

Abstract
Research in the human gut microbiome has bloomed with advances in next generation sequencing (NGS) and other high-throughput molecular profiling technologies. This has enabled the generation of multi-omics datasets which holds promises for big data–enabled knowledge acquisition in the form of understanding the normal physiological and pathological involvement of gut microbiomes. Ample evidence suggests that distinct microbial compositions in the human gut are associated with different diseases. However, the biological mechanisms underlying these associations are often unclear. There is a need to move beyond statistical associations to discover how changes in the gut microbiota mechanistically affect host physiology and disease development. This review summarises state-of-the-art big data and systems biology approaches for mechanism discovery.
Keywords: Metagenomic, Microbiome, Big data, Systems biology, Ecological modelling
Introduction
Traditional human physiology focuses on the dynamic homeostasis of human cells, human-derived molecules and their interactions. However, over the past two decades, it has become recognised that the diverse plethora of microorganisms not only reside but influence human physiology and pathology (Gilbert et al. 2018). These microorganisms include bacteria, fungi, viruses, protozoa and archaea. These microbes are highly abundant and diverse. With a focus on the human bacterial microbiome, there roughly exist a similar number of bacterial cells to human cells (3.0 × 1013) in the adult body (Sender et al. 2016). These microbes enrich the total number and diversity of protein coding genes. It has been estimated that the human microbiome contributes to more than five million genes per person (Huttenhower et al. 2012; Methé et al. 2012). With the majority of bacteria densely populated in the gut (Sender et al. 2016), these genes have been demonstrated to be integral to host physiology, with recent studies revealing their role in host metabolism (Gill et al. 2006), immune response (Chow et al. 2010) and pathophysiology leading to illnesses such as inflammatory bowel disease (IBD) (Gill et al. 2006), type-2 diabetes (Sato et al. 2014), hypertension (Yang et al. 2015; Nagao-Kitamoto et al. 2016), and autism spectrum disorder (Song et al. 2004; Adams et al. 2011). Therefore, it is important to consider the contribution of these microbial genes, pathways and cell-cell interactions when in the study of human physiology and pathophysiology.
The human gut microbial composition is highly heterogeneous between individuals. Metagenomic profiling reveals that generally the gut microbiome consists of a few highly abundance species, and a long tail of lowly abundant microbial species. No bacterial species is present universally across all individuals (Huttenhower et al. 2012), and it is difficult to define a consensus core gut microbiome (Aguirre de Cárcer 2018). This complicates the reductionist statistical association analysis between individual microbial abundance and host phenotypes. Therefore, it is informative to consider a systems biology approach which moves away from single taxa statistical association to developing holistic ecosystem models describing the functional relevance associated with each bacterial species simultaneously (van der Ark et al. 2017; Chong and Xia 2017). In this paper, we will review what is known about how the human gut microbiome affects human physiology and diseases, and the role of big data in modern gut microbiome research. We will then discuss the current research challenges and emerging solutions in this field.
What is in the gut microbiome?
The characterisation of a “core” gut microbiome becomes crucial when understanding physiological effects exerted from the human gut microbiome, and in identifying how perturbations to the microbiome may directly cause disease. However, defining such a core gut microbiome is not trivial because the exact composition of the human gut microbiome varies widely across individuals at the species level (Huttenhower et al. 2012). Longitudinal studies have observed a minority (~ 5%) to no species being common across timepoints (Caporaso et al. 2011; Halfvarson et al. 2017; Lloyd-Price et al. 2019). A cross-sectional study demonstrated only 18 species were found in all individuals from a cohort of 124 individuals, with the most common species (found in more than 90% of individuals) exhibiting large variations in their relative abundance across this cohort (2187 fold differences) (Qin et al. 2010). Despite this, the microbial composition of the gut microbiome appears uniquely individual with greater variation present in inter-individual profiles than intra-individual profiles (Huttenhower et al. 2012; Heintz-Buschart et al. 2016). Differences across individuals are also reduced within members of the same family, where family membership demonstrate stronger association to changes in the gut microbiome than other factors such as age or body mass index (Heintz-Buschart et al. 2016). This suggests individual compositional differences are possibly explained by various extrinsic and intrinsic factors such as genetics or lifestyle.
The nature of compositional change throughout time is another important feature required to be resolved when studying the gut microbiome. An individual’s gut microbiome is a dynamic system with its composition changing in response to a variety of intrinsic and extrinsic factors. The gut microbiome however demonstrates high plasticity where disturbances such as diet, infection or geographical migration can change the microbiome in a short time span (David et al. 2014a, b). Other intrinsic factors such as ageing and circadian rhythm have also been associated (Leone et al. 2015; Gilbert et al. 2018). Koeing et al. show the infant’s gut microbiome drastically and rapidly shifting over 2.5 years to attain functional characteristics found in an adult’s gut microbiome upon ingestion of solid foods (Koenig et al. 2011). Nonetheless, the overall composition is relatively stable over time (> 1 year) in comparison with microbiomes from other body sites or across individuals (Caporaso et al. 2011).
Another important characteristic of the human gut microbiome is the large number and different taxa (diversity) of microbes in each person’s gut. A high diversity of bacterial species in the gut is linked with healthy phenotypes, whereas low diversity is associated with diseases such as inflammatory bowel disease (IBD) (Ananthakrishnan et al. 2017), hypertension (Seungbum et al. 2020) and obesity (Turnbaugh et al. 2009). The mechanism underlying the association between microbial diversity and host physiology or diseases remains an active area of research.
A hypothesis is that microbial diversity is a deterrent for opportunistic and pathogenic organisms (Sokol et al. 2008). This forms the premise of faecal microbiota transplantation (FMT) (Van Nood et al. 2013). For example, FMT can be used as a successful treatment of Clostridium difficile infection as the increased diversity of commensal bacteria confer competitive growth and colonisation resistance (Khoruts and Sadowsky 2016; Ji et al. 2020). An appropriate reflection of this phenomenon is also present in ecology, in which a diverse ecosystem tends to be less susceptible to changes due to invasion (Naeem and Li 1997; Tilman et al. 2006; Dovčiak and Halpern 2010). This “insurance hypothesis” also explains the relationship between high microbial diversity and good health, whereby environmental perturbations are less likely to cause dysbiosis in a species-rich gut microbiome environment due to functional redundancy (Lozupone et al. 2012; Sommer et al. 2017). This hypothesis highlights the importance of elucidating the fundamental functional roles of species redundancy.
Another possible hypothesis is that the increase of abundance and diversity of specific metabolite-producing species can alter host physiology. Zhao et al. (2018) recently showed that increasing abundance and diversity of specific bacteria that produce short-chain fatty acids (SCFAs), promoted by high-fibre diet, can improve blood-glucose regulation (Zhao et al. 2018). This work suggests that targeted restoration of specific metabolite-producing gut bacteria may be a possible mechanism in which a diverse gut microbiome can affect host physiology.
Gut microbiome and host physiology
A possible mechanism that explains the gut microbiome’s influence on host physiology is through its production of secondary breakdown metabolites via the digestion of dietary fibres (Koenig et al. 2011; Oliphant and Allen-Vercoe 2019). These breakdown products such as acetate, ethanol, butyrate and lactate can directly act on the gut or permeate through the intestinal epithelial lining and enter the blood circulation, which allows interaction with various organs in the body (Turner 2009) (Fig. 1). This mechanism is regulated by the selectively permeable nature of the intestinal tract, with multiple factors contributing to the active absorption of dietary nutrients whilst maintaining a tight barrier to prevent the entry of microorganisms or microorganism-derived compounds which can adversely influence our health (Assimakopoulos et al. 2018). Host-derived mechanisms include the presence of mucus glycoproteins which form a physical barrier between the intestinal lumen (Turner 2009) and its underlying epithelial layer. Also, host immunological defences such as the secretion of immunoglobulin A block specific bacterial epitopes preventing binding and motility on the epithelial cell (Mantis et al. 2011). The regulation of these mechanisms is tightly associated with the microbial composition. An example of a positive association includes commensal bacteria eliciting epithelial protection and homeostasis through Toll-like receptor (TLR)–mediated host response (Rakoff-Nahoum et al. 2004). An adverse factor includes the reduction of Akkermansia spp. composition, which has been associated with the weakening of the mucosal layer leading to inflammation, higher susceptibility to infection and gut “leakiness” (Grander et al. 2018).
Fig. 1.

A summary of known host-microbial interactions at the gut epithelial lining, illustrating the complex dynamic between disease-associated pathways (right) and interactions promoting gut health (left). TLR Toll-like receptor. IL-10 interleukin-10. SCFA short-chain fatty acid. IBD inflammatory bowel disease. TMA trimethylamine. TMAO trimethylamine N-oxide. CVD cardiovascular disease. Bacteria are labelled in purple, and metabolites/proteins are labelled in turquoise
Bacterial metabolites can also elicit both negative and positive impacts on gut and systemic health. Trimethylamine (TMA) is produced by various gut bacteria from dietary amines, commonly found in animal-heavy diets (Rath et al. 2017). They absorb and move through the portal vein to the liver, which has been implicated in the onset of hepatic inflammation (Tripathi et al. 2018). TMA can be further oxidised in the liver to form trimethylamine N-oxide (TMAO) where increased levels of blood plasma TMAO is also largely associated with increased cardiovascular risk and accelerated atherosclerosis development (Roncal et al. 2019). The level of TMAO can be associated with the activity of specific microbial species (Koeth et al. 2013; Chen et al. 2016b; Al-Obaide et al. 2017) however is also highly associated with the intake of diets high in choline and l-carnitine (Koeth et al. 2013; Velasquez et al. 2016; Park et al. 2019).
Ethanol, another metabolite highly associated with human health, easily enters the blood stream by diffusion from the gut. It can be derived directly from exogenous intake of alcohol however can also be endogenously produced via fermentation of carbohydrates by gut bacteria such as Escherichia coli. Enterobacteriaceae such as E. coli can also further oxidise ethanol into acetaldehyde (Jokelainen et al. 1996) which has been implicated in the compromise of the gut barrier (Elamin et al. 2013), leading to increased translocation of bacterial metabolites and endotoxins into the blood (Bjarnason et al. 1984; Yan and Schnabl 2012), triggering inflammatory responses and eliciting immune responses (Ferrier et al. 2006). High levels of circulating blood ethanol and acetaldehyde and impaired gut barrier have been associated with liver disease (Zhu et al. 2013; López-Lázaro 2016).
Some microbe-derived gut metabolites also confer protective characteristics to the gut. Short-chain fatty acids (SCFAs), mainly butyrate, acetate and propionate, induce immune regulation (Corrêa-Oliveira et al. 2016) and anti-inflammatory effects (Vinolo et al. 2011) and strengthen epithelial lining tight junctions to prevent the entry of microorganisms (Ohata et al. 2005), as well as act as a primary energy source for colonocytes (Donohoe et al. 2011). The importance of butyrate-producing bacteria has also been mechanistically linked to the activity of intracellular butyrate sensor, peroxisome proliferator–activated receptor γ (PPAR-γ), which promotes colonocyte energy metabolism through β-oxidation allowing a selective hypoxic environment for SFCA-producing obligate anaerobes whilst limiting the expansion of opportunistic/pathogenic species such as Escherichia and Salmonella (Byndloss et al. 2017).
Probiotic bacteria such as the lactobacillus genus also confer beneficial impacts on gut health via several mechanisms. These include improving gut membrane integrity, preventing pathogenic infection (Mangell et al. 2002; Moorthy et al. 2009) and the production of antimicrobial metabolites such as reuterin (Cadieux et al. 2008) and lactic acid (Greifová et al. 2017) which are strong deterrents of opportunistic pathogens such as Salmonella (Abhisingha et al. 2018). Lactobacillus also demonstrate high acetaldehyde-metabolizing capacity (Nosova et al. 2000) which may assist reduce acetaldehyde’s damaging properties. The balance and dysbiosis of microbial flora and their metabolite products therefore become extremely important when considering the pathogenesis of diseases.
It should also be highlighted that the gut microbiome is also involved in the metabolism of drugs. An example includes metronidazole, an antibacterial agent against anaerobic infections (Löfmark et al. 2010). The activity of this drug involves both side-chain oxidation in the liver and ring-cleavage by Clostridium perfringens (Koch and Goldman 1979). On the other side, the gut microbiota can also sequester active drugs through its metabolism, such as dopaminergic drug levodopa, which can be sequestered by gut microbiota-induced decarboxylation (Goldin et al. 1973). For these reasons, the therapeutic response and efficacy of drug usage may be dependent on the unique profile of an individual’s gut microbiota.
The role of big data in gut microbiome analysis
A holistic systems biology approach to scrutinise the human gut microbiome was really only plausible with the development of next generation sequencing (NGS) which allowed the unbiased culture-independent screening of microbiomes (Gu et al. 2019). A pioneering study in 2004 led by J. Craig Venter characterised over one million novel genes and 148 novel bacterial phylotypes in the Sargasso Sea Metagenomic Survey, which considerably extended the breadth of existing protein database sequences by 10 fold (Venter et al. 2004). In 2007, the human microbiome project (HMP) pioneered human metagenomics as a collaborative effort to coordinate and facilitate multiple large-scale high-throughput human microbiome projects, setting the standard and direction of human microbiome research world-wide (Turnbaugh et al. 2007). The HMP successfully contributed to the expansion of reference microbial genome libraries, the development of bioinformatic tools (DeSantis et al. 2006; Lozupone et al. 2006) for short read assignment, the analysis of complex multivariate datasets and discovery of functional associations between the microbiome and host phenotype through comparative metagenomics (Gill et al. 2006; Turnbaugh et al. 2006). Although the primary framework of HMP focussed on elucidating the baseline microbiota of healthy individuals, it concluded that compositional data of the microbiome alone poorly correlate with host phenotypic metadata (Huttenhower et al. 2012). On the other hand, metabolic and functional pathway annotations predicted from metagenomic data are less variable and sporadic in cross-sectional surveys and associate better with host phenotypes (Morgan et al. 2012; Huttenhower et al. 2012).
A second phase of the HMP, the integrated Human Microbiome Project (iHMP), addressed many questions raised from the first and drew focus on the integration of various omics data across large and longitudinal cohorts. This demonstrated powerful capabilities in predicting individual risk for preterm birth (Fettweis et al. 2019), constructing large molecular interaction networks regarding IBD (Lloyd-Price et al. 2019), and the early detection of disease states such as type-2 diabetes (Zhou et al. 2019), metabolic disease, cardiovascular disease or cancers (Schüssler-Fiorenza Rose et al. 2019). These studies based their findings on multiple profiling technologies such as stool metagenomes, metatranscriptomes, metaproteomes, metabolomes, host transcriptomes, exomes, epigenomes, serological profiles and specific clinical laboratory measures such as oral glucose tolerance tests for diabetes studies (Proctor et al. 2019). The longitudinal aspect of these studies allowed the assessment of intra- and inter-individual variations, as well as defining changes from baseline compositions, the first temporal or healthy state measurement, in relation to clinical measurements regarding disease activity.
These multi-omics big data projects have identified many statistical associations between microbial compositions and diseases. Nonetheless, it still remains a challenge to discover the causal cellular and molecular mechanisms that underlie these statistical associations. Some important questions that need to be addressed include the importance of bacterial-bacterial interaction dynamics, the influence of demographic covariates such as ethnicity, diet and age, the direction of association and whether associations are sufficient or necessary for observed phenotypes. In the following sections, we will discuss some challenges and emerging solutions related to experimental designs and bioinformatics analysis that can be employed to human gut microbiome research.
Challenge 1: Multi-omic profiling does not necessarily provide mechanistic insight
The concept of integrative data analysis was recognised as a central stepping stone to move towards mechanistic inference with the close of the HMP. Performing integrative analysis requires adequate planning corresponding to the biological question to be investigated. This regards the type of host and microbial measurements collected, the type of samples used for omics measurement, cohort size and distribution and whether a longitudinal study design is required.
A cohort study in metabolic syndrome patients assesses the impact of fasting on the gut microbiome and blood pressure. The host immunome was utilised as an intermediate to associate changes in taxonomic/functional profiles with clinical metrics such as systolic blood pressure (Balogh et al. 2020). Through pairwise spearman correlation, network analysis identified clusters of cytokine-producing mucosa-associated invariant (MAIT) cells positively correlated with 24 h ambulatory systolic blood pressure and IL-2+TNFα+-producing MAIT cells with changes in Firmicutes and Bifidobacteriales abundances positively and negatively, respectively. This is significant as immune MAIT cells have not previously been associated with blood pressure, unlike other common T cells. Although no further mechanistic hypothesis was provided to link this correlation, it highlights the potential involvement the gut microbiome has with blood pressure and metabolic syndrome. A network diagram assisted visualise covarying groups across blood pressure measurements (systolic/diastolic blood pressure and mean arterial pressure), immune cell features and microbiome functional and taxonomic features, although is hard to interpret without hypothesis-driven analysis.
A study on pulmonary arterial hypertension (PAH) infers many functional mechanistic associations between the gut microbiome and PAH from shotgun metagenomics alone (Seungbum et al. 2020). This was achieved by synergistically assessing taxonomic and functional profiles, generated from KEGGs ontology databases. Predicting functional profiles from the metagenome have previously been demonstrated to be inconsistent in comparison with functional profiles predicted from metatranscriptome abundances (Heintz-Buschart et al. 2016). Metagenomics generally only represent a qualitative view of present genes but does not provide substantial quantitative resolution of their expression. This subsequently can only provide a probabilistic view of all potentially existing functional pathways. Despite this, through thorough validation of past literature, highly probable associations can still be enriched and suggestive in the new pathological context of PAH. The identified increase of Collinsella in the PAH cohort has been previously linked to increased gut permeability and thus inflammation and pathogenesis (Chen et al. 2016a). Functionally, Collinsella taxa contributes the most to the increase in genes for proline biosynthesis highlighting proline’s possible involvement in a metabolic pathway contributing to increased blood pressure. Collinsella along with other TMAO producers were found enriched in PAH microbiomes, with negatively correlated bacteria with TMAO being enriched in the reference cohort. This supports prior literature associating TMAO to cardiovascular disease risk (Roncal et al. 2019). In this study, the addition of the metatranscriptome would support enrich TMAO metabolism in PAH microbiomes; however, the challenge to establish new mechanistic relationships between the gut microbiome and PAH remains.
A major challenge in experimentally testing the causal relationship between specific gut microbial species and phenotype is that animal studies using germ-free animal are experimentally challenging, and it is uncertain whether results from animal models may be truly representative of human biology (Walter et al. 2020).
Challenge 2: Metagenomic microbial dark matter
Microbial “dark matter” can be defined as unannotated or under-characterised but annotated microbial species, which presumably are associated with species that are present at low abundance. Since the HMP, the goal to establish annotated bacterial genome libraries was a priority. This still remains a challenge in metagenomic analysis with the majority of molecular operational taxonomic units (mOTUs) originating from unannotated/unidentified species (Panek et al. 2018).
A cohort study assessing the link between familial type 1 diabetes and the gut microbiome utilises integrative multi-omics analysis by co-assembling metagenomic and metatranscriptomic raw reads through de novo genome assembly (Heintz-Buschart et al. 2016). This resulted in the generation of longer contigs and increased read usage in comparison to assembly based on the metagenome or metatranscriptome alone. They also demonstrated ~ 43% of detected microorganisms did not belong to mOTUs with sequenced isolate genomes, and a relative abundance of 9% unassigned at the phylum level (Heintz-Buschart et al. 2016). This de novo assemble approach enhances read usage to ~ 88%, although de novo methods are still inherently biased towards the most abundant species (Almeida et al. 2019). A recent study performing large-scale discovery of uncultured species generated 92,143 metagenome-assembled genomes (MAGs) from 11,850 human gut microbiomes. A total of 1952 where identified as uncultured when aligning with UniProtKB at a species level threshold. Furthermore, 26% of unclassified metagenomic species remained unassigned at the family level suggesting a sizeable proportion of isolates may belong to new families (Almeida et al. 2019).
The long tail of lowly abundant species found in metagenomic studies also remains uncharacterised at the functional level. This is a consistent feature of the gut microbiome, with only several species dominating the total composition. Many fundamental analysis methods will look at changes in dominant taxonomies such as the Firmicutes/Bacteroidetes ratio; however, the collective change in this tail may hold clues to important regulatory roles in the gut microbiome and host physiology. One approach that bypasses the statistical challenges faced when assessing changes of extremely small abundance quantification is to focus on the functional capability of the gut microbiome. This is generally enabled with metatranscriptomics, metaproteomics and metabolomics.
Challenge 3: Stool sample as a proxy for the gut microbiome
All gut metagenomic studies utilising stool samples as a proxy for the gut microbiome make the integral assumption that the measured microbial composition is similar or similar enough to the true environment of the gut. In this practice, we assume stool samples hold enough resolution to retain meaningful features of the gut microbiome. This however is a naïve approach and can be limiting when resolution is needed to distinguish changes in lowly expressed species or capturing subtle relationships with small effect size. Certain variables arising from stool sampling such as stool consistency have demonstrated the overrepresentation and underrepresentation of certain enterotypes. Harder stool is dominated by Ruminococcaceae-Bacteroides (RB) enterotype whilst softer stool samples reveal enrichment of Prevotella (Vandeputte et al. 2016).
A study of the chicken gut microbiota investigated the efficacy of using faecal samples as a proxy for the gut microbiota by comparing microbial diversity and abundance to metagenomic profiles generated from respective mucosal samples of the small intestine and cecum (Yan et al. 2019). Principal coordinates analysis (PCoA) on weighted/unweighted UniFrac distances revealed significant disparity between faecal communities to communities from other regions of the gut. The authors comment that the microbial community in faeces reflect an intermediate state between the ceca and small intestine. On comparison of observed OTUs across sampled sites, only 30.9% of total OTUs were present across all sites; however, it is promising to see that 96.5% of the faecal community consisted of this “core” microbiota. This could help improve the interpretation of stool samples as a representation of the intra-individual core microbiome.
A recent pilot study employs the use of 16S rRNA sequencing of the gut microbiome and metabolome from mucosal samples rather than stool samples (Wang 2020). The comparison of tumour tissue and adjacent normal tissue revealed differentially expressed compositions of bacteria with taxa’s such as Fusobacterium enriched in tumour tissue and whilst Lactobacillus, a probiotic family of bacteria, was reduced. Fusobacterium have previously been identified as a pathogenic gut microorganism (Mima et al. 2016). The overlay of metabolomic data revealed negative correlations with 4-HB abundance, a hydroxylated derivative of butyrate. This suggests Fusobacterium as an inhibitor of butyrate synthesis and the mechanistic driver for CRC, as butyrate confers links to anti-carcinogenic and anti-inflammatory properties (O’Keefe 2016).
Furthermore, associations with the epigenome reveal Fusobacterium’s correlations with DNA methylation status of the genes LSP1, FCRLA and PI16 which have previously been identified to contribute to cell apoptosis (Stone et al. 2015), mucosal immune response (Zhang et al. 2017) and decreased expression in colon cancer (Li et al. 2012), respectively. This hypothesises Fusobacterium’s inhibitory effect on butyrate synthesis prevents butyrate’s ability to regulate the DNA methylation of genes implicated in pathways relevant to CRC. The significant differential feature identification between normal and tumour tissue from the same patient suggests the importance of sample locality and how microenvironments of the gut microbiome exhibit different phenotypes/pathway behaviour. The use of stool samples would not be able to discern such differential associations at different sites in the gut. This highlights metagenomes, metatranscriptomes or metabolomes measured from stool samples will average microenvironment differences and will be insensitive to subtle changes relevant to disease. This is supported in iHMP’s IBD cohort study where taxonomic profiles only attained 1.9% (insignificant) of variation between disease states, and 16S biopsies attained 2.9% (significant, P ≤ 0.001) of variation for explaining disease states (Lloyd-Price et al. 2019).
Challenge 4: Uncertainty of microbiome abundance quantification and normalisation
The statistical analysis of metagenomic data inherently holds a caveat due to the inability to deduce the absolute quantification of microbial composition where the majority of quantitative analysis suffices with relative abundance. Performing differential abundance analysis using composition data under a sum 1-simplex has demonstrated to yield spurious results and false positive (Calle 2019). Even though read counts assigned to individual bacterial taxa are represented in Euclidean space, the true interpretation of these numbers is hard to assign as metagenomic samples represent a sub-population of the true environment. Especially in the case of stool samples, how accurately does the excreted microbial population represent the true gut microbiome must be questioned. In addition, which microenvironment of the gastrointestinal tract does it represent and if this location is relevant to the disease or phenotype of interest must all be considered.
A study in 2005 by Eckburg demonstrated through culture-independent analysis that the microbiome associated with the mucosa of the large intestine was significantly different to stool samples from the same individual (Eckburg et al. 2005). Although they did not investigate whether this difference was biologically significant, it would be unreasonable to interpret each read count as an individual bacterial organism, even in 16S long read sequencing where quantification is relatively more accurate than shotgun sequencing due to mOTU identification through long read alignment.
Standard normalisation methods such as, CSS, edgeR-TMM and DESeq-VS also demonstrate to poorly correlate number of reads and mean Bray-Curtis dissimilarity (BC) in samples, as well as BC between original community data and normalised data and resulted in spurious correlations. It was identified through simulated data that only normalising through proportions or rarefying achieved standardisation of read depth between samples, and subsequently acquired the highest BC scores, PCoAs and PERMANOVA results when comparing with the gold standard (McKnight et al. 2019). Although these insights should be reproduced in biological gold standard analysis, it raises the question of whether the interpretation of multi-omics quantitative data is valid, especially with the integrative effort for association.
Morton et al. demonstrate through the use of reference frames that direct statistical comparison between samples is not valid when utilising the compositional relative abundance of species. The bias introduced from differences in microbial load results in both false positives and negatives, as illustrated through a simulated dataset (Morton et al. 2019). By illustrating the idea of differential ranks, it is highlighted that statistical analysis on relative abundances accurately reflects results from analysis on absolute abundances. The process of selecting the right reference frame for differential comparison still remains an issue, as well as the fact that all conclusions are still relative and does not circumvent the issue of microbial load. Despite this, this method provides an intuitive and accurate interpretation of composition data and can prevent spurious conclusions.
Solution 1: Strategic integrative multi-omics
As described, “integrated multi-omics” studies of the human microbiome generally only systematically assess different modes of data and make inferences across two sets of data measurements, rather than simultaneously explore omics datasets as one integrated system. Although this generally helps demonstrate the reliability and consistency of identified associations which confers greater statistical power and confidence, it fails to grasp the possibility to identify subtle multivariate interactions that may not surpass statistical significance when univariately compared, or interactions that are dependent on multiple binary features (for example disease onset when components A, B and C are present beyond a threshold and when components E and D are not).
The multi-omics study of the gut microbial ecosystem in IBD as a part of the iHMP attempts to conduct this simultaneous integrative analysis by modelling an association map of host-microbiome interactions across 10 microbiome measurements (Lloyd-Price et al. 2019). The covariance of components across measurements was identified via a mixed-effects model which determines differential abundances though residualizing measurements whilst removing and controlling for patient-specific random effects and adjusting covariates: age, sex, diagnosis, dysbiosis status, antibiotic/immunosuppressant use and bowel surgery status. This in theory allows correlations between features to be assessed independent of inter-individual variation, and thus more accurately reflect a natural biological state and possible mechanism.
Through the visualisation as a network graph, tightly interconnected hubs and their associated components can be marked as potential mechanistic drivers of a dysbiosis state or clinical targets for diagnosis or treatment. This uncovered strong associations between F. prausnitzii abundances with functional profiles down-regulated in dysbiosis, and E. coli with upregulated functional profiles. Associations between metabolites, acylcarnitines and bile acids with multiple species associated with dysbiosis were also identified as hubs in the network. Although direct mechanistic pathway or the direction of causality between IBD and gut dysbiosis cannot be concluded through this association map, the characterisation of all significant connections exhaustively will help build on our understanding of the gut microbiome at a systems level.
A recent study strategically utilises an integrative multi-omics platform in an antibiotic perturbation experimental design (Kappel et al. 2020). Associated data collected included clinical measurements, serum metabolome, stool 16S metagenome, and the colon transcriptome. This allowed the identification of metabolic pathways responsible for antibiotic-mediated atherosclerosis. Weighted correlation network analysis of serum metabolites revealed 15 clusters of which 10 where highly impacted by antibiotic treatment. By correlating metabolite clusters to lesion size data, five clusters relating to tryptophan, lipid and secondary bile acid metabolism were isolated to be associated with atherosclerosis. These changes in metabolic pathways were then validated by predicting metabolic pathways using 16S microbial profiles. Similar to the IBD study, an integrative cross-omics model was developed to link serum metabolome with the mOTUs. The model hypotheses Clostridia, Lachnospiraceae and Ruminococcaceae as key classes associated with antibiotic-induced atherosclerosis, whilst also observing decreased tryptophan metabolites. The relationship was also evident through tryptophan supplementation which reverses antibiotic-induced atherosclerosis phenotype. This study design will thus support future biological follow-up studies to consolidate mechanism in a hypothesis-driven manner (Fig. 2).
Fig. 2.

Strategic integrative multi-omics pipeline. Multi-omics experimental design leverage existing literature such as pilot studies to determine cofounding factors, cohort selection and multi-omic measurements. Integration of multi-omics data can be performed through a network-based representation of association. The network structure can be used in conjunction with various public big data to identify high-priority hypothesis for molecular mechanism underlying the microbiome’s influence on host physiology
Solution 2: Harnessing intra-individual variability through longitudinal design
Large inter-individual variability poses challenges to standard case-control association studies. Longitudinal studies can bypass this limitation though the use of baseline compositional profiles as a means to control for inter-individual variability and enable intra-individual comparison. This process provides increased statistical power in case-control comparative analysis. Temporal changes in a study of IBD patients demonstrated microbiome compositional changes towards baseline characteristics with the inactivity of IBD (Lloyd-Price et al. 2019). It was also observed that the gut microbiome of IBD patients was more susceptible to compositional shifts than healthy individuals over time, where temporal changes of the gut microbiome in healthy individuals were rarely observed. The magnitude of variation in microbiome and immune response over the time-course of disease is also shown to be greater than clinical phenotypes cross-sectionally. This infers significant associations relevant to disease status are more detectable in longitudinal data due to larger comparable effect sizes (Morgan et al. 2012; Gevers et al. 2014). These observations may suggest difficulty or redundancy in directly comparing metagenomic profiles of the gut to derive universally applicable associations, although methods for co-variate adjustment can be performed. This strategy therefore addresses Challenge 4, where increased confidence in microbial abundance estimation and normalisation can be made through the comparison of longitudinal profiles of the same individual.
Solution 3: Bypassing microbial dark matter with functional profiles
Harnessing the increasingly comprehensive knowledge-base of pathways of many gut bacteria, we may be able to perform functional molecule profile analysis of the microbial community without needing to know the specific functional characteristic of each individual taxa. By doing so, this bypasses the challenge of microbial dark matter as discussed above (Challenge 2). Differentially expressed transcripts in TD1M individuals revealed functional differences in microbial cellulose usage and immune status (Heintz-Buschart et al. 2016). This was reflected in the metaproteome with proteins of similar functional group being the most differentially expressed. Microbial taxa exhibited these functional changes at the transcript and protein level however demonstrated no change in compositional abundance. This is consistent with iHMP’s IBD cohort study which revealed no significant change in bacterial species between case and control, but differences were identified at the metabolome level (Lloyd-Price et al. 2019). This suggests that metagenomic data alone can be uninformative when determining differences across disease states. The comparison of metagenome and metatranscriptome also revealed transcript abundance cannot be inferred from metagenomic data with weak correlation between metagenomic and metatranscriptomic depths of coverage for all identified ORFs (Heintz-Buschart et al. 2016). This suggests that studies that predict functional annotations exclusively from shotgun profiled metagenomics can result in spurious results. This was also true, with no correlation, when comparing functional profiles of the metagenome and metaproteome, whereas protein abundances exhibited greater association with transcript abundances (Heintz-Buschart et al. 2016). This suggests the possibility to extrapolate the metaproteome with metatranscriptomic data alone. Moreover, mOTU abundance profiles mapped from the metatranscriptome contain an almost identical set of mOTUs to mOTUs mapped from the metagenome. mOTUs also demonstrated strong quantification correlation between the metagenome and metatranscriptome. Given this, metatranscriptomics may be a good source for estimation of both metagenomic and metaproteomic data.
A study identifying early colorectal cancer pathogenesis demonstrates the efficacy of deriving functional insight from the metagenome and metabolome (Kim et al. 2020). The metabolome and metagenome demonstrated correlation between their first principle components through principal component analysis (PCA). Specific bacterial taxa and metabolites which contributed to this correlation were deduced through Spearman correlation between genus-level abundances and metabolic pathways. This revealed several genera from the Firmicute phylum and Actinobacteria phylum correlating negatively to more than ten pathways. These trends were also consistent when correlating microbial compositions directly with metabolite abundances. This contrasts previously mentioned findings that demonstrate metagenomics to poorly infer functional relevance. Despite this, the metabolome is known to couple tightly with diet which also strongly determines the gut microbiome composition (McHardy et al. 2013; Singh et al. 2017). Correlation may have been successful due to the use of principle components rather than linear regression on raw quantification. Regardless, this study still utilises the metabolome, rather than metagenome, to predict pathway-level profiles. The grouping of metabolites into functional pathways also provides a more intuitive understanding of their biological activity and role in physiology, thus making the link to disease clearer and parsimonious.
Solution 4: Modelling the gut microbiome as a dynamic ecosystem
The large population of gut microbes are subjected to competition for survival due to limited resources and spatial environment within the human gut. An ecological perspective of the gut microbiome moves the focus of how the gut microbiome influences the host to the interactions among host-bacteria and bacteria-bacteria. Foster describes the gut microbiome as an “ecosystem on a leash”, highlighting the complex dynamics of evolving microbe-microbe interactions bounded by evolving host-microbe interactions (Foster et al. 2017) (Fig. 3). The symbiotic evolution of bacterial communities with hosts and non-living environments has long pre-existed modern life. This allows highly adapted and evolved microbial communities to exist across environments which most likely represent mutualistic dependence necessary for their survival (Woese 2002). Therefore, the comparative analysis of these long evolved interaction observed in other environments may provide fundamental clues about how the gut microbiome form functional characteristics and respond to environmental pressures such as pH, salinity and oxidative stress (Lozupone et al. 2007; Lozupone and Knight 2007).
Fig. 3.
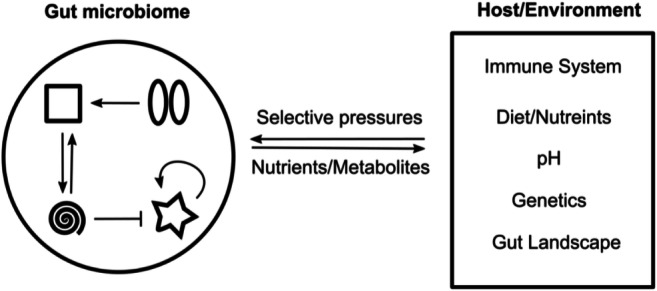
Modelling the gut microbiome as a dynamic ecosystem. The compositional state of the gut microbiome can be represented by three types of interactions: (1) host pressure that selects for microbes that are advantageous for survival, (2) the microbiome’s influence on the host via production of metabolites and digested compounds from the diet and (3) bacteria to bacteria interactions that include ecological relationships such as competition, commensalism, and mutualism
The initial bacterial colonisation of the gastrointestinal tract is thought to begin at birth and is influenced by milking methods as well as the method for delivery (Rinninella et al. 2019). The subsequence evolution of the gut microbiome through human development and growth displays macro-ecological characteristics seen in other ecological and economic systems (Ji et al. 2020). Both short- and long-term changes are found to follow robust quantitative scaling laws such as the Laplace distribution describing the short-term probability of change in species abundance (Ji et al. 2020). Diet also affected this phenomenon with smaller short-term variability in higher abundance species in low fat diets, and loss of niche diversity in high fat diets. Their comparative resemblance to other macro-ecological systems allows us to potentially adopt existing ecological models and analytical techniques to the analysis of the gut microbiome.
With this framework, a systems biology modelling approach can be taken by modelling three classes of interactions, host-bacteria, bacteria-host and bacteria-bacteria, in a stochastic process. Currently, such models exist as genome-scale metabolic models (GEMs) which are stoichiometric reconstructions of entire metabolic networks which can also include various data types such as gene, protein and metabolite data (Nielsen 2017; Altay et al. 2019). This can give rise to important observations such as covarying species, or the formation of metabolic functions (van der Ark et al. 2017; Das et al. 2019; Heinken et al. 2019). In order to accurately model this system and apply ecology theory, detailed and wholistic understanding of these three classes of interactions are required (Coyte et al. 2015), which can be enabled through integrative multi-omics (Hornung et al. 2018).
The hopes to accurately model an entire microbiome community within a GEM however face several complications other than phenotype prediction. As described by van der Ark et al., the construction of GEMs assumes a steady-state system where the rate of metabolite exchange reaches a constant (van der Ark et al. 2017). This neglects the dynamic nature of interaction within a real microbiome. In addition, the computational initiation of a species’ metabolic goal (i.e. optimisation function) will not hold true in the presence of multiple species with various ecological relationships i.e. symbionts and competition. A potential solution which may help reflect the accuracy of GEMs is through the use of gnotobiotic animals where monitoring controlled perturbations within an in vivo system would be a powerful tool for validation (Sartor 2018).
Utilising this systems biology approach to model the microbiome thus can draw potential in characterising functional annotations (Challenge 2) as well as derive insight into mechanistic pathways (Challenge 1).
Conclusion
This review summaries the limitation of traditional metagenomic studies and analytical approaches. A systems biology approach that involves well-designed multi-omics experiments, proper modelling and a machine learning–enabled analysis pipeline that fully utilises prior biological knowledge and existing published data sets is critical to discover cellular and molecular mechanisms underlying various observed statistical associations between microbial compositions and human phenotypes and diseases. We hope such systems biology approaches will drive the next wave of integrative microbiome studies.
Funding information
The work was supported in part by a Hong Kong PhD Fellowship and the Hong Kong Jockey Club Charities Trust.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abhisingha M, Dumnil J, Pitaksutheepong C. Selection of potential probiotic Lactobacillus with inhibitory activity against Salmonella and fecal coliform bacteria. Probiotics Antimicrob Proteins. 2018;10:218–227. doi: 10.1007/s12602-017-9304-8. [DOI] [PubMed] [Google Scholar]
- Adams JB, Johansen LJ, Powell LD, et al. Gastrointestinal flora and gastrointestinal status in children with autism - comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011;11:22. doi: 10.1186/1471-230X-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre de Cárcer D. The human gut pan-microbiome presents a compositional core formed by discrete phylogenetic units. Sci Rep. 2018;8:1–8. doi: 10.1038/s41598-018-32221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Mitchell AL, Boland M, et al. A new genomic blueprint of the human gut microbiota. Nature. 2019;568:499–504. doi: 10.1038/s41586-019-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Obaide M, Singh R, Datta P, et al. Gut microbiota-dependent trimethylamine-N-oxide and serum biomarkers in patients with T2DM and advanced CKD. J Clin Med. 2017;6:86. doi: 10.3390/jcm6090086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altay O, Nielsen J, Uhlen M, et al. Systems biology perspective for studying the gut microbiota in human physiology and liver diseases. EBioMedicine. 2019;49:364–373. doi: 10.1016/j.ebiom.2019.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan AN, Luo C, Yajnik V, et al. Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel diseases. Cell Host Microbe. 2017;21:603–610.e3. doi: 10.1016/j.chom.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimakopoulos SF, Triantos C, Maroulis I, Gogos C. The role of the gut barrier function in health and disease. Gastroenterol Res. 2018;11:261–263. doi: 10.14740/gr1053w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh A, Bartolomaeus H, Löber U et al (2020) Fasting alters the gut microbiome with sustained blood pressure and body weight reduction in metabolic syndrome patients. Medrvix:1–32. 10.1101/2020.02.23.20027029
- Bjarnason I, Ward K, Peters TJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;323:179–182. doi: 10.1016/S0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- Byndloss MX, Olsan EE, Rivera-Chávez F, et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357:570–575. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadieux P, Wind A, Sommer P, et al (2008) Evaluation of reuterin production in urogenital probiotic Lactobacillus reuteri RC-14. In: Applied and Environmental Microbiology. American Society for Microbiology, pp 4645–4649 [DOI] [PMC free article] [PubMed]
- Calle ML. Statistical analysis of metagenomics data. Genomics Inform. 2019;17:e6. doi: 10.5808/gi.2019.17.1.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wright K, Davis JM, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8:43. doi: 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Yi L, Zhang Y et al (2016b) Resveratrol attenuates trimethylamine-N-oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. MBio 7. 10.1128/mBio.02210-15 [DOI] [PMC free article] [PubMed]
- Chong J, Xia J (2017) Computational approaches for integrative analysis of the metabolome and microbiome. Metabolites 7 [DOI] [PMC free article] [PubMed]
- Chow J, Lee SM, Shen Y et al (2010) Host-bacterial symbiosis in health and disease. 10.1016/B978-0-12-381300-8.00008-3 [DOI] [PMC free article] [PubMed]
- Corrêa-Oliveira R, Fachi JL, Vieira A, et al. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol. 2016;5:e73. doi: 10.1038/cti.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- Das P, Babaei P, Nielsen J. Metagenomic analysis of microbe-mediated vitamin metabolism in the human gut microbiome. BMC Genomics. 2019;20:208. doi: 10.1186/s12864-019-5591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014;15:R89. doi: 10.1186/gb-2014-15-7-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Keller K, et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34:W394–W399. doi: 10.1093/nar/gkl244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Garge N, Zhang X, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovčiak M, Halpern CB. Positive diversity-stability relationships in forest herb populations during four decades of community assembly. Ecol Lett. 2010;13:1300–1309. doi: 10.1111/j.1461-0248.2010.01524.x. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, et al (2005) Diversity of the human intestinal microbial flora. Science (80- ) 308:1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed]
- Elamin EE, Masclee AA, Dekker J, Jonkers DM. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr Rev. 2013;71:483–499. doi: 10.1111/nure.12027. [DOI] [PubMed] [Google Scholar]
- Ferrier L, Bérard F, Debrauwer L, et al. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettweis JM, Serrano MG, Brooks JP, et al. The vaginal microbiome and preterm birth. Nat Med. 2019;25:1012–1021. doi: 10.1038/s41591-019-0450-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. The evolution of the host microbiome as an ecosystem on a leash. Nature. 2017;548:43–51. doi: 10.1038/nature23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naïve microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Blaser MJ, Caporaso JG, et al. Current understanding of the human microbiome. Nat Med. 2018;24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin BR, Peppercorn MA, Goldman P. Contributions of host and intestinal microflora in the metabolism of L dopa by the rat. J Pharmacol Exp Ther. 1973;86:160–166. [PubMed] [Google Scholar]
- Grander C, Adolph TE, Wieser V, et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2018;67:892–902. doi: 10.1136/gutjnl-2016-313432. [DOI] [PubMed] [Google Scholar]
- Greifová G, Májeková H, Greif G, et al. Analysis of antimicrobial and immunomodulatory substances produced by heterofermentative Lactobacillus reuteri. Folia Microbiol (Praha) 2017;62:515–524. doi: 10.1007/s12223-017-0524-9. [DOI] [PubMed] [Google Scholar]
- Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol Mech Dis. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfvarson J, Brislawn CJ, Lamendella R et al (2017) Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2. 10.1038/nmicrobiol.2017.4 [DOI] [PMC free article] [PubMed]
- Heinken A, Ravcheev DA, Baldini F, et al. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome. 2019;7:75. doi: 10.1186/s40168-019-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz-Buschart A, May P, Laczny CC et al (2016) Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat Microbiol 2. 10.1038/nmicrobiol.2016.180 [DOI] [PubMed]
- Hornung B, Martins dos Santos VAP, Smidt H, Schaap PJ. Studying microbial functionality within the gut ecosystem by systems biology. Genes Nutr. 2018;13:5. doi: 10.1186/s12263-018-0594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji BW, Sheth RU, Dixit PD, et al. Macroecological dynamics of gut microbiota. Nat Microbiol. 2020;5:768–775. doi: 10.1038/s41564-020-0685-1. [DOI] [PubMed] [Google Scholar]
- Jokelainen K, Siitonen A, Jousimies-Somer H, et al. In vitro alcohol dehydrogenase-mediated acetaldehyde production by aerobic bacteria representing the normal colonic flora in man. Alcohol Clin Exp Res. 1996;20:967–972. doi: 10.1111/j.1530-0277.1996.tb01932.x. [DOI] [PubMed] [Google Scholar]
- Kappel BA, De Angelis L, Heiser M, et al. Cross-omics analysis revealed gut microbiome-related metabolic pathways underlying atherosclerosis development after antibiotics treatment. Mol Metab. 2020;36:100976. doi: 10.1016/j.molmet.2020.100976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol. 2016;13:508–516. doi: 10.1038/nrgastro.2016.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Vogtmann E, Ahlquist DA et al (2020) Fecal metabolomic signatures in colorectal adenoma patients are associated with gut microbiota and early events of colorectal cancer pathogenesis. MBio:11. 10.1128/mBio.03186-19 [DOI] [PMC free article] [PubMed]
- Koch RL, Goldman P. The anaerobic metabolism of metronidazole forms N-(2-hydroxyethyl)-oxamic acid. J Pharmacol Exp Ther. 1979;208:406–410. [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108:4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone V, Gibbons SM, Martinez K, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17:681–689. doi: 10.1016/j.chom.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B-Q, Huang T, Liu L, et al. Identification of colorectal cancer related genes with mRMR and shortest path in protein-protein interaction network. PLoS One. 2012;7:e33393. doi: 10.1371/journal.pone.0033393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfmark S, Edlund C, Nord CE. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis. 2010;50:S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]
- López-Lázaro M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol. 2016;62:149–152. doi: 10.1016/j.oraloncology.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci U S A. 2007;104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Hamady M, Knight R (2006) UniFrac - an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7. 10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed]
- Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, et al. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangell P, Nejdfors P, Wang M, et al. Lactobacillus plantarum 299v inhibits Escherichia coli-induced intestinal permeability. Dig Dis Sci. 2002;47:511–516. doi: 10.1023/A:1017947531536. [DOI] [PubMed] [Google Scholar]
- Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011;4:603–611. doi: 10.1038/mi.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy IH, Goudarzi M, Tong M, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight DT, Huerlimann R, Bower DS, et al. Methods for normalizing microbiome data: an ecological perspective. Methods Ecol Evol. 2019;10:389–400. doi: 10.1111/2041-210X.13115. [DOI] [Google Scholar]
- Methé BA, Nelson KE, Pop M, et al. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima K, Nishihara R, Qian ZR, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–1980. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy G, Murali MR, Devaraj SN. Lactobacilli facilitate maintenance of intestinal membrane integrity during Shigella dysenteriae 1 infection in rats. Nutrition. 2009;25:350–358. doi: 10.1016/j.nut.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H et al (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment [DOI] [PMC free article] [PubMed]
- Morton JT, Marotz C, Washburne A, et al. Establishing microbial composition measurement standards with reference frames. Nat Commun. 2019;10:1–11. doi: 10.1038/s41467-019-10656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeem S, Li S. Biodiversity enhances ecosystem reliability. Nature. 1997;390:507–509. doi: 10.1038/37348. [DOI] [Google Scholar]
- Nagao-Kitamoto H, Shreiner AB, Gillilland MG, et al. Functional characterization of inflammatory bowel disease-associated gut dysbiosis in gnotobiotic mice. CMGH. 2016;2:468–481. doi: 10.1016/j.jcmgh.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. Systems biology of metabolism: a driver for developing personalized and precision medicine. Cell Metab. 2017;25:572–579. doi: 10.1016/j.cmet.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Nosova T, Jousimies-Somer H, Jokelainen K, et al. Acetaldehyde production and metabolism by human indigenous and probiotic Lactobacillus and Bifidobacterium strains. Alcohol Alcohol. 2000;35:561–568. doi: 10.1093/alcalc/35.6.561. [DOI] [PubMed] [Google Scholar]
- O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13:691–706. doi: 10.1038/nrgastro.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata A, Usami M, Miyoshi M. Short-chain fatty acids alter tight junction permeability in intestinal monolayer cells via lipoxygenase activation. Nutrition. 2005;21:838–847. doi: 10.1016/j.nut.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. 2019;7:1–15. doi: 10.1186/s40168-019-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek M, Čipčić Paljetak H, Barešić A et al (2018) Methodology challenges in studying human gut microbiota-effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci Rep 8. 10.1038/s41598-018-23296-4 [DOI] [PMC free article] [PubMed]
- Park JE, Miller M, Rhyne J, et al. Differential effect of short-term popular diets on TMAO and other cardio-metabolic risk markers. Nutr Metab Cardiovasc Dis. 2019;29:513–517. doi: 10.1016/j.numecd.2019.02.003. [DOI] [PubMed] [Google Scholar]
- Proctor LM, Creasy HH, Fettweis JM, et al. The integrative human microbiome project. Nature. 2019;569:641–648. doi: 10.1038/s41586-019-1238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rath S, Heidrich B, Pieper DH, Vital M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome. 2017;5:1–14. doi: 10.1186/S40168-017-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinninella E, Raoul P, Cintoni M, et al. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019;7:14. doi: 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncal C, Martínez-Aguilar E, Orbe J, et al. Trimethylamine-N-oxide (TMAO) predicts cardiovascular mortality in peripheral artery disease. Sci Rep. 2019;9:1–8. doi: 10.1038/s41598-019-52082-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB. Using gnotobiotic mice to discover and validate therapeutically active microbiota to maintain mucosal homeostasis and treat intestinal inflammation. Drug Discov Today Dis Model. 2018;28:73–77. doi: 10.1016/j.ddmod.2019.08.009. [DOI] [Google Scholar]
- Sato J, Kanazawa A, Ikeda F, et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care. 2014;37:2343–2350. doi: 10.2337/dc13-2817. [DOI] [PubMed] [Google Scholar]
- Schüssler-Fiorenza Rose SM, Contrepois K, Moneghetti KJ, et al. A longitudinal big data approach for precision health. Nat Med. 2019;25:792–804. doi: 10.1038/s41591-019-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. 10.1371/journal.pbio.1002533 [DOI] [PMC free article] [PubMed]
- Seungbum K, Rigatto K, Gazzana MB, et al. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension. 2020;62:147. doi: 10.1007/s00101-013-2141-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Chang HW, Yan D et al (2017) Influence of diet on the gut microbiome and implications for human health. J Transl Med 15 [DOI] [PMC free article] [PubMed]
- Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Anderson JM, Bharti R, et al. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15:630–638. doi: 10.1038/nrmicro.2017.58. [DOI] [PubMed] [Google Scholar]
- Song Y, Liu C, Finegold SM. Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol. 2004;70:6459–6465. doi: 10.1128/AEM.70.11.6459-6465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J, Thompson DJ, Dos Santos SI, et al. Novel associations between common breast cancer susceptibility variants and risk-predicting mammographic density measures. Cancer Res. 2015;75:2457–2467. doi: 10.1158/0008-5472.CAN-14-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilman D, Reich PB, Knops JMH. Biodiversity and ecosystem stability in a decade-long grassland experiment. Nature. 2006;441:629–632. doi: 10.1038/nature04742. [DOI] [PubMed] [Google Scholar]
- Tripathi A, Debelius J, Brenner DA, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:397–411. doi: 10.1038/s41575-018-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- van der Ark KCH, van Heck RGA, Martins Dos Santos VAP, et al. More than just a gut feeling: constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome. 2017;5:78. doi: 10.1186/s40168-017-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- Vandeputte D, Falony G, Vieira-Silva S, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62. doi: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez MT, Ramezani A, Manal A, Raj DS (2016) Trimethylamine N-oxide: the good, the bad and the unknown. Toxins (Basel) 8 [DOI] [PMC free article] [PubMed]
- Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3:858–876. doi: 10.3390/nu3100858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Armet AM, Finlay BB, Shanahan F. Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell. 2020;180:221–232. doi: 10.1016/j.cell.2019.12.025. [DOI] [PubMed] [Google Scholar]
- Wang Q (2020) Multi-omic profiling reveals associations between the gut mucosal microbiome , the metabolome , and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol [DOI] [PMC free article] [PubMed]
- Woese CR. On the evolution of cells. Proc Natl Acad Sci U S A. 2002;99:8742–8747. doi: 10.1073/pnas.132266999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan AW, Schnabl B. Bacterial translocation and changes in the intestinal microbiome associated with alcoholic liver disease. World J Hepatol. 2012;4:110–118. doi: 10.4254/wjh.v4.i4.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Sun C, Zheng J, et al. Efficacy of fecal sampling as a gut proxy in the study of chicken gut microbiota. Front Microbiol. 2019;10:2126. doi: 10.3389/fmicb.2019.02126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Santisteban MM, Rodriguez V, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng Q, Wang C, et al. Characterization of the B cell receptor repertoire in the intestinal mucosa and of tumor-infiltrating lymphocytes in colorectal adenoma and carcinoma. J Immunol. 2017;198:3719–3728. doi: 10.4049/jimmunol.1602039. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang F, Ding X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- Zhou W, Sailani MR, Contrepois K, et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature. 2019;569:663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]