

Abstract
Discovery of angiotensin converting enzyme 2 (ACE2) revealed that the renin angiotensin system (RAS) has two counterbalancing arms. ACE2 is a major player in the protective arm, highly expressed in lungs and gut with the ability to mitigate cardiopulmonary diseases such as inflammatory lung disease. ACE2 also exhibits activities involving gut microbiome, nutrition, and as a chaperone stabilizing the neutral amino acid transporter, B0AT1, in gut. But the current interest in ACE2 arises because it is the cell surface receptor for the novel coronavirus, SARS-CoV-2, to infect host cells, similar to SARS-CoV. This suggests that ACE2 be considered harmful, however because of its important other roles, it is paradoxically a potential therapeutic target for cardiopulmonary diseases including COVID-19, caused by SARS-CoV-2.
This review describes the discovery of ACE2, its physiological functions, and its place in the RAS. It illustrates new analyses of the structure of ACE2 that provides better understanding of its actions particularly in lung and gut, shedding of ACE2 by ADAM17 and role of TMPRSS2 in SARS-CoV-2 entry into host cells. Cardiopulmonary diseases are associated with decreased ACE2 activity and the mitigation by increasing ACE2 activity along with its therapeutic relevance are addressed. Finally, the potential use of ACE2 as a treatment target in COVID-19, despite its role to allow viral entry into host cells, is suggested.
Keywords: angiotensin-converting enzyme 2, SARS-COV-2, RAS, pulmonary hypertension, COVID-19, gut microbiome, brain-gut-lung axis, ADAM17
Introduction:
Discovery of angiotensin converting enzyme 2 (ACE2) was a major milestone proven to be paradigm-changing toward our understanding of the role of the renin-angiotensin system (RAS) in cardiovascular and pulmonary diseases. ACE2 is a transmembrane monopeptidyl carboxypeptidase that hydrolyzes angiotensin (Ang) I to Ang-(1–9) and Ang II to Ang-(1–7).1, 2 ACE2 together with Ang-(1–7) and the Mas receptor, comprise the vasoprotective axis of the RAS. This plays a key role in antagonizing the other RAS axis that has proliferative, hypertrophic, vasoconstrictive, inflammatory and apoptotic functions and consists of the angiotensin converting enzyme/angiotensin II/angiotensin type 1 receptor (ACE/Ang II/AT1 receptor).3,2,4,5 (Figure 1) A functional decrease in ACE2 has been implicated in development of cardiopulmonary diseases6,7,8,9 and other disorders impacting the cardiovascular (CV) system such as diabetes10,11 and hypertension.12,13 Therefore, increased expression or activation of ACE2 produces beneficial outcomes in these diseases.14,15,16,17,18,19,20
Figure 1:
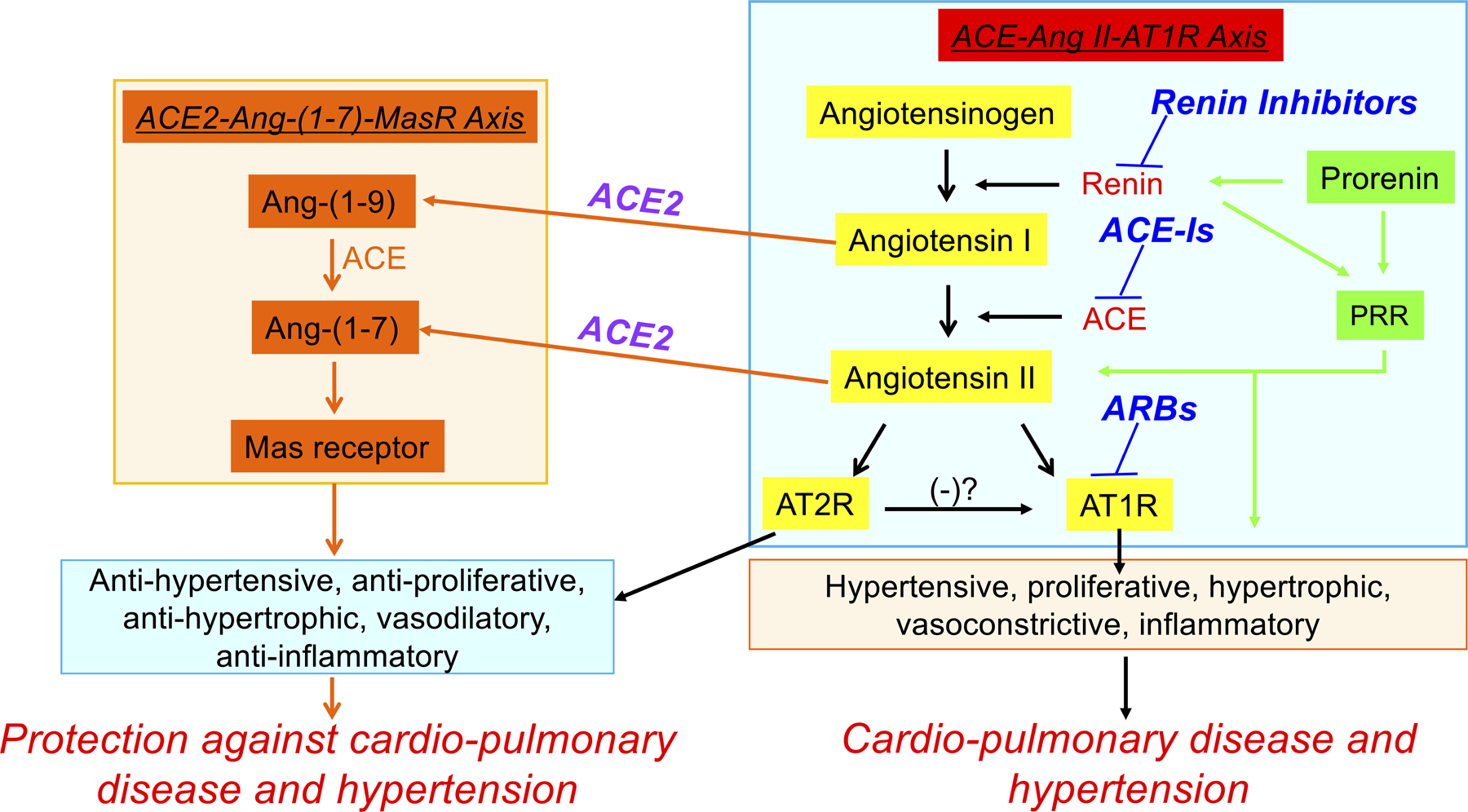
Vasodeleterious (ACE-AngII-AT1 receptor) and vasoprotective (ACE2-[Ang-(1–7]-Mas) axes of the renin-angiotensin system.
ACE2 gained widespread attention recently for its non-classical functions as a receptor for both severe acute respiratory syndromes caused by the viruses (SARS-CoV) and (SARS-CoV-2)21,22 and as stabilizing chaperone for the amino acid transporter B0AT1.23,24,4 Binding of SARS-CoV-2 to ACE2 and subsequent internalization of the complex are critical steps in infection initiation. As a result, plasma membrane ACE2 levels decreased leading to increases in the ACE/Ang II/AT1 receptor axis of the RAS and promotion of deleterious effects.25 Recent evidence supports this concept as circulating Ang II was elevated in novel coronavirus disease 19 (COVID-19) patients and positively correlated with lung injury and viral load.26 Conversely, patients dying from COVID-19 were deficient in ACE2 and reduced myocardial ACE2 expression was observed in post-mortem CV tissue from COVID-19 and SARS patients.25,4 More support comes from studies showing that treatment with catalytically active ACE2 ameliorated lung injury and fibrosis, vascular remodeling, rescued pulmonary hypertension (PH) and improved ventricular function in experimental models of human disease.20,17,8,15,18 These effects of ACE2 may be either direct actions on the lung or through the ACE2-dependant gut-lung axis. Therefore, ACE2’s beneficial outcomes on lung injury could be obtained by manipulating ACE2 or Ang-(1–7) to develop a therapeutic strategy against COVID-19 despite ACE2 on the epithelial surface of the lung being used by SARS-CoV-2 for entry into pulmonary cells.
Evidence is also accumulating that individuals with pre-existing conditions such as lung disease, CV disease (CVD), hypertension and/or diabetes are at particularly high risk for COVID-19.27,4 Additionally, abnormal gut, liver, renal and neurological function have been documented in COVID-19, suggesting multi-organ dysfunction is a critical manifestation of this disease.27,28,4 Experimental evidence supporting a central role of ACE2 in ameliorating the gut permeability and microbial dysbiosis in CVD29,30,31and lung diseases18,4,15,24 and neurobehavioral symptoms32 makes the ACE2/Ang-(1–7) axis an ideal target for managing COVID-19 pathophysiology. Here, we present a succinct understanding of the role of ACE2 and its product, Ang-(1–7), in cardiopulmonary diseases (CPDs), recent developments on involvement of ACE2 in SARS-CoV-2 infection, discuss various components of ACE2-mediated internalization of SARS-CoV-2 and propose mechanisms of the differing roles of ACE2 in this internalization in lungs and gut. Finally, we synthesize this information into a discussion of potential therapeutic targets for CPDs and COVID-19.
ACE2 structure and function:
In 2000, two seminal reports almost simultaneously described discovery of a gene encoding a protein with high similarity to well-known ACE but insensitive to its inhibitor, captopril.1,33 The enzyme was named ACE2 or ACEH (ACE homolog).33 The ACE2 gene contains 18 exons (Figure 2A) and is located on chromosome X (band: Xp22.2). The translated protein has a 740 amino acid extracellular N-terminal domain, a short single transmembrane domain, and a 44 amino acid cytosolic C-terminal tail (Figure 2A). ACE2 is a zinc metallocarboxyprotease in the cowrins family of metallopeptidases.34 The peptidase substrate binding pocket complexed with a specific inhibitor of ACE2, MLN-4760, was visualized using X-ray crystallography (Figure 2B).35 In contrast to ACE, ACE2 does not hydrolyze bradykinin.33 However, ACE2 can cleave apelin-13, apelin-17, Des-Arg-bradykinin, dynorphin A, neurotensin, and kinetensin.33,1,35 Functionally ACE2 converts angiotensin I to Ang-(1–9) and Ang II to Ang-(1–7).
Figure 2: A. Block diagrams of the ACE2 gene and ACE2, ADAM17 and TMPRSS2 proteins.
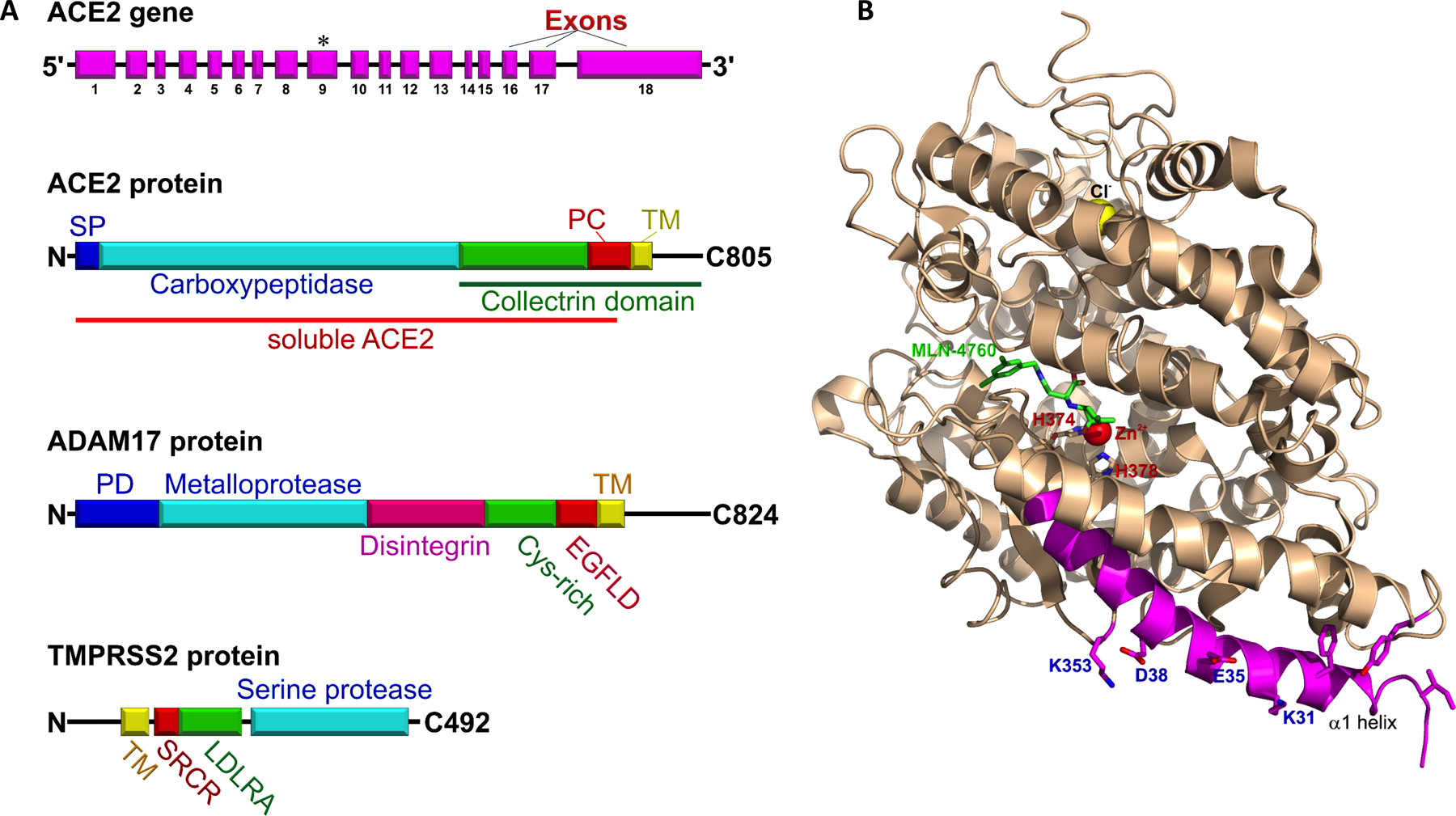
ACE2 gene, exons (magenta), asterisk indicates coding for zinc-binding site “HEMGH”. ACE2 protein comprised of N-terminal signal peptide (blue), catalytic carboxypeptidase domain (cyan), junctional segment of the collectrin domain (green), peptidase cleavage site (PC, red), transmembrane domain (TM, yellow), and the C-terminal tail. The distal part of ACE2 protein, (the protease cleavage site, the transmembrane domain and the C-terminal tail), is highly homologous to the classical collectrin domain (green line). The large proximal N-terminal ectodomain of the ACE2 protein (red line) can be shed after proteolytic cleavage by endopeptidases, such as ADAM17 and TMPRSS2. This shed segment of ACE2 is named soluble ACE2 (sACE2). sACE2 is a catalytically active serum protein. ADAM17 protein consists of the N-terminal prodomain (PD, blue), metalloprotease (cyan), disintegrin (magenta), cys-rich (green), epidermal growth factor-like (red), and transmembrane domains (yellow), and cytoplasmic tail. TMPRSS2 is a relatively short 492 amino acid protein that consists of a cytoplasmic N-terminal tail, transmembrane (yellow) and scavenger receptor cysteine-rich domains (SRCR, red), a low-density lipoprotein receptor A (LDLRA, green), and C-terminal serine protease domain (cyan).
B. A structure of human soluble ACE2 with its inhibitor (S,S)-2-[1-Carboxy-2-[3-(3,5-Dichloro-Benzyl)-3h-Imidazol-4-Yl]-Ethylamino]-4-Methyl-Pentanoic Acid (MLN-4760) at the presumed location of angiotensin I and angiotensin II catalytic site (pdb:1R4L). The cofactors Zn2+ and Cl− are shown as spheres (red and yellow, respectively). Histidine residues of the peptidase zinc-binding site HEMGH are labeled red (H374 and H378). The SARS-CoV and SARS-CoV-2 binding site is colored magenta, with key residues for binding S1-receptor binding domain of SARS-CoV-2 blue (K353, D38, E38, and K31).
ACE2 is membrane bound but can be shed extracellularly by several membrane-bound ectoproteases, such as a disintegrin and metallopeptidase domain 17 protein (ADAM17) and transmembrane serine proteases 2 (TMPRSS2) (Figure 2A).36,37 Several studies attempted to map cleavage sites of membrane-bound ACE2 attacked by ADAM17 and TMPRSS237,36 but no consensus has yet been reached. Further experiments will be necessary to resolve this.
ADAM17 sheds ACE2 in lung, heart, kidney and CNS, and this shedding may decrease ability of cells to counteract the deleterious effects of increased Ang II in COVID-19 (Figure 3). The soluble domain of ACE2 remains catalytically active and its recombinant form showed great potential in reducing circulating Ang II while alleviating associated dysfunction, including hypertension.5,38
Figure 3: ADAM17-mediated cleavage of monomeric ACE2 and release of soluble sACE2 ectodomain.
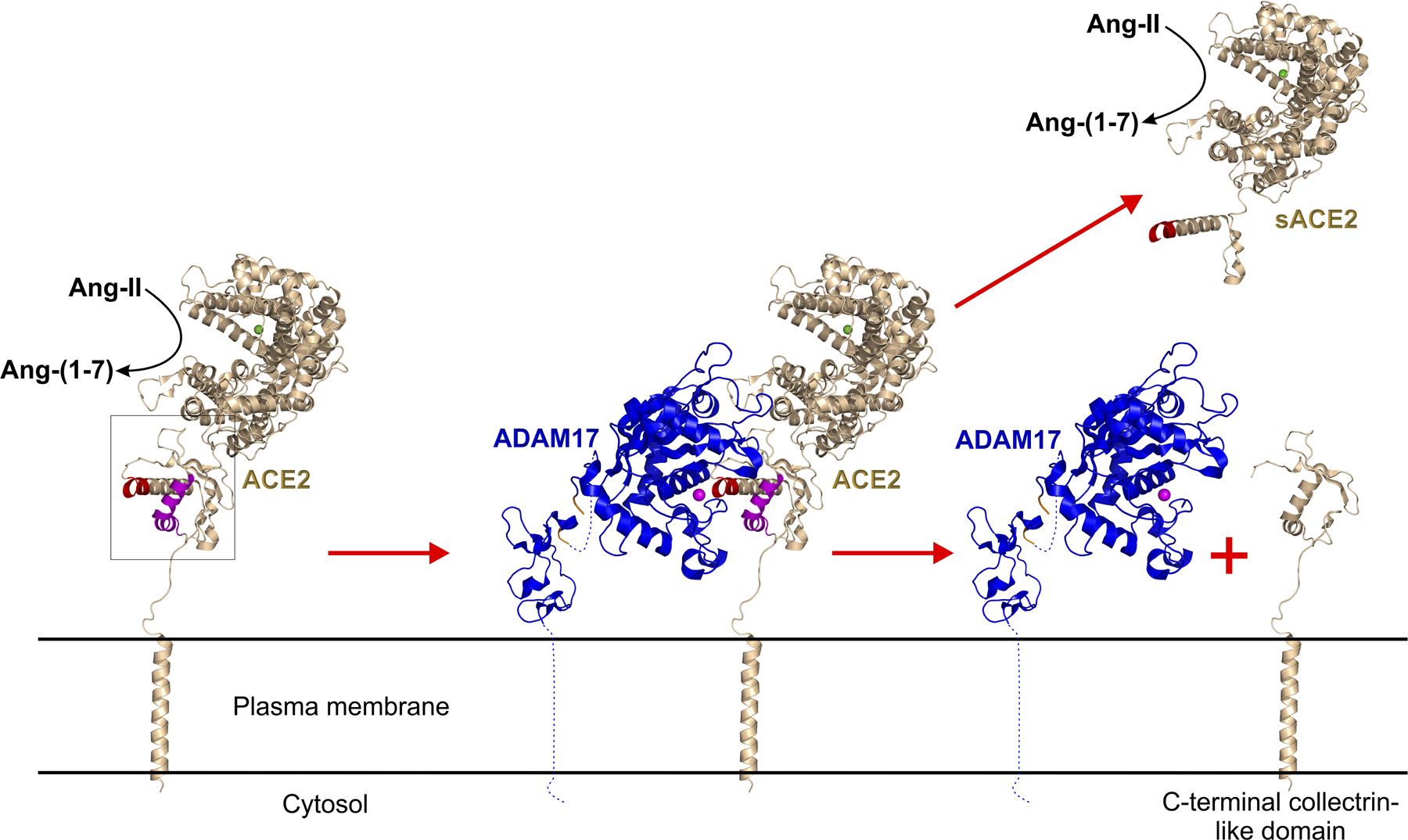
Left panel shows membrane-bound ACE2 (pdb: 6m17, gold) with box-outlined putative ADAM17 cleavage target site, red. The middle panel depicts ADAM17 (pdb:2m2f+2fv9) docking at its cleavage site within membrane-bound ACE2. The Zn2+ cofactor of ADAM17 is shown as a magenta sphere. The right panel shows the release of soluble sACE2 ectodomain following ADAM1 cleavage, leaving the membrane-bound C-terminal collectrin-like domain of ACE2. sACE ectodomain possess the enzymatic activity and the SARS-CoV-2 spike binding site.
Mechanism of ACE2-mediated SARS-CoV-2 entry into cells:
Human ACE2 (hACE2) is highly expressed in nasal and airway epithelium, lungs, small intestine, colon, kidneys and heart, with highest expression in intestines.4,39 The ACE2 expression pattern matters because both SARS-CoV and SARS-CoV-2 use membrane-bound ACE2 as a docking and anchoring site on the surface of epithelial cells,40 before viral RNA is internalized into the cytosol of victim cells. The SARS-CoV-2 ACE2-binding domain has a higher affinity for ACE2 versus SARS-CoV.41 The proteolytic cleavage-induced shedding of sACE2 is protective against SARS-CoV-2 virus infection of human epithelial cells in vitro.42 Therefore, sACE2 may exhibit therapeutic potential to alleviate COVID-19.
To date, it is well-established that TMPRSS2-mediated cleavage of the homotrimeric SARS-CoV-2 spike protein at its S1/S2 subunit junction is critical for virus entry into target cells (Figure 4).40 Spike protein subunits can exist in either open or closed (“down”) conformations.43 Cathepsin-mediated spike protein cleavage, independent from TMPRSS2 activity, has also been reported.40 Spike protein of SARS-CoV-2 additionally has a furin site at the same S1/S2 junction that is proteolytically cleaved in host cells during virus egress.43 The TMPRSS2-induced cleavage of S1 subunits from the SARS-CoV-2 spike protein allows the S2 subunit to initiate fusion of the viral and cellular membranes. Such fusion events were modeled in vitro.44 Alternatively, endocytosis of SARS-CoV-2 and reduction of viral entry by cathepsin L protease inhibition was reported.43,45 So the virus may use multiple pathways to access cells. After viral RNA enters the cytosol of host cells, initial translation is by the host ribosomes and thereafter by viral replication machinery. Viral replication occurs within double membrane vesicles whose presence in close proximity to the endoplasmic reticulum is characteristic of SARS-CoV-2 infected cells. Viral particle assembly involves the Golgi apparatus43 and viral RNA packaging into virions may be cytosolic.
Figure 4. SARS-CoV-2 interactions with TMPRSS2, monomeric lung ACE2 or tetrameric gut ACE2:B0AT1.
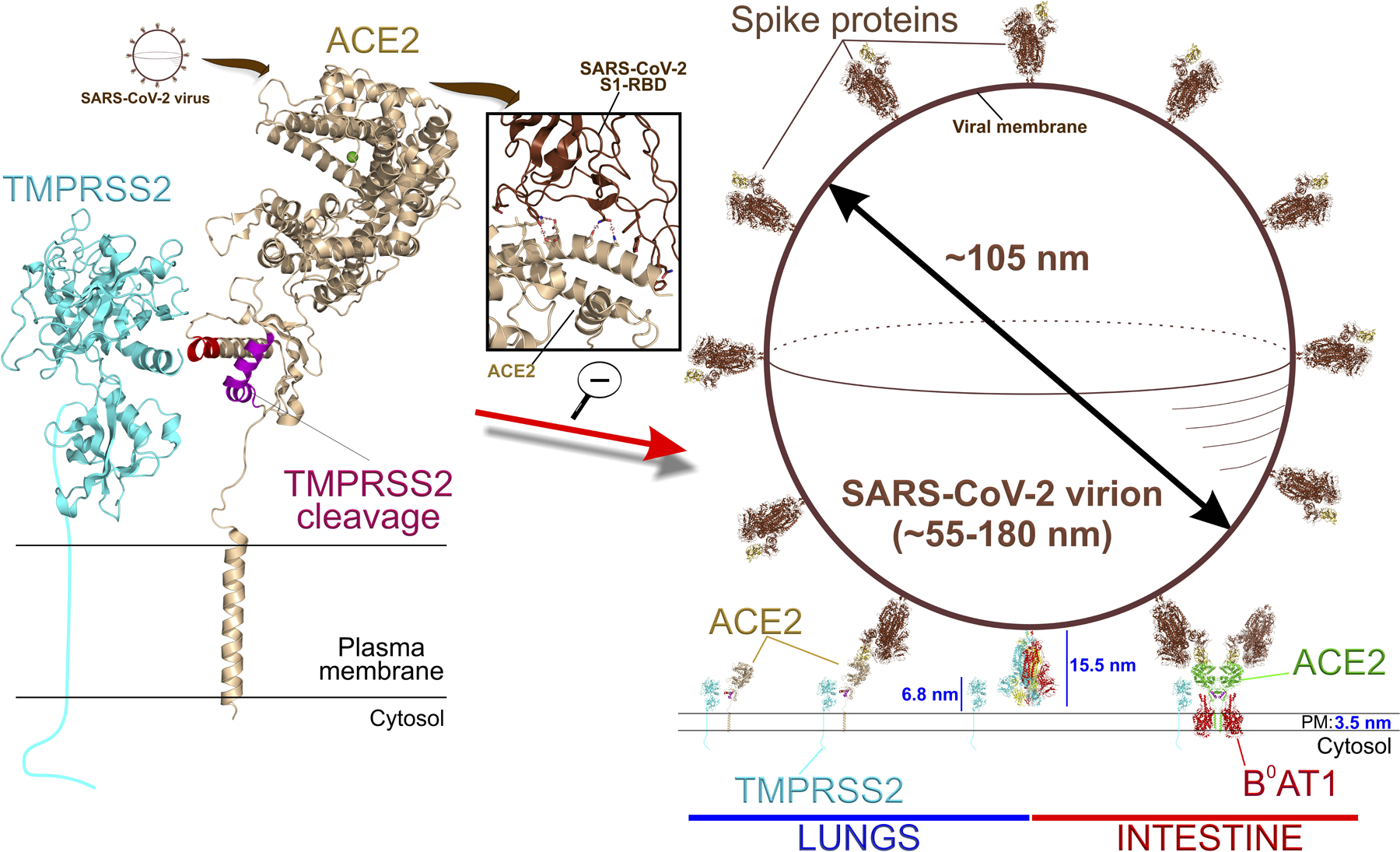
Upper left shows SARS-CoV-2 virion approaching ACE2, then binding its spike protein to the S1 receptor binding domain (RBD) of ACE2 ectodomain (enlarged center inset). TMPRSS2 (predicted structure; Swiss-Model:O15393, cyan) can putatively cleave off soluble sACE2 from a region overlapping with the ADAM17 cleavage site in lung monomer ACE2, but both proteases are sterically blocked by B0AT1 in gut ACE2 tetramer (see Figure 3). Right panel shows a scaled SARS-CoV-2 viral particle with spikes (brown) attached to either membrane-bound monomeric lung ACE2 or tetrameric intestinal ACE2:B0AT1 (pdb: 6m17; see Figure 5) (both organ events displayed side by side for graphical expediency). TMPRSS2 ultimately cleaves an adjacent free spike not attached to RBD (bottom center, multicolor15.5 nm long) at the S1/S2 junction, which primes SARS-CoV-2 for host cell fusion and entry.
The size of SARS-CoV-2 virions varies from 55 to 180 nm. Scaling a viral particle (~105 nm) and SARS-CoV-2 spike proteins (~15.5 nm) to the molecular sizes of ACE2 and TMPRSS2 proteins (~11nm and ~7 nm above the cell surface, respectively) reveals that TMPRSS2 is unlikely to cleave the spike protein anchored to an ACE2 protein (Figure 4). Rather, TMPRSS2 may cleave an adjacent spike protein complex, and this may occur even in adjacent spike proteins in the closed (“down”) conformation.
In the gastrointestinal tract (GI), ACE2 has enzymatic and non-enzymatic functions and both have implications in SARS-CoV-2 infection. First, as a member of the protective arm of the local GI RAS, ACE2 increases Ang-(1–7) and helps maintain gut barrier integrity against microbial dysbiosis by influencing glucose homeostasis via Na+-coupled SGLT1 and GLUT2, and release of GLP-1, GIP and CCK.46 Second, ACE2 chaperones its cytosolic trafficking partner subunit B0AT1, stabilized as a 2[ACE2:B0AT1] dimers of heterodimers (Figure 5).21 B0AT1 is the intestinal brush border membrane Na+-coupled neutral amino acid transport system discovered and characterized by Stevens and coworkers23, alternatively called NBB, B, B0. B0AT1 is the dominant pathway for gut uptake of tryptophan, leucine, glutamine and their metabolites implicated in dysfunctional gut-microbiome communication of many chronic diseases.47,29 Thus, physiological roles of gut B0AT1 are to 1) steer dietary protein digestion by absorption and processing of nutrient-sensor signaling by amino acids; 2) linking gut metabolism with the commensal gut microbiome by the metabolomic processing of luminal bioactive peptides in the context of humans as meta-organisms; 3) modulate whole-body glucose homeostasis by governing glucose uptake and incretin secretion; 4) steer Na+ and water absorption; and 5) defend against dysbiosis by mechanisms including innate immunity, acquired immunity, systemic inflammation and gut barrier integrity.
Figure 5: Diagrammatic representation of the role of the gut epithelial cell ACE2 in SARS-CoV-2 infection and gut pathology.
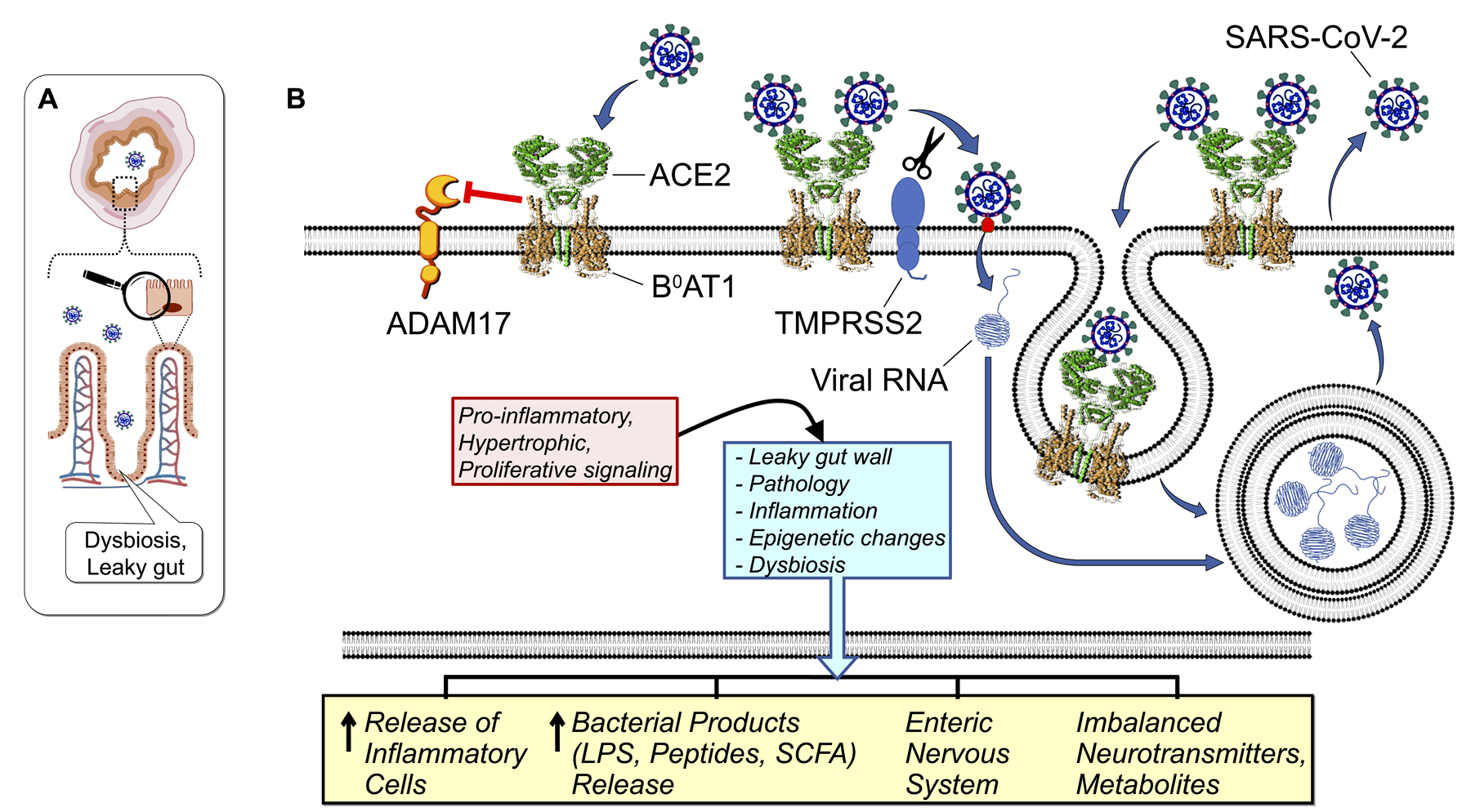
(A). The orientation of gut mucosa histology within a transverse cross-section of the intestine indicating virus in the lumen. Magnifying glass denotes the enlarged region of a single villus enterocyte, detailed in (B).
(B). Intestinal cell ACE2 chaperones the post-translational trafficking of B0AT1 to the apical membrane facing the lumen, forming a tetramer stabilized as a 2[ACE2:B0AT1] dimer of heterodimers (pdb: 6m17) limited primarily to gut cells of the body. Unlike lung monomeric ACE2 (see Figure 3), the intestinal ACE2 of the tetramer complex does not release soluble sACE2 ectodomains, because ADAM17 access is sterically blocked by the presence of B0AT1. SARS-CoV-2 spike S1 is activated (red) by integral membrane TMSRSS2 resulting in virus entry into host cells and replication within endosomes. Protease-damaged ACE2:B0AT1 is recycled by endosomal cathepsins. The disruption of gut ACE2 and B0AT1 initiates a complex signaling cascade leading to pathophysiological sequelae manifested locally within the gut and systemically. This includes the consequence of blunting B0AT1 transport of nutrient-signaling amino acids, e.g. tryptophan.
SARS-CoV-2 binding to intestinal apical membrane ACE2:B0AT1 decreases ACE2 activity disrupting the RAS, thereby inducing gut wall inflammation, barrier compromise and dysbiosis. Evidence for this includes: 1) significant number of COVID-19 patients have GI manifestations, sometimes prior to classic pulmonary symptoms.48; 2) COVID-19 patients exhibit altered fecal microbiomes.49 Interestingly, some of the altered bacteria have been associated with impaired ACE2.31,24; 3) disrupted gut nutrient-receptor signaling by B0AT1 transported amino acids, with attending disruption of glucose homeostasis and gut integrity.24; and 4) infectious SARS-CoV-2 virus is present in stool, and shed viral RNA is detected for weeks in sewage and toilet aerosols50 raising the potential for fecal transmission of SARS-CoV-2.
Gut-lung axis in COVID-19:
The pulmonary tropism of SARS-CoV-2, and attending respiratory pathophysiology and inflammation, may intimately be tied to gut tropism of SARS-CoV-2 and GI events of COVID-19. Firstly, active SARS-CoV-2 virus shed in feces50 and aerosolized from toilets likely contributes to pulmonary inoculation; this modality of transmission can inform the revisiting of public hygiene policy. Secondly, altered gut microbiome composition is reported in COVID-1949,51 consistent with involvement of an immune-gut dysbiosis-lung axis in COVID-19 progression.52 Although, impaired gut-lung communication in COVID-19 linked to ACE2 is presently an attractive concept, supporting evidence is rapidly emerging and further strengthened by evidence that gut and its microbiota are implicated in various pulmonary diseases including, chronic obstructive pulmonary disease (COPD)53, lung inflammatory diseases54 and pulmonary hypertension.18,55,56
Our studies were among the first demonstrating gut dysbiosis and gut pathology during PH in animal models.18,56 A link between gut dysbiosis and PH has been validated in patients with PH.55 This link is intriguing because the gut is a highly innervated and vascularized organ containing ~60% of the body’s immune cells. Furthermore, gut epithelium expresses high levels of ACE2.4 This raises questions of involvement of ACE2 in homeostasis of the gut epithelium and microbiota, also whether ACE2 influences PH pathophysiology. We have recent evidence indicating affirmative answers to these questions. Hypoxia-induced PH pathophysiology is attenuated in mice overexpressing ACE2.18 Moreover, fecal matter transfer from ACE2 overexpressing mice into wild type (WT) mice provides significant protection against PH.18 These studies demonstrate that gut microbiota composition alone has an impact on PH pathophysiology leading us to hypothesize gut-brain-lung axis dysfunction in PH (Figure 6). PH risk factors such as environmental toxins, smoking etc., can influence autonomic brain regions causing imbalanced sympathetic/parasympathetic activity resulting from microglia activation, increased pro-inflammatory pathways and neuroinflammation. Alternatively, PH risk factors may directly influence gut microbiota and epithelial-microbiota cross-talk resulting in leaky gut. This would permit release of inflammatory molecules, cells, microbiota metabolites into the blood to increase microglia activation and pathophysiological events in the lung and other cardiopulmonary-relevant organs. Together, they will perpetually influence overall PH pathology.
Figure 6: Hypothetical representation of dysfunctional gut-brain-lung axis in pulmonary hypertension.
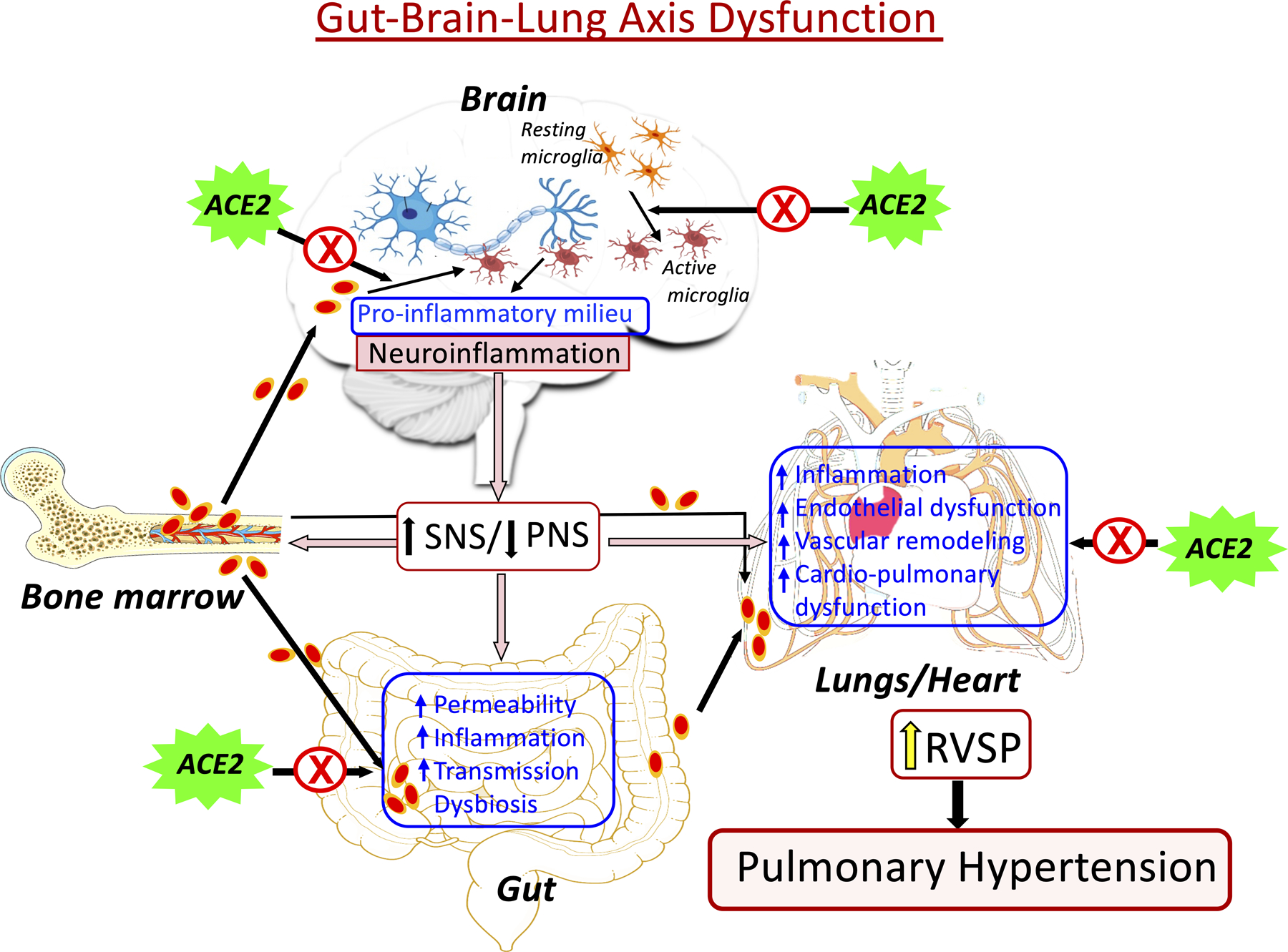
PH risk factors stimulate autonomic brain regions, initiating activation of microglia and neuroinflammation, altering neuron-microglia communication, and increased sympathetic nervous system activity. This sets up a sequence of events involving the gut, lung and bone marrow perpetuating the pathophysiological hallmarks of PH. ACE2 is proposed to act on the brain, gut, and lungs to interrupt this perpetual cycle and restore function to the axis.
Role of ACE2 in cardiopulmonary diseases (CPDs):
The most prevalent CPDs, with devastating health, economic and emotional impacts, include acute respiratory distress syndrome (ARDS), acute lung injury (ALI), and pulmonary hypertension (PH) and fibrosis (PF). The role and functions of RAS components, including ACE2, have been extensively studied in CPDs resulting in development of several therapeutic interventions. In this section, we review the physiological role of ACE2 and its involvement in the pathogenesis of various CPDs, including COVID-19.
1. Acute respiratory distress syndrome:
ARDS is a devastating inflammatory lung disease, commonly associated with multi-organ failure and high mortality. ARDS is a sudden injury to the lungs from infection, pathologically characterized by alveolar capillary leakage with diffuse alveolar damage. Clinical manifestations are severe hypoxemia and poor lung compliance. Mechanisms underlying ARDS are multifaceted making determination of associated risk factors challenging. Evidence strongly implicates RAS imbalance involving decreased ACE2 expression in the lung disorder pathophysiology.9,57 For instance, ACE2 deficient mice have severe ARDS and blocking the AT1 receptor prevents the syndrome.9,8,58 In other murine ARDS models, ACE2 activity is reduced with exaggerated neutrophil accumulation, augmented vascular permeability and aggravated pulmonary edema.8,9,57 Moreover, recombinant human ACE2 (rhACE2), purified from ACE2 transfected cell supernatant, improved pulmonary blood flow and oxygenation in the LPS-induced pig ARDS model.59
Lung ACE2 is primarily produced in alveolar epithelial (type II), Clara, endothelial and vascular smooth muscle cells.60 Due to epithelial cell injury during ARDS, ACE2 is decreased resulting in an increase in the pro-inflammatory, hypertrophic RAS axis9 Treatment of ACE2 deficient mice with ACEi or ARBs was beneficial, however ACEi and ARBs are not ideal because of potential to reduce systemic blood pressure. Instead, promising clinical trial results occurred with rhACE2.61
2. Acute lung injury
ALI is a milder form of ARDS with shared characteristics. Pulmonary ACE2 deficit and increased Ang II are associated with its development8 and ACE2 deficiency worsened virally-induced ALI.20 ACE2 null mice have more severe H5N1 influenza-induced ALI62 and ACE2 expression in lung was reduced in influenza induced by H5N162, H1N1 infection in WT mice.63 Furthermore, H5H1 influenza patients and mice with H5N1 influenza infection showed increased serum AngII.62 Thus, ACE2 supplementation and Ang II inhibition should ameliorate ALI64,15 and in fact rhACE2 normalized lung function in ACE2-deficient mice with H5N1 influenza-induced lung pathology.62 Therefore, rhACE2 was surmised as a novel therapeutic option for ALI.
Potential mechanisms underlying ALI amelioration by ACE2 supplementation is likely the counteracting of deleterious ACE/Ang II/AT1 receptor arm of the RAS. This blunts accumulation of inflammatory cells in the lungs8, reducing activation of LPS-TLR4 pathways65 and apoptosis of endothelial cells through suppression of SMAD phosphorylation.66 These data suggest an imbalance of RAS arms drives ALI, and ACE2 maintains RAS balance to protect lungs against ALI.
3. Pulmonary Fibrosis:
PF is a progressive lung disorder associated with high morbidity and mortality secondary to ALI/ARDS and in some cases as idiopathic pulmonary fibrosis (IPF). IPF is a fibrotic lung disease with distorted pulmonary architecture resulting in respiratory failure with right heart dysfunction and death. Early diagnosis can help manage symptoms, minimize inflammation and slow disease progression and with a healthy lifestyle may improve quality of life. There are few FDA-approved treatments such as pirfenidone (Esbret) and nintedanib (Ofev) antifibrotics that slow progression of lung damage.
Histopathologic characteristics of IPF include damaged alveolar epithelium and enlarged airspaces with thick fibrotic walls filled with mucin and inflammatory cells. Overproduction and deposition of collagen and extracellular matrix are also indicative of fibrotic lung.67 A determinant in the pathogenesis of IPF is local activation of deleterious RAS axis. This mediates production of Ang II and its precursor angiotensinogen (AGT) from apoptotic alveolar cells damaging lung tissue.68,69 Furthermore, ACE2 expression and activity are decreased in humans with IPF and in animal models of lung fibrosis.6,7,70 ACE2 null mice were more susceptible to bleomycin-induced lung fibrosis20,8 while ACE2 overexpression ameliorated bleomycin-induced lung fibrosis by reversing local ACE2 loss and degrading elevated lung Ang II and AGT.69 Intraperitoneal rhACE2 abrogates bleomycin-induced fibrosis and inflammation improving lung function, exercise capacity and survival in mice.20,8 ACE2 protects lungs by both degrading local Ang II and production of Ang-(1–7) which shifts RAS balance towards the anti-inflammatory, anti-fibrotic actions via the ACE2-Ang-(1–7)-Mas axis.
4. Pulmonary Hypertension
PH is a chronic and incurable CPD characterized by increased blood pressure (BP) in pulmonary arteries that progresses to right heart failure and death. Factors causing PH are heredity, drug-induced, HIV infection, schistosomiasis, portal hypertension and congenital heart disease. The pathophysiology is complex and multifactorial, involving smooth muscle cell proliferation and endothelial dysfunction leading to increased resistance, constriction, fibrosis and remodeled pulmonary vessels.71,72
Most PH treatments are directed towards the pulmonary vasculature. Thus, limited success in PH control and treatment of PH could be due to the dogma that it is a disease of the heart, lung and blood vessels. PH was recently suggested to be a systemic disorder18,73 because of the involvement of multiple organs (e.g. gut, brain, bone marrow).18 Activation of the ACE/Ang II/AT1 receptor arm of the RAS leads to endothelial dysfunction and vascular remodeling of PH. But discovery of ACE2 as a regulator of RAS launched investigations into its role in PH pathology and therapy. Animal models of PH demonstrated decreased ACE215 leading investigators to test the hypothesis that increased pulmonary and circulating ACE2 decrease PH-associated pathophysiology. Evidence supporting this concept includes: 1) Lentivirus-mediated ACE2 overexpression protected monocrotaline (MCT)-treated mice from increased right ventricular systolic pressure (RVSP), remodeling of pulmonary vessels and right ventricular hypertrophy (RVH).14 Overexpression also reversed established elevations of RVSP and muscularization of pulmonary vessels14, interestingly, without affecting systolic blood pressure. 2) Overexpression of Ang-(1–7) prevented bleomycin-induced pulmonary fibrosis and blocking Mas receptor inhibited this.7 3) Activation of ACE2 by diminazene aceturate (DIZE) both attenuated and arrested the progression of PH pathophysiology in multiple rat models involving angiogenic progenitor cells derived from bone marrow.16 Other ACE2 activators, resorcinolnapthalin and NCP2454, offered similar protection by improving endothelial function and suppressing intracellular inflammatory cascades.74 Oral delivery of ACE2 or Ang-(1–7) bioencapsulated in plant cells attenuated and arrested PH progression and pathophysiology.15 Bioencapsulation of a therapeutic protein like ACE2 for oral administration is a novel technology circumventing many limitations of delivery of ACE2 or Ang-(1–7) to preclinical testing.
Numerous preclinical studies have implicated benefits of ACE2 in PH. Decreased circulating ACE2 activity has been reported in PH patients.70,75 Others reported that although ACE2 concentration was higher, its enzymatic activity was reduced in PH patients versus control.75 This discrepancy was a result of more autoantibodies to ACE2 in PH patients versus healthy controls. Similarly, circulating Ang-(1–7) was reduced in PH patients with congenital heart disease.75 Finally, a small cohort study using rhACE2 supported the beneficial effect of ACE2 in PH.70 Taken together, decreased ACE2 is associated with PH pathophysiology and increasing it confirm that PH is associated with reduced ACE2 activity. Increasing ACE2 with rhACE2 is proposed to ameliorate oxidative stress, inflammatory status and disordered hemodynamics.
Autonomic nervous system imbalance has long been implicated in PH pathophysiology. Higher sympathetic activity and increased muscle sympathetic nerve activity correlated with increased heart rate in PH patients.76 Sympathetic stimulation contributes to pulmonary vasoconstriction while pulmonary artery denervation reduces pulmonary pressure.77 Furthermore, vagal nerve stimulation restores autonomic balance, preserves RV function and ameliorates vascular remodeling in PH animal models.78 This imbalance is associated with activation of microglia in autonomic brain regions, predominantly the paraventricular nucleus of the hypothalamus (PVN). This led us to propose that activation of microglia alters microglia-neuronal communication to induce neuroinflammation and increased sympathetic activity in PH.72 Evidence for this includes: (1) The number of activated microglia in the PVN positively correlates with RVSP.79 (2) Microglia in mice devoid of CX3CR1 remain at “resting state” under environmental effectors known to induce activation in WT mice.79 These mice were protected from hypoxia-induced PH.79 (3) Minocycline, an anti-inflammatory antibiotic that easily penetrates the blood brain barrier (BBB), inhibits activation of microglia. Treatment with minocycline attenuated activation of PVN microglia and ameliorated MCT-induced PH pathophysiology.72 ACE2, intrinsic to autonomic brain regions including the PVN, plays a critical role in neuroinflammation-driven PH pathophysiology. For example, global ACE2 overexpressing mice were protected from hypoxia-induced PH and failed to increase the number of activated PVN microglia.18
ACE2 as a target for SARS-CoV-2 Treatments:
(i). Increase ACE2:
The idea that increasing ACE2 would be beneficial is based on the decrease of plasma membrane ACE2 following internalization of SARS-CoV-2 complexed with it. This shifts the RAS towards the vasodeleterious axis increasing inflammatory, fibrotic, and hypertrophic actions of Ang II and the AT1 receptor, accounting for most of the symptoms of COVID-19 patients. Support is from data that ACE2 expression correlates with SARS-CoV spike protein-driven entry80 and is reduced in elderly subjects25, a group more susceptible to COVID-19. Furthermore, sACE2 combines with SARS-CoV-2 to decrease infectivity 1000–5000-fold in human blood vessel and kidney organoids.81 However, a recent study showed increased plasma ACE2 in COVID-19 patients by an ELISA assay.82 These counterintuitive data could be explained by a compensatory response or/and dimerization of ACE2, a configuration with little SARS-CoV-2 binding affinity.21 There are many potential mechanisms for the benefits of increased ACE2, including that circulating ACE2 may bind SARS-CoV-2 and remove it from circulation. Additionally, increased ACE2 could promote self-dimerization in cell membrane, reducing affinity for virus, internalization and propagation. Finally, increasing ACE2 would shift the RAS towards its vasoprotective functions. Multiple approaches and technologies are ready to be tested for therapeutic potential.
(ii). Recombinant human ACE2 protein.
Substantial evidence supports the validity of this approach. For example, treatment with rhACE2 is protective against vascular and pulmonary damage in many lung diseases.5,57,59,4 It reduces viral load and blocks SARS-CoV-2 infection in Vero E6 cells and organoids made from blood vessels and kidney.42 A phase II study is underway to test the efficacy of rhACE2 in COVID-19 patients (NCT04287686). Even though rhACE2 is safe and reduces systemic inflammation (NCT01597635) its stability, relatively short half-life (3.5 h) and high manufacturing costs need to be addressed.
(iii). ACE2 activators.
ACE2 activators like DIZE prevent and arrest progression of pulmonary pathophysiology induced by PH and alveolar remodeling16,64 and reduce MI-induced cardiac damage and stroke.83 DIZE is an anti-parasitic for the treatment of trypanosomiasis and commercially available (VERIBEN®and Berenil®).
(iv). Bioencapsulated ACE2 and ACE2/Ang-(1–7) probiotics.
Our group has established two oral delivery systems for ACE2, sACE2 and Ang-(1–7) to circumvent limitations to use of recombinant human proteins described above and inactivation of proteins by stomach acid and/or digestion. Engineering of plant chloroplasts by the transplastomic method generated orally deliverable ACE2 and Ang-(1–7) that increases ACE2 and Ang-(1–7) in blood. Feeding this material provided impressive protection against PH pathophysiology.15 In addition, pulmonary ACE2 levels were increased and AT1 receptor and pro-inflammatory cytokines (TNFα, TGFβ, TLR4) were decreased with improvement of right heart function. Additionally, feeding of ACE2 reduced endotoxin-induced uveitis.15 More recently, Lactobacillus paracasei expressing ACE284 was effective in diabetes-induced retinopathy.84 This probiotic strategy is easily scalable to produce large quantities of clinical grade shACE2 to sequester SARS-CoV-2 and potentially arrest pathophysiology of COVID-19. The probiotic delivery system has a potential additional advantage: Direct influence over gut B0AT1 transporter and gut epithelial-gut microbiota communication presumably relevant to the GI-related manifestations in COVID-19 patients.
(v). ACE inhibitors, angiotensin receptor blockers and biased AT1 receptor agonists.
Significant experimental evidence indicates that ACE-Is and ARBs increase ACE2.85,4 This led to predictions that COVID-19 patients with HTN taking these drugs would have worse outcomes. On the contrary, three recent retrospective studies demonstrate interesting observations. A Chinese study with 362 COVID-19 patients on ACE-Is or ARBs showed no difference in severity or mortality compared with patients without them86 and a second multicenter study involving 1128 patients reported significantly lower mortality.87 Finally, an UK study with 205 patients (not yet peer-reviewed), found reduced necessity for critical care and mortality when ACE-Is or ARBs were given 7 days before onset of COVID-19 symptoms.88 The REPLACE COVID trial is determining whether discontinuation of these drugs attenuate severity of the disease (NCT04338009). However, impacts of these drugs in high risk patients with HTN, diabetes and obesity on COVID-19 outcomes remain unknown.
A novel strategy of using β-arrestin-biased angiotensin receptor agonists has been proposed.89 This is based on the anti-inflammatory, anti-apototic and vasodilatory benefits of β-arrestin-mediated AT1 receptor signaling.89 Although, Ang II has both deleterious and protective effects, β-arrestin-biased ligands selectively activate only the β-arrestin pathway. Interestingly, Ang-(1–7) acts as a β-arrestin-biased ligand for the AT1 receptor.90 Therefore, these ligands, alone or in combination with ACE2, would be a worthy of consideration.
(vi). Agents that influence plasma membrane ACE2.
Two proteases, TMPRSS2 and ADAM17, critically regulate plasma membrane ACE2 and thus present interesting targets. TMPRSS2 is essential for entry of SARS-CoV-2 into cells by priming its spike protein.40 An inhibitor of this enzyme, camostat mesylate, demonstrably reduced SARS entry into cells40 and a clinical trial is evaluating its efficacy in COVID-19 patients (NCT04321096). ADAM17 sheds the extracellular domain of ACE2 and sACE2 has both enzyme activity and SARS-binding properties.37 It has been postulated that SARS-CoV increases both shedding and internalization to decrease cellular ACE2 which suggests that ADAM17 inhibitors would be beneficial. Therefore, the ADAM17 inhibitors, paricalcitol and synthetic vitamin D analogues need to be tested for clinical use.
(vii). Repurposing of anti-inflammatory drugs.
Inflammation and “cytokine storm” are characteristics of COVID-19. Additionally, neuroinflammation has been postulated in COVID-19 patients with neurological abnormalities91 However, the long-term overall health implications of neurological symptoms following recovery remain speculative. Therefore, it is prudent to consider neural effects of COVID-19 and test strategies for their control. Non-steroidal anti-inflammatory drugs (NSAIDs) have been tried but the latest WHO report indicates no resulting evidence of reduction in either severe adverse events, long-term survival or improvement to quality of life in COVID-19 patients. However, long-term outcomes cannot be evaluated at this early time point in the pandemic. This does not preclude testing of other classes of anti-inflammatory drugs that penetrate the BBB. For example, minocycline, a safe anti-inflammatory antibiotic in use for ≥40 years, easily passes through the BBB and attenuates neuro-inflammation.92 Our studies with experimental animal models have shown impressive effects of minocycline on neuroinflammation-driven HTN, PH and lung inflammation.72,93,94 Furthermore, minocycline’s antibiotic properties may rebalance gut microbiota and gut functions in COVID-19 patients with GI-related abnormalities.
Concluding Remarks:
We have discussed the importance of homeostasis of the RAS in maintaining lung function that, once perturbed in favor of the ACE/Ang II/AT1R axis predisposes to cardiopulmonary diseases such as ARDS, ALI, PF and PH, including its particular relevance to COVID-19. The imbalance can be ameliorated by reducing the ACE/AngII/AT1R or increasing the ACE2/Ang-(1–7)/MasR axes. There are effective means to increase the beneficial arm of the RAS, including infusing rhACE2, feeding bioencapsulated ACE2, using probiotics designed to release ACE2 or Ang-(1–7), drug-induced activation of ACE2, genetic manipulation and using beneficially biased agonists of the AT1R. These relieve cardiopulmonary disease. The RAS also has important effects in the gut, where imbalance similarly causes inflammation and, ACE2 in complex with B0AT1 as a dimer of heterodimers, effects neutral amino acid transport, dampening systemic and gut inflammation, and promoting healthy microbial-host interactions that impact the lung.
The novel coronavirus, SARS-CoV-2, the cause of pandemic COVID-19, has thrown the RAS into the spotlight. COVID-19 is a respiratory infection, spread mostly by respiratory droplets, but there is productive infection of the gut and shedding of virus in feces. ACE2, highly expressed in the lung and gut, is the receptor this virus (and SARS-CoV) uses to enter cells via viral spike protein binding to ACE2 and internalized with virus. We discuss how the cell is then doubly jeopardized by viral infection and imbalanced RAS, and how rebalancing the RAS as described above would bring benefits. We illustrate more complete understanding of virus binding with ACE2 and its interaction partners at the cell surface revealing other potential treatments. For example, ADAM17 cleaves ACE2 in the lung releasing sACE2; shedding is increased upon SARS-CoV infection suggesting ADAM17 inhibitors as possible candidates. The spike protein of the virus in either of its configurations is activated by TMPRSS2 to effect cell entry although it appears TMPRSS2 is unable to activate the spike protein bound to ACE2. Inhibitors of TMPRSS2 are in clinical trial for COVID-19. We suggest that while the world awaits development of an effective vaccine against COVID-19, there are available potentially beneficial treatments targeting the RAS, despite ACE2 being the receptor for SARS-CoV-2 entry into cells.
Acknowledgement:
Source of Funding: This study was supported by the National Institutes of Health grants HL102033 and HL132448 to M.K. Raizada.
Footnotes
Disclosure: None
References:
- 1.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–9. [DOI] [PubMed] [Google Scholar]
- 2.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. [DOI] [PubMed] [Google Scholar]
- 3.Simões e Silva AC, Silveira KD, Ferreira AJ and Teixeira MM. ACE2, angiotensin-(1–7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169:477–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB and Oudit GY. Angiotensin Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ Res. 2020;126:1456–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–28, 18 p following 728. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A and Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Díez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D et al. The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010;182:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wösten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic J, Florquin S and Bos AP. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–27. [DOI] [PubMed] [Google Scholar]
- 10.Patel VB, Bodiga S, Basu R, Das SK, Wang W, Wang Z, Lo J, Grant MB, Zhong J, Kassiri Z et al. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res. 2012;110:1322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tikellis C, Pickering R, Tsorotes D, Du XJ, Kiriazis H, Nguyen-Huu TP, Head GA, Cooper ME and Thomas MC. Interaction of diabetes and ACE2 in the pathogenesis of cardiovascular disease in experimental diabetes. Clin Sci (Lond). 2012;123:519–29. [DOI] [PubMed] [Google Scholar]
- 12.Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. 2006;116:2218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. [DOI] [PubMed] [Google Scholar]
- 14.Yamazato Y, Ferreira AJ, Hong KH, Sriramula S, Francis J, Yamazato M, Yuan L, Bradford CN, Shenoy V, Oh SP et al. Prevention of pulmonary hypertension by Angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54:365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shenoy V, Kwon KC, Rathinasabapathy A, Lin S, Jin G, Song C, Shil P, Nair A, Qi Y, Li Q et al. Oral delivery of Angiotensin-converting enzyme 2 and Angiotensin-(1–7) bioencapsulated in plant cells attenuates pulmonary hypertension. Hypertension. 2014;64:1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, Ferreira AJ, Fraga-Silva RA, Kearns P et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187:648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, Ostrov DA, Oh SP, Katovich MJ et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma RK, Oliveira AC, Yang T, Karas MM, Li J, Lobaton GO, Aquino VP, Robles-Vera I, de Kloet AD, Krause EG et al. Gut Pathology and Its Rescue by ACE2 (Angiotensin-Converting Enzyme 2) in Hypoxia-Induced Pulmonary Hypertension. Hypertension. 2020;76:206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH and Oudit GY. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J Am Coll Cardiol. 2017;69:805–819. [DOI] [PubMed] [Google Scholar]
- 20.Rey-Parra GJ, Vadivel A, Coltan L, Hall A, Eaton F, Schuster M, Loibner H, Penninger JM, Kassiri Z, Oudit GY et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J Mol Med (Berl). 2012;90:637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan R, Zhang Y, Li Y, Xia L, Guo Y and Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens BR, Ross HJ and Wright EM. Multiple transport pathways for neutral amino acids in rabbit jejunal brush border vesicles. J Membr Biol. 1982;66:213–25. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdecchia P, Cavallini C, Spanevello A and Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, Wang Z, Li J, Feng C, Zhang Z et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J, Wang H, Shen H, Qiu L, Li Z et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol. 2004;203:622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perlot T and Penninger JM. ACE2 - from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013;15:866–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qi Y, Kim S, Richards EM, Raizada MK and Pepine CJ. Gut Microbiota: Potential for a Unifying Hypothesis for Prevention and Treatment of Hypertension. Circ Res. 2017;120:1724–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan Y, Prasad R, Feng D, Beli E, Li Calzi S, Longhini ALF, Lamendella R, Floyd JL, Dupont M, Noothi SK et al. Bone Marrow-Derived Cells Restore Functional Integrity of the Gut Epithelial and Vascular Barriers in a Model of Diabetes and ACE2 Deficiency. Circ Res. 2019;125:969–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, de Kloet AD, Pati D, Hiller H, Smith JA, Pioquinto DJ, Ludin JA, Oh SP, Katovich MJ, Frazier CJ et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology. 2016;105:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G and Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43. [DOI] [PubMed] [Google Scholar]
- 34.Gomis-Rüth FX. Structure and mechanism of metallocarboxypeptidases. Crit Rev Biochem Mol Biol. 2008;43:319–45. [DOI] [PubMed] [Google Scholar]
- 35.Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279:17996–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O and Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88:1293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM and Turner AJ. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. 2005;280:30113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wysocki J, Ye M, Rodriguez E, González-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, Li Z, Cui X, Xiao J, Zhan J et al. Digestive system is a potential route of COVID-19: an analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut. 2020;69:1010–1018. [Google Scholar]
- 40.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A and Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020;181:905–913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT and Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong TP, Ho KY, Ng EK, Debnam ES and Leung PS. Upregulation of ACE2-ANG-(1–7)-Mas axis in jejunal enterocytes of type 1 diabetic rats: implications for glucose transport. Am J Physiol Endocrinol Metab. 2012;303:E669–81. [DOI] [PubMed] [Google Scholar]
- 47.Fairweather SJ, Bröer A, O’Mara ML and Bröer S. Intestinal peptidases form functional complexes with the neutral amino acid transporter B(0)AT1. Biochem J. 2012;446:135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian Y, Rong L, Nian W and He Y. Review article: gastrointestinal features in COVID-19 and the possibility of faecal transmission. Aliment Pharmacol Ther. 2020;51:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, Wan Y, Chung A, Cheung CP, Chen N et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology. 2020;S0016–5085:34701–6. doi: 10.1053/j.gastro.2020.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xing YH, Ni W, Wu Q, Li WJ, Li GJ, Wang WD, Tong JN, Song XF, Wing-Kin Wong G and Xing QS. Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J Microbiol Immunol Infect. 2020;53:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, Guo F, Zhang X, Luo R, Huang C et al. Alterations of the Gut Microbiota in Patients with COVID-19 or H1N1 Influenza. Clin Infect Dis. 2020; ciaa709 Online ahead of print, doi: 10.1093/cid/ciaa709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang AT and Marsland BJ. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 2019;12:843–850. [DOI] [PubMed] [Google Scholar]
- 53.Marsland BJ, Trompette A and Gollwitzer ES. The Gut-Lung Axis in Respiratory Disease. Ann Am Thorac Soc. 2015;12 Suppl 2:S150–6. [DOI] [PubMed] [Google Scholar]
- 54.Wang Q, Li F, Liang B, Liang Y, Chen S, Mo X, Ju Y, Zhao H, Jia H, Spector TD et al. A metagenome-wide association study of gut microbiota in asthma in UK adults. BMC Microbiol. 2018;18:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim S, Rigatto K, Gazzana MB, Knorst MM, Richards EM, Pepine CJ and Raizada MK. Altered Gut Microbiome Profile in Patients With Pulmonary Arterial Hypertension. Hypertension. 2020;75:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma RK, Oliviera A, Goel R, Aquino V, Lobaton G, Kim S, Richards EM, Katovich MJ and Raizada MK. Microbial Dysbiosis and Altered Gut Pathology in Monocrotaline-Induced Pulmonary Hypertension. B108. Pulm Hyper: Latest Findings. 2018; A4381–A4381. [Google Scholar]
- 57.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature medicine. 2005;11:875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H and Sun GY. LPS induces permeability injury in lung microvascular endothelium via AT(1) receptor. Arch Biochem Biophys. 2005;441:75–83. [DOI] [PubMed] [Google Scholar]
- 59.Treml B, Neu N, Kleinsasser A, Gritsch C, Finsterwalder T, Geiger R, Schuster M, Janzek E, Loibner H, Penninger J et al. Recombinant angiotensin-converting enzyme 2 improves pulmonary blood flow and oxygenation in lipopolysaccharide-induced lung injury in piglets. Crit Care Med. 2010;38:596–601. [DOI] [PubMed] [Google Scholar]
- 60.Wiener RS, Cao YX, Hinds A, Ramirez MI and Williams MC. Angiotensin converting enzyme 2 is primarily epithelial and is developmentally regulated in the mouse lung. J Cell Biochem. 2007;101:1278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, Hardes K et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zou Z, Yan Y, Shu Y, Gao R, Sun Y, Li X, Ju X, Liang Z, Liu Q, Zhao Y et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014;5:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X, Yang N, Tang J, Liu S, Luo D, Duan Q and Wang X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014;185:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rigatto K, Casali KR, Shenoy V, Katovich MJ and Raizada MK. Diminazene aceturate improves autonomic modulation in pulmonary hypertension. Eur J Pharmacol. 2013;713:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ye R and Liu Z. ACE2 exhibits protective effects against LPS-induced acute lung injury in mice by inhibiting the LPS-TLR4 pathway. Exp Mol Pathol. 2020;113:104350. [DOI] [PubMed] [Google Scholar]
- 66.Ji Y, Gao F, Sun B, Hao J and Liu Z. Angiotensin-Converting Enzyme 2 Inhibits Apoptosis of Pulmonary Endothelial Cells During Acute Lung Injury Through Suppressing SMAD2 Phosphorylation. Cell Physiol Biochem. 2015;35:2203–12. [DOI] [PubMed] [Google Scholar]
- 67.Wilson MS and Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uhal BD, Dang MT, Li X and Abdul-Hafez A. Angiotensinogen gene transcription in pulmonary fibrosis. Int J Pept. 2012;2012:875910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Wang Y, Yang T, Guo Y and Sun T. Angiotensin-Converting Enzyme 2 Attenuates Bleomycin-Induced Lung Fibrosis in Mice. Cell Physiol Biochem. 2015;36:697–711. [DOI] [PubMed] [Google Scholar]
- 70.Hemnes AR, Rathinasabapathy A, Austin EA, Brittain EL, Carrier EJ, Chen X, Fessel JP, Fike CD, Fong P, Fortune N et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51:170238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ward JP and McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol. 2009;9:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma RK, Oliveira AC, Kim S, Rigatto K, Zubcevic J, Rathinasabapathy A, Kumar A, Lebowitz JJ, Khoshbouei H, Lobaton G et al. Involvement of Neuroinflammation in the Pathogenesis of Monocrotaline-Induced Pulmonary Hypertension. Hypertension. 2018;71:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gurtu V and Michelakis ED. A Paradigm Shift Is Needed in the Field of Pulmonary Arterial Hypertension for Its Entrance Into the Precision Medicine Era. Circ Res. 2016;119:1276–1279. [DOI] [PubMed] [Google Scholar]
- 74.Haga S, Tsuchiya H, Hirai T, Hamano T, Mimori A and Ishizaka Y. A novel ACE2 activator reduces monocrotaline-induced pulmonary hypertension by suppressing the JAK/STAT and TGF-β cascades with restored caveolin-1 expression. Exp Lung Res. 2015;41:21–31. [DOI] [PubMed] [Google Scholar]
- 75.Sandoval J, Del Valle-Mondragón L, Masso F, Zayas N, Pulido T, Teijeiro R, Gonzalez-Pacheco H, Olmedo-Ocampo R, Sisniega C, Paez-Arenas A et al. Angiotensin Converting Enzyme 2 and Angiotensin (1–7) axis in Pulmonary Arterial Hypertension. Eur Respir J. 2020;1902416. [DOI] [PubMed] [Google Scholar]
- 76.Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R and van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004;110:1308–12. [DOI] [PubMed] [Google Scholar]
- 77.Le T, Makar C, Morway P, Hoftman N and Umar S. Pulmonary artery denervation: a novel treatment modality for pulmonary hypertension. J Thorac Dis. 2019;11:1094–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshida K, Saku K, Kamada K, Abe K, Tanaka-Ishikawa M, Tohyama T, Nishikawa T, Kishi T, Sunagawa K and Tsutsui H. Electrical Vagal Nerve Stimulation Ameliorates Pulmonary Vascular Remodeling and Improves Survival in Rats With Severe Pulmonary Arterial Hypertension. JACC Basic Transl Sci. 2018;3:657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oliveira AC, Sharma RK, Aquino V, Lobaton G, Bryant AJ, Harrison JK, Richards EM and Raizada MK. Involvement of Microglial Cells in Hypoxia-induced Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2018;59:271–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nie Y, Wang P, Shi X, Wang G, Chen J, Zheng A, Wang W, Wang Z, Qu X, Luo M et al. Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem Biophys Res Commun. 2004;321:994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang H, Zhou P, Wei Y, Yue H, Wang Y, Hu M, Zhang S, Cao T, Yang C, Li M et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann Intern Med. 2020;172:629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang H, Shang W, Liu Q, Zhang X, Zheng M and Yue M. Clinical characteristics of 194 cases of COVID-19 in Huanggang and Taian, China. Infection. 2020;1–8. Online ahead of print, doi: 10.1007/s15010-020-01440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qi Y, Zhang J, Cole-Jeffrey CT, Shenoy V, Espejo A, Hanna M, Song C, Pepine CJ, Katovich MJ and Raizada MK. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension. 2013;62:746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verma A, Xu K, Du T, Zhu P, Liang Z, Liao S, Zhang J, Raizada MK, Grant MB and Li Q. Erratum: Expression of Human ACE2 in Lactobacillus and Beneficial Effects in Diabetic Retinopathy in Mice. Mol Ther Methods Clin Dev. 2020;17:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferrario CM, Ahmad S and Groban L. Mechanisms by which angiotensin-receptor blockers increase ACE2 levels. Nat Rev Cardiol. 2020;17:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li J, Wang X, Chen J, Zhang H and Deng A. Association of Renin-Angiotensin System Inhibitors With Severity or Risk of Death in Patients With Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. 2020;e201624. doi: 10.1001/jamacardio.2020.1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, Liu YM, Zhao YC, Huang X, Lin L et al. Association of Inpatient Use of Angiotensin Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ Res. 2020;126:1671–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bean D, Kraljevic Z, Searle T, Bendayan R, Pickles A, Folarin A, Roguski L, Noor K, Shek A, o’gallagher K et al. ACE-inhibitors and Angiotensin-2 Receptor Blockers are not associated with severe SARS- COVID19 infection in a multi-site UK acute Hospital Trust. Eur J Heart Fail. 2020. Online ahead of print, DOI: 10.1002/ejhf.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manglik A, Wingler LM, Rockman HA and Lefkowitz RJ. β-Arrestin-Biased Angiotensin II Receptor Agonists for COVID-19. Circulation. 2020. Online ahead of print, doi: 10.1161/CIRCULATIONAHA.120.048723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teixeira LB, Parreiras-E-Silva LT, Bruder-Nascimento T, Duarte DA, Simões SC, Costa RM, Rodríguez DY, Ferreira PAB, Silva CAA, Abrao EP, Oliveira EB, Bouvier M, Tostes RC and Costa-Neto CM. Ang-(1–7) is an endogenous β-arrestin-biased agonist of the AT. Sci Rep. 2017;7:11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Montalvan V, Lee J, Bueso T, De Toledo J and Rivas K. Neurological manifestations of COVID-19 and other coronavirus infections: A systematic review. Clin Neurol Neurosurg. 2020;194:105921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garrido-Mesa N, Zarzuelo A and Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169:337–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharma RK, Yang T, Oliveira AC, Lobaton GO, Aquino V, Kim S, Richards EM, Pepine CJ, Sumners C and Raizada MK. Microglial Cells Impact Gut Microbiota and Gut Pathology in Angiotensin II-Induced Hypertension. Circ Res. 2019;124:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C and Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]