

Abstract
In eukaryotic cells, proteins that associate with RNA regulate its activity to control cellular function. To fully illuminate the basis of RNA function, it is essential to identify such RNA-associated proteins, their mode of action on RNA, and their preferred RNA targets and binding sites. By analyzing catalogs of human RNA-associated proteins defined by ultraviolet light (UV)-dependent and -independent approaches, we classify these proteins into two major groups: (i) the widely recognized RNA binding proteins (RBPs), which bind RNA directly and UV-crosslink efficiently to RNA, and (ii) a new group of RBP-associated factors (RAFs), which bind RNA indirectly via RBPs and UV-crosslink poorly to RNA. As the UV crosslinking and immunoprecipitation followed by sequencing (CLIP-seq) approach will be unsuitable to identify binding sites of RAFs, we show that formaldehyde crosslinking stabilizes RAFs within ribonucleoproteins to allow for their immunoprecipitation under stringent conditions. Using an RBP (CASC3) and an RAF (RNPS1) within the exon junction complex (EJC) as examples, we show that formaldehyde crosslinking combined with RNA immunoprecipitation in tandem followed by sequencing (xRIPiT-seq) far exceeds CLIP-seq to identify binding sites of RNPS1. xRIPiT-seq reveals that RNPS1 occupancy is increased on exons immediately upstream of strong recursively spliced exons, which depend on the EJC for their inclusion.
Keywords: CLIP-seq, RNA binding proteins, exon junction complex, UV crosslinking, formaldehyde crosslinking, pre-mRNA splicing
INTRODUCTION
As cells grow, divide, and respond to their environment, they critically depend on RNA-associated proteins to regulate RNA biogenesis and function. RNA-associated proteins participate in all aspects of RNA biology—they assemble RNA into ribonucleoprotein (RNP) machines (e.g., spliceosome, ribosome), membraneless RNP organelles (e.g., nuclear speckles, stress granules), and gene/chromatin regulatory complexes (e.g., Polycomb repressive complex 2). In the case of messenger RNA (mRNA), RNA-associated proteins control its processing, subcellular location, intracellular transport, translation into proteins, and its eventual degradation (Müller-McNicoll and Neugebauer 2013; Singh et al. 2015). Therefore, to fully comprehend the intricate workings of cellular processes, it is important to identify RNA-associated proteins and elucidate their functions.
The post-genomic era has witnessed a revolution in our ability to catalog RNA-associated proteins encoded in the human and other genomes. Early efforts to build catalogs of RNA-associated proteins mainly relied on sequence similarity to well-known RNA binding domains (Anantharaman et al. 2002). More recent efforts have taken advantage of the ability of RNA and proteins in direct physical contact to form “zero-length” covalent bonds when exposed to shortwave ultraviolet (UV) light (Hockensmith et al. 1993). Such covalent crosslinking “freezes” dynamic intermolecular RNA:protein interactions as they occur in situ to enable their biochemical analysis. This property of direct RNA:protein contacts has been exploited to identify the protein interactome of polyadenylated RNA (Baltz et al. 2012; Castello et al. 2012; Hentze et al. 2018), and more recently to unveil the proteins that come in contact with all cellular RNA (Queiroz et al. 2019; Trendel et al. 2019; Urdaneta et al. 2019). These studies conservatively estimate that the human genome encodes more than 1200 proteins that directly contact RNA, and hence function as RNA binding proteins (RBPs). The UV reactivity of the RNA:protein contacts has also transformed our understanding of global-scale RNA cargoes of individual RBPs. UV crosslinking-immunoprecipitation (CLIP)-based methods allow purification of an RBP of interest and its crosslinked RNAs, which can then be identified via high-throughput sequencing (Lee and Ule 2018). Advantageously, the protein adducts on crosslinked RNA can be leveraged to map RNA positions directly in contact with RBPs to obtain a single-nucleotide resolution view of in vivo RNA:protein interactions.
While UV crosslinking-based approaches have illuminated many areas of RNA biology, like any other method, they too come with limitations. For example, the UV crosslinking ability of an RBP is likely to be influenced by sequence composition of RNA and protein at binding interfaces (Smith 1969; Hockensmith et al. 1986), and also by the strength and duration of interactions. Importantly, UV crosslinking is limited in applicability to proteins that are in direct contact with RNA (i.e., RBPs) and will be ill-suited to study proteins that interact with RNA indirectly via RBPs (RBP-associated factors, RAFs). Although we currently lack full understanding of the prevalence of RAFs, a closer examination of cellular RNPs reveals many proteins that play a critical role in RNA biology without directly contacting RNA (e.g., nuclear export factors GLE1 and NXT1; EIF4E binding proteins). RAFs can function along with RBPs either as their regulators or as subunits of multiprotein complexes that act on RNA, or both. It is therefore important to gain insights into the prevalence, properties, and functions of RAFs. Thus, UV crosslinking-independent methods are critical to investigate interactions of RAFs with RNA inside the cells.
Our perspective on RBPs and RAFs is shaped by our investigation of the exon junction complex (EJC), a multiprotein complex that mainly assembles ∼24 nt upstream of exon–exon junctions during pre-mRNA splicing (Boehm and Gehring 2016; Le Hir et al. 2016; Woodward et al. 2017). The EJC contains both RBPs (EIF4A3) and RAFs (MAGOH, RBM8A) within its core. The EJC core also interacts with several peripheral proteins that link it to various steps in post-transcriptional mRNA regulation. We recently showed that two peripheral proteins, RNPS1 and CASC3, interact with the EJC in a mutually exclusive and sequential manner (Mabin et al. 2018). To identify RNAs bound to EJC inside human cells, we have devised a UV crosslinking-independent tandem purification approach termed RNA IP in tandem (RIPiT) (Singh et al. 2012, 2014). To investigate binding sites of more transient and/or labile complexes, such as the EJCs containing alternate factor RNPS1, we have also combined RIPiT with formaldehyde-based chemical-crosslinking (Singh et al. 2014). We and others have also successfully applied RIPiT-seq with and without formaldehyde crosslinking to investigate binding profiles of RNA-associated proteins beyond the EJC, that is, Staufen1 (Ricci et al. 2014) and WDR5 (Yang et al. 2014).
Formaldehyde is a small, cell permeable, rapid, and reversible crosslinker that can covalently link proteins to nucleic acids and other proteins when they exist in close proximity (Hoffman et al. 2015). Thus, formaldehyde is an attractive alternative to UV to crosslink both RBPs and RAFs within cellular RNPs. The utility of formaldehyde crosslinking prior to RIP to enrich RBP-bound RNAs was first shown nearly two decades ago (Niranjanakumari et al. 2002). Ever since, formaldehyde crosslinking has been utilized to capture RNA cargoes of RNA-associated proteins from diverse eukaryotic systems (e.g., Huang and Hopper 2015; Hendrickson et al. 2016; Chatterjee et al. 2017). More recently, formaldehyde crosslinking has been combined with a CLIP-seq workflow to identify binding sites of DROSHA, a double-stranded RBP that poorly UV-crosslinks with RNA (Kim and Kim 2019). Despite such general use, several fundamental issues regarding formaldehyde crosslinking remain to be tested: its degree of influence on specificity of RIP signal, its performance in comparison to UV crosslinking, and its applicability to RBPs versus RAFs, to name a few.
Here we describe a comparative analysis of published catalogs of human RNA-associated proteins that were defined based on UV crosslinking ability of proteins to RNA or protein:protein interaction networks of annotated RBPs (a UV crosslinking-independent approach). This analysis enables us to categorize human RNA-associated proteins into RBPs and RAFs. We find that RAFs are prevalent in all steps of RNA metabolism and display a wide array of molecular functions and biochemical activities. This analysis also confirms the classification of EJC factors into RBPs (EIF4A3, CASC3) and RAFs (MAGOH, RNPS1). To investigate binding sites of EJC proteins, we have previously used RIPiT-seq either from uncrosslinked human cells (nRIPiT-seq) (Singh et al. 2012; Mabin et al. 2018) or from formaldehyde-crosslinked cells (xRIPiT-seq) (Mabin et al. 2018). Other groups have instead used CLIP-seq to map binding sites of RBPs and RAFs within EJCs (Saulière et al. 2012; Hauer et al. 2016). Here we use these existing nRIPiT-seq, xRIPiT-seq, and CLIP-seq data sets for CASC3 and RNPS1 as a case study to systematically evaluate and compare the efficacy of UV and formaldehyde crosslinking methods for enriching RBP/RAF binding sites. We show that formaldehyde crosslinking significantly improves RIPiT-seq signal for both CASC3 and RNPS1. Further, xRIPiT-seq is comparable to CLIP-seq for identifying CASC3 binding sites but is far superior to CLIP-seq for finding RNPS1 occupancy sites. Finally, RNPS1 binding to RNA measured via xRIPiT-seq led us to uncover increased RNPS1 occupancy on exons preceding recursively spliced exons, which were previously shown to depend on the EJC and RNPS1 for their inclusion in mRNA.
RESULTS
Proteins that poorly UV-crosslink to RNA are widespread in RNA metabolism
Within the EJC core, RBM8A and MAGOH bind RNA indirectly via EIF4A3 and do not UV-crosslink to RNA, in all likelihood due to their lack of direct contact with RNA (Andersen et al. 2006; Bono et al. 2006). We hypothesized that many more RAFs like RBM8A and MAGOH must exist that interact indirectly with RNA via RBPs to control RNA function. We therefore sought to systematically categorize RNA-associated proteins encoded in the human genome into RBPs and RAFs. Such a classification can be made by comparing a set of proteins that efficiently UV-crosslink to RNA to a set where RNA-associated proteins are defined without a requirement for UV crosslinking. For a set of proteins that efficiently UV-crosslink to RNA, we identified 830 proteins that are present in at least two of the seven poly(A) RNA interactome capture (RIC) data sets compiled by Hentze et al. (2018). For a set of proteins defined as RNA-associated proteins independent of their UV crosslinking ability, we combined two data sets reported by Brannan et al. (2016). The first subset is comprised of 1786 annotated human RBPs that were used as a training set for predicting RBPs via SONAR, a computational approach that analyzes large-scale affinity purification-mass spectrometry protein–protein interactomes of annotated RBPs to predict RNA binding activity (Brannan et al. 2016). The second subset consisted of 1923 proteins that were predicted as RBPs by SONAR based on RBP classification score >0.79 in Brannan et al. These two subsets were combined to obtain a set of 2784 unique proteins that we refer to as the human “annotated and predicted RNA-associated proteins” (Supplemental Table S1). The UV-crosslinkable poly(A) RIC RBP set shows almost complete (∼94%) overlap with the annotated and predicted RNA-associated proteins (Fig. 1A). However, only ∼30% of proteins among the annotated and predicted RNA-associated proteins represent the poly(A) UV-crosslinkable proteins. The remaining two-thirds of the RNA-associated proteins likely include proteins that do not efficiently UV-crosslink to RNA. Consistently, RBM8A and MAGOH are among this group. Further, the alternate EJC factor RNPS1 is also among the non-UV-crosslinkable RBPs. In contrast, EIF4A3, the RNA anchor of the EJC, and CASC3, the alternate EJC factor that directly contacts RNA, are detected in three out of seven RIC data sets. These observations validate our classification approach.
FIGURE 1.

RNA binding proteins that UV-crosslink poorly to RNA are widespread in RNA metabolism. (A) Venn diagram showing the overlap between RBPs defined based on UV crosslinking-dependent RNA interactome capture (RIC, from Hentze et al. 2018) and those defined by protein:protein interaction network of annotated RBPs (annotated and predicted RNA-associated proteins, from Brannan et al. 2016). (B) Venn diagram as in A showing overlap between integrated human RBPome defined based on UV-crosslinkability (ihRBPome, from Trendel et al. 2019) and annotated and predicted RNA-associated proteins (from Brannan et al. 2016) based on protein:protein interaction network of annotated RBPs. (C) Top sixteen gene ontology terms (biological process) enriched among RAFs. Legend on the right indicates the number of genes in each term. Some redundant GO terms were removed. (D) GO terms (cellular compartment) enriched among RAFs as in C. (E) GO terms (molecular function) enriched among RAFs as in C. (F) Major processes in the life-cycle of eukaryotic messenger RNAs (left) along with some examples of RAFs involved in each of the steps. Also shown are some examples of RAFs that are components of key membraneless RNP compartments or large RNP complexes (right).
Among the annotated and predicted RNA-associated proteins that do not overlap with RIC RBPs, it is likely that a subset may interact with non-poly(A) RNA and are thus not represented in the RIC data set. To test this idea, we compared the human annotated and predicted RNA-associated proteins to the integrated human RBPome (ihRBPome) defined by Trendel et al. (2019) based on proteins that UV-crosslink to all cellular RNA. In this comparison, indeed the number of UV-crosslinkable RBPs from the annotated and predicted RNA-associated proteins goes up to ∼40% (Fig. 1B). Still, a large group of the annotated and predicted RNA-associated proteins remain undetected among the UV-crosslinkable proteins, which are likely to contain RAFs. MAGOH and RNPS1 are again within this RAF category. Unexpectedly, RBM8A is detected in the ihRBPome along with EIF4A3 and CASC3 suggesting that it can bind RNA directly, perhaps in an EJC-independent fashion. Overall, this analysis suggests that a sizable fraction of the RNA-associated proteins encoded in the human genome are RAFs. Notably, the ihRBPome contains 638 proteins that are absent from the annotated and predicted RNA-associated proteins set (Fig. 1B), suggesting that no one approach is sufficient to define all RNA-associated proteins encoded in a genome.
The comparison of the annotated and predicted RNA-associated proteins to the ihRBPome provides a refined list of RAFs (Supplemental Table S1). A search for functionally related groups of genes among the RAFs revealed that these proteins function in diverse biological processes involving coding as well as noncoding RNAs (Fig. 1C). They are constituents of discrete RNP complexes (e.g., the spliceosome) and of the various nuclear and cytoplasmic phase-separated membraneless RNP organelles (e.g., nuclear speckles, Cajal body, chromatoid body, processing bodies) (Fig. 1D). RAFs within these processes and cellular compartments function as RNA modifying and degrading enzymes (Fig. 1E). In the case of mRNA metabolism, RAFs are key factors within all major nuclear mRNA processing steps (capping, pre-mRNA splicing, cleavage and polyadenylation), mRNA export into the cytoplasm as well as translation and mRNA degradation in the cytoplasm (some examples are listed in Fig. 1F). Among other notable RAFs are three of the four protein subunits of the polycomb repressive complex (EED, EZH1, and EZH2), whose chromatin modification function is guided by binding to several long noncoding RNAs (Margueron and Reinberg 2011). Conspicuously underrepresented are ribosomal proteins suggesting that most protein subunits of this RNP machine are RBPs. These observations indicate that the RAFs function within all major steps of RNA metabolism.
Chemical crosslinking stabilizes RBPs and RAFs within RNPs
The above analysis suggests that CLIP-seq–based approaches are ill-suited to identify RNA cargoes and binding sites of RAFs. Instead, UV crosslinking-independent RNA immunoprecipitation (RIP)-based approaches such as digestion-optimized RIP (DO-RIP-seq) (Nicholson et al. 2017) or RIPiT-seq (Singh et al. 2014) may be more suitable for this purpose. However, these RIP-based strategies lack the covalent protein attachment to RNA, which is the main advantage of CLIP. The absence of direct attachment between protein and RNA leads to two challenges in applying RIP to RAFs. First, due to the dynamic nature of molecular assemblies, RAFs may dissociate from their RNA:protein complexes and/or reassort during purification under native conditions (Mili and Steitz 2004). Second, native conditions during RIP, as compared to stringent conditions used for CLIP, can lead to copurification of nonspecific interactors. The former issue is highlighted in our attempts to increase EJC purification stringency during immunoprecipitation (IP) of stably expressed FLAG-tagged MAGOH from human embryonic kidney (HEK293) cells. As seen in Figure 2A, RNPS1 (and its associated factors) dissociate from the EJC core as the ionic strength of the IP reaction is increased (compare lanes 4–6 to lane 3). Thus, to use RIP to faithfully capture binding sites of RAFs such as RNPS1 with high specificity, it will be important to stabilize their in vivo interactions prior to cell lysis. To achieve this, we have systematically evaluated chemical crosslinking of RNPs using formaldehyde.
FIGURE 2.

Formaldehyde crosslinking stabilizes an RAF-EJC interaction. (A) Western blots showing proteins on the right in the total extract (TE) or FLAG immunoprecipitation (IP) fractions of HEK293 Flp-In cells expressing FLAG-MAGOH. The presence or absence of RNase A and the amount of NaCl present in the extracts during the IP are indicated above each lane. The dotted line indicates where gel images were spliced together. Data are representative of two biological replicates. (B) Western blots showing proteins (labeled on the right) in the insoluble pellet, soluble extract, and anti-FLAG immunoprecipitate fractions (indicated on the top) of HEK293 Flp-In cells. The lysis and IP conditions are also indicated on the top. Also indicated above each lane is the expression of FLAG-RNPS1 (+) or FLAG epitope only (−) in the cells, and the formaldehyde concentration used for in vivo crosslinking. Data are representative of three biological replicates.
A wide range of formaldehyde concentrations (0.1% to 3%) has been used in previous studies to crosslink macromolecular complexes (Fabre et al. 2013; Ricci et al. 2014; Chu et al. 2015). Excessive formaldehyde treatment can crosslink nonspecific interactions, and can also crosslink complexes to cellular structures thereby rendering them insoluble. We found that a 10-min treatment of HEK293 cells with 1% or more formaldehyde followed by sonication-mediated cell disruption under stringent lysis conditions (in the presence of 0.1% sodium dodecyl sulfate and 0.1% sodium deoxycholate) leads to extremely poor solubility of EJC proteins (data not shown). Therefore, we evaluated three formaldehyde concentrations below this threshold (0.03%, 0.1%, and 0.3%) to identify an optimal balance between RNP crosslinking and solubility. We find that crosslinking cells with 0.3% formaldehyde traps a significant fraction of the tested proteins in the insoluble fraction, which pellets along with cellular debris at 15,000g (Fig. 2B, lane 5). In comparison, cells treated with 0.1% formaldehyde show much better protein solubility (Fig. 2B, lane 4), which is comparable to protein solubility under native lysis conditions (Fig. 2B, lane 6). We also find that crosslinking cells with 0.1% formaldehyde sufficiently stabilizes interaction of FLAG-RNPS1 with the EJC core such that EIF4A3 and MAGOH co-IP with FLAG-RNPS1 even under stringent conditions (Fig. 2B, compare lanes 14 and 16). Thus, 0.1% formaldehyde sufficiently crosslinks FLAG-RNPS1 containing RNPs while maintaining their solubility.
Formaldehyde crosslinking enhances RIPIT-seq signal over background
While formaldehyde has long been utilized for RNP crosslinking during RIP, the quantitative influence of formaldehyde crosslinking on the efficiency and specificity of RIP signal remains yet to be evaluated. We therefore decided to test the effect of formaldehyde crosslinking on enrichment of RBP and RAF binding sites via RIPiT-seq. This two-step purification approach is an extension of RIP and involves sequential IP of a pair of proteins to enrich an RNP of a particular composition, whose RNA footprints can then be identified by high-throughput sequencing (Supplemental Fig. S1; Singh et al. 2012, 2014; Woodward et al. 2019). Previously, we used RIPiT-seq to obtain footprints of EJCs containing CASC3 and RNPS1 to describe the mutually exclusive nature of the two complexes (Mabin et al. 2018). These CASC3 and RNPS1 RIPiTs were performed from formaldehyde-crosslinked HEK293 cells under stringent conditions (xRIPiT-seq) as well as from noncrosslinked cells under native conditions (nRIPiT-seq), although in the previous work we exclusively focused on the xRIPiT-seq data to study the two mutually exclusive complexes. We remapped these data sets to the human genome (see Materials and Methods and Supplemental Table S2) to compare nRIPiT and xRIPiT side-by-side to assess the effect of formaldehyde crosslinking on enrichment of CASC3 and RNPS1 footprints. As previously reported, RIPiT-seq yields highly reproducible EJC binding across protein coding genes (Supplemental Fig. S2). Therefore, for all further analyses, we combined the biological replicates (where available, i.e., all xRIPiT-seq experiments) for each unique RIPiT-seq experiment.
First, we compared the ability of nRIPiT and xRIPiT to reveal the major binding site of CASC3, which was purified by sequential IP of FLAG-CASC3 and EIF4A3 under the two conditions. A meta-exon plot of CASC3 footprint densities at exon ends show that while both approaches yield highest read densities ∼24 nt upstream of exon junctions, xRIPiT shows a much more striking enrichment of reads at this position (Fig. 3A). Next, we quantified the ability of the two RIPiT formats to obtain CASC3-EJC footprints on two different “regions” of spliced protein-coding transcripts: canonical EJC sites (cEJC; −39 to −9 nt from exon ends) and noncanonical regions (ncEJC; exon start to −50 nt from exon ends). To estimate background binding of the EJC proteins to RNA, we also quantified CASC3 footprints on intronless protein-coding transcripts as the EJC is not expected to bind to intronless transcripts. To compare CASC3 footprint densities (RIPiT-seq reads per kilobase per million or RPKMRIPiT-seq) across the entire gene expression range, we divided spliced transcripts or intronless transcripts into twenty bins such that each bin contained transcripts that fall within twofold expression levels based on RNA-seq. Figure 3B shows that CASC3 footprint detection by either RIPiT-seq format is directly proportional to RNA expression levels. Further, as expected, at all expression levels, each RIPiT format shows highest signal on canonical sites, followed by noncanonical regions and lowest signal on intronless transcripts. Importantly, across the entire gene expression range, xRIPiT shows higher signal as compared to nRIPiT at canonical sites except the last expression bin where detection by the two RIPiT formats is comparable. In the case of noncanonical regions, differences in CASC3 detection via xRIPiT and nRIPiT are much smaller, and insignificant in the higher expression bins. We conclude that formaldehyde crosslinking boosts CASC3 footprint enrichment via RIPiT, possibly by freezing dynamic RNPs and preventing their dissociation after lysis.
FIGURE 3.
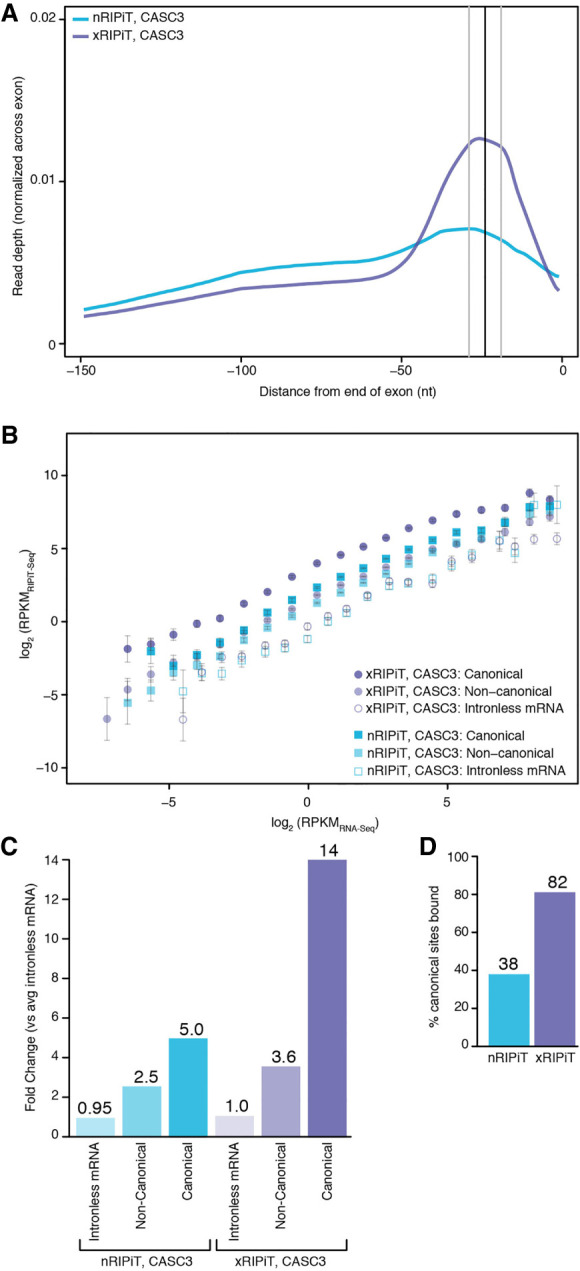
Formaldehyde crosslinking enhances RIPiT-seq signal for CASC3. (A) A meta-exon plot showing nRIPiT and xRIPiT read densities in the 150 nt window from the end of exons of protein-coding genes (excluding final exons). (B) Comparison of gene-level CASC3 read density (RPKMRIPiT-seq) in native RIPiT (nRIPiT, squares) and formaldehyde-crosslinked RIPiT (xRIPiT; circles) for canonical (darker-shaded shapes) and noncanonical regions (lighter-shaded shapes), and for intronless genes (empty shapes). Along the x-axis, genes are binned into twenty bins where each bin contains exons from genes within a twofold expression level range based on RNA-seq. Error bars represent the standard error of the mean signal in each bin. (C) A comparison of linear fit coefficients (or intercepts, in log space) of the six classes in B. Classes are labeled on the bottom. The coefficient for the average of the two intronless classes was set to 1 and all intercepts were adjusted accordingly. The fold-change as compared to the average of the two intronless classes is shown above each bar. (D) Percentage of all canonical EJC regions where read count is greater than or equal to twofold as compared to read counts on intronless genes of similar expression level.
To quantitatively compare CASC3 binding site enrichment via native and crosslinked RIPiTs, we obtained a single summary statistic that represents overall CASC3 binding in each of the canonical regions, noncanonical regions and intronless transcripts. Assuming a direct relationship between RIPiT enrichment (RIPiT-seq) and RNA expression level (RNA-seq), we fitted a linear trendline with slope equal to one to each binned-data distribution in Figure 3B (see Supplemental Fig. S3). The y-intercept of each fit in the log space corresponds to its slope in the untransformed space. That is, the y-intercept of each trendline reflects in a relative sense how much a given RNA region is detected in CASC3 footprints relative to RNA-seq. To normalize the signal within native and crosslinked RIPiTs to CASC3 binding to intronless transcripts, the average signal for intronless transcripts in the two RIPiT conditions was set to one, and all intercepts were shifted accordingly. The fit coefficients thus computed when compared within the nRIPiT-seq data set show that, as compared to intronless transcripts, CASC3 footprints are enriched approximately fivefold in canonical and ∼2.5-fold in noncanonical regions (Fig. 3C). CASC3 enrichment is much more pronounced within xRIPiT-seq data sets, which shows 14-fold and ∼3.6-fold enrichment of CASC3 footprints in canonical and noncanonical regions, respectively (Fig. 3C). Thus, in comparison to uncrosslinked nRIPiT, formaldehyde crosslinking during xRIPiT leads to a nearly threefold enhancement of CASC3 footprint signal at cEJC sites (Fig. 3C). This overall increase in CASC3 enrichment via xRIPiT over nRIPiT is likely due to greater detection at individual binding sites. To examine this idea, we limited our analysis to highly expressed genes (top 20% expression corresponding to >5.9 RPKM). We find that a greater fraction of individual canonical regions (Supplemental Fig. S4A) as well as genes (Supplemental Fig. S4B) show higher CASC3-EJC read densities in xRIPiT as compared to nRIPiT, as shown by the rightward shift of the scatter plots.
We predict that enhanced CASC3 RNA binding site enrichment observed in xRIPiT over nRIPiT also leads to significant enrichment of CASC3 footprints on a greater number of cEJC sites. To test this idea, we identified the canonical regions on which CASC3 footprint signal is greater than or equal to twofold as compared to the signal on intronless transcripts at similar expression level. As seen in Figure 3D, we observe an increase in significantly enriched cEJC sites from 38% in nRIPiT to 82% in xRIPiT. The increase in cEJC sites significantly bound by CASC3 in xRIPiT over nRIPiT is observed over almost the entire expression range sampled (Supplemental Fig. 4C), suggesting that xRIPiT enhances CASC3 detection irrespective of gene expression level. Therefore, we conclude that formaldehyde crosslinking of cells before performing RIPiT-seq enhances the capture of in situ CASC3-EJCs to yield higher signal over background at canonical and noncanonical sites. Such an effect consequently also leads to more robust detection of CASC3 at a greater proportion of the expected binding sites.
We have previously shown that translation inhibition impacts CASC3-EJC occupancy (Mabin et al. 2018). We argued that translation inhibition, when combined with formaldehyde crosslinking, will lead to further preservation of CASC3-containing EJCs on their in vivo binding sites. To test this prediction, we compared CASC3 enrichment via xRIPiT-seq with and without pretreatment with translation elongation inhibitor, cycloheximide (CHX). As expected, CHX treatment leads to ∼2.7-fold increase in CASC3 detection on cEJC sites (Supplemental Fig. S4D,E). This CHX-dependent enhanced CASC3 enrichment leads to detection of significant CASC3 footprints at 90% of all possible cEJC binding sites as compared to detection on 82% of sites under normal translation conditions (Supplemental Fig. S4F). These data show that translation impacts CASC3-EJC occupancy, and further highlights the quantitative ability of our approach to compare protein binding site enrichment.
xRIPIT-seq is comparable to CLIP-seq in revealing binding sites of CASC3
CASC3 is an RBP that can be photo-crosslinked to RNA. To compare the ability of chemical and photo-crosslinking methods to enrich CASC3 binding sites, we compared CASC3 xRIPiT-seq with the CASC3 CLIP-seq data set of Hauer et al. (2016). This CASC3 binding profile was obtained using the individual nucleotide resolution CLIP-seq variation (Huppertz et al. 2014), which we refer to simply as CLIP-seq. The raw CLIP-seq and the corresponding RNA-seq data sets were aligned to the human reference genome using the same parameters as the xRIPiT-seq data sets. The CLIP-seq data are from HeLa cells and xRIPiT-seq from HEK293 cells. To minimize the effect of gene expression differences in the two cell lines on our analysis, we limited the analysis to only the subset of protein-coding genes whose expression levels are within 1.5-fold (based on RNA-seq RPKM) in the two cell lines (Supplemental Fig. S5). Importantly, both data sets compared were obtained from cycloheximide treated cells. Similar to the findings in Figure 3, both CLIP-seq and xRIPiT-seq for CASC3 strongly enrich canonical EJC binding sites (Fig. 4A). Further, across the gene expression bins, both approaches detect the highest CASC3 binding at canonical sites followed by noncanonical sites and then by intronless transcripts (Fig. 4B). Some deviation from this trend is observed in the four highest expression bins where both CLIP and xRIPiT signal is more variable in the three regions. Still, even in these bins, the highest CASC3 signal is detected at the canonical sites. When xRIPiT-seq is compared to CLIP-seq, both methods detect similar CASC3 binding throughout the gene expression range (Fig. 4B). The few exceptions are noted in the medium expression range where xRIPiT-seq signal is significantly higher than CLIP-seq in both canonical as well as noncanonical regions. When intercepts from the linear fits for the six classes are compared, the two methods show a robust detection of CASC3 binding at canonical regions (30-fold enrichment in CLIP-seq and 56-fold enrichment in xRIPiT-seq over intronless mRNA signal; Fig. 4C). Lower but appreciable CASC3 binding is detected in the noncanonical regions (Fig. 4C). Notably, xRIPiT shows nearly twofold higher signal as compared to CLIP-seq in both canonical and noncanonical regions (Fig. 4C). This increased detection of CASC3 binding by xRIPiT over CLIP is also evident at a larger fraction of individual canonical sites (Supplemental Fig. S4G) and genes (Supplemental Fig. S4H). The robust detection of CASC3 binding by both xRIPiT and CLIP leads to greater than or equal to twofold enrichment of CASC3 signal on a slightly larger number of canonical sites as compared to background detection on intronless transcripts (84% of all cEJC sites are enriched in CLIP as compared to 90% with xRIPiT; Fig. 4D; see also Supplemental Fig. S4C). Finally, the majority of the sites enriched by xRIPiT and CLIP are shared between the two approaches, and also with nRIPiT (Fig. 4E). Overall, we conclude that xRIPiT-seq is comparable to, or even slightly more efficient than, CLIP-seq to uncover binding sites of an RBP such as CASC3.
FIGURE 4.
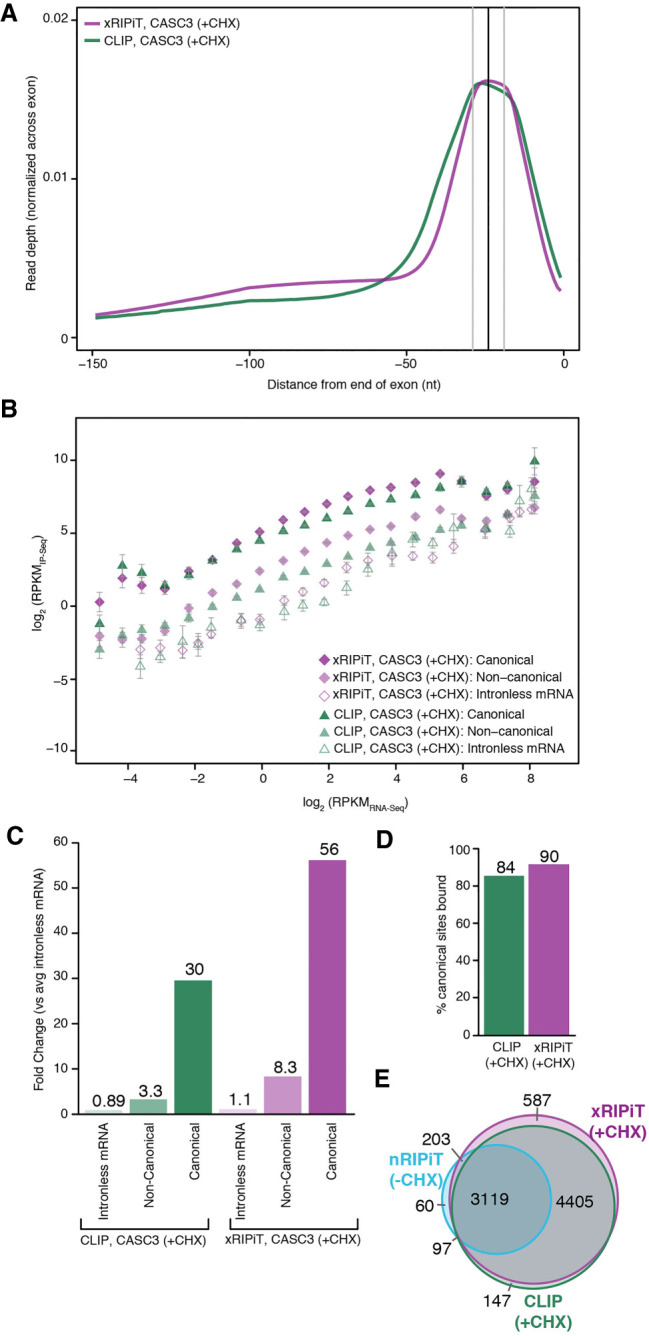
xRIPiT-seq and CLIP-seq are robust and comparable approaches to identify CASC3 binding sites. (A) A meta-exon plot showing xRIPiT and CLIP read counts in the 150 nt window at exon ends. Read normalization was carried out as in Figure 3A. (B) Comparison of gene-level CASC3 read density (RPKMIP-seq) in xRIPiT (diamonds) and CLIP (triangles) for canonical (darker-shaded shapes) and noncanonical regions (lighter-shaded shapes), and for intronless genes (empty shapes). Gene binning and error bars are as in Figure 3B. (C) Comparison of the linear fit coefficients (or intercepts, in log space) of the six classes in B. Classes are labeled on the bottom. (D) Percentage of all canonical EJC regions where read depth is greater than or equal to twofold as compared to intronless gene read counts in the indicated data sets. (E) Venn diagram showing counts of canonical regions from the top 20% expressed genes where CASC3 footprint read depth in nRIPiT, xRIPiT and CLIP is greater than or equal to twofold as compared to intronless gene read counts.
xRIPiT-seq is superior to nRIPiT-seq and CLIP-seq for identifying RNPS1-EJC binding sites
We next compared the ability of the three different approaches (nRIPiT-seq, xRIPiT-seq, and CLIP-seq) to enrich binding sites of RNPS1, an RAF within the EJC. A direct comparison of nRIPiT and xRIPiT shows that, as in the case of CASC3, formaldehyde crosslinking dramatically improves identification of the major RNPS1 binding site, which corresponds to the canonical EJC position (Fig. 5A). Further, at low and medium expression levels xRIPiT yields better enrichment of RNPS1 binding sites than nRIPiT both in canonical as well as noncanonical regions (Fig. 5B,C) although among the five highest expression bins, xRIPiT efficacy drops to the level of nRIPiT (Fig. 5B). These trends are observed at both canonical and noncanonical positions. Still, xRIPiT leads to increased detection of RNPS1 over nRIPiT across individual canonical sites (Supplemental Fig. S6A) and also at the individual gene level (Supplemental Fig. S6B). Furthermore, within the top 20% most expressed genes, xRIPiT also boosts RNPS1 detection at a greater percentage of canonical positions (92%) as compared to nRIPiT (47%) (Fig. 5D). In this group of genes, xRIPiT detects increased RNPS1 binding sites in each of the ten expression bins (Supplemental Fig. S6C). Thus, as compared to nRIPiT, xRIPiT leads to an overall enhanced capture of RNPS1-EJC bound to its target sites.
FIGURE 5.

xRIPiT-seq outperforms nRIPiT-seq and CLIP-seq to detect RNPS1 binding sites. (A) A meta-exon plot showing RNPS1 nRIPiT and xRIPiT footprint read counts at each position in the 150 nt window from exon ends. (B) Comparison of gene-level RNPS1 read density (RPKM) in nRIPiT (squares) and xRIPiT (circles) for canonical (darker-shaded shapes) and noncanonical regions (lighter-shaded shapes), and for intronless genes (empty shapes). Gene bins along the x-axis and error bars are as in Figure 3B. (C) Comparison of linear fit coefficients (or intercepts, in log space) of the six classes in B, which are labeled on the bottom. (D) Percentage of all canonical EJC regions where read depth is greater than or equal to twofold as compared to intronless read counts in the indicated data sets. (E) A meta-exon plot showing RNPS1 xRIPiT and CLIP footprint read counts at each position in the 150 nt window from exon ends. (F) Comparison of normalized read density (RPKM) in xRIPiT (diamonds) and CLIP (triangles) for canonical (darker-shaded shapes) and noncanonical regions (lighter-shaded shapes), and for intronless genes (empty shapes). The bins of genes along the x-axis and the error bars are as in Figure 3B. (G) Comparison of linear fit coefficients (or intercepts, in log space) of the six data sets in F. (H) Percentage of all canonical EJC regions where read depth is greater than or equal to twofold as compared to intronless gene read counts. (I) Venn diagram showing counts of canonical regions from the top 20% expressed genes where RNPS1 footprint read depth in nRIPiT, xRIPiT, and CLIP is greater than or equal to twofold as compared to intronless gene read counts.
We next compared the detection of RNPS1 binding on RNA by CLIP and xRIPiT. The RNPS1 CLIP-seq data from Hauer et al. (2016) were mapped and compared to xRIPiT-seq data as in the case of CASC3 above. Importantly, xRIPiT reveals a clear preference for RNPS1 binding to canonical EJC sites near exon 3′ ends whereas RNPS1 CLIP-seq reads show only a modest enrichment at this site (Fig. 5E). RNPS1 xRIPiT signal in canonical and noncanonical regions on spliced transcripts is consistently, and in many expression bins, significantly higher than RNPS1 CLIP signal (Fig. 5F). Further, in xRIPiT, the RNPS1 binding observed on canonical and noncanonical regions is significantly higher than its binding to intronless mRNAs with the exception of the top four bins (Fig. 5F). In comparison, RNPS1 CLIP-seq signal on canonical and noncanonical regions of spliced transcripts is indistinguishable from signal on intronless mRNAs, except for some of the medium expression bins (Fig. 5F). Comparisons of coefficients of linear fits show that detection of RNPS1 binding is much higher in xRIPiT as compared to CLIP at both canonical sites (approximately sevenfold higher) as well as noncanonical positions (∼4.5-fold higher, Fig. 5G). The superiority of xRIPiT over CLIP for RNPS1 binding detection is evident at individual canonical sites (Supplemental Fig. S6D) and at individual genes (Supplemental Fig. S6E). Consequently, xRIPiT detects RNPS1 binding at more than ten times the number of canonical EJC positions as compared to CLIP (cEJC sites detected among the top 20% of all expressed genes: xRIPiT—82%, CLIP—7%; Fig. 5H). It is noteworthy that even nRIPiT detects RNPS1 binding on a greater number of canonical positions when compared to CLIP (Fig. 5I, also compare Fig. 5D,H). As expected based on our previous work (Mabin et al. 2018), unlike CASC3 occupancy, RNPS1 occupancy does not show an increase when formaldehyde crosslinking is combined with cycloheximide treatment (Supplemental Fig. S6F–H). In fact, RNPS1 binding slightly decreases at both canonical and noncanonical sites upon translation inhibition as compared to normal conditions (Supplemental Fig. S6G,H). Overall, we conclude that RIPiT in general, and xRIPiT in particular, is a much more suitable and sensitive approach than CLIP to identify RNAs and specific sites bound by RAFs such as RNPS1.
xRIPiT-seq is more efficient than CLIP-seq to detect increased RNPS1 occupancy on exons preceding recursively spliced exons
The end goal of approaches that map RBP/RAF binding sites is to obtain insights into the functions of these proteins. We wanted to determine if increased detection of RNPS1 binding to EJC sites via xRIPiT-seq can shed light on the biological roles of RNPS1 and the EJC. Blazquez et al. (2018) recently showed that the presence of an EJC on an exon–exon junction inhibits recursive splicing (RS) of the downstream exon when this exon begins with a sequence resembling a 5′-splice-site (Fig. 6A). Although RNPS1 and EJC core proteins were shown to be critical for repression of 5′-splice-site usage at RS exon junctions, it remains unknown if such splicing-regulatory activity of RNPS1 is specifically dependent on its increased binding on exons preceding RS exons. To test this idea, we identified 1912 exons in the human transcriptome with RS scores higher than the threshold defined by Blazquez et al. and compared RNPS1 and CASC3 binding signal from xRIPiT-seq and CLIP-seq on these RS exons and up to three preceding exons (Fig. 6A). As a control, we similarly compared RNPS1 and CASC3 binding around an equal number of exons with the lowest RS scores (non-RS exons). Regardless of the experimental method, we detected increased binding of CASC3 on the upstream exon of RS junctions as well as one exon further upstream as compared to similar exons upstream of non-RS junctions (Fig. 6B). This increased CASC3 occupancy, which likely signifies increased EJC core deposition, is less prominent on the exon further upstream and on the RS exon itself. Consistent with the results in Figure 5, we detected greater RNPS1-EJC signal using xRIPiT-seq as compared to CLIP-seq on all exons regardless of their position relative to the RS exon (Fig. 6C). Importantly, xRIPiT-seq finds significantly higher RNPS1-EJC binding on all exons preceding RS junctions compared to those preceding non-RS junctions with the most significantly increased RNPS1 binding observed on the exon upstream of the RS junction. In comparison, CLIP-seq finds a smaller and less significant increase in RNPS1 binding on this exon when it precedes an RS versus a non-RS junction, and no significant difference in RNPS1 binding on other exons. This increase in RNPS1 binding on exons upstream of RS junctions is also evident at individual genes containing RS exons where EJC deposition was previously shown by Blazquez et al. to suppress RS (Fig. 6D). Finally, we also observed that entire transcripts that contain high-scoring RS junctions show increased RNPS1 and CASC3 binding as compared to transcripts that contain only low-scoring RS junctions (Fig. 6E). Again, as compared to CLIP-seq, xRIPiT-seq detected much higher and more significantly increased RNPS1 binding to transcripts containing RS junctions whereas both CLIP-seq and xRIPiT-seq performed similarly in the case of CASC3. These results further highlight xRIPiT-seq's ability to uncover biologically relevant features of RAF binding as compared to CLIP-seq. We also conclude that EJC and RNPS1 deposition on upstream junctions may play a role in suppressing downstream recursive exon splicing, possibly by stabilizing EJC and/or RNPS1 binding on downstream RS junctions (Fig. 6A).
FIGURE 6.

Comparison of xRIPiT-seq and CLIP-seq signal for RNPS1 and CASC3 occupancy on high- and low-scoring RS exons and their neighboring exons. (A) A schematic of recursively spliced (RS) exon and its neighboring exons. Empty rectangles: constitutive exons; shaded rectangle: RS exon; black line: intron; dotted lines: possible exon splicing patterns; shaded ovals: RNPS1-EJC; RNPS1-EJCs upstream of RS junction suppresses RS, whereas the complex on one exon further upstream (shown on shaded background) stabilizes the downstream complex. The number below each exon represents the number of exons for which data is presented in panels B and C. (B) Box plots showing CASC3 xRIPiT-seq and CLIP-seq read densities on high-scoring versus low-scoring RS exon and its three preceding exons. Each set of four boxplots is arranged directly below the RS exon or the one of its three preceding exons it corresponds to in A. (Top) Wilcoxon rank-sum test P-values. (C) Box plots as in B showing RNPS1 xRIPiT-seq and CLIP-seq exonic read densities. (D) Integrated genome viewer tracks showing read coverage (normalized for library size) on TMA16’s RS exon and its three preceding exons. (E) Box plots showing RNPS1 and CASC3 xRIPiT-seq and CLIP-seq genic read densities on genes that contain a high-scoring RS exon (n = 5001) and those containing only low-scoring RS exons (n = 5001). (Top) Wilcoxon rank-sum test P-values.
DISCUSSION
In the quest to define functions of RNA-associated proteins, their different modes of interactions with RNA necessitate orthogonal approaches to “freeze” RNA-associated proteins in action on cellular RNAs. Here we use the EJC as a test case to show that while UV crosslinking works well to capture RNA interaction sites of proteins that bind RNA directly (CASC3, an RBP), chemical crosslinking using formaldehyde is a far superior option for identifying binding sites of proteins that act on RNA via other proteins (RNPS1, an RAF). Our results suggest that xRIPiT-seq, or similar RIP-seq approaches from formaldehyde-crosslinked cells, can be generally applicable to all RNA-associated proteins, in particular to RAFs, to interrogate their cellular sites of action (Fig. 7).
FIGURE 7.

A schematic summarizing suitable approaches for identification of binding sites of RBPs versus RAFs. RNA (dark line) is shown bound by RBPs (lower two ovals) and an RAF (upper oval). Methods suitable for binding site enrichment of the two classes of RNA-associated proteins are on the right.
xRIPiT-seq reveals binding profile and functions of RNPS1, an RAF within EJC
We have previously developed and applied RIPiT-seq to investigate functions of EJC factors in post-transcriptional gene regulation (Singh et al. 2012, 2014). A key advantage of RIPiT is the two-step purification, which enables enrichment of an RNP containing a pair of proteins. The tandem purification strategy boosts signal specificity by reducing enrichment of nonspecific RNA interactors. Combination of formaldehyde crosslinking with RIPiT-seq, that is, xRIPiT-seq (Mabin et al. 2018), further enhances several-fold the specificity of capturing binding sites of RNA-associated proteins (Figs. 3, 5). Essentially, xRIPiT is an extension of well-tested formaldehyde-crosslinked RIP approaches described in the past (Niranjanakumari et al. 2002), and is analogous to the fCLIP-seq approach described recently (Kim and Kim 2019), with the added advantage of tandem purification. Formaldehyde crosslinking, being agnostic to direct versus indirect binding of RNA-associated proteins to RNAs, makes xRIPiT-seq more broadly applicable than CLIP-seq approaches. Consistent with this, as compared to CLIP-seq, xRIPiT-seq yields robust signal for both CASC3 and RNPS1 binding to EJC sites (Figs. 4, 5).
The enhanced detection of RNA binding sites via xRIPiT-seq is particularly striking in the case of RNPS1 (Fig. 5E–I), providing new insights into its binding and functions within the EJC. The poor UV crosslinking of RNPS1 to canonical EJC sites and yet its strong enrichment at these positions during xRIPiT suggests that RNPS1 binds to EJC mainly via protein:protein interactions. Such a view is consistent with the current understanding of RNPS1 interaction with the EJC core via other peripheral EJC factors such as ACIN1 and/or PNN (Boehm et al. 2018; Wang et al. 2018). Importantly, as compared to the CASC3-EJC binding sites, the RNPS1-EJC occupancy sites revealed by xRIPiT are enriched in degenerate sequences that resemble SR-protein binding motifs, for example, GA-rich sequences (Mabin et al. 2018). Notably, numerous SR and SR-like proteins copurify with RNPS1-EJC but not with CASC3-EJC (Mabin et al. 2018). Thus, while RNPS1 does not contact RNA on its own, xRIPiT-seq faithfully captures RNA sites that are associated with RNPS1-EJCs. xRIPiT-seq also reveals that the RNPS1-EJC has an increased abundance upstream of exons that depend on RNPS1 for their splicing (Fig. 6). This increased RNPS1 occupancy on neighboring exons could be important for its splicing-regulatory function on RS exons (Blazquez et al. 2018). Possibly, akin to its function in promoting splicing of neighboring introns in Drosophila piwi transcripts (Malone et al. 2014), RNPS1 binding on neighboring exon-junctions can promote increased EJC deposition on RS exon junction thereby suppressing RS exon splicing (Fig. 6A).
In addition to illuminating EJC functions, our work has broader implications for investigating RNA–protein interactions using chemical versus photo-crosslinking approaches. Therefore, it is important to consider factors that can influence UV- and formaldehyde crosslinking abilities of RNA-associated proteins.
Factors that impact UV crosslinking ability of RNA-associated proteins
Several factors can negatively impact a protein's ability to efficiently crosslink to RNA with UV light, resulting in their classification as RAFs (Fig. 1). The most obvious factor is the indirect RNA binding mode of proteins where they act on RNA from a distance through RBPs. Such RAFs can function as regulators of RBPs as in the case of RBM8A and MAGOH, which interact exclusively with the RNA-clamped form of EIF4A3 (Andersen et al. 2006; Bono et al. 2006). Other RAFs can serve as adapters to connect an RBP and its bound RNA to other cellular machineries (e.g., mRNA export factors NXT1, GLE1). RAFs can also serve as components of multisubunit assemblies where they may take a structural or regulatory role (e.g., EIF3 subunits—3F, 3I, 3K; nuclear exosome noncatalytic subunits—EXOSC5, 7, 8). For such proteins, which are physically away from RNA while in action, only chemical crosslinking methods can immobilize them in their native in vivo complexes.
The chemistry at the RNA:protein interaction interface also influences UV crosslinking of proteins to RNA. Among the nucleobases in single-stranded DNA polymers, polypurine oligomers are much less reactive than polypyrimidine oligomers for protein photo-crosslinking (Hockensmith et al. 1986). Among the amino acids, most robust UV induced crosslinking to uracil is observed for amino acids with aromatic side-chains that can engage in stacking interactions with nucleobases (F and Y), and amino acids with positively charged side-chains that can form electrostatic interactions with the negatively charged phosphate backbone (K, R, and H) (Smith 1969). Confirming these biases, in UV crosslinked RBP:RNA complexes identified by XRNAX, uracil is the most frequently crosslinked base, and phenylalanine, lysine and glycine are the top three amino acids in the uracil-crosslinked peptides (Trendel et al. 2019). EIF4A3 is an example of an RBP that inefficiently UV-crosslinks to RNA possibly due to the chemical nature of the RNA:protein interface. EIF4A3 binds RNA by exclusively contacting the ribose-phosphate backbone and lacks specific interactions with bases (Andersen et al. 2006; Bono et al. 2006). We have previously shown in human cells that this protein UV-crosslinks to RNA very inefficiently as compared to a sequence-specific RBP HNRNPA1 (Singh et al. 2014). Recently, this poor in vivo UV crosslinking ability of EIF4A3 was also reported in Drosophila adult animals (Obrdlik et al. 2019). Instead, chemical crosslinking using dithio(bis-) succinimidyl propionate was found to stabilize EIF4A3 within EJCs for identification of Drosophila EJC binding sites. Many other DEAD-box proteins bind RNA in a sequence-independent manner similar to EIF4A3, and some of these RBPs may lack readily UV-crosslinkable functional groups at RNA:RBP interfaces. Despite being “true” RBPs, such proteins are more likely to be classified as RAFs and can benefit from xRIPiT-seq over CLIP-seq.
Surprisingly, many proteins that interact with the RNA 7-methyl-guanosine cap are classified among RAFs, for example, nuclear cap binding protein NCBP2, all three cytoplasmic cap binding proteins EIF4E1, EIF4E2, and EIF4E3, and decapping proteins DCP2, NUDT16, and DCPS. Notably, these proteins interact with the cap via stacking interactions between aromatic amino acid side chains and the methylated guanosine base of the cap (Marcotrigiano et al. 1997). Despite such direct and specific contacts between these proteins and the RNA cap structure, they are apparently poorly UV-crosslinked to RNA in vivo. A previous report that chemical crosslinking is superior to UV crosslinking to detect EIF4E-cap interactions (Kahvejian et al. 2005) further suggests that the arrangement of functional groups at the RBP:RNA interface of these proteins could be suboptimal for UV crosslinking. RAFs also include many RNA decay enzymes: 19 different RNases (e.g., RNASE1, RNASE2), cytoplasmic RNA exosome catalytic subunit DIS3L, polyuridylated RNA specific 3′–5′ exonuclease DIS3L2, and major cytoplasmic deadenylases PAN2 and PAN3. While these proteins likely act on RNA directly, their engagement with RNA in vivo is likely not amenable to efficient UV crosslinking, possibly due to their mode of interaction with RNA and also due to the transient nature of their interactions. The latter view is supported by the observation that a much stronger PAR-CLIP signal is obtained when nucleolytic activity of DIS3 is mutated (Szczepińska et al. 2015), which possibly traps the protein on RNA for more efficient photo-crosslinking. Overall, the above examples highlight several conditions where UV crosslinking can prove ineffective, and chemical crosslinking with formaldehyde (or other crosslinkers) is a more viable approach to trap RBPs/RAFs in action.
Formaldehyde as an alternative to UV for in vivo RBP/RAF crosslinking
Formaldehyde is a bifunctional, electrophilic molecule that reacts with two nucleophilic groups in sequential steps to link the two groups via a methylene bridge (Hoffman et al. 2015). In the first step, a nucleophilic group, such as an amino or imino group from a protein or nucleic acid, reacts with formaldehyde to form a Schiff's base. In the second step, the Schiff's base reacts with a second nucleophilic group resulting in a methylene bridge. If the two attacking nucleophilic groups are on two different molecules, their reaction with formaldehyde forms an intermolecular crosslink. Due to the small size of formaldehyde, the two groups to be crosslinked must be no more than ∼2 Å apart, thus making it suitable to study molecules that are in close proximity. Formaldehyde-mediated protein:protein crosslinking stabilizes macromolecular complexes in vivo (for review, see Sutherland et al. 2008). Mild formaldehyde crosslinking stabilizes dynamic interactions between the core and regulatory particles of the proteasome enabling purification of a catalytically active proteasome (Fabre et al. 2013). It is this protein:protein crosslinking ability of formaldehyde that makes it suitable for stabilizing RAFs such as RNPS1 within their functional RNPs (Fig. 2B), allowing enrichment of sites where it is bound to the EJC core in vivo (Fig. 5; Supplemental Fig. S6). Formaldehyde crosslinking can also stabilize RBPs such as CASC3 within RNPs (Figs. 3, 4; Supplemental Fig. S4). This can occur due to protein–protein crosslinks between an RBP and other proteins within an RNP, which can stabilize an RBP:RNA interaction. Consistent with this, it was shown that formaldehyde crosslinking but not UV crosslinking enhances capture of AGO:siRNA interactions (Au et al. 2014). Similarly, formaldehyde crosslinking stabilized double-stranded RBP DROSHA within pri-microRNPs to allow mapping of DROSHA cleavage sites with a CLIP-seq workflow (Kim and Kim 2019). Similar to UV light, the stabilizing effect of formaldehyde on RBPs can also result from its ability to form protein:nucleic acid crosslinks, which makes it a widely popular crosslinker of choice in chromatin IP studies (Collas 2010; Hoffman et al. 2015). Future studies should evaluate formaldehyde's ability to immobilize protein:protein and protein:RNA interactions, and relative contributions of these two types of linkages to RNP stabilization.
For formaldehyde crosslinking of in vivo RNPs, crosslinker concentration is an important consideration. We find that a relatively low formaldehyde concentration (0.1%) provides a good balance between protein crosslinking and solubility (Fig. 2B). Possibly, at higher concentrations, formaldehyde can crosslink nonspecific interactions trapping macromolecular complexes in their local cellular environments. Indeed, a previous study showed that formaldehyde concentrations of 0.2% or higher leads to inappropriate detection of cytoplasmic and mitochondrial proteins in the nuclear fraction (Fabre et al. 2013). Interestingly, the xRIPiT signal shows a dip in the top quarter of expression bins while such a drop is not seen in nRIPiT samples (e.g., Fig. 5B). Possibly, this decrease in signal results from nonspecific, over-crosslinking of RNPs to cellular structures, which may particularly affect more abundant RNPs. The formaldehyde concentrations we use here are an order of magnitude lower than those in ChIRP-MS approaches used to illuminate lncRNA proteomes (Chu et al. 2015). Perhaps, high formaldehyde concentrations can induce crosslinks within a larger fraction of an individual RNP to improve signal as in the case of single RNP interactomes. However, low concentrations of formaldehyde like we use here are likely to be more critical to preserve the relative abundance of mRNPs, which range several orders of magnitude in expression.
While chemical crosslinking with formaldehyde provides a robust alternative to UV crosslinking to study in vivo RNA–protein interactions, unlike UV crosslinking, it is unable to discriminate between direct and indirect interactions. Also, unlike CLIP, so far formaldehyde crosslinking has not been exploited to identify the direct sites of contacts between an RNA and a protein. It is noteworthy that formaldehyde crosslinking has been used to map points of protein contact on DNA (Solomon and Varshavsky 1985), and can be similarly applied to RNA–protein complexes as well. Nevertheless, our current and previous work (Singh et al. 2014; Mabin et al. 2018; Woodward et al. 2019) shows that formaldehyde crosslinking combined with RIPiT-seq is a powerful approach to isolate and analyze RNA footprints of multisubunit RNP assemblies, which are often heterogeneous in nature.
MATERIALS AND METHODS
Source data for classification of RBPs and RAFs
Sets of UV-crosslinkable human RBPs were from two sources: (i) Proteins detected in RNA interactome capture (RIC) experiments were obtained from Hentze et al. (2018) (Supplemental Table S2). For inclusion in our analysis, we required the proteins to be detectable in at least two of the seven RIC data sets. (ii) Proteins that were defined as the “integrated human RBPome” or “ihRBPome” based on protein-crosslinked RNA extraction (XRNAX) were obtained from Trendel et al. (2019) (Supplemental Table S2). For a comprehensive set of UV crosslinking independent human RBPs, we relied on the analysis of Brannan et al. (2016). A list of 1786 annotated RBPs (Supplemental Table S3; Brannan et al. 2016) was combined with 1923 proteins that achieved the RBP classification score of greater than 0.79 as predicted by SONAR (Supplemental Table S4; Brannan et al. 2016) resulting in a set of 2784 unique proteins that we defined as “annotated and predicted RNA-associated proteins.” All protein lists are provided in Supplemental Table S1 of this publication.
Gene ontology analysis
DAVID gene ontology tool (Huang et al. 2009) was used to determine terms enriched in RAFs with all human genes as a background. Only nonredundant terms with lowest P-value (with Benjamini-Hochberg correction) are reported.
Cell lines and cell culture
Human embryonic kidney (HEK293) Flp-In TRex cell lines expressing EJC factors have been described previously (Singh et al. 2012; Mabin et al. 2018). HEK293 Flp-In TRex cells were cultured under 5% carbon dioxide in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Formaldehyde crosslinking and FLAG immunoprecipitation
Formaldehyde crosslinking was performed as previously described (Singh et al. 2014). Briefly, HEK293 cells were cultured in 10 or 15 cm plates. The culture medium was removed and cells were washed with room temperature Phosphate Buffered Saline (PBS). Cells were scraped in 10–30 mL PBS and transferred to a 15 or 50 mL conical. Formaldehyde (37%) was added to the cell suspension at the desired concentration (v/v). Following 10-min incubation at room temperature, 1–3 mL (10% volume of cell suspension) of the quenching buffer [2.5 M Glycine, 25 mM Tris-base] was added to the suspension, and incubation at room temperature was continued for 5 min. Cells were pelleted at 400g at 4°C and resuspended in denaturing lysis buffer [20 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate, 1× Sigma protease inhibitor cocktail, 1 mM PMSF]. Cells were sonicated on ice bath at 20% amplitude using a microtip for 10 sec in 2 sec bursts separated by 5 sec pauses. Cell debris was pelleted at 15,000g at 4°C for 10 min. The clear lysate was mixed with 80 µL (per 10 cm plate) of prewashed anti-FLAG M2 agarose beads, and incubated with gentle mixing for 1–2 h at 4°C. FLAG-RNP complexes were washed two times in denaturing wash buffer [20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate] followed by two washes in Isotonic Wash Buffer [IsoWB, 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40] and then treated with 125 µg/mL RNase A. After four more washes in IsoWB, complexes were eluted in 80 µL of IsoWB supplemented with 125 µg/mL FLAG peptide. For anti-FLAG IP under native conditions, HEK293 cells stably expressing FLAG-tagged EJC protein were lysed in ice-cold hypotonic lysis buffer (HLB) [20 mM Tris-HCl pH 7.5, 15 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.1% Triton X-100, 1× Sigma protease inhibitor cocktail, 1 mM PMSF]. Lysates were sonicated, supplemented with NaCl to a final concentration of 150 mM and RNase A was added to 125 µg/mL. Complexes were then combined with anti-FLAG agarose beads and incubated with gentle mixing for 1–2 h at 4°C. FLAG beads were washed eight times with IsoWB and complexes were eluted with IsoWB supplemented with FLAG peptide as above.
RNase I conditions during formaldehyde-crosslinked RIPiT-seq (xRIPiT-seq)
The general steps and reaction conditions during xRIPiT-seq were previously described in Mabin et al. (2018). RNase I digestion conditions during xRIPiT were optimized by performing a titration where the enzyme concentration was increased in threefold increments (Supplemental Fig. S1B). RNase I concentration that yielded the greatest signal in the 30–80 nt range was used in all subsequent experiments.
High-throughput DNA sequencing data processing and alignment
Adaptor trimming and PCR duplicate removal
All RNA-seq and RIPiT-seq data sets were previously described in Mabin et al. (2018) (GEO: GSE115977), and all data processing was carried out as described therein. Briefly, demultiplexed fastq files containing unmapped reads were first trimmed using Cutadapt (Martin 2011). A 12-nt sequence on read 5′ ends consisting of a 5-nt random sequence (UMI), 5-nt identifying barcode, and a CC was removed with the random sequence saved for each read for identifying PCR duplicates down the line. Next, as much of the 3′-adapter (miR-Cat22) sequence TGGAATTCTCGGGTGCCAAGG was removed from the 3′ end as possible. Any reads less than 20 nt in length after trimming were discarded.
Alignment and removal of multimapping reads
Trimmed RNA and RIPiT-seq reads and the CLIP-seq data from Hauer et al. (2016) downloaded from ArrayExpress: E-MTAB-4215), which already had adapters trimmed and thus were not further preprocessed by us, were aligned with HISAT2 v2.1.0 (Kim et al. 2015) using 28 threads to Genome Reference Consortium Human Build 38 (GRCh38). After alignment, only reads with a mapping score >50, indicative of unique mapping, were retained, and remaining reads (likely multimapped) were discarded. RNA-seq (HEK293 cells) and RIPiT-seq reads, which contained a 5 nt unique molecular identifier (UMI), further underwent a PCR duplicate removal step, in which reads with identical sequences aligned to the same location with the same UMI had all but one copy discarded. The CLIP-seq data had PCR duplicates previously removed by Hauer et al.
Removal of stable RNA and mitochondrial DNA mapping reads
Reads mapping to stable RNAs were counted and removed as follows. All reads were checked for overlap against GRCh38 annotations for miRNA, rRNA, tRNA, scaRNA, snoRNA, and snRNA using bedtools function: intersect (Quinlan and Hall 2010). Any read that overlapped with these RNA sequences by more than 50% was filtered out. Reads aligned to chrM (mitochondrial DNA) were also counted and removed.
Data analysis and quantification
Human reference transcriptome and annotations
Reference annotations, for example, for protein coding transcripts, were retrieved from the Ensembl BioMart (GRCh38.p12). All analyses were done using the APPRIS Principal 1 transcript variant for each gene (Rodriguez et al. 2013).
Read distribution assignment
Fractions of reads corresponding to exonic and canonical EJC and noncanonical EJC regions were computed as follows. All analyses were limited to exons 100 nt or longer in order to have sufficiently long noncanonical EJC regions. Furthermore, for canonical and noncanonical EJC annotations we excluded the last exon, which lacks a canonical EJC deposition site. For the canonical EJC regions the 30 nt surrounding the center of the primary EJC binding site at −24 (−39 to −9) were considered, while for the noncanonical EJC region all nucleotides from the start of each exon to −50 were used. The single exon gene annotation was similarly limited to exons (genes) at least 100 nt in length. Bedtools function “intersect” was used to compare reads against these annotations and reads which overlapped the annotation by more than 50% were counted.
Read density calculation
After read assignments, reads from replicates of each experiment were combined to compute read densities (RPKMRIPiT-seq or RPKMIP-seq) per protein-coding gene as follows. For each calculation, the total aligned reads were used as the scaling factor and the length (in kb) of the canonical or noncanonical regions of a gene, or length of the intronless genes were used for the base adjustment.
Quantification of EJC binding in canonical and noncanonical regions, and on intronless genes
Only protein-coding genes with RNA-seq RPKM greater than zero were included in the analysis. For comparing EJC protein binding at different expression levels, the full range of RNA-seq gene log2 RPKM values was covered by 20 reference points equidistant in logarithmic space. Each point was associated with a bin containing all genes within twofold of the central RPKM value. This was done to ensure the same number of bins, with the same expression range (in terms of fold change) between experiments—bins may therefore overlap to different degrees depending on the comparison. Log2 of mean experimental RPKM values in each bin (RPKMRIPiT-seq or RPKMIP-seq), which provide estimates of EJC protein binding on all canonical or noncanonical regions within a gene, or on intronless genes, were plotted against RNA-seq RPKM values. In tests comparing RIPiT-seq and CLIP-seq, analyses were limited to the set of genes with RPKM values within 1.5-fold in RNA-seq libraries from each cell line. The average between these two libraries was then used as an estimate of gene expression (RPKMRNA-seq) to compare xRIPiT and CLIP-seq data sets in Figures 4, 5E–I. Linear fits were then produced for each experiment in log2-space with a set slope of 1. That is, a linear agreement between experimental and RNA-seq RPKMs was assumed. The calculated intercepts therefore correspond to the exponent of the slope in linear space: log2(F(x)) = log2(x) + b in log2-space becomes F(x) = x*2b in linear space. In the bar plots that compare the intercepts calculated from linear fits to the scatter plots, the y-axis is 2 to the power of each intercept, which is exactly the slope in linear space and therefore the total fold-change of each experiment versus RNA-seq.
Quantification of individual EJC binding sites
For analysis of individual binding sites, we first limited our search to genes in the top 20% expression range in RNA-seq, corresponding to an RPKM > 5.9, and normalized canonical region counts by intronless gene levels as detailed above. Canonical regions between experiment types were then compared (Supplemental Figs. S4A,B,G,H, S6A,B,D,E) to observe overall trends. Individual canonical sites were then considered to be observed binding sites if the expression in those regions was twofold higher than extrapolated intronless gene expression on the same genes. Venn diagrams showing the overlap between individual binding sites were then constructed to show overlap between experimental methods (Figs. 4E, 5I) and similarly all binding sites were plotted in deciles as a function of their RPKM range to show discovered binding sites by experiment, based on gene expression levels (Supplemental Figs. S4C, S6C).
Quantification of EJC binding on recursively spliced (RS) exons and their neighboring exons
MaxEntScan splice site scoring software (Yeo and Burge 2004) was first used to find 5′ splice site (5′ss) scores of all exon–exon junctions using sequences obtained from Ensembl BioMart (GRCh38.p12). For all exon–exon junctions with the 5′ss score above the threshold of 5.52, which was defined previously by Blazquez et al. (2018), the downstream exons were classified as RS exons. The same number of exons that showed the lowest 5′ss score at their start were used as a non-RS exon control. CASC3 or RNPS1 occupancy at each exon was estimated by determining total exon coverage (CLIP or RIPiT RPKM for a particular exon) normalized to gene coverage, with gene coverage as the average of the two RNA-seq data sets within 1.5-fold of each other, as detailed above. Only exons with an RPKM > 0 were considered in the analysis. Such normalized exon coverage was determined for the RS exon itself, as well as the three preceding exons. For gene level analysis genes were classified by the highest exon–exon 5′ss score within a given gene, with RS genes defined by the same 5.52 threshold. Similarly, an equal number of non-RS genes were selected to have the lowest maximum 5′ss score within a gene.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by an allocation from the Ohio Supercomputer Center. Funding for this work was provided by the National Institutes of Health (R01-GM120209) to G.S.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.074856.120.
Freely available online through the RNA Open Access option.
REFERENCES
- Anantharaman V, Koonin EV, Aravind L. 2002. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res 30: 1427–1464. 10.1093/nar/30.7.1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CBF, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CLP, Pedersen JS, Séraphin B, Le Hir H, Andersen GR. 2006. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 313: 1968–1972. 10.1126/science.1131981 [DOI] [PubMed] [Google Scholar]
- Au PCK, Helliwell C, Wang M-B. 2014. Characterizing RNA-protein interaction using cross-linking and metabolite supplemented nuclear RNA-immunoprecipitation. Mol Biol Rep 41: 2971–2977. 10.1007/s11033-014-3154-1 [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. 10.1016/j.molcel.2012.05.021 [DOI] [PubMed] [Google Scholar]
- Blazquez L, Emmett W, Faraway R, Pineda JMB, Bajew S, Gohr A, Haberman N, Sibley CR, Bradley RK, Irimia M, et al. 2018. Exon junction complex shapes the transcriptome by repressing recursive splicing. Mol Cell 72: 496–509.e9. 10.1016/j.molcel.2018.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm V, Gehring NH. 2016. Exon junction complexes: supervising the gene expression assembly line. Trends Genet 32: 724–735. 10.1016/j.tig.2016.09.003 [DOI] [PubMed] [Google Scholar]
- Boehm V, Britto-Borges T, Steckelberg A-L, Singh KK, Gerbracht JV, Gueney E, Blazquez L, Altmüller J, Dieterich C, Gehring NH. 2018. Exon junction complexes suppress spurious splice sites to safeguard transcriptome integrity. Mol Cell 72: 482–495.e7. 10.1016/j.molcel.2018.08.030 [DOI] [PubMed] [Google Scholar]
- Bono F, Ebert J, Lorentzen E, Conti E. 2006. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126: 713–725. 10.1016/j.cell.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Brannan KW, Jin W, Huelga SC, Banks CAS, Gilmore JM, Florens L, Washburn MP, Van Nostrand EL, Pratt GA, Schwinn MK, et al. 2016. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol Cell 64: 282–293. 10.1016/j.molcel.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. 10.1016/j.cell.2012.04.031 [DOI] [PubMed] [Google Scholar]
- Chatterjee K, Majumder S, Wan Y, Shah V, Wu J, Huang H-Y, Hopper AK. 2017. Sharing the load: Mex67-Mtr2 cofunctions with Los1 in primary tRNA nuclear export. Genes Dev 31: 2186–2198. 10.1101/gad.305904.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. 2015. Systematic discovery of Xist RNA binding proteins. Cell 161: 404–416. 10.1016/j.cell.2015.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P. 2010. The current state of chromatin immunoprecipitation. Mol Biotechnol 45: 87–100. 10.1007/s12033-009-9239-8 [DOI] [PubMed] [Google Scholar]
- Fabre B, Lambour T, Delobel J, Amalric F, Monsarrat B, Burlet-Schiltz O, Bousquet-Dubouch M-P. 2013. Subcellular distribution and dynamics of active proteasome complexes unraveled by a workflow combining in vivo complex cross-linking and quantitative proteomics. Mol Cell Proteomics 12: 687–699. 10.1074/mcp.M112.023317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer C, Sieber J, Schwarzl T, Hollerer I, Curk T, Alleaume A-M, Hentze MW, Kulozik AE. 2016. Exon junction complexes show a distributional bias toward alternatively spliced mRNAs and against mRNAs coding for ribosomal proteins. Cell Rep 16: 1588–1603 10.1016/j.celrep.2016.06.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Kelley DR, Tenen D, Bernstein B, Rinn JL. 2016. Widespread RNA binding by chromatin-associated proteins. Genome Biol 17: 28 10.1186/s13059-016-0878-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T. 2018. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19: 327–341. 10.1038/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- Hockensmith JW, Kubasek WL, Vorachek WR, von Hippel PH. 1986. Laser cross-linking of nucleic acids to proteins. Methodology and first applications to the phage T4 DNA replication system. J Biol Chem 261: 3512–3518. [PubMed] [Google Scholar]
- Hockensmith JW, Kubasek WL, Vorachek WR, von Hippel PH. 1993. Laser cross-linking of proteins to nucleic acids. I. Examining physical parameters of protein-nucleic acid complexes. J Biol Chem 268: 15712–15720. [PubMed] [Google Scholar]
- Hoffman EA, Frey BL, Smith LM, Auble DT. 2015. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem 290: 26404–26411. 10.1074/jbc.R115.651679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-Y, Hopper AK. 2015. In vivo biochemical analyses reveal distinct roles of β-importins and eEF1A in tRNA subcellular traffic. Genes Dev 29: 772–783. 10.1101/gad.258293.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, König J, Ule J. 2014. iCLIP: protein-RNA interactions at nucleotide resolution. Methods 65: 274–287. 10.1016/j.ymeth.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahvejian A, Svitkin YV, Sukarieh R, M'Boutchou M-N, Sonenberg N. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev 19: 104–113. 10.1101/gad.1262905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Kim VN. 2019. fCLIP-seq for transcriptomic footprinting of dsRNA-binding proteins: lessons from DROSHA. Methods 152: 3–11. 10.1016/j.ymeth.2018.06.004 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FCY, Ule J. 2018. Advances in CLIP technologies for studies of protein-RNA interactions. Mol Cell 69: 354–369. 10.1016/j.molcel.2018.01.005 [DOI] [PubMed] [Google Scholar]
- Le Hir H, Saulière J, Wang Z. 2016. The exon junction complex as a node of post-transcriptional networks. Nat Rev Mol Cell Biol 17: 41–54. 10.1038/nrm.2015.7 [DOI] [PubMed] [Google Scholar]
- Mabin JW, Woodward LA, Patton RD, Yi Z, Jia M, Wysocki VH, Bundschuh R, Singh G. 2018. The exon junction complex undergoes a compositional switch that alters mRNP structure and nonsense-mediated mRNA decay activity. Cell Rep 25: 2431–2446.e7. 10.1016/j.celrep.2018.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Mestdagh C, Akhtar J, Kreim N, Deinhard P, Sachidanandam R, Treisman J, Roignant J-Y. 2014. The exon junction complex controls transposable element activity by ensuring faithful splicing of the piwi transcript. Genes Dev 28: 1786–1799. 10.1101/gad.245829.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. 1997. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89: 951–961. 10.1016/S0092-8674(00)80280-9 [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Mili S, Steitz JA. 2004. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10: 1692–1694. 10.1261/rna.7151404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-McNicoll M, Neugebauer KM. 2013. How cells get the message: dynamic assembly and function of mRNA–protein complexes. Nat Rev Genet 14: 275–287. 10.1038/nrg3434 [DOI] [PubMed] [Google Scholar]
- Nicholson CO, Friedersdorf M, Keene JD. 2017. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 23: 32–46. 10.1261/rna.058115.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26: 182–190. 10.1016/S1046-2023(02)00021-X [DOI] [PubMed] [Google Scholar]
- Obrdlik A, Lin G, Haberman N, Ule J, Ephrussi A. 2019. The transcriptome-wide landscape and modalities of EJC binding in adult Drosophila. Cell Rep 28: 1219–1236.e11. 10.1016/j.celrep.2019.06.088 [DOI] [PubMed] [Google Scholar]
- Queiroz RML, Smith T, Villanueva E, Marti-Solano M, Monti M, Pizzinga M, Mirea D-M, Ramakrishna M, Harvey RF, Dezi V, et al. 2019. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat Biotechnol 37: 169–178. 10.1038/s41587-018-0001-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci EP, Kucukural A, Cenik C, Mercier BC, Singh G, Heyer EE, Ashar-Patel A, Peng L, Moore MJ. 2014. Staufen1 senses overall transcript secondary structure to regulate translation. Nat Struct Mol Biol 21: 26–35. 10.1038/nsmb.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink J-J, Lopez G, Valencia A, Tress ML. 2013. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res 41: D110–D117. 10.1093/nar/gks1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulière J, Murigneux V, Wang Z, Marquenet E, Barbosa I, Le Tonquèze O, Audic Y, Paillard L, Roest Crollius H, Le Hir H. 2012. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat Struct Mol Biol 19: 1124–1131. 10.1038/nsmb.2420 [DOI] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764. 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Ricci EP, Moore MJ. 2014. RIPiT-Seq: a high-throughput approach for footprinting RNA:protein complexes. Methods 65: 320–332. 10.1016/j.ymeth.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Pratt G, Yeo GW, Moore MJ. 2015. The clothes make the mRNA: past and present trends in mRNP fashion. Annu Rev Biochem 84: 325–354. 10.1146/annurev-biochem-080111-092106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KC. 1969. Photochemical addition of amino acids to 14C-uracil. Biochem Biophys Res Commun 34: 354–357. 10.1016/0006-291X(69)90840-7 [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Varshavsky A. 1985. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc Natl Acad Sci 82: 6470–6474. 10.1073/pnas.82.19.6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BW, Toews J, Kast J. 2008. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom 43: 699–715. 10.1002/jms.1415 [DOI] [PubMed] [Google Scholar]
- Szczepińska T, Kalisiak K, Tomecki R, Labno A, Borowski LS, Kulinski TM, Adamska D, Kosinska J, Dziembowski A. 2015. DIS3 shapes the RNA polymerase II transcriptome in humans by degrading a variety of unwanted transcripts. Genome Res 25: 1622–1633. 10.1101/gr.189597.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendel J, Schwarzl T, Horos R, Prakash A, Bateman A, Hentze MW, Krijgsveld J. 2019. The human RNA-binding proteome and its dynamics during translational arrest. Cell 176: 391–403.e19. 10.1016/j.cell.2018.11.004 [DOI] [PubMed] [Google Scholar]
- Urdaneta EC, Vieira-Vieira CH, Hick T, Wessels H-H, Figini D, Moschall R, Medenbach J, Ohler U, Granneman S, Selbach M, et al. 2019. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat Commun 10: 990 10.1038/s41467-019-08942-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ballut L, Barbosa I, Le Hir H. 2018. Exon junction complexes can have distinct functional flavours to regulate specific splicing events. Sci Rep 8: 9509 10.1038/s41598-018-27826-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward LA, Mabin JW, Gangras P, Singh G. 2017. The exon junction complex: a lifelong guardian of mRNA fate. Wiley Interdiscip Rev RNA 8: e1411 10.1002/wrna.1411 [DOI] [PubMed] [Google Scholar]
- Woodward L, Gangras P, Singh G. 2019. Identification of footprints of RNA:protein complexes via RNA immunoprecipitation in tandem followed by sequencing (RIPiT-Seq). J Vis Exp 149: e59913 10.3791/59913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YW, Flynn RA, Chen Y, Qu K, Wan B, Wang KC, Lei M, Chang HY. 2014. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife 3: e02046 10.7554/eLife.02046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.