

Abstract
Supramolecular hydrogels have great potential as biomaterials for sustained delivery of therapeutics. While peptide-based supramolecular hydrogels have been developed that show promise for drug delivery applications, the high cost of production has limited their widespread adoption. Low molecular weight (LMW) supramolecular hydrogels are emerging as attractive and inexpensive alternatives to peptide-based hydrogels. We recently reported novel cationic fluorenylmethyloxycarbonyl-modified phenylalanine (Fmoc-Phe) hydrogels for localized and sustained in vivo release of an anti-inflammatory agent for functional pain remediation. In an effort to further elucidate design principles to optimize these materials for delivery of a variety of molecular agents, we herein report a systematic examination of electrostatic effects on the release of cargo molecules from Fmoc-Phe derived hydrogels. Specifically, we interrogate the release of cationic, anionic, and neutral cargo molecules from a series of cationic and anionic Fmoc-Phe derived hydrogels. We observed that cargo was readily released from the hydrogels except when the cargo and hydrogel network had complementary charges, in which case the cargo was highly retained in the network. These results demonstrate that the electrostatic characteristics of both the hydrogel network and the specific cargo are critical design parameters in the formulation of LMW supramolecular hydrogel systems in the development of next-generation materials for drug delivery applications.
Graphical Abstract

Introduction
Innovative and cost-effective strategies for the controlled delivery of therapeutics are in demand.1–3 Conventional systemic drug delivery methods often require increased or repeated dosing, especially with payloads that suffer from poor solubility, low bioavailability, and easy degradation.2–5 In contrast, systems for localized and sustained delivery of therapeutics minimize dosage requirements and systemic toxicity, and materials can be tailored to protect payloads, especially fragile biologics such as oligonucleotides and proteins.2–5 Hydrogels are commonly considered for localized drug delivery because they are highly porous, water-filled networks with material properties that can be tuned for the type of cargo or rate of release that is desired.4–7 Supramolecular hydrogels offer distinct advantages to traditional polymeric materials because the noncovalently assembled network is inherently more dynamic and stimuli responsive, allowing direct encapsulation of the cargo and noninvasive syringe delivery.8–13
Peptide-based supramolecular hydrogel systems are attractive as drug delivery vehicles.10–16 Synthetic peptides are biocompatible and offer precise control over emergent material properties, including viscoelasticity and shear-responsive self-healing, by modification of the primary amino acid sequence.9–19 Self-assembled peptide systems have demonstrated promise for controlled release of many therapeutics in vitro and in vivo, including opioids,20–21 anti-inflammatory drugs,22–23 chemotherapeutics,24–27 insulin,28–29 and immunotherapies.30–31 However, a major impediment for the widespread adoption of peptide-based materials is the high cost of production.32–33 In many cases, the cost of the peptide hydrogel required for delivery exceeds the cost of the therapeutic agent.10 To address this problem, there has been increasing interest in inexpensive, low molecular weight (LMW) supramolecular hydrogels as alternatives to peptide hydrogels for drug delivery.34–38
Self-assembling amino acid derivatives and dipeptides have shown early potential as LMW supramolecular hydrogels that exhibit many of the requisite viscoelastic and shear-response properties needed for drug delivery at significantly lower cost than peptide-derived hydrogels.32–34,38–41 In particular, phenylalanine (Phe) derivatives have dominated this area of study due to their high propensity to self-assemble into a variety of defined nanostructures capable of forming hydrogel networks.42–45 Phe-based gelators typically have an aromatic N-terminal capping group, most commonly the fluorenylmethyloxycarbonyl (Fmoc) group, to promote self-assembly through additional π–π stacking interactions.46–48 Several Phe-based materials have been developed for controlled release applications.49–55 Sustained release of small molecules has been observed from hydrogels of diphenylalanine with Fmoc and other aromatic capping groups,52–53 and from modified amino acid derivatives.55
We recently reported the application of supramolecular hydrogels of cationic Fmoc-Phe derivatives modified with diaminopropane (DAP) (1a–3a, Figure 1A) for functional in vivo delivery of an anti-inflammatory drug diclofenac in a mouse model for chronic pain.56 Gelation of these derivatives does not occur immediately in water due to intermolecular electrostatic repulsion, but addition of saline masks charge repulsion and initiates formation of a self-supporting hydrogel in seconds.57 These hydrogels are shear-responsive, meaning the hydrogel network can temporarily withstand the shear forces of syringe delivery and spontaneously reform after exiting the syringe.56 We found that injection of an Fmoc-F5-Phe-DAP (3a) hydrogel containing diclofenac into the ankles of mice resulted in diclofenac release and pain mitigation for nearly two weeks.56 While characterizing the in vitro release profile of diclofenac from these hydrogels, we noted that the maximum amount released over approximately three days was less than 2% of the total diclofenac loaded into the hydrogel. Diclofenac is negatively charged at physiological pH, so we hypothesized that the cationic hydrogel network could be retaining most of the diclofenac through electrostatic ion pairing. Schneider and coworkers have reported similar phenomena with release of proteins from their charged, self-assembled β-hairpin peptide hydrogels, noting that only a small amount of protein was released when the protein and peptide hydrogel had complementary charges.58–60 Determining how interactions between cargo and the hydrogel network affect cargo release is critical to understand the limitations of the system and to enable optimization of these systems for different cargo.
Figure 1.
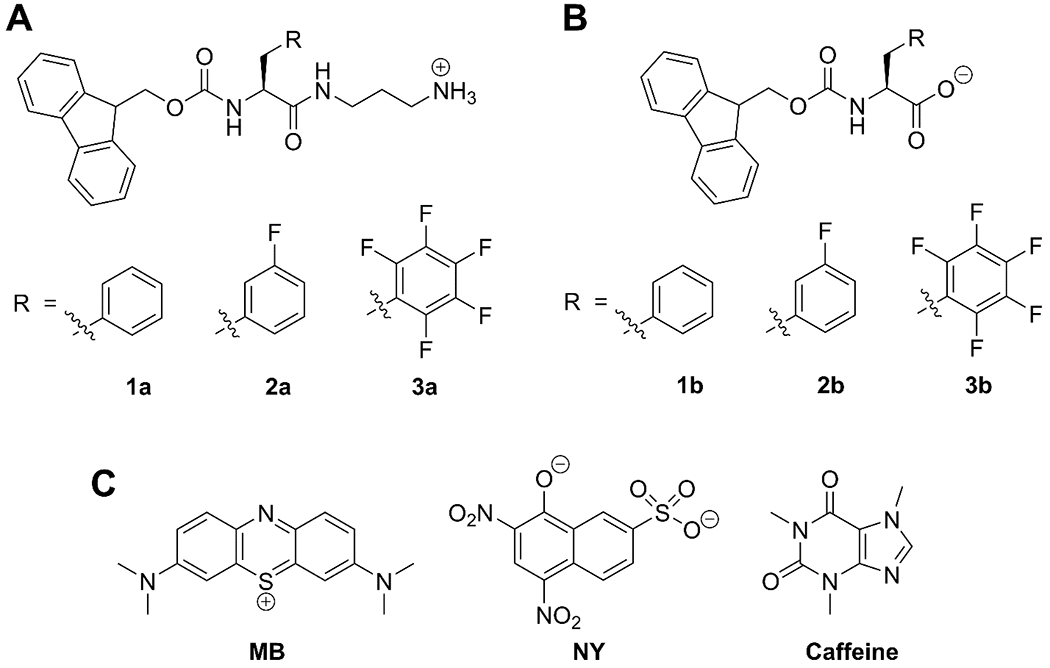
(A) Fmoc-Phe-DAP derivatives: Fmoc-Phe-DAP (1a), Fmoc-3F-Phe-DAP (2a), and Fmoc-F5-Phe-DAP (3a). (B) Fmoc-Phe derivatives: Fmoc-Phe (1b), Fmoc-3F-Phe (2b), Fmoc-F5-Phe (3b). (C) Small molecule cargo for release: methylene blue (MB), naphthol yellow S (NY), and caffeine.
Accordingly, we herein report a systematic study of the effects of the charge of supramolecular hydrogel networks and the charge of small molecule cargo on release of cargo from these networks. To investigate the effect of electrostatic interactions on the release of charged cargo from LMW supramolecular hydrogels, two hydrogel systems of opposing charge and three model small molecule cargo were chosen. The Fmoc-Phe-DAP derivatives 1a–3a were used to form cationic hydrogels while their parent amino acids 1b-3b were adopted as corresponding anionic hydrogels (Figure 1A, B). Methylene blue (MB), caffeine, and naphthol yellow S (NY) were chosen as cationic, neutral, and anionic cargo, respectively (Figure 1C). The release of each cargo molecule from each type of hydrogel was characterized. Strikingly, when the charge of the cargo and network were complementary, the cargo was highly retained by the hydrogel and, in these examples, there was no detectable amount of cargo released for the duration of the study. In contrast, neutral cargo and cargo that had the same charge as the hydrogel network was readily released from the network. This study demonstrates the importance of considering the physicochemical properties of both cargo and hydrogel as well as the emergent properties of the hydrogel network in the design of supramolecular hydrogels for drug delivery applications.
Experimental
Materials.
Reagents and organic solvents were purchased commercially and used without further purification. Fmoc-amino acids 1b–3b were purchased at the highest available quality and used directly in gelation experiments without further purification. Compounds 1a–3a were synthesized by a modified version of the previously reported method.57 Detailed synthetic protocols are in the Supplementary Information. Water used for gelation was purified using a nanopure filtration system (Barnstead NANOpure, 0.2 μm filter, 18.2 MΩ-cm).
Hydrogelation Conditions.
All hydrogels were prepared with a final gelator concentration of 20 mM, final cargo concentration of 0.5 mM, and total volume of 1 mL. To make cationic hydrogels with no cargo, gelators 1a–3a were dissolved in 800 μL of water by sonication in a 70 ºC water bath until fully solubilized. Then, 200 μL of 570 mM aqueous NaCl was added to give a final NaCl concentration of 114 mM. Immediately after addition of NaCl, the vial was briefly agitated by vortex mixer and a hydrogel formed within seconds. To make cationic hydrogels loaded with caffeine or naphthol yellow S, gelators 1a–3a were dissolved in 750 μL of water by the method stated above, then 50 μL of a 10 mM solution of caffeine or naphthol yellow S was added to bring the total volume to 800 μL before adding salt. To make cationic hydrogels loaded with methylene blue, gelators 1a–3a were dissolved in 733.3 μL of water by the method stated above, then 66.7 μL of 7.5 mM methylene blue was added to bring the total volume to 800 μL before adding salt as detailed above. Images of all variations of hydrogels are in the Supplementary Information (Figures S1–S3).
To make anionic hydrogels with no cargo, gelators 1b–3b were dissolved in 4.8 mL of water using 1.2 mL of 0.1 M aqueous NaOH (one molar equivalent) to a final gelator concentration of 20 mM in a 6 mL solution. A fresh aqueous stock solution of glucono-δ-lactone (GdL) was prepared at a concentration of 100 mg/mL (561 mM) immediately prior to gelation experiments. To trigger gelation, 8.9 μL (0.25 molar equivalent, gelator 1b) or 26.7 μL (0.75 molar equivalent, gelators 2b and 3b) of the GdL stock solution was added to a glass vial containing 1 mL of the 20 mM gelator solution and the vial was agitated by vortex mixer for five seconds. The vials were left undisturbed overnight as the hydrogels formed over the course of several hours. To make anionic hydrogels loaded with caffeine or naphthol yellow S, gelators 1b–3b were dissolved in 4.5 mL of water using 1.2 mL of 0.1 M aqueous NaOH (one molar equivalent), and then 0.3 mL of a 10 mM solution of caffeine or naphthol yellow S was added for a final gelator concentration of 20 mM and final cargo concentration of 0.5 mM in a 6 mL solution. GdL was added by the procedure stated above to 1 mL aliquots of these solutions. To make anionic hydrogels loaded with methylene blue, gelators 1b–3b were dissolved in 4.4 mL of water using 1.2 mL of 0.1 M aqueous NaOH (one molar equivalent), and then 0.4 mL of a 7.5 mM solution of methylene blue was added for a final gelator concentration of 20 mM and a final cargo concentration of 0.5 mM in a 6 mL solution. GdL was added by the procedure stated above to 1 mL aliquots of these solutions. Images of all variations of hydrogels are in the Supplementary Information (Figures S4–S6).
Transmission Electron Microscopy.
TEM images were obtained using a Hitachi 7650 transmission electron microscope with an accelerating voltage of 80 kV. Aliquots of hydrogels (10 μL) were applied directly onto 100 mesh carbon-coated copper grids and allowed to stand for 1 min. Excess sample was carefully removed by capillary action using filter paper and then the grids were stained with uranyl acetate (8 μL) for 2 min. Excess stain was removed via capillary action and the grids were allowed to air-dry for 10 min. Dimensions of nanostructures were determined using ImageJ software and are reported as the average of at least 100 independent measurements with error reported as the standard deviation about the mean value for these measurements (Table S1 and S2). TEM images for all hydrogel variations are in the Supplementary Information (Figures S7–S12).
Oscillatory Rheology.
Rheological measurements were obtained using a TA Instruments Discovery HR-2 rheometer. A 20 mM parallel plate geometry was used for the experiments. Hydrogels were formed at a 1 mL volume in 1.5 mL plastic microcentrifuge tubes. Immediately prior to rheological characterization, the plastic tube containing the hydrogel was cut at the 0.5 mL line using a razor blade and the cylindrical hydrogel was placed directly on the rheometer stage for characterization. Experiments were performed using a 1.2 mm gap size operating in oscillatory mode. Strain sweep experiments were performed from 0.01 to 100% strain at a frequency of 6.283 rad s−1 to determine the linear viscoelastic region for each hydrogel. Frequency sweep experiments were performed from 0.1 to 100 rad s−1 at a constant strain of 0.2%, which falls within the linear viscoelastic region for all hydrogels examined. Reported values for storage and loss moduli (G′ and G″, respectively) are the average of at least three distinct measurements on separate hydrogels with the error reported as the standard deviation about the mean (Table S3). See the Supplementary Information for strain sweep data and frequency sweep data not included in the main text (Figures S13–S24).
Sustained Release Assays.
Hydrogels containing cargo were prepared as described above in 2-dram glass vials to form a 1 mL hydrogel with 20 mM gelator and 0.5 mM cargo. The next day, 1.5 mL of an isotonic solution (114 mM NaCl for cationic gels and water for anionic gels) was slowly deposited over the top of the hydrogel and the vial was sealed. Aliquots of the layered solution (100 μL) were removed at 2 min, 20 min, 40 min, 60 min, 2 h, 3 h, 4 h, 6 h, 8 h, 24 h, 48 h, 72 h, and 96 h from the time the buffer was initially layered on top of the gel. After removing each aliquot, the volume of removed buffer solution was immediately replaced by an equal volume of isotonic solution. The concentration of methylene blue or naphthol yellow S in each aliquot was determined by measurement of the absorbance at the λmax of the dye (666 nm for methylene blue and 428 nm for naphthol yellow S) using a Tecan Infinite M1000 microplate reader and correlation of the absorbance to a standard concentration curve. The concentration curves were constructed by recording the absorbance of serial dilutions of a solution of known dye concentration (Figure S25). The concentration of caffeine in each aliquot was determined by injection onto an analytical HPLC instrument (Shimadzu 2010A) equipped with a Phenomenex Gemini 5 μm C18 column (250 × 4.6 mm) and correlation of the integrated peak area of caffeine to a standard concentration curve. A gradient of water and acetonitrile, both containing 0.05% trifluoroacetic acid, was used as the mobile phase at a flow rate of 1 mL/min and UV detection was monitored at 215 nm. The concentration curve was constructed by injection of serial dilutions of a solution of known caffeine concentration onto the HPLC under the defined mobile phase conditions (Figure S25). These methods enabled interpolation of the amount of cargo released into the 1.5 mL solution at each time point by conversion of concentration of cargo into μmol of cargo. The diffusion constant was determined using Equation 1, a non-steady state diffusion model equation, where Mt/M∞ (unitless) is the ratio of molecules of cargo released to the total molecules of cargo in the system, t is the time (min), λ is gel thickness (height, m) and D is the diffusion constant (m2 min−1).15,55,61
| Equation 1. |
For each timepoint, the concentration of cargo in the aliquot was used to calculate the total amount of cargo present in the 1.5 mL layered solution in μmol. For the first timepoint, this value was used directly as Mt, but for subsequent timepoints the amount calculated for Mt was adjusted to include the amount of cargo removed in prior aliquots so that Mt reflected the total amount of cargo released from the hydrogel from the start time to time t. The data were collected in triplicate and plotted as Mt/M∞ vs. t (min) with the error reported as the standard deviation about the mean (Figure 3). A second plot was constructed by plotting Mt/M∞ vs. t1/2 (min1/2) from the initial linear section of the first plot (consisting of the first 120 minutes of the release study) (Figure 4). Equation 1 can be rearranged to yield a linear relationship between Mt/M∞ and t1/2 (s1/2) as follows:
Figure 3.
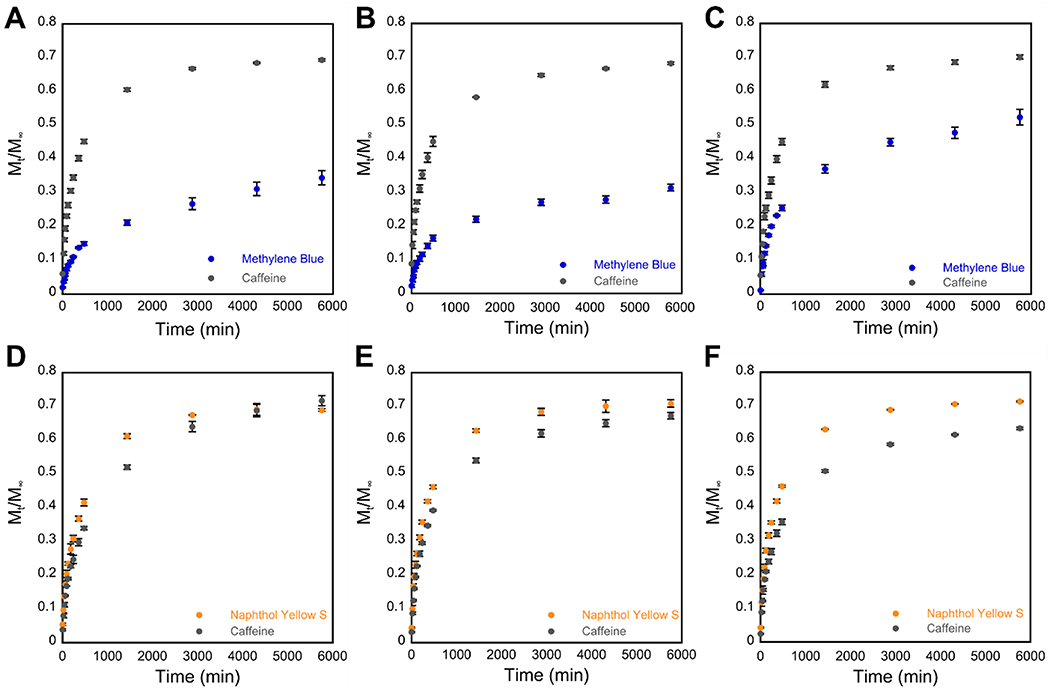
Cumulative release profiles indicating the fraction of cargo released (Mt/M∞) vs. time in minutes from 20 mM hydrogels over 96 hours. Total amount of cargo released after 96 hours is tabulated in Table 1. (A–C) Release of MB (blue) and caffeine (gray) from cationic hydrogels of (A) gelator 1a, (B) gelator 2a, and (C) gelator 3a. (D–F) Release of NY (yellow) and caffeine (gray) from anionic hydrogels of (D) gelator 1b, (E) gelator 2b, and (F) gelator 3b.
Figure 4.
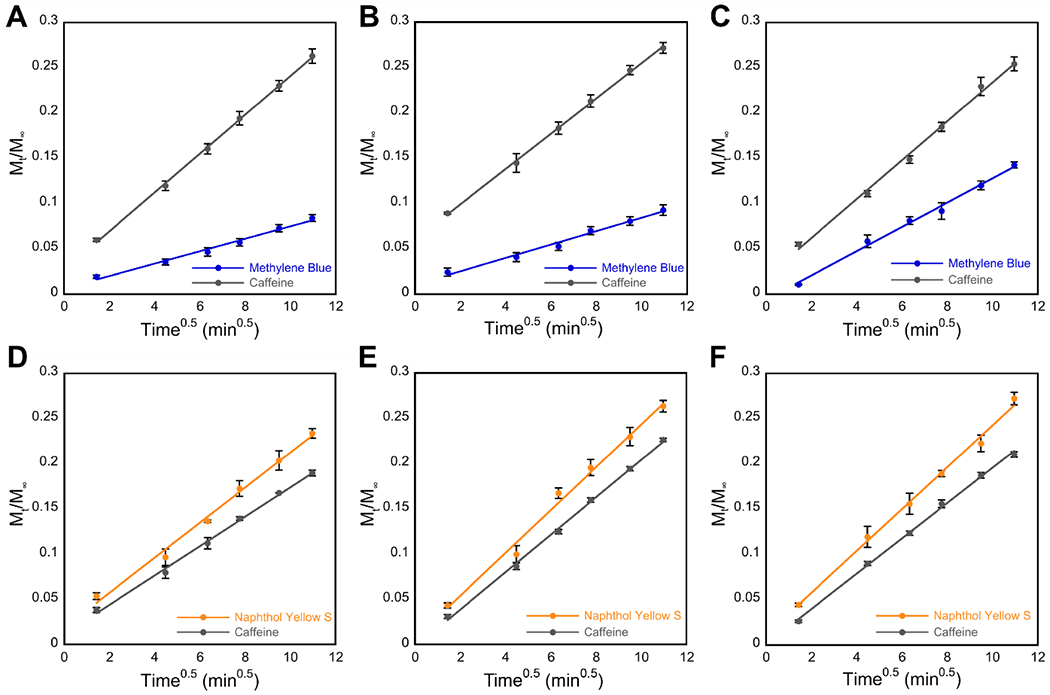
A linear correlation is observed when Mt/M∞ is plotted against t1/2 during the first 120 minutes of cargo release from hydrogels. The slopes obtained were used to calculate the diffusion coefficients found in Table 2. (A–C) Release of MB (blue) and caffeine (gray) from cationic hydrogels of (A) gelator 1a, (B) gelator 2a, and (C) gelator 3a. (D–F) Release of NY (yellow) and caffeine (gray) from anionic hydrogels of (D) gelator 1b, (E) gelator 2b, and (F) gelator 3b.
Thus, the diffusion coefficient, D (m2 s−1), was determined by measuring the slope of Mt/M∞ vs. t1/2 (s1/2) from the second plot and setting this value equal to the coefficient of t1/2 (s1/2) above in order to solve for the value of D (m2 s−1).
For convenience, these release studies were conducted at 25 °C. We have previously shown that hydrogels of 1a–3a release the NSAID diclofenac efficiently at 37 °C and are viable for delivery of this drug in vivo over a period of several weeks.56 Since the purpose of this study is to determine the effects of charge on the release of model molecules from these hydrogels in vitro we chose to conduct these comparative studies at room temperature. Increasing the temperature does not affect the comparative release trends, but simply slightly increases the rate of release.
Results and Discussion
Hydrogel Formulation.
Cationic Fmoc-Phe-DAP gelators 1a–3a were synthesized by coupling 1,3-diaminopropane to the C-terminus of Fmoc-Phe derivatives 1b–3b via amide bond formation by a modified version of our prior synthesis.57 We have previously shown that meta-fluorination or perfluorination of the phenyl ring of Phe-derived gelators yields hydrogels with superior viscoelastic properties.57,62–63 Thus, we chose to examine cargo release from Fmoc-Phe-DAP (1a), Fmoc-3F-Phe-DAP (2a), and Fmoc-F5-Phe-DAP (3a) cationic hydrogels (Figure 1A). These gelators are soluble in water due to the positively charged amine and remain mostly unassembled due to the electrostatic repulsion of monomeric units. Addition of saline to the gelator solution masks the positive charge between monomeric units, triggering rapid self-assembly and gelation. Detailed protocols for gelation of these derivatives may be found in the Experimental. We hypothesized that the positively charged hydrogel network should repel cationic cargo, facilitating its release from the network, while anionic cargo should be released to a lesser extent due to Coulombic attraction between the gel network and cargo.
As a comparison to the cationic hydrogels, we surmised that we could use the parent amino acids Fmoc-Phe (1b), Fmoc-3F-Phe (2b), and Fmoc-F5-Phe (3b) (Figure 1B) as a model anionic hydrogel system. Hydrogels of 1b–3b were prepared using the glucono-δ-lactone (GdL) pH switch method of gelation originally described by Adams and coworkers.64 Detailed protocols for gelation of these derivatives may be found in the Experimental. Briefly, the gelators were dissolved in water using an equimolar amount of 0.1 M NaOH to fully deprotonate the C-terminal carboxylic acid. Then, GdL was added to the high pH solution of gelator to trigger self-assembly and gelation over several hours. GdL is slowly hydrolyzed under basic conditions, releasing a steady stream of protons to reprotonate the C-terminus of the gelator and induce gelation. We hypothesized that the hydrogel network should still retain an overall negative charge, since gelation occurs when the pH is near the apparent pKa of the gelator and some of the C-termini remain deprotonated.65–66 Thus, anionic cargo should be repelled and released from hydrogels of 1b–3b, while cationic cargo should be released to a much lesser extent.
We chose a uniform gelator concentration of 20 mM for all hydrogels based on the upper solubility limits of anionic gelators 1b–3b. Water-soluble dyes methylene blue (MB) and naphthol yellow S (NY) were chosen as the charged cargo for ease of hydrogel loading and quantification by UV-vis spectroscopy (Figure 1C). For further comparison, we expanded our cargo set to include a neutral molecule, caffeine, that would not interact via electrostatics with any of the hydrogel networks. All cargo molecules were encapsulated in the various hydrogel networks by adding them to each gelator solution prior to triggering gelation by the addition of NaCl or GdL for the cationic and anionic hydrogels, respectively. Detailed protocols for formulation of all cargo loaded hydrogels may be found in the Experimental. Self-supporting hydrogels were formed for all combinations of gelator and cargo as indicated by stability to vial inversion (Figures S1–S6).
We also determined the pH of each hydrogel in order to maintain, as much as possible, constant pH conditions for each system. To accommodate efficient gelation conditions for the majority of the gelators, hydrogels were formulated at approximately pH 6 in all cases except for gelator 1b. Though gelators 1a–3a can tolerate gelation under a range of pH values as previously reported,57 it has been well established that anionic gelators such as 1b–3b form hydrogels in a much narrower pH range dictated by the identity of the gelator.41,55,64 Adams and coworkers have previously reported the tendency of Fmoc-Phe (1b) to precipitate from the gel state at lower pH values, which we also observed for samples of 1b at pH 6.41,55 Thus, hydrogels of 1b had to be formulated at pH 7 to ensure stable gels were formed. Hydrogels of 2b and 3b were found to be most stable at pH 6. To verify that the small difference in pH would not significantly affect our ability to compare subsequent release studies, hydrogels of gelator 3a were loaded with MB at pH 6 and pH 7. No significant differences in MB release from these gels were observed, giving confidence that release in these pH ranges (6–7) is not significantly impacted by pH. It should also be noted, that even though these gels were not formulated at physiological pH in this study, we have previously shown that gels of 1a–3a are stable under physiological conditions in in vivo drug delivery studies.56
Characterization of Cargo Release Kinetics from Hydrogel Networks.
Cationic and anionic hydrogels were first loaded with MB and NY to determine the effect of network charge on the release of charged cargo. Isotonic solutions of aqueous NaCl for cationic gels and nanopure water for anionic gels were used for layering to avoid any destabilization or erosion of the hydrogel network during prolonged contact with the layering solution. Upon layering cationic hydrogels of 1a–3a containing cationic MB dye with 114 mM NaCl solution, the layering solution appeared visibly blue within the first 30 minutes of surface contact and continued to darken over several days until the dye saturated the solution (Figure 2A, B). Strikingly, cationic hydrogels containing anionic NY dye did not show visible evidence of release from the cationic network into the liquid layering solution, even after several weeks in contact with the layering solution (Figure 2A, B). No measurable concentration of NY was detected by absorbance in an aliquot of this solution, confirming our qualitative observation that the NY was completely retained by the cationic network. Conversely, the opposite was observed for release of cargo molecules from anionic hydrogels of 1b-3b into layered solutions of water. Visible release of anionic NY from hydrogels of 1b-3b into the layering solution was observed, while no cationic MB was visibly present in the water layer after several weeks (Figure 2C, D). Again, no measurable amount of MB was detected in the layering solution, confirming that cationic MB was retained by the anionic network.
Figure 2.

Representative images of hydrogels layered with isotonic solution after a saturating release study. (A) Hydrogels of 2a loaded with MB (left) and NY (right) that were layered with 114 mM NaCl, showing release of MB and retention of NY. (B) Inverted vials from Panel A. (C) Hydrogels of 2b loaded with MB (left) and NY (right) that were layered with water, showing release of NY and retention of MB. (D) Inverted vials from Panel C.
Based on these qualitative observations, we pursued a quantitative characterization of the release kinetics for each cargo/hydrogel combination that exhibited release. Caffeine was included as a neutral cargo for release studies to have a direct point of comparison between the cationic hydrogels and anionic hydrogels, since only dyes with the same charge as the hydrogel were released. The release of each cargo at 25 °C was monitored for 96 hours after layering the hydrogels with an isotonic solution (detailed release study protocols may be found in the Experimental). Aliquots of the layered solution were removed at predetermined timepoints and the amount of cargo released was determined from the concentration of the molecule in each aliquot. The ratio of the amount of cargo released after t minutes to the total amount of cargo loaded in the hydrogel (Mt/M∞) was plotted against time (t, min) to characterize the cumulative release (Figure 3, Table 1).55,61
Table 1.
Cumulative release of cargo from hydrogels after 96 hours.
| Gelator | MB Released (%) | NY Released (%) | Caffeine Released (%) |
|---|---|---|---|
| 1a | 34.3 ± 2.1 | - | 69.1 ± 0.2 |
| 2a | 31.4 ± 1.0 | - | 68.3 ± 0.4 |
| 3a | 52.3 ± 2.2 | - | 70.1 ± 0.6 |
| 1b | - | 68.9 ± 0.4 | 71.7 ± 1.5 |
| 2b | - | 70.9 ± 1.1 | 67.2 ± 0.9 |
| 3b | - | 71.4 ± 0.2 | 63.5 ± 0.4 |
The cumulative release of caffeine was consistent for all six hydrogels. Between 63–72% of the total amount of encapsulated caffeine was released from the gels over 96 h (Figure 3, Table 1). This indicates that the neutral caffeine molecules, which do not form charge-based interactions with either gel type, interact with the cationic and anionic hydrogel systems in a similar fashion. Similarly, about 70% of the anionic NY dye was released from all three anionic hydrogels of 1b–3b (Figure 3D–F, Table 1), indicating that the NY release is not strongly influenced by differences in the phenyl ring fluorination patterns. However, only about 35% of the cationic MB dye was released from cationic gels of 1a and 2a, while 50% of the MB loaded into gels of 3a was released (Figure 3A–C, Table 1). The reason for lower overall release of MB compared to the other two cargo is not clear, but it could be due to increased noncovalent interaction with the network. MB has a more extensive aromatic ring system than caffeine or NY, so MB molecules could have stronger π-π interactions with the aromatic regions of the gelators, causing more MB to be retained in the network. Additionally, the MB release data suggest that specific cargo/gelator interactions may be important for the release of MB from cationic hydrogels. The phenyl side chain of Fmoc-F5-Phe-DAP (3a) has an altered quadrupole moment compared to Fmoc-Phe-DAP (1a) and Fmoc-3F-Phe-DAP (2a), which could result in less favorable interactions between the gelator network and MB, resulting in the higher amount of MB release from gels of 3a over the course of the experiment.
The rate of diffusion for each cargo/hydrogel system was also determined from the release study data. The diffusion coefficient (D, m2 s−1) for a system can be determined using Equation 1, derived from Fick’s second law of diffusion (see Experimental). A plot of Mt/M∞ vs. t1/2 was linear for the first 120 minutes of each release study, indicating Fickian diffusion, and the average diffusion coefficient D for each hydrogel/cargo combination was calculated from the slope of this plotted data (Figure 4, Table 2).55,61 Similar trends can be seen for D values as were observed for the cumulative release numbers. Caffeine and NY released from gels had an average diffusion coefficient near 1 × 10−10 m2 s−1, but for MB it was about half that value when releasing from gels of 3a, and an order of magnitude lower when releasing from gels of 1a and 2a (Table 2). The cargo/network interactions discussed previously likely are responsible for the decreases in the diffusion coefficient for MB compared to NY and caffeine, since they broadly trend the same as the decreases in cumulative release.
Table 2.
Diffusion coefficient (D, 10−10 m2 s−1) of cargo releasing from hydrogels calculated from the slope of Mt/M∞ vs. t1/2 for the first 120 minutes of release.
| Gelator | MB (10−10 m2 s−1) | NY (10−10 m2 s−1) | Caffeine (10−10 m2 s−1) |
|---|---|---|---|
| 1a | 0.146 ± 0.003 | - | 1.42 ± 0.05 |
| 2a | 0.16 ± 0.01 | - | 1.17 ± 0.05 |
| 3a | 0.64 ± 0.09 | - | 1.35 ± 0.11 |
| 1b | - | 1.23 ± 0.10 | 0.86 ± 0.03 |
| 2b | - | 1.83 ± 0.16 | 1.40 ± 0.05 |
| 3b | - | 1.74 ± 0.16 | 1.24 ± 0.03 |
Viscoelastic Properties of Unloaded vs. Loaded Hydrogel Systems.
To determine if loading cargo into the hydrogels had any effect on their emergent properties, every hydrogel/cargo combination was characterized by oscillatory rheology and transmission electron microscopy (TEM). First, the nanomorphology of each network was examined by TEM, both after gelation and after 96 hours of interfacial contact with a layering solution (Figures S7–S12, Table S1 and S2). The different gelators assemble to produce distinct nanostructures that vary in size and shape dependent on the identity of the gelator, but we did not observe a significant difference in morphology when comparing unloaded and loaded hydrogels of the same gelator. Small differences were observed only among samples of Fmoc-Phe-DAP (1a) hydrogels (Figure S7). We have previously noted that cationic gelators 1a–3a self-assemble in a hierarchical process by first forming thin fibrils that subsequently laminate to form flat nanotapes or nanoribbons, which can further twist to ultimately form nanotubes.56–57 The amount of time this process takes is dependent on pH, temperature, and gelator identity, and we have previously observed that 1a seems to undergo the transition to nanotapes and nanotubes more readily than its fluorinated counterparts 2a and 3a.56–57 A similar pattern was observed here in the images taken at the later timepoint, with hydrogels of 1a comprised of a heterogeneous mixture of fibrils, nanotapes, and nanotubes, hydrogels of 2a comprised primarily of flat ribbons of laminated fibrils, and hydrogels of 3a comprised of small bundles of fibrils (Figures S7–S9, Table S1). For the anionic gelators 1b–3b, thin, high-aspect ratio fibrils were observed in all cases as has been previously reported (Figures S10–S12, Table S2).62–63 In a few cases, some larger structures or bundles of fibrils were also observed in hydrogels that were visually more opaque, but fibrils were still the dominant species in these samples (Figures S11, S12).
Hydrogel viscoelasticity was examined by oscillatory rheology for all cargo/hydrogel combinations (see Experimental for detailed protocols). Hydrogels were formulated with and without cargo and strain sweep measurements were performed on each gel in order to characterize the linear viscoelastic region for each gel prior to the performance of frequency sweep experiments. A strain value of 0.2% strain was found to be within the linear viscoelastic region for all gels (Figures S13–S18). Frequency sweep experiments were then performed within the linear viscoelastic region to measure the storage moduli (G′) and the loss moduli (G″) as a function of angular frequency for all cargo/hydrogel combinations (Figure 5, Table S3). Values of G′ exceeded G″ by an order of magnitude and were independent of frequency for all gelators except 1b, indicating structurally robust materials.67 Hydrogels of 1b exhibited a modest frequency dependence, indicating a more fluid-like dynamic network than the other materials studied (Figure 5D).67 Only hydrogels of 1a showed high variation in G′ and G″ values when comparing loaded and unloaded gels, ranging from 23.5–164.9 kPa and 4.8–23.9 kPa, respectively (Figure 5A, Table S3). As noted above, hydrogels of 1a were made up of a heterogeneous mixture of fibrils, nanotapes, and nanotubes. The exact composition of this dynamic mixture might vary between gels loaded with different cargo due to cargo participation in the hierarchical assembly process, and the distribution of fibrils, nanotapes, and nanotubes likely influences the overall viscoelastic properties of the material. As a result, the variability in storage and loss moduli is much higher for hydrogels of 1a than for any of the other hydrogels.
Figure 5.
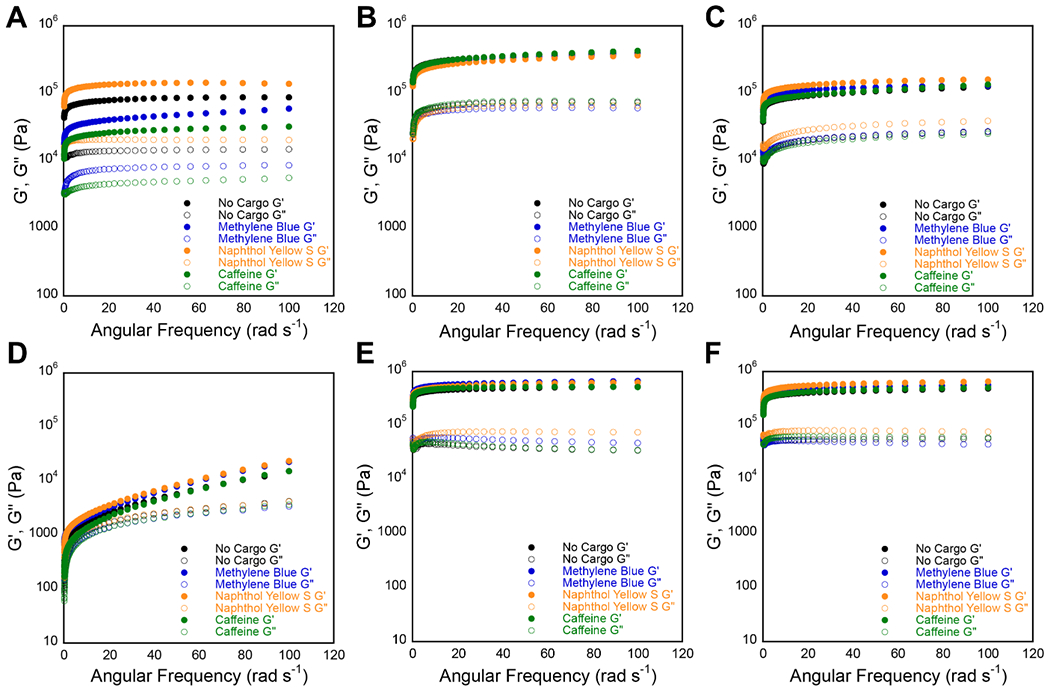
Representative frequency sweep data collected via oscillatory rheology for 20 mM unloaded hydrogels (black) and hydrogels loaded with MB (blue), NY (yellow), and caffeine (green). G′ and G″ values (Pa) are represented by closed circles and open circles, respectively. (A-C) Frequency sweeps for cationic hydrogels of (A) gelator 1a, (B) gelator 2a, and (C) gelator 3a. (D-F) Frequency sweeps for anionic hydrogels of (D) gelator 1b, (E) gelator 2b, and (C) gelator 3b.
The remaining hydrogels both with and without cargo were highly consistent in viscoelastic character. Hydrogels of 2a and 3a across all load variations had average G′ and G″ values of 397.5 ± 50.2 kPa and 73.9 ± 8.0 kPa (2a), and 138.5 ± 21.9 kPa and 32.6 ± 8.9 kPa (3a), respectively (Figure 5B, C, Table S3). The hydrogels of 2a had the highest moduli of the Fmoc-Phe-DAP series as we have reported previously.56–57 Anionic hydrogels had average G′ and G″ values of 19.2 ± 6.0 kPa and 3.6 ± 0.4 kPa (1b), 554.8 ± 71.2 kPa and 46.0 ± 22.1 kPa (2b), and 545.3 ± 92.3 kPa and 59.5 ± 14.6 kPa (3b), respectively (Figure 5E, F, Table S3). The fluorinated gelators had similar but superior viscoelastic strength when compared to the nonfluorinated Fmoc-Phe gelator 1b, as we have previously reported.62–63,68 The reasons that Fmoc-Phe (1b) forms inferior hydrogels to fluorinated derivative (2b and 3b) to is, as yet, not completely understood. However, subtle differences in hydrophobicity may contribute to these properties. Structural studies from our group have also suggested that side chain fluorination enhances intermolecular dipolar interactions within the self-assembled fibrils, which also contributes to the observed differences in emergent viscoelasticity, although the correlation between self-assembly propensity and emergent gel rigidity is still a mystery.69–70 Overall, the presence of cargo did not observably affect the emergent properties of the hydrogels, indicating that these materials can tolerate small molecule cargo in a predictable manner.
Discussion.
We have demonstrated that the charge of LMW supramolecular hydrogel networks significantly affects the release profile of charged small molecule cargo. Under the conditions examined here, pairing a hydrogel system and cargo molecule with complementary charges effectively prohibited release of the cargo. It is important to recognize, however, that this is a function of the gelator/cargo concentrations used in this study (0.5 mM cargo to 20 mM gelator) and that at different gelator/cargo ratios, release of cargo with complementary charge to the hydrogel network is possible. For example, in a recently published study, we demonstrated release of the anionic drug diclofenac from cationic Fmoc-Phe-DAP hydrogels.56 However, in this previously published work, the concentration of diclofenac in the hydrogels was 15.7 mM in 33.7 mM gelator, which is over 30 times higher than the amount of cargo loaded into hydrogels in the work reported herein. Even at such high concentrations of diclofenac, the rate of release was several orders of magnitude slower than was observed for like-charged and neutral cargo release in the present study; the maximum amount of diclofenac released was only 1.4% after 72 hours, while between 28–48% of like charged cargo was released over the same time frame in the present study. Taken together, these observations confirm that release of cargo from hydrogels is at least very inefficient when the cargo and the hydrogel bear complementary charge. Slow release, however, may be advantageous under conditions where sustained delivery of cargo is required over long periods of time. In our aforementioned diclofenac study, the slow release of diclofenac enabled functional pain mitigation in a mouse model for nearly 2 weeks upon injection of the hydrogel. Thus, the charge of molecular cargo and of the hydrogel network are critical design elements in the formulation of hydrogels for drug delivery applications.
The release profile of cationic MB from cationic hydrogels also bears further discussion. Specifically, we observed that after 96 hours, 35–50% of cationic MB was released from cationic hydrogels. Over the same time period, approximately 70% of the total amount of anionic NY was released from anionic hydrogels and approximately 70% of the total amount of neutral caffeine was released from both neutral and cationic hydrogels. In addition, the rate of release of MB was an order of magnitude slower that the rate of release for NY and caffeine. There are several reasons that can potentially explain the less efficient release of MB from cationic hydrogels relative to the corresponding anionic and neutral cargo release profiles. First, we can consider that the density of charge may differ between the cationic and anionic hydrogels due to the different methods of gelation, which would be expected to influence cargo release rates. The cationic Fmoc-Phe-DAP hydrogels (1a–3a) effectively display 20 mM cationic ammonium groups at 20 mM gelator since the pH of the hydrogels are well below the effective pKa of the ammonium functionality. In contrast, the anionic Fmoc-Phe hydrogels (1b–3b) undergo gelation under conditions in which the pH is gradually lowered as a function of GdL hydrolysis. The final pH of these anionic hydrogels is similar to the apparent pKa of the Fmoc-Phe carboxylic acid groups in the context of the self-assembled material, resulting in an effective anionic charge density of approximately 10 mM carboxylate at gelator concentrations of 20 mM (roughly 50% of the carboxyl groups are deprotonated in the assembled state). Thus, cationic cargo in hydrogels of 1a–3a effectively interact with twice as many positive charged groups in the hydrogel network compared to the negative charged groups that anionic cargo experience in hydrogels of 1b–3b. Thus, we would expect that the rate of release of MB from cationic hydrogels should be faster than the rate of release of NY from anionic hydrogels. This is the opposite of the observed trends.
However, it is not obvious from the data that charge plays a significant role in the release of cargo from hydrogels of like charge, even though the effect of complementary charge on cargo retention has been clearly demonstrated. The rate of release of anionic NY from anionic hydrogels was only slightly faster than the release of neutral caffeine from the same hydrogels, even though it would be reasonable to predict that NY would have released at a much higher rate due to repulsion between it and the anionic network. Furthermore, in hydrogels of 1a slightly less NY was released by the end of the study than caffeine, the opposite of what would be expected if repulsive forces were important. Thus, there does not appear to be a strong repulsive effect between like charged cargo and hydrogel networks that results in any notable expulsion of the cargo from the network. Based on these observations, it is not likely that variations in the charge density of the cationic and anionic networks would have been the cause of such a dramatic difference in the release of MB compared to the other cargo.
Since charge effects cannot adequately explain this puzzling disparity in the comparative release profiles MB from cationic and NY from anionic hydrogels, we must consider that specific effects that arise from hydrogel network morphologies must also influence release of cargo from the hydrogels. The Fmoc-Phe-DAP series of gelators (1a–3a) assemble to form structures with varying morphology depending on the identity of the gelator. Hydrogels of 1a consist of flat nanotapes and nanotubes, hydrogels of 2a consist of ribbons of 2–6 laminated fibrils, while hydrogels of 3a are composed of fibril bundles devoid of wider nanoribbon structures. Release of MB from hydrogels of 3a is more efficient (approximately 50% of total MB release over 96 hours) than from hydrogels of 1a and 2a (approximately 30% total MB release over 96 hours). It is possible that MB may have increased interactions with the flatter nanostructures, possibly through surface π-π or cation-π interactions, resulting in higher retention of MB in hydrogels of 1a and 2a than in hydrogels of 3a. However, this phenomenon does not appear to be general, as caffeine release was unaffected by the different network morphologies in the cationic hydrogel networks. These fascinating observations invite further study into the specific non-electrostatic interactions that influence release of cargo from hydrogel networks. These future studies will include efforts to correlate the effects of the structure of molecular cargo and the structural morphology of the hydrogel network to release profiles of cargo from the networks, which will provide even greater insight into design parameters for the formulation of supramolecular hydrogels that are tuned to specific molecular cargo for drug delivery.
Overall, the emergent viscoelastic properties of these hydrogels appear to have a very subtle impact on release of small molecule cargo. No strong correlations between viscoelastic properties and release were observed for any combination of cargo and hydrogel. The release profiles of MB, NY, or caffeine from the respective hydrogel series appear to be independent of G′ and G″, which can vary by an order of magnitude across the materials reported herein. For example, even though caffeine and MB released at different rates and cumulative amounts from cationic hydrogels of 2a or 3a, the viscoelasticity of the caffeine and MB loaded hydrogels was very similar. Since all hydrogels were kept at a constant 20 mM concentration of gelator it is possible that the differences in network viscoelasticity are not significant enough to impact cargo release. Comparison of hydrogels in which gelator concentration is varied more dramatically to give wider ranges of storage and loss moduli would be expected to impact release profiles of cargo molecules more significantly. As such, modification of emergent viscoelastic character of hydrogels should not be overlooked as another tool to further tune release rates from LMW supramolecular hydrogels for drug delivery.
Conclusion
Herein, we have demonstrated the significant impact of electrostatic interactions on the release of charged cargo from charged supramolecular hydrogels. Notably, cargo was highly retained within a network of complementary charge, but readily released from a network with like charge. More subtle effects were observed when comparing release profiles of MB to those of NY and caffeine, which may be due to specific molecular interactions between MB and the cationic network. These data underscore the fact that one type of supramolecular hydrogel may not be effective as an all-purpose drug delivery vehicle. While LMW Phe-derived supramolecular hydrogels are innovative, inexpensive alternatives to peptide hydrogels, it is yet paramount to understand the principles that influence cargo release. When designing supramolecular hydrogels for drug delivery, special consideration must be given to the charge of both the target cargo and the hydrogel network. Appropriately incorporating these chemical characteristics into the design of drug delivery materials will enable precise tuning of release profiles of the cargo molecules for applications in which sustained release is desired. Novel hydrogel delivery systems that are precisely tuned to deliver molecular cargo over a specified time period are highly desirable, and the work reported herein elucidates important design principles for the next generation of LMW supramolecular hydrogels for the drug delivery.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation (DMR-1148836), the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01HL138538), and a University of Rochester University Research Award. BLA was supported by a University of Rochester Robert L. and Mary L. Sproull Fellowship. EST was supported by the University of Rochester Discover Undergraduate Research Grant. NJT was supported by a McNair Scholar Fellowship from the David T. Kearns Center at the University of Rochester. We gratefully acknowledge Karen Bentley in the URMC Electron Microscope Shared Resource for her assistance in TEM imaging experiments.
Footnotes
Conflict of Interest
There are no conflicts to declare.
Electronic Supplementary Information
The Electronic Supplementary Information (ESI) is available free of charge. ESI includes digital images of hydrogels, TEM images, additional rheological information, concentration curves, and UV-vis spectra.
References
- 1.Park K Controlled drug delivery systems: past forward and future back. J. Control. Release 2014, 190, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fenton OS; Olafson KN; Pillai PS; Mitchell MJ; Langer R Advances in Biomaterials for Drug Delivery. Adv. Mater. 2018, e1705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tibbitt MW; Dahlman JE; Langer R Emerging Frontiers in Drug Delivery. J. Am. Chem. Soc 2016, 138, 704–717. [DOI] [PubMed] [Google Scholar]
- 4.Li J; Mooney DJ Designing hydrogels for controlled drug delivery. Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimatteo R; Darling NJ; Segura T In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Delivery Rev 2018, 127, 167–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayanaswamy R; Torchilin PV Hydrogels and Their Applications in Targeted Drug Delivery. Molecules 2019, 24, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thambi T; Li Y; Lee DS Injectable hydrogels for sustained release of therapeutic agents. J. Controlled Release 2017, 267, 57–66. [DOI] [PubMed] [Google Scholar]
- 8.Mayr J; Saldías C; Díaz Díaz D Release of small bioactive molecules from physical gels. Chem. Soc. Rev 2018, 47, 1484–1515. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J; Li J; Du X; Xu B Supramolecular biofunctional materials. Biomaterials 2017, 129, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sis MJ; Webber MJ Drug Delivery with Designed Peptide Assemblies. Trends Pharmacol. Sci 2019, 40, 747–762. [DOI] [PubMed] [Google Scholar]
- 11.Li Y; Wang F; Cui H Peptide-based supramolecular hydrogels for delivery of biologics. Bioeng. Transl. Med 2016, 1, 306–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C; Zhang Q; Zhu S; Liu H; Chen J Preparation and applications of peptide-based injectable hydrogels. RSC Adv. 2019, 9, 28299–28311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J; Xing R; Bai S; Yan X Recent advances of self-assembling peptide-based hydrogels for biomedical applications. Soft Matter 2019, 15, 1704–1715. [DOI] [PubMed] [Google Scholar]
- 14.Moore AN; Hartgerink JD Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration. Acc. Chem. Res 2017, 50, 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koutsopoulos S; Unsworth LD; Nagai Y; Zhang S Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4623–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukunaga K; Tsutsumi H; Mihara H Self-Assembling Peptides as Building Blocks of Functional Materials for Biomedical Applications. Bull. Chem. Soc. Jpn 2018, 92, 391–399. [Google Scholar]
- 17.Yu Z; Cai Z; Chen Q; Liu M; Ye L; Ren J; Liao W; Liu S Engineering β-sheet peptide assemblies for biomedical applications. Biomater. Sci 2016, 4, 365–374. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q; Jiang N; Fu B; Huang F; Liu J Self-assembling peptide-based nanodrug delivery systems. Biomater. Sci 2019, 7, 4888–4911. [DOI] [PubMed] [Google Scholar]
- 19.Fan T; Yu X; Shen B; Sun L Peptide Self-Assembled Nanostructures for Drug Delivery Applications. J. Nanomater 2017, 2017, 4562474. [Google Scholar]
- 20.Martin C; Dumitrascuta M; Mannes M; Lantero A; Bucher D; Walker K; Van Wanseele Y; Oyen E; Hernot S; Van Eeckhaut A; Madder A; Hoogenboom R; Spetea M; Ballet S Biodegradable Amphipathic Peptide Hydrogels as Extended-Release System for Opioid Peptides. J. Med. Chem 2018, 61, 9784–9789. [DOI] [PubMed] [Google Scholar]
- 21.Martin C; Oyen E; Mangelschots J; Bibian M; Ben Haddou T; Andrade J; Gardiner J; Van Mele B; Madder A; Hoogenboom R; Spetea M; Ballet S Injectable peptide hydrogels for controlled-release of opioids. MedChemComm 2016, 7, 542–549. [Google Scholar]
- 22.Webber MJ; Matson JB; Tamboli VK; Stupp SI Controlled release of dexamethasone from peptide nanofiber gels to modulate inflammatory response. Biomaterials 2012, 33, 6823–6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altunbas A; Lee SJ; Rajasekaran SA; Schneider JP; Pochan DJ Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials 2011, 32, 5906–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei L; He S; Liu Z; Xu K; Zhong W Co-assembled supramolecular hydrogels of doxorubicin and indomethacin-derived peptide conjugates for synergistic inhibition of cancer cell growth. Chem. Commun 2019, 55, 4411–4414. [DOI] [PubMed] [Google Scholar]
- 25.Mei L; Xu K; Zhai Z; He S; Zhu T; Zhong W Doxorubicin-reinforced supramolecular hydrogels of RGD-derived peptide conjugates for pH-responsive drug delivery. Org. Biomol. Chem 2019, 17, 3853–3860. [DOI] [PubMed] [Google Scholar]
- 26.Tao M; Liu J; He S; Xu K; Zhong W In situ hydrogelation of forky peptides in prostate tissue for drug delivery. Soft Matter 2019, 15, 4200–4207. [DOI] [PubMed] [Google Scholar]
- 27.Raza F; Zhu Y; Chen L; You X; Zhang J; Khan A; Khan MW; Hasnat M; Zafar H; Wu J; Ge L Paclitaxel-loaded pH responsive hydrogel based on self-assembled peptides for tumor targeting. Biomater. Sci 2019, 7, 2023–2036. [DOI] [PubMed] [Google Scholar]
- 28.Fu M; Zhang C; Dai Y; Li X; Pan M; Huang W; Qian H; Ge L Injectable self-assembled peptide hydrogels for glucose-mediated insulin delivery. Biomater. Sci 2018, 6, 1480–1491. [DOI] [PubMed] [Google Scholar]
- 29.Roth-Konforti ME; Comune M; Halperin-Sternfeld M; Grigoriants I; Shabat D; Adler-Abramovich L UV Light–Responsive Peptide-Based Supramolecular Hydrogel for Controlled Drug Delivery. Macromol. Rapid Commun 2018, 39, 1800588. [DOI] [PubMed] [Google Scholar]
- 30.Miller SE; Yamada Y; Patel N; Suárez E; Andrews C; Tau S; Luke BT; Cachau RE; Schneider JP Electrostatically Driven Guanidinium Interaction Domains that Control Hydrogel-Mediated Protein Delivery In Vivo. ACS Cent. Sci 2019, 5, 1750–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leach DG; Dharmaraj N; Piotrowski SL; Lopez-Silva TL; Lei YL; Sikora AG; Young S; Hartgerink JD STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 2018, 163, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao K; Levin A; Adler-Abramovich L; Gazit E Fmoc-modified amino acids and short peptides: simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev 2016, 45, 3935–3953. [DOI] [PubMed] [Google Scholar]
- 33.Panda JJ; Chauhan VS Short peptide based self-assembled nanostructures: implications in drug delivery and tissue engineering. Polym. Chem 2014, 5, 4418–4436. [Google Scholar]
- 34.Ni M; Zhuo S Applications of self-assembling ultrashort peptides in bionanotechnology. RSC Adv. 2019, 9, 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu Q; Li H; Wang L; Gu H; Fan C DNA Nanotechnology-Enabled Drug Delivery Systems. Chem. Rev 2019, 119, 6459–6506. [DOI] [PubMed] [Google Scholar]
- 36.Khan F; Bera D; Palchaudhuri S; Bera R; Mukhopadhyay M; Dey A; Goswami S; Das S Dual release kinetics in a single dosage from core–shell hydrogel scaffolds. RSC Adv. 2018, 8, 32695–32706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friggeri A; Feringa BL; van Esch J Entrapment and release of quinoline derivatives using a hydrogel of a low molecular weight gelator. J. Control. Release 2004, 97, 241–248. [DOI] [PubMed] [Google Scholar]
- 38.Draper ER; Adams DJ Low-Molecular-Weight Gels: The State of the Art. Chem 2017, 3, 390–410. [Google Scholar]
- 39.Ryan DM; Nilsson BL Self-assembled amino acids and dipeptides as noncovalent hydrogels for tissue engineering. Polym. Chem 2012, 3, 18–33. [Google Scholar]
- 40.Chakraborty P; Gazit E Amino Acid Based Self-assembled Nanostructures: Complex Structures from Remarkably Simple Building Blocks. ChemNanoMat 2018, 4, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Draper ER; Morris KL; Little MA; Raeburn J; Colquhoun C; Cross ER; McDonald TO; Serpell LC; Adams DJ Hydrogels formed from Fmoc amino acids. CrystEngComm 2015, 17, 8047–8057. [Google Scholar]
- 42.Marchesan S; Vargiu VA; Styan EK The Phe-Phe Motif for Peptide Self-Assembly in Nanomedicine. Molecules 2015, 20, 19775–19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das T; Haring M; Haldar D; Díaz Díaz D Phenylalanine and derivatives as versatile low-molecular-weight gelators: design, structure and tailored function. Biomater. Sci 2018, 6, 38–59. [DOI] [PubMed] [Google Scholar]
- 44.Diaferia C; Morelli G; Accardo A Fmoc-diphenylalanine as a suitable building block for the preparation of hybrid materials and their potential applications. J. Mater. Chem. B 2019, 7, 5142–5155. [DOI] [PubMed] [Google Scholar]
- 45.German HW; Uyaver S; Hansmann UHE Self-Assembly of Phenylalanine-Based Molecules. J. Phys. Chem. A 2015, 119, 1609–1615. [DOI] [PubMed] [Google Scholar]
- 46.Fleming S; Debnath S; Frederix PWJM; Tuttle T; Ulijn RV Aromatic peptide amphiphiles: significance of the Fmoc moiety. Chem. Commun 2013, 49, 10587–10589. [DOI] [PubMed] [Google Scholar]
- 47.Shi J; Gao Y; Yang Z; Xu B Exceptionally small supramolecular hydrogelators based on aromatic–aromatic interactions. Beilstein J. Org. Chem 2011, 7, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh V; Snigdha K; Singh C; Sinha N; Thakur AK Understanding the self-assembly of Fmoc–phenylalanine to hydrogel formation. Soft Matter 2015, 11, 5353–5364. [DOI] [PubMed] [Google Scholar]
- 49.Thota CK; Yadav N; Chauhan VS “A novel highly stable and injectable hydrogel based on a conformationally restricted ultrashort peptide”. Sci. Rep 2016, 6, 31167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao S; Fu X; Wang N; Wang H; Yang Y Release behavior of salicylic acid in supramolecular hydrogels formed by l-phenylalanine derivatives as hydrogelator. Int. J. Pharm 2008, 357, 95–99. [DOI] [PubMed] [Google Scholar]
- 51.Nanda J; Biswas A; Banerjee A Single amino acid based thixotropic hydrogel formation and pH-dependent morphological change of gel nanofibers. Soft Matter 2013, 9, 4198–4208. [Google Scholar]
- 52.Mahler A; Reches M; Rechter M; Cohen S; Gazit E Rigid, Self-Assembled Hydrogel Composed of a Modified Aromatic Dipeptide. Adv. Mater 2006, 18, 1365–1370. [Google Scholar]
- 53.Aldilla VR; Martin AD; Nizalapur S; Marjo CE; Rich AM; Ho KKK; Ittner LM; Black DS; Thordarson P; Kumar N Glyoxylamide-based self-assembly hydrogels for sustained ciprofloxacin delivery. J. Mater. Chem. B 2018, 6, 6089–6098. [DOI] [PubMed] [Google Scholar]
- 54.Silva RF; Araújo DR; Silva ER; Ando RA; Alves WA l-Diphenylalanine Microtubes As a Potential Drug-Delivery System: Characterization, Release Kinetics, and Cytotoxicity. Langmuir 2013, 29, 10205–10212. [DOI] [PubMed] [Google Scholar]
- 55.Sutton S; Campbell NL; Cooper AI; Kirkland M; Frith WJ; Adams DJ Controlled Release from Modified Amino Acid Hydrogels Governed by Molecular Size or Network Dynamics. Langmuir 2009, 25, 10285–10291. [DOI] [PubMed] [Google Scholar]
- 56.Raymond DM; Abraham BL; Fujita T; Watrous MJ; Toriki ES; Takano T; Nilsson BL Low-Molecular-Weight Supramolecular Hydrogels for Sustained and Localized in Vivo Drug Delivery. ACS Appl. Bio Mater 2019, 2, 2116–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajbhandary A; Raymond DM; Nilsson BL Self-Assembly, Hydrogelation, and Nanotube Formation by Cation-Modified Phenylalanine Derivatives. Langmuir 2017, 33, 5803–5813. [DOI] [PubMed] [Google Scholar]
- 58.Nagy-Smith K; Yamada Y; Schneider JP Protein release from highly charged peptide hydrogel networks. J. Mater. Chem. B 2016, 4, 1999–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Branco MC; Pochan DJ; Wagner NJ; Schneider JP The effect of protein structure on their controlled release from an injectable peptide hydrogel. Biomaterials 2010, 31, 9527–9534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Branco MC; Pochan DJ; Wagner NJ; Schneider JP Macromolecular diffusion and release from self-assembled beta-hairpin peptide hydrogels. Biomaterials 2009, 30, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagai Y; Unsworth LD; Koutsopoulos S; Zhang S Slow release of molecules in self-assembling peptide nanofiber scaffold. J. Control. Release 2006, 115, 18–25. [DOI] [PubMed] [Google Scholar]
- 62.Ryan DM; Anderson SB; Nilsson BL The influence of side-chain halogenation on the self-assembly and hydrogelation of Fmoc-phenylalanine derivatives. Soft Matter 2010, 6, 3220–3231. [Google Scholar]
- 63.Ryan DM; Anderson SB; Senguen FT; Youngman RE; Nilsson BL Self-assembly and hydrogelation promoted by F5-phenylalanine. Soft Matter 2010, 6, 475–479. [Google Scholar]
- 64.Adams DJ; Butler MF; Frith WJ; Kirkland M; Mullen L; Sanderson P A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter 2009, 5, 1856–1862. [Google Scholar]
- 65.Ryan DM; Doran TM; Anderson SB; Nilsson BL Effect of C-Terminal Modification on the Self-Assembly and Hydrogelation of Fluorinated Fmoc-Phe Derivatives. Langmuir 2011, 27, 4029–4039. [DOI] [PubMed] [Google Scholar]
- 66.Cross ER; Adams DJ Probing the self-assembled structures and pKa of hydrogels using electrochemical methods. Soft Matter 2019, 15, 1522–1528. [DOI] [PubMed] [Google Scholar]
- 67.Dawn A; Kumari H Low Molecular Weight Supramolecular Gels Under Shear: Rheology as the Tool for Elucidating Structure–Function Correlation. Chem. Eur. J 2017, 24, 762–776. [DOI] [PubMed] [Google Scholar]
- 68.Abraham BL; Liyanage W; Nilsson BL Strategy to Identify Improved N-Terminal Modifications for Supramolecular Phenylalanine-Derived Hydrogelators. Langmuir 2019, 35, 14939–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liyanage W; Nilsson BL Substituent Effects on the Self-Assembly/Coassembly and Hydrogelation of Phenylalanine Derivatives. Langmuir 2016, 32, 787–799. [DOI] [PubMed] [Google Scholar]
- 70.Liyanage W; Brennessel WW; Nilsson BL Spontaneous Transition of Self-assembled Hydrogel Fibrils into Crystalline Microtubes Enables a Rational Strategy To Stabilize the Hydrogel State. Langmuir 2015, 31, 9933–9942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.