

Abstract
During aging, muscle mass decreases, leading to sarcopenia, associated with low-level chronic inflammation (inflammaging), which induces sarcopenia by promoting proteolysis of muscle fibers and inhibiting their regeneration. Patients with a variety of pathologic conditions associated with sarcopenia, including rheumatoid arthritis (RA), have systemically elevated TNFα serum levels, and transgenic mice with TNFα over-expression (TNF-Tg mice, a model of RA) develop sarcopenia between adolescence and adulthood before they age. However, if and how TNFα contributes to the pathogenesis of sarcopenia during the normal aging process and in RA remains largely unknown. We report that TNFα levels are increased in skeletal muscles of aged WT mice, associated with muscle atrophy and decreased numbers of satellite cells and Type IIA myofibers, a phenotype that we also observed in adult TNF-Tg mice. Aged WT mice also have increased numbers of myeloid lineage cells in their skeletal muscles, including macrophages and granulocytes. These cells have increased TNFα expression, which impairs myogenic cell differentiation. Expression levels of TNF receptor-associated factor 6 (TRAF6), an E3 ubiquitin ligase, which mediates signaling by some TNF receptor (TNFR) family members, is elevated in skeletal muscles of both aged WT mice and adult TNF-Tg mice. TRAF6 binds to TNFR2 in C2C12 myoblasts and mediates TNFα-induced muscle atrophy through NF-κB-induced transcription of the muscle-specific E3 ligases, Atrogen1 and Murf1, which promote myosin heavy chain degradation. Haplo-deficiency of TRAF6 prevents muscle atrophy and the decrease in numbers of satellite cells, Type IIA myofibers, and myogenic regeneration in TRAF6+/−;TNF-Tg mice. Our findings suggest that pharmacologic inhibition of TRAF6 signaling in skeletal muscles during aging could treat/prevent age- and RA-related sarcopenia by preventing TNFα-induced proteolysis and inhibition of muscle fiber regeneration.
Keywords: Muscle atrophy, sarcopenia, inflammaging, muscle environmental cells, NF-κB, TNFα, TRAF6, rheumatoid arthritis
Introduction
Age-related sarcopenia is a leading health concern for the elderly, characterized by progressive loss of muscle mass, strength and function, and associated with low-level chronic inflammation, known as inflammaging (1). Elderly individuals with sarcopenia are at higher risk of adverse outcomes, including fall-related fractures and injuries, physical disability, hospitalization, and ultimately increased mortality (2). The systemic, low-level continuous inflammation of aging is characterized by elevated levels of circulating pro-inflammatory mediators, including TNFα, IL-1β, IL-6 and C-reactive protein (CRP) (3). Sarcopenia also affects around 20% of patients with rheumatoid arthritis (RA) (4), a chronic inflammatory disease in which levels of TNFα, IL-1β and CRP (5,6) are increased to levels much higher than those during aging, and also associated with whole-body protein catabolism (6). Two current clinical interventions for sarcopenia are increased physical activity and nutritional supplementation, but neither is able to fully reverse age-related muscle wasting (7). Given the high prevalence of sarcopenia and the high costs of the associated health care, it is critical to elucidate the underlying molecular mechanisms of inflammaging- and RA-associated muscle atrophy so that they can be targeted to optimize current therapeutic approaches and/or develop novel ones.
Skeletal muscles are composed of multinucleated cells that have numerous myofibers comprised mainly of two different types, according to specific metabolism patterns. Type 1 slow-twitch myofibers use oxidative metabolism and type 2 fast-twitch myofibers generally use glycolytic metabolism. The reduction in total myofiber number in sarcopenia is mainly due to atrophy of the type 2 fast-twitch glycolytic myofibers (8). Various factors have been implicated in the pathogenesis of sarcopenia, including epigenetic, hormonal and cellular alterations, poor nutritional conditions and metabolic changes (9,10). However, an increasing body of evidence supports the thesis that chronic inflammation plays a critical pathogenetic role in sarcopenia (3,11) and may be the most important contributor to sarcopenia in the elderly.
Population-based studies reported that plasma/serum TNFα and IL-6 levels are higher in the elderly than in young adults (3,11). TNFα is a pivotal inflammatory cytokine produced by various cell types, including lymphocytes, macrophages and fibroblasts (12), and functions as a “master regulator” to promote expression of other inflammatory factors. TNFα significantly reduces expression levels of myosin heavy chain (MHC), creatine kinase (CK) activity and myotube cell surface area (13), all parameters of muscle cell functions. TNFα binds to TNF receptors, R1 and R2, to activate NF-κB signaling via the RIP1/TRAF2/IKK (14) and TRAF2/NIK pathways (14,15), respectively. NF-κB activation inhibits myogenic differentiation and promotes degradation of myosin heavy chains (MyHCs) (16) by activating the muscle-specific E3 ligases, Atrogen-1 and Murf-1, which ubiquitinate myogenic proteins and promote their degradation (17,18).
TNF receptors lack intrinsic kinase activity and recruit TNF receptor-associated factors (TRAFs) to mediate downstream signaling. TRAF6 is an important adaptor protein that positively regulates signaling of some TNF receptor family members, including RANK (19), but to date, we are unaware of any reports of TRAF6 binding to or regulating TNF receptor signaling in any cell type. TRAF6 expression is increased in skeletal muscle in a number of pathologic conditions, including cancer cachexia-associated muscle wasting, denervation-induced muscle atrophy, and type I diabetes-related muscle atrophy (20). TRAF6, along with TRAF2, can function as an E3 ligase and associate with the dimeric ubiquitin-conjugating enzyme, Ubc13/Uev1A, to promote Lys-63-, but not Lys-48-, linked poly-ubiquitination of proteins targeted for degradation (21,22). Unlike other TRAFs, TRAF6 interacts with the scaffold protein, p62/sequestosome 1, and thus regulates autophagy and proteasomal degradation (23). TRAF6 plays crucial roles in activation of several receptor-mediated signaling pathways, including Nuclear factor κB (NF-κB), MAPK and PI3K/Akt, in response to cytokines and bacterial products (24,25). Stimuli that induce muscle wasting upregulate the expression and autoubiquitination of TRAF6, leading to downstream activation of major catabolic pathways in skeletal muscle (26). Inhibition of TRAF6 blocks the expression of components of the ubiquitin-proteasome system and autophagosome formation in skeletal muscles undergoing atrophy (26). However, it is not known if TRAF6 is involved in or mediates TNFα-induced sarcopenia during aging.
In this study, we examined changes in non-myogenic cells in muscles of mice during aging and found that macrophages and granulocytes accumulate among muscle fibers where they have increased expression of TNFα, which we show inhibits myotube formation in-vitro. Notably, because we found that TRAF6 levels were increased in skeletal muscles from both aged mice and adult TNFα transgenic (TNF-Tg) mice and that TRAF6 associated with TNFR2 and promoted degradation of MyHC, we generated TNF-Tg/TRAF6 double mutant (DM) mice. We examined the phenotype of muscles from these double mutant, TNF-Tg, TRAF6+/− and WT mice with/without injury-induced muscle damage and regeneration and the mechanisms involved.
Materials and Methods
Animals.
The TRAF6+/− mouse line was generated on a C57BL/6 background by Naito et al. (27). The TNF-transgenic (TNF-Tg) mouse (3647 line in C57BL/6 background) carries a human TNF transgene that was modified by replacing the 3’-region of the TNF gene with that of the human α-globin gene. The TNF-Tg mice we used for in vivo studies were all male because female TNF-Tg mice of this mouse line develop joint lesions earlier and die at around 6 months of age due to lung inflammation (28). 24-m-old WT male mice that were supplied by the National Institute on Aging (NIA) at the NIH. WT, TNF-Tg, TRAF6+/− and TNF-Tg;TRAF6+/− littermates were generated by crossing TRAF6+/− with TNF-Tg mice, and were sacrificed at 5-m-old for assessment of any muscle atrophy phenotype. By 5-m-old, the TNF-Tg male mice have typically developed various degrees of arthritis and weight loss, and inflammation has developed in other tissues, including joints (29) and lungs (28), TRAF6+/− mice are healthy, TRAF6+/−;TNF-Tg mice are in better health than TNF-Tg mice, but still are weaker than WT and TRAF6+/− mice. They were also used in an experimental muscle injury model in which mice were first given injections of Buprenorphine (0.1 mg/kg) and anesthetized with an i.p. injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). They were then given an injection of 50 μl of a 1.2% solution of barium chloride (BaCl2) in saline with a 30-gauge needle into the tibialis anterior (TA) muscle. After 5 days, the injured TA muscles from the mice were harvested for histomorphometric analysis. No random allocation was performed because specific genotypes of mice were required for our studies. 5 mice or fewer were housed in individual cages in a one-way MIT (pathogen free) room and fed with a laboratory autoclaved diet (LabDiet; #5010) and potable, uncontaminated drinking water (Hydropac) according to NIH Guidelines (ILAR 1996). All animal procedures were conducted in accordance with institutional guidelines approved by the University Committee on Animal Resources, University of Rochester Medical Center.
Reagents.
Information about all reagents is listed on line in Supporting Information.
Primary myogenic cell isolation.
Muscles were removed and chopped around 300 times until a fine slurry was generated and then digested in 7.5 ml 0.2% collagenase type II (in DMEM containing 10% FBS) and shaken gently at 37 ℃ for 60 min. After one wash with 15 ml F10 medium, the muscle slurry was chopped 100 times with a sterile knife, and then digested in 10 ml 0.01% collagenase type II/0.04% dispase digestion solution and shaken gently at 37 ℃ for 60 min. After the 2nd digestion, the cells were collected after being filtered using a 40 μm strainer. For staining, 2 μl biotin-conjugated anti-CD45, -CD11b, -CD31 and -Sca1 Abs were added to 106 cells in 100 μl 0.5% BSA-PBS and incubated at 4℃ for 30 min, followed by staining with 5 μl streptavidin-conjugated microbeads for every 106 cells and incubated at 4℃ for 20 min. Stained cells were passed through a Miltenyi LS magnetic column (Miltenyi; # 130-042-401) and the isolated negative cells were called myogenic cells (MCs). See Supporting Information for full description.
Induction of myotube formation.
2×104 isolated myogenic cells (CD45−CD11b−CD31−Sca1−) were seeded in each well of 8-well chamber Permanox plastic slides (Nunc™ Lab-Tek™ Chamber Slide System; Cat #:177445) in 300 μl basal medium (10% horse serum, 1% Pen/Strep, 1% HEPES and 5 ng/ml FGF-basic in DMED) for 2 d, then changed to 300 μl growth medium (20% FBS, 1% Pen/Strep, 1% HEPES and 5 ng/ml FGF-basic in F10 culture medium) for 4–6 d until cell confluency reached ~70%. Then the cells were induced with differentiation medium (3% horse serum, 1% Pen/Strep and 1% HEPES in DMEM) for 36 h, and half of the differentiation medium was changed every 12 h. In co-culture experiments, positively-selected non-myogenic cells or CD11b+ cells were freshly isolated from muscles and added to myogenic cells after they reached ~70% confluence in co-culture chamber wells. These co-cultures were maintained in the myogenic differentiation medium described above for 36 h before cell fixation, and half of the differentiation medium was changed gently every 12 h. See Supporting Information for full description.
Cryosection and myotube staining.
Cryosections (10 μm thick) were blocked with 10% normal goat serum in PBST for 30 min at RT and with 3% affinipure Fab fragment anti-mouse IgG (H+L)/anti-mouse IgM (Cat #: 115-007-003/115-006-020; Jackson ImmunoResearch Laboratories) for 1 h at RT before Ab incubation. Primary Abs anti-MyHC-IIA (Cat #: SC-71; DSHB) and -IIB (Cat #: BF-F3; DSHB), and anti-Pax7 (Cat #: Pax7; DSHB) diluted at 1:40, anti-laminin (Cat #: L9393; Sigma-Aldrich) diluted at 1:1500, were incubated at 4℃ overnight. On the second day, Alexa flour 488/568-conjugated secondary Abs (Cat #: A-21042/A-21124; Invitrogen) diluted at 1:400 incubated for 1 h at RT. For myotube staining, primary Ab anti-MyHC Ab (Cat #: MAB4470; R&D) diluted at 1:100 was incubated at 4℃ overnight. On the 2nd day, Alexa Flour 488-conjugated goat anti-mouse IgG (Cat #: A11001; Invitrogen) diluted at 1:200 was incubated at RT for 1 h. The stained slides were mounted with Vectashield medium with DAPI (Cat #: H-1200; Vectashield) and imaged using a Zeiss Axio Observer A1 inverted microscope. See Supporting Information for full description.
Skeletal muscle regeneration assay.
TA muscles were harvested at 5 d post BaCl2-induced injury and sectioned for MyHC and laminin IF and H&E staining. The area of inflammation/regeneration in H&E-stained sections of each TA muscle was photographed in 3–6 microscopic fields (with a 10× objective lens), and the cross-sectional area (CSA) of 250–300 CNFs in each TA was measured using ImageJ. An average value was generated to represent the CSA size of each TA. Representative images from each group are illustrated in the lower panel of Figure 5A. The MyHC+ area in anti-MyHC and laminin dual-stained sections was assessed in 5 independent images (with a 20× objective lens) taken from the muscle regeneration area of each TA muscle. The CSA of 15–20 MyHC+ CNFs in 5 representative images of each TA muscle was counted, and the mean value for each TA was calculated.
Figure 5. Haplo-deficiency of TRAF6 partially prevents TNFα-induced inhibition of muscle regeneration.
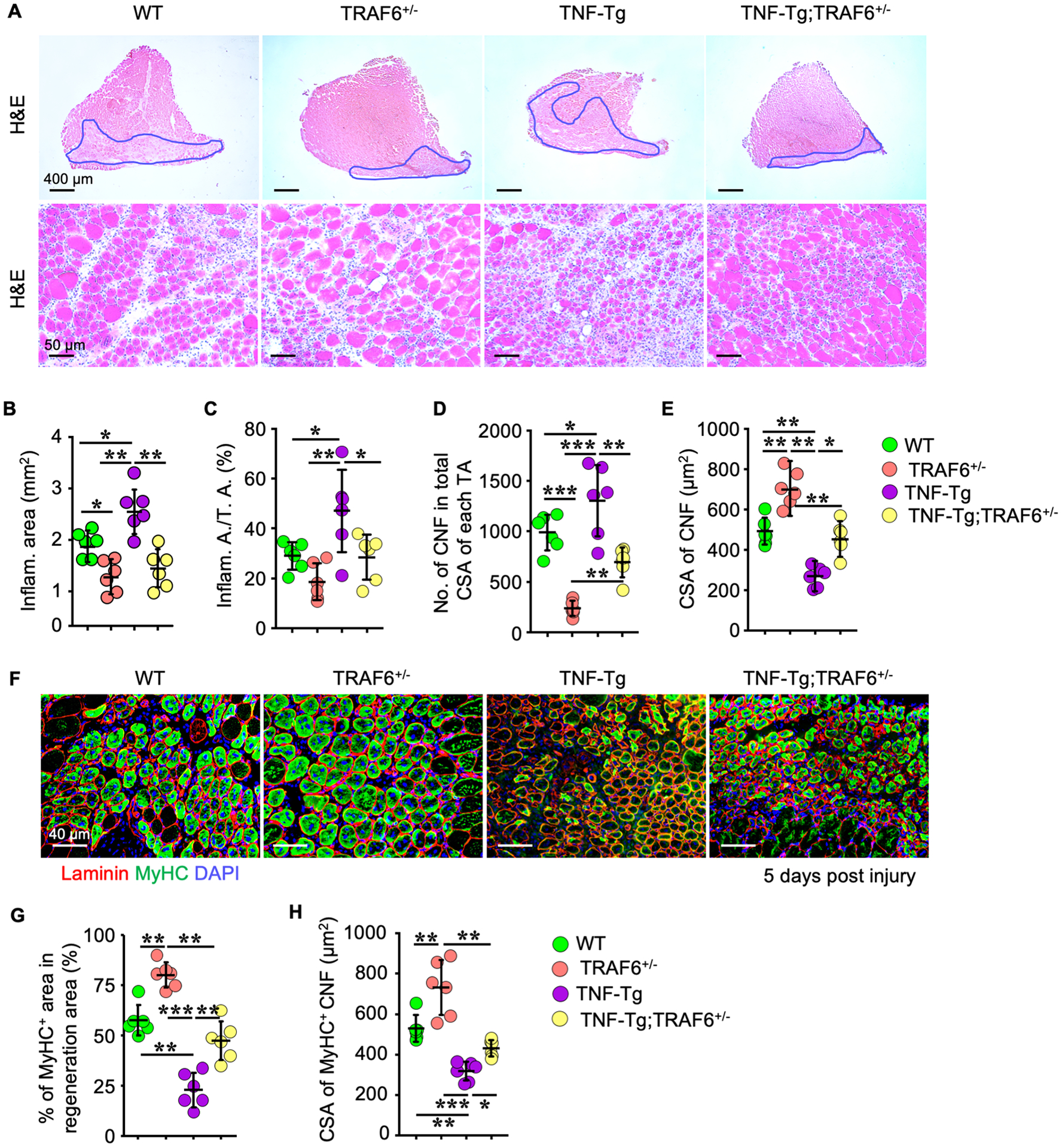
(A) 10 μm-thick H&E-stained TA cryosections of 5-m-old WT, TNF-Tg, TRAF6+/− and TNF-Tg;TRAF6+/− mice (n = 6/group) injected with BaCl2, with areas of injury and inflammatory cell infiltration outlined in blue (upper panel) and at higher magnification (lower panel). (B-E) Parameters of muscle injury and regeneration in H&E-stained TA muscle sections. n = 6/group. (F) IF images of regeneration areas in cryosections of TA muscles stained for MyHC and laminin. (G) Percentage of MyHC+ areas in regeneration areas of each TA muscle, and (H) cross-sectional area of MyHC+ central nucleated fibers in IF-stained sections in (F). n = 6/group; *p < 0.05, **p < 0.01, ***p < 0.001.
Western Blot analysis.
To test potential TRAF6 association with TNFR and RANK, 2 μg anti-TNFR1, TNFR2 or RANK Abs were added to 500 μg whole cell protein lysates, and the precipitated proteins were subjected to WB for analysis of TRAF6. For ubiquitination assay, 500 μg of protein lysates were incubated with anti-TRAF6 Ab, and precipitated proteins were Western-blotted using an anti-Ub Ab. 10–20 μg of protein lysates were loaded in 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes, which were incubated with primary Abs, such as anti-mouse TRAF6, MyHC, GAPDH and MyoD, with 1:500 dilution at 4℃ overnight, followed by incubation with horseradish peroxidase-linked secondary Ab (Bio-Rad) at RT for 2 h. The membranes were exposed to ECL substrate and signals were detected by a Bio-Rad imaging system and analyzed using Image Lab 5.1 software. See Supporting Information for full description.
Quantitative real-time PCR.
1 μg of total RNA extracted from sorted EVCs and MCs, as well as gastrocnemius muscles from WT, TNF-Tg, TRAF6+/− and TRAF6+/−;TNF-Tg mice was reverse transcribed to cDNA in a 20 μl reaction system using an iSCRIPT cDNA Synthesis kit (Cat #: 170–8891; Bio-Rad). The mRNA expression levels of Atrogen1, Murf1, LC3B, Traf6, RelA, RelB, p50, p52 and Gapdh were measured using an iCycler real-time PCR machine (Bio-Rad) with iQ SYBR SuperMix (Bio-Rad), according to the manufacturer’s instructions. Primer sequences are listed on line in Supporting Information.
Flow cytometry.
2 μl Biotin-conjugated anti-CD45, -CD11b, -CD31 and -Sca1 Abs, and 1 μl of each Ab, including anti-CD11b-PE-Cy5, anti-F4/80-Percp-Cy5.5, anti-Ly6G-APC-Cy7, anti-Ly6C-APC, anti-B220-PE, anti-CD3e-APC, anti-CD31-PE, anti-CD11c-BV785 and anti-TNFα-PE-Cy7, were added to 106 cells in 100 μl FACS buffer (2% FBS in PBS) and incubated at 4℃ for 30 min, followed by incubation with APC-R700-conjugated streptavidin at 4℃ for another 20 min. Stained cell samples were acquired using a flow cytometer (FACS LSR II; BD Biosciences). FCS Express 6 software (De Novo Software) was used for data analysis. See Supporting Information for full description.
Data availability.
All relevant data are available from the authors upon reasonable request.
Statistics.
All results are the mean ± S.D. Variance was similar between groups for most parameters assessed. All comparisons between groups were analyzed using one-way ANOVA with Tukey’s post-hoc test with the exception of those in Figs. 2D&E, 3A,C,D&E and 6A,C&F in which Student t-test used. p values <0.05 were considered statistically significant. Each experiment was repeated at least twice with similar results. The sample size for in vivo experiments is based on an un-paired t-test power analysis carried out by our statistician using SigmaStat Statistical Software: 5–8 mice were needed in each group where muscle parameters were assessed to detect significant differences from controls with an alpha error of 5%. The power is 0.98, i.e. there is 98% chance of detecting a specific effect with 95% confidence when alpha=0.05. No data were excluded from the analyses, which were carried out by investigators blinded to the identity of the various groups.
Figure 2. TNFα is expressed by subsets of myeloid cells that accumulate in muscles of WT mice during aging and inhibit myotube formation.
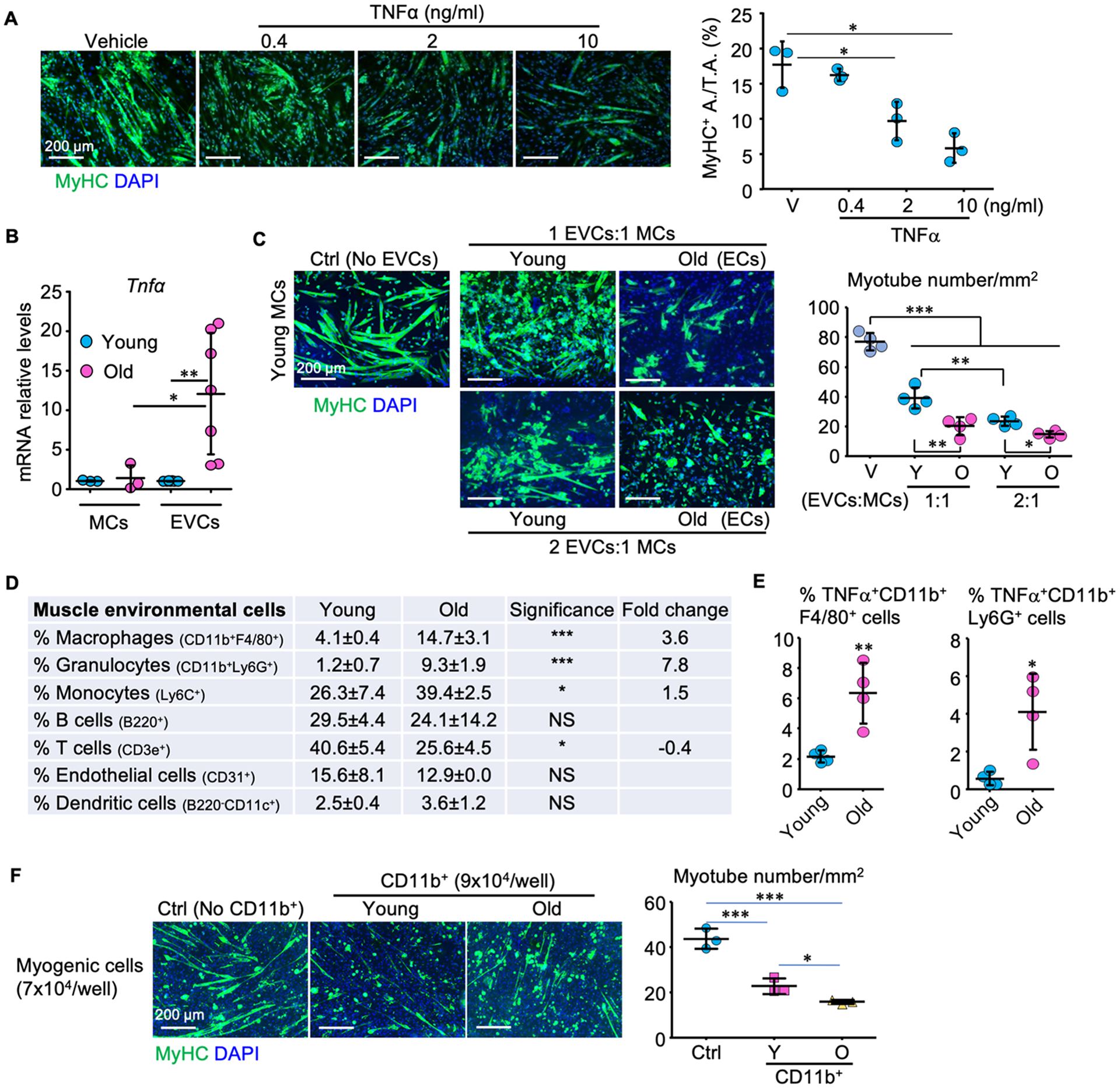
(A) Effects of TNFα on myotube formation (MyHC-positive area/total area) from primary myogenic cells (MCs; CD45−CD31−CD11b−Sca1−) isolated from skeletal muscles of 3-m-old C57B6 mice. (B) TNFα mRNA levels in MCs and environmental cells (EVCs; CD45+;CD31+;CD11b+;Sca1+ cells) from skeletal muscles of 3- and 22-m-old C57B6 mice. MCs: n = 3, EVCs: n = 7 samples. (C) Myotube formation from primary myogenic cells from young mice co-cultured for 24 h with different ratios (MCs:EVCs −1:1 and 1:2) of EVCs from young or old C57B6 mice. n=4 samples. (D) Percentages of various muscle environmental cells from 3– 22-m-old C57B6 mice. n = 4 mice/group. (E) Percentages of TNFα+ macrophages and granulocytes in muscle EVCs from 3- and 22-m-old C57B6 mice in (D). (F) Myotube formation from primary myogenic cells from young mice co-cultured with CD11b+ EVCs from young or old C57B6 mice for 36 h. n=3 samples/group; *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 3. TRAF6 expression is increased in muscles from aged mice and mediates TNFα-induced reduction in myotube formation.
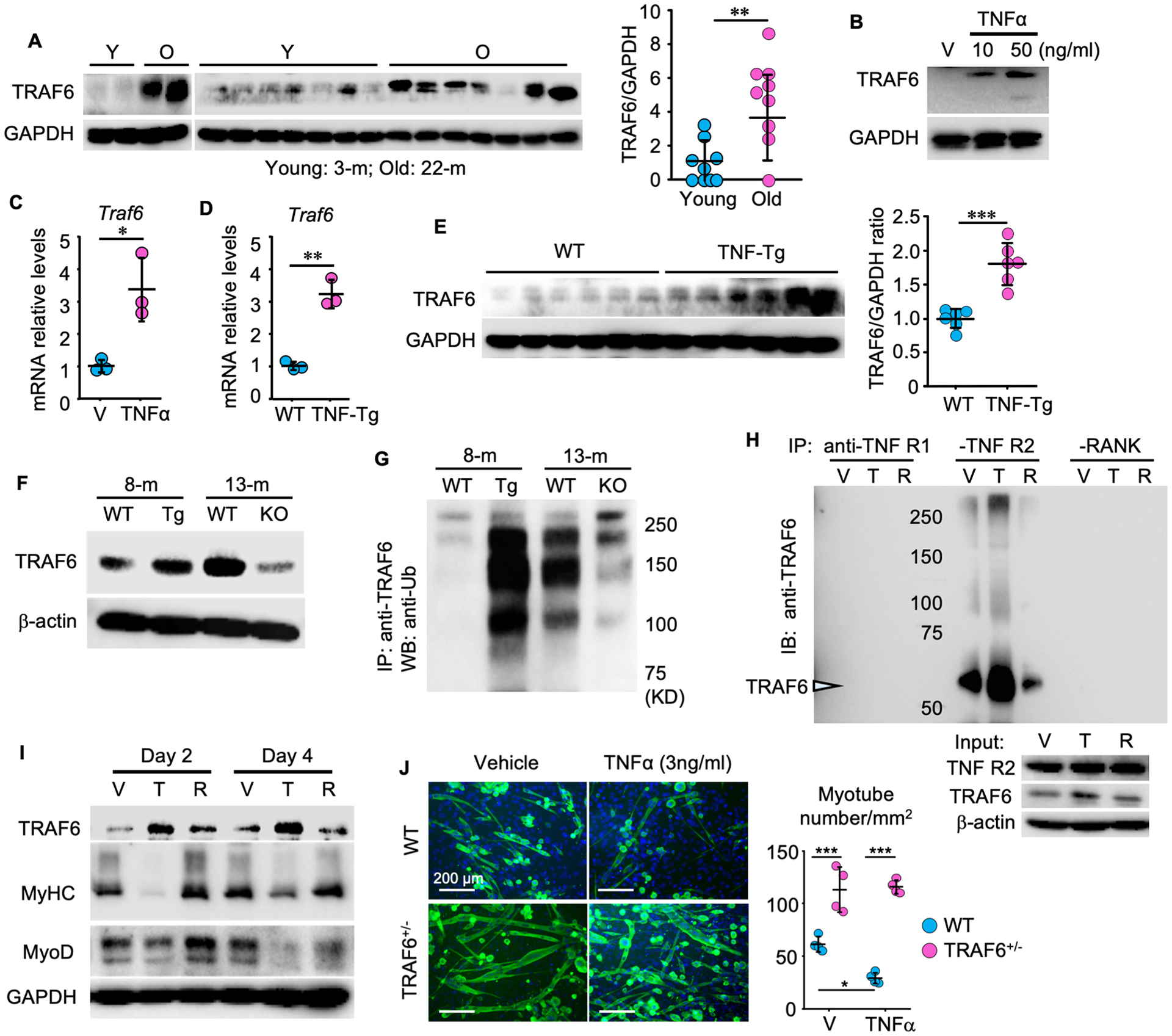
(A) WBs of TRAF6 and GAPDH proteins in gastrocnemius lysates from 3- and 22-m-old male C57B6 mice. n = 9/group. (B) WBs of C2C12 cell lysates after treatment with vehicle or TNFα for 8 h. (C-D) TRAF6 mRNA levels (C) in C2C12 cells treated with TNFα (10 ng/ml) for 24 h and (D) in gastrocnemius muscles from 5-m-old WT and TNF-Tg mice. n = 3/group. (E) TRAF6 protein levels in gastrocnemius lysates from 8-m-old WT and TNF-Tg male mice. n = 6/group. (F) TRAF6 total and (G) ubiquitinated protein levels in gastrocnemius from 8-m-old WT and TNF-Tg (Tg) male mice and 13-m-old WT and TNFα−/− (KO) male mice. (H) C2C12 cells treated with TNFα (T, 10 ng/ml) or RANKL (R,10 ng/ml) for 8 h. 300 μg cell protein lysate (IP using anti-TNFR1, -TNFR2 or -RANK Ab, and immunoblotted using anti-TRAF6 Ab. (I) Protein levels of TRAF6, MyHC, MyoD and GAPDH in C2C12 cells treated with Vehicle (V), TNFα (T, 10 ng/ml) or RANKL (R, 10 ng/ml) for 2 and 4 days. (J) Myotube formation from primary myogenic cells from 4-m-old WT and TRAF6+/− male mice and treated with vehicle or TNFα for 24 h. n = 4/group. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 6. TRAF6 mediates NF-κB activation, E3 ligase transcription and MyHC degradation induced by TNFα.
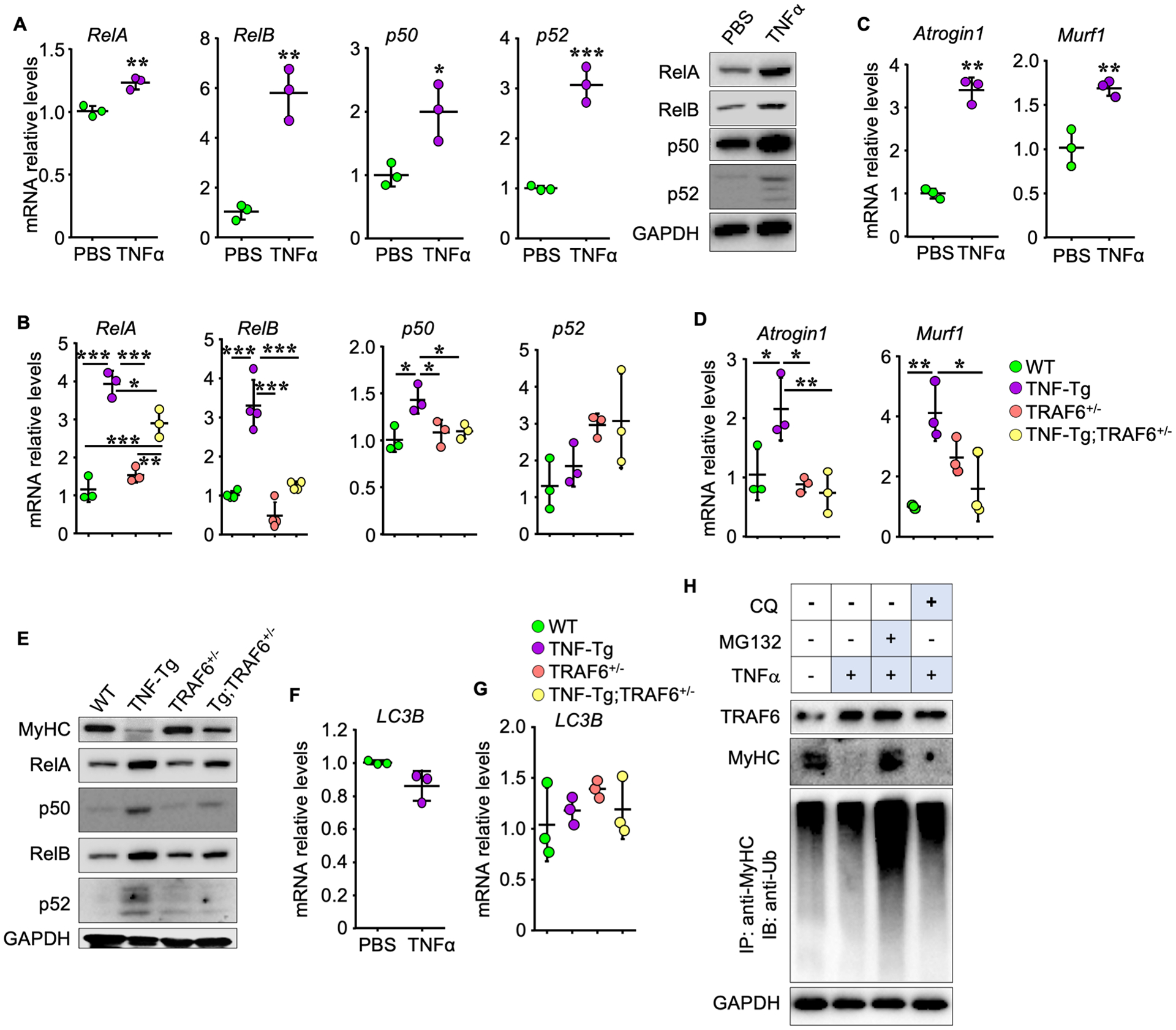
(A) NF-κB mRNA and protein levels in C2C12 cells treated with TNFα (10 ng/ml) for 24 h. (B) NF-κB mRNA levels in gastrocnemius muscles in 5-m-old WT, TNF-Tg, TRAF6+/− and TNF-Tg;TRAF6+/− mice. n = 3/group in (A) and 3 (n = 4 for RelB) mice in (B). (C-D) mRNA levels of Atrogen1 and Murf1 in (C) C2C12 cells treated with TNFα (10 ng/ml) for 24 h and (D) gastrocnemius muscles from the mice in (B). n = 3/group in (C) and 3 mice in (D). (E) WBs of proteins in gastrocnemius muscles from mice in (B) and (D). (F-G) LC3B mRNA levels in (F) C2C12 cells treated with TNFα, as in (A) and (C), and (G) gastrocnemius muscles from mice as in (B) and (D). n = 3/group in (F) and 3 mice in (G). (H) C2C12 cells treated with vehicle or 10 ng/ml TNFα plus 5 μM MG132 or 20 μM chloroquine (CQ) for 8 h. WB of MyHC, TRAF6 and GAPDH and MyHC ubiquitination following IP with anti-MyHC Ab and IB with anti-Ub Ab. *p < 0.05; **p < 0.01; ***p < 0.001.
Results
TNFα induces muscle atrophy in aged WT and adult TNF-transgenic mice.
To examine the role of TNFα in age-related sarcopenia, we first compared circulating mouse and human TNFα protein levels in adult WT and TNF-Tg (8-m-old) and aged WT (24-m-old) male mice and found that the mean levels were higher in aged WT and adult TNF-Tg mice than in adult WT mice (Supplementary Fig. 1). We then compared mouse TNFα protein levels in gastrocnemius muscles from young (3-m-old) and aged (22-m-old) WT male mice, as well as human TNFα protein levels in gastrocnemius muscles from adult (8-m-old) WT and TNF-Tg mice. We found that the mean levels of TNFα were higher in samples from the aged than from young mice and from adult TNF-Tg than from control mice (Fig. 1A). We next compared the skeletal muscle phenotypes of adult (8-m-old) and aged (24-m-old) male WT mice and found that the lean mass of the tibialis anterior (TA), soleus and gastrocnemius muscles was lower in the aged WT mice (Fig. 1B). The paired box transcription factor, Pax7, is an established muscle stem cell (MuSC) marker and plays a critical role in regulating MuSC proliferation. We found that the Pax7+ cell numbers and muscle fiber size in TA muscles were lower in aged than in adult WT mice (Fig. 1C). Of note, characterization of myofiber type by IF staining showed that the number of MyHC IIA+ and MyHC IIB+ fibers in TA muscles was lower in aged than adult WT mice, with no significant changes detected in MyHC IIX+ fiber number. To determine if prolonged exposure to TNFα during adolescence and early adulthood in mice affects the above muscles adversely, we examined muscle samples from 8-m-old male TNF-Tg mice, which have high serum TNFα levels and begin to develop TNFα-induced inflammatory arthritis from ~4 months after birth (28). We found that the TNF-Tg mice had similar reductions in muscle mass, muscle fiber size, Pax7+ MuSCs and MyHC IIA+ fibers as the aged WT mice, but they had higher numbers of MyHC IIB+ fibers than the 8-m-old WT mice (Fig 1B, 1C and 1D).
Figure 1. TNFα induces muscle atrophy in aged WT and adult TNF-transgenic mice.
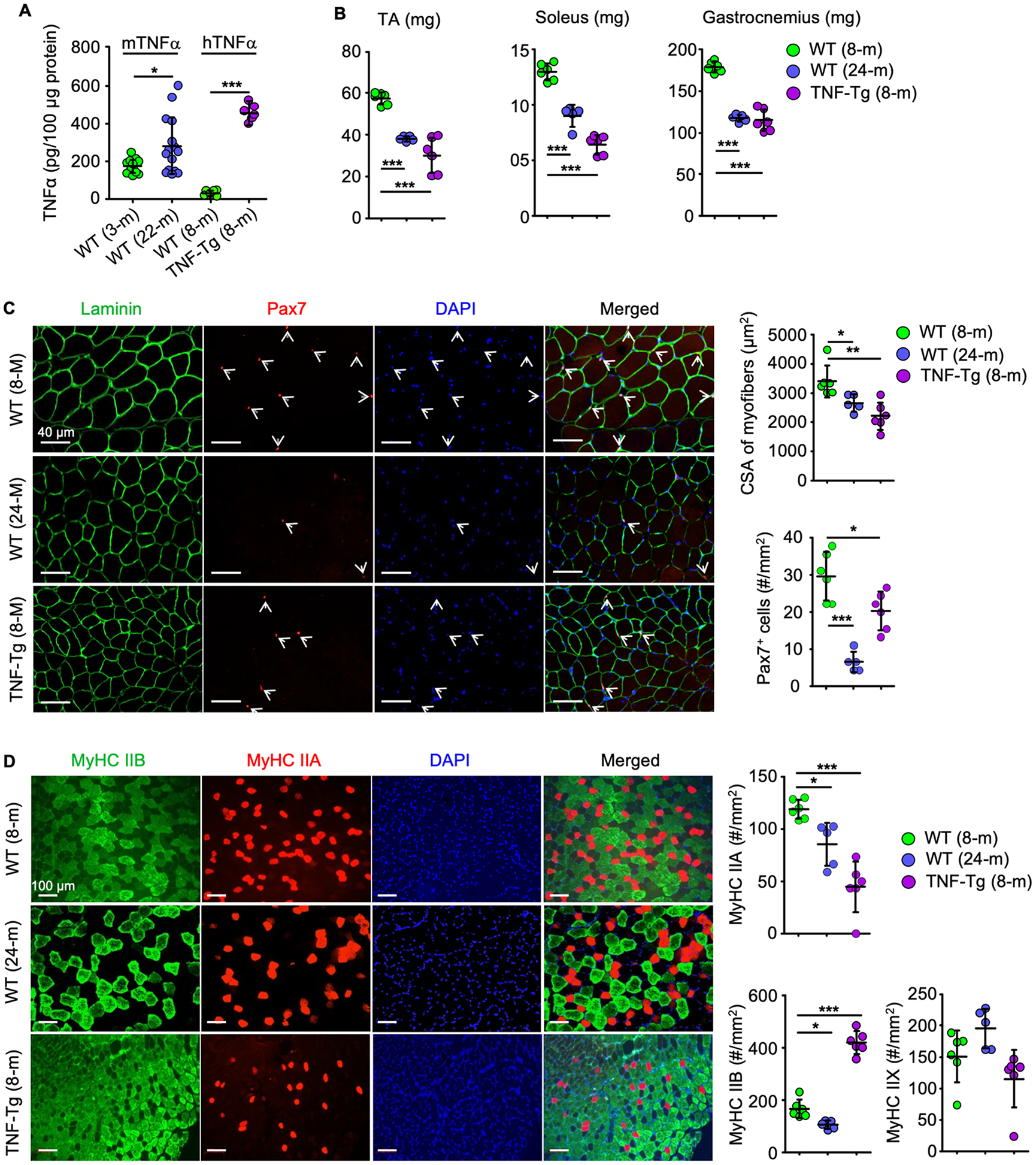
(A) Murine (m) and human (h) TNFα levels in protein lysates from gastrocnemius from 14 young and 15 old male C57BL6/J mice and 6 WT and 6 TNF-Tg male mice. *p < 0.05, ***p < 0.001. (B-D) Muscle phenotypes of 6 adult and 5 old WT and 6 adult TNF-Tg mice. (B) Lean weights of tibialis anterior (TA), soleus and gastrocnemius muscles. (C) 10 μm-thick cryosections of TA muscles stained for laminin (green) and Pax7 (red) expression to assess the cross-sectional area (CSA) of myofibers and the numbers of Pax7+ satellite cells (white arrows). (D) 10 μm-thick cryosections of TA muscles IF-stained for myosin heavy chain (MyHC) IIA and IIB expression and the numbers of type IIA (red), IIB (green) and IIX (unstained) fibers. *p < 0.05, **p < 0.01, ***p < 0.001.
TNFα is expressed by subsets of myeloid cells that accumulate in muscles of WT mice during aging and inhibit myotube formation.
We next performed magnetic-activated cell sorting (MACS) and cultured primary myogenic cells (MCs) from young WT mice. We found that TNFα induced a small, but significant increase in cell death of primary myogenic cells, but not of C2C12 cells (Supplementary Fig. 2), but significantly inhibited myotube formation from these cells in a dose-dependent manner (Fig. 2A), consistent with the development of muscle atrophy in aged WT and adult TNF-Tg mice, associated with increased serum TNFα levels. To investigate the potential sources of TNFα in skeletal muscle during aging, we MACS-sorted MCs (CD45−CD11b−CD31−Sca1−) and environmental cells (EVCs; (CD45+; CD11b+; CD31+; Sca1+) separately from skeletal muscles of young and aged WT mice. EVCs include circulating white blood cells, endothelial and dendritic cells (Fig 2D). We found that TNFα mRNA levels were significantly higher in EVCs, but not in MCs, from muscles from aged than from young WT mice (Fig. 2B). Of note, EVCs from young mice co-cultured with MCs from young mice significantly inhibited myotube formation from the MCs. This inhibitory effect was increased significantly when EVCs from aged mice were co-cultured with the MCs from young mice (Fig. 2C). We next sought to determine which EVCs might be the source of the increased TNF and if the numbers of cells comprising these EVCs changed in muscles during aging. We MACS-sorted EVCs from the TA, EDL, gastrocnemius, soleus and quadriceps muscles from young and aged WT mice and found that the samples from aged mice had higher percentages of macrophages (CD11b+F4/80+), granulocytes (CD11b+Ly6G+) and monocytes (Ly6C+) and a lower percentage of T cells than those from young mice, with no differences in the percentages of B cells, endothelial or dendritic cells (Fig. 2D). Notably, significantly higher percentages of macrophages (CD11b+F4/80+) and granulocytes (CD11b+Ly6G+) sorted from muscles from aged mice expressed TNFα than those sorted from young mice (Fig. 2E), suggesting that myeloid subsets with increased TNFα expression accumulate in skeletal muscles during aging. We next investigated the effects of CD11b+ cells isolated from muscles from both young and aged mice on myotube formation from MCs of young mice and found that they inhibited myotube formation (Fig. 2F) and that this inhibitory effect was greater with the CD11b+ cells from aged mice (Fig. 2F).
TRAF6 expression is increased in muscles from aged mice and mediates TNFα-induced reduction in myotube formation.
Since TRAF6 expression is increased in atrophic skeletal muscle in a number of pathologic conditions (20), we measured TRAF6 protein levels in gastrocnemius muscles and found that they were significantly higher in samples from old (22-m) than from young (3-m) WT mice (Fig. 3A; Supplementary Fig. 3A), raising the possibility that TRAF6 may be involved in TNFα signaling in age-related muscle atrophy. We next treated C2C12 cells with TNFα and found that it increased TRAF6 protein (Fig. 3B) and mRNA (Fig. 3C) levels. Consistent with these findings, TRAF6 mRNA (Fig. 3D) and protein (Fig. 3E; Supplementary Fig. 3B) levels were significantly higher in gastrocnemius muscle samples from 5-m-old TNF-Tg than from WT mice. In comparison, TRAF6 protein levels were lower in gastrocnemius muscles from 13-m-old TNF global knockout (KO) than from WT littermates (Fig. 3F). The level of TRAF6 auto-ubiquitination correlates positively with TRAF6 signaling activity in cells (22). Thus, we measured the levels of TRAF6 ubiquitination and found that they were higher in gastrocnemius muscles from 8-m-old TNF-Tg mice than WT littermates, but they were lower in samples from 13-m-old TNFα KO mice (Fig. 3G). We next examined the effects of TNFα on binding of TRAF6 to the TNF and RANKL receptors in C2C12 cells and compared that with the effects of RANKL. The cells were treated with these cytokines for 8 hours, following pre-treatment with MG132 or chloroquine for 4 h to prevent proteasomal and lysosomal degradation, respectively (30,31). We found that treatment of the cells with TNFα resulted in increased binding of TRAF6 to TNF receptor II, but not to TNF receptor I or RANK (Fig. 3H), while treatment with RANKL reduced this binding of TRAF6 to TNF receptor II, presumably because of competitive attraction of TRAF6 by RANKL signaling.
To further examine the effects of TRAF6-mediated TNF signaling on myotube differentiation, we treated C2C12 cells and primary myogenic cells from WT and TRAF6+/− mice with TNFα and measured protein levels of myogenic markers and myotube formation. We used TRAF6+/−, rather than TRAF6−/− mice to further examine the role of TRAF6 in these experiments because TRAF6−/− mice die ~2–3 weeks after birth with splenomegaly, thymic atrophy and failure of lymph node development, which do not occur in the TRAF6+/− mice (27). We found that TNFα increased TRAF6 expression levels and inhibited MyHC and MyoD expression (Fig. 3I). In addition, TNFα inhibited MyHC+ myotube formation, which did not occur in cells derived from TRAF6+/− mice (Fig. 3J).
Haplo-deficiency of TRAF6 partially prevents muscle atrophy in TNF-Tg mice.
To determine if haplo-deficiency of TRAF6 can prevent TNFα-induced muscle atrophy in vivo, we crossed TNF-Tg with TRAF6+/− mice to generate TNF-Tg;TRAF6+/− double mutant mice and compared muscle cells from them with those from TRAF6+/−, TNF-Tg and WT mice. We found that TRAF6 mRNA and protein levels in gastrocnemius muscles from TNF-Tg;TRAF6+/− mice were similar to those from TRAF6+/− mice, but lower than in WT mice, as anticipated (Fig. 4A&B). The weights of TA, EDL, soleus, gastrocnemius and quadriceps muscles from TNF-Tg mice were significantly lower than those from WT mice, while those from TNF-Tg;TRAF6+/− mice were similar to the values in WT mice (Fig. 4C). The numbers of Pax7+ satellite cells (Fig. 4D), cross-sectional area (CSA) of myofibers (Fig. 4D) and MyHC IIA+ fibers (Fig.4E) in TA muscles from TNF-Tg mice were significantly lower than those in WT mice, similar to our findings in Fig. 1C&D, while the values in TNF-Tg;TRAF6+/− mice were similar to those in WT mice and significantly higher than those in TNF-Tg mice. However, we detected no improvement in the severity of the arthritis induced by TNF in the TNF-Tg;TRAF6+/− mice, which had values for mean inflammatory area (Fig. 4F) and osteoclast numbers (Fig. 4G) in foot joints similar to those in TNF-Tg mice.
Figure 4. Haplo-deficiency of TRAF6 partially prevents muscle atrophy in TNF-Tg mice.

(A) TRAF6 mRNA levels in muscle samples from the listed mice. n = 3/group; *p < 0.05, **p < 0.01. (B) WBs of TRAF6 in gastrocnemius. (C) Lean muscle weight of TA, EDL, soleus (Sol), gastrocnemius (Gast) and quadriceps (Quad). (WT: n = 7, TNF-Tg: n = 6, TRAF6+/−: n = 5, TNF-Tg;TRAF6+/−: n = 6; *p < 0.05, **p < 0.01, compared with TNF-Tg group). (D-E) 10 μm-thick cryosections of TA muscles IF-stained for laminin (green) and Pax7 (red) expression, and (D) CSA of myofibers and numbers of Pax7+ satellite cells (white arrows) and (E) MyHC type IIA (red) myofiber numbers. (WT: n = 8, TNF-Tg: n = 8, TRAF6+/−: n = 6, TNF-Tg;TRAF6+/−: n = 6 in (D); n = 5, 5, 7 and 7, respectively, in (E). (F-G) H&E and TRAP staining paraffin sections of footpads and (F) inflammatory tissue (blue arrows) area and (G) OC (yellow arrows) numbers. n = 5, 5, 7 and 7, respectively. B: bone. *p < 0.05; **p < 0.01; ***p < 0.001.
Haplo-deficiency of TRAF6 partially prevents TNFα-induced inhibition of muscle regeneration.
To determine if TRAF6 mediates TNF-induced inhibition in muscle regeneration, we used a standard muscle injury model in which BaCl2 is injected into TA muscles of WT, TNF-Tg, TRAF6+/− and TNF-Tg;TRAF6+/− mice. We found that 5 d post-injury, the inflammatory/regenerating area in H&E-stained cross sections of TA muscles was lower in TRAF6+/− and higher in TNF-Tg than in WT mice, with the increase in the TNF-Tg mice being partially prevented in TNF-Tg;TRAF6+/− mice (Fig. 5A). The area with inflammatory cell infiltration and the percentage of this area/total TA cross sectional area were comparable between TRAF6+/− and WT mice, but were significantly higher in TNF-Tg than WT mice. Of note, these parameters in TNF-Tg;TRAF6+/− mice were lower than in TNF-Tg mice and were similar to the levels in WT mice (Fig. 5B and 5C). The numbers of newly formed regenerating central nucleated fibers (CNF) in TRAF6+/− TA muscles were less, but the cross-sectional area of CNF was larger than in TA muscles from WT mice (Fig. 5D and 5E). In contrast, the CNF number in TNF-Tg TA muscles was higher, but the CNF cross-sectional area was lower than that in WT TA muscles. Notably, compared with TNF-Tg mice, TNF-Tg;TRAF6+/− mice had fewer CNF in their TA, but the fibers were larger (Fig. 5D and 5E). MyHC IF staining of the area of regeneration in TA muscles showed that the MyHC+ area in TNF-Tg was lower (Fig. 5F and 5G) and the cross-sectional area of MyHC+ CNF was lower than in WT mice (Fig. 5F and 5H), while these value were higher in TA muscles from the TNF-Tg;TRAF6+/− than from TNF-Tg mice (Fig. 5F, 5G and 5H).
TRAF6 mediates NF-κB activation, E3 ligase transcription and MyHC degradation induced by TNFα.
NF-κB activation promotes expression of the E3 ligases, Atrogin1 and MURF1, in muscle cells. To investigate if TRAF6 mediates TNFα-induced NF-κB activation and MyHC reduction in muscle, we first treated C2C12 cells with TNFα and found that TNFα stimulated transcription and translation of NF-κB RelA, RelB, p50 and p52 genes in C2C12 cells (Fig. 6A). Similar to this, TNF-Tg mice had higher mRNA levels of RelA, RelB and p50, but not of p52, and higher protein levels of RelA, RelB, p50 and p52, in gastrocnemius muscles than WT mice (Fig. 6B & E). mRNA and protein levels of these genes in gastrocnemius muscles from TRAF6+/− mice were similar to those in WT mice. Notably, TNF-Tg;TRAF6+/− mice had significantly lower mRNA and protein levels of RelA, RelB and p50 in gastrocnemius muscles than TNF-Tg mice (Fig. 6B & E). In response to TNFα, gene transcription levels of Atrogin1 and Murf1 were upregulated in C2C12 cells (Fig. 6C). TNF-Tg mice had higher transcription levels of Atrogin1 and Murf1 than WT mice, while this increase was significantly lower in gastrocnemius muscles of TNF-Tg;TRAF6+/− mice (Fig. 6D). Consistent with this, MyHC protein levels in gastrocnemius samples from TNF-Tg;TRAF6+/− mice were largely similar to levels in WT mice (Fig. 6E). To further investigate the mechanism whereby TNFα degraded MyHC, we measured mRNA levels of LC3B, a key gene in autophagosomal degradation, but they were unchanged in TNFα-treated C2C12 cells (Fig. 6F) and in TNF-Tg mouse gastrocnemius muscles (Fig. 6G). The MyHC reduction induced by TNFα was blocked by the proteasome inhibitor, MG132, but not by the lysosome inhibitor, chloroquine (Fig. 6H), suggesting that MyHC is degraded mainly through the proteasome pathway following ubiquitin modification.
Discussion
This is the first report of an important regulatory role of TRAF6 in the pathogenesis of muscle atrophy induced by TNFα during aging. As illustrated in Fig. 7, we conclude that TNFα−expressing myeloid cells accumulate in skeletal muscle in mice during aging and inhibit myotube formation, which is mediated by TRAF6 expressed by myogenic cells. TNFα stimulates TRAF6 gene transcription and ubiquitination, which promotes TRAF6 signaling activity and its association with TNF receptor II, thereby accelerating expression of Murf-1 and Atrogen-1, through NF-κB signaling. MuRF1 and MAFbx/Atrogin-1 are muscle-specific E3 ligases that bind to MyHC to mediate its ubiquitination and degradation through the ubiquitin-proteasome system in myogenic cells (32). Both are heavily involved in skeletal muscle loss in vivo. For example, genetic deletion or shRNA-mediated knockdown of MuRF1 effectively restored MyHC protein levels and prevented muscle loss induced by acute lung injury (33) and dexamethasone (34). Thus, we propose that elevated expression of Atrogen-1 and Murf-1, in response to TRAF6-mediated NF-κB activation, increases MyHC ubiquitination and degradation, leading to muscle atrophy and inhibition of muscle fiber regeneration. In addition, during aging and in adult TNF-Tg mice, increased levels of TNFα induce loss of Pax7+ satellite cells and of MyHC IIA+ myofibers through TRAF6 and thus promote muscle atrophy and insufficient regeneration.
Figure 7. Model of TNFα-induced muscle loss during aging through TRAF6-mediated NF-κB-induced activation of E3 ligases.
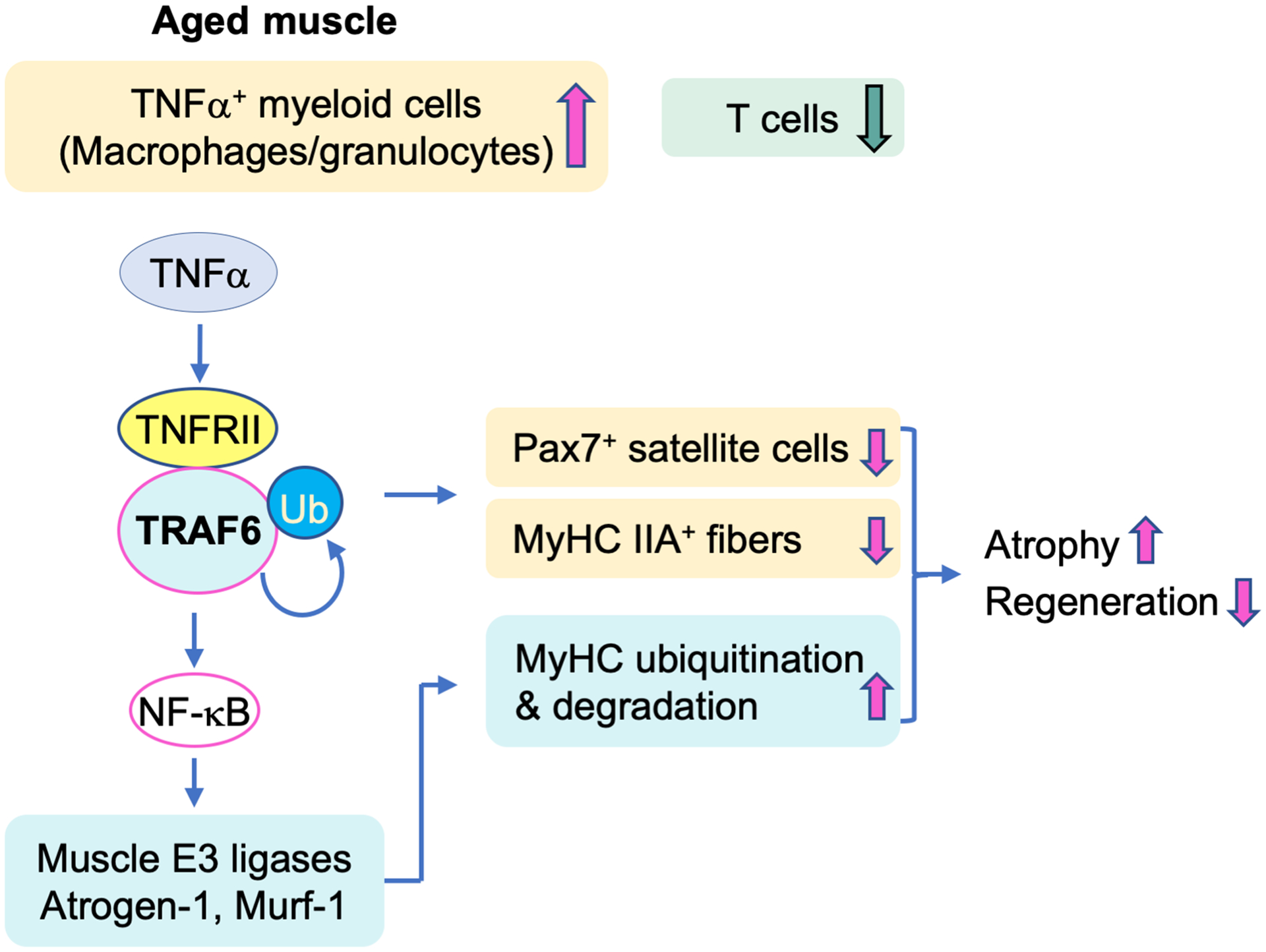
During aging, myeloid lineage cells, including macrophages and granulocytes, increase in skeletal muscle, while T cell numbers decrease, associated with increased expression of TNFα and reduced muscle mass. TNFα induces TRAF6 gene transcription and protein ubiquitination and its binding to TNF receptor II, thereby activating NF-κB-mediated transcription of the muscle-specific E3 ligases, Atrogen-1 and Murf-1. These ubiquitinate MyHC proteins and promote their degradation. By this mechanism, TRAF6 positively regulates TNFα-induced loss of Pax7+ satellite cells, reduction of MyHC IIA+ myofibers, and increased MyHC degradation, leading to muscle atrophy and impaired regeneration.
Low-level chronic inflammation accompanies aging, associated with increased levels of TNFα, IL-6 and C-reactive protein in the circulation, plays a causative role in the pathogenesis of various age-related diseases, including diabetes (35), atherosclerosis (36), Alzheimer’s disease (37), arthritis (38), osteoporosis (19,39) and sarcopenia (10,40). Many in vivo and in vitro studies support a predominant pathogenic role for TNFα in low-level chronic inflammation age-related diseases. By activating NF-κB signaling, TNFα stimulates expression of a wide range of inflammatory mediators, such as IL-6 (41). We used adult TNF transgenic mice, which have progressively increasing circulating levels of TNFα as they age (42), as a model to mimic the effects of increasing levels of TNFα, which increase later in WT mice during aging (43) and to explore the mechanisms whereby sarcopenia occurs in patients with RA (4). Similarities between adult TNFα transgenic mice and aged WT mice include myofiber atrophy, reduction in the muscle stem cell pool and of MyHC IIA fibers, and an increase in TNFα expression accompanied by local aggregation of TNFα+ myeloid cells. These findings suggest that this TNFα transgenic mouse may be an appropriate model to study the role of TNFα in muscle atrophy during aging. Compared with adult and aged WT mice, adult TNF-Tg mice have higher numbers of MyHC IIB+ myofibers per unit area in their TA muscles. This might be a compensatory effect following the loss of IIA+ myofibers and significant reduction in myofiber size and is consistent with adult TNF-Tg mice having higher TNFα levels in serum and muscles than adult and aged WT mice. However, we recognize that TNF-Tg mice also develop inflammation in other tissues, including joints (29) and lungs (28), which limit their utility as a model of aging-associated diseases.
We found that significant upregulation of TRAF6 levels in skeletal muscles of aged mice and adult TNF-Tg mice supports TRAF6 involvement in mediating TNFα signaling, and that inhibitory effects of TNFα in myogenic cells are mediated by TRAF6, which is supported by our finding that haplo-deficiency of TRAF6 rescues the muscle atrophy and impaired regeneration observed in TNFα transgenic mice. These findings suggest that TRAF6 mediates TNFα-induced muscle loss during aging and in this mouse model of RA, and that targeting TRAF6 may help to treat age- and RA-related muscle loss. Further studies are underway to determine if TRAF6 haplo-deficiency protects TRAF6 heterozygous mice from TNFα-induced muscle loss and impaired regeneration as they age.
Skeletal muscles contain resident immune cells, and the types and abundance of these cells change in various pathologic conditions, such as inflammatory myopathies (44). It is well established that cross-talk between immune cells, especially macrophages, and myogenic cells, plays a fundamental role in post-injury muscle regeneration (45). However, there is a large gap in current understanding of how immune cell functions change and contribute to sarcopenia during aging. A recent study from Tidball and colleagues (46) reported that transplantation of bone marrow cells from old into young mice decreases the number of satellite cells and promotes a shift toward a fibrogenic phenotype, which contribute to sarcopenia, suggesting that during aging the immune system modulates sarcopenia (46). We found that various types of myeloid lineage cells, including macrophages, granulocytes and monocytes, were significantly increased, while T lymphocytes were reduced in skeletal muscle during aging. TNFα overproduction by macrophages and granulocytes in muscle during aging may result from: 1) preferential myeloid commitment of hematopoietic stem cells in bone marrow; 2) age-related accumulation of these immune cells in muscle, as shown in Fig. 2D&2E; and 3) increased TNFα expression by these cells, as supported by Fig. 2F. However, the molecular mechanisms whereby these myeloid cells accumulate in muscle and produce high levels of TNFα remain unclear. Age-related redox imbalance may be involved, since increased reactive oxygen species production from various cellular sources (47) activates NF-κB by stimulating inflammasomes (48), which are intracellular danger-sensing multi-protein platforms (49). NF-κB activation promotes the transcription of TNFα (50) as well as chemokines, including CCL5 and CXCL8 (51), which may attract myeloid cells (macrophages and granulocytes) to the muscle microenvironment. However, further studies will be required to fully elucidate the mechanisms underlying the aggregation of myeloid cells in muscles during aging.
To our knowledge, this is the first report of TRAF6 positively mediating TNFα signaling in any cell type. TRAF6 is broadly expressed in mammalian tissues and mediates signaling by several cytokines through binding to the intracellular domains of transmembrane receptors, such as TGFβR1/ALK5 (25) and TNF receptor superfamily members, including CD40 and RANK (52), or to signaling receptors indirectly, such as the TLR/IL-1R superfamily (52) and the IL-17R family (53). It is not completely understood how TRAF6 mediates downstream signal activation. Generally, self-ubiquitinated TRAF6 recruits the downstream kinases, TAK1 and IKK, to form a molecular complex that activates IKK and TAK1 kinase activity (54). Polyubiquitination of TRAF6 is typically used as a read-out of its activation (54) and mediates NF-κB activation. TRAF6 is a vital downstream mediator of RANKL/RANK signaling in osteoclastogenesis (55), but it is dispensable for RANKL-independent osteoclast formation induced by TNFα (56). Interestingly, we found that TNFα, but not RANKL, upregulates TRAF6 expression and stimulates TRAF6 recruitment to TNFR2 in myoblasts in vitro, and haplo-deficiency of TRAF6 partially prevents TNFα-induced muscle atrophy, but not TNFα-induced osteoclastogenesis in vivo. These findings suggest that TRAF6 transduces TNFα signaling, but not RANKL signaling in myogenic cells, while it mediates RANKL-, but not TNFα-, induced osteoclastogenesis.
Systemic increases in levels of TNFα, a hallmark of inflammaging, are involved in the pathogenesis of various age-related diseases. Exercise reduces systemic TNFα levels in elderly humans and aged mice (57). It also downregulates TRAF6 expression and inhibits NF-κB activation in cardiomyocytes (58). These findings suggest that TNFα/TRAF6/NF-κB signaling would be reduced in exercised muscles and activated in muscles during disuse. TNF-Tg mice and aged WT mice have reduced mobility (59), consistent with our findings that these mice have elevated TNFα and TRAF6 expression levels in their muscles. Thus, the elevated TNFα/TRAF6/NF-κB signaling we report in aged mice in this study may in part be caused by their reduced mobility.
TRAF6 has previously been reported to be involved in various pathological conditions affecting muscles, including injury (60), cancer-related cachexia (20), denervation, and type I diabetes (20). In this study, we found that TRAF6 is upregulated in skeletal muscles from TNF-Tg and aged mice. Thus, elevated levels of TRAF6 may be a broad indicator of pathology in various conditions affecting muscles. Untreated TRAF6+/− mice appear to have normal skeletal muscle phenotypes. However, TRAF6 haplo-deficiency prevents the reduction in the muscle stem cell pool and muscle mass induced in transgenic mice with TNFα overexpression and accelerates recovery from muscle regeneration post-injury, reflecting the broad and vital role of TRAF6 in muscle diseases. Therefore, potential therapeutic approaches that inhibit TRAF6 expression or activity could help individuals with sarcopenia.
Supplementary Material
Acknowledgements:
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR43510 (to BFB.), P30 AR069655 (to Edward Schwarz), AR063650 (to LX) and by the National Institute for Aging under award number R01AG049994 (to BFB and ZY), and 1S10RR027340 (to BFB) from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interests: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1:S4–9. [DOI] [PubMed] [Google Scholar]
- 2.Bunout D, de la Maza MP, Barrera G, Leiva L, Hirsch S. Association between sarcopenia and mortality in healthy older people. Australas J Ageing. 2011;30(2):89–92. [DOI] [PubMed] [Google Scholar]
- 3.Schaap LA, Pluijm SM, Deeg DJ, Visser M. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am J Med. 2006;119(6):526 e9–17. [DOI] [PubMed] [Google Scholar]
- 4.Barone M, Viggiani MT, Anelli MG, Fanizzi R, Lorusso O, Lopalco G, et al. Sarcopenia in Patients with Rheumatic Diseases: Prevalence and Associated Risk Factors. J Clin Med. 2018;7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson-Hughes B, et al. Rheumatoid cachexia: cytokine-driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest. 1994;93(6):2379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rall LC, Roubenoff R. Rheumatoid cachexia: metabolic abnormalities, mechanisms and interventions. Rheumatology (Oxford). 2004;43(10):1219–23. [DOI] [PubMed] [Google Scholar]
- 7.Chhetri JK, de Souto Barreto P, Fougere B, Rolland Y, Vellas B, Cesari M. Chronic inflammation and sarcopenia: A regenerative cell therapy perspective. Exp Gerontol. 2018;103:115–23. [DOI] [PubMed] [Google Scholar]
- 8.Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, et al. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp Gerontol. 2013;48(5):492–8. [DOI] [PubMed] [Google Scholar]
- 9.Livshits G, Gao F, Malkin I, Needhamsen M, Xia Y, Yuan W, et al. Contribution of Heritability and Epigenetic Factors to Skeletal Muscle Mass Variation in United Kingdom Twins. J Clin Endocrinol Metab. 2016;101(6):2450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalle S, Rossmeislova L, Koppo K. The Role of Inflammation in Age-Related Sarcopenia. Front Physiol. 2017;8:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bian AL, Hu HY, Rong YD, Wang J, Wang JX, Zhou XZ. A study on relationship between elderly sarcopenia and inflammatory factors IL-6 and TNF-alpha. Eur J Med Res. 2017;22(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Libert C Cytokine anniversary: TNF trailblazers five centuries apart. Nature. 2015;523(7559):158. [DOI] [PubMed] [Google Scholar]
- 13.De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, et al. TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle. 2012;2(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011;278(6):862–76. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Liu B, Liang C, Li Y, Song YH. Cytokine Signaling in Skeletal Muscle Wasting. Trends Endocrinol Metab. 2016;27(5):335–47. [DOI] [PubMed] [Google Scholar]
- 16.Jo E, Lee SR, Park BS, Kim JS. Potential mechanisms underlying the role of chronic inflammation in age-related muscle wasting. Aging Clin Exp Res. 2012;24(5):412–22. [DOI] [PubMed] [Google Scholar]
- 17.Wu CL, Cornwell EW, Jackman RW, Kandarian SC. NF-kappaB but not FoxO sites in the MuRF1 promoter are required for transcriptional activation in disuse muscle atrophy. Am J Physiol Cell Physiol. 2014;306(8):C762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl). 2008;86(10):1113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyce BF, Li J, Xing L, Yao Z. Bone Remodeling and the Role of TRAF3 in Osteoclastic Bone Resorption. Front Immunol. 2018;9:2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191(7):1395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315(5809):201–5. [DOI] [PubMed] [Google Scholar]
- 22.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem. 2007;282(6):4102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280(42):35625–9. [DOI] [PubMed] [Google Scholar]
- 24.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325(5944):1134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31(6):918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul PK, Kumar A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy. 2011;7(5):555–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4(6):353–62. [DOI] [PubMed] [Google Scholar]
- 28.Bell RD, Wu EK, Rudmann CA, Forney M, Kaiser CRW, Wood RW, et al. Selective Sexual Dimorphisms in Musculoskeletal and Cardiopulmonary Pathologic Manifestations and Mortality Incidence in the Tumor Necrosis Factor-Transgenic Mouse Model of Rheumatoid Arthritis. Arthritis Rheumatol. 2019;71(9):1512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun W, Meednu N, Rosenberg A, Rangel-Moreno J, Wang V, Glanzman J, et al. B cells inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nat Commun. 2018;9(1):5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A, et al. TGFbeta-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun. 2019;10(1):2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab. 2014;307(6):E469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Files DC, D’Alessio FR, Johnston LF, Kesari P, Aggarwal NR, Garibaldi BT, et al. A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wasting. Am J Respir Crit Care Med. 2012;185(8):825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6(5):376–85. [DOI] [PubMed] [Google Scholar]
- 35.Akash MSH, Rehman K, Liaqat A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J Cell Biochem. 2018;119(1):105–10. [DOI] [PubMed] [Google Scholar]
- 36.Libby P Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(9):2045–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(10):580–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ilich JZ, Kelly OJ, Kim Y, Spicer MT. Low-grade chronic inflammation perpetuated by modern diet as a promoter of obesity and osteoporosis. Arh Hig Rada Toksikol. 2014;65(2):139–48. [DOI] [PubMed] [Google Scholar]
- 40.Beyer I, Mets T, Bautmans I. Chronic low-grade inflammation and age-related sarcopenia. Curr Opin Clin Nutr Metab Care. 2012;15(1):12–22. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Wang N, Brodt P. Metastatic cells can escape the proapoptotic effects of TNF-alpha through increased autocrine IL-6/STAT3 signaling. Cancer Res. 2012;72(4):865–75. [DOI] [PubMed] [Google Scholar]
- 42.Li P, Schwarz EM. The TNF-alpha transgenic mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25(1):19–33. [DOI] [PubMed] [Google Scholar]
- 43.Hayward MD, Jones BK, Saparov A, Hain HS, Trillat AC, Bunzel MM, et al. An extensive phenotypic characterization of the hTNFalpha transgenic mice. BMC Physiol. 2007;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reidy PT, McKenzie AI, Mahmassani ZS, Petrocelli JJ, Nelson DB, Lindsay CC, et al. Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am J Physiol Endocrinol Metab. 2019;317(1):E85–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juhas M, Abutaleb N, Wang JT, Ye J, Shaikh Z, Sriworarat C, et al. Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat Biomed Eng. 2018;2(12):942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Wehling-Henricks M, Welc SS, Fisher AL, Zuo Q, Tidball JG. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 2019;33(1):1415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res. 2018;122(6):877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richmond A Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2(9):664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418(6896):443–7. [DOI] [PubMed] [Google Scholar]
- 53.Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12(9):844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamothe B, Campos AD, Webster WK, Gopinathan A, Hur L, Darnay BG. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J Biol Chem. 2008;283(36):24871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13(8):1015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005;202(5):589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greiwe JS, Cheng B, Rubin DC, Yarasheski KE, Semenkovich CF. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J. 2001;15(2):475–82. [DOI] [PubMed] [Google Scholar]
- 58.Abdullah M, Berthiaume JM, Willis MS. Tumor necrosis factor receptor-associated factor 6 as a nuclear factor kappa B-modulating therapeutic target in cardiovascular diseases: at the heart of it all. Transl Res. 2018;195:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hess A, Axmann R, Rech J, Finzel S, Heindl C, Kreitz S, et al. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A. 2011;108(9):3731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hindi SM, Kumar A. TRAF6 regulates satellite stem cell self-renewal and function during regenerative myogenesis. J Clin Invest. 2016;126(1):151–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors upon reasonable request.