


Abstract
Small molecule-based modulation of a triple helix in the long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been proposed as an attractive avenue for cancer treatment and a model system for understanding small molecule:RNA recognition. To elucidate fundamental recognition principles and structure–function relationships, we designed and synthesized nine novel analogs of a diphenylfuran-based small molecule DPFp8, a previously identified lead binder of MALAT1. We investigated the role of recognition modalities in binding and in silico studies along with the relationship between affinity, stability and in vitro enzymatic degradation of the triple helix. Specifically, molecular docking studies identified patterns driving affinity and selectivity, including limited ligand flexibility, as observed by ligand preorganization and 3D shape complementarity for the binding pocket. The use of differential scanning fluorimetry allowed rapid evaluation of ligand-induced thermal stabilization of the triple helix, which correlated with decreased in vitro degradation of this structure by the RNase R exonuclease. The magnitude of stabilization was related to binding mode and selectivity between the triple helix and its precursor stem loop structure. Together, this work demonstrates the value of scaffold-based libraries in revealing recognition principles and of raising broadly applicable strategies, including functional assays, for small molecule–RNA targeting.
INTRODUCTION
The identification (1) and functional annotation (2) of mammalian non-protein-coding RNA (ncRNA) transcripts have led to a new appreciation of RNA molecules as important drivers of cellular function and targets for therapeutic intervention in addition to proteins (3). The resulting ‘RNA revolution’ has identified ncRNA molecules as critical regulators of cancer (4), neurodegenerative disease (5), heart disease (6) and other pathologies (7). For example, the overexpression of the long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been implicated in a variety of cancers (8), and its knockdown was shown to decrease oncogenic processes (9,10), among other phenotypes (11–13). The specific mechanisms and interactions through which this regulation is achieved are still being elucidated (14). Small molecule-based modulation of such phenotypes is of specific interest, as small molecules have the potential to serve as spatiotemporal probes for mechanistic pathway and structural motif investigations, in addition to serving as leads for drug development (15).
MALAT1 is particularly well suited for small molecule-based targeting as the structural motif proposed to be responsible for high transcript accumulation, namely a 3′-triple helix, has been characterized functionally in cell-based assays (16) and structurally through X-ray crystallography (17). This structure is thought to sequester the otherwise single-stranded adenine (A)-rich 3′-tail through base pairing with a uridine (U)-rich stem loop, resulting in the protection of MALAT1 from exonucleolytic degradation. Importantly, an oligonucleotide-based approach aimed at targeting the triple helix was unsuccessful, possibly either due to low sequence complexity of the A-rich tract or due to the unavailability of the triplex-forming sequence for binding (18), emphasizing the need for small molecule-based modulation of this motif. Furthermore, recent research has supported a role for local conformational dynamics, in addition to overall stability, in triplex-mediated transcript protection (19), and small molecules can modulate RNA conformational landscapes as well as global stability (20–23).
Our lab and others have subsequently started to search for small molecules that can specifically recognize the MALAT1 triple helix and modulate its stability. For example, Le Grice and co-workers recently identified two small molecules from a microarray-based high-throughput screen that were found to globally modulate MALAT1 triple helix thermal stability (20). These ligands led to a decrease in MALAT1 levels and branching in breast cancer organoid models, supporting the promise of small molecule-based modulation as a strategy to regulate oncogenic ncRNA levels and related cancer processes. Many of the molecular details behind small molecule–triple helix recognition, however, remain unknown. The knowledge of small molecule properties that differentially modulate binding, thermal stability and effects on MALAT1 triple helix degradation would greatly expedite the discovery of probes for MALAT1 structure–function studies and therapeutic leads for this target.
Scaffold-based small molecule libraries are well poised to provide important insights for emerging targets such as the MALAT1 triple helix, as these libraries enable strategic tuning of relevant small molecule properties. For example, the para-substituted diphenylfuran (DPF) diamidine called furamidine, which originates from the promiscuous nucleic acid binder pentamidine (24–27), has known literature precedence for binding to a variety of nucleic acid structures and has enabled structure–activity relationship studies for therapeutically relevant RNAs (28–32). Importantly, the affinity and selectivity of DPF derivatives for nucleic acids can be modulated through varying amidine positioning and substituent incorporation through the amidine moieties (28,31). Additionally, the intrinsic fluorescence of this scaffold enables its simultaneous use as both a chemical probe and a handle, allowing for non-competitive binding assays to be conducted (33,34).
In previous work, we leveraged the tunability of the parent furamidine DPF scaffold to achieve differential recognition of the MALAT1 triple helix and stem loop structures and identified the first high-affinity, triplex-binding ligand DPFp8 that displayed no binding toward the stem loop structure, the proxy for the unfolded triple helix (Figure 1A) (34). Computational analysis using principal moments of inertia (PMI) found that this ligand has a distinguishing rod-like character, indicating the potential importance of 3D shape recognition for this target. Interestingly, rod-like shapes were also reported to be enriched in general bioactive RNA-binding ligands (35) as well as in ligandable pockets of structurally characterized RNAs (36). It remains unclear, however, what specific advantages are conferred by rod-like ligand shapes, both for the MALAT1 triple helix and for other RNA targets.
Figure 1.
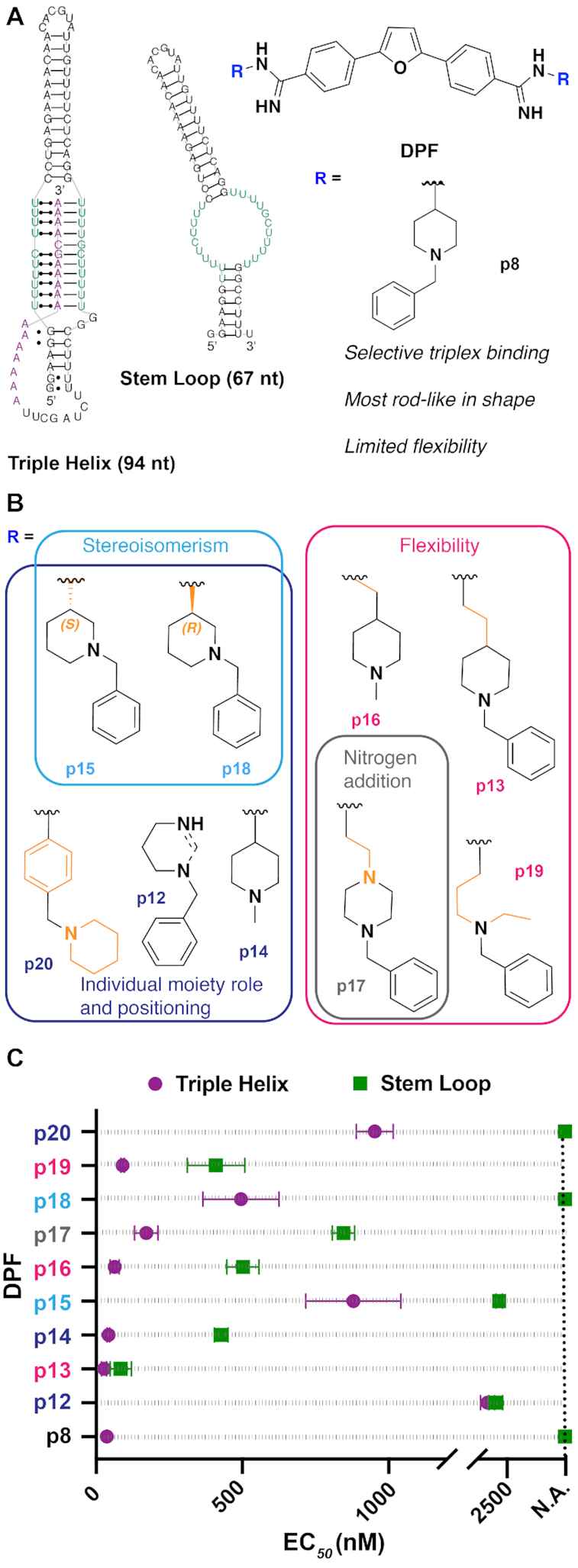
(A) Summary of the previous MALAT1-targeted study (34), in which DPF scaffold-based molecules were evaluated against the MALAT1 triple helix and stem loop structures, leading to the identification of DPFp8 as a triplex-selective ligand. nt = nucleotide. The RNA stem loop secondary structure was predicted using the default settings in the RNAstructure web server. (B) Design of the current study, in which DPFp8 analogs were designed to explore various aspects of the R group. R group number scheme reflects the expansion upon the original para-sublibrary containing subunits p1–p11. (C) EC50 values of DPFp8 analogs for binding to the MALAT1 triple helix and stem loop RNA as measured by monitoring the change in fluorescence of DPF ligands with increasing concentrations of RNA. See the ‘Materials and Methods’ section for EC50 value calculation. Values represent averages of three independent experiments ± standard deviation. Data points at dashed line indicate a not available (N.A.) value where little to no change in fluorescence of DPF (1 μM) was observed at the RNA concentrations used in the experiment (0–5 μM), leading to ambiguous curve fits.
Herein, we synthesize novel rod-shaped DPFp8 analogs with altered recognition properties to explore the unknown determinants of binding strength and selectivity as well as determinants of thermal stability and resistance to exonucleolytic degradation. The combined binding and molecular docking studies of the MALAT1 target as well as a detailed selectivity screen against other nucleic acid constructs provided a rationale for the preference of rod-like shape in MALAT1 ligands. Namely, we found that high-affinity MALAT1-binding ligands retain rod-like shapes to fit the preferred DPF binding pocket, indicating that ligand preorganization in the privileged 3D shape for the specific RNA target may be important. Furthermore, we established a relationship between thermal stabilization and in vitro exonucleolytic degradation of the MALAT1 triple helix and discovered ligands that confer resistance to triplex degradation. Preliminary insights into factors driving these events revealed that ligand binding mode and selectivity, but not affinity, modulate the magnitude of stabilization and consequent degradation resistance. As the first reported ligands with the ability to stabilize and prevent degradation of the MALAT1 triple helix in vitro, these small molecules are complementary to previously reported molecules that disrupt this structure (20). Collectively, this work raises important considerations for future studies in small molecule-based regulation of RNA stability and dynamics and further signifies that scaffold-based libraries have great potential to elucidate guidelines for targeting novel RNA structures and contribute to the toolbox of RNA-targeted ligands (37–40).
MATERIALS AND METHODS
Synthesis of diphenylfurans p12–p20
The synthesis of DPFp8 was reported previously (34). General procedures for the synthesis of precursor dinitrile scaffold and DABAL-Me3-assisted amidine synthesis with primary amine subunits (DPFp12–DPFp19) can be found in the same report (34). The synthetic scheme for ligands in this work can be found in Supplementary Scheme S1. DPFp20 was synthesized using toluene as solvent due to reported side product formation of tetrahydrofuran with aniline subunits (41). All compounds were purified through silica column chromatography using a dichloromethane/methanol/28% ammonium hydroxide gradient (95:4:1 to 90:8:2), with the exception of DPFp12, which was additionally purified using a 1:1 mixture of ethyl acetate:(acetonitrile:methanol:water). DPFp20 was additionally recrystallized in a 9:1 acetonitrile:methanol mixture. All compounds were isolated as bright yellow solids. Compound characterization, 1H- and 13C-NMR spectra, and HPLC chromatograms can be found in the Supplementary Information.
RNA preparation
All surfaces were cleaned with RNase ZaP (Ambion) prior to handling reagents to avoid RNase contamination. All DNA sequences, including the AT-rich DNA duplex, were purchased from Integrated DNA Technologies (IDT). Yeast tRNA was purchased from Invitrogen (Thermo Fisher Scientific). MALAT1 stem loop and triple helix RNA were prepared through polymerase chain and in vitro transcription reactions as described previously (34). The procedure for the preparation of Rev response element (RRE) and NEAT1 RNA constructs can be found in the Supplementary Information, and the sequences used for their preparation in Supplementary Table S15. Sequence length and purity of the MALAT1 and NEAT1 RNA constructs were determined using 8% PAGE-urea gels, while those of RRE RNA and DNA duplex were determined using 15% PAGE-urea gels (Invitrogen) (Supplementary Figure S1). Sequences of the RNA and DNA used in this study are listed in Supplementary Table S1.
Fluorescence binding experiments
All RNA sequences were folded at 50 μM concentration by heating at 95°C for 3 min, snap cooling on ice for 10 min and leaving the solution at room temperature for 1.5 h. The concentration of each RNA or DNA was calculated by measuring absorbance at 260 nm and using the extinction coefficient as predicted by the IDT OligoAnalyzer Tool, with the exception of yeast tRNA, for which the average predicted molecular weight of 25 kDa was used to calculate concentration. Serial dilutions of RNA or DNA in phosphate screening buffer (10 mM NaH2PO4, pH 7.3, 25 mM NaCl, 4 mM MgCl2, 0.5 mM EDTA) or low-ionic buffer (20 mM HEPES–KOH, pH 7.4, at 25°C, 52 mM KCl, 0.1 mM MgCl2) were conducted as described previously (34). Endpoint fluorescence readings were recorded in a CLARIOstar plate reader (BMG Labtech) at 360 nm excitation/460 nm emission for all new ligands with 15 nm/20 nm slits, respectively. For DPFp20, a 380 nm excitation wavelength was used (16 nm slit), and an emission spectrum from 410 to 600 nm (10 nm slit) was recorded for the RNA titration (Supplementary Figure S2). A gain adjustment was conducted prior to all measurements for the wells with no RNA at 90% target value, and a focal height of 11.3 mm was used for all measurements. EC50 values were calculated for three independent experiments in GraphPad Prism version 8.0.2 for Macintosh (GraphPad Software, La Jolla, CA, USA, www.graphpad.com) by fitting to the [Agonist] versus response – variable slope model using Equation (1):
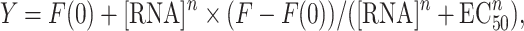 |
(1) |
where Y is normalized % change in fluorescence intensity, [RNA] is RNA concentration in M, n is the Hill slope, F(0) is fluorescence % change of the well with DPF and no RNA or DNA, and F is fluorescence % change of the well with DPF and the highest RNA or DNA concentration. EC50 is defined as the dosage at which 50% of the DPF fluorescence change is observed as measured by fluorescence emission at the appropriate λmax.
Reported values are averages of three independent experiments ± standard deviation. Normalized % fluorescence change was calculated as shown in Equation (2):
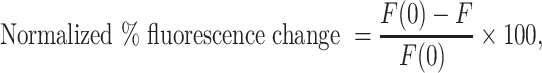 |
(2) |
where F(0) is the fluorescence intensity of the well with DPF and no RNA or DNA, and F is the fluorescence intensity of the well with DPF and RNA or DNA. Binding curves and EC50 values can be found in Supplementary Figures S3–S9 and Supplementary Tables S2, S3 and S14.
Statistical analysis of the binding selectivity screen
Statistical analysis of the selectivity screen was conducted by entering EC50 values of individual experiments and performing one-way ANOVA followed by Tukey’s multiple comparison test using GraphPad Prism version 8.0.2 for Macintosh (GraphPad Software, La Jolla, CA, USA, www.graphpad.com). Test results and associated box–whisker plots can be found in Supplementary Figures S10–S20.
Conformational search and PMI calculations
Tautomer- and protonation-corrected SMILES strings at pH 7.4 (Supplementary Table S4), low-energy ensembles and PMI calculations were generated, performed and analyzed as previously reported (34). Normalized PMI coordinates are listed in Supplementary Table S5 and plotted in Supplementary Figure S21. Detailed procedures on Boltzmann average calculations can be found in (35).
Intramolecular interactions
Intramolecular interactions were analyzed for the low-energy conformation (ΔE = 0) as described previously (34). Molecular Operating Environment (v2018.01) software (42) was used to view ligands and identify hydrogen bonds, cation–π, NH–π and CH–π interactions. For identifying face-to-face or edge-to-face π–π interactions, manual inspection was performed following distances and angles as defined by Schrödinger (43). Interactions are described in Supplementary Table S6.
Molecular docking
Molecular docking was performed as previously reported using ICM-Pro (Molsoft) (44). The X-ray determined structure of MALAT1 was retrieved from the PDB database (PDB ID: 4PLX). Of the three RNA molecules deposited in the PDB, only the most ordered molecule A was considered. The molecule was initially converted to an ICM object with the default settings (remove water and optimize the hydrogens). Binding pocket prediction was performed by using the icmPocketFinder with tolerance set to 4.6 to provide six binding pockets with arbitrarily assigned numbers 1–6 (Supplementary Figure S22).
The low-energy ligand conformation (ΔE = 0) obtained from the Boltzmann-weighted average in PMI calculations was uploaded to ICM as a .pdb file and converted to ICM objects retaining the original geometry. Flexible ligand docking was performed three times for each ligand and each pocket using docking effort equal to 5 and flexible ring sampling level equal to 2. All the ligands showed lower grid energies when docked to the same binding pocket (pocket 1, Supplementary Figure S22); therefore, only the energetic values generated for this pocket were used for analysis (Supplementary Tables S7 and S8). Root-mean-square deviation (RMSD) values between the starting and docked conformation were calculated by using the RMSD function in ICM after superposition of the two conformations and are reported in Supplementary Table S9. Docking of all ligands to the preferred binding pocket was also repeated by using the extended ICM-Pro-VLS suite. However, the docking score functions provided by this version of the software showed less agreement with the experimental data. For this reason, grid docking energies are reported.
Differential scanning fluorimetry
The procedure was adapted from Hengesbach and co-workers (45). All experiments were performed in white 96-well plates in a LightCycler® 480 Instrument II (Roche) using degassed high-ionic buffer, Buffer 1 (20 mM HEPES–KOH, pH 7.4, at 25°C, 152.6 mM KCl, 1 mM MgCl2), or low-ionic buffer, Buffer 2 (20 mM HEPES–KOH, pH 7.4, at 25°C, 52 mM KCl, 0.1 mM MgCl2), which were reported to differentially stabilize the triple helix architecture (19). In a typical experiment, 4 μM MALAT1 triple helix RNA in degassed buffer was annealed by heating at 95°C for 3 min, snap cooling on ice for 10 min and leaving the solution at room temperature for 1.5 h. A total of 42 μl of this solution was aliquoted to wells and 3.6 μM of ligand was added. The mixture was left to incubate at room temperature for 20 min, after which 3 μl of a 20 μM Quant-iT™ RiboGreen™ (Thermo Fisher Scientific) dye stock solution in appropriate buffer was added to each well. The 96-well plate was then sealed with an optically clear foil, shook for 5 min and centrifuged for 1 min at 4000 rpm prior to being placed in the instrument. In the melting curve method, fluorescence intensity was monitored using the SYBR Green I/HRM dye filter combination (465–510 nm) from 20 to 95°C at a ramp rate of 0.01°C/s with 75 acquisitions per °C. Melting profiles were obtained by Tm analysis in the LightCycler® 480 software (release 1.5.1.62) and can be found in Supplementary Tables S10 and S11 and Supplementary Figures S23 and S24. All melting profiles were biphasic, which is considered representative of the triple helix melting profile as previously reported (46), where the first peak is considered to be the triplex-melting peak (Tm1) and the second peak represents the remainder of the structure (Tm2).
RNase R exonucleolytic degradation experiments
The procedure was adapted from Baird and co-workers (19). SM5 (N-(3-methoxybenzyl)-5-(4-methoxyphenyl)-1-methyl-1H-imidazol-2-amine) and SM16 (5-amino-1-cyclopentyl-4-(6-methyl-1H-benzo[d]imidazol-2-yl)-1,2-dihydro-3H-pyrrol-3-one) were purchased from ChemDiv and ChemBridge, respectively (see Supplementary Figure S25 for structures). For a typical 20 μl reaction, 0.2 μM of MALAT1 triple helix or RRE RNA was annealed in low-ionic buffer, Buffer 2 (20 mM HEPES–KOH, pH 7.4, at 25°C, 52 mM KCl, 0.1 mM MgCl2), by heating at 95°C for 3 min, snap cooling on ice for 10 min and leaving the solution at room temperature for 1.5 h. Next, the RNA was mixed with 0.2 μM of a GC-rich DNA duplex (see Supplementary Table S1 for sequence) and 0.2 μM DPF or 1 μM SM5 or SM16, and incubated at room temperature for 20 min. Following the incubation, 5 units of RNase R (Lucigen) was added and the reaction was incubated at 37°C for 5 h in a thermal cycler with a heated lid (Eppendorf). A total of 3 μl of time point aliquots was taken at 0, 30 and 300 min, 3 μl of 2× RNA loading dye was added to each aliquot (Thermo Fischer Scientific) and samples were stored at −80°C prior to gel analysis. Reactions were analyzed on pre-cast 15% PAGE-urea gels (Invitrogen) in 1× TBE buffer (100 mM Tris base, 100 mM borate, 2 mM EDTA). Gels were run for 45 min at 180 V, stained with Diamond™ Nucleic Acid Dye (Promega) for 20 min and analyzed on a ChemiDoc™ Imaging System (Bio-Rad). Gel band intensity of the MALAT1 triple helix or RRE RNA and DNA loading control was quantified using ImageJ software (47). To quantify time-dependent exonucleolytic degradation for each sample, the intensity was normalized using Equation (3), and fold difference was calculated as shown in Equation (4).
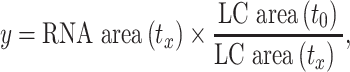 |
(3) |
where y is normalized RNA area for a specific time point and the sample (tx), RNA area is the raw band intensity for the RNA in the lane corresponding to time point x for that sample, LC (loading control) area (t0) is the raw band intensity for the DNA in the lane corresponding to the 0 min time point for the sample and LC area (tx) is the raw band intensity for the DNA in the lane corresponding to time point x for that sample.
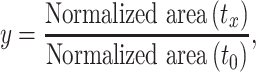 |
(4) |
where y is fold difference of the exonucleolytic degradation for the specific sample, with normalized areas for its specific time point x and time point 0 calculated as described in Equation (3). Experimental results are listed in Supplementary Figure S26 and Supplementary Table S12. To ensure that the measured time points were within the linear decay range, a control experiment was conducted in which a 40 μl reaction was set up using the same procedure as described, but with 0, 15, 30, 60, 90 and 300 min time points (Supplementary Figure S27 and Supplementary Table S13). Unprocessed gel images can be found in Supplementary Figures S37 and S38.
SYBR Green II fluorescence indicator displacement assay
The protocol was adapted from a previously reported procedure by Le Grice and co-workers (20). A serial dilution of small molecule (0, 1, 5, 10, 25, 50, 100, 150, 250, 300, 400, 500, 600, 700, 800, 900 and 1000 μM) was performed in phosphate buffer (10 mM NaH2PO4, 25 mM NaCl, 4 mM MgCl2, 0.5 mM EDTA, pH 7.3) or low-ionic buffer, Buffer 2 (20 mM HEPES–KOH, pH 7.4, at 25°C, 52 mM KCl, 0.1 mM MgCl2), in a 96-well plate in triplicate. A total of 8 μl of each dilution was transferred to a 384-well plate, followed by 8 μl of a 202 nM solution of MALAT1 RNA and 4× SYBR Green II dye solution (Invitrogen™). The RNA was previously annealed by heating to 95°C for 5 min and cooling to 0°C on ice for 30 min. The 384-well plates were shaken for 5 min, centrifuged at 4000 rpm for 1 min and incubated in the dark for 20 min. The plates were excited at 496 nm (8 nm slit) and emission was read at 525 nm (8 nm slit, focal height 11.3 nm) using a CLARIOstar plate reader (BMG Labtech). Percent fluorescence indicator displacement (% FID) was calculated by subtracting and, subsequently, dividing by the blank wells with RNA–dye complex and no small molecule as shown in Equation (5):
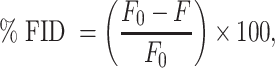 |
(5) |
where F0 is the fluorescence of the blank well with RNA + dye and no DPF, and F is the fluorescence of the well with all three components (RNA + dye + DPF).
Each technical triplicate was averaged and the resulting FID values were averaged between three independent experiments. Binding curves and CD50 values were obtained by using a non-linear fit curve for agonist versus response with variable slope at four parameters (GraphPad Prism Software version for Windows 7.04, La Jolla, CA, USA, www.graphpadprism.com) as shown in Equation (6):
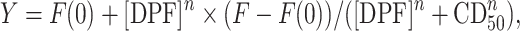 |
(6) |
where Y is % FID as calculated in Equation (5), [DPF] is DPF concentration in M, n is the Hill slope, F(0) is fluorescence of the well with RNA + dye and no DPF, and F is fluorescence of the well with all three components and the highest DPF concentration. CD50 is defined as the competitive dosage required for displacing 50% of SYBR Green II from the SYBR Green II:RNA complex, as measured by SYBR Green II fluorescence emission at 525 nm.
RESULTS AND DISCUSSION
Design and synthesis of next-generation DPF library
In our previous study (34), we found that DPFp8 displayed exclusive selectivity for the triple helix over the stem loop structure and increased triplex stability as measured by UV–Vis thermal denaturing experiments. Its benzylpiperidine subunit, which was the only subunit containing both alkyl and aryl moieties, conferred a distinguishing rod-like character (Figure 1A). To gain detailed insights into which features of DPFp8 confer triplex affinity, selectivity and stability, we designed nine novel ligands with altered molecular recognition properties by modifying moiety orientation, positioning, flexibility and hydrogen bonding pattern while retaining the rod-like shape. These designs included the incorporation of stereoisomer subunits that are regioisomers of p8, addition of nitrogen atoms and flexible alkyl chains, and reversal or inclusion of the individual alkyl and aryl moieties (Figure 1B). Utilizing the two-step route that was optimized for the synthesis of the original para-substituted library members (p1–p11) (34), nine proposed analogs were synthesized (Supplementary Scheme S1).
DPF library evaluation against MALAT1 targets
We next evaluated the binding of the new analogs to MALAT1 triple helix and stem loop targets (Figure 1C) by monitoring the change in DPF fluorescence upon increasing RNA concentrations as reported previously (34). The stem loop RNA functions as a proxy of the non-triplex version of our target, as this structure is proposed to be an intermediate in the triplex folding pathway (48). Among these derivatives, the parent compound DPFp8 remains the ligand with highest affinity and selectivity for the triple helix over stem loop structure. We also found that increasing alkyl linker length (DPFp13, DPFp16 and DPFp19) or nitrogen atom incorporation in the alkyl ring system (DPFp17) did not impact triple helix affinity but did reduce selectivity as compared to DPFp8 (Figure 1B and Supplementary Table S2). The role of limited flexibility is exemplified by DPFp16, which contains the shortest alkyl linker in this series and has improved selectivity compared to other flexible subunits. As the closest analog to DPFp16, DPFp14 further supports this trend given the absence of alkyl linker and further improved selectivity for the triple helix (Figure 1B and Supplementary Table S2). The selectivity of DPFp14 was still not as significant as DPFp8, which contains the additional aryl moiety. These trends are in line with our previous study, where we observed that increased flexibility, along with the number of positive charges, led to promiscuous binding behavior (34). In the case of p8 specifically, it appears that rigidifying the alkyl moieties is necessary for increased triple helix selectivity over the stem loop structure but that exclusive triplex binding is achieved through additional aryl ring incorporation.
In line with this observation, regioisomers of the p8 subunit with different stereochemistry (DPFp15 and DPFp18) or positioning (DPFp20) of the aryl and piperidine moieties appear to preserve exclusive triplex selectivity but display lower affinity for the triple helix (Figure 1B and Supplementary Table S2). These observations imply that both aryl and nitrogen-containing alkyl ring systems are required for selectivity, but that their arrangement relative to each other may be important for affinity trends. In the future, other types of aliphatic and aromatic ring systems may be explored for their ability to bind and discriminate between these two targets.
DPFp12 is the weakest and least selective binder of our next-generation ligands, and computational analysis predicted that the subunits on this ligand form π–π interactions with the phenyl rings on the DPF scaffold (Supplementary Figure S28 and Supplementary Table S5). This observation underscores the trend from the original study, in which ligands that displayed lower triplex affinity and selectivity were predicted to form intramolecular interactions (34). The original study also found that intramolecular interactions increased with the distance from the rod vertex, a trend that is difficult to assess for these new analogs as they all populate the rod sub-triangle in PMI calculations (Supplementary Figure S21 and Supplementary Table S4). Given that changes in parameters that affect subunit molecular recognition did not lead to significant differences in binding profiles of the analogs, and that they all retain the rod-like shape of DPFp8, these observations reinforce the notion that triple helix recognition may be shape-driven and that rod-shaped DPFs are privileged to bind the triple helix with high selectivity and affinity.
Molecular docking study
To gain further insights into binding trends for these ligands and understand the role of 3D shape recognition, we took advantage of the 3.1 Å MALAT1 triple helix X-ray diffraction structure reported by Steitz and co-workers (17) and conducted molecular docking studies. We selected ligands from the original para-DPF library (34) and from this study to cover a spectrum of high (DPFp8 and DPFp13; EC50 ∼ 30 nM) and intermediate-to-low (DPFp12, DPFp15, DPFp18 and DPFp20; EC50 ∼ 500–1500 nM) binding affinities, as well as non-binders (DPFp7; EC50 not available). Each docking run was conducted three times, starting with the low-energy ligand structure from the conformational search. All analogs showed preferential binding, as measured by lower grid docking energies (Edock), for the same binding pocket in the narrow region of the major groove at the base of the triple helix (Figure 2A and Supplementary Figure S22). In comparison, one small molecule identified by Le Grice and co-workers was predicted to occupy a similar binding site while the other occupied the minor groove, though a different docking program was utilized in that study (20). Comparison of EC50 values and the average Edock values revealed a reasonable correlation between the experimentally measured binding affinities and grid docking energies (Figure 2B) (R2 = 0.77). Specifically, the non-binder (DPFp7) was found to have the least favorable docking energy (∼5 kcal/mol), while the highest affinity binders (DPFp8 and DPFp13) had the most favorable grid docking energies (<−50 kcal/mol) (Figure 2B and Supplementary Table S7). Intermediate- to low-affinity binders in the series (DPFp12, DPFp15, DPFp18 and DPFp20) had intermediate Edock values between −20 and −40 kcal/mol. Although their relative EC50 values did not perfectly rank with the predicted Edock values, it is not unusual for docking programs to have limited utility in distinguishing subtle differences in affinity, particularly with modest affinity ligands (49). These ligands may also have other binding modes or interact with RNA conformations that are not accounted for by the docking program (50) or by the crystallized triple helix construct, which represents a mutated and truncated version of the 94-nt RNA transcript utilized in the fluorescence binding experiments.
Figure 2.

(A) Example of docked structure of DPFp8 (red) binding at the base of the MALAT1 triple helix (PDB ID: 4PLX). (B) Comparison of the average ‘Edock’ values and experimentally measured EC50 values. Edock error bars represent standard deviations of three individual docking runs, and EC50 error bars represent standard deviations from three independent experiments. R2 value was obtained through linear regression analysis in GraphPad Prism v8.0.2 software. (C) Comparison of the average ‘Edock’ value and average RMSD of the starting ligand conformations from PMI calculations versus the final bound conformation in the binding pocket displayed in (A). RMSD error bars represent standard deviations from three individual docking runs. R2 value was obtained through linear regression analysis in GraphPad Prism v8.0.2 software.
Given the generally promising correlation between the experimental results and computational predictions, we were interested in identifying particular parameters that could be used to explain high-affinity binding events. While hydrogen bonding was observed for amidine and piperidine nitrogen atoms of the DPF scaffold with both backbone phosphates and purine or uridine bases (Supplementary Table S8), the number and the contribution of these contacts, as well as other individual parameters that comprise the Edock value (Supplementary Table S7), did not reveal any significant trends. While this lack of correlation may suggest that hydrogen bonding is not the driving binding interaction, it is worth noting that hydrogen bonding strength might not be accurately assessed in docking programs, especially for charge-assisted interactions such as the one observed with the negative oxygen of the phosphates. We next assessed the change in ligand conformation by measuring the RMSD of ligands in their apo (after Boltzmann-averaged conformational search) versus bound states (Supplementary Table S9). Interestingly, this analysis provided a favorable correlation between Edock and RMSD values (Figure 2C, R2 = 0.75). In particular, high-affinity binders (DPFp8 and DPFp13) had a smaller average conformational rearrangement (RMSD values ∼1–2 Å) between the free and bound conformations, while intermediate–low (DPFp12) and non-binders (DPFp7) had the largest conformational change (RMSD values ∼3–4 Å) (Figure 2C and Supplementary Table S9). Together, these data suggest that a certain degree of ligand preorganization may be necessary for high-affinity binding, further supporting a role for shape complementarity in triplex recognition.
Selectivity screening of next-generation DPFs
In order to test selectivity between other nucleic acid structures and to compare the ligands synthesized herein to those reported previously (34), we conducted a selectivity screen of all next-generation ligands against four additional nucleic acids. These included an AT-rich DNA duplex, tRNA and the HIV-1 RRE stem loop IIB, all of which have been previously shown to bind DPF derivatives (25,28,33). Additionally, we tested these DPFs for binding to the triple helix found in the lncRNA NEAT1, whose triplex-forming sequence is similar to that of MALAT1, though its small molecule binding potential remains unexplored (46).
The selectivity screen revealed several trends supporting the previous finding that rod-shaped DPFs are particularly well poised for high-affinity recognition of both the MALAT1 triple helix (34) and other helical RNA structures. For example, we previously showed that the most rod-shaped ligand, DPFp8, displayed varying affinities for the tRNA and RRE structures but retained selectivity for the MALAT1 triple helix. Herein, we learned that DPFp8 also binds to the structurally similar NEAT1 triple helix, albeit with lower affinity compared to the MALAT1 triple helix (Supplementary Table S3). Similarly, we observed that the next-generation high-affinity binders of both the MALAT1 triple helix and stem loop structures generally bind other RNA structures tested herein to some extent but show no measurable binding of the AT-rich DNA duplex (Supplementary Table S3). Ligands that displayed moderate- to low-affinity binding to the MALAT1 targets such as DPFp12, DPFp15 and DPFp20 as well as the non-binder from the previous study (DPFp7) displayed binding of the DNA duplex (Supplementary Table S3). Notably, DPFp7 displayed no binding to any of the RNA constructs tested herein, and was previously found to have the most disc-like character in the PMI analysis of para-DPF ligands (34). Together, these data further confirm that nucleic acid selectivity of DPF ligands can indeed be modulated through tuning of the subunits on this scaffold, and that a driving factor of this tunability may be the distinct 3D shapes and conformations conferred by the different subunits.
Studying the effect of next-generation ligands on MALAT1 thermal stability and exonucleolytic degradation
Studies conducted by Wilusz et al. (16,18) and Steitz and co-workers (17,51) provided first evidence toward the role of the triple helix as a ‘molecular knot’ that prevents exonucleolytic degradation, showing that mutations expected to disrupt the triple helix structure led to decreased transcript accumulation. More recent work has shown that small inorganic and organic ligands can modulate the stability of this structure. For example, Baird and co-workers revealed that high- and low-ionic concentrations have stabilizing and destabilizing effects on triple helix thermal stability, respectively, thus differentially protecting the transcript from in vitro degradation by the RNase R exonuclease (19). The aforementioned study by Le Grice and co-workers identified two small molecules with the ability to globally modulate MALAT1 triple helix thermal stability landscapes in the different ionic environments as measured by differential scanning FRET (DS-FRET) experiments (20). While these experiments showed that the molecules induced stabilizing and destabilizing DS-FRET profiles across a range of ionic conditions tested, both caused diminished levels of MALAT1 in breast cancer organoids. Additionally, their direct effect on triple helix exonucleolytic degradation was not evaluated. These observations prompted us to study the effect of our ligands on triple helix thermal stability in both low- and high-ionic conditions and to include the two small molecules identified by Le Grice and co-workers in our RNase R exonucleolytic studies for comparison. We envisioned that this direct comparison of in vitro stabilization and in vitro degradation profiles would clarify the relationship between ligand-induced phenotype changes that are relevant for modulating triple helix function.
We utilized differential scanning fluorimetry (DSF) to rapidly assess the effect of DPFp8 and its novel analogs on global triple helix stability under high- and low-ionic concentrations. This method uses the RiboGreen™ dye, which differentially senses single- versus double-stranded RNA, and overcomes the low-throughput capabilities of UV–Vis spectroscopy by enabling screening in 96-well plates using a qPCR instrument (45). DSF has been successfully utilized to identify small molecule stabilizers of a precursor microRNA-21 (52) as well as to evaluate MALAT1 triple helix global stability under various ionic conditions (19). However, DSF was not previously used to evaluate effects of small molecules on MALAT1 triple helix stability. We observed that the new DPF ligands displayed stabilizing effects on the triple helix to varying extents in both triplex-stabilizing and destabilizing conditions via DSF. Stabilization was more pronounced in the low-ionic, destabilizing condition (Table 1 and Supplementary Tables S10 and S11). The effects ranged from negligible stabilization induced by ligands such as DPFp7 and DPFp12 to the highest increase in stability of ∼6°C by DPFp20 (Table 1). Negligible stabilization was also observed for pentamidine, which was included as a control (Supplementary Tables S10 and S11). We envision that the use of DSF for high-throughput screening (HTS) holds promise for the discovery of ligands with varying effects on triple helix stability as well as the elucidation of small molecule properties that impart distinct stability effects for this target.
Table 1.
DSF experiments measuring the effect of DPF ligands on MALAT1 triplex stability under low-ionic buffer conditions
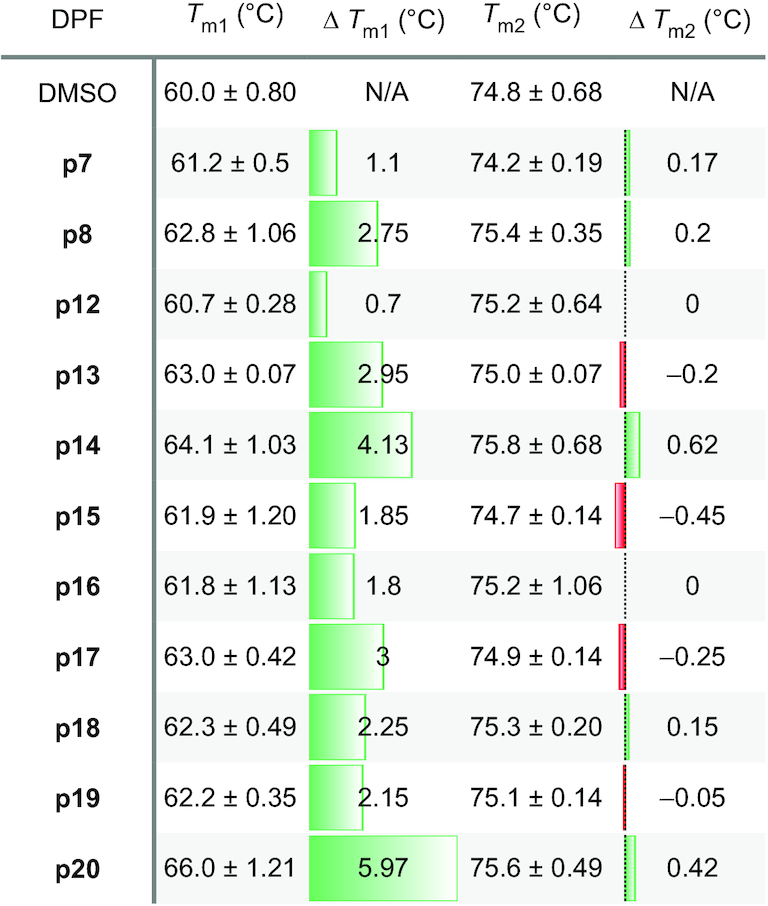
|
T m represents melting temperature as obtained from the negative first derivative graph of a DSF melting profile. Tm1 indicates shift of the first peak of the melting profile, which is proposed to be the triple helix-forming strand, and Tm2 represents melting of the remainder of the structure. Green and red colors represent an increase and decrease in stability, respectively, upon DPF ligand addition. ΔTm errors were calculated through standard error propagation from three independent experiments and are listed in Supplementary Tables S10 and S11. N/A = Not applicable.
We next studied the relationship between the extent to which these ligands stabilize the triple helix and its susceptibility to exonucleolytic degradation. As our ligands displayed stabilizing effects, we expected to observe ligand-dependent prevention of degradation, a mechanism that has not been reported to date for the MALAT1 triple helix. Utilizing the RNase R exonuclease assay, we found that DPF ligands indeed had a protective effect on triple helix degradation in the low-ionic, triplex-destabilizing conditions (Supplementary Figure S26 and Supplementary Table S12). On the other hand, triplex-destabilizing ligands reported by Le Grice and co-workers accelerated the degradation of this target (Supplementary Figure S26 and Supplementary Table S12). The extent of protection against RNase R correlated with the ΔTm values as measured in DSF experiments, with DPFp20 providing the most significant resistance to degradation (Figure 3 and Supplementary Figure S29). An exception to this trend was DPFp7, which provided a minimal stabilizing effect in DSF experiments but prevented degradation by RNase R (Supplementary Figures S29 and S30). We suspected that this effect may be a result of RNase R inhibition instead of triple helix binding and conducted a control degradation experiment with RRE RNA, an RNA from the selectivity screen where no binding was observed with DPFp7 or DPFp20 (Supplementary Table S3). We observed slower RRE degradation with DPFp7 relative to DMSO but not with DPFp20, suggesting that the outlier behavior of DPFp7 may, indeed, be due to RNase R inhibition (Supplementary Figure S31).
Figure 3.
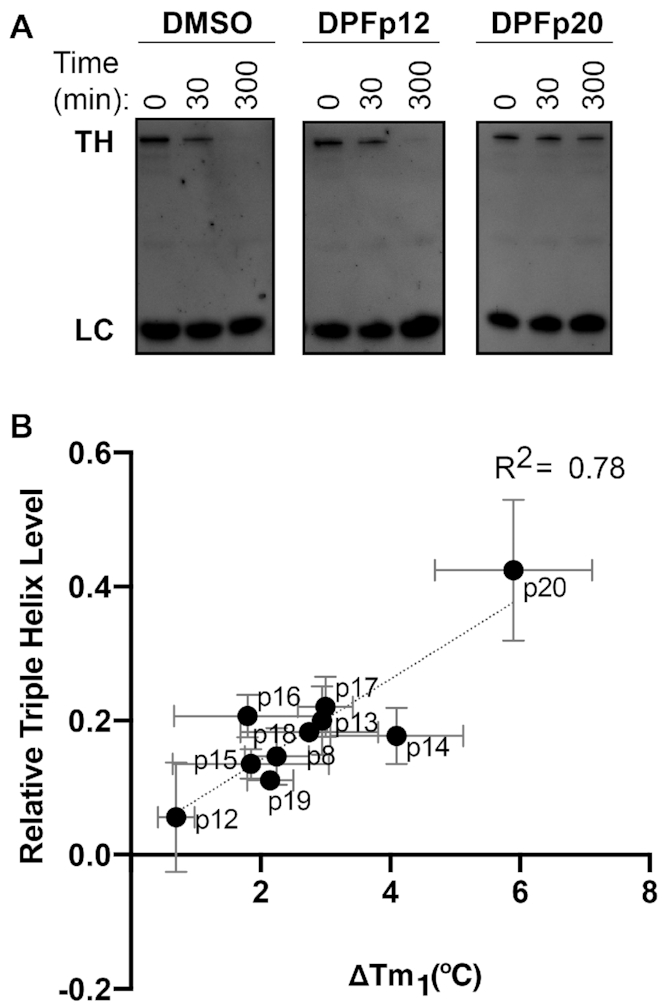
(A) RNase R experiments with DMSO control, DPFp12 and DPFp20 assessed by denaturing gel electrophoresis in low-ionic buffer conditions utilizing 5 U RNase R, 0.2 μM RNA and 0.2 μM DPF at 0, 30 and 300 min time points. TH = triple helix; LC = loading control, a GC-rich DNA hairpin. (B) Correlation between melting temperature shifts as measured by DSF experiments in low-ionic buffer conditions and level of MALAT1 triple helix at the 300 min time point during an RNase R incubation in low-ionic buffer conditions as measured by denaturing gel electrophoresis and quantified in ImageJ software. The value was measured relative to the loading control and the 0 min time point sample. R2 value was obtained through linear regression analysis in GraphPad Prism v8.0.2 software. Error bars represent standard deviation from three independent experiments.
Interestingly, however, the comparison of EC50 values measured as change of intrinsic fluorescence and the degree of stabilization by these ligands did not yield a clear trend (Supplementary Figure S32). DPFp8, for example, only had a modest effect on stability, despite being a high-affinity binder. On the other hand, DPFp20 was found to have the most significant stabilizing effect (Table 1), despite having modest affinity for the triple helix (Figure 1B and Supplementary Table S2). When binding experiments were conducted in the low-ionic buffer that was used for DSF and RNase R experiments, we observed similar trends in binding, although with weaker affinities (Supplementary Figure S33 and Supplementary Table S14).
The unexpectedly high degree of stabilization by DPFp20, combined with modest affinity, suggested a different binding mode for this ligand. Of note, DPFp20 has a distinct chemical architecture from other DPF ligands synthesized to date in our group: it is the only molecule with an aniline subunit instead of a primary amine. We reasoned that the extended aromatic surface area conferred by this subunit may enable a more stabilizing binding mode through favorable π-stacking interactions. Additionally, DPFp20 is the only ligand that displayed a shift in the fluorescence emission peak upon addition of RNA (Supplementary Figure S2). Given that this phenomenon has been previously observed for ligands with an intercalating binding mode (53–55), the computational prediction of this binding mode is notoriously challenging (56) and was not seen in our docking study (Supplementary Figure S34), we reasoned that this ligand may be acting through intercalation.
To test this hypothesis, we conducted a fluorescent intercalator displacement experiment utilizing a common RNA-intercalating ligand SYBR Green II. In this assay, DPFp20 yielded a significantly lower CD50 value (5.7 ± 0.6 μM) compared to DPFp8 (65.9 ± 2.5 μM) in the buffer utilized in binding studies, indicating that DPFp20 may bind through an intercalative mode to a greater extent relative to DPFp8 (Figure 4). This trend remained unchanged when the experiment was conducted using the low-ionic buffer utilized in thermal denaturation and RNase R studies (Supplementary Figure S35). In contrast, Le Grice and co-workers used the same assay to eliminate intercalation as a binding mode for small molecules that decrease MALAT1 levels in organoid assays (20). DPFp20 thus represents a complementary probe to the triplex-destabilizing ligands reported by Le Grice and co-workers.
Figure 4.
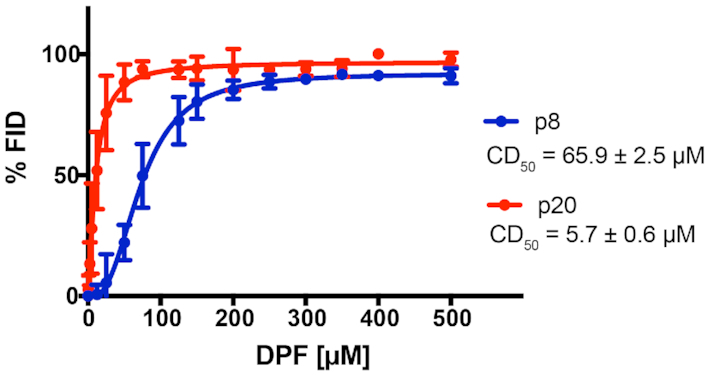
FID assay of intercalative ligand SYBR Green II and increasing concentrations of DPFp8 and DPFp20. Error bars represent the standard deviation of three technical replicates. See the ‘Materials and Methods’ section for CD50 value calculation. CD50 values represent averages of three independent experiments ± standard deviation.
Given that π-stacking interactions increase base pairing stability, this preliminary experiment suggested that DPFp20 confers the high stabilization and resistance to exonucleolytic degradation by enhancing the stability of base triples and consequently disfavoring the single-stranded state through intercalating to a higher extent than DPFp8. It is worth noting, however, that changes in fluorescence measured in indicator displacement assays may be a result of other factors (57). For example, displacement could be caused through a non-intercalative binding mode of ligands, and multiple binding modes may be occurring simultaneously. Indeed, DPF derivatives were previously reported to display distinct binding modes (31). These modes include traditional intercalation, in which the aromatic moieties bind between nucleic acid base pairs using mainly π interactions, groove binding in which ligand complementarity is used to ‘read’ the hydrogen bond acceptor and donor patterns, and threading intercalation in which intercalation and groove binding modes are combined to achieve additional affinity and selectivity (58,59). Studies to test for these binding modes with our analogs and target are currently underway.
We also noted a relationship between fold selectivity for the triple helix versus stem loop structures and the extent of stabilization. Specifically, ligands with higher measured selectivity for the triple helix had a greater stabilizing effect as measured by DSF experiments (Supplementary Figure S36). This observation suggests that the protective function of DPFs, as observed in RNase R experiments, may be enabled by their ability to selectively recognize and stabilize the triple helix conformation over the stem loop, a representative proxy for the ‘unfolded’ triplex conformation in this equilibrium, as well as to confer a favorable binding mode by enhancing stacking interactions. Conversely, it is possible that ligands with selective and stabilizing effects on the stem loop conformation, if identified, may have the reverse effect, leading to a decrease in Tm and increase in exonucleolytic degradation. This effect would be similar to that observed in studies of ligand-induced protein stabilization, where binding to the native versus unfolded state can impact the predicted Tm shifts (60).
CONCLUSION
The MALAT1 triple helix is an RNA structural motif with established therapeutic relevance but poorly understood mechanism of action and underexplored potential for small molecule targeting. In this study, we synthesized analogs of the first reported MALAT1 triple helix binder, DPFp8, to gain insights into the drivers of affinity and selectivity as well as stability and resistance to in vitro degradation. Through tuning the substituent composition and individual moiety order or placement, we learned that exclusive triplex selectivity is enabled by the presence of less flexible aliphatic as well as aromatic moieties in the subunits. Affinity, on the other hand, is driven by the positioning of the individual moieties relative to each other, which was clarified by docking. Specifically, this study revealed that high-affinity ligands undergo little conformational change between apo and bound states in the predicted binding pocket, suggesting the importance of ligand preorganization and preservation of rod-likeness for triple helix recognition. Thermal melting studies revealed that next-generation ligands display a range of stabilizing effects on the MALAT1 triple helix, enabling different degrees of resistance to exonucleolytic degradation. These first-in-class small molecules that prevent triple helix degradation represent complementary probes to the triplex-disrupting ligands previously reported (20). The most stabilizing ligand, DPFp20, was found to be triplex-selective, and our results suggested it might act through an intercalative binding mode.
The discoveries reported herein represent initial steps toward understanding the driving forces behind ligand-based modulation of MALAT1 stability and also raise a number of future considerations and directions for this target and the field in general. For example, our analyses suggest that future development of ligands for MALAT1 should consider ligand shape preorganization for achieving high affinity but that additional factors such as ligand selectivity and binding modes should be taken into account for interpreting trends in functional studies. Additionally, we emphasize the importance of conducting both binding and functional studies, as we have shown that the two may not be correlated. Given that rod-like molecules appear to have privileged shapes for triple helix and other RNA molecules targeted to date (35), it will be interesting to test whether this principle holds for a broader range of structurally diverse RNA as well as DNA molecules. Recently, a PMI analysis of ligandable RNA pockets conducted by Schneekloth and co-workers found that the majority of RNA pockets are rod-like shaped themselves, including the MALAT1 triple helix (36).
Furthermore, the HTS potential of the DSF method described herein and elsewhere (19,52) offers the possibility of evaluating a large number of ligands in search for differential modulators of MALAT1 triple helix stability, which in turn correlates with susceptibility to in vitro degradation. By utilizing methods such as DSF to first evaluate the impact of small molecules on global triplex stability, and conducting additional in vitro studies to reveal the currently elusive small molecule properties that enable these effects, we can obtain well-characterized mechanistic probes to interrogate the structure and function of the MALAT1 triple helix. As the DPF ligands developed herein are first-in-class ligands with a stabilizing effect on MALAT1 thermal stability, testing their effect on MALAT1 levels as well as MALAT1-dependent activities in cells should provide insight into MALAT1 biology and targeting approaches. We consider this to be of particular importance, as the spatiotemporal regulation and cellular interactome of the MALAT1 triple helix in both healthy and disease states remain understudied and expanding the MALAT1-targeting toolbox with differential small molecule modulators could unravel new regulatory pathways and therapeutic strategies. Toward these goals, however, it will be necessary not only to establish the impact of triplex-targeting small molecules on MALAT1 levels in immortalized cells that has not been reported to date, but also to measure changes in transcript half-life and variation between cell lines, as well as to conduct global expression profiling. These experiments are currently ongoing in our laboratory.
Lastly, as recent studies have shown that both MALAT1 stability and dynamics have a role in preventing degradation (19,51), it will be important to test whether small molecules can modulate the lifetimes of meta-stable structures in between the fully folded triplex and unfolded stem loop species. These studies are currently underway in our laboratory and can contribute to the emerging strategy of leveraging RNA dynamics for trapping inactive or non-functional conformations (61–63). As the field is only beginning to understand the mechanisms of ligand-based modulation of RNA stability and dynamics, we have shown that focused small molecule libraries such as the one developed herein continue to provide novel insights into the fundamental principles for these interactions. We anticipate that these discoveries and their implications have the potential to unlock novel strategies for studying and controlling RNA-associated disease pathways.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Hargrove Lab members, and especially to Emily McFadden, for their feedback and helpful discussions regarding experimental design and data interpretation.
Notes
Present address: Amanda E. Hargrove, Department of Chemistry, Duke University, Durham, NC 27708, USA.
Contributor Information
Anita Donlic, Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27708, USA.
Martina Zafferani, Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27708, USA.
Giacomo Padroni, Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27708, USA.
Malavika Puri, Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27708, USA.
Amanda E Hargrove, Department of Chemistry, Duke University, 124 Science Drive, Durham, NC 27708, USA; Department of Biochemistry, Duke University School of Medicine, Durham, NC 27710, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM124785 to A.E.H.]; National Science Foundation [CAREER1750375 to A.E.H.]; Duke University [Joe Taylor Adams Fellowship to A.D. and Independent Study Grant to M.P.]. Funding for open access charge: National Institutes of Health, National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. The ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 2. Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C. et al.. The transcriptional landscape of the mammalian genome. Science. 2005; 309:1559–1563. [DOI] [PubMed] [Google Scholar]
- 3. Cech T.R., Steitz J.A.. The noncoding RNA revolution: trashing old rules to forge new ones. Cell. 2014; 157:77–94. [DOI] [PubMed] [Google Scholar]
- 4. Ling H., Vincent K., Pichler M., Fodde R., Berindan-Neagoe I., Slack F.J., Calin G.A.. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene. 2015; 34:5003–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salta E., De Strooper B.. Noncoding RNAs in neurodegeneration. Nat. Rev. Neurosci. 2017; 18:627–640. [DOI] [PubMed] [Google Scholar]
- 6. Poller W., Dimmeler S., Heymans S., Zeller T., Haas J., Karakas M., Leistner D.-M., Jakob P., Nakagawa S., Blankenberg S. et al.. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur. Heart J. 2017; 39:2704–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011; 12:861–874. [DOI] [PubMed] [Google Scholar]
- 8. Amodio N., Raimondi L., Juli G., Stamato M.A., Caracciolo D., Tagliaferri P., Tassone P.. MALAT1: a druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018; 11:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gutschner T., Hämmerle M., Eißmann M., Hsu J., Kim Y., Hung G., Revenko A.S., Arun G., Stentrup M., Groß M. et al.. The non-coding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2012; 73:1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arun G., Diermeier S., Akerman M., Chang K.C., Wilkinson J.E., Hearn S., Kim Y., MacLeod A.R., Krainer A.R., Norton L. et al.. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016; 30:34–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arun G., Spector D.L.. MALAT1 long non-coding RNA and breast cancer. RNA Biol. 2019; 16:860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J., Piao H.L., Kim B.J., Yao F., Han Z., Wang Y., Xiao Z., Siverly A.N., Lawhon S.E., Ton B.N. et al.. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018; 50:1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kwok Z.H., Roche V., Chew X.H., Fadieieva A., Tay Y.. A non-canonical tumor suppressive role for the long non-coding RNA MALAT1 in colon and breast cancers. Int. J. Cancer. 2018; 143:668–678. [DOI] [PubMed] [Google Scholar]
- 14. Li Z.-X., Zhu Q.-N., Zhang H.-B., Hu Y., Wang G., Zhu Y.-S.. MALAT1: a potential biomarker in cancer. Cancer Manag. Res. 2018; 10:6757–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donlic A., Hargrove A.E.. Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA. 2018; 9:e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilusz J.E., JnBaptiste C.K., Lu L.Y., Kuhn C.D., Joshua-Tor L., Sharp P.A.. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012; 26:2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown J.A., Bulkley D., Wang J., Valenstein M.L., Yario T.A., Steitz T.A., Steitz J.A.. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014; 21:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilusz J.E., Freier S.M., Spector D.L.. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008; 135:919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ageeli A.A., McGovern-Gooch K.R., Kaminska M.M., Baird N.J.. Finely tuned conformational dynamics regulate the protective function of the lncRNA MALAT1 triple helix. Nucleic Acids Res. 2018; 47:1468–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abulwerdi F.A., Xu W., Ageeli A.A., Yonkunas M.J., Arun G., Nam H., Schneekloth J.S., Dayie T.K., Spector D., Baird N. et al.. Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1. ACS Chem. Biol. 2019; 14:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stelzer A.C., Frank A.T., Kratz J.D., Swanson M.D., Gonzalez-Hernandez M.J., Lee J., Andricioaei I., Markovitz D.M., Al-Hashimi H.M.. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011; 7:553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davis B., Afshar M., Varani G., Murchie A.I.H., Karn J., Lentzen G., Drysdale M., Bower J., Potter A.J., Starkey I.D. et al.. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic “hot spots”. J. Mol. Biol. 2004; 336:343–356. [DOI] [PubMed] [Google Scholar]
- 23. Matsumoto S., Caliskan N., Rodnina M.V., Murata A., Nakatani K.. Small synthetic molecule-stabilized RNA pseudoknot as an activator for –1 ribosomal frameshifting. Nucleic Acids Res. 2018; 46:8079–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu L., Wang F., Tong Y., Li L.F., Liu Y., Gao W.Q.. Pentamidine inhibits prostate cancer progression via selectively inducing mitochondrial DNA depletion and dysfunction. Cell Prolif. 2019; 53:e12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun T., Zhang Y.. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res. 2008; 36:1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Warf M.B., Nakamori M., Matthys C.M., Thornton C.A., Berglund J.A.. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl Acad. Sci. U.S.A. 2009; 106:18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Li Z., Pilch D.S., Leibowitz M.J.. Pentamidine inhibits catalytic activity of group I intron Ca.LSU by altering RNA folding. Nucleic Acids Res. 2002; 30:2961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chaires J.B., Ren J., Hamelberg D., Kumar A., Pandya V., Boykin D.W., Wilson W.D.. Structural selectivity of aromatic diamidines. J. Med. Chem. 2004; 47:5729–5742. [DOI] [PubMed] [Google Scholar]
- 29. Gelus N., Bailly C., Hamy F., Klimkait T., Wilson W.D., Boykin D.W.. Inhibition of HIV-1 Tat–TAR interaction by diphenylfuran derivatives: effects of the terminal basic side chains. Bioorg. Med. Chem. 1999; 7:1089–1096. [DOI] [PubMed] [Google Scholar]
- 30. Xiao G., Kumar A., Li K., Rigl C.T., Bajic M., Davis T.M., Boykin D.W., Wilson W.D.. Inhibition of the HIV-1 rev–RRE complex formation by unfused aromatic cations. Bioorg. Med. Chem. 2001; 9:1097–1113. [DOI] [PubMed] [Google Scholar]
- 31. Zhao M., Ratmeyer L., Peloquin R.G., Yao S., Kumar A., Spychala J., Boykin D.W., Wilson W.D.. Small changes in cationic substituents of diphenylfuran derivatives have major effects on the binding affinity and the binding mode with RNA helical duplexes. Bioorg. Med. Chem. 1995; 3:785–794. [DOI] [PubMed] [Google Scholar]
- 32. Yang W.-Y., Gao R., Southern M., Sarkar P.S., Disney M.D.. Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat. Commun. 2016; 7:11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ratmeyer L., Zapp M.L., Green M.R., Vinayak R., Kumar A., Boykin D.W., Wilson W.D.. Inhibition of HIV-1 Rev–RRE interaction by diphenylfuran derivatives. Biochemistry. 1996; 35:13689–13696. [DOI] [PubMed] [Google Scholar]
- 34. Donlic A., Morgan B.S., Xu J.L., Liu A., Roble C. Jr, Hargrove A.E.. Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew. Chem. Int. Ed. Engl. 2018; 57:13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morgan B.S., Forte J.E., Culver R.N., Zhang Y., Hargrove A.E.. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew. Chem. Int. Ed. Engl. 2017; 56:13498–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hewitt W.M., Calabrese D.R., Schneekloth J.S. Jr. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorg. Med. Chem. 2019; 27:2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Angelbello A.J., Chen J.L., Childs-Disney J.L., Zhang P., Wang Z.F., Disney M.D.. Using genome sequence to enable the design of medicines and chemical probes. Chem. Rev. 2018; 118:1599–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Connelly C.M., Moon M.H., Schneekloth J.S. Jr. The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol. 2016; 23:1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di Giorgio A., Duca M.. Synthetic small-molecule RNA ligands: future prospects as therapeutic agents. MedChemComm. 2019; 10:1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hermann T. Small molecules targeting viral RNA. Wiley Interdiscip. Rev. RNA. 2016; 7:726–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee D.S., Amara Z., Poliakoff M., Harman T., Reid G., Rhodes B., Brough S., McInally T., Woodward S.. Investigating scale-up and further applications of DABAL-Me-3 promoted amide synthesis. Org. Process Res. Dev. 2015; 19:831–840. [Google Scholar]
- 42. Chemical Computing Group, Inc. 2018; Montreal, Canada: Chemical Computing Group Inc; Molecular Operating Environment, v2018.01. [Google Scholar]
- 43. Schrödinger, LLC In the ligand interaction diagram, what are the maximum lengths of hydrogen bonds, pi–pi stacking interactions, pi–cation interactions and metal coordination interactions that are identified and displayed?. 2018; NY. [Google Scholar]
- 44. Abagyan R., Totrov M., Kuznetsov D.. ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994; 15:488–506. [Google Scholar]
- 45. Silvers R., Keller H., Schwalbe H., Hengesbach M.. Differential scanning fluorimetry for monitoring RNA stability. ChemBioChem. 2015; 16:1109–1114. [DOI] [PubMed] [Google Scholar]
- 46. Brown J.A., Valenstein M.L., Yario T.A., Tycowski K.T., Steitz J.A.. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl Acad. Sci. U.S.A. 2012; 109:19202–19207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yonkunas M.J., Baird N.J.. A highly ordered, nonprotective MALAT1 ENE structure is adopted prior to triplex formation. RNA. 2019; 25:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Patwardhan N.N., Ganser L.R., Kapral G.J., Eubanks C.S., Lee J., Sathyamoorthy B., Al-Hashimi H.M., Hargrove A.E.. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. MedChemComm. 2017; 8:1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ganser L.R., Lee J., Rangadurai A., Merriman D.K., Kelly M.L., Kansal A.D., Sathyamoorthy B., Al-Hashimi H.M.. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol. 2018; 25:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brown J.A., Kinzig C.G., DeGregorio S.J., Steitz J.A.. Hoogsteen-position pyrimidines promote the stability and function of the MALAT1 RNA triple helix. RNA. 2016; 22:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matarlo J.S., Krumpe L.R.H., Heinz W.F., Oh D., Shenoy S.R., Thomas C.L., Goncharova E.I., Lockett S.J., O’Keefe B.R.. The natural product butylcycloheptyl prodiginine binds pre-miR-21, inhibits Dicer-mediated processing of pre-miR-21, and blocks cellular proliferation. Cell Chem. Biol. 2019; 26:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pakravan P., Masoudian S.. Study on the interaction between isatin-β-thiosemicarbazone and calf thymus DNA by spectroscopic techniques. Iran. J. Pharm. Res. 2015; 14:111–123. [PMC free article] [PubMed] [Google Scholar]
- 54. Kozurkova M., Sabolova D., Paulikova H., Janovec L., Kristian P., Bajdichova M., Busa J., Podhradsky D., Imrich J.. DNA binding properties and evaluation of cytotoxic activity of 9,10-bis-N-substituted (aminomethyl)anthracenes. Int. J. Biol. Macromol. 2007; 41:415–422. [DOI] [PubMed] [Google Scholar]
- 55. Shahraki S., Mansouri-Torshizi H., Sori Nezami Z., Ghahghaei A., Yaghoubi F., Divsalar A., Saboury A.-A., H Shirazi F.. The effects of extending of co-planarity in a series of structurally relative polypyridyl palladium(II) complexes on DNA-binding and cytotoxicity properties. Iran. J. Pharm. Res. 2014; 13:1279–1294. [PMC free article] [PubMed] [Google Scholar]
- 56. Gilad Y., Senderowitz H.. Docking studies on DNA intercalators. J. Chem. Inf. Model. 2014; 54:96–107. [DOI] [PubMed] [Google Scholar]
- 57. Wicks S.L., Hargrove A.E.. Fluorescent indicator displacement assays to identify and characterize small molecule interactions with RNA. Methods. 2019; 167:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilson W.D., Tanious F.A., Ding D., Kumar A., Boykin D.W., Colson P., Houssier C., Bailly C.. Nucleic acid interactions of unfused aromatic cations: evaluation of proposed minor-groove, major-groove, and intercalation binding modes. J. Am. Chem. Soc. 1998; 120:10310–10321. [Google Scholar]
- 59. Morgan B.S., Hargrove A.E.. Synthetic Receptors for Biomolecules: Design Principles and Applications. 2015; London: The Royal Society of Chemistry; 253–325. [Google Scholar]
- 60. Waldron T.T., Murphy K.P.. Stabilization of proteins by ligand binding: application to drug screening and determination of unfolding energetics. Biochemistry. 2003; 42:5058–5064. [DOI] [PubMed] [Google Scholar]
- 61. Ganser L.R., Chu C.-C., Bogerd H.P., Kelly M.L., Cullen B.R., Al-Hashimi H.M.. Probing RNAConformational Equilibria within the Functional Cellular Context. Cell Rep. 2020; 30:2472–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ganser L.R., Kelly M.L., Herschlag D., Al-Hashimi H.M.. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 2019; 20:474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hermann T. Strategies for the design of drugs targeting RNA and RNA–protein complexes. Angew. Chem. Int. Ed. 2000; 39:1890–1904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.