

SUMMARY
The clinical spectra of irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) intersect to form a scantily defined overlap syndrome, termed pre-IBD. We show that increased Enterobacteriaceae and reduced Clostridia abundance distinguish the fecal microbiota of pre-IBD patients from IBS patients. A history of antibiotics in individuals consuming a high-fat diet was associated with the greatest risk for pre-IBD. Exposing mice to these risk factors resulted in conditions resembling pre-IBD and impaired mitochondrial bioenergetics in the colonic epithelium thereby triggering dysbiosis. Restoring mitochondrial bioenergetics in the colonic epithelium with 5-amino salicylic acid, a PPAR-γ agonist that stimulates mitochondrial activity, ameliorated pre-IBD symptoms. As with patients, mice with pre-IBD exhibited notable expansions of Enterobacteriaceae that exacerbated low-grade mucosal inflammation, suggesting that remediating dysbiosis can alleviate inflammation. Thus, environmental risk factors cooperate to impair epithelial mitochondrial bioenergetics, thereby triggering microbiota disruptions that exacerbate inflammation and distinguish pre-IBD from IBS.
Keywords: Inflammatory bowel disease, Irritable bowel syndrome, high-fat diet, antibiotics, microbiota
Graphical Abstract
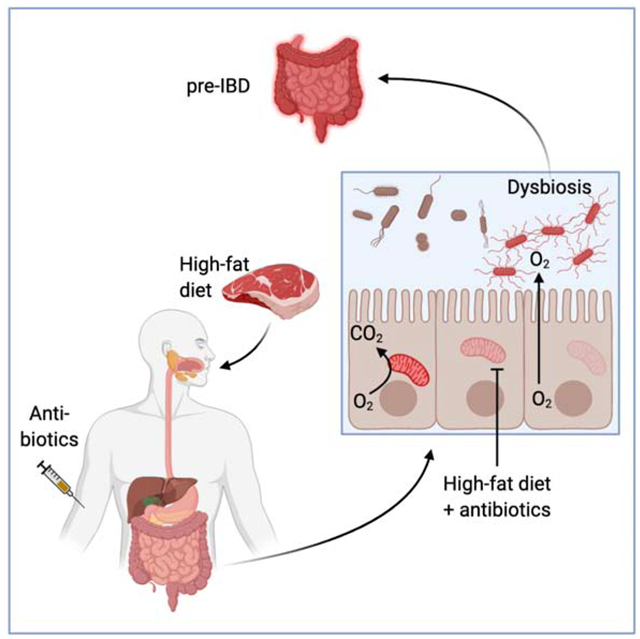
eTOC blurb
Lee et al find that antibiotics and high-fat diet cooperate to increase the risk for pre-IBD, characterized by low-grade mucosal inflammation and changes in the microbiota composition (dysbiosis). In a mouse model of pre-IBD, antibiotics and high-fat diet jointly impair epithelial mitochondrial function to drive dysbiosis that exacerbates mucosal inflammation.
INTRODUCTION
Inflammatory bowel disease (IBD) is thought to result from inappropriate activation of the mucosal immune system driven by the microbiota(Podolsky, 2002). During homeostasis, the fecal microbiota is dominated by obligate anaerobic bacteria belonging to the classes Clostridia (phylum Firmicutes) and Bacteroidia (phylum Bacteroidetes)(Human Microbiome Project, 2012). IBD is associated with an imbalance in the fecal microbiota (dysbiosis), characterized by an elevated relative abundance of facultative anaerobic Enterobacteriaceae (phylum Proteobacteria) and a reduced relative abundance of Clostridia(Rigottier-Gois, 2013; Rizzatti et al., 2017; Shin et al., 2015). An expansion of Enterobacteriaceae in the gut microbiota has been linked to an exacerbation of colitis in mouse models of IBD(Garrett et al., 2010; Zhu et al., 2018). Changes in the gut microbiota induced by chemically induced colitis can exacerbate inflammation, but only after transfer to genetically susceptible mice(Schulfer et al., 2018).
In addition to dysbiosis, IBD is linked to genetic risk factors, as indicated by an increased relative risk for first-degree relatives of individuals affected by IBD, and to environmental factors, as suggested by a rising incidence in countries with historically low rates(Xavier and Podolsky, 2007). Potentially reversible environmental factors associated with an increased risk for IBD include smoking, antibiotics and a Western-style diet(Albenberg et al., 2012; Frolkis et al., 2013; Hildebrand et al., 2008; Hviid et al., 2011; Lewis, 2014; Zou et al., 2020). A palm-oil based high-fat diet accelerates disease onset in mice genetically susceptible to IBD(Gruber et al., 2013; Paik et al., 2013) and maternal high-fat diet consumption exacerbates chemically induced colitis in genetically susceptible murine offspring(Bibi et al., 2017). However, the links between exposure to environmental risk factors and dysbiosis remain incompletely understood.
Symptoms originating from persistent pathological changes associated with IBD are difficult to distinguish from functional bowel disorders, such as irritable bowel syndrome (IBS)(Colombel et al., 2019). IBS is characterized by recurring episodes of abdominal pain, bloating, and altered bowel habits without endoscopic lesions, which affects approximately 11% of the general population worldwide(Lovell and Ford, 2012). Low-grade mucosal inflammation contributes to the pathophysiology in a subset of IBS patients(Banerjee et al., 2015; Chang et al., 2014; David et al., 2015; Emmanuel et al., 2016; Gwee et al., 2003; Hod et al., 2016; Kristjansson et al., 2004; Melchior et al., 2017). IBS patients with low grade mucosal inflammation form a subgroup located at the intersection between the clinical spectra of IBS and IBD (Colombel et al., 2019; Spiller and Lam, 2011; Spiller and Major, 2016). Names proposed for this overlap syndrome include pre-IBD(Abdul Rani et al., 2016) and subclinical IBD(Berrill et al., 2013). The former term will be used throughout this study. Pathophysiology and risk factors for pre-IBD remain poorly defined, because individuals with this syndrome are excluded from studies on IBD patients with current or previous endoscopic lesions. Here we identified changes in the microbiota composition and potential risk factors in patients with pre-IBD. We then developed an animal model to study how environmental risk factors alter the microbiota composition and whether dysbiosis exacerbates inflammation in pre-IBD.
RESULTS
High-fat diet and antibiotics increase the risk for pre-IBD
We enrolled 43 adult healthy subjects and 49 adult patients diagnosed with IBS, who had normal findings from colonoscopy or barium enema performed within five years or one year prior to enrollment, respectively (Table S1 and S2). To identify patients with low-grade mucosal inflammation (i.e. pre-IBD), we measured fecal calprotectin, a non-invasive biomarker for intestinal inflammation(Costa et al., 2003), which is elevated in a subset of IBS patients (Banerjee et al., 2015; Chang et al., 2014; David et al., 2015; Emmanuel et al., 2016; Melchior et al., 2017). Fecal calprotectin was elevated (> 50 μg/g feces) in a subgroup of 19 IBS patients, henceforth categorized as pre-IBD patients, compared to the remaining 30 IBS patients with normal fecal calprotectin and 43 healthy controls (Fig. 1A). Fecal myeloperoxidase levels in pre-IBD patients were not elevated compared to other groups, suggesting that neutrophils were not the main source of elevated fecal calprotectin (Fig. 1B). Distress due to bowel symptoms and related changes in quality of life were more severe in the pre-IBD group (p=0.04, 0.03, respectively, Table S3). Typing patients into pre-IBD and IBS did not relate to traditional IBS subtyping by stool form, which distinguishes a diarrhea-predominant (IBS-D) type, a constipation-predominant (IBS-C) type, and a mixed (IBS-M) type with alternating periods of diarrhea and constipation(Longstreth et al., 2006) (Fig. 1C). A history of acute gastrointestinal infection prior to onset of IBS, known as postinfectious (PI)-IBS(Cremonini and Talley, 2005), is associated with elevated fecal calprotectin levels(David et al., 2015). A total of 14 patients (8 in IBS and 6 in pre-IBD, p=0.48; Table S3) qualified as PI-IBS, but there was no difference in fecal calprotectin levels between patients with or without history of infection (Fig. 1D).
Figure 1: Subtyping patients using the biomarker fecal calprotectin identifies risk factors for pre-IBD.
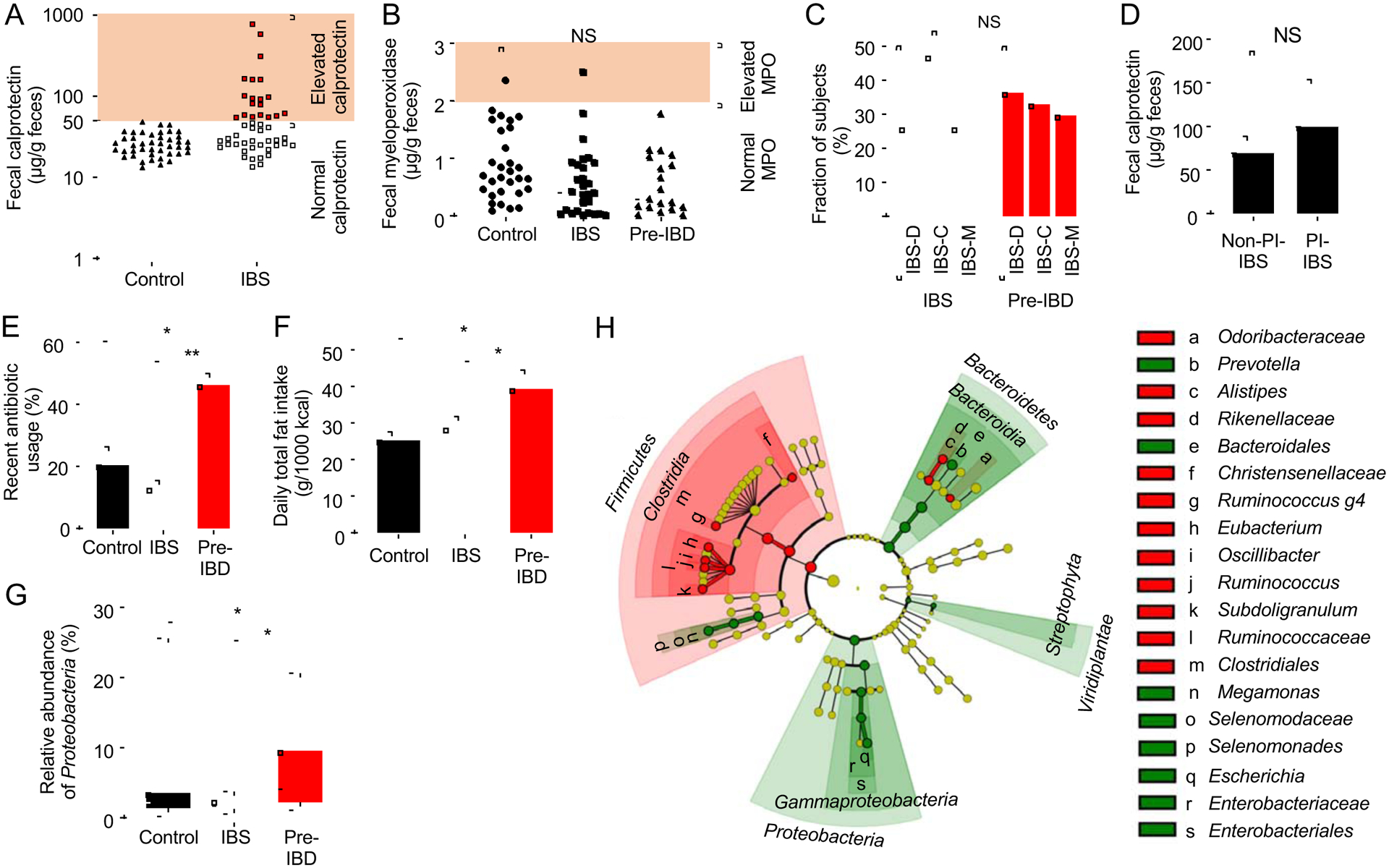
(A) Levels of fecal calprotectin were measured in healthy subjects (control) and subjects with symptoms of IBS. IBS participants were sub-grouped into those with normal fecal calprotectin levels (IBS) and elevated fecal calprotectin levels (pre-IBD). (B) Levels of fecal myeloperoxidase (MPO) was measured in patients by ELISA. (C) The graph shows the percentage of IBS patients or pre-IBD patients that had stool forms consistent with IBS-C, IBS-D or IBS-M. (D) Fecal calprotectin levels in patients with PI-IBS and in patients without a history of infection (Non-PI-IBS). (E) Fraction of subjects with a history of antibiotic usage within one year. (F) Daily fat intake was determined by calculating nutrient density and expressed as intake / 1000kcal. (G) Relative abundance of Proteobacteria was determined by microbiota profiling from feces. (H) The cladogram shows differences in taxa composition determined by microbiota profiling of feces of IBS patients and pre-IBD patients. Green, elevated in pre-IBD compared to IBS; Red, reduced in pre-IBD compared to IBS. (A and B) Each symbol represents data from one individual subject. (C-F) Bars represent mean ± standard deviation. (G) The boxes in the Whisker blot represent the 1st to 3rd quartile ranges and the horizontal lines represent the median value. The bars in the whisker blot represent the minimum and maximum value in each group. *, P<0.05; NS, P>0.05. P values were calculated by one-way ANOVA followed by Bonferroni multiple comparison test (F), by Kruskal–Wallis rank test followed by Dunn’s test (A-B, D, G) or by chi square test for categorical data (C,E). See also Figure S1, S2 and S3 and Table S1, S2, S3, S4, S5 and S6
Notably, a higher proportion of pre-IBD patients had a history of recent (within the past year) antibiotic usage (p = 0.02, Fig. 1E) or reported higher total fat intake (p = 0.01, Fig. 1F) or higher saturated fat intake (p = 0.03, Fig. S1A) than healthy subjects or IBS patients. A history of recent antibiotic usage or reported higher total fat intake was not associated with elevated fecal calprotectin in healthy subjects (Fig. S1B and S1C). Next, we divided study participants into three groups based on the amount of fat consumption (Table S4). Multivariate logistic regression analysis revealed that participants with the highest fat consumption were about 2.8-times more likely to have pre-IBD than participants with the lowest fat intake, after adjusting for other clinical parameters (i.e. age, gender, body mass index [BMI], alcohol consumption and exercise). Further subdivision revealed that a history of recent antibiotic usage was associated with an approximately 3.9-times higher likelihood of having pre-IBD after adjusting for other clinical parameters (Table S4). To test whether the effect of antibiotics on having pre-IBD was dependent on fat intake, we performed an interaction test, which revealed a statistically significant interaction between fat intake and a history of antibiotic usage (p = 0.03) (Table S5). Therefore, we divided study participants into four groups based on the amount of fat consumption (1st and 2nd tercile versus 3rd tercile of fat consumption) and their history of antibiotic usage (no history versus usage within the past year). A multivariate logistic regression analysis revealed that high-fat diet in combination with antibiotic usage was associated with 8.6-times higher risk for having pre-IBD (Table S6). Collectively, these data suggested that a history of antibiotic usage in individuals consuming a high-fat diet is a risk factor for developing pre-IBD.
Next, we wanted to determine whether changes in the fecal microbiota composition associated with pre-IBD were similar to those observed in IBS patients (Jeffery et al., 2012; Rodino-Janeiro et al., 2018) or in IBD patients(Rigottier-Gois, 2013; Rizzatti et al., 2017; Shin et al., 2015). Consistent with previous reports, 16S ribosomal RNA gene amplicon sequencing (microbiota profiling) of fecal samples from all study participants revealed that the microbiota composition of IBS patients was characterized by a reduced relative abundance of the genera Bifidobacterium (phylum Actinobacteria) and Lactobacillus (phylum Firmicutes) compared to healthy subjects(Malinen et al., 2005) (Fig. S2A). Notably, a reduced relative abundance of members of the class Clostridia and an elevated relative abundance of the family Enterobacteriaceae distinguished the fecal microbiota composition in patients with pre-IBD from that in patients with IBS (Fig. 1G, 1H, S1D, S2B and S3). Dysbiosis in pre-IBD thus featured shifts in taxa composition that were similar to those observed in IBD, which is associated with reduced Clostridia and elevated Enterobacteriaceae abundance(Rigottier-Gois, 2013; Rizzatti et al., 2017; Shin et al., 2015).
Exposing mice to risk factors models aspects of pre-IBD
To further investigate the link between risk factors and signs of disease, we wanted to develop an animal model that recapitulates aspects of pre-IBD. To this end, we used mice from Jackson laboratories (C57BL/6J), because animals from this vendor do not carry endogenous Enterobacteriaceae(Velazquez et al., 2019), thus offering experimental control over the presence of this taxon. To recapitulate high-fat intake, we chose a lard-based diet with a similar profile of saturated fatty acids (35%), monosaturated fatty acids (39.7%) and poly-unsaturated fatty acids (25.3%) as human Western-style diet(Hintze et al., 2012), but with a lower cholesterol content. To mimic risk factors identified in patients (Fig. 1E, 1F and S1A), mice reared and maintained throughout the experiment on a high-fat diet (45% fat) or a low-fat diet (10% fat) were mock-treated or received a single dose of streptomycin by oral gavage to generate a history of antibiotic usage. One day after streptomycin treatment, mice were inoculated with strains of E. coli, because this species exhibited an increased relative abundance in patients with pre-IBD (Fig. S1D). Four weeks later, mice were considered to have a “history” of antibiotic treatment (STAR Methods section). A single dose of streptomycin did not increase body weight or visceral fat weight in mice on a low-fat diet (Fig. 2A, 2B, S4A and S4B), which is in contrast to observations in mice receiving antibiotics for 40 days(Suarez-Zamorano et al., 2015). However, mice exposed to pre-IBD risk factors (i.e. a history of antibiotic treatment in mice on a high-fat diet) exhibited exacerbated gains in visceral fat weight.
Figure 2: A history streptomycin treatment in mice on a high-fat diet induces signs of pre-IBD.
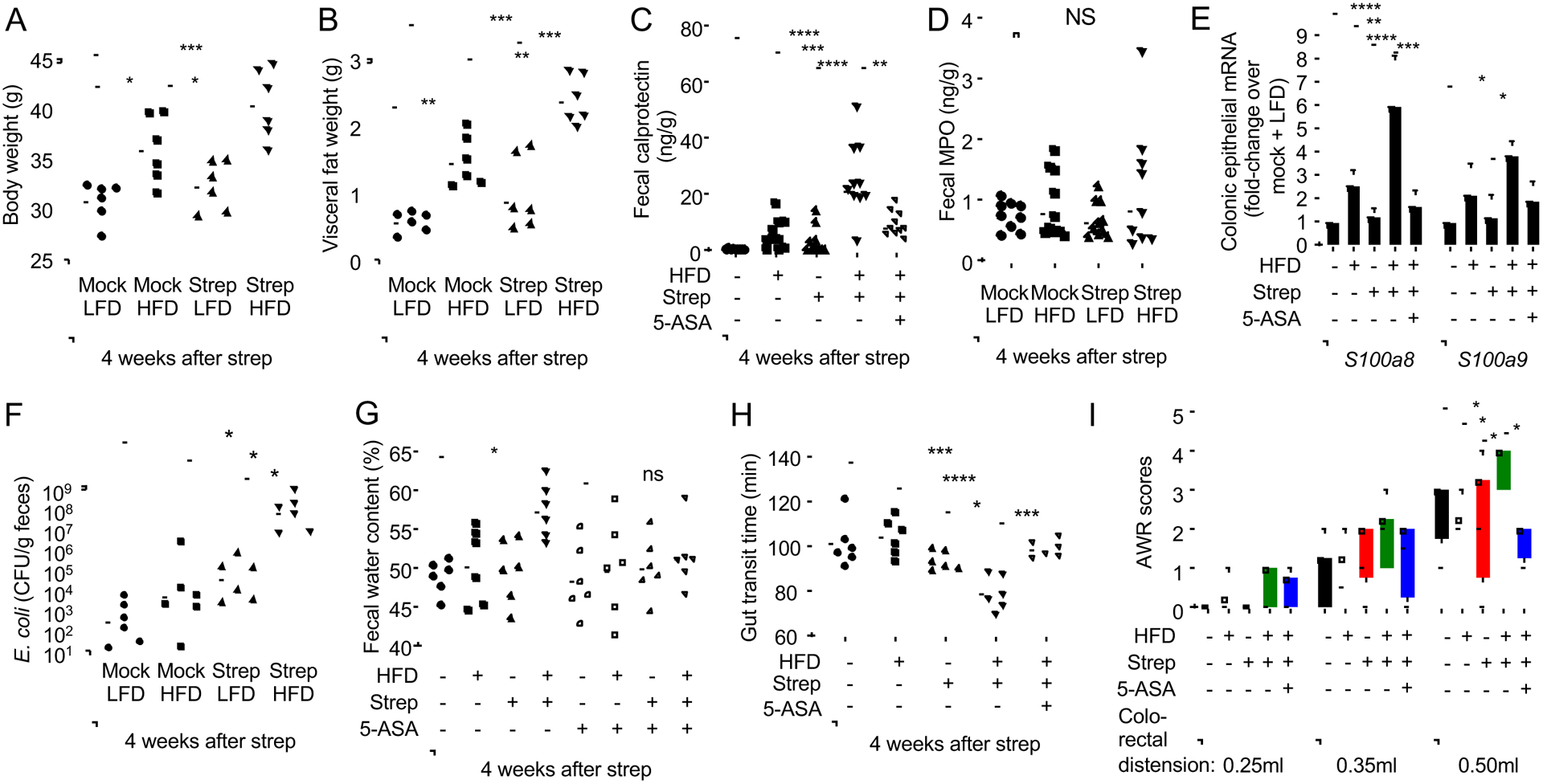
Groups of male (A-G and I) or female (H) mice (N = 6) were reared on a high-fat diet (HFD, 45% fat) or on a low-fat diet (LFD, 10% fat) and were mock treated or treated with a single dose of streptomycin (Strep) four weeks before necropsy. Mouse body weight (A) and visceral fat weight (B) were determined during necropsy. (C, E and G-I) Chow was supplemented with 5-ASA (5-ASA: +) or did not contain supplementation (5-ASA: −). (C) Fecal calprotectin levels were determined by ELISA. (D) Fecal myeloperoxidase (MPO) levels were determined by ELISA. (E) Transcript levels of the indicated genes were determined by quantitative real-time PCR in RNA isolated from colonocyte preparations. (F) Mice were inoculated with E.coli and numbers in the feces determined 4 weeks later. (G and H) The water content of mouse feces (G) and gut transit time (H) were measured. (I) Abdominal withdrawal reflex (AWR) scores at different levels of colorectal distension were measured. (A-B and E-H) Symbols represent data from individual animals and black bars represent geometric mean. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; NS, P>0.05. (C-G) Data were transformed logarithmically before analysis. P values were calculated by one-way ANOVA followed by Tukey’s multiple-comparison tests (A-H) or by a one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test (I). See also Figure S4, Figure S5 and Table S2.
Consistent with the development of low-grade intestinal inflammation, mice exposed to pre-IBD risk factors exhibited a reduced colon length (Fig. 3A and S4C) and a heightened expression of inflammatory markers in the colonic mucosa (Fig. 3B–E and S4D) and lower numbers of Alcian blue-positive (goblet) cells in the colonic epithelium (Fig. S5A). Histologically, exposure to pre-IBD risk factors produced a very mild infiltration of the intestinal wall by inflammatory cells (Fig. 3F and 3G). Notably, exposure to pre-IBD risk factors significantly elevated levels of fecal calprotectin (Fig. 2C and S4E), thus recapitulating observations in pre-IBD patients (Fig. 1A). Exposure to pre-IBD risk factors did not result in elevated fecal myeloperoxidase levels (Fig. 2D), suggesting that neutrophils were not the main source of fecal calprotectin in this model. Since intestinal epithelial cells release calprotectin upon cytokine stimulation(Lee et al., 2012; Raffatellu et al., 2009), we investigated expression of S100a8 and S100a9, the genes encoding the two subunits of calprotectin, in mRNA isolated from preparations of the colonic epithelium. Exposure to pre-IBD risk factors elevated epithelial expression of S100a8 and S100a9 (Fig. 2E and S4F), suggesting that the colonic epithelium was a possible source of fecal calprotectin in our model.
Fig. 3: Pre-IBD risk factors induce low-grade mucosal inflammation.
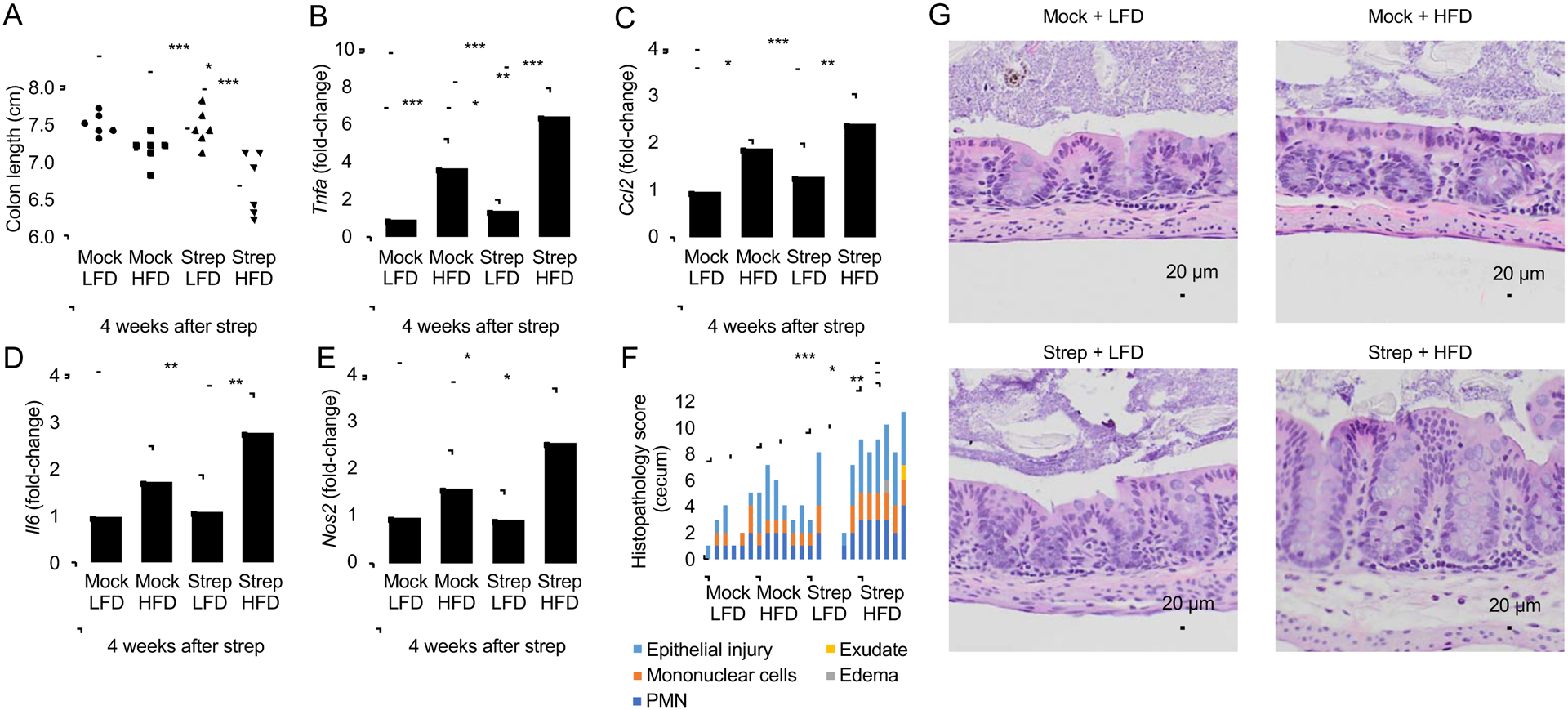
Groups of male mice (N = 6) were reared on a high-fat diet (HFD, 45% fat) or on a low3 fat diet (LFD, 10% fat) and were mock treated or treated with a single dose of streptomycin (Strep) four weeks before necropsy. (A) Colon length was determined during necropsy. (B-E) RNA was isolated from preparations of colonic epithelial cells (colonocytes) and transcript levels of Tnfa (B), Ccl2 (C), Il6 (D) and Nos2 (E) were determined by quantitative real-time PCR. Bars represent geometric mean± standard deviation. (F) A veterinary pathologist scored histopathological changes in blinded sections of the cecum. (G) Representative images of Histopathological changes in H&E stained sections of the cecal mucosa. No inflammation or alteration of the mucosal architecture in sections of mock-treated mice on a low-fat diet (Mock + LFD). No inflammation or alteration of the mucosal architecture is present in streptomycin-treated mice on a low-fat diet (Strep + LFD), but occasionally, mild neutrophil infiltration of the lamina propria is observed. In mock-treated mice on a high-fat diet (Mock + HFD) there is mild infiltration of the lamina propria by neutrophils, the mucus layer is diminished, and the surface epithelium is multifocally attenuated with frequent apoptotic cells and occasional loss of cellular adhesion with sloughing. In streptomycin-treated mice on a High-fat diet (Strep + HFD) there is mild to moderate infiltration of the lamina propria and submucosa by neutrophils. The submucosa is mildly expanded by edema. The mucus layer is diminished. The surface epithelium is multifocally attenuated with frequent apoptotic cells and occasional loss of cellular adhesion with sloughing. *, P<0.05; **, P<0.01; ***, P<0.001. (B-E) Data were transformed logarithmically before analysis. P values were calculated by one-way ANOVA followed by Tukey’s multiple-comparison tests (A-E) or by a one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test (F). See also Table S3.
The total numbers of E. coli recovered from the feces of streptomycin-treated mice were elevated by approximately three orders of magnitude compared to mock-treated controls early (5 days) after antibiotic treatment, which was observed independently of diet (Fig. S5B). E. coli numbers gradually declined in streptomycin-treated mice on a low-fat diet (Fig. S5C and 2F), illustrating that effects of antibiotic treatment are short-lived when studied in isolation. However, four weeks after antibiotic treatment, streptomycin-treated mice on a high-fat diet (i.e. mice exposed to pre-IBD risk factors) shed E. coli with the feces at significantly higher numbers than other treatment groups (Fig. 2F and S4G). Thus, exposure of mice to risk factors for pre-IBD recapitulated signs of disease observed in patients, including elevated levels of fecal calprotectin and an E. coli expansion in the gut microbiota.
To assess whether our model recapitulated general features commonly observed in IBS and pre-IBD, such as changes in the form of stool, altered bowel movement or abdominal pain, we assessed fecal water content, gut transit times and visceral hypersensitivity, respectively. Compared to mock-treated mice, mice exposed to pre-IBD risk factors exhibited a significant increase in fecal water content (Fig. 2G and S4H), a surrogate for diarrhea in humans(Woo et al., 2008). Changes in gut transit times were observed in female mice exposed to pre-IBD risk factors (Fig. 2H), but not in male mice (Fig. S5D). Finally, mice exposed to pre-IBD risk factors had elevated abdominal withdraw reflex (AWR) scores compared to other experimental groups (Fig. 2I and S4I), indicative of abdominal pain. In conclusion, exposure to risk factors for pre-IBD produced signs of disease in mice that recapitulated general features of pre-IBD in humans.
Risk factors for pre-IBD impair mitochondrial bioenergetics in colonocytes
An increased abundance of E. coli within the gut microbiota has recently been linked to increased epithelial oxygenation in the colon(Byndloss et al., 2017). Visualization of epithelial hypoxia using the exogenous hypoxic marker pimonidazole(Terada et al., 2007) revealed that exposure of mice to risk factors for pre-IBD diminished hypoxia in colonic epithelial cells (colonocytes) (Fig. 4A and 4B), which was indicative of increased epithelial oxygenation. To determine whether elevated epithelial oxygenation increased the bioavailability of oxygen in the intestinal lumen, mice received a 1:1 mixture of E. coli indicator strains that were either proficient in aerobic respiration (E. coli Nissle 1917 wild type) or deficient for aerobic respiration under microaerophilic conditions (E. coli Nissle 1917 cydA mutant). The competitive index (i.e. the ratio of wild type over mutant recovered from feces) was determined four weeks after streptomycin treatment. Mice exposed to risk factors for pre-IBD shed the aerobic respiration-proficient E. coli strain at markedly higher numbers than the aerobic respiration-deficient strain, suggesting increased oxygen bioavailability drove an E. coli expansion in the lumen of the colon (Fig. 4C and S4J).
Figure 4: A history streptomycin treatment in mice on a high-fat diet alters epithelial energy metabolism and increases oxygen bioavailability in the colon.
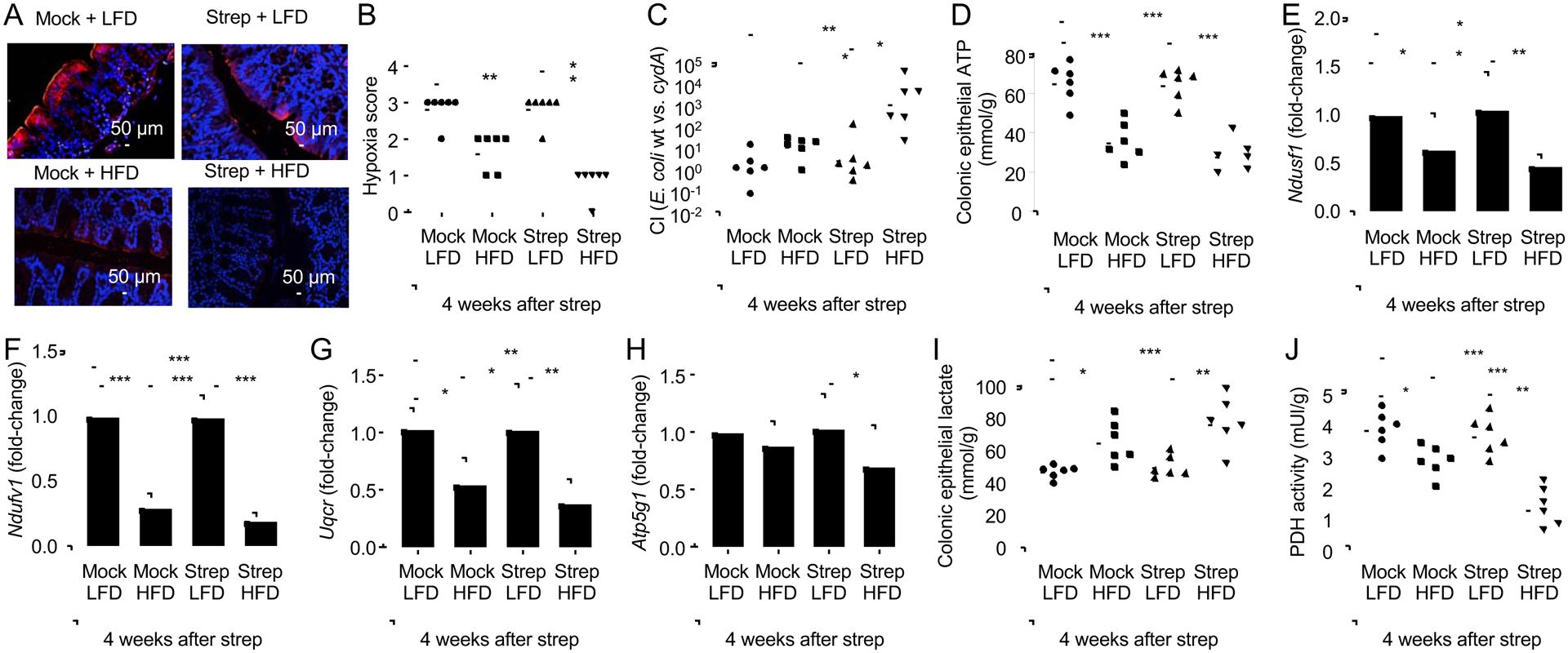
Groups of male mice (N = 6) were reared on a high-fat diet (HFD, 45% fat) or on a low-fat diet (LFD, 10% fat) and were mock treated or treated with a single dose of streptomycin (Strep) four weeks before necropsy. (A-B) Binding of pimonidazole was detected using hypoxyprobe-1 primary antibody and a Cy-3 conjugated goat anti-mouse secondary antibody (red fluorescence) in the sections of proximal colon that were counterstained with DAPI nuclear stain (blue fluorescence). (A) Representative images are shown. (B) Pimonidazole staining was quantified by scoring blinded sections of proximal colon. (C) Mice were inoculated with a 1:1 mixture of E.coli wild type (wt) and an isogenic respiration-deficient (cydA) mutant and the competitive index (CI) in colon contents was determined 4 weeks after streptomycin treatment. (D-J) Colonocytes were isolated from the colonic mucosa to measure cytosolic concentrations of ATP (D). (E-H) Transcript levels of genes encoding components of the electron transport chain, including Ndufs1 (E), Ndufv1 (F), Uqcr (G) and Atp5g1 (H) were determined by quantitative real-time PCR in RNA isolated from colonocytes. Lysates of colonocytes were used to measure cytosolic concentrations of lactate (I) and pyruvate dehydrogenase activity (PDH) (J). (B-D, I and J) Dots represent data from individual animals and bars represent geometric mean. (E-H) Bars represent geometric mean± standard deviation. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. (C, E-H) Data were transformed logarithmically before analysis. P values were calculated by a one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test (B) or by one-way ANOVA followed by Tukey’s multiple-comparison tests (C-J and F). See also Table S2.
Epithelial hypoxia is maintained by high mitochondrial oxygen consumption through oxidative phosphorylation (Zheng et al., 2015), whereas epithelial oxygenation is increased when the energy metabolism shifts to a conversion of glucose into lactate(Byndloss et al., 2017; Furuta et al., 2001). Consistent with impaired mitochondrial bioenergetics, exposure to pre-IBD risk factors reduced epithelial ATP levels (Fig. 4D) and epithelial expression of genes encoding components of the electron transport chain, including Ndufs1 (Fig. 4E), encoding NADH:ubiquinone oxidoreductase core subunit S1, Ndufv1 (Fig. 4F), encoding NADH:ubiquinone oxidoreductase core subunit V1, Uqcr (Fig. 4G), encoding a subunit of ubiquinol-cytochrome c reductase and Atp5g1, which encodes a subunit of mitochondrial ATP synthase (Fig. 4H). In line with a switch to a glycolytic metabolism, colonic epithelial cells of mice exposed to pre-IBD risk factors exhibited an increased concentration of intracellular lactate (Fig. 4I) and reduced levels of pyruvate dehydrogenase activity (Fig. 4J). Collectively, these data supported the idea that pre-IBD risk factors drove an expansion of E. coli by reducing mitochondrial bioenergetics in colonocytes.
5-ASA ameliorates signs of disease in mice exposed to pre-IBD risk factors
Since our data suggested that an aerobic respiration-driven E. coli expansion (Fig. 4C) was secondary to diminished mitochondrial bioenergetics in colonocytes (Fig. 4D–4H), we wanted to explore whether signaling pathways that activate mitochondrial activity could be targeted to remediate dysbiosis. 5-amino salicylic acid (5-ASA, the active ingredient in mesalazine) is a PPAR-γ agonist(Rousseaux et al., 2013) that acts topically on the colonic epithelium(Zhou et al., 1999). Consistent with a previous report(Byndloss et al., 2017), epithelial expression of the PPAR-γ-regulated gene Angptl4 was reduced in the colon five days after streptomycin treatment, but normal Angptl4 expression was restored by four weeks after antibiotic administration (Fig. 5A), suggesting that antibiotics alone only produced short-lived changes in epithelial gene expression. Similarly, high-fat diet alone did not reduce Angptl4 expression. In contrast, exposure to pre-IBD risk factors (i.e. a history of antibiotic treatment in mice on a high-fat diet) caused a prolonged reduction in epithelial Angptl4 expression, which could be ameliorated by 5-ASA treatment (Fig. 5A).
Figure 5: 5-ASA treatment restores mitochondrial activity and limits the bioavailability of oxygen in the colon of streptomycin-treated mice on a high-fat diet.
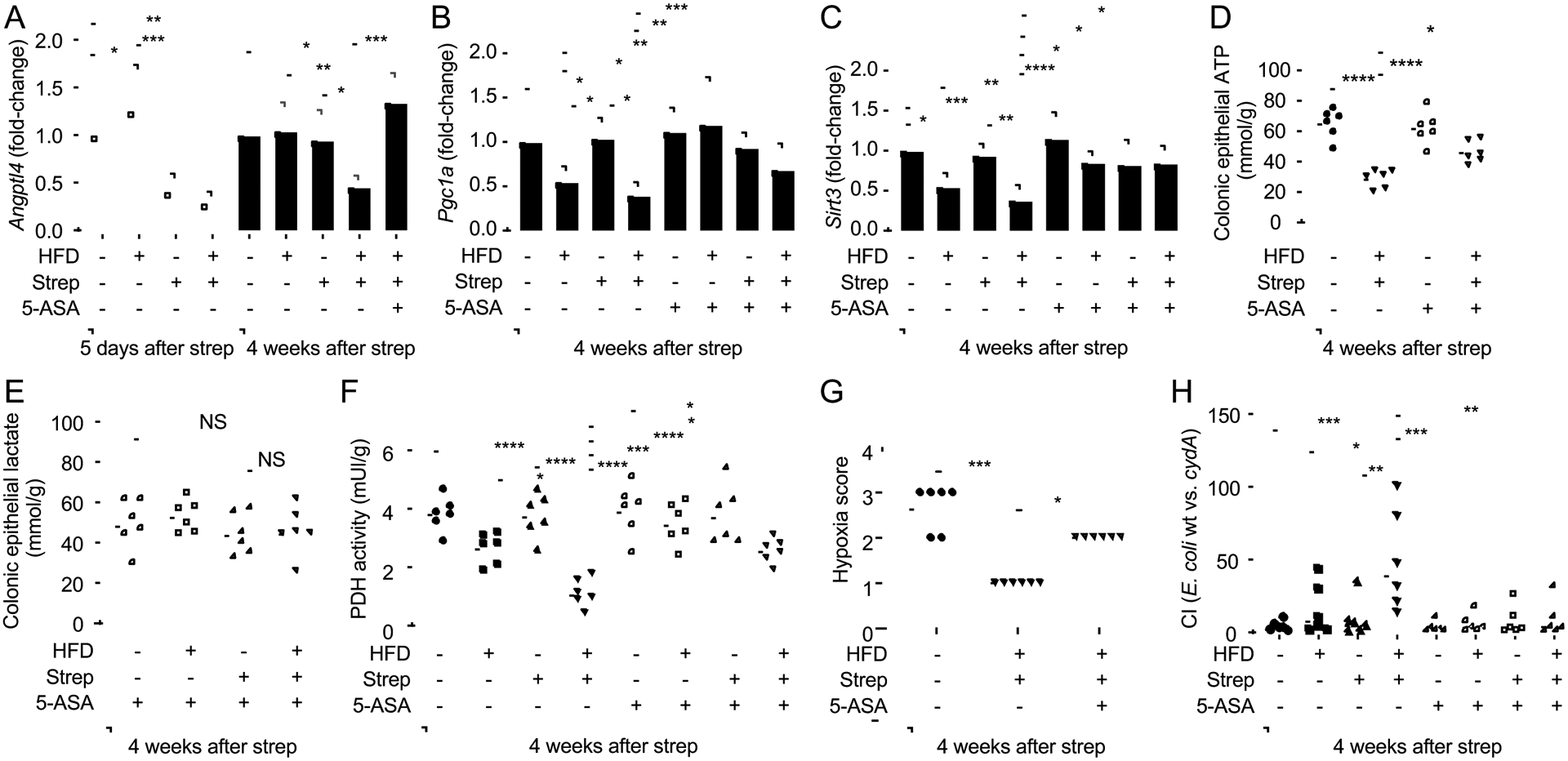
Groups of male mice (N = 6) were reared on a high-fat diet (HFD: +) or on a low-fat diet (HFD: −) and were mock treated (Strep: −) or treated with a single dose of streptomycin (Strep: +) four weeks before necropsy. Chow was supplemented with 5-ASA (5-ASA: +) or did not contain supplementation (5-ASA: −). (A-F) Colonocytes were isolated from the colonic mucosa for analysis. Transcript levels of Angptl4 (A), Pgc1a (B) and Sirt3 (C) were determined by quantitative real-time PCR from RNA isolated from colonocytes. Lysates of colonocytes were used to measure concentrations of ATP (D), lactate (E) and pyruvate dehydrogenase activity (PDH) (F). (G) Pimonidazole staining was quantified by scoring blinded sections of proximal colon. (H) Mice were inoculated with a 1:1 mixture of E.coli wild type (wt) and an isogenic respiration-deficient (cydA) mutant and the competitive index (CI) in colon contents was determined 4 weeks later. (A-C) Bars represent geometric mean± standard deviation. (D-H) Dots represent data from individual animals and bars represent geometric mean. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; NS, P>0.05. (A-C, H) Data were transformed logarithmically before analysis. P values were calculated by one-way ANOVA followed by Tukey’s multiple-comparison tests (A-F, H) or by one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test (G). See also Table S2.
PPAR-γ engages the transcription coactivator PGC-1α (Peroxisome Proliferator-activated Receptor-gamma Coactivator-1 alpha) to activate transcription of the Sirt3 gene, which encodes a protein deacetylase termed Sirtuin 3 (SIRT3)(Giralt et al., 2011). SIRT3 is localized to the mitochondria, where it deacetylates and activates a number of enzymes involved in oxidative phosphorylation(Ahn et al., 2008), the tricarboxylic acid (TCA) cycle(Finley et al., 2011) and β-oxidation of fatty acids(Hirschey et al., 2010). Exposure to pre-IBD risk factors reduced expression of Pgc1a (Fig. 5B) and Sirt3 (Fig. 5C) in the colonic epithelium of mice, which could be ameliorated by treatment with 5-ASA. Consistent with a restoration of mitochondrial activity, 5-ASA treatment prevented a drop in colonic epithelial ATP levels in mice exposed to pre-IBD risk factors (Fig. 5D). Furthermore, 5-ASA treatment blocked an increase in the concentration of intracellular lactate in the colonic epithelium (Fig. 5E), blunted a decline in epithelial pyruvate dehydrogenase activity (Fig. 5F), restored epithelial hypoxia (Fig. 5G) and prevented an aerobic respiration-mediated expansion of E. coli (Fig. 5H and S4J) in the colon of mice exposed to pre-IBD risk factors. Collectively, these data suggested that 5-ASA treatment blunted or abrogated a weakening in mitochondrial activity in the colonic epithelium that was triggered by exposure to pre-IBD risk factors.
Next, we wanted to determine whether 5-ASA was effective in ameliorating other signs of disease in mice exposed to pre-IBD risk factors. One of the changes in the microbiota composition associated with pre-IBD patients was a reduced relative abundance of members of the class Clostridia (Fig. S2B). High-fat diet was not associated with a reduction in the relative abundance of Clostridia (Fig 6A), which was consistent with a previous report(Verdam et al., 2013), but did not match our observations in patients with pre-IBD (Fig. 1H and S2B). Streptomycin caused a marked reduction in the abundance of Clostridia two days after administration, but the abundance of this taxon rebounded by 5 days after administration and was normal at four weeks after antibiotic treatment (Fig 6A). Notably, animals exposed to pre-IBD risk factors (i.e. a history of antibiotic treatment in mice on a high-fat diet) was the only treatment group that recapitulated a lasting Clostridia depletion (Fig 6A), thus recapitulating the reduced relative abundance of Clostridia observed in patients with pre-IBD (Fig. 1H and S2B). Thus, a combination of high-fat diet and a history of antibiotic treatment triggered changes in the microbiota composition that were not observed with either treatment alone. Importantly, 5-ASA treatment restored a normal abundance of Clostridia in streptomycin-treated mice on a high-fat diet (Fig 6A). 5-ASA treatment did not alter the body weight (Fig. 6B), but diminished gains in visceral fat weight in mice exposed to pre-IBD risk factors (Fig. 6C). Furthermore, 5-ASA treatment abrogated signs of low-grade mucosal inflammation, as indicated by measuring fecal calprotectin (Fig. 2E and S4E) and colon length (Fig 6D), scoring histopathology (Fig. 6E), and quantifying expression of Tnfa (Fig 6F and S4D), S100a8 and S100a9 (Fig. 2E and S4F) in the colonic mucosa of mice exposed to pre-IBD risk factors. Additionally, 5-ASA treatment ameliorated visceral hypersensitivity in mice exposed to risk factors for pre-IBD (Fig. 2I and S4I). Finally, treatment with 5-ASA prevented an increase in fecal water content in mice exposed to pre-IBD risk factors (Fig. 2G and S4H). Thus, 5-ASA treatment blunted or abrogated most signs of disease produced by exposing mice to pre-IBD risk factors.
Figure 6: 5-ASA treatment abrogates colonic signs of disease in streptomycin-treated mice on a high-fat diet.
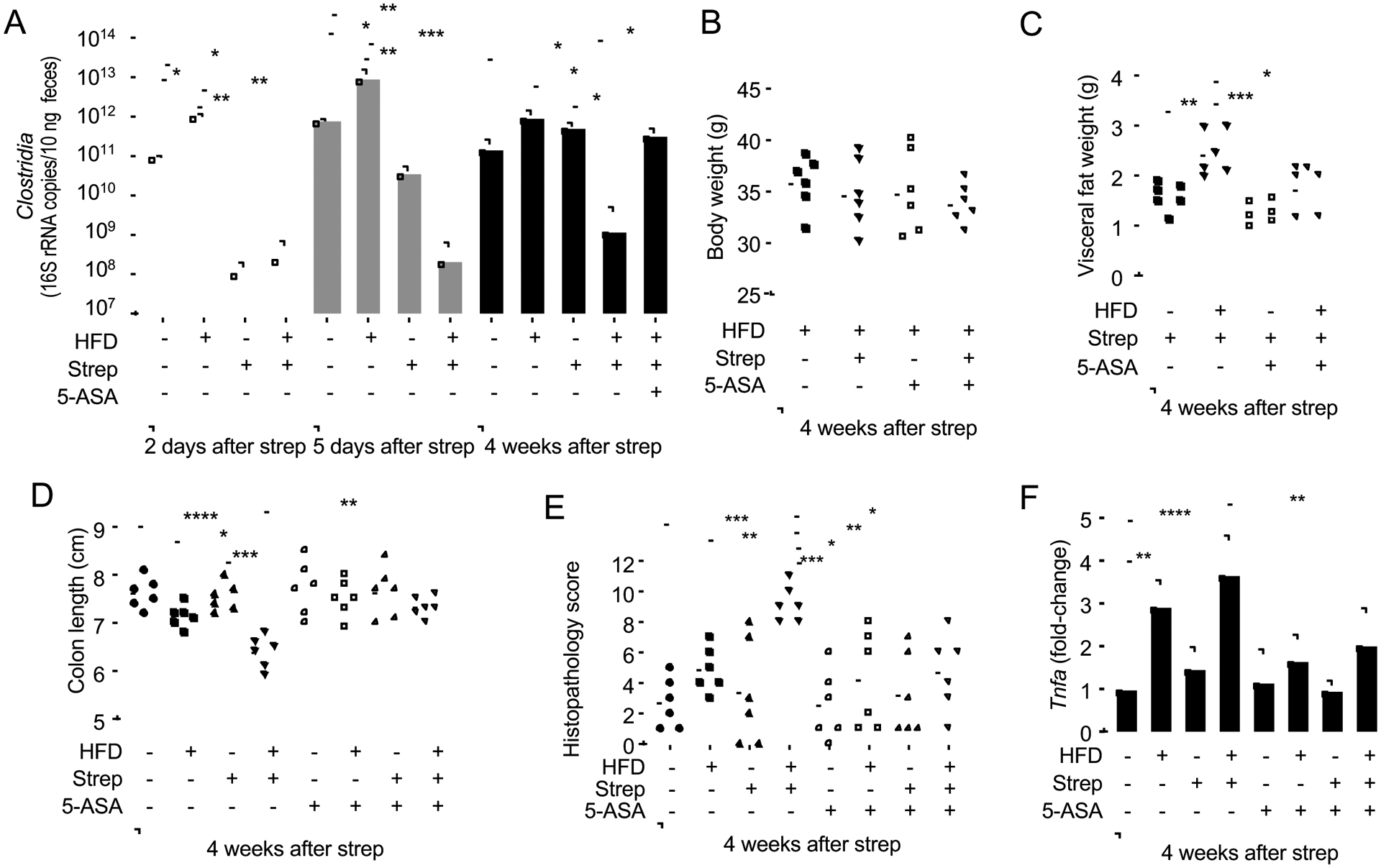
Groups of male mice (N = 6) were reared on a high-fat diet (HFD: +) or on a low-fat diet (HFD: −) and were mock treated (Strep: −) or treated with a single dose of streptomycin (Strep: +) four weeks before necropsy. Chow was supplemented with 5-ASA (5-ASA: +) or did not contain supplementation (5-ASA: −). (A) The abundance of Clostridia in fecal samples collected was determined by quantitative real-time PCR using class specific primers for the 16S rRNA gene. Mouse body weight (B), visceral fat weight (C) and colon length (D) were determined during necropsy. (E) A veterinary pathologist scored histopathological changes in blinded sections of the cecum. (F) Colonocytes were isolated from the colonic mucosa and transcription levels of Tnfa determined by quantitative real-time PCR. (A and F) Bars represent geometric mean± standard deviation. (B-E) Dots represent data from individual animals and bars represent geometric mean. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. (F) Data were transformed logarithmically before analysis. P values were calculated by one-way ANOVA followed by Tukey’s multiple-comparison tests (A-D, F) or by one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test (E). See also Table S2.
A dysbiotic expansion of Enterobacteriaceae exacerbates mucosal inflammation in pre-IBD
Finally, we wanted to determine whether dysbiosis triggered by combining high-fat diet with a history of antibiotic treatment adversely effected the host. Endogenous Enterobacteriaceae exacerbate inflammation in mouse models of IBD(Garrett et al., 2010; Zhu et al., 2018) and exhibited an increased relative abundance in pre-IBD patients (Fig. 1I and S1D). To study whether a dysbiotic expansion of this taxon elevates markers of mucosal inflammation, C57BL/6J mice (Jackson Laboratories), which were confirmed to be Enterobacteriaceae-free, were inoculated one day after streptomycin treatment with three endogenous Enterobacteriaceae (E. coli, Klebsiella oxytoca and Proteus vulgaris) isolated from C57BL/6NTac mice (Taconic Farms) (Velazquez et al., 2019). Exposure to pre-IBD risk factors resulted in an approximately 1,000-fold increase in the fecal recovery of Enterobacteriaceae 4 weeks after streptomycin treatment compared to control mice (Fig. 7A). Enterobacteriaceae did not alter levels of lactate or expression of S100a8 and S100a9 in colonocytes (Fig. 7B and 7C). The presence of Enterobacteriaceae in control mice (i.e. in mice on a low-fat diet with no history of antibiotic treatment) did not alter colon length (Fig. 7D) or markers of mucosal inflammation (Fig. 7E and 7F). In contrast, a dysbiotic expansion of Enterobacteriaceae in mice exposed to pre-IBD risk factors (Fig. 7A) exacerbated a shortening of the colon (Fig. 7D) and expression of inflammatory markers in the colonic mucosa (Fig. 7E and 7F). These data suggested that dysbiosis can further exacerbate low-grade mucosal inflammation observed in pre-IBD.
Figure 7: Endogenous Enterobacteriaceae exacerbate signs of disease in streptomycin-treated mice on a high-fat diet.
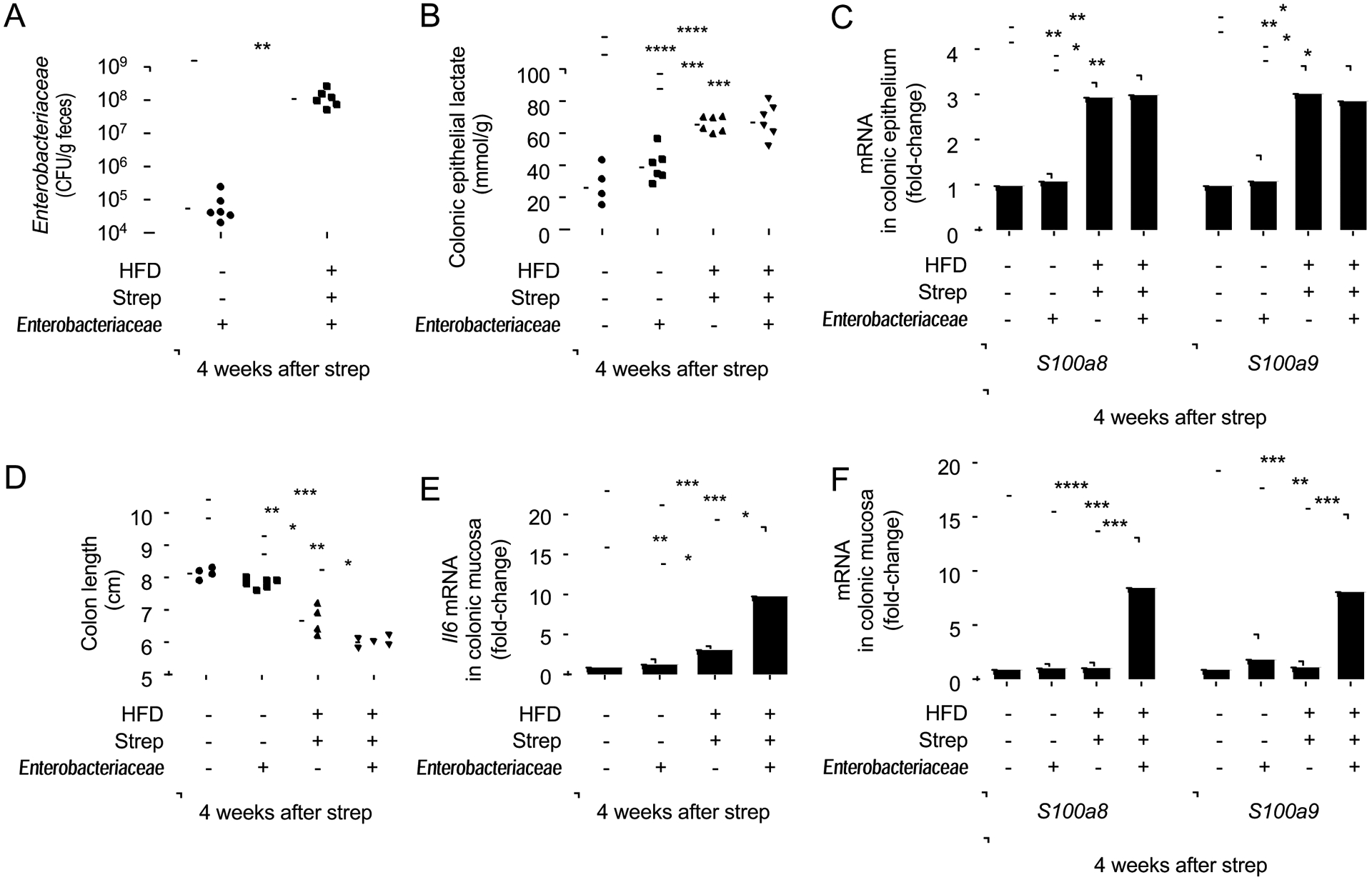
Groups of male Enterobacteriaceae-free mice (N = 6) were reared on a high-fat diet (HFD: +) or on a low-fat diet (HFD: −) and were mock treated (Strep: −) or treated with a single dose of streptomycin (Strep: +) four weeks before necropsy. One day after streptomycin treatment, mice were mock inoculated (Enterobacteriaceae: −) or received commensal isolates of E. coli, K. oxytoca and P. vulgaris (Enterobacteriaceae: +). (A) Colony-forming units (CFU) of Enterobacteriaceae in the feces were determined. (B) Lysates of colonocytes were used to measure concentrations of lactate. (C) Transcript levels of the indicated genes were determined by quantitative real-time PCR in RNA isolated from colonocyte preparations. (D) Colon length was determined during necropsy. (E and F) Transcript levels of Il6 (E), S100a8 and S100a9 (F) were determined by quantitative real-time PCR in RNA isolated from the colonic mucosa. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001. (A,C,E,F) Data were transformed logarithmically before analysis. P values were calculated by Student’s t test (A) or by one-way ANOVA followed by Tukey’s multiple-comparison tests (B-F). See also Table S2.
DISCUSSION
Elevated fecal calprotectin (>250 μg/g) is a surrogate marker for endoscopic lesions in IBD(D’Haens et al., 2012). Fecal calprotectin levels between 200 and 250 μg/g are predictive of endoscopic remission(Kawashima et al., 2017), whereas fecal calprotectin levels <50 μg/g point towards a non-IBD etiology(Banerjee et al., 2015; Chang et al., 2014; David et al., 2015). However, levels between 50 and 250 μg/g are difficult to interpret, because mild calprotectin elevation may be seen with non-specific low-grade inflammation(Bjarnason, 2017), which is observed in a fraction of IBS patients with normal colonic histology(Banerjee et al., 2015; Chang et al., 2014). Low-grade mucosal inflammation in patients with symptoms of IBS represents a syndrome at the intersection of the clinical spectra of IBS and IBD(Abdul Rani et al., 2016; Berrill et al., 2013; Colombel et al., 2019; Spiller and Lam, 2011; Spiller and Major, 2016). Although the pathophysiology remains unknown, current thinking classifies this overlap syndrome as pre-IBD(Abdul Rani et al., 2016). In support of this acronym, changes in the microbiota composition of patients with pre-IBD included elevated Enterobacteriaceae and reduced Clostridia abundances, which matched observations made in IBD patients(Rigottier-Gois, 2013; Rizzatti et al., 2017; Shin et al., 2015), but was distinct from changes in the microbiota composition observed in IBS patients with no mucosal inflammation.
Comparison of patient groups revealed that high-fat diet and antibiotics, two known environmental risk factors for IBD(Albenberg et al., 2012; Frolkis et al., 2013; Lewis, 2014), also increased the risk for developing pre-IBD. Interestingly, patients exposed to both risk factors exhibited the highest risk for acquiring pre-IBD, suggesting that environmental risk factors might cooperate to drive disease. We developed a mouse model of pre-IBD to investigate this phenomenon. In choosing our mouse diet, we aimed at matching the increase in saturated fatty acid intake observed in patients, but a potential caveat is that other parameters, such as contents of cholesterol and cornstarch, differed from human diet. We first examined epithelial energy metabolism, because colonic epithelial cells from ulcerative colitis patients exhibit lower epithelial PPAR-γ synthesis(Giralt et al., 2011) and reduced mitochondrial bioenergetics compared to healthy controls(Roediger, 1980). Notably, high-fat diet and streptomycin treatment produced a long-term reduction in mitochondrial bioenergetics in the colonic epithelium of mice that was not observed with exposure to either environmental factor individually. A likely reason for this cooperative effect is that high-fat diet and streptomycin treatment both reduce mitochondrial activity, but through different mechanisms. By depleting microbiota-derived butyrate, a PPAR-γ agonist, antibiotic treatment causes a short-term impairment of mitochondrial bioenergetics in the colonic epithelium by transiently reducing PPAR-γ signaling (Byndloss et al., 2017). In contrast, diet-impaired mitochondrial bioenergetics are due to induction of mitochondrial hydrogen peroxide by saturated fatty acids(Cardoso et al., 2013; Kakimoto et al., 2015). Thus, the cooperative effect of high-fat diet and antibiotics is likely due to the fact that both factors impair the same host cell function, thereby explaining the marked long-term reduction in epithelial mitochondrial bioenergetics that was seen with neither high-fat diet nor streptomycin treatment alone.
Mitochondrial activity is important for maintaining the colonic surface in a state of physiological hypoxia, which limits the amount of oxygen emanating from the mucosal surface, thereby checking aerobic growth of facultative anaerobic bacteria(reviewed in(Litvak et al., 2018)). Conversely, reduced mitochondrial bioenergetics lower epithelial oxygen consumption, thereby increasing epithelial oxygenation and the diffusion of oxygen into the intestinal lumen(Byndloss et al., 2017; Furuta et al., 2001; Kelly et al., 2015). It has been hypothesized that an expansion of facultative anaerobic bacteria observed in the fecal microbiota of IBD patients might be triggered by a disruption in anaerobiosis in the colon, a concept known as the “oxygen hypothesis”(Rigottier-Gois, 2013). Our results provide experimental support for the idea that increased oxygen bioavailability is responsible for an elevated abundance of Enterobacteriaceae during pre-IBD, because an E. coli expansion in the gut microbiota could be blunted by genetic ablation of cytrochrome bd oxidase-mediated oxygen respiration.
The idea that changes in the microbiota composition were secondary to changes in epithelial energy metabolism suggested that treating mitochondrial dysfunction in the colon would be a potential strategy for remediating dysbiosis. To test this hypothesis, we used the PPAR-γ agonist 5-ASA(Rousseaux et al., 2013), because this drug acts topically on the colonic epithelium (Zhou et al., 1999) to activate transcription of Sirt3(Giralt et al., 2011), which encodes a protein deacetylase that activates mitochondrial activity(Ahn et al., 2008; Finley et al., 2011; Hirschey et al., 2010). Remarkably, 5-ASA treatment ameliorated signs of disease in mice with pre-IBD and normalized the microbiota composition by restoring epithelial hypoxia. Although these results show that dysbiosis was secondary to impaired mitochondrial bioenergetics, a dysbiotic expansion of Enterobacteriaceae exacerbated intestinal inflammation in our model of pre-IBD, which was similar to observations made in mouse models of chemically or genetically induced colitis(Garrett et al., 2010; Zhu et al., 2018). These data suggest that the ability of 5-ASA to reduce the abundance of Enterobacteriaceae in the fecal microbiota(Xu et al., 2018) likely contributes to its anti-inflammatory activity.
STAR METHODS
Resource Availability
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andreas J. Baumler (ajbaumler@ucdavis.edu).
Materials Availability.
The study did not generate new unique reagents except 16S rRNA gene amplicon sequencing data, which are available through the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
Data and Code Availability.
16S rRNA gene amplicon sequencing data have been deposited with links to BioProject accession number PRJNA541572 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
Experimental Model and Subject Details
Human studies.
All subjects participated in the study voluntarily, and written informed consent was obtained from each participant. The study complied with the Declaration of Helsinki, and the Institutional Review Board of CHA Bundang Medical Center (protocol number 2016-06-055) and Yonsei University College of Medicine (protocol number 4-2015-0608) approved this study. Subjects fulfilling ROME III criteria for IBS were recruited from the Department of Family Medicine, CHAUM Hospital, Seoul, Korea between June 2016 and July 2017. To rule out the presence of other gastrointestinal (GI) diseases with structural alterations (e.g. colorectal cancer) or chronic inflammatory diseases (e.g. Crohn’s disease or ulcerative colitis), we enrolled IBS patients that had undergone clinical investigation and colonoscopy or barium enema of GI tract less than 5 years before participating or barium enema of GI tract less than 1 year before participating. We excluded subjects with histories of any types of cancer, abdominal surgery and inflammatory bowel disease. Patients who had a history of taking steroidal agents or none-steroidal anti-inflammatory drugs (NSAIDs) within one year were also excluded. Follow up of the enrolled participants with IBS symptoms for one year revealed that no patients were subsequently diagnosed with organic GI diseases, such as colorectal cancer or inflammatory bowel disease. Healthy control subjects without any GI symptoms were recruited by advertising. Each healthy control subject completed a questionnaire about GI symptoms to confirm the absence thereof. We excluded subjects with histories of any types of cancer, abdominal surgery and inflammatory bowel disease. All subjects completed a screening blood test and subjects with abnormal liver function, abnormal kidney function, abnormal blood glucose level or abnormal C reactive protein (CRP) level were excluded. Subjects with a history of taking antibiotics or probiotics within 4 weeks before the participation were also excluded. Information about the participants including age and gender of the study participants are provided in the Table S3.
Mouse Experiments.
The Institutional Animal Care and Use Committee at the University of California at Davis approved all animal experiments in this study. Male and female C57BL/6J mice, aged 6 weeks were obtained from The Jackson Laboratory. Mice were reared and maintained on a 10% control Diet (Teklad Diet, #TD110675) or a 45% fat diet (HFD) (Teklad Diet, #TD06415) from weaning at the age of 6 weeks until the end of the experiment. For 5-aminosalicyclic acid (5-ASA) treatment, 5-asa powder (Sigma-Aldrich) was mixed directly into the control diet or HFD at 1650mg/kg/day by Envigo Inc. Mice were treated with 20mg/animal streptomycin or mock treated via oral gavage. And after 24 hours later, mice were orally inoculated with 1×109 CFU/100uL of a 1:1 mixture of indicated E.coli Nissle 1917 strain mixtures in Luria-Bertani (LB) broth (BD Biosciences #244620). Alternatively, mice were inoculated with 1×109 CFU/100uL of an equal mixture of three commensal Enterobacteriaceae (K. oxytoca, P.vulgaris and E. coli) isolated from C57BL/6NTac mice. Colonization of bacteria in feces was determined by homogenizing feces in 1 mL of sterile PBS, diluting the samples serially and then plating on plates of LB containing appropriate antibiotics. Samples were collected at 4 weeks after infection (12 weeks old).
Bacterial culture.
E. coli strains (wild-type E. coli Nissle 1917 and E. coli Nissle 1917 cydAB mutant (Byndloss et al., 2017) and murine Enterobacteriaceae isolates were routinely grown aerobically at 37 °C in LB broth, on LB agar plates or on MacConkey agar plates. When necessary, 0.1mg/mL of carbenicillin or 0.05mg/mL of kanamycin were added. When growing for competitive infection, strains were grown in a hypoxia chamber with 0.5% of oxygen at 37°C in LB broth.
Method details
Human subjects.
Subjects with symptoms of IBS were classified into three subtypes by predominant stool pattern: IBS with diarrhea (IBS-D), IBS with constipation (IBS-C) and mixed IBS (IBS-M) according to the Rome III criteria. IBS patients with a history of acute gastroenteritis prior to the onset of IBS symptoms were classified as post-infectious IBS.
Questionnaire in human subjects.
All human subjects completed a questionnaire about lifestyle factors, including exercise, physical activity, cigarette smoking, alcohol consumption, underlying medical conditions and medications. The frequency of antibiotic treatment within a year was recorded. Each participant with IBS also completed a questionnaire for measuring the severity of bowel symptom, quality of life related with IBS and mental health at the baseline assessment. Patients were questioned about a possible history of acute gastroenteritis prior to the onset of IBS suggestive of post-infectious IBS and about a possible worsening of IBS symptoms due to stress. The Bowel Symptom Severity Scales (BSSS) (Boyce et al., 2000) were used to measure the severity of bowel symptom. The IBS-QOL questionnaire (Patrick et al., 1998) was used to assess the quality of life for patients with IBS.
Anthropometric measurement of human subjects.
Participants were asked to rest for more than 10 minutes before measuring blood pressure. Blood pressure was measured in the sitting position and the systolic and diastolic blood pressures were recorded. Body mass index (BMI) was calculated as weight (kg) divided by height squared (m2). Bio-electrial impedance analysis using InBody U20 (Biospace, Seoul Korea) was used to estimate the percentage of body fat and lean body mass (gram).
Nutrition assessment in human subjects.
The nutrition assessment of this study was estimated based on the 1 day 24-hour recall diet diary. A standardized form to record every food item consumed, including the amount, ingredients of foods and times within the previous 24 hours were given to each participant. Consumed nutrients including total calorie intakes, total carbohydrate intakes, total fiber intakes, total protein intakes, total fat intakes and intakes of various fatty acids were analyzed using Can-Pro software 5.0; developed by the Korean Nutrition Society and the energy adjustment was performed by calculating nutrient density and expressed as total intake/1000kcal.
Human fecal calprotein (FCP) analysis.
Human FCP level was measured using a commercial enzyme-linked immunosorbent assay kit (EK-CAL, BUHLMANN, Schonenbunch, Switzerland) according to the manufacturer’s instruction. Elevated FCP level was defined as >50μg/g.
Human fecal Myeloperoxidase (MPO) analysis.
Human fecal MPO level was measured using a commercial ELISA kit (MPO ELISA Kit, IDK) according to the manufacturer’s instruction. High MPO level was defined as >2μg/g according to the manufacturer’s instruction.
Isolation of endogenous Enterobacteriaceae from mice.
Ten-fold dilutions of fecal homogenates from C57BL6/NTac mice (Taconic Farms) were spread on MacConkey agar and incubated at 37°C overnight. Single colonies with different morphologies were picked and streaked for isolation. Each colony was identified biochemically using EnteroPluri Test (BD Biosciences) using the manufacturer’s instruction.
Bacterial Community Analysis.
Genomic DNA extraction from 400mg of human stool samples were added to 15 mL of DNA extraction lysis buffer (SDS 4%, Tris-HCL 50mM, EDTA 50mM, NaCl 5000mM) followed by vigorous homogenization by vortexing for 1 minute. 1.4mL of homogenized fecal suspensions were transferred to a 2mL eppendorf tube and followed by bead beating for 50 seconds and centrifuging at 14,000g for 10 minutes. 200μL of supernatants were transferred to 96 well plates and 1μL of supernatants were dissolved in 29 μL of nuclease free water. The plates were kept at −25°C until performing PCR. The extracted DNA was amplified using the primers targeting from V3 to V4 region of bacterial 16s rRNA gene using the primers of 341F (5’-TCGTCGGCAGCGTC-AGATGTGTATAAGAGACAG-CCTACGGGNGGCWGCAG-3’; underlining sequence indicates the target region primer) and 805R (5’-GTCTCGTGGGCTCGG-AGATGTGTATAAGAGACAG-GACTACHVGGGTATCTAATCC-3’). The PCR reaction were performed under the following conditions: initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 95 °C or 30 sec, primer annealing at 55 °C for 30 sec, and extension at 72 °C for 30 sec, with a final elongation at 72 °C for 5 min. Then, secondary amplification for attaching the Illumina NexTera barcode was performed with i5 forward primer (5’-AATGATACGGCGACCACCGAGATCTACAC-xrefXX-TCGTCGGCAGCGTC-3’; X indicates the barcode region) and i7 reverse primer (5’-CAAGCAGAAGACGGCATACGAGAT-xrefXX-AGTCTCGTGGGCTCGG-3’). The condition of secondary amplification is equal to the former one except the amplification cycle was set to 8. The PCR product was confirmed by using 2% agarose gel electrophoresis and the amplified products were purified using QIAquick PCR purification kit (Qiagen, Valencia, CA, USA) and the equal concentrations of purified products were pooled together and short fragments (non-target products) were removed with Ampure beads kit (Agencourt Bioscience, MA, USA). The product size and the quality were assessed using Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA) using a DNA 7500 chip. Mixed amplicons from different samples were pooled and subjected to pyrosequencing. The process of sequencing was performed by Chunlab, Inc. (Seoul, South Korea) using Illumina Miseq Sequencing system (Illumina, USA) according to the manufacturer’s instructions. The data have been deposited with links to BioProject accession number PRJNA541572 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
Pyrosequencing data analysis: Reads obtained from the different samples were sorted by unique barcodes of each PCR product. Then, the sequences of the barcode, linker and primers were removed from the original read of sequences. Among the sequencing reads, reads containing two or more ambiguous nucleotides, reads with low quality scores (average <25) or reads shorter than 300 bp were all discarded. Bellerophone method that compares the BLASTN search results between the forward half and reverse half sequences were used for detecting potential chimeric sequences (Huber et al., 2004). After removing the chimeric sequences, each read was assigned to a taxonomic classification in the EzTaxon-e database (http://eztaxon-e.ezbiocloud.net) (Kim et al., 2012). This database contains the 16s rRNA gene sequence of type strains with valid published names and representative species level phylotypes from either cultured or uncultured entries in the GeneBank database with complete hierarchical taxonomic classification from the phylum to species. To compare the abundance of taxa among the different groups, the LDA Effect Size (LEfSe; Linear Discriminant Analysis Effect Size) algorithm was used with online interface Galaxy (http://huttenhower.sph.harvard.edu/lefse/). LEfSe analysis was performed using a Kruskal-Wallis sum-rank test with an alpha value of <0.5, followed by the Wilcoxon-rank sum test with an alpha score <0.05 and a one-against-all strategy for multi-class analysis. An effect size >2 (on a log scale) were considered significant in this study.
Mouse IBS phenotype assays.
To determine the water content of the mouse feces, fecal pellets of each mouse were collected and weighed. The pellets were dried in a 60°C oven for 24 hours then weighed again and the percentage of water was calculated. To measure the total intestinal transit time, mice were orally gavaged with 0.2mL of semi-liquid solution containing 5% Evans blue in sterile water with 5% gum Arabic and placed individually into clean empty cages for monitoring. The time for expulsion of the first blue pellet was recorded (McLaughlin et al., 2017). The tests for measuring mouse GI function were performed during the last week of the experiment. Abdominal withdrawal reflex (AWR) in response to colorectal distension was measured to assess visceral sensitivity as previously described (Yang et al., 2015). The test was performed during the last week of the experiment. On the day the test was performed, mice were briefly sedated with isoflurane while a balloon catheter (6-Fr, 2mm external diameter) was inserted intra-rectally into the descending colon of mice. After waking up and adapting for 1 hour in a single cage, colorectal distension was performed in a stepwise fashion. Each 20-second distention was followed by a 30 second resting period. Each level of distention (0.25, 0.35 and 0.5mL) was repeated twice and the AWR score was assigned as follows: 0, no behavioral response to colorectal distention; 1, brief head movement followed by immobility; 2, contraction of abdominal muscles; 3, lifting of abdomen; 4, body arching and lifting of pelvic structures. The tests for measuring mouse GI function were performed during the last week of the experiment.
Colonocyte isolation.
The colon was opened lengthwise and cut into 2–4cm pieces, placed in a 50 mL falcon tube filled with ice-cold 1X RPMI 1640 (Gibco) buffer. The tissue was transferred into another 50 mL falcon tube filled with 20mL ice-cold DPBS (Gibco) for cleaning. After inverting the tubes gently four times, the tissues were transferred to sterile petri dishes and moved into the dissociation reagent #1 (30mM EDTA, 1.5mM DTT, diluted into 1X DPBS) and incubated on ice for 20 minutes. Then, tissues were placed into a pre-warmed (37°C) dissociation reagent #2 (30mM EDTA, diluted into 1X DPBS) and incubated at 37°C for 10 minutes. After the incubation period, tubes with tissues were shaken vigorously for 30 seconds to detach the epithelium from the basement membrane. After removing remnant intestinal tissue, the cell suspensions were transferred to sterile 15mL conical tubes and pelleted by centrifugation at 800g for 10 minutes at 4 °C. Supernatants were removed and the cell pellets were resuspended in 1mL of Tri Reagent (Molecular Research Center) for subsequent RNA extraction or flash frozen in liquid nitrogen for subsequent Lactate, ATP and PDH activity colorimetric assay.
Lactate measurements.
For measuring intracellular lactate levels, primary colonocytes were isolated as described above and then deproteinized by using Deproteinizing Sample Preparation Kit (Biovision, Milpitas, CA) following manufacturer’s instructions. Lactate measurement in colonocyte lysates were performed by using a Lactate Colorimetry Assay Kit 2 (Biovision, Milpitas, CA) according to the manufacturer’s instructions.
ATP measurements.
For measuring intracellular ATP levels, primary colonocytes were isolated as described above and then deproteinized by using Deproteinizing Sample Preparation Kit (Biovision, Milpitas, CA) following manufacturer’s instructions. Lactate measurement in colonocyte lysates were performed by using ATP Colorimetry Assay Kit (Biovision, Milpitas, CA) according to the manufacturer’s instructions.
PDH activity measurements.
To measure PDH activity, primary colonocytes were isolated as described above and then lysed with RIPA buffer and cellular debris were removed by centrifugation for 5 minutes at 13,000rpm at room temperature. PDH activity was measured with supernatants by using the PDH activity assay kit (Biovision, Milpitas, CA) according to the manufacturer’s instructions.
Real time PCR analysis.
RNA was isolated from colonocyte with Tri-reagent (Molecular Research Center) according to the manufacturer’s instructions. A reverse transcriptase reaction was performed to prepare complementary DNA (cDNA) using TaqMan reverse transcription reagents (Applied Biosystems). In a total volume of 25μL volume of each real-time PCR reaction, 4μL of cDNA was used as the template. SYBR green (Applied Biosystems) and appropriate primer sets (Table S7) were used for performing Real-time PCR. Data were analyzed using the comparative threshold method (Applied Biosystems). Transcript levels of target genes were normalized to mRNA levels of the housekeeping gene Act2b, encoding beta-actin. For measuring Clostridia copy numbers in feces, stool DNA was extracted using the Powersoil DNA Isolation kit according to the manufacturer’s instructions. SYBR-Green (Applied Biosystems) based real-time PCR was performed using the listed primers (Table S1). A standard curve ranging from 108 to 101 copies/mL of plasmid carrying the cloned gene diluted in a 0.02mg/mL of yeast RNA (Sigma Aldrich) were generated to calculate the copy numbers. SYBR PCR mix and the appropriate primer sets (Table S1) were used and the absolute values were calculated using a plasmid carry the cloned gene to generate a standard curve ranging from 108 to 101 copies/mL in a 0.02mg/mL yeast RNA solution.
Alcian blue stain.
Colon tissue was fixed in 10% phosphate-buffered formalin and 5 uM sections of tissue were stained with Alcian Blue staining by UCD VMTH Histopathology laboratory. Pictures were taken on Axiocam Microscope (Zeiss) with 2.2μm*2.2μm pixel distance dimensions. Quantification of mature Alcian blue-positive cells were calculated based on comet like features of the cells with dense blue staining on the apical side of colonic crypt within the frame. Each measurement was conducted based on 5 pictures per mouse on at 40x magnification.
Mouse FCP and MPO measurements.
A murine fecal pellet was homogenized in 1 mL of sterile PBS, centrifuged at 3000g for 5 minutes and then the supernatants were collected. Fecal calprotectin levels were determined by using mouse S100A8/S100A9 heterodimer enzyme-linked immunosorbent assay kit (R&D biosystem, USA) and fecal MPO levels were determined by MPO Mouse ELISA kit (Biovision, Milpitas, CA) according to the manufacturer’s instruction.
Histopathology.
Tissue of proximal colon and cecum was fixed in 10% phosphate-buffered formalin and 5um sections of tissue were stained with hematoxylin and eosin. Scoring of blinded tissue sections was performed by a veterinary pathologist based on the criteria listed (Table S8). Representative images were taken using an Olympus BX41 microscope. Detailed information about Alcian blue staining and hypoxia staining can be found in the supplementary methods details.
Hypoxia staining.
For detection of hypoxia, mice were injected with 60mg/kg of pimonidaxole HCL i.p. (Hypoxyprobe TM-1 kit, Hypoxyprobe) one hour before euthanasia. Colon tissues were fixed in 10 % buffered phosphate-buffered formalin and paraffin-embedded tissue was probed with mouse anti-pimonidazole monoclonal IgG1 (MAb 4.3.11.3). Then the slide was stained with Cy-3 conjugated goat anti-mouse antibody (Jackson Immuno Research Laboratories) followed by counterstaining with DAPI using SlowFade Gold mountant (Molecular Probes). Then, scoring of slides was performed based on the degree of colonic epithelial hypoxia (0:no hypoxia; 1: mild focal hypoxia; 2: moderate multifocal hypoxia; 3: intense diffuse hypoxia). Representative images were obtained using a Zeiss Axiovert 200M fluorescent microscope and brightness adjusted.
Quantification and Statistical Analysis.
For human studies, normally distributed continuous data are expressed as the mean ± standard deviation (SD) and non-normally distributed data are expressed as median and interquartile range. Categorical data are represented as number (%). A Kolmogorov–Smirnov goodness-of-fit test was performed to determine the population normality of each group. The characteristics among the healthy control, IBS and pre-IBD groups were compared with a one-way ANOVA, followed by Bonferroni multiple comparison test for normally distributed data, by Kruskal–Wallis rank test followed by the Dunn’s test for multiple comparison for non-normally distributed data or by chi square test for categorical data. The participants were categorized into three groups based on the inter-tercile ranges of total fat intake (1st, 2nd and 3rd tercile group of total fat intake) and categorized into two groups (no antibiotics treatment within a year, more than one antibiotics treatment within a year). The odds ratio and 95% confidence intervals (CIs) for the prevalence of IBS with high FCP according to fat intake and a history of antibiotic use were calculated by multivariate logistic regression analysis after adjustment for confounding variables including age, gender, BMI, regular alcohol consumption and regular exercise. To determine the combined effect of fat intake and antibiotics use on the prevalence of pre-IBD, interaction analyses for pre-IBD were performed between fat intake and antibiotics use using logistic regression analysis. The interaction between fat intake and antibiotics use was tested at a significance level of 0.10. A likelihood-ratio test was performed to compare the goodness of fit. Since the interaction terms of fat intake and antibiotics were significant, the participants were further stratified into 4 groups: (i) No high fat intake/no antibiotics, (ii) High fat diet/No antibiotics, (iii) No high fat diet/Antibiotics, (iv) High fat diet/Antibiotics. A logistic regression was used to compare the prevalence of pre-IBD among the 4 groups. For mouse experiments, fold changes of ratios (Bacterial CI and mRNA levels) and bacterial numbers were transformed logarithmically before analysis to normalized the data. One-way ANOVA followed by Tukey’s multiple-comparison tests were used to determine the differences among the groups. Significance of differences in histopathology, Scores of Alcian Blue or hypoxia score and AWR scores was determined by a one-tailed non-parametric test (Kruskal Wallis test) followed by Dunn’s multiple-comparison test. We performed all statistical analyses using the Statistical Package for Social Science version 20.0 (SPSS INC., Chicago, IL, USA) and GraphPad Prism 8.0 Software. Statistical significance was defined as p<0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-pimonidazole monoclonal antibody MAb1 | Hydroxyprobe | Hypoxyprobe™−1 Kit, HP1–1000, lot 06242014 |
| Alexa fluor 546 goat anti-mouse IgG antibody | Life Technologies | Cat#:A11003 |
| Bacterial and Virus Strains | ||
| Escherichia coli Nissle 1917 | (Spees et al., 2013) | |
| Escherichia coli Nissle 1917 cydAB | (Byndloss et al., 2017) | |
| Escherichia coli isolated from C57BL/6NTac | This study | |
| Proteus vulgaris isolated from C57BL/6NTac | This study | |
| Klebsiella oxytoca isolated from C57BL/6NTac | This study | |
| Biological Samples | ||
| Human feces microbiota | This paper | |
| Mouse primary colonocytes | This paper | |
| Mouse large intestine microbiota | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma Aldrich | Cat#: D9542 |
| ImmuMount mounting reagent | Thermo Scientific | Cat#: 9990402 |
| TRI-Reagent | Molecular Research Center, Inc. | Cat#: TR118 |
| SYBR green | Life Technologies | Cat#: 4309155 |
| 5-aminosalicyclic acid | Sigma Aldrich | Cat#: A3537 |
| Streptomycin sulfate | Fluka | Cat#: 85884 |
| Critical Commercial Assays | ||
| PowerSoil DNA isolation kit | Quagen | Cat#: 12888–100 |
| Lactate colorimetric assay kit II | Biovision | Cat#: K627 |
| ATP colorimetric Assay kit | Biovision | Cat#: K354 |
| PDH activity assay kit | Biovision | Cat#: K679 |
| Myeloperoxidase (MPO) (Stool, Urine), ELISA | Immunodiagnostik | Cat#: K6630 |
| BÜHLMANN fCAL® ELISA | BÜHLMANN | Cat#: EK-Cal |
| Mouse S100A8/S100A9 heterodimer DuoSet ELISA | R&D Biosystem | Cat#: DY8596–05 |
| MPO (Mouse) Elisa Kit | Biovision | Cat#: E4580 |
| Deposited Data | ||
| 16S ribosomal RNA gene sequencing data | NCBI Biosample | PRJNA541572 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratories | Strain 000664 |
| Swiss Webster germ free mice | Bred inhouse, original breeders from Taconic | Original breeders #GF-B6 |
| Oligonucleotides | ||
| See Table S7 | ||
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| Prism 8.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| EzTaxon-e database | Kim et al. 2012 | http://eztaxon-e.ezbiocloud.net |
| Statistical Package for Social Science version 20.0 | SPSS Inc. | https://www.ibm.com/analytics/spss-statistics-software |
| Other | ||
| Mouse 10% Fat Diet | Teklad Diet | #TD110675 |
| Mouse 45% Fat Diet | Teklad Diet | #TD06415 |
Highlights.
A combination of antibiotics and high-fat diet increases the risk for pre-IBD
Antibiotics and high-fat diet jointly impair epithelial mitochondrial function
Impaired mitochondrial bioenergetics drive an Enterobacteriaceae expansion
An Enterobacteriaceae expansion exacerbates mucosal inflammation in pre-IBD
ACKNOWLEDGEMENTS
The process of 16S profiling of the microbial community in human feces has supported by ChunLab Inc., Seoul, South Korea. The authors declare no other conflict of interest.
This work was supported in part by the National Research Foundation of Korea (NRF-2017R1C1B5016190) grant to J.-Y.L.. Work in A.J.B.’s laboratory was supported by USDA/NIFA award 2015-67015-22930 and by Public Health Service Grants AI044170, AI096528, AI112445 and AI112949. S.A.C. was supported by Public Health Service Grant AI060555. E.E.O. was supported by Public Health Service Grant 5TL1R001861.
16S rRNA gene amplicon sequencing data have been deposited with links to BioProject accession number PRJNA541572 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST
The authors declare no competing interests.
REFERENCES
- Abdul Rani R, Raja Ali RA, and Lee YY (2016). Irritable bowel syndrome and inflammatory bowel disease overlap syndrome: pieces of the puzzle are falling into place. Intest Res 14, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, and Finkel T (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A 105, 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albenberg LG, Lewis JD, and Wu GD (2012). Food and the gut microbiota in inflammatory bowel diseases: a critical connection. Curr Opin Gastroenterol 28, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Srinivas M, Eyre R, Ellis R, Waugh N, Bardhan KD, and Basumani P (2015). Faecal calprotectin for differentiating between irritable bowel syndrome and inflammatory bowel disease: a useful screen in daily gastroenterology practice. Frontline Gastroenterol 6, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrill JW, Green JT, Hood K, and Campbell AK (2013). Symptoms of irritable bowel syndrome in patients with inflammatory bowel disease: examining the role of sub-clinical inflammation and the impact on clinical assessment of disease activity. Aliment Pharmacol Ther 38, 44–51. [DOI] [PubMed] [Google Scholar]
- Bibi S, Kang Y, Du M, and Zhu MJ (2017). Maternal high-fat diet consumption enhances offspring susceptibility to DSS-induced colitis in mice. Obesity (Silver Spring) 25, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnason I (2017). The Use of Fecal Calprotectin in Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y) 13, 53–56. [PMC free article] [PubMed] [Google Scholar]
- Boyce P, Gilchrist J, Talley NJ, and Rose D (2000). Cognitive-behaviour therapy as a treatment for irritable bowel syndrome: a pilot study. Aust N Z J Psychiatry 34, 300–309. [DOI] [PubMed] [Google Scholar]
- Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y et al. (2017). Microbiota-activated PPAR-g signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AR, Kakimoto PA, and Kowaltowski AJ (2013). Diet-sensitive sources of reactive oxygen species in liver mitochondria: role of very long chain acyl-CoA dehydrogenases. PLoS One 8, e77088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MH, Chou JW, Chen SM, Tsai MC, Sun YS, Lin CC, and Lin CP (2014). Faecal calprotectin as a novel biomarker for differentiating between inflammatory bowel disease and irritable bowel syndrome. Mol Med Rep 10, 522–526. [DOI] [PubMed] [Google Scholar]
- Colombel JF, Shin A, and Gibson PR (2019). AGA Clinical Practice Update on Functional Gastrointestinal Symptoms in Patients With Inflammatory Bowel Disease: Expert Review. Clin Gastroenterol Hepatol 17, 380–390 e381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa F, Mumolo MG, Bellini M, Romano MR, Ceccarelli L, Arpe P, Sterpi C, Marchi S, and Maltinti G (2003). Role of faecal calprotectin as non-invasive marker of intestinal inflammation. Dig Liver Dis 35, 642–647. [DOI] [PubMed] [Google Scholar]
- Cremonini F, and Talley NJ (2005). Irritable bowel syndrome: epidemiology, natural history, health care seeking and emerging risk factors. Gastroenterol Clin North Am 34, 189–204. [DOI] [PubMed] [Google Scholar]
- D’Haens G, Ferrante M, Vermeire S, Baert F, Noman M, Moortgat L, Geens P, Iwens D, Aerden I, Van Assche G et al. (2012). Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm Bowel Dis 18, 2218–2224. [DOI] [PubMed] [Google Scholar]
- David LE, Surdea-Blaga T, and Dumitrascu DL (2015). Semiquantitative fecal calprotectin test in postinfectious and non-postinfectious irritable bowel syndrome: cross-sectional study. Sao Paulo Med J 133, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanuel A, Landis D, Peucker M, and Hungin AP (2016). Faecal biomarker patterns in patients with symptoms of irritable bowel syndrome. Frontline Gastroenterol 7, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, and Haigis MC (2011). Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6, e23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolkis A, Dieleman LA, Barkema HW, Panaccione R, Ghosh S, Fedorak RN, Madsen K, Kaplan GG, and Alberta IBDC (2013). Environment and the inflammatory bowel diseases. Can J Gastroenterol 27, e18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, and Colgan SP (2001). Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN et al. (2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell host & microbe 8, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Hondares E, Villena JA, Ribas F, Diaz-Delfin J, Giralt M, Iglesias R, and Villarroya F (2011). Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem 286, 16958–16966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber L, Kisling S, Lichti P, Martin FP, May S, Klingenspor M, Lichtenegger M, Rychlik M, and Haller D (2013). High fat diet accelerates pathogenesis of murine Crohn’s disease-like ileitis independently of obesity. PLoS One 8, e71661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwee KA, Collins SM, Read NW, Rajnakova A, Deng Y, Graham JC, McKendrick MW, and Moochhala SM (2003). Increased rectal mucosal expression of interleukin 1beta in recently acquired post-infectious irritable bowel syndrome. Gut 52, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand H, Malmborg P, Askling J, Ekbom A, and Montgomery SM (2008). Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand J Gastroenterol 43, 961–966. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Benninghoff AD, and Ward RE (2012). Formulation of the Total Western Diet (TWD) as a basal diet for rodent cancer studies. J Agric Food Chem 60, 6736–6742. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hod K, Ringel-Kulka T, Martin CF, Maharshak N, and Ringel Y (2016). High-sensitive C-Reactive Protein as a Marker for Inflammation in Irritable Bowel Syndrome. J Clin Gastroenterol 50, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber T, Faulkner G, and Hugenholtz P (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20, 2317–2319. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project C (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hviid A, Svanstrom H, and Frisch M (2011). Antibiotic use and inflammatory bowel diseases in childhood. Gut 60, 49–54. [DOI] [PubMed] [Google Scholar]
- Jeffery IB, O’Toole PW, Ohman L, Claesson MJ, Deane J, Quigley EM, and Simren M (2012). An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut 61, 997–1006. [DOI] [PubMed] [Google Scholar]
- Kakimoto PA, Tamaki FK, Cardoso AR, Marana SR, and Kowaltowski AJ (2015). H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol 4, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima K, Ishihara S, Yuki T, Fukuba N, Sonoyama H, Kazumori H, Yamashita N, Tada Y, Kusunoki R, Oka A et al. (2017). Fecal Calprotectin More Accurately Predicts Endoscopic Remission of Crohn’s Disease than Serological Biomarkers Evaluated Using Balloon-assisted Enteroscopy. Inflamm Bowel Dis 23, 2027–2034. [DOI] [PubMed] [Google Scholar]
- Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A et al. (2015). Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 17, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BS, Kim JN, Yoon SH, Chun J, and Cerniglia CE (2012). Impact of enrofloxacin on the human intestinal microbiota revealed by comparative molecular analysis. Anaerobe 18, 310–320. [DOI] [PubMed] [Google Scholar]
- Kristjansson G, Venge P, Wanders A, Loof L, and Hallgren R (2004). Clinical and subclinical intestinal inflammation assessed by the mucosal patch technique: studies of mucosal neutrophil and eosinophil activation in inflammatory bowel diseases and irritable bowel syndrome. Gut 53, 1806–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Lee JK, Choi JW, Lee CS, Sim JH, Cho CH, Lee KH, Cho IH, Chung MH, Kim HR et al. (2012). Interleukin-6 induces S100A9 expression in colonic epithelial cells through STAT3 activation in experimental ulcerative colitis. PLoS One 7, e38801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD (2014). A review of the epidemiology of inflammatory bowel disease with a focus on diet, infections and antibiotic exposure. Nestle Nutr Inst Workshop Ser 79, 1–18. [DOI] [PubMed] [Google Scholar]
- Litvak Y, Byndloss MX, and Baumler AJ (2018). Colonocyte metabolism shapes the gut microbiota. Science 362, pii: eaat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, and Spiller RC (2006). Functional bowel disorders. Gastroenterology 130, 1480–1491. [DOI] [PubMed] [Google Scholar]
- Lovell RM, and Ford AC (2012). Global prevalence of and risk factors for irritable bowel syndrome: a meta-analysis. Clin Gastroenterol Hepatol 10, 712–721 e714. [DOI] [PubMed] [Google Scholar]
- Malinen E, Rinttila T, Kajander K, Matto J, Kassinen A, Krogius L, Saarela M, Korpela R, and Palva A (2005). Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am J Gastroenterol 100, 373–382. [DOI] [PubMed] [Google Scholar]
- McLaughlin PJ, Jagielo-Miller JE, Plyler ES, Schutte KK, Vemuri VK, and Makriyannis A (2017). Differential effects of cannabinoid CB1 inverse agonists and antagonists on impulsivity in male Sprague Dawley rats: identification of a possibly clinically relevant vulnerability involving the serotonin 5HT1A receptor. Psychopharmacology (Berl) 234, 1029–1043. [DOI] [PubMed] [Google Scholar]
- Melchior C, Aziz M, Aubry T, Gourcerol G, Quillard M, Zalar A, Coeffier M, Dechelotte P, Leroi AM, and Ducrotte P (2017). Does calprotectin level identify a subgroup among patients suffering from irritable bowel syndrome? Results of a prospective study. United European Gastroenterol J 5, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J, Fierce Y, Treuting PM, Brabb T, and Maggio-Price L (2013). High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a−/− male mice. J Nutr 143, 1240–1247. [DOI] [PubMed] [Google Scholar]
- Patrick DL, Drossman DA, Frederick IO, DiCesare J, and Puder KL (1998). Quality of life in persons with irritable bowel syndrome: development and validation of a new measure. Dig Dis Sci 43, 400–411. [DOI] [PubMed] [Google Scholar]
- Podolsky DK (2002). Inflammatory bowel disease. N Engl J Med 347, 417–429. [DOI] [PubMed] [Google Scholar]
- Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T et al. (2009). Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigottier-Gois L (2013). Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J 7, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzatti G, Lopetuso LR, Gibiino G, Binda C, and Gasbarrini A (2017). Proteobacteria: A Common Factor in Human Diseases. Biomed Res Int 2017, 9351507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodino-Janeiro BK, Vicario M, Alonso-Cotoner C, Pascua-Garcia R, and Santos J (2018). A Review of Microbiota and Irritable Bowel Syndrome: Future in Therapies. Adv Ther 35, 289–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roediger WE (1980). The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet 2, 712–715. [DOI] [PubMed] [Google Scholar]
- Rousseaux C, El-Jamal N, Fumery M, Dubuquoy C, Romano O, Chatelain D, Langlois A, Bertin B, Buob D, Colombel JF et al. (2013). The 5-aminosalicylic acid antineoplastic effect in the intestine is mediated by PPARgamma. Carcinogenesis 34, 2580–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulfer AF, Battaglia T, Alvarez Y, Bijnens L, Ruiz VE, Ho M, Robinson S, Ward T, Cox LM, Rogers AB et al. (2018). Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat Microbiol 3, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin NR, Whon TW, and Bae JW (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33, 496–503. [DOI] [PubMed] [Google Scholar]
- Spiller R, and Lam C (2011). The shifting interface between IBS and IBD. Curr Opin Pharmacol 11, 586–592. [DOI] [PubMed] [Google Scholar]
- Spiller R, and Major G (2016). IBS and IBD - separate entities or on a spectrum? Nat Rev Gastroenterol Hepatol 13, 613–621. [DOI] [PubMed] [Google Scholar]
- Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, Stevanovic A, Veyrat-Durebex C, Tarallo V, Rigo D, Germain S et al. (2015). Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med 21, 1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada N, Ohno N, Saitoh S, and Ohno S (2007). Immunohistochemical detection of hypoxia in mouse liver tissues treated with pimonidazole using “in vivo cryotechnique”. Histochem Cell Biol 128, 253–261. [DOI] [PubMed] [Google Scholar]
- Velazquez EM, Nguyen H, Heasley KT, Saechao CH, Gil LM, Rogers AWL, Miller BM, Rolston MR, Lopez CA, Litvak Y et al. (2019). Endogenous Enterobacteriaceae underlie variation in susceptibility to Salmonella infection. Nat Microbiol. doi: 10.1038/s41564-019-0407-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, and Rensen SS (2013). Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21, E607–615. [DOI] [PubMed] [Google Scholar]
- Woo H, Okamoto S, Guiney D, Gunn JS, and Fierer J (2008). A model of Salmonella colitis with features of diarrhea in SLC11A1 wild-type mice. PLoS One 3, e1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier RJ, and Podolsky DK (2007). Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen N, Wu Z, Song Y, Zhang Y, Wu N, Zhang F, Ren X, and Liu Y (2018). 5-Aminosalicylic Acid Alters the Gut Bacterial Microbiota in Patients With Ulcerative Colitis. Front Microbiol 9, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zhou X, and Lan C (2015). Changes of cytokine levels in a mouse model of post-infectious irritable bowel syndrome. BMC Gastroenterol 15, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Kelly CJ, and Colgan SP (2015). Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol 309, C350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SY, Fleisher D, Pao LH, Li C, Winward B, and Zimmermann EM (1999). Intestinal metabolism and transport of 5-aminosalicylate. Drug Metab Dispos 27, 479–485. [PubMed] [Google Scholar]
- Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Buttner L, de Lima Romao E, Behrendt CL, Lopez CA et al. (2018). Precision editing of the gut microbiota ameliorates colitis. Nature 553, 208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Wu L, Xu W, Zhou X, Ye K, Xiong H, Song C, and Xie Y (2020). Correlation between antibiotic use in childhood and subsequent inflammatory bowel disease: a systematic review and meta-analysis. Scand J Gastroenterol 55, 301–311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene amplicon sequencing data have been deposited with links to BioProject accession number PRJNA541572 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).