independent effect(s).
Given that sex is linked to the initiation and progression of atherosclerosis.27 It is important to probe whether the effect of tranilast on atherosclerosis is sex‐dependent or ‐independent. To address this important issue, we employed equal amounts of male and female mice for each treatment at the onset of each study. One group, which was depicted in Figure 4, could not be subjected to sex‐specific analysis because of unexpected death of mice during this study. Remarkably, the other 3 treatments revealed that the atheroprotective role of tranilast is sex‐independent in both the ApoE−/− (Figure S2) and Ldlr−/− (data not shown) mice.
It is believed that macrophage‐mediated inflammatory responses play critical roles throughout all stages of atherosclerosis.2, 45, 46 Particularly, atherosclerosis is characterized by the accumulation of cholesterol‐laden macrophages,1 culminating in NLRP3 inflammasome activation.2 We found that tranilast treatment significantly reduced the accumulation and activation of macrophages in the atherosclerotic lesions. In contrast, a previous study described that tranilast had no effect on the macrophage content in the plaques although tranilast repressed to some extent the initiation of atherosclerosis in Watanabe heritable hyperlipidemic rabbits.47 The discrepancy between the previous study and ours is unknown. A simple explanation is that different animal models were utilized in the 2 studies. Given that IL‐1β is sufficient to recruit monocytes/macrophages into the atherosclerotic lesions,2 we assumed that tranilast performed its atheroprotective function through acting on macrophages. On the other hand, while the previous study explored only the role of tranilast in the initiation of atherosclerosis,47 our study demonstrated that tranilast prevents not only the initiation of atherosclerosis but also retards the progression of this disease, further highlighting the translational perspective of tranilast in atherosclerosis treatment. To the best of our knowledge, the evidence for the significance for the NLRP3 inflammasome in the rabbit model of atherosclerosis is lacking. In this regard, the 2 mouse models are far more reliable in the mechanistic study of atherosclerosis. By virtue of the fundamentally important role for the NLRP3 inflammasome in driving atherosclerosis,2 it is highly possible that our work uncovers the major mechanism behind tranilast fueling atherosclerosis. To fully substantiate that tranilast conducts its atheroprotective role primarily through targeting NLRP3, further study should be performed with Ldlr−/− or ApoE−/− mice deficient for NLRP3.
We found that tranilast reduces macrophage release of IL‐1β and ASC. IL‐1β is a primordial cytokine that is closely implicated in a broad spectrum of inflammatory diseases including atherosclerosis.2 IL‐1β is the apical inflammatory mediator that is crucial for mounting the proinflammatory response by strongly inducing diverse types of cells to elaborate secondary inflammatory mediators including IL‐6, an important atherosclerotic cytokine.2 IL‐1β acts via the autocrine, paracrine, or endocrine mechanisms.2 Importantly, it has been reported that tranilast reduced the levels of cytokines such as IL‐1β in the blood.36 Extracellular ASC and its oligomers function to propagate inflammation.35 Thus, tranilast is a potent inhibitor of the NLRP3 inflammasome and inflammation. During the course of our study, a group reported that tranilast blunted NLRP3 inflammasome activation in WT macrophages and macrophage cell line.36 However, the authors did not address whether tranilast can conduct a similar function in atheroprone macrophages. Moreover, the mechanism behind tranilast regulation of the NLRP3 inflammasome was not completely defined in the aforementioned study.36 Our work showed that tranilast exerts its inflammasome‐inhibiting function not only in WT macrophages but also in ApoE−/− and Ldlr−/− macrophages. Moreover, our work sheds new light on the mechanism of tranilast action.
NLRP3 is ubiquitinated in resting macrophages to prevent improper NLRP3 activation.28 More important, elimination of K63‐linked polyubiquitin chains from NLRP3 was found to promote the assembly and activation of the NLRP3 inflammasome.28, 29 There is little, if any, evidence about the pharmacologic manipulation of K63‐linked ubiquitination of NLRP3. We demonstrated that tranilast boosts K63‐linked ubiquitination of NLRP3, which heightens the threshold of the assembly and activation of the NLRP3 inflammasome. Of note, this effect of tranilast does not alter the abundance of the NLRP3 protein. Instead, tranilast suppressed oligomerization of NLRP3, ensuing the assembly of the NLRP3 inflammasome, accounting for its effect on NLRP3 inflammasome activation. Our study provides a novel mechanistic insight into the role for tranilast in antagonizing the activation of the NLRP3 inflammasome. Collectively, our study significantly extends the previous finding.36
Conclusions
Our study demonstrated that tranilast promotes K63‐linked ubiquitination of NLRP3, enhancing the threshold of NLRP3 inflammasome assembly and activation. In support of this proposition, tranilast antagonizes oligomerization of NLRP3 and thereby the assembly of the NLRP3 inflammasome, accounting for its suppressive role in NLRP3 inflammasome activation. Our study provides a previously unidentified mechanistic insight into the role of tranilast in inhibiting the activation of the NLRP3 inflammasome and inflammation. This is the first study to discover a pharmacologic enhancer of NLRP3 ubiquitination and further uncover its inhibitory role in both initiation and progression of atherosclerosis. Thus, pharmacologic manipulation of NLRP3 ubiquitination provides a new avenue to therapeutic targeting of vascular inflammation and atherosclerosis. Our research also highlights that a pharmacologic approach has great advantage to substantiate the causal role of the NLRP3 inflammasome in the initiation and progression of atherosclerosis.
To date, low‐density lipoprotein cholesterol–lowering drugs remain the mainstay for the treatment of atherosclerosis.2 Targeting residual inflammation holds promise for the treatment of atherosclerotic cardiovascular disease.3 CANTOS further demonstrates that reduction in IL‐1β reduces inflammation risk.4 Our study and others assign tranilast novel functions in regulating NLRP3 ubiquitination and atherosclerosis. Remarkably, tranilast is an old drug with great safety in the clinic. From a translational point of view, application of tranilast combined with other lipid‐lowering therapies is beneficial in effective atherosclerosis therapy.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81872381) and from the Health Commission of Hubei Province Scientific Research Projects (WJ2019Z003 and WJ2019M047) to Fu and Jin. This work was also supported by startup funds from the Hubei University of Medicine and the Hubei University of Medicine Renmin Hospital to Fu and Jin.
Disclosures
None.
Supporting information
Figures S1–S2
Acknowledgments
We are grateful to Dr Lian Li in the Department of Laboratory Medicine at Renmin Hospital for her assistance with the profiling of plasma lipids and lipoproteins.
Author contributions: Jin and Fu conceived the experiments. Chen, Wang, Pan, Liu, Zheng, Ding, Mu, Yuan, and Li performed the experiments. Yuan and Song provided technical support for the experiments. Chen, Wang, Mu, Jin, and Fu analyzed the data. Jin and Fu wrote the article. Jin and Fu secured funding.
(J Am Heart Assoc. 2020;9:e015513 DOI: 10.1161/JAHA.119.015513.)
For Sources of Funding and Disclosures, see page 17.
Contributor Information
Ying Jin, Email: jdyj0001@163.com.
Jian Fu, Email: jianfu0001@yahoo.com.
References
- 1. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jin Y, Fu J. Novel insights into the NLRP 3 inflammasome in atherosclerosis. J Am Heart Assoc. 2019;8:e012219 DOI: 10.1161/JAHA.119.012219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation. 2018;138:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 5. Voronov E, Apte RN. Targeting the tumor microenvironment by intervention in interleukin‐1 biology. Curr Pharm Des. 2017;23:4893–4905. [DOI] [PubMed] [Google Scholar]
- 6. Py BF, Kim MS, Vakifahmetoglu‐Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–338. [DOI] [PubMed] [Google Scholar]
- 7. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. [DOI] [PubMed] [Google Scholar]
- 8. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in ApoE‐deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Getz GS, Reardon CA. Do the Apoe‐/‐ and Ldlr‐/‐ mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol. 2016;36:1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freeman L, Amar MJ, Shamburek R, Paigen B, Brewer HB Jr, Santamarina‐Fojo S, Gonzalez‐Navarro H. Lipolytic and ligand‐binding functions of hepatic lipase protect against atherosclerosis in LDL receptor‐deficient mice. J Lipid Res. 2007;48:104–113. [DOI] [PubMed] [Google Scholar]
- 12. Mezdour H, Jones R, Dengremont C, Castro G, Maeda N. Hepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E‐deficient mice. J Biol Chem. 1997;272:13570–13575. [DOI] [PubMed] [Google Scholar]
- 13. Song L, Schindler C. IL‐6 and the acute phase response in murine atherosclerosis. Atherosclerosis. 2004;177:43–51. [DOI] [PubMed] [Google Scholar]
- 14. Schieffer B, Selle T, Hilfiker A, Hilfiker‐Kleiner D, Grote K, Tietge UJ, Trautwein C, Luchtefeld M, Schmittkamp C, Heeneman S, et al. Impact of interleukin‐6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110:3493–3500. [DOI] [PubMed] [Google Scholar]
- 15. Ditiatkovski M, Toh BH, Bobik A. GM‐CSF deficiency reduces macrophage PPAR‐gamma expression and aggravates atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2337–2344. [DOI] [PubMed] [Google Scholar]
- 16. Subramanian M, Thorp E, Tabas I. Identification of a non‐growth factor role for GM‐CSF in advanced atherosclerosis: promotion of macrophage apoptosis and plaque necrosis through IL‐23 signaling. Circ Res. 2015;116:e13–e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res. 2015;91:15–28. [DOI] [PubMed] [Google Scholar]
- 18. Fu J, Taubman MB. EGLN3 inhibition of NF‐kappaB is mediated by prolyl hydroxylase‐independent inhibition of IkappaB kinase gamma ubiquitination. Mol Cell Biol. 2013;33:3050–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu J, Menzies K, Freeman RS, Taubman MB. EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J Biol Chem. 2007;282:12410–12418. [DOI] [PubMed] [Google Scholar]
- 20. Fu J, Taubman MB. Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF‐kappaB‐dependent pathway. J Biol Chem. 2010;285:8927–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu J. Catalytic‐independent inhibition of cIAP1‐mediated RIP1 ubiquitination by EGLN3. Cell Signal. 2016;28:72–80. [DOI] [PubMed] [Google Scholar]
- 22. Fu J, Jin Y, Arend LJ. Smac3, a novel Smac/DIABLO splicing variant, attenuates the stability and apoptosis‐inhibiting activity of X‐linked inhibitor of apoptosis protein. J Biol Chem. 2003;278:52660–52672. [DOI] [PubMed] [Google Scholar]
- 23. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan W, Jin Y, Stanger B, Kiernan AE. Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc Natl Acad Sci USA. 2010;107:15798–15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan W, Jin Y, Chen J, Rottier RJ, Steel KP, Kiernan AE. Ectopic expression of activated notch or SOX2 reveals similar and unique roles in the development of the sensory cell progenitors in the mammalian inner ear. J Neurosci. 2013;33:16146–16157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu F, Wu L, Wu G, Wang C, Zhang L, Tomlinson S, Qin X. Targeted mouse complement inhibitor CR2‐Crry protects against the development of atherosclerosis in mice. Atherosclerosis. 2014;234:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia‐Cardena G, Lusis AJ, Owens AP III, Rosenfeld ME, Virmani R, et al. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 28. Juliana C, Fernandes‐Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palazon‐Riquelme P, Worboys JD, Green J, Valera A, Martin‐Sanchez F, Pellegrini C, Brough D, Lopez‐Castejon G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018;19:e44766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73. [DOI] [PubMed] [Google Scholar]
- 31. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. [DOI] [PubMed] [Google Scholar]
- 32. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH. Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell. 2014;156:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al‐Amoudi A, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, Deng X, Liang G, Zhang H, Jiang W, et al. Tranilast directly targets NLRP3 to treat inflammasome‐driven diseases. EMBO Mol Med. 2018;10:e8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–1542. [DOI] [PubMed] [Google Scholar]
- 39. Wu D, Ren P, Zheng Y, Zhang L, Xu G, Xie W, Lloyd EE, Zhang S, Zhang Q, Curci JA, et al. NLRP3 (nucleotide oligomerization domain‐like receptor family, pyrin domain containing 3)‐caspase‐1 inflammasome degrades contractile proteins: implications for aortic biomechanical dysfunction and aneurysm and dissection formation. Arterioscler Thromb Vasc Biol. 2017;37:694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E‐deficient mice‐brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461. [DOI] [PubMed] [Google Scholar]
- 41. Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH, Lee YH, Lim SJ, Lee SH, Moon JY. Inflammasome‐independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol. 2018;9:2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kostadinova E, Chaput C, Gutbier B, Lippmann J, Sander LE, Mitchell TJ, Suttorp N, Witzenrath M, Opitz B. NLRP3 protects alveolar barrier integrity by an inflammasome‐independent increase of epithelial cell adherence. Sci Rep. 2016;6:30943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, et al. Inflammasome‐independent NLRP3 augments TGF‐beta signaling in kidney epithelium. J Immunol. 2013;190:1239–1249. [DOI] [PubMed] [Google Scholar]
- 44. Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J, Hu R. Inflammasome‐independent NLRP3 is required for epithelial‐mesenchymal transition in colon cancer cells. Exp Cell Res. 2016;342:184–192. [DOI] [PubMed] [Google Scholar]
- 45. Tang RZ, Zhu JJ, Yang FF, Zhang YP, Xie SA, Liu YF, Yao WJ, Pang W, Han LL, Kong W, et al. DNA methyltransferase 1 and Kruppel‐like factor 4 axis regulates macrophage inflammation and atherosclerosis. J Mol Cell Cardiol. 2019;128:11–24. [DOI] [PubMed] [Google Scholar]
- 46. Vallee A, Vallee JN, Lecarpentier Y. Metabolic reprogramming in atherosclerosis: opposed interplay between the canonical WNT/beta‐catenin pathway and PPARgamma. J Mol Cell Cardiol. 2019;133:36–46. [DOI] [PubMed] [Google Scholar]
- 47. Matsumura T, Kugiyama K, Sugiyama S, Ota Y, Doi H, Ogata N, Oka H, Yasue H. Suppression of atherosclerotic development in Watanabe heritable hyperlipidemic rabbits treated with an oral antiallergic drug, tranilast. Circulation. 1999;99:919–924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2