

SUMMARY
Despite its clear impact on Alzheimer’s disease (AD) risk, apolipoprotein (apo) E4’s contributions to AD etiology remain poorly understood. Progress in answering this and other questions in AD research has been limited by an inability to model human-specific phenotypes in an in vivo environment. Here we transplant human induced pluripotent stem cell (hiPSC)-derived neurons carrying normal apoE3 or pathogenic apoE4 into human apoE3 or apoE4 knockin mouse hippocampi, enabling us to disentangle the effects of apoE4 produced in human neurons and in the brain environment. Using single-nucleus RNA sequencing (snRNA-seq), we identify key transcriptional changes specific to human neuron subtypes in response to endogenous or exogenous apoE4. We also find that Aβ from transplanted human neurons forms plaque-like aggregates, with differences in localization and interaction with microglia depending on the transplant and host apoE genotype. These findings highlight the power of in vivo chimeric disease modeling for studying AD.
Graphical Abstract
In Brief
Najm et al. use a chimeric disease model that reveals the differential effect of neuronally produced apoE4 versus the apoE4 brain environment on excitatory and inhibitory human neurons. This model identifies changes in human neuron transcription, Aβ deposition, and mouse microglia responses depending on isoform and source of apoE.
INTRODUCTION
Alzheimer’s disease (AD) is thought to be caused by complex interactions among multiple genetic, epigenetic, and environmental factors, making its pathogenic mechanisms difficult to fully understand. Apolipoprotein (apo) E4 is the major genetic risk factor for AD, and it gene-dose-dependently increases the risk and lowers the age of disease onset (Huang and Mucke, 2012; Long and Holtzman, 2019; Najm et al., 2019; Yamazaki et al., 2019). ApoE4 carriers represent 20%–25% of the human population; however, 60%–75% of AD patients harbor at least one APOE4 allele, highlighting the importance of apoE4 in AD pathogenesis (Farrer et al., 1997; Ward et al., 2012). In the central nervous system (CNS), apoE is primarily produced by astrocytes (Pitas et al., 1987). However, in response to aging, injury, or stress, neurons also produce apoE (Wang et al., 2018; Xu et al., 1996, 2006), and apoE produced in various cell types of the CNS plays different roles in AD pathogenesis (Huang et al., 2004; Najm et al., 2019).
AD is characterized by three major pathological hallmarks: extracellular plaques composed primarily of β-amyloid (Aβ) peptides, intracellular neurofibrillary tangles (NFTs) composed primarily of hyperphosphorylated tau protein, and a neuroinflammatory response marked by gliosis (Huang and Mucke, 2012; Long and Holtzman, 2019). These three pathologies are influ enced by apoE expression and apoE isoform. For example, apoE both assists in Aβ clearance, with varying efficacy based on isoform (apoE2 > apoE3 > apoE4), and enhances Aβ deposition, with a decrease of apoE expression resulting in less Aβ deposition in amyloid mouse models (Bales et al., 1999; Bien-Ly et al., 2012; Holtzman et al., 2000; Kim et al., 2012; Ma et al., 2018). ApoE also increases Aβ clearance by promoting migration and activating phagocytosis of microglia, wherein again, apoE3 is more effective than apoE4 (Baitsch et al., 2011; Cudaback et al., 2011; Zhu et al., 2012). We have previously shown that apoE4 facilitates Ab production in human induced pluripotent stem cell (hiPSC)-derived neurons and that neuronally expressed apoE4 increases tau phosphorylation, particularly in inhibitory neurons, both in apoE knockin (apoEKI) mice and in hiPSC-derived neurons in vitro (Andrews-Zwilling et al., 2010; Wang et al., 2018).
Mouse models traditionally have been used in AD research, and although they have the benefit of capturing the complexity of the in vivo environment, they lack some fundamental hallmarks of the disease that are specific to humans (De Strooper and Karran, 2016; Najm et al., 2019). More recent in vitro techniques, such as hiPSC-derived models of CNS cells, have revealed important insights into some human-specific aspects of apoE4 toxicity in AD pathogenesis (Lin et al., 2018; Meyer et al., 2019; Wadhwani et al., 2019; Wang et al., 2018). However, the in vitro hiPSC model systems lack crucial features of the in vivo environment, such as cell-type heterogeneity, vasculature, and neuroinflammatory responses, limiting the scope and translatability of these systems.
To model apoE4 toxicity in human neurons in an in vivo environment, we used a chimeric disease modeling system (Espuny-Ca-macho et al., 2017; Hasselmann et al., 2019; Mancuso et al., 2019), in which hiPSC-derived neurons were transplanted into mouse hippocampi and then maintained for 7 months. To establish this system, we generated apoE4/4 (E4/4)-hiPSC and isogenic apoE3/3 (iE3/3)-hiPSC lines, differentiated them into a mixed population of both excitatory and inhibitory human neurons, and then transplantedthese neurons into eitherapoE3/3-KI(E3KI)or apoE4/4-KI (E4KI) mice. By including both apoE genotype-concordant transplants (E4/4 human neurons into anE4KI mouse and iE3/3 human neurons into an E3KI mouse) and apoE genotype-discordant transplants (E4/4 human neurons into anE3KI mouse and iE3/3 human neurons into an E4KI mouse), we were able to examine the differential effects of neuronally expressed (endogenous) apoE4 and host environment (exogenous) apoE4 on the transplanted human neurons. Single-nucleus RNA sequencing (snRNA-seq) of the transplanted human neurons identified the cell-autonomous (endogenous) and non-cell-autonomous (exogenous) effects of apoE4 on human excitatory and inhibitory neurons. By integrating snRNA-seq analysis with immunohistochemistry, we identified gene expression patterns that likely contribute to apoE4-related molecular pathologies in an apoE-source-dependent and neuron-subtype-specific manner. This in vivo chimeric AD modeling system also revealed an apoE-isoform-dependent microglial functional difference in mouse brains, pointing to a potential mechanism contributing to increased amyloid accumulation in human AD patients carrying APOE4.
RESULTS
In Vitro Generation of hiPSC-Derived Functional Neurons with Different apoE Genotypes
An E4/4-hiPSC line was previously generated from an AD patient (Wang et al., 2018) using established methods (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). From this E4/4-hiPSC line, we generated an iE3/3-hiPSC line by gene editing, without identifiable off-target effects, as previously reported (Wang et al., 2018). To trace human neurons within mouse brains for electrophysiological and morphological analyses, we engineered the isogenic pair of hiPSCs to constitutively express mCherry by knocking in an mCherry expression cassette driven by the CAG promoter into the human AAVS1 locus using CRISPR-Cas9 technology (Chen et al., 2016) (Figure S1A). Integration of the reporter construct was confirmed by PCR, and homozygous clones were selected from the pair of hiPSC lines (Figures S1B and S1C). E4/4-mCherry and iE3/3-mCherry hiPSC lines maintained pluripotency, expression of classical hiPSC markers, and a normal karyotype (Figures S1D–S1Q).
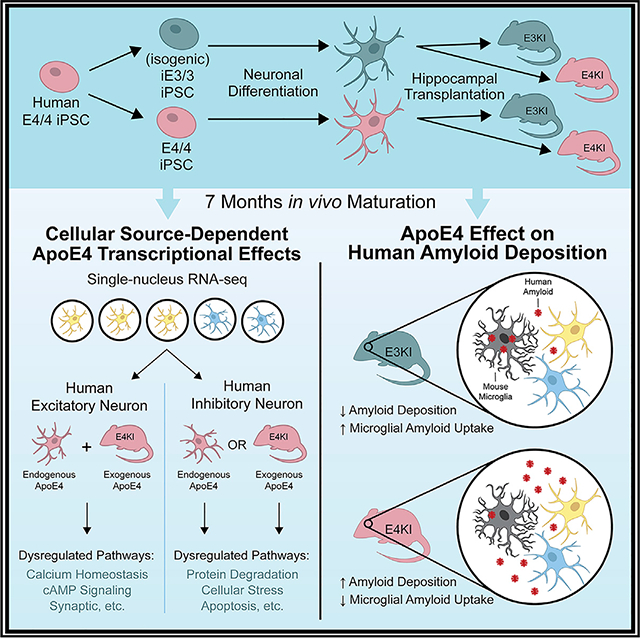
We differentiated both E4/4-mCherry and iE3/3-mCherry hiPSC lines into neurons using established methods (Chambers et al., 2009; Wang et al., 2018) (Figure S2A). We first generated neural progenitors expressing high levels of the forebrain marker FOXG1, the dorsal telencephalic marker PAX6, and—to a lesser extent—the developing inhibitory neuron marker NKX2.1 (Kessaris et al., 2014), indicating that E4/4-mCherry and iE3/3-mCherry hiPSC-derived neural progenitors contained a mix of predominantly dorsal forebrain (excitatory) and a minority of ventral (inhibitory) lineages (Figures S2B–S2G). At the time of transplantation (+1 week to +2 weeks) (Figure S2A), hiPSC-derived neurons expressed neuronal markers MAP2 and TUJ1, as well as the cortical neuron marker TBR1, the excitatory neuron marker vGlut1, or the inhibitory neuron marker g-aminobutyric acid (GABA) (Figures 1A–1J).
Figure 1. Validation of Lineage Commitment and Functionality of iE3/3-and E4/4-hiPSC-Derived Neurons In Vitro.
(A–J) Immunocytochemical staining of hiPSC-derived neurons (at the time of transplantation) for classical neuronal (MAP2 and TUJ1) markers (A, B, F, and G), excitatory (TBR1 and vGlut1) markers (C, D, H, and I), and inhibitory (GABA) markers (E and J). Scale bar, 100 μm.
(K–M) Whole-cell patch-clamp recordings of iE3/3 neurons (3 weeks of maturation with astrocytes) demonstrating the capability of iE3/3 neurons to fire action potentials (K) and to receive sPSCs from both excitatory and inhibitory inputs, which were deactivated upon incubation with CNQX, APV, and gabazine, respectively (L). The average sPSC frequency was quantified from multiple iE3/3 neurons (n = 5) (M). Holding potential was −70 mV.
(N–P) Whole-cell patch-clamp recordings of E4/4 neurons (3 weeks of maturation with astrocytes) demonstrating the capability of E4/4 neurons to fire action potentials (N) and to receive sPSCs from both excitatory and inhibitory inputs, which were deactivated upon incubation with CNQX, APV, and gabazine, respectively (O). The average sPSC frequency was quantified from multiple E4/4 neurons (n = 6) (P). Holding potential was −70 mV.
(Q) Quantification of sPSC frequency recorded in iE3/3- and E4/4-hiPSC-derived neurons in vitro (iE3/3, n = 5; E4/4, n = 6).
(R) Quantification of the percentage of inhibitory sPSCs remaining after CNQX and APV wash-in, recorded in iE3/3- and E4/4-hiPSC-derived neurons in vitro (iE3/3, n = 5; E4/4, n = 6).
We next investigated whether the hiPSC-derived neurons were electrically active in vitro, an indicator of neuronal maturity. To promote functional synapse formation, we cocultured the hiPSC-derived neurons with primary rat astrocytes. After three weeks of coculture, patch-clamp recordings revealed that these neurons were capable of firing action potentials and were receiving synaptic inputs (spontaneous post-synaptic currents [sPSCs]) (Figures 1K, 1L, 1N, and 1O). To functionally characterize the neuronal subtypes present in hiPSC-derived neuronal cultures, neurons were treated with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and D-(−)-2-amino-5-phosphonopentanoic acid (APV), which are α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptor antagonists, respectively, to block excitatory inputs into the recorded cells. Approximately 60% of sPSCs were blocked upon treatment with CNQX and APV. The remaining sPSCs were eliminated upon further treatment with gabazine, a GABAA receptor blocker, indicating that both excitatory and inhibitory neurons in culture were functionally connected (Figures 1L, 1M, 1O, and 1P). Quantification of the frequency of sPSCs and of the percentage of inhibitory sPSCs revealed no significant difference between iE3/3 and E4/4 neurons in this 3-week in vitro culture condition (Figures 1Q and 1R). Because our study focuses on in vivo chimeric AD modeling, we did not pursue long-term in vitro culture experiments.
Transplanted iE3/3 and E4/4 Neurons Survive and Functionally Integrate into the Mouse Hippocampus
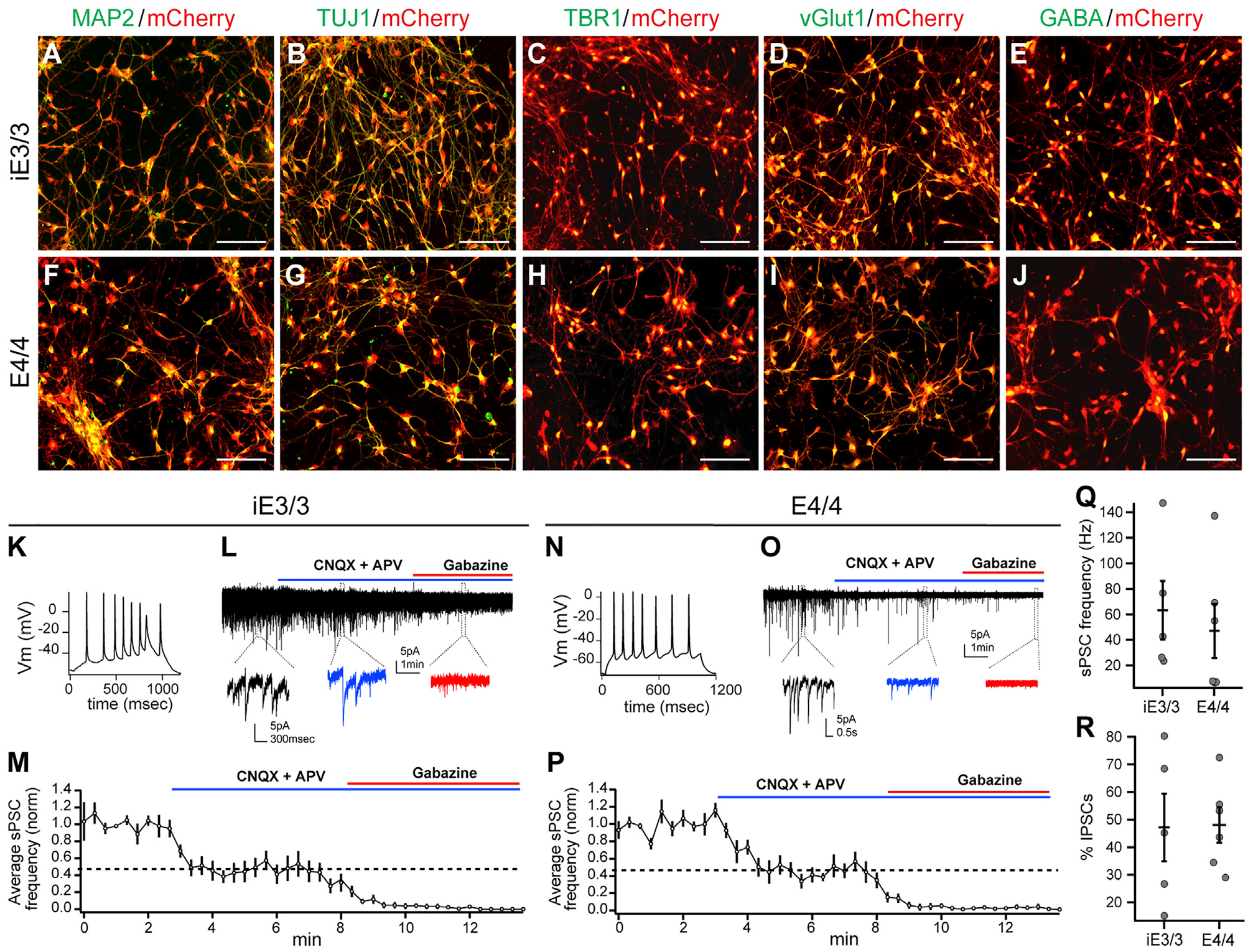
After validating the lineage and functionality of iE3/3 and E4/4 neurons in vitro, we transplanted them bilaterally into the hippocampi of 7-month-old E3KI and E4KI mice. An immunosuppressant cocktail was administered immediately after transplantation (day 0), as well as 2, 4, and 6 days post-transplantation to block the host immune response and to enhance the survival of transplanted human cells in the host mouse brains (Pearl et al., 2011). At 7 months post-transplantation (MPT), we sacrificed and transcardially perfused the transplanted mice. One hemisphere of the brain was frozen at −80°C for snRNA-seq analysis, and the other under went immunohistochemical or electrophysiological analysis to determine whether transplanted human neurons had survived and were capable of receiving synaptic inputs. Transplanted iE3/3 and E4/4 neurons were positive for mCherry, human nuclear antigen (HNa) (Figures 2A and 2D), the neuronal nuclear marker NeuN (Figures 2B and 2E), and human synaptophysin (SYN) (Figures 2C and 2F). We noted that the transplanted human neurons did not migrate significantly from the transplant core (Figures 2A–2F), as reported in other studies (Cunningham et al., 2014; Espuny-Camacho et al., 2017; Liu et al., 2013). However, they did display extensive axonal projections throughout the hippocampus (Figures 2G and 2H). At 7 MPT, patch-clamp recordings on transplanted iE3/3 and E4/4 neurons in ex vivo brain slices revealed that they were capable of generating action potentials, as well as receiving sPSCs (Figures 2I and 2J). Altogether, these data demonstrate that transplanted hiPSC-derived iE3/3 and E4/4 neurons survived, integrated into mouse hippocampus, and are capable of firing action potentials, as well as receiving synaptic inputs.
Figure 2. The Transplanted iE3/3 and E4/4 Neurons Survive and Functionally Integrate into the Mouse Hippocampus at 7 MPT.
(A–F) Representative immunohistochemical staining of transplanted iE3/3 and E4/4 neurons for HNa (A and D), NeuN (B and E), and human SYN (C and F). Scale bar, 25 μm.
(G and H) Representative immunohistochemical staining for mCherry+ (red) displayed distal projections emanating from the transplant core (G) (scale bar, 500 μm), with a magnified image of the inset (H) (scale bar, 50 μm).
(I) Whole-cell patch-clamp recordings in ex vivo slices of transplanted iE3/3 neurons (left) and E4/4 neurons (right) demonstrating capability to fire action potentials.
(J) Whole-cell patch clam recordings in ex vivo slices of transplanted iE3/3 neurons (top) and E4/4 neurons (bottom) demonstrating capability to receive sPSCs. Holding potential is −70 mV.
Transcriptional Characterization of the Transplanted hiPSC-Derived Human Neurons
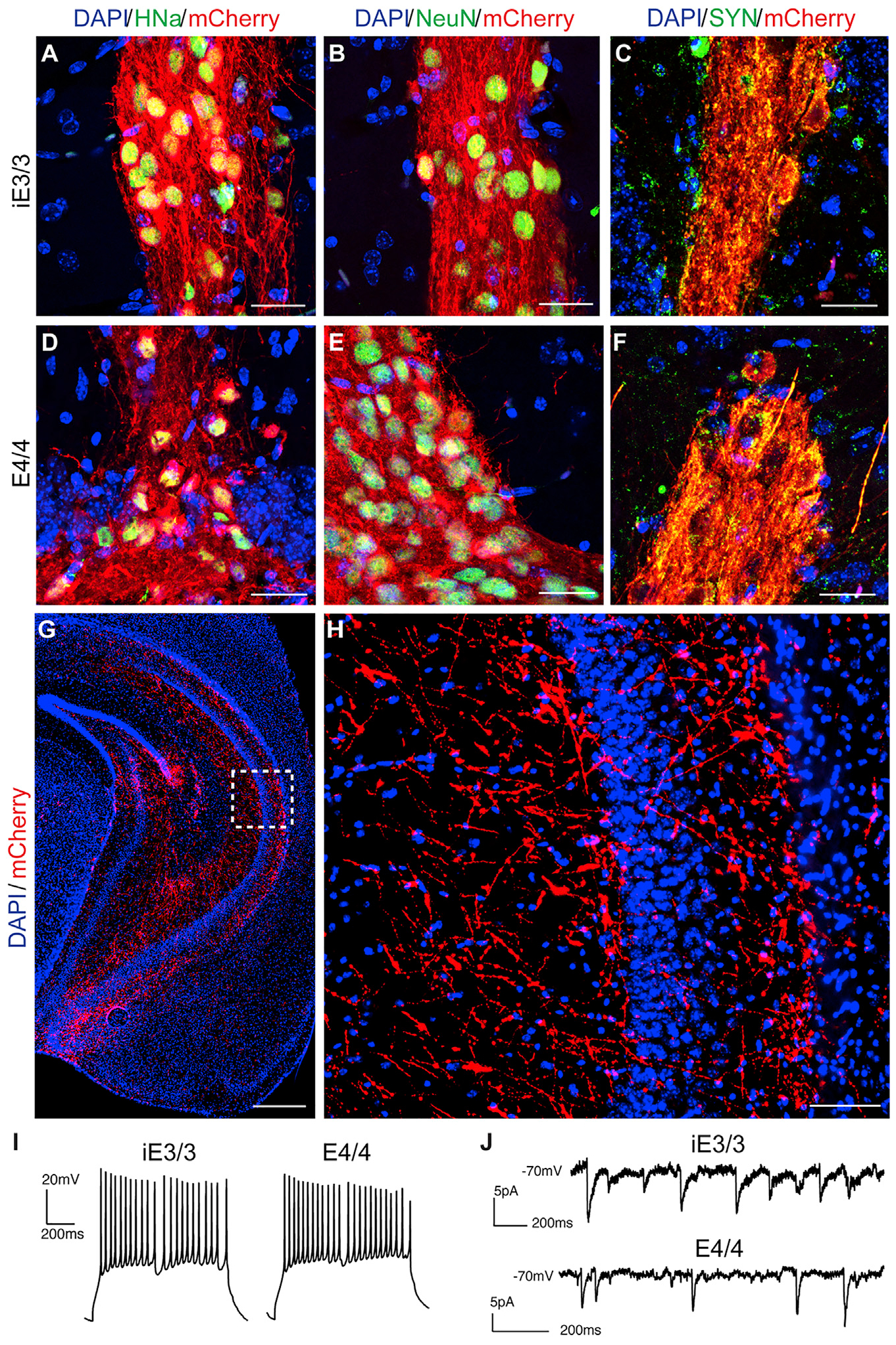
Having established that transplanted hiPSC-derived neurons survived and integrated in the mouse hippocampus, we next performed snRNA-seq on human transplants. Single nuclei were isolated from frozen mouse hippocampi containing the human neuron transplants at 7 MPT. Because of limitations in single-nucleus sequencing technology, we could not reasonably sequence single nuclei from the entire mouse hippocampus and use solely computational methods to select the human cells. Instead, we generated a single-nucleus suspension as previously described, enriched for mCherry+ human neurons within the mouse brain using fluorescence-activated cell sorting (FACS) and then performed snRNA-seq library preparation on the sorted nuclei (Figure 3A) (Krishnaswami et al., 2016). In our analysis, we first clustered the human cells and checked for batch effects that may have skewed our interpretation of the snRNA-seq results (Figures S3A–S3D). All major clusters showed similar proportions of cells contributed from each experimental condition, apart from cluster 9, which we identified as oligodendrocytes (Figure S3D). However, further investigation is needed to better understand the reason for this phenomenon and whether apoE4 and variability in lineage commitment or survival of oligodendrocytes are connected.
Figure 3. Transcriptional Characterization of hiPSC-Derived Human Neurons at 7 MPT.
(A) Experimental design for collecting snRNA-seq data from transplanted human neurons. E3KI mice (iE3/3 transplants, N = 6; E4/4 transplants, N = 6). E4KI mice (iE3/3 transplants, N = 6; E4/4 transplants, N = 5).
(B) Feature plots of classical panneuronal (RBFOX3 and SYN1), excitatory neuron (NEUROD6 and SLC17A7), and inhibitory neuron (DLX1 and GAD1) markers.
(C) t-distributed stochastic neighbor embedding (tSNE) clustering of transplanted hiPSC-derived neurons.
Next, we identified different cell types within the snRNA-seq data (Table S1) by analyzing marker genes defining individual clusters and cross-referencing those genes with other published datasets (Lake et al., 2016; Mathys et al., 2019; Mayer et al., 2018; Nowakowski et al., 2017; Pollen et al., 2015; Yao et al., 2017). From the transplanted human nuclei, we identified neurons by the expression of RBFOX3, SYN1, SYT1, NRXN3, and SNAP25 (Figures 3B, S3E, and S3F; Table S1). Within this panneuronal population, we distinguished excitatory neurons through expression of NEUROD6 and SLC17A7 and inhibitory neurons by their expression of DLX1 and GAD1 (Figures 3B, S3E, and S3F). Within the inhibitory neuron cluster (cluster 1), we also saw expression of genes specific to various inhibitory neuron subtypes, including PVALB and SST, indicating that this population consisted of various subtypes of more mature inhibitory neurons. Within the excitatory neuron clusters (clusters 2–5), the pattern of gene expression indicated that these cells were also made up of various subtypes attributed to different brain regions. For example, we observed expression of the upper-layer marker SATB2 (clusters 2–5), the deeper-layer cortical markers FEZF2 and BCL11B (CTIP2) (clusters 4 and 5), and the hippocampal granule cell marker PROX1 (cluster 3) (Figure S3E; Table S1). Within non-neuronal cell clusters, we identified a small proportion as neuronal precursors by EBF1 and ZIC1 expression, as well as low expression of other neuronal genes (clusters 6–8), oligodendrocytes by PLP1 and MBP (cluster 9), and astrocytes by GFAP, APOE, AQP4, and SLC1A2 expression (cluster 10) (Figures S3E and S3F; Table S1). We determined that 87.6% of transplanted human cells were neurons, 4.5% were oligodendrocytes, 4.3% were neuronal precursors, and 3.6% were astrocytes (Figures S3G), without differences across donor and recipient apoE genotypes (Figure S3H).
Having characterized the lineage of transplanted human neurons in vivo and separated them broadly into either excitatory or inhibitory lineages, we went on to ask the following questions: (1) How does the environmental (exogenous) apoE4 affect the transcriptional profile of human neurons? (2) How does neuronally expressed (endogenous) apoE4 affect the transcriptional profile of human neurons? (3) Is there an interactive effect between endogenous and exogenous apoE4 on the transcriptional profile of human neurons? (4) Do these effects differ between excitatory and inhibitory neuron subtypes?
Differential Responses of iE3/3 and E4/4 Human Excitatory Neurons to Environmental (Exogenous) ApoE4
We first examined how exogenous apoE4 in the host brain environment affects the transcriptional profile of transplanted human excitatory neurons, which comprised 78.2% of the total trans planted human cells (Figures 4A–4C; Table S2). E4/4 human excitatory neurons exposed to the apoE4 environment displayed significant dysregulation of 24 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, many of which were synaptic (Figures 4D and 4E). In contrast, iE3/3 excitatory neurons exposed to the apoE4 environment displayed significant dysregulation of just 6 pathways, representing more fundamental cellular processes, such as ribosome, spliceosome, and protein processing (Figures 4D and 4E). This disparity suggests that the effect of the E4KI brain environment on the transplanted human excitatory neurons depends strongly on whether the neurons express apoE4, an effect that could not be uncovered without this in vivo chimeric disease modeling system.
Figure 4. Differential Responses of iE3/3 and E4/4 Human Excitatory Neurons to Environmental (Exogenous) ApoE4.
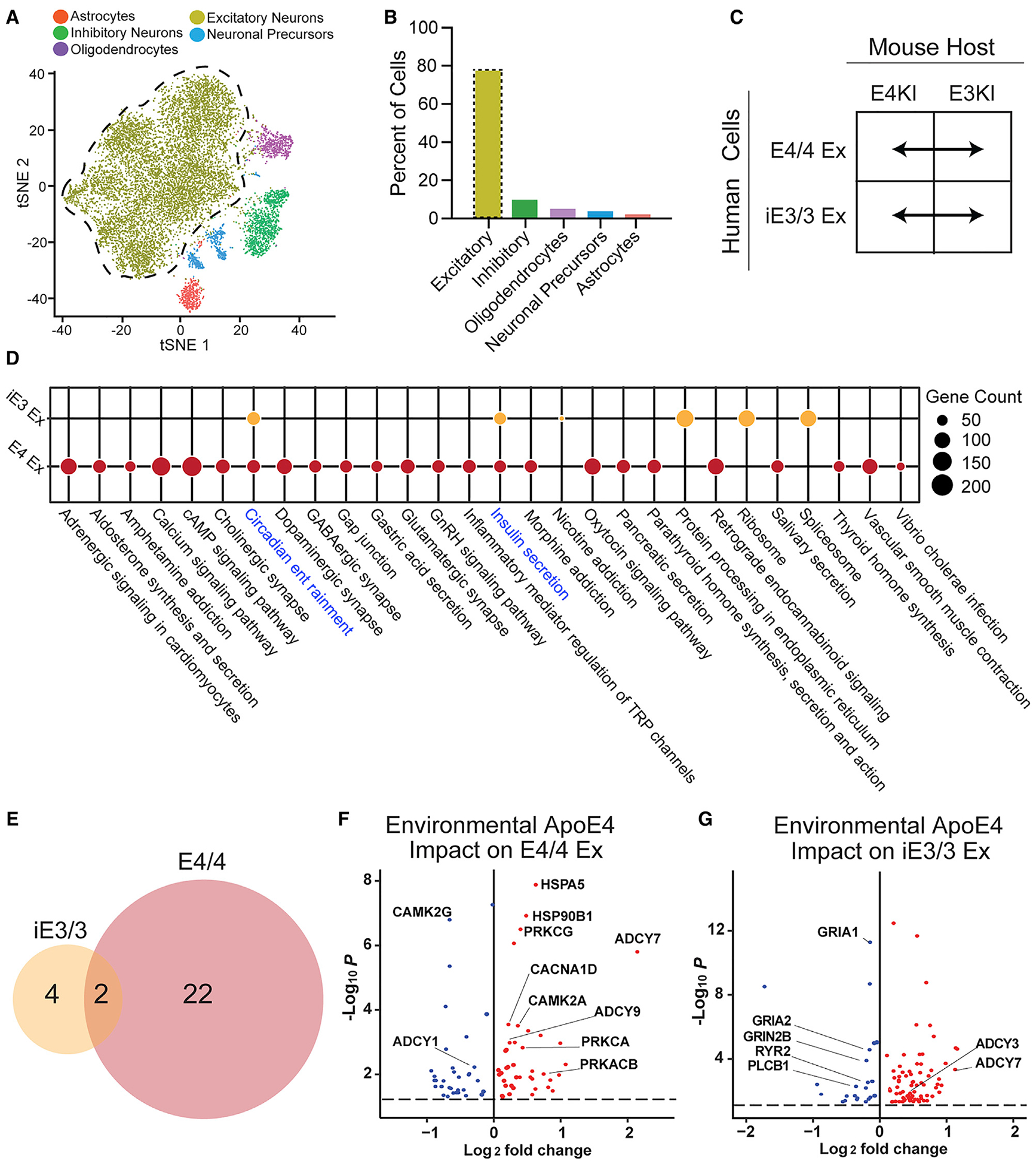
(A) tSNE clustering indicating a human excitatory neuron cluster.
(B) Quantification of cell-type percentages represented in isolated transplanted human neurons (excitatory neurons outlined by the dotted line).
(C) These comparisons were made for apoE-genotype-matched hiPSC-derived excitatory neurons across E4KI and E3KI mouse hosts.
(D) Dotplot displaying the pathways dysregulated by the E4KI mouse host environment in E4/4-hiPSC-derived excitatory neurons and iE3/3-hiPSC-derived excitatory neurons. Node size indicates the number of genes dysregulated within the pathway. Shared pathways are highlighted in blue.
(E) Many more pathways were dysregulated by the E4KI mouse host environment in E4/4-hiPSC-derived excitatory neurons (right) than in iE3/3-hiPSC-derived excitatory neurons (left).
(F and G) Genes dysregulated by the E4KI mouse environment in E4/4-hiPSC-derived excitatory neurons (F). Genes dysregulated by the E4KI mouse environment in iE3/3-hiPSC-derived excitatory neurons (G). Red points represent an increase in log2 fold change and blue points represent a decrease in log2 fold change in response to the E4KI brain environment. Only genes with a Benjamini-Hochberg corrected p < 0.05 are shown. The p values are assigned per cell.
In (C), (D), (F), and (G), Ex denotes excitatory neurons.
In addition to the clear dysregulation of synaptic gene expression of E4/4 neuronal transplants in the E4KI brain, these excitatory neurons displayed transcriptional dysregulation of calcium homeostasis and cyclic AMP (cAMP) signaling (Figure 4D)—processes frequently implicated in AD and, in particular, in synaptic dysfunction linked to Aβ pathology. For example, pathological accumulation of Aβ can disrupt calcium signaling through protein kinase A (PKA)/protein kinase C (PKC) and the cAMP response-element binding protein (CREB), all of which can lead to synaptic dysfunction and loss (Alkon et al., 2007; Bastrikova et al., 2008; Bezprozvanny and Mattson, 2008; Chapman et al., 1999; Demuro et al., 2010; Hermes et al., 2010; Kamat et al., 2016; Kashyap et al., 2019; Mattson, 2004; Morrison and Baxter, 2012; Popugaeva et al., 2017; Pozueta et al., 2013; Ricciarelli and Fedele, 2018; Selkoe, 2002; Sun and Alkon, 2014; Tong et al., 2018). When we further examined the genes that were most common across pathways dysregulated in human E4/4 excitatory neurons in response to the E4KI brain environment, we saw an upregulation of genes along multiple layers of this signaling cascade, including the Ca2+/calmodulin-dependent protein kinase IIA (CAMK2A), multiple adenylate cyclases (ADCY7 and ADCY9), and multiple isoforms of PKC (including PRKCA, PRKCAB, and PRKCG) (Figure 4F; Table S2).
The iE3/3 human excitatory neurons transplanted into E4KI mouse brains, in contrast to the E4/4 human excitatory neuron transplants, displayed few alterations in calcium homeostasis (Figure 4D). Instead, we observed evidence of compensatory processes that would rescue potential calcium dyshomeostasis in these cells. For example, the ryanodine receptor RYR2, which releases calcium from the endoplasmic reticulum; phospholipase C (PLCB1), which enhances inositol triphosphate (IP3)-mediated calcium influx from the extracellular space; and the NMDA receptor subunit (GRIN2B), which can modulate calcium influx, were all downregulated (Figure 4G; Table S2) (Abid et al., 2019; Del Prete et al., 2014; Gibson and Thakkar, 2017; Kadamur and Ross, 2013; Santulli et al., 2018; Zhang et al., 2016). Altogether, the contrast between the response of E4/4 and that of iE3/3 human excitatory neurons suggests that iE3/3 excitatory neurons avoid gene expression changes indicative of calcium dyshomeostasis and synaptic dysfunction observed in E4/4 human excitatory neurons in the apoE4 environment.
Differential Responses of Human Excitatory Neurons to Neuronally Produced (Endogenous) ApoE3 and ApoE4
We next investigated gene expression changes in transplanted excitatory neurons that result from neuronally expressed apoE4 (Figure S4A; Table S3). We again saw evidence for a strong interactive effect between endogenous and exogenous apoE4 on the transplanted excitatory neurons (Figures S4B and S4C). In the apoE4 host context, 17 pathways were dysregulated between transplanted E4/4 and transplanted iE3/3 neurons, many of them synaptic. In contrast, in the apoE3 host context, only two pathways were dysregulated in E4/4 compared with iE3/3 transplants (Figures S4B and S4C). These findings again suggest that in excitatory neurons, both the diathesis of endogenous apoE4 expression and the stress of the apoE4 environment are necessary to induce the synaptic dysfunction typically associated with apoE4-expressing neurons in AD.
Analysis of the genes and pathways dysregulated by neuronal apoE4 expression in the E4KI brain context again revealed genes involved in cAMP synthesis and calcium homeostasis (Figures S4C and S4D). Among them were PRKCA and PRKCG, which were represented in nearly all dysregulated pathways and were all significantly upregulated. CAMK2A, CAMK2G, and ADCY9, all of which affect most dysregulated pathways, were also altered in the same direction as in the environmental (exogenous) apoE4 comparison (Figures 4F and S4D). Remarkably, in the E3KI host brain, all significantly dysregulated pathway genes encoded ribosomal subunits, and these ribosomal genes were consistently downregulated by neuronal apoE4 in the E3KI brain context, potentially indicating a downregulation of translational activity (Figure S4E; Table S3).
Differential Responses of iE3/3 and E4/4 Human Inhibitory Neurons to Environmental (Exogenous) ApoE4
We next looked at how environmental or neuronally expressed apoE4 affects gene expression in transplanted iE3/3 or E4/4 human inhibitory neurons, which comprised 9.5% of the total transplanted cells (Figures 5A and 5B). We first investigated the impact of the apoE4 environment on iE3/3 or E4/4 human inhibitory neurons (Figure S5A; Table S4). Both iE3/3 and E4/4 human inhibitory neurons displayed significant KEGG pathway dysregulation when exposed to the E4/4 environment in host E4KI mouse brains. Unlike in excitatory neurons, exogenous apoE4 alone was sufficient to induce substantial pathway dysregulation in inhibitory neurons (Figures S5B and S5C). Both iE3/3 and E4/4 human inhibitory neurons had 27 dysregulated pathways in response to the apoE4 environment (Figure S5C). This result suggests a selective vulnerability of inhibitory neurons, regardless of their apoE genotype, to exogenous apoE4 and the toxic milieu of the E4KI brain environment.
Figure 5. Differential Responses of Human Inhibitory Neurons to Neuronally Produced (Endogenous) ApoE3 and ApoE4.
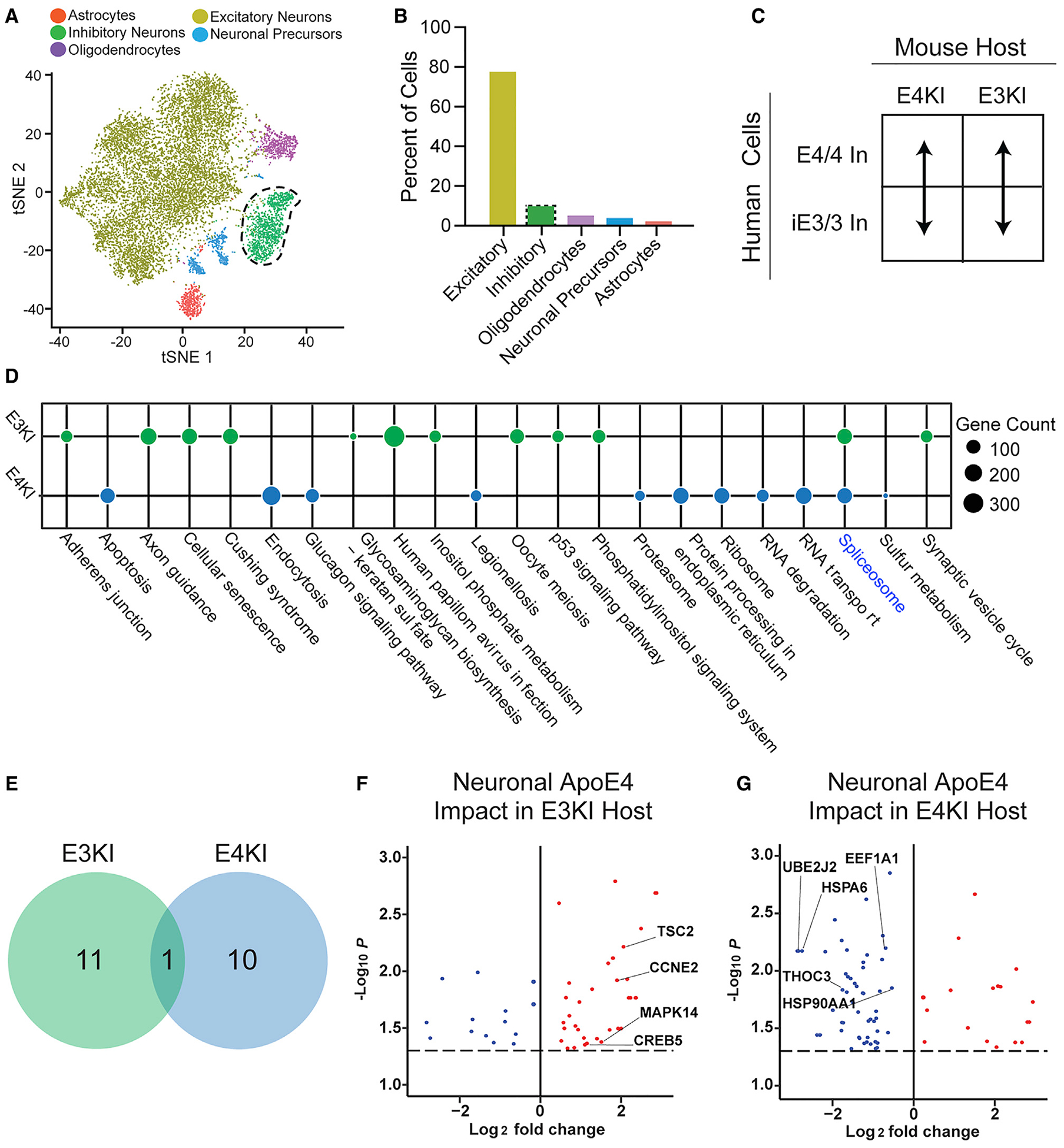
(A) tSNE clustering indicating a human inhibitory neuron cluster.
(B) Quantification of cell-type percentages represented in isolated transplanted human neurons (inhibitory neurons outlined by the dotted line).
(C) These comparisons were made between E4/4- and iE3/3-hiPSC-derived inhibitory neurons within apoE-genotype-matched mouse hosts. In denotes inhibitory neurons.
(D) Dotplot displaying the pathways dysregulated in transplanted E4/4 human inhibitory neurons relative to transplanted iE3/3 human inhibitory neurons in the E4KI mouse host environment and the E3KI mouse host environment. Node size indicates the number of genes dysregulated within the pathway. Shared pathways are highlighted in blue.
(E) Number of pathways dysregulated by the transplanted human inhibitory neuron apoE4 genotype were similar in the E4KI and E3KI mouse environment, with little overlap between pathways.
(F and G) Genes dysregulated by the transplanted human inhibitory neuron apoE4 genotype in the E3KI mouse environment (F). Genes dysregulated by the transplanted human inhibitory neuron apoE4 genotype in the E4KI mouse environment (G). Red points represent an increase in log2 fold change and blue points represent a decrease in log2 fold change in response to neuronal apoE4. Only genes with a Benjamini-Hochberg corrected p < 0.05 are shown. The p values are assigned per cell.
In examining these dysregulated KEGG pathways, we observed that iE3/3 human inhibitory neurons exposed to the E4KI brain displayed dysregulation of many pathways involved in cell stress, including mitophagy, autophagy, and apoptosis, as well as neurodegenerative diseases such as Parkinson’s disease and Huntington’s disease. The gene implicated in the largest number of dysregulated pathways is MAPK10 (aka JNK3) (Figure S5D; Table S4), which is activated by cellular stressors and is known to participate in the development of NFTs (Yarza et al., 2016). SRC, a non-receptor tyrosine kinase, was also implicated in multiple pathways (Figure S5D; Table S4), and Src family tyrosine kinases have been implicated as genetic risk factors in AD, possibly because of their tau phosphorylation activities (Lee et al., 1998; Scales et al., 2011). Finally, we noted an upregulation in multiple heat shock proteins (HSPs), including HSP90AA1, HSP90B1, and HSPA6, which are known to respond to cellular stress and misfolded proteins in general and relate to tau aggregation in neurodegenerative disease (Figure S5D; Table S4) (Campanella et al., 2018; Lackie et al., 2017; Mok et al., 2018; Young et al., 2016). These proteins are uniquely upregulated in iE3/3 human inhibitory neurons, but not E4/4 human inhibitory neurons (Figure S5E; Table S4), in response to the E4KI brain environment, which suggests the intriguing possibility that iE3/3 human inhibitory neurons more successfully initiate adaptive responses to stress than do E4/4 human inhibitory neurons, leaving those E4/4 human inhibitory neurons especially vulnerable to environmental apoE4.
In E4/4 human inhibitory neurons within the E4KI brain, we again saw dysregulated pathways indicative of cell stress, including autophagy and the longevity regulating pathway, but there were additionally multiple dysregulated pathways related to protein degradation and secretion (Figure S5C). The transcription factors ATF4 and CREB5 were dysregulated in most altered pathways in E4/4 human inhibitory neurons by environmental apoE4 and were substantially downregulated in the E4KI brain context (Figure S5E; Table S4). Both ATF4 and CREB are essential to synaptic plasticity and memory formation and have been shown to be dysregulated in AD models and human patients (Ameri and Harris, 2008; Baleriola et al., 2014; Costa-Mattioli et al., 2005; Ma et al., 2013; Oliveira and Lourenco, 2016; Ron and Harding, 2012). Given the essential role of inhibitory neuron input in regulating synaptic plasticity and memory formation, it is particularly fascinating that ATF4 and CREB signaling is uniquely dysregulated in E4/4 human inhibitory neurons in the E4KI host context, suggesting an interactive effect of endogenous and exogenous apoE4 on these pathways.
Differential Responses of Human Inhibitory Neurons to Neuronally Produced (Endogenous) ApoE3 and ApoE4
Finally, we examined gene expression changes in human inhibitory neurons caused by neuronally expressed apoE4 across the E3KI or E4KI host (Figure 5C; Table S5). In contrast to the effect of neuronal apoE4 expression on human excitatory neurons, where we found a strong dependence of host apoE genotype, we found in human inhibitory neurons that neuronal apoE4 alone is sufficient to induce extensive pathway dysregulation, regardless of exposure to the E3KI or E4KI environment (Figures 5D and 5E). When inspecting the effect of neuronal apoE4 on human inhibitory neurons in the E3KI mouse brain, we again found dysregulation in protein degradation pathways and pathways involved in fundamental cellular processes, such as p53 signaling, apoptosis, and cellular senescence. In particular, we saw multiple dysregulated genes that have been previously implicated in AD in most dysregulated pathways. These included CCNE2, which is a member of the cyclin family that contributes to cell-cycle control (Absalon et al., 2013; Ludwig et al., 2019; Nguyen et al., 2002); TSC2, which among other functions, contributes to cell-cycle control and has been tied to the mTOR pathway (Li et al., 2018; Maiese, 2014; Shang et al., 2013); and MAPK14, a member of the ERK signaling cascade (Figure 5F; Table S5) (Alam and Scheper, 2016; Bito et al., 1996; Kamat et al., 2016; Khan and Alkon, 2006; Querfurth and LaFerla, 2010; Silver et al., 2012; Sun and Alkon, 2014; Vitolo et al., 2002).
When we looked at the effect of neuronal apoE4 on human inhibitory neurons in the E4KI host context, we noted that many dysregulated pathways were involved in fundamental cell processes, such as RNA degradation, the proteasome, and apoptosis (Figure 5D). When we looked at the genes most heavily represented in these dysregulated pathways, we found significant downregulation of several genes related to misfolded proteins or the unfolded protein response (UPR), including both HSPA6 and HSP90AA1; the HSP70 transcription elongation factor EEF1A (Vera et al., 2014); the protein transporter THOC3, which is necessary for export of HSP mRNAs (Guria et al., 2011; Katahira et al., 2009); and UBE2J2, which plays an essential role in the ubiquitin UPR (Figure 5G; Table S5) (Kumar et al., 2015; Scheper and Hoozemans, 2015; Upadhya and Hegde, 2007). HSP family members have been heavily implicated in the turnover and pathological aggregation of tau (Campanella et al., 2018; Lackie et al., 2017; Mok et al., 2018; Young et al., 2016), which has previously been shown to be increased in E4/4 inhibitory neurons (Andrews-Zwilling et al., 2010; Knoferle et al., 2014; Wang et al., 2018). Thisdownregulation of multiple HSP family members by neuronal apoE4 in human inhibitory neurons (Figure 5G) is in sharp contrast to the adaptive response to host apoE4 that was seen in iE3/3 human inhibitory neurons (Figure S5D). The coordinated downregulation of multiple genes involved in chaperoning proper protein folding, as well as the UPR, suggests that neuronal apoE4 results in a deficit in the proper folding of proteins and the handling and degradation of misfolded proteins in human inhibitory neurons.
Human E4/4 Neurons Produce More Aβ Aggregates in E4KI Than in E3KI Mouse Brains
Based on gene expression perturbations revealed from the transcriptomic analysis, which were reminiscent of a response to Aβ toxicity in excitatory neurons, we next investigated whether Aβ is produced and aggregates in or around the transplant area and whether there are apoE-genotype-dependent effects. There is evidence that human neurons in vitro produce Aβ and that E4/4-hiPSC-derived neurons produce significantly more Aβ than E3/3-hiPSC-derived neurons (Choi et al., 2014; Wang et al., 2018). However, in traditional 2D in vitro models, Aβ produced by human neurons does not aggregate, presumably because the Aβ diffuses into the culture media (Choi et al., 2014; Wang et al., 2018). Importantly, without the addition of familial AD (FAD) mutations, neither E3KI nor E4KI mice produce Aβ aggregates in the mouse brain (Huang, 2010). Therefore, we asked whether human neurons transplanted into mouse brains develop Aβ pathology as a result of apoE4 exposure. We found 3D6+ Aβ aggregates present inside and near iE3/3 and E4/4 human neuron transplants in both E3KI and E4KI mice (Figures 6A–6E). Colocalization of Aβ staining with Thioflavin-S staining demonstrated that 3D6+ Aβ aggregates were fibrillar and amyloid plaque-like (Figures 6F–6J). Although 3D6 staining is not exclusive to Aβ produced by human cells, we did see a highly significant difference between the number of 3D6+ aggregates inside and the number outside a 100 μm perimeter of the transplants (Figure 6K). Moreover, when we looked for 3D6+ aggregates in transplanted mouse brains away from the transplant area in regions with no mCherry+ cells, we saw no 3D6+ staining (Figures S6A–S6D). Altogether, these data strongly indicate that 3D6+ Aβ aggregates were produced by the human neurons. When we quantified the number of 3D6+ Aβ aggregates per square micrometer within 100 μm of the transplants, we saw a significantly higher number of aggregates in E4/4 transplants within E4KI mouse brains than in E4/4 transplants within E3KI mouse brains (Figure 6L), implicating a stimulatory effect of exogenously expressed apoE4 in Aβ aggregation. There was also a trend toward higher numbers of 3D6+ Aβ aggregates per square micrometer in E3/3 transplants within E4KI mouse brains compared with those in E3/3 transplants within E3KI mouse brains (Figure 6L). Therefore, environmental apoE4 appears more critical than neuronal apoE4 in promoting human-neuron-derived Aβ aggregation in mouse brains.
Figure 6. Aβ from Human E4/4 Neurons Forms More Aggregates in E4KI Than in E3KI Mouse Brains.
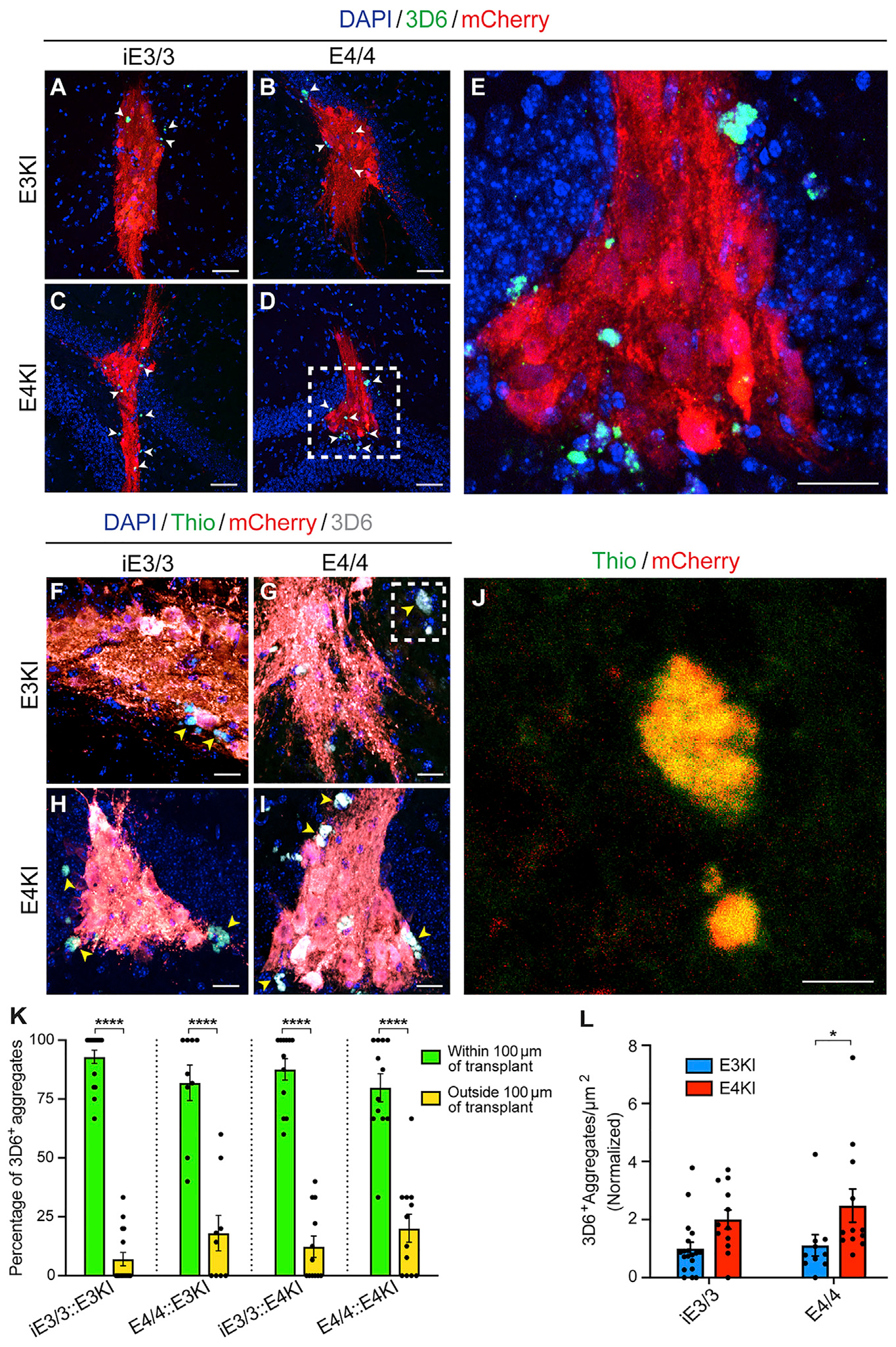
(A–D) 3D6+ Aβ aggregates present within and immediately surrounding human neuronal transplants (arrowheads). Scale bar, 50 μm.
(E) Magnified image of the inset in (D). Scale bar, 25 μm.
(F–I) Thioflavin (green) costaining with 3D6+ aggregates (gray, arrowheads) demonstrates the plaque-like nature of Aβ aggregates. Scale bar, 25 μm.
(J) Magnified image of the inset in (G). Scale bar, 10 βm.
(K) Quantification of percentage of 3D6+ aggregates within or outside a 100 μm perimeter of human neuronal transplants. E3KI (iE3/3 transplants, n = 16, N = 6; E4/4 transplants, n = 9, N = 5). E4KI (iE3/3 transplants, n = 12, N = 5; E4/4 transplants, n = 12, N = 4). Data are represented as mean ± SEM, two-way ANOVA with Sidak’s multiple comparison test: ****p < 0.0001.
(L) Quantification of the number of 3D6+ aggregates within a 100 μm perimeter per transplant area. E3KI (iE3/3 transplants, n = 19, N = 6; E4/4 transplants, n = 10, N = 5). E4KI (iE3/3 transplants, n = 12, N = 5; E4/4 transplants, n = 12, N = 4). Values are normalized to iE3/3 neurons transplanted into the E3KI host. Data are represented as mean ± SEM, two-way ANOVA with Sidak’s multiple comparison test, *p < 0.05.
Mouse ApoE4 Microglia Contain Fewer Human-Neuron-Derived Aβ Aggregates Than Mouse ApoE3 Microglia
Because environmental apoE is largely produced by astrocytes and microglia, we stained for the microglial marker IBA1 and the astrocytic marker GFAP to examine differences in the glial response to human neuronal transplants with different APOE genotypes. At 7 MPT, staining revealed no significant differential infiltration of microglia (Figures S6E–S6I) or astrocytes (Figures S6J–S6N) into the transplants or toward their perimeters, regardless of host or transplant apoE genotype. Costaining of IBA1 with 3D6 demonstrated that 3D6+ Aβ aggregates often colocalized with mouse microglia (Figures 7A–7D), suggesting that they were within microglia (Figure 7E). When we measured colocalization of IBA1+ microglia and 3D6+ Aβ aggregates, we found that there was a significant decrease in the association of Aβ aggregates with microglia in E4KI mouse hippocampus compared with E3KI mouse hippocampus, independent of the apoE genotype of human neuronal transplants (Figure 7F). These data suggest that the apoE4 microglia in the E4KI mouse brain might have impaired phagocytosis of Aβ aggregates, leading to increased accumulation of Aβ aggregates in E4KI compared with E3KI mouse hippocampus.
Figure 7. Mouse ApoE4 Microglia Contain Fewer Human Neuronal Transplant-Derived Aβ Aggregates Than Mouse ApoE3 Microglia, Regardless of the Transplant ApoE Genotypes.
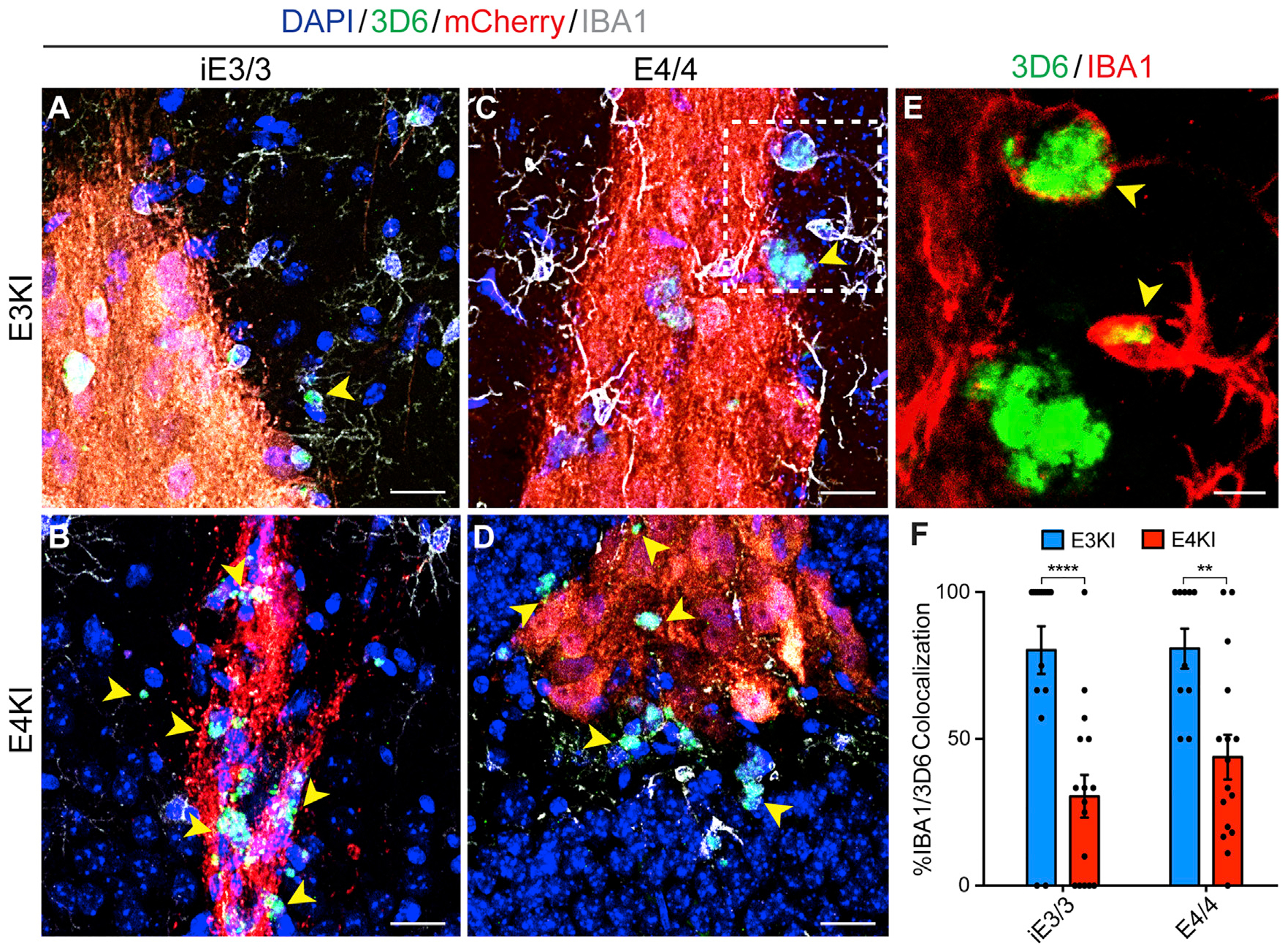
(A–D) IBA1+ microglia (gray) and 3D6+ Aβ aggregates (green) within and immediately surrounding transplants. The 3D6+ Aβ aggregates not colocalized with microglia (arrowheads) are dramatically increased in transplants into the E4KI brain (B and D). Scale bar, 25 μm.
(E) Magnified image of the inset in (C). Two arrows indicate that 3D6+ Aβ aggregates are inside two IBA1+ microglia. Scale bar, 10 μm.
(F) Quantification of the percentage of 3D6+ Aβ aggregates that colocalize with host IBA1+ microglia. E3KI (iE3/3 transplants, n = 17, N = 5; E4/4 transplants, n = 10, N = 4). E4KI (iE3/3 transplants, n = 16, N = 4; E4/4 transplants, n = 16, N = 5). Data are represented as mean ± SEM, two-way ANOVA with Sidak’s multiple comparison test, **p < 0.01, ****p < 0.0001.
DISCUSSION
Here we establish via an in vivo chimeric AD modeling system that exogenously expressed apoE4 and endogenously expressed apoE4 interact to generate distinct gene expression changes and differential molecular pathologies in human excitatory and inhibitory neurons. We first demonstrated that human excitatory neurons require both the neuronally expressed apoE4 and the pathological environment of the apoE4-expressing brain for the most pronounced gene expression changes to occur. These transcriptomic changes highlight well-characterized pathways linked to neurodegeneration, including synaptic dysfunc tion and dysregulation of calcium signaling via the PKC cascade and cAMP. These gene expression changes, unique to excitatory neurons, could result from increased intraneuronal Aβ accumulation, which is known to increase intracellular Ca2+ from both extracellular and intracellular sources (Alkon et al., 2007; Kamat et al., 2016; Kashyap et al., 2019; Popugaeva et al., 2017; Small, 2009). Alternatively, these gene expression changes could be a signature of the neuronal response to extracellular events, such as the observed increase of Ab aggregates, and/or to dysfunctional microglia in their surroundings. Others have shown that in response to neuronally expressed apoE4, hiPSC-derived excitatory neurons in vitro dysregulated genes involved in synaptic function, which were similar to apoE4-induced gene expression changes in humans (Lin et al., 2018). Importantly, the chimeric disease modeling system allows us to go one step further in understanding how the source of apoE4 may affect synaptic gene expression in vivo. In the chimeric system, we showed clearly that the most pronounced gene expression dysregulation in excitatory neurons occurred only when apoE4 was produced both endogenously and exogenously, reaffirming the importance of incorporating the in vivo environment into experiments.
Our data also suggest that inhibitory neurons are more susceptible to apoE4-mediated gene expression changes than are excitatory neurons, and both exogenous and endogenous apoE4 substantially alter their transcription. Furthermore, their transcriptional response to apoE4 was distinct to that uncovered in excitatory neurons. Inhibitory neuron responses were enriched for the UPR, oxidative stress, and RNA degradation, whereas excitatory neuron responses were dominated by calcium homeostasis and synaptic function, together suggesting a neuron-subtype-specific response to apoE4 toxicity. This dysregulation in the UPR could make human inhibitory neurons more vulnerable to stressors and injuries, pointing to a potential mechanism explaining the increased susceptibility of inhibitory neurons to apoE4’s detrimental effects in AD pathogenesis, as reported previously (Andrews-Zwilling et al., 2010; Najm et al., 2019; Wang et al., 2018). Alternatively, this dysregulation of the UPR could be a downstream effect of some other action that makes inhibitory neurons especially vulnerable to apoE4. Multiple groups have shown that certain neuron lineages are selectively vulnerable in the context of various neurodegenerative disorders, and investigating this hypothesis is an intriguing line of questioning for the future (Fu et al., 2018).
Using this in vivo chimeric AD modeling system, we also demonstrated that hiPSC-derived neurons without FAD gene mutations mimic what we see in human late-onset AD patients and generate Aβ aggregates in vivo and that the E4KI brain environment leads to increased Aβ aggregate accumulation through increased deposition, decreased clearance, or a combination of the two. Traditional in vitro 2D models of AD using hiPSC-derived neurons do not generate Aβ aggregates or plaques. Likewise, E4KI mouse models without FAD gene mutations do not develop Aβ plaques, causing challenges for the in vivo study of apoE4’s roles in Aβ production, accumulation, and clearance in sporadic, late-onset AD. By taking advantage of this in vivo chimeric modeling paradigm, we demonstrate that hiPSC-derived neurons can generate Aβ aggregates and plaques without mutations in the amyloid processing pathway, and that this process is significantly exacerbated by the apoE4 brain environment. We do not see clear degeneration of transplanted human neurons when exposed to Aβ, as shown elsewhere (Espuny-Camacho et al., 2017). However, this is likely an effect of the difference in model systems. More specifically, the amount of Aβ produced in this chimeric disease model in the context of apoE4 is substantially less than the amount of Aβ produced in APPFAD transgenic mice used in the other study, in which the former is more representative of sporadic, late-onset AD driven by apoE4 and the latter is more representative of early-onset FAD. Our apoE4-specific in vivo chimeric AD model will allow more in-depth studies on human Aβ production, seeding, aggregation, and clearance and future therapeutic development, especially for late-onset AD.
Finally, this in vivo chimeric AD modeling system provides insight into the roles of microglia in human-neuron-derived Aβ aggregate formation and/or accumulation. We showed both that amyloid plaques are increased in the E4KI host context and that E4KI microglia colocalize less with Aβ aggregates produced by human neurons. These findings may indicate a deficit in the phagocytic function of apoE4 microglia, as previously reported (Jiang et al., 2008; Lee and Landreth, 2010; Lin et al., 2018; Yamazaki et al., 2019). Alternatively, Aβ might form aggregates inside microglia that are released into the extracellular space upon microglial death, which may seed plaque formation, as suggested in some studies (Baik et al., 2016; Shi and Holtzman, 2018; Sosna et al., 2018; Spangenberg et al., 2019). In this case, microglia play a role in generating Aβ aggregates, and apoE4 may predispose microglia to this pathological process. In the future, microglial ablation experiments, in combination with hiPSC-derived neuronal transplants, would give us the opportunity to dissect these two possibilities.
In summary, we have shown that in vivo chimeric disease modeling using hiPSC-derived neuron subtypes, in combination with snRNA-seq and immunohistochemistry, can be a powerful tool for better understanding complex and human-specific disease mechanisms of AD. This in vivo chimeric AD modeling system enables investigation into how diverse brain cell types interact with human neurons in modulating AD pathologies in an apoE-genotype-dependent manner in the in vivo environment. Future experiments using this system will continue to shed light on human-specific aspects of AD pathogenesis and potentially lead to novel therapeutics that are only discoverable by examining human-specific effects of AD pathogenesis in the in vivo environment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yadong Huang (yadong.huang@gladstone.ucsf.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated during this study have been deposited to the NCBI Gene Expression Omnibus with the accession code GSE152867. In addition, processed data are available in the Supplemental Information. The KEGG pathway database used for pathway analysis is available at https://www.genome.jp/kegg/kegg1.html (Kanehisa and Goto, 2000).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice with human apoE3 or apoE4 knocked-in at the mouse Apoe locus on a C57BL/6 background were originally obtained from Taconic (Sullivan et al., 2004). All animals were bred in-house using trio breeding producing 8–10 pups per litter on average, which were weaned at 28 days. Female littermates at 6–7 months of age were randomly assigned to experimental groups. Animals were housed in a pathogen-free barrier facility on a 12hr light cycle (lights on at 7 am and off at 7 pm) at 19–23°C and 30%–70% humidity. Animals were identified by ear punch under brief isofluorane anesthesia and genotyped by polymerase chain reaction (PCR) of a tail clipping at both weaning and perfusion. All animals otherwise received no procedures except those reported in this study. All animal experiments were conducted in accordance with the guidelines and regulations of the National Institutes of Health, the University of California, and the Gladstone Institutes under IACUC protocol AN117112.
Cell Lines
All hiPSC lines were derived from human skin fibroblasts from female donors and reprogrammed as previously described (Wang et al., 2018) and were maintained at 37°C with 5% humidity. All hiPSC lines were characterized for normal pluripotency gene expression, apoE genotypes, karyotypes, and capability of differentiating into neural stem cells as well as different types of neurons in culture. All hiPSC lines were tested negative for mycoplasma.
METHOD DETAILS
Generation of mCherry-expressing hiPSC lines
The E4/4 hiPSC line was generated as described (Takahashi and Yamanaka, 2006; Takahashi et al., 2007) from skin fibroblasts of a subject with an APOE4 genotype. The isogenic E3/3 hiPSC line was generated from this parental E4/4 hiPSC line as previously described (Wang et al., 2018). hiPSCs were maintained under feeder-free conditions in mTeSR1 medium (STEMCELL Technologies) and routinely passaged 1:10–1:15 by brief treatment with Accutase (Millipore). The hiPSC protocol was approved by the Committee on Human Research at the University of California San Francisco. To achieve constitutive mCherry expression, we knocked in an mCherry expression cassette into the AAVS1 locus of both the E4/4 hiPSC line and the isogenic E3/3 hiPSC line. pCas9-GFP (Addgene #44719) (Ding et al., 2013), and gRNA_AAVS1_T2 (Addgene #41818) (Mali et al., 2013) were used to target the AAVS1 locus and insert the CAG-mCherry expression cassette (Addgene #80946) (Chen et al., 2016). Integration of the construct at the proper locus was analyzed by PCR similarly to previous methods (Mandegar et al., 2016). A Junction PCR spanning the 3’ end of the wild-type sequence and the 5’ site of the construct was used to assess proper targeting. Primers for the 3’ end of the wild-type sequence and 5’ end of the construct were 5’-CGGTTAATGTGGCTCTGGTT-3’ and 5’-GTGGGCTTGTACTCGGTCAT-3’ respectively (expected product size = 1068bp). Additionally, homozygosity of mCherry integration was tested by PCR spanning the wild-type sequence. Successfully homozygous integration would result in no band in the PCR gel as the expected sequence was over 8kb and therefore too large for a successful standard PCR reaction. Primers used included (WT-AAV-F) 5’-CGGTTAATGTGGCTCTGGTT-3’ and (WT-AAV-R) 5’-AGGATCCTCTCTGGCTCCAT-F’.
Generation of hiPSC-derived neurons
Mixed hiPSC-derived neuronal populations were derived based on methods developed previously from our lab (Wang et al., 2018). Three 10cm-dishes (Corning) of 85% confluent hiPSCs were dissociated with a 5–8 min treatment of warm Accutase (Millipore). Accutase was then neutralized by addition of equal volume of N2B27 medium (50% DMEM/F12 (GIBCO), 50% Neurobasal Medium (GIBCO), 1% NEAA (GIBCO), 1% Glutamax (GIBCO), 0.5% Pen/Strep (GIBCO), 1X N2 Supplement (GIBCO), 1X B27 Supplement (GIBCO)). The dissociated hiPSCs were centrifuged and re-suspended in N2B27 medium with the addition of 10 μM SB (Stemgent) and 0.25 μM LDN (Stemgent) as well as 10 μM Rock Inhibitor (Tocris) in a T-175 flask (Thermo Scientific). Flasks were briefly shaken by hand every hour for the first 3 h after dissociation to aid in the formation of small spheres and prevent large sheets of cells aggregating. After 24 h, hiPSCs formed embryoid bodies (EBs) and were centrifuged at 200 g for 3 min, medium was aspirated and spheres were resuspended in N2B27 medium containing 10 μM SB and 0.25 μM LDN only. This was repeated every 48 h for 10 days. On day 10, the EBs were plated down onto two 10cm dishes coated with growth factor reduced Matrigel (Corning) and allowed to form neuronal rosettes. The rosettes were sustained in N2B27 media alone and half of the media was replaced every 48–72 h depending on confluency and media consumption. On day 21, rosettes were dissociated with Accutase for 8–10 min. Accutase was then neutralized with an equal volume of N2B27 medium, and cells were collected and centrifuged at 200 g for 3 min. The cells were then resuspended in N2B27 medium supplemented with bFGF (5 ng/mL) (Peprotech) and EGF (20 ng/mL) (Peprotech) and allowed to form neurospheres in suspension in a T-175 flask. The neurospheres were maintained for 14 more days in suspension with cells centrifuged and re-suspended in N2B27 medium with 5 ng/mL bFGF (Peprotech) and 20 ng/mL EGF (Peprotech) every 48–72 h depending on media consumption. On Day 35, neurospheres were collected by centrifugation (200 g for 3 min) and resuspened in Accutase (Millipore) for 8–10 min at 37 C for dissociation with brief, gentle pipetting, as needed. Once spheres were disassociated, medium was added to bring the total volume of the cell suspension up to 40mL, which was filtered through a 40 mm strainer (Fisher) to ensure a single cell suspension. The cells were then collected by centrifugation, resuspended in N2B27 medium, and counted to determine cell concentration. Once appropriate cell numbers were determined, they were plated at 30,000 cell/cm2 onto tissue culture plates, or glass coverslips (Corning) for immunocytochemistry, that were pre-coated with Poly-L-Lysine (Sigma, 0.1 mg/mL) overnight at 37°C, washed 3X with DPBS (GIBCO), and then further coated with Laminin (GIBCO, 6 mg/mL) overnight at 37°C. During neuronal differentiation, the medium was supplemented with 10 μM DAPT (Tocris), 10 ng/mL BDNF (Peprotech), and 10 ng/mL GDNF (Peprotech) for one week, after which DAPT was removed and 10 ng/mL BDNF and 10 ng/mL GDNF were maintained. 75% of the culture medium was replaced on maturing neurons every 3–4 days.
Immunocytochemistry
Cells plated on glass coverslips were fixed in 4% paraformaldehyde for 15 min and washed three-times for 5 min/wash with 1X-PBS. Cells were treated with 0.5% Triton-X in PBS for 5 min followed by 1 h blocking with 10% Normal Donkey Serum, 0.5% Triton-X in PBS before incubation with primary antibodies diluted in antibody dilution buffer (3% Donkey Serum in PBS) overnight at 4°C. Cells were then washed three-times for 5 min/wash with 1X PBS and incubated for 1-hour at room temperature with fluorescently conjugated secondary antibodies diluted in antibody dilution buffer (See Key Resources Table). Cells were mounted with Vectashield Gold containing DAPI for nuclear staining. Images were collected with an inverted epifluorescence microscope (Keyence).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-mCherry | Abcam | Cat# ab167453; RRID:AB_2571870 |
| Mouse anti-mCherry | Abcam | Cat# ab125096; RRID:AB_11133266 |
| Rat anti-mCherry | Thermo Fisher Scientific | Cat# M11217 RRID:AB_2536611 |
| Mouse anti-Human Nuclear Antigen-PE | Abcam | Cat#ab215755 RRID: N/A |
| Mouse anti-MAP2 | Santa Cruz Biotechnologies | Cat# sc-32791; RRID:AB_627948 |
| Mouse anti-TUJ1 | Promega | Cat# G7121; RRID:AB_430874 |
| Mouse anti-vGlut1 | Millipore Sigma | Cat#MAB5502 RRID:AB_262185 |
| Rabbit anti-TBR1 | Abcam | Cat# ab31940; RRID:AB_2200219 |
| Rabbit anti-GABA | Sigma Aldrich | Cat# a2052; RRID:AB_477652 |
| Mouse anti-OCT-3/4 | Santa Cruz Biotechnologies | Cat# sc-5279; RRID:AB_628051 |
| Rabbit anti-Nanog | Stemgent | Cat# 09-0020; RRID:AB_2298294 |
| Mouse anti-TRA-1-81 | Millipore Sigma | Cat# MAB4381; RRID:AB_177638 |
| Mouse anti-TRA-1-60 | Millipore Sigma | Cat# MAB4360; RRID:AB_2119183 |
| Mouse anti-HNA | Millipore Sigma | Cat# MAB1281; RRID:AB_94090 |
| Guinea Pig anti-NeuN | Millipore Sigma | Cat# ABN90; RRID:AB_11205592 |
| Rabbit anti-NKX2.1 | Santa Cruz Biotechnologies | Cat# sc-13040; RRID:AB_793532 |
| Moues anti-PAX6 | Millipore Sigma | Cat# MAB5554; RRID:AB_570718 |
| Rabbit anti-FOXG1 | Santa Cruz Biotechnologies | Cat# sc-48788; RRID:AB_2231897 |
| Mouse anti-3D6 | Elan Pharmaceuticals | N/A |
| Rabbit anti-IBA1 | Wako | Cat#019-19741 RRID:AB_839504 |
| Mouse anti Human Synaptophysin | Millipore | Cat# MAB5555 RRID:AB_10553797 |
| Donkey anti-mouse 488nm | Thermo Fisher Scientific | Cat#A-21202; RRID:AB_141607 |
| Donkey anti-rabbit 488nm | Thermo Fisher Scientific | Cat#A-21206; RRID:AB_2535792 |
| Donkey anti-mouse 594nm | Thermo Fisher Scientific | Cat#R37115; RRID:AB_2556543 |
| Donkey anti-rabbit 594nm | Thermo Fisher Scientific | Cat#A-21207; RRID:AB_141637 |
| Donkey anti-rat 594nm | Thermo Fisher Scientific | Cat#A-21209; RRID:AB_2535795 |
| Donkey anti-guinea pig 647nm | Jackson ImmunoResearch | Cat#706-605-148; RRID:AB_2340476 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hibernate (HEB) medium | BrainBits LLC | Cat#HAB 500 |
| MACS Smart Strainer | Miltenyi Biotech | Cat#130-110-915 |
| 24-well Coverslips | Corning | Cat#354085 |
| Mouse-on-Mouse Blocking Reagent | Vector Labs | Cat#MKB-2213; |
| Normal Donkey Serum | Jackson Labs | Cat#017-000-121 |
| Triton-X | Millipore Sigma | Cat#T8787-250mL |
| Tween-20 | Millipore Sigma | Cat#P2287-500mL |
| Accutase | Millipore Sigma | Cat#SCR005 |
| NEAA | GIBCO | Cat#11140-050 |
| Glutamax | GIBCO | Cat#35050-061 |
| Pen/Strep | GIBCO | Cat#15140-122 |
| SB | Stemgent | Cat#04-0010-10 |
| LDN | Stemgent | Cat#04-0074 |
| Rock Inhibitor | Tocris | Cat#1254 |
| bFGF | Peprotech | Cat#100-18C |
| EGF | Peprotech | Cat# AF-100-15 |
| Poly-L-Lysine | Sigma | Cat#4707 |
| PBS | Thermo Fisher Scientific | Cat# 14190250 |
| Laminin | GIBCO | Cat#23017-015 |
| DMEM/F12 | GIBCO | Cat#11330-032 |
| Neurobasal | GIBCO | Cat# 21103-049 |
| DAPT | Tocris | Cat#2634 |
| BDNF | Peprotech | Cat#450-02 |
| GDNF | Peprotech | Cat#450-10 |
| Matrigel hESC Qualified | Corning | Cat#354277 |
| Matrigel Growth Factor Reduced | Corning | Cat#354230 |
| N2 Supplement | Thermo Fisher Scientific | Cat#17502048 |
| mTESR | Stemcell technologies | Cat#85850 |
| B27 Supplement | Thermo Fisher Scientific | Cat#17504044 |
| Thioflavin-S | Sigma Aldrich | Cat#T-1892 |
| DNaseI | Roche | Cat#04536282001 |
| Ketamine | Henry Schein | Cat#1049007 |
| Xylazine (Anased) | Henry Schein | Cat#1311139 |
| Isofluorane | Henry Schein | Cat#029405 |
| Buprenorphine | Henry Schein | Cat#055175 |
| Ketofen | Henry Schein | Cat#005487 |
| Avertin (2,2,2-Tribromoethanol) | Millipore Sigma | Cat#T48402 |
| ProLong Gold with DAPI | Thermo Fisher Scientific | Cat#P36931 |
| InVivoMab anti-mouse CD40L (CD154) Clone MR-1 | BioXCell | Cat#BE0017-1 |
| InVivoMab anti-mouse CTLA-4 (CD152) Clone 9D9 | BioXCell | Cat#BE0164 |
| InVivoMab anti-mouse LFA-1a (CD11a) Clone M17/4 | BioXcell | Cat#BE0006 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3′ Library/Gel Bead kit v3 | 10x Genomics | Cat#1000092 |
| Deposited Data | ||
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | Kanehisa and Goto, 2000 | https://www.genome.jp/kegg/kegg1.html |
| Raw snRNA-seq data | This manuscript | GEO: GSE152867 |
| Processed snRNA-seq data | This manuscript | Tables S1, S2, S3, S4, and S5 |
| Experimental Models: Cell Lines | ||
| Human: Isogenic ApoE3/3 iPSC line | Wang et al., 2018 | N/A |
| Human: ApoE4/4 iPSC line | Wang et al., 2018 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: ApoE4-KI: B6.129P2-Apoetm3(APOE-4)Mae n8 | Taconic | Cat#1549-F |
| Mouse: ApoE4-KI: B6.129P2-Apoetm3(APOE*4)Mae N8 | Taconic | Cat#1548-F |
| Oligonucleotides | ||
| Primer: Junction Forward CGGTTAATGTGGCTCTGGTT | Mandegar et al., 2016 | N/A |
| Primer: Junction Reverse GTGGGCTTGTACTCGGTCAT | Mandegar et al., 2016 | N/A |
| Primer: WT-AAV Forward CGGTTAATGTGGCTCTGGTT | Mandegar et al., 2016 | N/A |
| Primer: WT-AAV Reverse AGGATCCTCTCTGGCTCCAT | Mandegar et al., 2016 | N/A |
| Recombinant DNA | ||
| Plasmid: pCas9-GFP | Ding et al., 2013 | Addgene Plasmid Cat# 44719 RRID:Addgene_44719 |
| Plasmid: gRNA_AAVS1_T2 | Mali et al., 2013 | Addgene Plasmid Cat# 41818 RRID:Addgene_41818 |
| Plasmid: CAG-mCherry | Chen et al., 2016 | Addgene Plasmid Cat# 80946 RRID:Addgene_80946 |
| Software and Algorithms | ||
| Seurat v2.3.4 | Butler et al., 2018; Stuart et al., 2019 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Rmagic v1.0.0 | van Dijk et al., 2018 | https://cran.r-project.org/web/packages/Rmagic/index.html |
| limma v3.34.9 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| gplots v3.0.1 | CRAN | https://cran.r-project.org/web/packages/gplots/index.html |
| Cellranger v2.0.1 | 10x Genomics | https://github.com/10XGenomics/cellranger |
| R v.3.4.2 | R Core Team, 2019 | http://www.R-project.org/ |
| Axon pCLAMP | Molecular Devices | https://mdc.custhelp.com/app/answers/detail/a_id/18779/~/axon%E2%84%A2pclamp%E2%84%A2-10-electrophysiology-data-acquisition-%26-analysis-software-download |
| IGOR Pro | WaveMetrics | https://www.wavemetrics.com/ |
| Other | ||
| BD FACSAria-II | BD Biosciences | http://www.bdbiosciences.com/documents/BD_FACSAria_II_cell_sorter_brochure.pdf |
| Chromium Controller & Next GEM Accessory Kit | 10x Genomics | Cat#120223 |
| Nucleofector II | Amaxa | Cat# AAB-1001 |
| Illumina NovaSeq 6000 | Illumina | https://www.illumina.com/systems/sequencing-platforms/novaseq.html |
Transplantation
hiPSC-derived neurons (D42–49; i.e., +1 to +2 weeks of maturation, see Figure S2A) were washed in 1X PBS which was then aspirated and replaced with warm Accutase (Millipore) for 15 min or until neurons dissociated with gentle tapping. Accutase (Millipore) was neutralized with N2B27 medium to bring total volume > 30 mL and then cells were filtered through a 40 μm strainer (Fisher) to ensure a single cell suspension. Single cells were then centrifuged and resuspended to concentration of 1000 cells/nL in 1X HBSS (GIBCO) supplemented with 10 ng/mL BDNF (Peprotech), 10 ng/mL GDNF (Peprotech) and 100 ng/mL DNaseI (Roche) and kept at 4°C until transplantation. ApoE4-KI and apoE3-KI mice were anesthetized with an intraperitoneal injection of ketamine (60 mg/kg) and xylazine (30 mg/kg) and maintained on 0.8%–1.0% isofluorane (Henry Schein). The head was secured with earbars and a tooth bar in a stereotaxic alignment system (Kopf Instruments). Fur was removed from the scalp, which was then sterilized with alternating swabs of chlorhexidine and 70% ethanol. The scalp was opened and sterilized with 3% hydrogen peroxide. Cell suspensions (~1000 cells/nL) were loaded into ~60 μm tip diameter, 30° beveled glass micropipette needles (Nanoject, Drummon Scientific Company). Bilateral rostral and caudal stereotaxic sites were drilled with a 0.5 mm microburr (Foredom, Fine Science Tools), and the coordinates used for hilar transplantation were X = ± 1.65, Y = 2.00, Z = 1.7 and X = ± 2.90, Y = 3.20, Z = 2.2, with Z measured from the surface of the brain (David Kopf Instruments). At each transplantation site, ~20nL (~20,000 cells) were injected and allowed to diffuse for 3 min. There were two hippocampal transplantation sites on each hemisphere (a total of four transplantation sites per mouse). For recovery, mice were sutured with 6–0 monofilament nonabsorbent nylon sutures (Ethicon), administered analgesics ketophen (5 mg/kg subcutaneously) and buprenorphine (0.0375 mg/kg intraperitoneally), and monitored on a heating pad until ambulatory. Immunosuppressants were administered immediately after transplantation (day 0) via intraperitoneal injection followed by injections on day 2, 4, and 6 post transplantation. Immunosuppressants were comprised of a cocktail of anti-mouse CD40L (CD154) (BioXCell), anti-mouse CTLA-4 (CD152) (BioXCell), and anti-mouse LFA-1a (CD11a) (BioXcell), and all were used at a concentration of 20 mg/kg.
Immunohistochemistry
At 7 months post transplantation (MPT), animals were deeply anesthetized with avertin and perfused with 0.9% saline solution. Mouse brains were then collected and followed by 48 h incubation in 4% PFA at 4°C, 24 h incubation in 1X PBS, and 48 h in 30% Sucrose (Sigma) or until sunk. Coronal sections were cut on a microtome at a thickness of 30 μm and stored in cryoprotectant solution (30% Ethylene Glycol, 30% Glycerol, 40% 1X PBS) at −20°C. For immunohistochemical staining, every tenth section was first transferred to a 24-well plate in PBS and were then washed 3×10min in PBS to remove cryoprotectant solution and another 2×15min in PBS-T (PBS + 0.1% Tween-20 (Sigma)). Slices were then transferred to blocking solution (10% NDS, 0.2% Gelatin (Sigma), 0.5% Triton-X (Sigma) in PBS) for 1 h at room temperature. After blocking, slices were washed 1×10min in PBS. PBS was then aspirated and slices were incubated in Mouse on Mouse Blocking Buffer (M.O.M Vectashield kit (1 drop M.O.M IgG/5mL PBS) for 1 h at room temperature. After M.O.M block, slices were incubated in primary antibody diluted in M.O.M antibody dilution buffer ([1:12.5] M.O.M Protein Concentrate) at 4°C overnight. After primary antibody incubation, slices were washed 3×10 min in PBS-T (PBS + 0.1% Tween-20 (Sigma)) and then incubated in fluorescent secondary antibodies diluted in M.O.M antibody dilution buffer for 1 h at room temperature protected from light. After secondary antibody incubation, slices were washed 3×10 min in PBS and mounted onto microscope slides and mounted with Vectashield with DAPI (Vector Laboratories). When performing Thioflavin-S staining, floating sections were stained with 0.015% thioflavin-S in 50% ethanol diluted in PBS for 10 min, washed three-times for 5 min/wash with 1X PBS before mounting. Images of transplants were collected with an LSM 880 confocal microscope (Zeiss)
Electrophysiology
Transplanted mice were deeply anesthetized with isoflurane and decapitated. The brain was rapidly removed from the skull and placed in the 4°C slicing solution comprised of 110 mM Choline Chloride, 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 2 mM CaCl2, 1.3 mM Na Pyruvate, 1 mM L-Ascorbic Acid, and 10 mM dextrose. 300mm sagittal sections were cut using a vibratome (VT 1200s, Leica). Following slicing, the slices were transferred into a vapor interface chamber aerated with 95% O2/5% CO2 gas mixture and allowed to recover at 34°C for 1 h before recording. Cultured cells or brain slices were placed into a submerged recording chamber at 34°C perfused at 10 mL/min with oxygenated aCSF solution comprised of 124 mM NaCl, 26 mM NaHCO3, 10 mM Glucose, 1.25 mM NaH2PO4, 2.5 mM KCl, 1.25 mM MgCl2, and 1.5 mM CaCl2. Patch pipettes were filled with potassium-gluconate based solution containing 122.5 mM K-gluconate, 8 mM KCl, 10 mM HEPES, 2 mM MgCl2, 0.2 mM EGTA, 2 mM ATPNa, and 0.3 mM GTPNa. For synaptic current recordings in cultured cells, cesium-based intracellular solution was used containing 130 mM CsCl, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES-NaOH, 0.2 mM EGTA-KOH, 2.5 mM Na2ATP and 0.5 mM Na2GTP. Neurons were imaged using a modified Olympus BXW-51 microscope with a 60x objective (Scientifica Inc, Great Britain). Patch-clamp recordings were performed using a Multiclamp 700B amplifier (Molecular Devices, CA, USA). The signals were sampled at 10 kHz, low-pass filtered at 0.2 kHz and digitized using a Digidata 1550B with Axon pCLAMP software (Molecular Devices, CA, USA). Data analysis was performed using custom software in IgorPRO (Wavemetrics Inc, USA). For cultured cells’ synaptic current recordings, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 mM), D-(−)-2-amino-5-phosphonopentanoic acid (APV, 50 μM) were applied in the ACSF to selectively block excitatory synaptic currents, and SR95531 (Gabazine, 25 μM) to block GABAA receptor-mediated currents. Spontaneous post-synaptic currents (PSCs) were recorded in voltage-clamp configuration at a holding potential of −70mV. For recordings in ex-vivo brain slices, target neurons were identified by mCherry fluorescence; spontaneous post-synaptic currents (sPSCs) were recorded in voltage-clamp mode at a holding potential of −70mV
Single nuclei FACS sorting of transplanted human neurons from mouse hippocampi
Hippocampi were isolated from single hemispheres of the transplanted mice at the time of euthanasia, immediately frozen in dry ice and stored at −80°C. The relative contribution of each transplant condition to the FACS sorting was as follows: E3KI::iE3/3 transplants N = 6; E3KI::E4/4 transplants N = 6;E4KI::iE3/3 transplants N = 6; E4KI::E4/4 transplants N = 5. For isolation of human nuclei, hippocampi were thawed and incubated in HEB (BrainBits LLC)(1mL/hippocampi) and kept on ice. 2mL chilled lysis buffer was then added to the tissue and was homogenized with 21G needle. Tissue was lysed on ice for 15 min and swirled to mix. This was repeated 3 times during incubation. Lysed hippocampi were titrated 5–7 times with 1mL pipette. Lysate was then washed in 30 μm MACS Smart Strainer to remove large clumps and then centrifuged at 500 g for 5 min are 4°C. Supernatant was aspirated and pellet was washed with 1mL Nuclei Wash and Resuspension Buffer. Nuclei were centrifuged at 500 g for 5 min at 4°C and supernatant was then aspirated. The nuclear pellet was resuspended in 500 mL staining buffer (0.5% RNase-free BSA and 0.2 μg/mL RNasin Plus RNase inhibitory in RNase-free PBS) and incubated for 15 min on ice to allow for blocking of non-specific antibody binding. 100 μL of samples were removed for negative control staining. After blocking, anti-human nuclear antigen antibody conjugated to phycoerythrin was added to the nuclei solution [1:100] and incubated at 4°C for 30 min rotating. After incubation, samples were washed by adding 500 μL of staining buffer to each tube and inverting several times. Samples were then collected by centrifugation for 5 min at 400 g at 4°C. Supernatant was aspirated, and nuclei were resuspended in 500 μL staining buffer for FACS sorting, with DAPI (0.1 μg/mL) added directly before sorting on Aria FACS sorter (BD Biosciences).
Human cDNA library preparation and sequencing
cDNA libraries were prepared using the Chromium Single Cell 3’ Library and Gel Bead kit v3 (10x Genomics: 1000092) according to the manufacturer’s instructions. Libraries were sequenced on an Illumina NovaSeq 6000 sequencer at the UCSF CAT Core.
Pre-processing and clustering of human-nucleus RNA sequencing samples
The samples were aligned with Cellranger v.2.0.1 to a custom reference genome built from GRCh38–3.3.0 that includes introns, as nuclear pre-mRNA includes intronic portions. Each of the filtered UMI count matrices was loaded into Seurat v.2.3.4 (Butler et al., 2018; Stuart et al., 2019). Data were filtered to include only protein coding genes. Cells were filtered to include only cells with 100–1000 genes detected, 100–2000 UMI, and < 0.05% mitochondrial reads. This quality assurance process resulted in a final matrix of 33,538 genes by 13,648 nuclei. The gene expression matrices were then log-normalized with a scale of factor of 10,000. Highly variable genes were selected by filtering for an average expression range of 0.25 to 8 and a minimum dispersion of 1.25, resulting in a list of 2,430 genes. Principal components (PCs) were calculated and visually examined as an elbow plot. The shared-nearest neighbor graph was constructed using the first 16 PCs and a resolution of 0.2 resulting in a set of 10 distinct clusters. Visualization of clusters was performed with a t-stochastic neighbors embedding (tSNE), again using the first 16 PCs.
Cell-type assignment of hiPSC-derived neurons from transplants
Data visualization by tSNE revealed clusters where transplanted samples were intermingled, with no discernable evidence of batch effects by donor or host. Marker genes for each cluster were calculated using the FindAllMarkers function in Seurat, using the Wilcoxon rank sum test, with the parameters logfc.threshold = 0.25 and min.pct = 0.1. Broad cell classes, such as excitatory and inhibitory neurons, astrocytes, and oligodendrocytes were identified on the basis of canonical markers, and markers derived from previous RNA sequencing data on sorted cell types.
Image analysis
Confocal fluorescent images (z stacks) were acquired using a Zeiss LSM880 Confocal microscope. Image analysis was performed using custom macros written in the open source Fiji (ImageJ) software. Quantification of 3D6+ puncta was performed by manually counting each puncta and measuring its distance from the edge of the transplant in order to determine if it was within a 100 μm perimeter. Analyses of 3D6+ puncta, 3D6/IBA1 colocalization, as well as Iba-1+ and GFAP+ cell counts were conducted manually, but blinded to samples, again to exclude the possibility of bias.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where applicable, all statistical details of the experiments including tests used, value of n, definition of center, and dispersion measures can be found in the corresponding figure legend. Additional description of statistical methods used is detailed on a per-experiment basis below.
MAGIC imputation
The raw counts data were filtered to include only genes with >100 reads across all nuclei, library size and square-root normalized as previously described (van Dijk et al., 2018). Data were then imputed with the magic function, allowing the function itself to set the optimal parameters. All feature plots are derived from the Magic imputed dataset.
Gene expression network analysis
Proportion of shared genes in KEGG pathways was calculated using custom software in R, with KEGG annotations from the limma package.
Groupwise statistics on immunohistochemical analysis
All immunohistochemical statistics were conducted as a two-way ANOVA in Graphpad Prism 7 (Graphpad Software Inc.). When a central value is plotted it is always ± SEM, as indicated in figure legends. In all cases, n represents the number of transplant images which were used for quantification and N represents the number of mice from which the transplant images were gathered. All image quantification was based on number of transplant images. Significance was established as p < 0.05. No data were excluded based on statistical tests. p values displayed are from Sidak’s multiple comparisons post hoc tests.
Supplementary Material
Highlights.
In vivo chimeric Alzheimer’s disease modeling of apoE4 toxicity
Excitatory and inhibitory neurons have distinct transcriptional responses to apoE4
Human iPSC (hiPSC)-derived neurons generate Aβ aggregates in vivo
Mouse apoE4 microglia are deficient in clearing Aβ aggregates made by human neurons
ACKNOWLEDGMENTS
This work was supported in part by grants R01AG048017, RF1AG048030, and R01AG055682 to Yadong Huang from the National Institutes of Health (NIH). The Gladstone Flow Cytometry Core FACSAria cell sorter is supported by NIH grant S10 RR028962 and by the James B. Pendleton Charitable Trust. We thank the lab of Li Gan, specifically Yungui Zhou, for providing primary rat astrocytes for in vitro electrophysiological studies; Eric Chow and the staff at the University of California, San Francisco (UCSF) Center for Advanced Technology (CAT) core for advice and support with RNA sequencing (RNA-seq); Natasha Carli, Jim McGuire, and Po-Lin So of the Gladstone Genomics Core for advice on sample preparation and sequencing; Nandhini Raman of the Gladstone Flow Cytometry Core for sorting the nuclei; Steve Belunek and Wil Maguire of Gladstone Information Technology for server support; Julia Roudabush and Veronica Viray for contract support; Sidney Oduah and Linda Hagimori for purchasing; and Theodora Pak for editorial assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107962.
DECLARATION OF INTERESTS
Yadong Huang is a cofounder and scientific advisory board member of EScape Bio, Inc.; GABAeron, Inc.; and Mederon Bio, LLC. Other authors declare no competing financial interests.
REFERENCES
- Abid NB, Naseer MI, and Kim MO (2019). Comparative Gene-Expression Analysis of Alzheimer’s Disease Progression with Aging in Transgenic Mouse Model. Int. J. Mol. Sci 20, 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Absalon S, Kochanek DM, Raghavan V, and Krichevsky AM (2013). MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci 33, 14645–14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J, and Scheper W (2016). Targeting neuronal MAPK14/p38a activity to modulate autophagy in the Alzheimer disease brain. Autophagy 12, 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkon DL, Sun M-K, and Nelson TJ (2007). PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci 28, 51–60. [DOI] [PubMed] [Google Scholar]
- Ameri K, and Harris AL (2008). Activating transcription factor 4. Int. J. Biochem. Cell Biol 40, 14–21. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, and Huang Y (2010). Apolipoprotein E4 causes age-and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 30, 13707–13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik SH, Kang S, Son SM, and Mook-Jung I (2016). Microglia contributes to plaque growth by cell death due to uptake of amyloid b in the brain of Alzheimer’s disease mouse model. Glia 64, 2274–2290. [DOI] [PubMed] [Google Scholar]
- Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, and Nofer JR (2011). Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol 31, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, and Hengst U (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, et al. (1999). Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 96, 15233–15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, and Dudek SM (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. USA 105, 3123–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, and Mattson MP (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 31, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, and Huang Y (2012). Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci 32, 4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, and Tsien RW (1996). CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, Buscemi S, and Palumbo Piccionello A (2018). Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci 19, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TVP, Hyman BT, et al. (1999). Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci 2, 271–276. [DOI] [PubMed] [Google Scholar]
- Chen Y, Xiong M, Dong Y, Haberman A, Cao J, Liu H, Zhou W, and Zhang S-C (2016). Chemical Control of Grafted Human PSC-Derived Neurons in a Mouse Model of Parkinson’s Disease. Cell Stem Cell 18, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. (2005). Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature 436, 1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudaback E, Li X, Montine KS, Montine TJ, and Keene CD (2011). Apolipoprotein E isoform-dependent microglia migration. FASEB J. 25, 2082–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M, Cho J-H, Leung A, Savvidis G, Ahn S, Moon M, Lee PKJ, Han JJ, Azimi N, Kim K-S, et al. (2014). hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell 15, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- Del Prete D, Checler F, and Chami M (2014). Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I, and Stutzmann GE (2010). Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem 285, 12463–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, and Musunuru K (2013). Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I, Arranz AM, Fiers M, Snellinx A, Ando K, Munck S, Bonnefont J, Lambot L, Corthout N, Omodho L, et al. (2017). Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 93, 1066–1081.e8. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, and van Duijn CM; APOE and Alzheimer Disease Meta Analysis Consortium (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278, 1349–1356. [PubMed] [Google Scholar]
- Fu H, Hardy J, and Duff KE (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci 21, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, and Thakkar A (2017). Interactions of Mitochondria/Metabolism and Calcium Regulation in Alzheimer’s Disease: A Calcinist Point of View. Neurochem. Res 42, 1636–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guria A, Tran DDH, Ramachandran S, Koch A, El Bounkari O, Dutta P, Hauser H, and Tamura T (2011). Identification of mRNAs that are spliced but not exported to the cytoplasm in the absence of THOC5 in mouse embryo fibroblasts. RNA 17, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, McQuade A, Kolahdouzan M, Echeverria K, Claes C, et al. (2019). Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 103, 1016–1033.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes M, Eichhoff G, and Garaschuk O (2010). Intracellular calcium signalling in Alzheimer’s disease. J. Cell. Mol. Med 14, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, and Paul SM (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 97, 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y (2010). Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol. Med 16, 287–294. [DOI] [PubMed] [Google Scholar]
- Huang Y, and Mucke L (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Weisgraber KH, Mucke L, and Mahley RW (2004). Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J. Mol. Neurosci 23, 189–204. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CYD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, et al. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadamur G, and Ross EM (2013). Mammalian phospholipase C. Annu. Rev. Physiol 75, 127–154. [DOI] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, and Tyagi N (2016). Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol 53, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap G, Bapat D, Das D, Gowaikar R, Amritkar RE, Rangarajan G, Ravindranath V, and Ambika G (2019). Synapse loss and progress of Alzheimer’s disease—A network model. Sci. Rep 9, 6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Inoue H, Hurt E, and Yoneda Y (2009). Adaptor Aly and co-adaptor Thoc5 function in the Tap-p15-mediated nuclear export of HSP70 mRNA. EMBO J. 28, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Magno L, Rubin AN, and Oliveira MG (2014). Genetic programs controlling cortical interneuron fate. Curr. Opin. Neurobiol 26, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan TK, and Alkon DL (2006). An internally controlled peripheral biomarker for Alzheimer’s disease: Erk1 and Erk2 responses to the inflamma-tory signal bradykinin. Proc. Natl. Acad. Sci. USA 103, 13203–13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, and Holtzman DM (2012). Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J. Exp. Med 209, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, Bien-Ly N, and Huang Y (2014). Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci 34, 14069–14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, Linker SB, Pham S, Erwin JA, Miller JA, et al. (2016). Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc 11, 499–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Jha NK, Jha SK, Ramani K, and Ambasta RK (2015). Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: clinical relevance in Alzheimer’s disease. J. Alzheimers Dis 43, 341–361. [DOI] [PubMed] [Google Scholar]
- Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy W-Y, Duennwald ML, Prado VF, and Prado MAM (2017). The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci 11, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung H-L, Chen S, et al. (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CYD, and Landreth GE (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. (Vienna) 117, 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, and Panchamoorthy G (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci 111, 3167–3177. [DOI] [PubMed] [Google Scholar]
- Li H, Ren Y, Mao K, Hua F, Yang Y, Wei N, Yue C, Li D, and Zhang H (2018). FTO is involved in Alzheimer’s disease by targeting TSC1-mTOR-Tau signaling. Biochem. Biophys. Res. Commun 498, 234–239. [DOI] [PubMed] [Google Scholar]
- Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, et al. (2018). APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1141–1154.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Weick JP, Liu H, Krencik R, Zhang X, Ma L, Zhou GM, Ayala M, and Zhang S-C (2013). Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol 31, 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig N, Fehlmann T, Kern F, Gogol M, Maetzler W, Deutscher S, Gurlit S, Schulte C, von Thaler A-K, Deuschle C, et al. (2019). Machine Learning to Detect Alzheimer’s Disease from Circulating Non-coding RNAs. Genomics Proteomics Bioinformatics 17, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, and Klann E (2013). Suppression of eIF2a kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci 16, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, Verghese PB, Herz J, Holtzman DM, and Zlokovic BV (2018). Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-b42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K (2014). Taking aim at Alzheimer’s disease through the mammalian target of rapamycin. Ann. Med 46, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, et al. (2019). Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci 22, 2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, and Satija R (2018). Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Feldman HM, Lu T, Drake D, Lim ET, Ling K-H, Bishop NA, Pan Y, Seo J, Lin Y-T, et al. (2019). REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer’s Disease. Cell Reports 26, 1112–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S-A, Condello C, Freilich R, Gillies A, Arhar T, Oroz J, Kadavath H, Julien O, Assimon VA, Rauch JN, et al. (2018). Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol 25, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, and Baxter MG (2012). The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat. Rev. Neurosci 13, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm R, Jones EA, and Huang Y (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol. Neurodegener 14, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Mushynski WE, and Julien J-P (2002). Cycling at the interface between neurodevelopment and neurodegeneration. Cell Death Differ. 9, 1294–1306. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MM, and Lourenco MV (2016). Integrated Stress Response: Connecting ApoE4 to Memory Impairment in Alzheimer’s Disease. J. Neurosci 36, 1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl JI, Lee AS, Leveson-Gower DB, Sun N, Ghosh Z, Lan F, Ransohoff J, Negrin RS, Davis MM, and Wu JC (2011). Short-term immuno-suppression promotes engraftment of embryonic and induced pluripotent stem cells. Cell Stem Cell 8, 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, and Mahley RW (1987). Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 917, 148–161. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. (2015). Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Pchitskaya E, and Bezprozvanny I (2017). Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun 483, 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, and Shelanski ML (2013). Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251, 51–65. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, and LaFerla FM (2010). Alzheimer’s disease. N. Engl. J. Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- R Core Team (2019). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing; ). https://www.R-project.org/. [Google Scholar]
- Ricciarelli R, and Fedele E (2018). cAMP, cGMP and Amyloid β: Three Ideal Partners for Memory Formation. Trends Neurosci. 41, 255–266. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Harding HP (2012). Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb. Perspect. Biol 4, a013177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Lewis D, des Georges A, Marks AR, and Frank J (2018). Ryanodine Receptor Structure and Function in Health and Disease In Membrane Protein Complexes: Structure and Function, Harris JR and Boekema EJ, eds. (Springer; Singapore: ), pp. 329–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales TM, Derkinderen P, Leung K-Y, Byers HL, Ward MA, Price C, Bird IN, Perera T, Kellie S, Williamson R, et al. (2011). Tyrosine phosphor-ylation of tau by the SRC family kinases lck and fyn. Mol. Neurodegener 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper W, and Hoozemans JJM (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Shang YC, Chong ZZ, Wang S, and Maiese K (2013). Tuberous sclerosis protein 2 (TSC2) modulates CCN4 cytoprotection during apoptotic amyloid toxicity in microglia. Curr. Neurovasc. Res 10, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Holtzman DM (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol 18, 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver M, Janousova E, Hua X, Thompson PM, and Montana G; Alzheimer’s Disease Neuroimaging Initiative (2012). Identification of gene pathways implicated in Alzheimer’s disease using longitudinal imaging phenotypes with sparse regression. Neuroimage 63, 1681–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH (2009). Dysregulation of calcium homeostasis in Alzheimer’s disease. Neurochem. Res 34, 1824–1829. [DOI] [PubMed] [Google Scholar]
- Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, and Glabe CG (2018). Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun 10, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Mace BE, Maeda N, and Schmechel DE (2004). Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience 124, 725–733. [DOI] [PubMed] [Google Scholar]
- Sun M-K, and Alkon DL (2014). The ‘‘Memory Kinases’’: Roles of PKC Iso-forms in Signal Processing and Memory Formation. Prog. Mol. Biol. Transl. Sci 122, 31–59. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Tong BC-K, Wu AJ, Li M, and Cheung K-H (2018). Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res 1865, 1745–1760. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, and Hegde AN (2007). Role of the ubiquitin proteasome system in Alzheimer’s disease. BMC Biochem. 8 (Suppl 1), S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera M, Pani B, Griffiths LA, Muchardt C, Abbott CM, Singer RH, and Nudler E (2014). The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. eLife 3, e03164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, and Shelanski M (2002). Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 99, 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwani AR, Affaneh A, Van Gulden S, and Kessler JA (2019). Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann. Neurol 85, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med 24, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, and Arrighi HM (2012). Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology 38, 1–17. [DOI] [PubMed] [Google Scholar]
- Xu P-T, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu H-L, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, and Gilbert JR (1996). Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol. Dis 3, 229–245. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, and Huang Y (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci 26, 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, and Bu G (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 15, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Mich JK, Ku S, Menon V, Krostag A-R, Martinez RA, Furchtgott L, Mulholland H, Bort S, Fuqua MA, et al. (2017). A Single-Cell Road-map of Lineage Bifurcation in Human ESC Models of Embryonic Brain Development. Cell Stem Cell 20, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza R, Vela S, Solas M, and Ramirez MJ (2016). c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol 6, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ZT, Rauch JN, Assimon VA, Jinwal UK, Ahn M, Li X, Dunyak BM, Ahmad A, Carlson GA, Srinivasan SR, et al. (2016). Stabilizing the Hsp70-Tau Complex Promotes Turnover in Models of Tauopathy. Cell Chem. Biol 23, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li P, Feng J, and Wu M (2016). Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci 37, 1039–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW, and LaDu MJ (2012). APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study have been deposited to the NCBI Gene Expression Omnibus with the accession code GSE152867. In addition, processed data are available in the Supplemental Information. The KEGG pathway database used for pathway analysis is available at https://www.genome.jp/kegg/kegg1.html (Kanehisa and Goto, 2000).