


Abstract
MicroRNAs (miRNAs) are important regulators of eukaryotic gene expression and their dysfunction is often associated with cancer. Alongside the canonical miRNA biogenesis pathway involving stepwise processing and export of pri- and pre-miRNA transcripts by the microprocessor complex, Exportin 5 and Dicer, several alternative mechanisms of miRNA production have been described. Here, we reveal that the atypical box C/D snoRNA U3, which functions as a scaffold during early ribosome assembly, is a miRNA source. We show that a unique stem–loop structure in the 5′ domain of U3 is processed to form short RNA fragments that associate with Argonaute. miR-U3 production is independent of Drosha, and an increased amount of U3 in the cytoplasm in the absence of Dicer suggests that a portion of the full length snoRNA is exported to the cytoplasm where it is efficiently processed into miRNAs. Using reporter assays, we demonstrate that miR-U3 can act as a low proficiency miRNA in vivo and our data support the 3′ UTR of the sortin nexin SNX27 mRNA as an endogenous U3-derived miRNA target. We further reveal that perturbation of U3 snoRNP assembly induces miR-U3 production, highlighting potential cross-regulation of target mRNA expression and ribosome production.
INTRODUCTION
Modulation of gene expression is critical during development and cellular adaptation, and can be achieved via a variety of different mechanisms at the transcriptional, post-transcriptional and translational levels. MicroRNAs (miRNAs) have emerged as key post-transcriptional regulators of their messenger RNA (mRNA) targets through their roles in inducing degradation and/or translational repression of specific transcripts (see, e.g. (1–3)). As a result of this important function, dysregulation of miRNA expression or biogenesis is often associated with disease, especially cancer (4–6).
Canonical miRNAs are transcribed by RNA polymerase II (Pol II) as long pri-miRNAs that are 5′ m7G capped and 3′ polyadenylated, and contain multiple step-loop structures, each harbouring a miRNA sequence. Pri-miRNAs undergo cleavage by the Microprocessor complex, which contains the RNase III-like endonuclease Drosha and its cofactor DGCR8, to release short hairpin RNAs of ∼65 nucleotides (nt) in length (7,8). These pre-miRNAs are then recognised by Ran-GTP-bound Exportin 5 (XPO5), translocate through the nuclear pore complex and are released into the cytoplasm upon GTP hydrolysis (9–12). There, they undergo further processing by another RNase III-like enzyme, Dicer, to generate a ∼22 nt RNA duplex with 2 nt overhangs at the 3′ end of each strand (13–15). This duplex is loaded into a pre-miRNA-induced silencing complex (pre-miRISC) composed of Dicer, TRBP, HSC70, HSP90 and an Argonaute protein (AGO1–4 in humans) (16,17). The RNA duplex is rapidly unwound and the ‘guide’ and ‘passenger’ strands are identified based on the thermodynamic stability of their 3′ ends. While the passenger strand is degraded, the guide strand is retained in the mature miRISC complex. Basepairing between the miRNA seed region (nt 2–7) and complementary sequences in the 3′ untranslated regions (UTRs) of mRNAs directs the miRISC complex to specific target mRNAs, where either the endonucleolytic activity of AGO2 mediates mRNA cleavage leading to degradation or other miRISC-associated proteins regulate the fate of the mRNA e.g. by inducing translational repression.
Alongside this canonical biogenesis pathway, miRNAs also can be produced from other genomic sources and by alternative maturation pathways (reviewed in (1)). A Dicer-independent maturation pathway has been described for miR-451; pri-miR-451 is cleaved by Drosha to produce a pre-miRNA with an atypically short stem that is directly loaded onto AGO2 where the 3′ strand undergoes cleavage and processing to yield a mature miRISC complex. Furthermore, several Drosha- and DGCR8-independent miRNA biogenesis pathways have been described. Some pre-miRNA sequences are present in ‘mirtron loci’ within the introns of protein-coding pre-mRNAs and are excised by the action of the spliceosome. Alternatively, pre-miRNAs can be directly transcribed and therefore bypass the requirement for processing by Drosha. In contrast to canonically produced miRNAs, these 5′-capped miRNAs are exported to the cytoplasm by Exportin 1 (XPO1, also known as CRM1). Interestingly, pre-miRNAs can also be produced in a Drosha- and DGCR8-independent manner from other small RNA species, such as transfer RNAs (tRNAs) that function as adaptors between ribosome-associated mRNAs and amino acids (18,19), and small nucleolar/small Cajal body-associated RNAs (sno/scaRNAs), which typically guide modifications in ribosomal RNAs (rRNAs) and small nuclear RNAs (snRNAs) respectively (20–22). For example, a 3′ extended, pre-tRNAIle transcript can form a non-cloverleaf stem–loop structure that is processed by Dicer to generate miR-1983 (18), while tRNAGly(GCC) serves are a source of the CU1276 miRNA that targets the RPA1 mRNA thereby modulating the DNA damage response (19). The H/ACA box scaRNA, ACA45 (SCARNA15), which is predicted to guide pseudouridylation of uridine 37 of the U2 snRNA, is the best characterised example of a sno/scaRNA-derived miRNA (20). Complementary 20–22 nt fragments corresponding to the sequences that form a hairpin at the 3′ end of ACA45 were identified in small RNA-seq analysis of AGO1/2-associated RNAs. The ability of the predicted guide strand to function as a miRNA was demonstrated using luciferase reporter assays and the 3′UTR of the CDC2L6 mRNA, which encodes a protein component of the mediator complex that plays an important role in Pol II-mediated transcription, was identified as a target (20). The stem–loop structure of ACA45 differs from the characteristic Drosha substrates and generation of the ACA45-derived fragments was found to be independent of Drosha and DGCR8. Instead, it is suggested that a portion of the full-length scaRNA is exported to the cytoplasm where is it directly processed by Dicer (20). Deep sequencing of small RNA libraries from a variety of different human cell types has revealed the presence of various other snoRNA-derived fragments that have been suggested to act as miRNAs (23–25). However, in most cases, experimental evidence of functionality is lacking.
In this work, we demonstrate that the box C/D snoRNA U3, which does not guide rRNA modification but rather coordinates folding of the initial precursor ribosomal RNA (pre-rRNA) transcript, is processed to produce miRNAs. Compared to canonical box C/D snoRNAs, U3 contains an additional 5′ stem–loop region, which we show is processed to form short RNA fragments that associate with Argonaute. The biogenesis pathway of U3-derived fragments is independent of the microprocessor component Drosha, but requires the cytoplasmic action of Dicer. We demonstrate the ability of the U3-derived guide fragment to function as a low proficiency miRNA in vivo and identify the SNX27 mRNA as a cellular target of miR-U3. Our data expand the repertoire of characterised snoRNA-derived miRNAs and highlight a means for co-regulation of ribosome production and miRNA-mediated regulation of gene expression.
MATERIALS AND METHODS
Cell culture
Wild-type human HCT116 cells and HCT116 cells carrying genomic alterations to knock-out expression of particular genes were obtained from the Korean Collection for Type Cultures (BP1230983–BP1230988) and cultivated in McCoy's 5A medium (26). HEK293 cells were obtained from American Type Culture Collection (ATCC-CRL1573) and were cultivated in Dulbecco's modified Eagle's Medium (DMEM). Cell culture media were supplemented with 10% foetal bovine serum and 1% antibiotics (Penicillin and Streptomycin). All cell lines were maintained and cultured according to standard protocols in humidified incubators at 37°C with 5% CO2.
DNA, RNA and LNA transfections
For siRNA treatments (miR-U3 mimic and scrambled, and miRNA outcompeting), cells were transfected with 30 nM of siRNA (Supplementary Table S1) using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions and harvested after 72 h. DNA transfections (pLKO.1 (Addgene plasmid #10878)-based plasmids for expression of shRNAs, YFP-CFP miRNA activity reporter constructs and pFRT/TO/FLAG/HA-DEST DICER (Addgene plasmid #19881)) were performed on ∼300 000 cells using 2 μg DNA and Lipofectamine 2000 (Thermo Fisher Scientific). Cells were harvested 72 h after transfection. 30 nM LNA miRNA inhibitors (QIAGEN) (Supplementary Table S1), were co-transfected with appropriate plasmid DNAs using Lipofectamine 2000 (Life Technologies) and cells were harvested after 72 h.
RNA extraction and small RNA enrichment
Total RNA was isolated using Trizol (Thermo Fisher Scientific) according to the manufacturer's instructions unless stated otherwise. RNA concentrations and purity were measured on an Eppendorf BioSpectrometer basic (Eppendorf). For northern blot analysis, small RNAs (>200 nt) were enriched from 90 μg of total RNA using the mirVana™ miRNA isolation kit (AM1561-Life Technologies) following manufacturer's instructions.
Small RNA-seq
Small RNA-seq was performed as described previously (27). In brief, small RNAs were enriched from 2 μg of total RNA using the Magnetic bead cleanup module (Life Technologies). Libraries were then prepared using the Ion Total RNA-Seq Kit v2 (Life Technologies) and analysed via BioAnalyzer HS Chip (Agilent CA, USA). Afterwards, libraries (18 pM each) were clonally amplified by emulsion PCR in the IonTorrent OneTouch System (Life Technologies) using the Ion PGM Template OT2 200 Kit (Life Technologies) according to manufacturer′s protocol. Sequencing was performed in an IonTorrent Personal Genome Machine using an IonTorrent 316 Chip (Life Technologies).
Cellular fractionation
Cells from a confluent 10 cm dish were incubated in lysis buffer (10 mM Tris pH 8.4, 140 mM NaCl 1.5 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 0.5% NP40) on ice for 3 min and centrifuged at 5900 g at 4°C for 5 min. The supernatant (cytoplasmic fraction) was collected and mixed with homogenization buffer (50 mM Tris pH 8.0, 1% SDS, 50 mM NaCl, 0.5 mM EDTA) in a ratio 1:0.87. The pellet (nuclear fraction) was resuspended in lysis buffer supplemented with CaCl2 at a final concentration of 1.5 mM, and centrifuged at 5900 g at 4°C for 5 min. Nuclear RNA was extracted using TRI Reagent (Sigma Aldrich) following the manufacturer's instructions, while cytoplasmic RNA was extracted with phenol:chloroform:isoamylalchol (25:24:1), precipitated with isopropanol and resuspended in nuclease free water.
Quantitative PCR analysis of mRNA and sn(o)RNAs
2.5 μg of RNA was reverse transcribed using the Sensifast cDNA Synthesis Kit (Bioline), which was used for quantitative analysis on an ABI StepOnePlus system (Applied Biosystems) using the SensiMix SYBR Hi-ROX Kit and the primers listed in Supplementary Table S1. Cytoplasmic and nuclear RNA levels of the U3 snoRNA, U6 snRNA and GAPDH mRNA were normalized against total cellular RNA as follows: 2(Ct total RNA – Ct RNA fraction) and these values were then used for calculating the cytoplasmic to nuclear (C/N) ratio.
LNA-based quantitative PCR of miRNAs
Total RNA (5 μg) was reverse transcribed using the miRCURY LNA RT Kit (QIAGEN). Poly(A) tails were added to mature miRNA templates, and cDNA synthesized using a 5′ universal tag and 3′ degenerate anchor. The resultant cDNA was used for LNA-based RNA expression detection using a miRCURY LNA SYBR Green PCR Kit (QIAGEN) on an ABI StepOnePlus system (Applied Biosystems). U3-derived miRNA relative expression was calculated by ΔΔCT-method Ct with U6 as reference using custom made primers synthesized by QIAGEN. The amplified products were also visualized by electrophoresis using the E-Gel Precast Agarose Electrophoresis System (Thermo Fischer Scientific).
Northern blotting
Total and fractionated RNAs, and small RNAs were separated on 10% or 15% TBE-urea gels (Thermo Fischer Scientific), and then transferred to Hybond-N+ membrane (GE Healthcare). RNAs were crosslinked to membranes using UV light (total RNA) or chemically with N-(3-dimethylaminopropyl)-N′-ethylcarbodimide hydrochloride and 1-methylimidazole (Sigma-Aldrich) (small RNAs). Membranes were hybridized with 32P-labelled DNA oligonucleotides antisense to each target (Supplementary Table S1) overnight at 37°C in (0.5 M sodium phosphate pH 7.2, 7% SDS, 1 mM EDTA pH 8.0). After thorough washing steps, northern blots were exposed to phosphorimager screens before documentation using a Typhoon FLA 9500 (GE Healthcare). The acquired images were analysed and quantified using te Image Studio 5.2.5 software (LI-COR).
Western blotting
Cells were lysed in RIPA buffer (10 mM Tris–HCl pH 8.0, 140 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton-X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and 1 mM phenylmethylsulfonyl fluoride (PMSF)). Proteins were precipitated by addition of 12.5% trichloroacetic acid (TCA) and washed with cold acetone (–20°C). Protein pellets were resuspended in loading buffer (125 mM Tris pH 6.8, 5% SDS, 0.004 Bromophenol Blue, 1.4 M β-mercaptoethanol,10% glycerol) and 50 μg of protein per sample were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. The membrane was incubated with blocking solution (5% milk in TBS plus 0.1% Tween-20) and incubated with a primary antibody (Supplementary Table S3) overnight at 4°C. After thorough washing, membranes were incubated with IRDye LI-COR secondary antibodies (Supplementary Table S3) for 1 h at room temperature. After further washing steps, membranes were scanned using an Odyssey CLx infrared scanner (LI-COR Biosciences) then images were analysed and quantified using the Image Studio 5.2.5 software (LI-COR).
Analytical flow cytometry for miRNA activity
Small RNA sequences were derived from miRBase (www.mirbase.org) or NGS data analysis to design oligonucleotides encoding relevant miRNA targets (Supplementary Table S1). Oligonucleotides with NotI and XhoI overhangs were annealed and ligated into p.UTA.2.0 Empty (Addgene plasmid #82446). miRNA activity at single cell level was detected as previously (27,28). In brief, HEK293 cells or HTC-116 (50,000 cells/well) were transfected with appropriate plasmids and, where appropriate, siRNA (Supplementary Table S1) as described above, and incubated for 72 h. Flow cytometry was performed using a BD LSR II instrument, using the 550LP-BP575/26 and BP450/50 filters for YFP and CFP, respectively. YFP positive cells were selected using the FACSdiva™ software and FCS files were exported for analysis in R using scripts reported previously (28).
Experimental measurements of analytical flow cytometry were fitted to a biochemical model of miRNA-mediated regulation described before (27,29). In brief, this model describes miRNA posttranscriptional regulation using a set of coupled, first-order, ordinary differential Equations (1 and 2) and the conserved relation for miRNA (3):
 |
(1) |
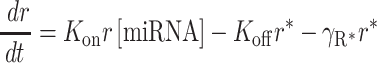 |
(2) |
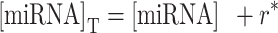 |
(3) |
By solving these equations for the steady state, the following equation is given:
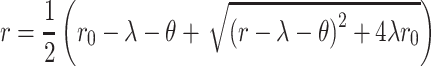 |
where:
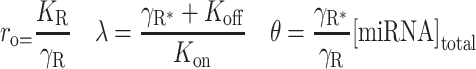 |
Using non-linear (weighted) least squares, the parameters λ and θ were calculated for each sample.
Bioinformatic analyses
Raw reads from Ion Torrent small RNA libraries (27) were trimmed and quality filtered employing the Ion-Torrent Suite Software (Life Technologies). Publicly available next-generation sequencing datasets (Supplementary Table S2) were retrieved from the Gene Expression Omnibus (GEO) using the SRA-toolkit and transformed from binaries to FASTQ using the fastq-dump tool. All the downloaded libraries were analysed with FASTQC and only data fitting the original publication descriptions were used for further analysis. Depending on the sequencing platform and library preparation strategy used, demultiplexing and adaptor trimming were applied as necessary using the Flexbar FASTX-toolkit (http://hannonlab.cshl.edu/). FASTQ files were aligned to the human genome GRCh38.p10 using STAR and results were contrasted with reported analyses as control. Afterwards all the FASTQ filtered outputs were analysed uniformly: they were converted to FASTA format and collapsed using FASTX-toolkit (http://hannonlab.cshl.edu/). Next, collapsed FASTA files were aligned to miRbase v 21 and snoRNA base v3 with Bowtie2. The SAM output files were converted to Blast format using sam2blast, and significant hits, according to E-value, were selected using mtophits_blast and duplicates were removed with remove_duplicate_hits_blast.pl using scripts adapted from Grzegorz Kudla (https://github.com/gkudla). Plots and additional analyses were performed using custom made R scripts, and data was normalized using the DEseq2 Package.
Statistical analyses
Statistical analysis of the data was performed using R and R-Studio. For qPCR measurements of miRNA expression levels and expression levels of other RNAs in cytoplasmic and nuclear fractions, sample groups were compared using unpaired two-samples t-test. For protein and RNA relative expression from northern and western blot quantifications, the sample groups were tested for normality of distribution using Shapiro-Wilk test. Since data followed normal distribution, samples were compared to wild-type (NB) or Mock (WB) using the one-sample t-test. For analytical flow cytometry, CFP intensity distributions were compared using the Mann-Whitney-Wilcoxon test. For all tests, the significance threshold was set at P < 0.05.
RESULTS
Small RNA profiling identifies box C/D snoRNA-derived fragments with miRNA-like characteristics
Building on the discovery of various small RNAs derived from non-coding RNA species, we set out to identify and characterise small RNA fragments derived from box C/D snoRNAs that may function as miRNAs. Small RNA sequencing (small RNA-seq) was performed on libraries (L1–3) prepared from HEK293 cells and subjected to Ion Torrent deep sequencing. The data obtained from the three library preparations showed good correlation (L001–L002 R2 = 0.972, L001–L003 R2 = 0.967) (Supplementary Figure S1A) and the majority of reads corresponded to RNAs or RNA fragments of <40 nt in length (Supplementary Figure S1B). Analysis of the distribution of sequence reads between different genome features showed that the majority mapped to annotated miRNA or sno/scaRNA sequences, and of these, a significant proportion of the reads (average 17.7%) mapped to box C/D snoRNA sequences while only few were derived from scaRNAs or box H/ACA snoRNAs (Supplementary Figure 2A). Closer inspection revealed that the reads mapped to 19 different sno/scaRNAs, including the known miRNA precursor, ACA45. To effectively function as miRNAs, small RNA fragments must be present in cells at sufficient levels. We therefore determined the relative numbers of reads mapping to each snoRNA sequence as a measure of expression level, and compared the novel candidates to the ACA45-derived fragments, which are known to be functional miRNAs (Figure 1A and Supplementary Figure S1C). From this, we selected nine candidates whose expression level was greater than (U78, U44, snR39B) or similar to (U45A, U24, U3, U74, U27) that of the ACA45 fragments for further analysis. Another key criterium for the identification of pre-miRNAs is the presence of a distinct read distribution pattern in which homogeneous populations of reads corresponding to potential guide and/or passenger strands of <24 nt can be observed. Hierarchical clustering analysis (HCA), an algorithm that groups similar objects, was therefore used to investigate the distribution of sequence reads mapping to each of the identified sno/scaRNAs (Figure 1B), and the average read length of clusters corresponding to similar fragments was determined (Figure 1C). For ACA45, as anticipated from previous data (20), two sequence clusters corresponding to nt 65–86 and 104–125 (average length 21 nt) were detected in the small RNA libraries (Figure 1B; middle left panel). In the case of U45A, U24 and U27, significant heterogeneity was observed in the fragment lengths (Figure 1B and C), implying that these fragments likely represent RNA degradation products rather than specifically processed, functional RNAs. While more homogeneous populations of reads mapping to U78, U44, snR39B and U74 were detected (Figure 1B), the average lengths of these fragments spanned >24 nt (Figure 1C), implying that they do not correspond to miRNA sequences. However, analysis of the distribution of reads derived from the U3 snoRNA revealed three relatively homogeneous clusters corresponding to nt 45–64, 75–94 and 195–214 (Figure 1B; lower left panel). These fragments are all within the length threshold and the fragments corresponding to nt 45–64 and 75–94 are separated at distance (11 nt) that would be suitable for the loop region of a pre-miRNA, raising the possibility that this region of the U3 snoRNA is processed to form miRNAs.
Figure 1.
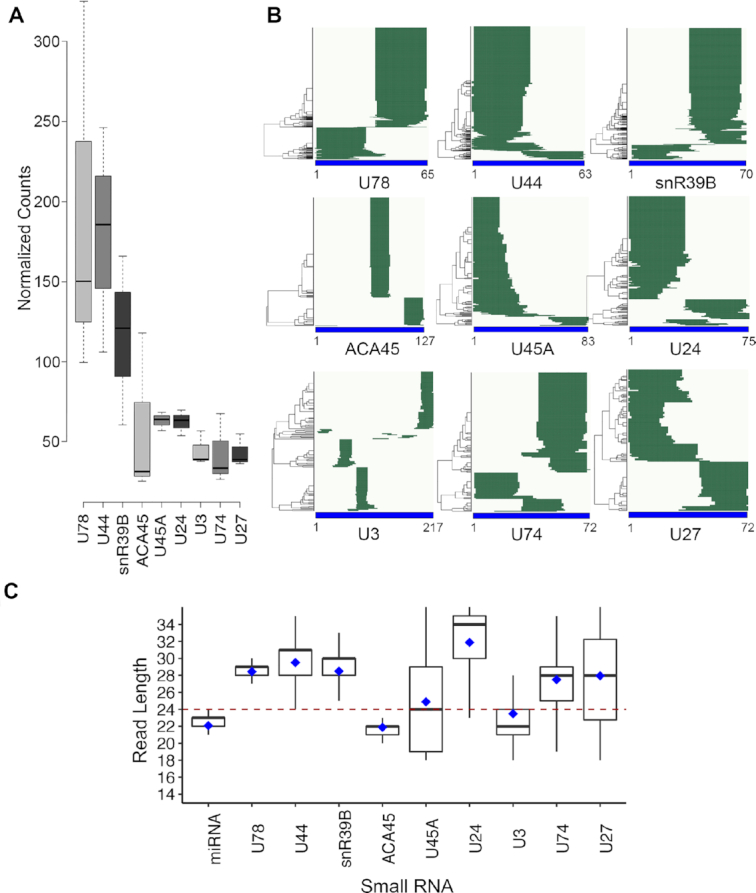
miRNA-like small RNA fragments are produced from the U3 box C/D snoRNA. (A) Small RNA-seq was performed on total RNA extracted from HEK293 cells and the normalised numbers of reads of <40 nt in length mapping to different snoRNAs is shown as boxplots. Error bars represent the variation between three independent library preparations. (B) Hierarchical clustering was performed to analyse the distribution of reads mapping to selected snoRNA sequences in the small RNA-seq libraries. Nucleotide positions are indicated below the x-axis. (C) The distribution of sequence lengths of snoRNA-derived fragments and known miRNAs are shown as box blots. The black line represents the median and blue diamond shows the mean.
Small RNA fragments originating from the 5′ end of the U3 snoRNA associate with components of the miRNA biogenesis machinery
During their maturation and function, miRNA sequences are contacted by various proteins, including the microprocessor complex components Drosha and DGCR8, as well as Dicer and Argonaute. To determine if the U3-derived fragments could function as miRNAs, we sought evidence of physical interactions between these RNA sequences and miRNA biogenesis factors. Previously, crosslinking and immunoprecipitation (CLIP) analyses have been performed to obtain transcriptome-wide inventories of the cellular RNAs bound by Drosha (30), DGCR8 (31), Dicer (32) and Argonaute (33–35) (Supplementary Table S2). To determine if the U3-derived fragments associate with components of the miRNA biogenesis machinery, we therefore mined these publicly available datasets. For comparison, available datasets obtained from CLIP analyses of the core box C/D snoRNP proteins, NOP56, NOP58 and Fibrillarin (36) were also included. As all these datasets have previously been subjected to independent bioinformatic analyses, to enable the RNA targets of each of these proteins to be compared directly, the datasets were first re-analysed using a common pipeline. Consistent with the initial analyses, this confirmed the enrichment of miRNAs in the Drosha, DGCR8, Dicer and Argonaute datasets as well as the co-purification of box C/D snoRNAs with NOP56, NOP58 and Fibrillarin (Supplementary Figure S2B and C).
While analysis of the distribution of the sequencing reads present in the datasets derived from the core box C/D snoRNP proteins on the U3 snoRNA confirmed their binding primarily to the 3′ stem–loop region containing the C/C’ and D/D’ box elements common to all snoRNAs (Figure 2A, upper panel) (37), this was in stark contrast to the binding profiles observed for the miRNA biogenesis factors. The 5′ region of the U3 snoRNA (approx. nt 40–90) from which the small RNA fragments were derived (Figures 1B and 2A, lower panel; ‘small RNA-seq’) was found to be enriched in the DGCR8, Dicer and Argonaute datasets (Figure 2A, lower panel), further supporting the potential role of these fragments as miRNAs. Notably, these sequences were not crosslinked by Drosha (Figure 2A) and no clear crosslinking sites of any of the miRNA biogenesis factors on the other snoRNAs detected in the small RNA profiling analyses were observed (data not shown).
Figure 2.
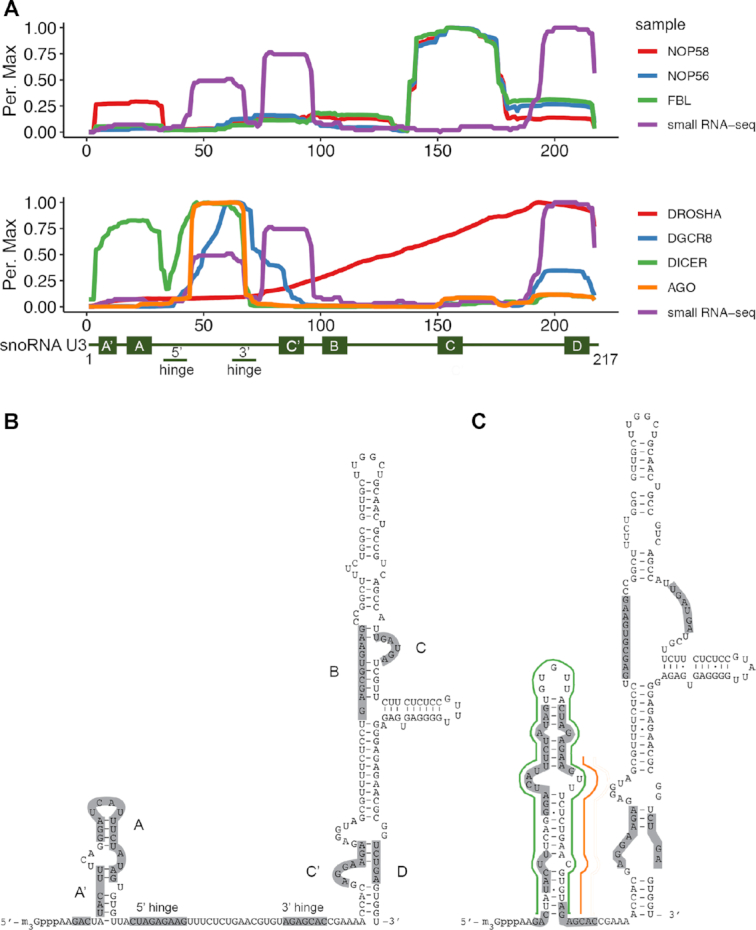
U3-derived small RNA fragment sequences are bound by components of the miRNA biogenesis machinery. (A) The normalised number of reads mapping to each nucleotide of the U3 sequence in (PAR-)CLIP analysis of snoRNP proteins (upper panel; FBL - fibrillarin) and miRNA-associated proteins (lower panel; AGO - Argonaute) is shown as a percentage of the maximum (Per. Max) above a schematic model of the U3 snoRNA. (B) Schematic secondary structure of the U3 snoRNA within the U3 snoRNP. Evolutionarily conserved features of the snoRNA are indicated. (C) Schematic secondary structure of the U3 snoRNA within the U3 snoRNP. Evolutionarily conserved features of the snoRNA. The hairpin containing of Dicer-associated sequences present in the small RNA-seq/CLIP libraries is marked in green and the guide strand of the putative miRNA duplex is indicated in orange.
Interestingly, the U3 snoRNA has been proposed to form alternative secondary structures, depending on whether it is associated with the box C/D snoRNP proteins or not (38). In the context of the U3 snoRNP, the U3 snoRNA is suggested to possess a small stem–loop containing the box A and A’ sequences while the 5′ and 3′ hinge regions, which basepair with the pre-rRNA upon pre-ribosome binding, remain unbasepaired (Figure 2B) (39). However, in the absence of proteins, both thermodynamic modelling (40) and in vitro structure probing analyses (38) indicate that the 5′ domain of U3 folds into an alternative stem–loop conformation (Figure 2C). In this conformation, a hairpin containing the U3 RNA sequences identified in the small RNA profiling (Figure 1B) is observed, and a potential miRNA guide strand can be identified based on the binding profile of Argonaute (Figure 2C).
U3-derived RNA fragments can act as low proficiency miRNAs in cells
We next aimed to demonstrate the functionality of the U3-derived RNA fragments as miRNAs. While luciferase-based reporter assays have extensively been used to analyse miRNA activity in mammalian cells (41), the discovery that some miRNAs, such as miR-23 and the ACA45-miRNA, act with only low proficiency has necessitated the development of alternative, more sensitive reporters (42). The seed pairing stability (SPS) and the abundance of predicted targets (TA) contribute significantly to the proficiency of a given miRNA, and can be used in bioinformatic analyses to predict the relative efficiencies of different sequences (42). To enable a suitable reporter system to be selected, we first computationally determined the SPS and TA of the U3-derived guide fragment and compared them with those of other miRNAs. This revealed that the U3-derived guide sequence has a relatively high TA and low SPS, with values comparable to those of miR-23 and the ACA45-miRNA (Supplementary Figure S3), suggesting that the U3-derived fragment would likely function as a low proficiency miRNA. Based on this result, a well-established fluorescence-based reporter system coupled to flow cytometry detection that allows highly sensitive, single cell analysis (27), was employed to demonstrate the action of the U3-derived sequence as a miRNA. The reporter construct enables expression of yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) from TK and SV40 promoters respectively (Figure 3A). Target sequences perfectly complementary to the high proficiency miRNA miR-92a, the ACA45-derived miRNA, the putative U3-derived guide sequence or a non-cognate sequence that is not targeted by any known miRNA were introduced within the 3′ UTR of the gene encoding CFP to estimate their AGO2-induced slicing activity. In the case of the ACA45-derived and potentially U3-derived miRNA target sites, either one or three copies of the target site were inserted to enable dosage-dependent effects to be monitored. Comparison of the relative YFP (control) and CFP (target) expression in the cell population then enables the efficiency of each miRNA to be assessed. It has previously been shown with this system, that low proficiency miRNAs decrease the expression of CFP relative to YFP, while maintaining a linear relationship between expression of the two fluorophores, leading to only a subtle shift in the flow cytometry profile (27). In contrast, the action of high proficiency miRNAs leads to a non-linear relationship between CFP and YFP expression, and formation of strongly shifted, curved profiles. After excluding the possibility that differences in CFP levels arise by chance, obtained data are fitted using a previously described mathematical model of miRNA-mediated regulation of mRNA targets to confirm that the observed differences arise due to miRNA activity rather than another form of post-transcriptional regulation. Mathematical modelling of the obtained data to an established model of miRNA-mediate regulation of mRNA targets (27,29) then enables the significance of differences between the observed profiles to be calculated.
Figure 3.
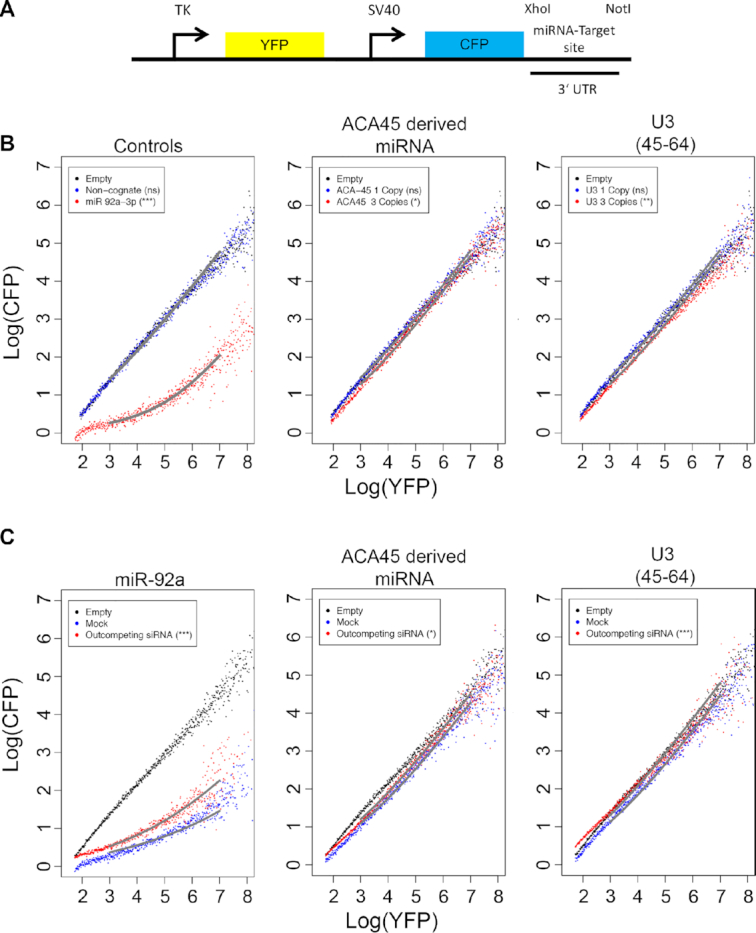
The U3-derived fragments behave like low proficiency miRNAs in reporter assays. (A) Schematic overview of the reporter construct used for analysing miRNA function in human cells. (B) The relative expression of CFP and YFP in individual cells (dots) was determined by flow cytometry in cells containing constructs encoding no miRNA target site (Empty), a sequence not targeted by any known human miRNA (Non-cognate) or the indicated miRNA target sites in one or three copies, within the 3′ UTR of the CFP gene. Grey lines indicate fitting to a previously described model of miRNA-mediated regulation (27,29). The significance of differences between cells transfected with the empty construct and those carrying the miRNA target sites indicated was calculated using the Mann-Whitney-Wilcox test (ns – not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). (C) The relative expression of CFP and YFP in individual cells (dots) was determined by flow cytometry, as a measure of AGO2 slicing activity, in cells containing empty reporter constructs (Empty) or reporter constructs encoding three copies of the target sites of the (putative) miRNAs indicated above the panels within the 3′ UTR of the CFP gene and which had been mock transfected (Mock) or treated with specific siRNAs to outcompete the relevant miRNA (siRNA). Fitting and statistical analysis were performed as in (B).
Expression of the reporter cassette encoding the non-cognate sequence did not alter the relative expression of CFP and YFP compared to cells transfected with a control plasmid (Empty), whereas inclusion of the target sequence of miR-92a lead to a significant decrease in the relative expression of CFP in the cell population confirming the high proficiency of this miRNA (Figure 3B, left panel; Supplementary Table S4). In contrast, expression of a single copy of the ACA45-derived miRNA target site lead to a mild, linear shift in the profile, which was increased upon expression of three target sites, confirming the action of this sequence as a low proficiency miRNA (Figure 3B, middle panel; Supplementary Table S4) (20). Similar to ACA45, expression of a sequence complementary to the putative U3-derived miRNA guide strand lead to a linear decrease in CFP expression relative to YFP, which was more prominent when three copies of the potential target site were incorporated (Figure 3B, right panel; Supplementary Table S4), implying that the U3-derived fragment can impair expression of a target gene. To further consolidate this finding, experiments were performed in which cells expressing the reporter constructs were co-transfected with siRNA to outcompete the endogenous miRNA or mock transfected. Cells expressing the target sites of miR-92a, the ACA45-derived miRNA or the U3-derived fragment with showed strong (miR-92a) or weak (ACA45- or U3-derived fragments) decreases in the expression of CFP relative to YFP as previously. In all cases, treatment with a competing siRNA (partially) rescued CFP expression (Figure 3C; Supplementary Table S4). The mild effect of the competing siRNA on the functionality of the high proficiency miR-92a is expected as the titration model of miRNA effectiveness predicts that alterations in concentration will not strongly influence a miRNA with a high seed region pairing energy. Taken together, these data support the action of the U3-derived small RNA fragments as bona fide low proficiency miRNAs in vivo.
Production and function of U3-derived miRNA is Dicer-dependent and Drosha-independent
To understand more about the production of the U3-derived miRNA (miR-U3), we next explored the requirement for components of the classical miRNA biogenesis pathway for its production and function. For this, we utilised HCT116 cells in which the genes encoding Drosha, Dicer or XPO5 had been disrupted by CRISPR-Cas genome editing leading to a lack of protein expression ((26); Supplementary Figure S4A). To first verify the levels of miRNAs produced by different pathways in these cells, we analysed available small RNA-seq data (Supplementary Table S2; (26)). As anticipated, the levels of the canonical miRNAs miR-19a, miRNA-19b and miR-92a were markedly reduced in the Dicer and Drosha knockout (KO) cell lines compared to the WT cells, but were largely unaffected by lack of XPO5 (Supplementary Figure S4B; Figure 4A). Also, in line with previously published findings, the levels of the 5′ capped/XPO1-dependent miRNAs miR-320a, miR-484, miR-320b and miR-3615, as well as the mirtron miR-877, were markedly increased in cells lacking Drosha and to a lesser extent in the XPO5 KO cells but were clearly reduced in the absence of Dicer (Supplementary Figure S4B, Figure 4A). Analysis of the levels of miR-U3 in these small RNA-seq libraries revealed, similar to the 5′ capped/XPO1-dependent miRNA miR-320a, but in contrast to the canonical miRNA miR-92a, accumulation in the Drosha KO cells, little effect of lack of XPO5 and a clear reduction in the absence of Dicer (Figure 4A). These observations are further supported by LNA-based quantitative PCR with no amplification of miR-U3 detected in the Dicer KO cell lines and a significant increase in the amount detected in the Drosha KO cell line compared to the wild-type control cells (Figure 4B). In addition, to avoid potential artefacts arising from reverse transcription bias during cDNA synthesis, northern blotting of RNAs extracted from the different KO cell lines was performed. This confirmed that the amount of the full length U3 snoRNA was not affected by lack of any of the miRNA biogenesis factors. However, although miR-U3 was present at a low level in wild-type cells and was largely unaffected by lack of XPO5, it was more abundant in cells lacking Drosha and could not be detected in Dicer KO cells (Figure 4C). These data, together with the observation that Dicer and Argonaute, but not Drosha, associate with the 5′ region of the U3 snoRNA (Figure 2A), indicate that production of miR-U3 is Drosha-independent.
Figure 4.

Production of the U3-derived miRNA requires Dicer but not Drosha. (A) The relative proportions of sequencing reads derived from the indicated miRNAs in small RNA-seq datasets derived from wild-type HCT116 cells (WT) or HCT116 cells lacking Drosha, XPO5 or Dicer (KO) is shown. (B) Total RNA from wild-type HCT116 cells (WT) or the knockout (KO) cell lines indicated was reverse transcribed and the levels of the U3-derived miRNA and the U6 snRNA were determined using LNA-based qPCR. Data from three independent experiments is shown as mean ± standard deviation. Amplified products were separated by gel electrophoresis and detected using Ethidium bromide. (C) Total RNA was purified from wild-type HCT116 cells (WT) or HCT116 cells lacking Drosha, XPO5 or Dicer (KO) and small RNAs (<200 nt) were enriched. Total and small RNAs were separated by denaturing polyacrylamide gel electrophoresis and transferred to a nylon membrane where they were detected by northern blotting using probes against the U3 and U6 snRNAs (total RNA) and the U3-derived miRNA (small RNAs). (D) The relative expression of CFP and YFP in individual cells (dots) was determined by flow cytometry in wild-type HCT116 cells (WT) or cells lacking Drosha or Dicer (KO) containing constructs encoding no miRNA target site (Empty), or the indicated miRNA target sites in one or three copies, within the 3′ UTR of the CFP gene. Significance was calculated using the Mann–Whitney–Wilcox test; ns – not significant, *P < 0.05, **P < 0.01, *** P ≤ 0.001.
The finding that miR-U3 is produced in a Drosha-independent manner raises the possibility that in Drosha KO cells, where canonical miRNAs are lacking, the abundance of miRISC complexes containing the U3-derived miRNA may be increased leading to stronger effects on gene expression. To test this hypothesis, fluorescence-based reporter assays were performed in wild-type HCT116 cells and cells lacking Drosha or Dicer. We first verified this system by analysing the behaviour of the canonical miRNA miR-92a, the Dicer-independent miRNA miR-21 (26), and the Drosha-independent miRNA miR-320a (43). Inclusion of the perfectly complementary target sequences of each of these miRNAs into the 3′ UTR of the CFP gene within the reporter construct reduced the expression of CFP relative to YFP to greater or lesser extents in wild-type cells demonstrating the activity of these miRNAs (Figure 4D, upper left panel; Supplementary Table S5). As anticipated, no activity of miR-92a or miR-21 was observed in cells lacking Drosha while the influence of miR-320 was still apparent (Figure 4D, upper middle panel; Supplementary Table S5). Likewise, while in the absence of Dicer the repressive activity of miR-21 was only minimally affected, no effects on CFP expression were observed when the target sequences of miR-92a or miR-320 were incorporated into the reporter construct (Figure 4D, upper right panel; Supplementary Table S5). We then analysed the effects of Dicer or Drosha knockout on CFP expression using the reporter constructs encoding either one or three copies of the U3-derived miRNA target sequence. As previously, in wild-type cells, the U3-derived fragment behaved as a low proficiency miRNA (Figure 4D, lower panels; Supplementary Table S5). However, the relative expression of CFP to YFP was markedly reduced in the Drosha KO cells and these effects were stronger when three copies of the U3-derived miRNA target sequence were present. These data not only confirm the activity of miR-U3 in the absence of Drosha, but also demonstrate its increased efficiency when the canonical miRNA biogenesis pathway is impaired.
A portion of the U3 snoRNA is exported to the cytoplasm where it is efficiently processed to generate miRNAs
Many snoRNAs are encoded within pre-mRNA introns, however, some, including U8, U13 and U3, are independently transcribed by RNA polymerase II (21). Similar to the U1, U2, U4 and U5 small nuclear RNAs (snRNAs), after their synthesis, these non-intronic snoRNAs are 5′ m7G capped, and associate with the cap-binding complex (CBC) as well as the trafficking proteins, XPO1 and PHAX (44). While the U1, U2, U4, U5 snRNAs and the U8 and U13 snoRNAs have been shown to undergo a cytoplasmic phase during their biogenesis (45,46), whether this is also the case for U3 has remained unclear. The finding that production of the U3-derived miRNA is Dicer-dependent and Drosha-independent suggests that they may be produced via an XPO1 (CRM1)-dependent pathway. On the one hand, this is in line with the known association of the U3 snoRNA with XPO1, and on the other hand suggests that U3 may be efficiently processed into miRNAs by Dicer upon export to the cytoplasm. While only little or no U3 snoRNA has previously been detected in the cytoplasm (44,47), this model suggests that the amount of cytoplasmic U3 would be increased in cells lacking Dicer. To test this hypothesis, wild-type HCT116 cells or those lacking Dicer ((26); Supplementary Figure S4A) were subjected to nuclear-cytoplasmic fractionation, and the levels of the U3 snoRNA were determined using northern blotting and/or qRT-PCR. The U6 snRNA as well as the tRNAMet or the GAPDH mRNA were also monitored as representatives of predominantly nuclear and cytoplasmic RNAs respectively. As expected, lack of Dicer did not affect the sub-cellular distribution of the U6 snRNA or tRNAMet. However, the northern blot analysis demonstrated the presence of a portion of the U3 snoRNA in the cytoplasm and revealed an increase in its cytoplasmic to nuclear ratio in the absence of Dicer (Figure 5A). These finding were confirmed by qRT-PCR analysis, which also demonstrated the lack of significant effects of the Dicer KO on the nuclear-cytoplasmic distribution of the U6 snRNA and the GAPDH mRNA (Figure 5B). To further consolidate the role of Dicer in regulating the U3 snoRNA level in the cytoplasm, Dicer KO cells were transfected with a plasmid for overexpression of Flag tagged Dicer (Supplementary Figure S5A). Northern blot analysis of nuclear and cytoplasmic RNA derived from wild-type cells, cells lacking Dicer and cells where Dicer was overexpressed revealed that while lack of Dicer increases the amount of cytoplasmic U3 snoRNA, Dicer overexpression reduces the cytoplasmic level of U3 compared to that observed in wild-type cells (Supplementary Figure S5B).
Figure 5.
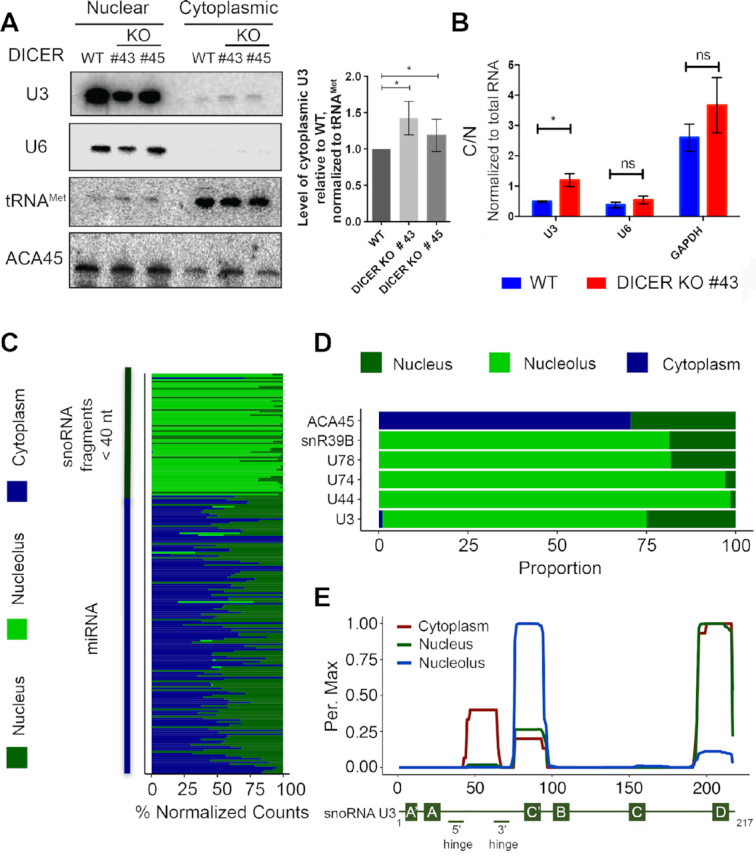
A portion of the U3 snoRNA is exported to the cytoplasm where it is processed to form miRNAs. (A) Wild-type HCT116 cells (WT) or HCT116 cells lacking Dicer (KO #43 and #45) were fractionated into nucleoplasm and cytoplasm, and total RNA was extracted. RNAs were separated by denaturing polyacrylamide gel electrophoresis and transferred to a nylon membrane. Northern blotting was performed using radiolabelled probes against the indicated RNAs. The levels of cytoplasmic U3 in the knockout cells lines relative to the wild type, normalised to tRNAMet, were determined in three independent experiments and data are shown as mean ± standard error. Significance was calculated using the one sample t-test; *P < 0.05. (B) Total RNA prepared from cells as in (A) was reverse transcribed and quantitative PCR was performed to monitor the levels of the U3 snoRNA, the U6 snRNAs and the GAPDH mRNA. The relative amounts of each RNA in cytoplasm and nucleus was calculated. * indicates P < 0.05, ns indicates non-significant (two sample t-test). (C) The relative proportions of reads mapping to sno/scRNAs and miRNAs in small RNA-seq data from HeLa cell nucleoli, nuclei and cytoplasm is shown. (D) Selected examples of the data presented in (C) are shown. (E) The normalised number of reads mapping to each nucleotide of the U3 sequence in the datasets described in (C) is shown as a percentage of the maximum (Per. Max) above a schematic model of the U3 snoRNA.
Next, to determine if all regions of the U3 snoRNA are present in the cytoplasm in equal amounts, we performed bioinformatic analysis of small RNA-seq data derived from the nucleolar, nucleoplasmic and cytoplasmic fractions of HeLa cells (48). This confirmed the global enrichment of sno/scaRNA-derived fragments of <40 nt and miRNAs in the nucleolus/nucleus and cytoplasm/nucleus respectively (Figure 5C). However, closer inspection of the distribution of sequencing reads mapping to selected snoRNAs in the different datasets show that while fragments originating from non-miRNA encoding snoRNAs, such as snR39B, U78, U74 and U44 are predominantly nucleolar, the ACA45-derived miRNA is present in the cytoplasm and nucleus (Figure 5D). Moreover, these data demonstrate that small RNA fragments derived from U3 are detected not only in the nucleolar fraction, but are also relatively enriched in the nucleoplasm compared to the fragments of most other snoRNAs and some U3 snoRNA-derived small RNA species, likely representing the U3-derived miRNA are also present in the cytoplasm (Figure 5D). Analysis of the distribution of the reads mapping to the U3 sequence in the three datasets confirmed that the miR-U3 sequence (nt 45–64) is specifically enriched in the cytoplasmic fraction (Figure 5E). Taken together, these data support the model that after nuclear export of a portion of the full length U3 snoRNA, it is efficiently processed by Dicer to generate a U3-derived miRNA.
The SNX27 mRNA is an endogenous target of the U3-derived miRNA
The finding that the U3-derived fragments exhibit low proficiency miRNA-like activity in human cells suggests the potential existence of endogenous mRNA target(s). Mapping the human miRNA-interactome was recently approached using the crosslinking, ligation and sequencing of hybrids (CLASH) method to search for associated miRNA and mRNA sequences bound by AGO1 (49). We therefore used the Hyb pipeline (50) to bioinformatically probe the dataset generated by this study for hybrid sequence reads including the miR-U3 sequence. This recovered chimeras containing the miR-U3 sequence together with sequences from the mRNAs encoding the box C/D snoRNP component NOP58, the serine/threonine protein kinase LMTK2, and the trafficking proteins ATXN2 and SNX27. Alignment of the recovered sequences with the full-length mRNA transcripts revealed that in the case of the SNX27 mRNA, the hybrids contained a sequence present in the 3′ UTR (Figure 6A). Analysis of the potential miR-U3 target site using the TargetScan algorithm (51) showed that it conforms to the definition of a canonical 7mer-m8 site (Figure 6A). The detection of complementary U3-derived and mRNA 3′ UTR sequences associated with AGO1 in human cells strongly supports the SNX27 mRNA as a target of the U3-derived miRNA. As miRNAs and their targets sites are subjected to evolutionary pressure (3,52), to further consolidate the likely functionality of the identified sequences, their conservation within metazoans was analysed. Both the U3-derived sequence and its SNX27 mRNA target site are conserved among placental mammals, but not to amphibia and oviparous animals (Supplementary Figure S6). This suggests selection pressure on these sequences only recently during metazoan evolution, which is in line with the observed low proficiency of the U3-derived miRNA in our reporter assays.
Figure 6.

The U3-derived miRNA targets the SNX27 mRNA. (A) Schematic representation of the chimeric sequence reads containing sequence of the SNX27 mRNA (upper, green) and the U3-derived miRNA-like fragment (lower, yellow) identified in CLASH analysis of AGO1. The seed sequence of the U3-derived miRNA-like fragment is shown in white, italics and underlined, and the target site (mRNA) is shown in bold, italics and underlined. ‘(‘ and ‘)’ indicate nucleotides that can form basepairing interactions. (B) Wild-type HCT116 cells (WT) and DICER KO cells were mock transfected (Mock) or transfected with a siRNA mimic of the U3-derived miRNA (U3 mimic) or a scrambled sequence (Scramble). The levels of SNX27 and GAPDH proteins were determined by western blotting and the amount of SNX27 in the transfected cells compared to the mock, normalised according to the GAPDH loading control is shown as a bar graph. Data from three biological and two technical replicates is shown as mean ± standard error. Significance was calculated using the one sample t-test; ns – not significant, *P < 0.05. (C) HCT116 cell lacking Drosha (DROSHA KO) were transfected with plasmids for expression of a short-hairpin RNA (shRNA) targeting U3–55K or mock transfected (Mock). Protein levels were determined by western blotting using the indicated antibodies (upper panels). Total RNA was extracted and small RNAs (<200 nt) were enriched. Total and small RNAs were separated by denaturing polyacrylamide gel electrophoresis and transferred to nylon membranes where they were detected by northern blotting using probes against the U3 and U6 snRNAs (total RNA) and the U3-derived miRNA (small RNAs). (D) HCT116 cell lacking Drosha (DROSHA KO) were mock transfected (Mock) or transfected with plasmids for expression of a short-hairpin RNA (shRNA) targeting U3–55K or a scrambled sequence and were co-transfected with a miR-U3 anti-miR (U3) or control anti-miR (neg). Protein levels were determined by western blotting using the indicated antibodies (middle panel). The levels of SNX27 and GAPDH proteins were determined and the amount of SNX27 in the transfected cells compared to the mock, normalised according to the GAPDH loading control is shown as a bar graph (right panel). Data from three biological and two technical replicates is shown as mean ± standard error. Significance was calculated using the one sample t-test; ns – not significant, *P < 0.05, ***P < 0.001.
We next aimed to demonstrate the functionality of the U3-derived miRNA and confirm SNX27 as a target. To analyse the effect of overexpression of miR-U3, a siRNA mimic of miR-U3 was transfected into wild-type cells. As miR-U3 acts as a low proficiency miRNA, the effect on target expression would not be expected to be strong, therefore, to increase the formation of miR-U3 mimic-containing miRISC complexes by preventing production of most endogenous miRNAs, the U3-derived miRNA mimic was also transfected into Dicer KO cells. Analysis of proteins from wild-type cells confirmed a mild, but significant, effect on the SNX27 level, whereas in the Dicer KO background, treatment with the miR-U3 mimic lead to a strong reduction in the SNX27 protein level, confirming the SNX27 mRNA as cellular target of the U3 miRNA (Figure 6B).
Impaired U3 snoRNP assembly induces miR-U3 production
The finding that, beyond its well characterised role in the early nucleolar stages of ribosome assembly, a portion of the U3 snoRNA is exported to the cytoplasm where it serves as a miRNA source suggests that the distribution of U3 snoRNA between these two functions may be regulated. We therefore set out to explore the relationship between the amounts of the U3 snoRNP and the U3-derived miRNA present in the cell. Compared to other box C/D snoRNPs, the U3 snoRNP contains an additional protein, U3–55K, and it has previously been shown that lack of U3–55K leads to a reduced level of the U3 snoRNA/P (53). To determine whether reduction of the U3 snoRNP leads to increased miR-U3 production, we analysed the effect of U3–55K depletion on the levels of full-length U3, miR-U3 and the miR-U3 target SNX27. To increase the availability of miRISC components and thereby enhance the phenotypic effect of miR-U3, this analysis was performed in Drosha KO cells where production of most miRNAs, but not miR-U3, is inhibited. Comparison of RNAs and proteins from mock transfected cells and cells transfected with a plasmid for expression of a short-hairpin RNA (shRNA) targeting U3-55K confirmed the efficient depletion of U3-55K and the previously observed decrease in the full-length U3 snoRNA level. Strikingly, the amount of miR-U3 was strongly increased in cells lacking U3-55K and consistent with this, the SNX27 protein level was also markedly reduced (Figure 6C). To demonstrate that the effect on SNX27 arises due to the miR-U3, an analogous experiment was performed in the presence of a specific anti-miR that should block the effect of miR-U3 or a non-target anti-miR (Neg) that served as a negative control. Cells transfected with a plasmid for expression of a control shRNA (shRNA scramble) showed no alterations in the levels of U3-55K or SNX27 in the presence of either anti-miR compared to the mock transfected control (Figure 6D). In contrast, while cells depleted of U3-55K and carrying the control anti-miR had a reduced SNX27 level, those transfected with the anti-miR-U3 had a normal SNX27 level (Figure 6D), demonstrating that the effect of U3-55K depletion on SNX27 is indeed mediated by miR-U3. Together, these data reveal crosstalk between the canonical function of the U3 snoRNA in ribosome assembly and its additional role as a miRNA source.
DISCUSSION
The wide-spread application of deep sequencing techniques has revealed the presence of numerous small RNAs and small RNA fragments in diverse cell types and tissues. While some of these transcripts likely represent degradation intermediates of longer RNAs, others have functional roles in regulating gene expression. Bioinformatic analyses of small RNA-seq datasets have previously predicted miRNA-like functions for sno/scaRNA-derived fragments (24), however, to date, the only characterized example of a sno/scaRNA that is processed to produce a functional miRNA is the H/ACA box scaRNA, ACA45 (20). Indeed, it has been suggested that sno/scaRNA- and tRNA-derived miRNA-like fragments represent ‘leakage’ from the normal snoRNA/tRNA biogenesis pathways and that proteins, such as the chaperone La, serve as gatekeepers to minimise the generation of such Argonaute-associated complexes (34,54).
Here, we demonstrate that the unique 5′ region of the box C/D snoRNA U3 acts as a miRNA source. For ribosome assembly, the U3 snoRNA is associated with the core box C/D snoRNP proteins fibrillarin, NOP56, NOP58 and 15.5K as well as the U3-specific protein U3–55K, which bind to the common C/C’ and D/D’ elements towards the 3′ end of the snoRNA (37), while the specialised 5′ end of the snoRNA basepairs with specific pre-rRNA sequences. Interestingly, binding of the core snoRNP proteins to the 3′ end of the snoRNA and/or association of the snoRNA with pre-ribosomes has been suggested to influence folding of the 5′ region such that in its non-protein/pre-ribosome-bound form, the 5′ end of U3 forms an extended hairpin structure, reminiscent of a pre-miRNA duplex (38). Consistent with this, we show association of Dicer and Argonaute with the 5′ region of the U3 snoRNA in vivo. Notably, the region of the U3 snoRNA that is processed to form miRNAs is distinct from the C/C’ and D/D’ elements that are common to other box C/D snoRNAs. Consistent with this, although small RNA-seq analysis revealed RNA fragments derived for other box C/D snoRNAs, these were either present at very low levels and/or exceeded the threshold length for miRNAs, further supporting the notion that processing of box C/D snoRNAs to miRNAs is likely not a wide-spread phenomenon.
Analogous to the ACA45-derived miRNAs (20), processing of U3 to produce miRNAs requires Dicer, but is independent of Drosha. This conclusion is supported by the enrichment of sequences corresponding to the U3-derived miRNA in crosslinking analyses of Dicer but not Drosha (Figure 2A). Notably, although sequences derived from this region of the snoRNA were also detected in CLIP data from the Drosha-associated protein DGCR8, structural analysis of a DGCR8-Drosha complex does not support this region of U3 as a miRNA substrate. As DGCR8 also functions as an adaptor for the exosome complex (31,55), it is likely that the crosslinking of DGCR8 to U3 rather reflects its role in snoRNA degradation. Based on the known association of the U3 snoRNA with the nuclear export factor XPO1 (44), it is probable that a portion of the full-length snoRNA is exported to the cytoplasm in an XPO1-dependent manner, where it is efficiently processed by Dicer to produce miRNAs. Although the association of both precursor and mature forms of the U3 snoRNA with XPO1 was demonstrated more than a decade ago (44), it has remained unclear whether the U3 snoRNA undergoes a cytoplasmic phase during its maturation. On the one hand, it is possible that, similar to other independently transcribed, m7G-capped small RNAs (U1, U2, U4 and U5 snRNAs, and U8 and U13 snoRNAs), XPO1-bound U3 is normally exported to the cytoplasm, where the extended 5′ stem–loop structure of a small fraction of the snoRNAs is recognised and cleaved by Dicer. Interestingly, it remains unknown whether the fraction of the U3 snoRNA exported to the cytoplasm is associated with the core box C/D snoRNP proteins. It may be that only aberrant U3 snoRNAs lacking associated proteins are processed by Dicer while protein-bound U3 snoRNPs are re-imported into the nucleus/nucleolus where they fulfil their function in ribosome assembly. Alternatively, it is also possible that the lack of pre-ribosome association can, in some cases, render the 5′ end of the U3 snoRNA sufficiently accessible for export and subsequent Dicer-mediated cleavage. On the other hand, it has been previously suggested that the binding of XPO1 to the U3 snoRNA contributes to nucleolar localisation of the snoRNA (47) and therefore, potentially, only a sub-population of U3 translocates to the cytoplasm where it is processed into miRNAs. This suggests an equilibrium between the proportions of the U3 snoRNA involved in ribosome assembly in the nucleolus and miRNA production in the cytoplasm.
The evolution of miRNA sequences embedded within other transcripts, e.g. pre-mRNA introns and sno/scaRNAs, and the discovery that miRNAs can be generated by non-canonical biogenesis pathways suggests additional layers of regulation of miRNA production as well as the potential for co-regulation of miRNA production with other cellular processes. The finding that the U3 snoRNA, which is one of only few essential snoRNAs and serves as a scaffold within early pre-ribosomal complexes, is a miRNA source highlights the potential for cross-regulation of target mRNAs with ribosome biogenesis or function. Indeed, our data demonstrates that depletion of U3-55K, which leads to a lower level of the U3 snoRNP and impairs the early steps of ribosome biogenesis (53,56), increases miR-U3 production leading to a decrease of its target SNX27 strongly support this model. Ribosome production has emerged as a central hub that co-ordinates protein production with cellular proliferation (reviewed in (57)) and it is tempting to speculate that the ribosome assembly pathway and miRNA-regulated expression of specific mRNAs is co-ordinated to induce particular cellular responses to certain conditions. Indeed, during serum starvation, when the requirement for protein synthesis and therefore ribosome production is reduced, miRNA biogenesis shifts towards the non-canonical XPO1-dependent transport (58), and in mice, the cytoplasmic fraction of the U3 snoRNA has been observed to increase (59), strongly supporting this model. Interestingly, upon exposure to hypertonic stress, U3–55K is hyperacetylated leading to decreased levels of the U3 snoRNA (60), and the level of the U3 snoRNA is reduced during differentiation and elevated during tumorigenesis (53,61–62). In the future, it will therefore be interesting to analyse miR-U3 levels and functions in these conditions to determine the contribution of miR-U3-regulated mRNA expression to the cellular stress response and cancer development.
Analysis of basepaired RNAs associated with AGO1 in human cells revealed the association of the miR-U3 with a conserved sequence in the 3′ UTR of the SNX27 mRNA. A decreased SNX27 level upon overexpression of a siRNA mimic of miR-U3 or an increase of endogenous miR-U3 induced by lack of U3-55K confirmed the SNX27 mRNA as an endogenous miRNA target. Interestingly, SNX27 is a member of the sortin nexin family of proteins, which plays a central role in recycling internalized transmembrane receptor proteins from endosomes to the plasma membrane (63). More specifically, SNX27 is highly expressed in brain tissue where it plays an important role in synaptic function by modulating glutamate receptor recycling and alterations in SNX27 levels have been observed in neurodegenerative disorders and Down's syndrome (64). SNX27 binds in excess of 400 PDZ domain-containing proteins involved in diverse cellular processes including signal transduction and metabolite transport (65). Given the broad interactome of SNX27, fine-tuning of SNX27 expression by the miR-U3 has the potential to influence diverse aspects of cellular function.
DATA AVAILABILITY
GEO database (http://www.ncbi.nlm.nih.gov/geo/): identifier GSE136745.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank A. Schuder and N. Kleiber for experimental assistance, A. Backhaus for administrative assistance, Anita Kriško for the XPO5 and SNX27 antibody, fruitful discussions and comments on the manuscript, and Dr S. Schneider, Prof. R. Lührmann and Dr H. Shcherbata for helpful discussions.
Notes
Present address: Jens Gruber, Destiny GmbH, Bosestraße 4, 04109 Leipzig, Germany.
Present address: Rafael Rinaldi Ferreira, University of Sao Paulo Ribeiro Preto Medical School, Avenida Bandeirantes, 3900 Riberão Preto, Sao Paulo Brazil.
Contributor Information
Nicolas Lemus-Diaz, Department of Molecular Biology, University Medical Center Göttingen, Humboldtallee 23, 37073 Göttingen, Germany; Junior Research Group Medical RNA Biology, German Primate Center, Leibniz Institute for Primate Research, Kellnerweg 4, 37077 Göttingen, Germany.
Rafael Rinaldi Ferreira, Junior Research Group Medical RNA Biology, German Primate Center, Leibniz Institute for Primate Research, Kellnerweg 4, 37077 Göttingen, Germany.
Katherine E Bohnsack, Department of Molecular Biology, University Medical Center Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Jens Gruber, Junior Research Group Medical RNA Biology, German Primate Center, Leibniz Institute for Primate Research, Kellnerweg 4, 37077 Göttingen, Germany.
Markus T Bohnsack, Department of Molecular Biology, University Medical Center Göttingen, Humboldtallee 23, 37073 Göttingen, Germany; Göttingen Center for Molecular Biosciences, Georg-August University, Göttingen, Justus-von-Liebig-Weg 11, 37077 Germany; Cluster of Excellence “Multiscale Bioimaging: from Molecular Machines to Networks of Excitable Cells” (MBExC).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CNPq [CNPq234389/2014-1 to R.R.]; Deutsche Forschungsgemeinschaft [SFB1190 to M.T.B., K.E.B.]; Göttingen Graduate School for Neuroscience, Biophysics, and Molecular Biosciences DFG Grant [GSC 226/2 to N.L.]; Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy [EXC 2067/1-390729940 to M.T.B.]. Funding for open access charge: Göttingen University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ha M., Kim V.N.. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014; 15:509–524. [DOI] [PubMed] [Google Scholar]
- 2. Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedman R.C., Farh K.K.-H., Burge C.B., Bartel D.P.. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Di Leva G., Garofalo M., Croce C.M.. MicroRNAs in cancer. Annu. Rev. Pathol. 2014; 9:287–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng Y., Croce C.M.. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016; 1:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paul P., Chakraborty A., Sarkar D., Langthasa M., Rahman M., Bari M., Singha R.S., Malakar A.K., Chakraborty S.. Interplay between miRNAs and human diseases. J. Cell. Physiol. 2018; 233:2007–2018. [DOI] [PubMed] [Google Scholar]
- 7. Denli A.M., Tops B.B.J., Plasterk R.H.A., Ketting R.F., Hannon G.J.. Processing of primary microRNAs by the microprocessor complex. Nature. 2004; 432:231–235. [DOI] [PubMed] [Google Scholar]
- 8. Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S. et al.. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003; 425:415–419. [DOI] [PubMed] [Google Scholar]
- 9. Bohnsack M.T., Czaplinski K., Gorlich D.. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004; 10:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lund E., Guttinger S., Calado A., Dahlberg J.E., Kutay U.. Nuclear export of microRNA precursors. Science. 2004; 303:95–98. [DOI] [PubMed] [Google Scholar]
- 11. Leisegang M.S., Martin R., Ramirez A.S., Bohnsack M.T.. Exportin t and Exportin 5: tRNA and miRNA biogenesis - and beyond. Biol. Chem. 2012; 393:599–604. [DOI] [PubMed] [Google Scholar]
- 12. Yi R., Qin Y., Macara I.G., Cullen B.R.. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003; 17:3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J.. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001; 409:363–366. [DOI] [PubMed] [Google Scholar]
- 14. Hutvagner G., McLachlan J., Pasquinelli A.E., Balint E., Tuschl T., Zamore P.D.. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001; 293:834–838. [DOI] [PubMed] [Google Scholar]
- 15. Zhang H., Kolb F.A., Jaskiewicz L., Westhof E., Filipowicz W.. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004; 118:57–68. [DOI] [PubMed] [Google Scholar]
- 16. Hammond S.M., Boettcher S., Caudy A.A., Kobayashi R., Hannon G.J.. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001; 293:1146–1150. [DOI] [PubMed] [Google Scholar]
- 17. Chendrimada T.P., Gregory R.I., Kumaraswamy E., Norman J., Cooch N., Nishikura K., Shiekhattar R.. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005; 436:740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R.. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008; 22:2773–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maute R.L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R.. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ender C., Krek A., Friedlander M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G.. A human snoRNA with microRNA-like functions. Mol. Cell. 2008; 32:519–528. [DOI] [PubMed] [Google Scholar]
- 21. Watkins N.J., Bohnsack M.T.. The box C/D and H/ACA snoRNPs: Key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA. 2012; 3:397–414. [DOI] [PubMed] [Google Scholar]
- 22. Bohnsack M.T., Sloan K.E.. Modifications in small nuclear RNAs and their roles in spliceosome assembly and function. Biol. Chem. 2018; 399:1265–1276. [DOI] [PubMed] [Google Scholar]
- 23. Scott M.S., Avolio F., Ono M., Lamond A.I., Barton G.J.. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput. Biol. 2009; 5:e1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brameier M., Herwig A., Reinhardt R., Walter L., Gruber J.. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic. Acids. Res. 2011; 39:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ono M., Scott M.S., Yamada K., Avolio F., Barton G.J., Lamond A.I.. Identification of human miRNA precursors that resemble box C/D snoRNAs. Nucleic. Acids. Res. 2011; 39:3879–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim Y.K., Kim B., Kim V.N.. Re-evaluation of the roles of DROSHA, Exportin 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E1881–E1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lemus-DIaz N., Böker K.O., Rodriguez-Polo I., Mitter M., Preis J., Arlt M., Gruber J.. Dissecting miRNA gene repression on single cell level with an advanced fluorescent reporter system. Sci. Rep. 2017; 7:45197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lemus-Diaz N., Tamon L., Gruber J.. Dual fluorescence reporter based analytical flow cytometry for miRNA induced regulation in mammalian cells. Bio-Protocol. 2018; 8:e3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mukherji S., Ebert M.S., Zheng G.X.Y., Tsang J.S., Sharp P.A., Van Oudenaarden A.. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011; 43:854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim B., Jeong K., Kim V.N.. Genome-wide mapping of DROSHA cleavage sites on primary MicroRNAs and noncanonical substrates. Mol. Cell. 2017; 66:258–269. [DOI] [PubMed] [Google Scholar]
- 31. Macias S., Plass M., Stajuda A., Michlewski G., Eyras E., Caceres J.F.. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 2012; 19:760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rybak-Wolf A., Jens M., Murakawa Y., Herzog M., Landthaler M., Rajewsky N.. A variety of dicer substrates in human and C. elegans. Cell. 2014; 159:1153–1167. [DOI] [PubMed] [Google Scholar]
- 33. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M.J., Jungkamp A.-C., Munschauer M. et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Flores O., Kennedy E.M., Skalsky R.L., Cullen B.R.. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic. Acids. Res. 2014; 42:4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kishore S., Gruber A.R., Jedlinski D.J., Syed A.P., Jorjani H., Zavolan M.. Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biol. 2013; 14:R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Granneman S., Kudla G., Petfalski E., Tollervey D.. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parker K.A., Steitz J.A.. Structural analysis of the human U3 ribonucleoprotein particle reveal a conserved sequence available for base pairing with pre-rRNA. Mol. Cell. Biol. 1987; 7:2899–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Granneman S., Vogelzangs J., Luhrmann R., van Venrooij W.J., Pruijn G.J.M., Watkins N.J.. Role of pre-rRNA base pairing and 80S complex formation in subnucleolar localization of the U3 snoRNP. Mol. Cell. Biol. 2004; 24:8600–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steinkraus B.R., Toegel M., Fulga T.A.. Tiny giants of gene regulation: experimental strategies for microRNA functional studies. Wiley Interdiscip. Rev. Dev. Biol. 2016; 5:311–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P.. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011; 18:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xie M., Li M., Vilborg A., Lee N., Shu M. Di, Yartseva V., Šestan N., Steitz J.A.. Mammalian 5′-capped microRNA precursors that generate a single microRNA. Cell. 2013; 155:1568–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Watkins N.J., Lemm I., Ingelfinger D., Schneider C., Hossbach M., Urlaub H., Luhrmann R.. Assembly and maturation of the U3 snoRNP in the nucleoplasm in a large dynamic multiprotein complex. Mol. Cell. 2004; 16:789–798. [DOI] [PubMed] [Google Scholar]
- 45. Watkins N.J., Lemm I., Luhrmann R.. Involvement of nuclear import and export factors in U8 box C/D snoRNP biogenesis. Mol. Cell. Biol. 2007; 27:7018–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fischer U., Englbrecht C., Chari A.. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. RNA. 2011; 2:718–731. [DOI] [PubMed] [Google Scholar]
- 47. Boulon S., Verheggen C., Jady B.E., Girard C., Pescia C., Paul C., Ospina J.K., Kiss T., Matera A.G., Bordonne R. et al.. PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol. Cell. 2004; 16:777–787. [DOI] [PubMed] [Google Scholar]
- 48. Bai B., Liu H., Laiho M.. Small RNA expression and deep sequencing analyses of the nucleolus reveal the presence of nucleolus-associated microRNAs. FEBS Open Bio. 2014; 4:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Travis A.J., Moody J., Helwak A., Tollervey D., Kudla G.. Hyb: a bioinformatics pipeline for the analysis of CLASH (crosslinking, ligation and sequencing of hybrids) data. Methods. 2014; 65:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agarwal V., Bell G.W., Nam J.-W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fromm B., Billipp T., Peck L.E., Johansen M., Tarver J.E., King B.L., Newcomb J.M., Sempere L.F., Flatmark K., Hovig E. et al.. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 2015; 49:213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Knox A.A., McKeegan K.S., Debieux C.M., Traynor A., Richardson H., Watkins N.J.. A weak C’ box renders U3 snoRNA levels dependent on hU3-55K binding. Mol. Cell. Biol. 2011; 31:2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hasler D., Lehmann G., Murakawa Y., Klironomos F., Jakob L., Grässer F.A., Rajewsky N., Landthaler M., Meister G.. The lupus autoantigen La prevents Mis-channeling of tRNA fragments into the human MicroRNA pathway. Mol. Cell. 2016; 63:110–124. [DOI] [PubMed] [Google Scholar]
- 55. Macias S., Cordiner R.A., Gautier P., Plass M., Cáceres J.F.. DGCR8 acts as an adaptor for the exosome complex to degrade double-stranded structured RNAs. Mol. Cell. 2015; 60:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sloan K.E., Bohnsack M.T., Schneider C., Watkins N.J.. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA. 2014; 20:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bohnsack K.E., Bohnsack M.T.. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019; 38:e100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Martinez I., Hayes K.E., Barr J.A., Harold A.D., Xie M., Bukhari S.I.A., Vasudevan S., Steitz J.A., DiMaio D.. An Exportin-1-dependent microRNA biogenesis pathway during human cell quiescence. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E4961–E4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sienna N., Larson D.E., Sells B.H.. Altered subcellular distribution of U3 snRNA in response to serum in mouse fibroblasts. Exp. Cell Res. 1996; 227:98–105. [DOI] [PubMed] [Google Scholar]
- 60. Chen S., Blank M.F., Iyer A., Huang B., Wang L., Grummt I., Voit R.. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat. Commun. 2016; 7:10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Langhendries J.-L., Nicolas E., Doumont G., Goldman S., Lafontaine D.L.J.. The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget. 2016; 7:59519–59534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Valleron W., Ysebaert L., Berquet L., Fataccioli V., Quelen C., Martin A., Parrens M., Lamant L., de Leval L., Gisselbrecht C. et al.. Small nucleolar RNA expression profiling identifies potential prognostic markers in peripheral T-cell lymphoma. Blood. 2012; 120:3997–4005. [DOI] [PubMed] [Google Scholar]
- 63. Worby C.A., Dixon J.E.. Sorting out the cellular functions of sorting nexins. Nat. Rev. Mol. Cell Biol. 2002; 3:919–931. [DOI] [PubMed] [Google Scholar]
- 64. Wang X., Zhao Y., Zhang X., Badie H., Zhou Y., Mu Y., Loo L.S., Cai L., Thompson R.C., Yang B. et al.. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat. Med. 2013; 19:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clairfeuille T., Mas C., Chan A.S.M., Yang Z., Tello-Lafoz M., Chandra M., Widagdo J., Kerr M.C., Paul B., Merida I. et al.. A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat. Struct. Mol. Biol. 2016; 23:921–932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GEO database (http://www.ncbi.nlm.nih.gov/geo/): identifier GSE136745.