

Abstract
DEAD-box proteins (DBPs) are RNA remodeling factors associated with RNA helicase activity that are found in nearly all organisms. Despite extensive studies on the mechanisms used by DBPs to regulate RNA function, very little is known about how DBPs themselves are regulated. In this work, we have analyzed the expression and regulation of DeaD/CsdA, the largest of the DBPs in Escherichia coli (E. coli). We show that deaD transcription initiates 838 nt upstream of the start of the coding region. We have also found that DeaD is autoregulated through a negative feedback mechanism that operates both at the level of deaD mRNA stability and Rho-dependent transcription termination, and this regulation is dependent upon its mRNA 5′ untranslated region (5′ UTR). These findings suggest that DeaD might be regulating the conformation of its own mRNA through its RNA helicase activity to facilitate ribonuclease and Rho access to its 5′ UTR.
Keywords: RNA helicase, autoregulation, transcription termination, Rho factor
INTRODUCTION
DEAD-box proteins (DBPs) represent a major class of RNA remodeling factors that have been implicated in a variety of RNA regulatory processes in both prokaryotic and eukaryotic organisms (Rocak and Linder 2004; Linder and Jankowsky 2011). The characteristic features of DBPs are a set of >13 conserved motifs distributed over two RecA-like domains, which jointly bind to RNA and ATP to mediate ATP-dependent RNA or ribonucleoprotein remodeling. Escherichia coli contains five members of the DBP family: DbpA, DeaD, RhlB, RhlE, and SrmB, which have been implicated in translation regulation, mRNA stability and ribosome biogenesis (Iost et al. 2013; Redder et al. 2015). In particular, an absence of either DeaD or SrmB results in cold-sensitive growth, which is associated with an accumulation of ribosomal precursors (Charollais et al. 2003, 2004).
In an earlier study, we showed that whereas all five DBPs could be simultaneously removed without loss of viability, the cold-sensitivity of the derivative strain could be largely accounted by the phenotype of the singly mutated ΔdeaD strain (Jagessar and Jain 2010). DeaD is also highly induced during cold shock when cells growing at 37°C are rapidly transferred to 10°C–16°C, and an absence of this factor impedes the recovery of growth during the acclimatization phase (Jones et al. 1996). Additionally, during cold-shock, DeaD has been found to partially replace RhlB as the helicase component of the E. coli degradosome, an RNA degrading complex, suggesting a role in RNA degradation (Prud'homme-Genereux et al. 2004). Model studies using a chloramphenicol acetyl transferase (cat) reporter mRNA showed that DeaD can alleviate the inhibitory effects of secondary structures that overlap the cat mRNA ribosome binding site (Butland et al. 2007). Similarly, DeaD has been implicated in the regulation of the uvrY mRNA via a translational mechanism (Vakulskas et al. 2014). Additionally, by using cross-linking and immunoprecipitation (CLIP), DeaD was found to cross-link to 39 transcripts in vivo, many of which are also regulated by DeaD (Vakulskas et al. 2014). Thus, DeaD appears to have multifaceted roles in gene regulation even though a detailed knowledge of DeaD function in the cell is still incomplete.
Despite the progress made toward defining E. coli DBP function, virtually nothing is known about how these factors themselves are regulated. Moreover, any potential regulatory elements in their mRNA 5′ UTR are unknown because their transcription start sites (TSSs) have not been defined or validated. Here we show that deaD mRNA transcription starts 838 nucleotides (nt) upstream of the coding region, resulting in one of the longest 5′ UTRs known for any E. coli transcript. A potential regulatory role for the 5′ UTR is confirmed by the observation that DeaD regulates its own expression, and sequences within the 5′ UTR appear to be important for this process. We show that a high degree of autoregulatory control is mediated by the joint effects of DeaD activity on mRNA stability and transcription elongation. With respect to the latter, the Rho termination factor is shown to play a key role in determining the amount of full-length deaD mRNA synthesized inside the cell.
RESULTS
The deaD mRNA contains an unusually long 5′ UTR
To study regulatory aspects of deaD expression, we first investigated the evidence for deaD transcription. A deaD mRNA TSS mapping to 173 nt upstream of the coding region was previously identified by using primer extension, although evidence for such a start site was based on RNA prepared from cells containing a multicopy deaD plasmid (Toone et al. 1991). In contrast, high-throughput sequencing studies have shown no evidence for this TSS. Specifically, a recent genome-wide study to map mRNA 5′ ends revealed only three significant TSSs within the first 1000 nt upstream of the deaD coding region, of which reads starting 834 nt upstream of the coding region were the most prevalent (Fig. 1A; Ettwiller et al. 2016). A second study found evidence for only two TSSs starting 834 and 838 nt upstream of the coding region (Thomason et al. 2015). To characterize the deaD transcript, Northern blot analysis was performed on RNA samples derived from wild-type (WT) and ΔdeaD strains, and a band of ∼2.7 kilo-base (kb) could be visualized in the former strain (Fig. 1B). With a coding region of 1.89 kb and an expected 3′-UTR of 70 nt, the observed size of the deaD mRNA is consistent with transcription initiating ∼0.8 kb upstream of the coding region. Interestingly, we noticed the deaD transcript runs as a diffuse band, possibly because of its close migration with the abundant 2.9 kb 23S ribosomal RNA. To define the deaD TSS more precisely, we analyzed the deaD mRNA by rapid amplification of cDNA ends (RACE), which revealed that the deaD TSS starts 838 nt upstream of the coding region, rather than 834 nt (Fig. 1C).
FIGURE 1.

The deaD mRNA contains a long 5′ UTR. (A) Schematic description of TSSs upstream of the deaD coding region identified by 5′ RNA-seq. The deaD coding region is depicted by a red rectangle and the upstream region by a red line. Transcription start sites identified by Ettwiller et al. (2016), are denoted by rightward arrows with relative read scores (RRS) indicated at the top. Only sites with RRS > 5 are depicted. Dashed lines and numbers at the bottom indicate the distance between the transcription initiation positions and the start of the coding region. (B) Northern-blot analysis of total RNA isolated from wild-type and ΔdeaD strains. The RNA was analyzed using a radiolabeled riboprobe complementary to the deaD coding region. The deaD mRNA is marked by an arrow. The sizes of molecular weight RNA markers, in nucleotides, run alongside the samples, are shown on the right. A 16S rRNA loading control is also shown. (C) TSS determination by RACE. A reverse transcription product generated using total RNA from a WT strain and a primer complementary to the deaD 5′ UTR was ligated to a linker, amplified, cloned and sequenced. The resulting trace shows the reverse complement sequence at the junction of deaD 5′ UTR and linker sequences (shaded). The sequence of the first ten 5′ nt of the deaD mRNA, starting 838 nt upstream of the start codon, is shown on the right. (D) β-galactosidase assays. Plasmids containing a deaD-lacZ fusion, a derivative that contains mutations in the putative −10 sequence of the deaD promoter, or a 50 bp deletion spanning the −10 and −35 regions, were transformed into a ΔlacZ strain and β-galactosidase activity was measured. Regions that correspond to deaD and lacZ sequences are shown in red and blue, with noncoding and coding regions depicted by lines and rectangles, respectively. β-galactosidase activity was measured after growth at 37°C in LB medium to mid-log phase, in LB medium to stationary phase, in minimal M9 medium to mid-log phase, or after transfer of cultures for 1 h to 16°C following an initial growth to mid-log phase in LB medium at 37°C (cold-shock).
Next, we were interested in determining the fraction of transcription that originates from the −838 TSS. For these purposes, a plasmid-based deaD-lacZ translation fusion and a derivative containing multiple mutations in the putative −10 sequence (TATGAT > CCCCCC) were constructed. Transformation of these plasmids into a ΔlacZ strain resulted in significant β-galactosidase expression for the wild-type construct, but only ∼10% residual expression with the mutated construct for cultures grown to mid-log phase in LB medium (Fig. 1D), suggesting that nearly 90% of deaD mRNA transcription is directed by a promoter that initiates transcription 838 nt upstream of the coding region. Additionally, as most of the activity could be abolished due to mutation, it confirmed that the mapped 5′ end of the deaD mRNA corresponds to a genuine TSS, rather than to a processed transcript. A second construct that contained a deletion spanning from 38 nt upstream of the TSS to 12 nt downstream, including the −35 region of the promoter, yielded a similar decrease, suggesting that the residual transcription in each mutant construct arises from start sites that are not close to the main TSS. To determine the contribution of the −838 TSS to deaD transcription under additional conditions, the β-galactosidase activity of the wild-type and mutant deaD-lacZ constructs were measured in stationary phase, after growth in minimal medium to mid-log phase or after cold-shock at 16°C. In each case, transcription from the −838 TSS was found to account for most of the signal, indicating that the majority of deaD mRNA transcription under different conditions is directed by a single TSS over 800 nt upstream of the coding region.
The expression of DeaD is autoregulated
The presence of a long 5′ UTR in the deaD mRNA suggested that DeaD expression could be regulated at multiple levels. One type of regulation, observed for proteins with important physiological cellular roles, is autoregulation, which refers to their ability to negatively regulate their synthesis to maintain homeostasis. To determine whether DeaD is autoregulated, mutations that alter conserved motifs in DeaD were introduced into the genomic deaD locus. Three different DBP motifs, I, II, or VI were targeted, of which motif I and II are also referred to as Walker A and B motifs. Thereafter, RNA was prepared from strains expressing wild-type DeaD, as well as the indicated variants, and analyzed by Northern blot (Fig. 2A). Significantly, each mutant strain was found to show elevated levels of deaD mRNA. Specifically, expression of the deaD mRNA was increased by threefold in a strain containing a R332Q mutation in motif VI, whereas a higher increase (19-fold) was observed when the first aspartic acid residue in the motif II D-E-A-D sequence was changed to alanine. The further addition of a motif I mutation (K56N) to the D156A mutant increased expression marginally. The strain containing the K56N + D156A mutants was selected for further analysis and is hereafter referred to as the DM strain.
FIGURE 2.
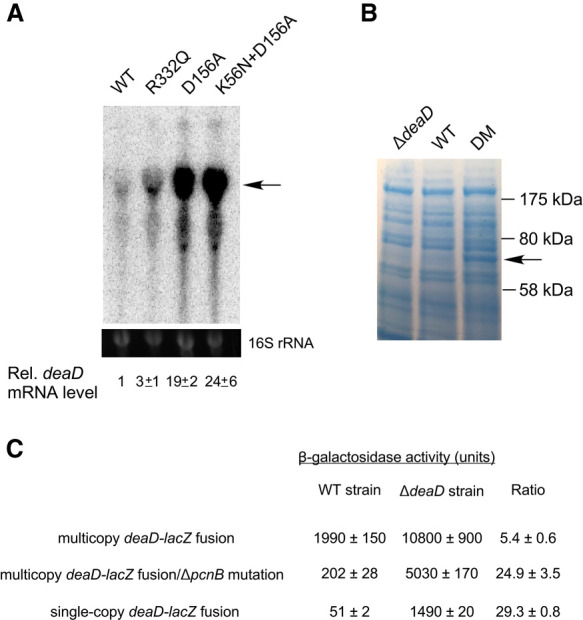
Expression of DeaD is autoregulated. (A) Expression of deaD mRNA in WT strains and in derivatives containing mutations in conserved motifs of DeaD. The position of the full-length deaD mRNA is indicated by an arrow. The normalized levels of deaD mRNA, along with their associated standard errors, are shown below. (B) Protein analysis. Total cellular proteins were separated by gel electrophoresis and the gels were dye-stained to visualize proteins. The band corresponding to DeaD is indicated by an arrow. The sizes of molecular weight markers run alongside the proteins is shown on the right. (C) β-galactosidase assays. Assays were performed in WT or ΔdeaD strains containing a multicopy deaD-lacZ fusion plasmid, in plasmid-containing strains that contained an additional ΔpcnB mutation, or in strains containing a single copy chromosomal deaD-lacZ fusion.
To determine whether the increased expression of deaD mRNA observed in the DM strain leads to similar increases at the protein level, total cellular proteins were directly analyzed by dye staining (Fig. 2B). No DeaD-specific protein band could be observed for the WT strain in comparison with the ΔdeaD strain, suggesting that the endogenous levels of DeaD are too low to be detected directly. However, in the DM strain, a 70 kilodalton (kDa) band compatible in size with the 629 amino acids DeaD protein could be clearly identified, consistent with the significant degree of deaD mRNA over-expression observed in the DM strain.
Finally, the multicopy deaD-lacZ plasmid was transformed into ΔlacZ derivatives of the WT and ΔdeaD strains and β-galactosidase activity was measured (Fig. 2C). Increased expression of the fusion protein was observed in the ΔdeaD strain suggesting that the deaD determinants present in the fusion construct, which include the 5′ UTR and the first 100 base pairs (bp) of the coding region, confer DeaD-dependent regulation. The extent of DeaD-mediated regulation (fivefold) was lower than the increase of mRNA levels in the DM strain (∼25-fold, Fig. 2A), suggesting that some regulatory determinants might be missing in this fusion construct. However, we noticed that the plasmid-transformed ΔdeaD strain exhibited impaired growth, suggesting an alternate possibility that this strain may be unable to express the fusion protein at high levels. To distinguish between these possibilities, we made ΔpcnB derivatives of the WT and ΔdeaD strains to reduce the plasmid copy number, as ColE1 plasmids, such as the one containing the deaD-lacZ fusion, propagate at lower copy number in ΔpcnB strains (Xu et al. 2002). As expected, when the plasmid was transformed into the ΔpcnB strain, lacZ expression was reduced by ∼10-fold. However, relative to its expression in the ΔpcnB strain, the activity of the deaD-lacZ fusion in the ΔdeaDΔpcnB strain was found to be increased by 25-fold, suggesting that a significantly higher extent of regulation can be achieved when the expression of the fusion construct is reduced by lowering the plasmid copy number. To corroborate these results, the deaD-lacZ fusion was transferred to the chromosome in single copy. As was observed with the ΔpcnB strains, the expression of the fusion in the ΔdeaD strain background was nearly 30-fold higher than in the WT strain. Based on the inhibited expression of the deaD-lacZ fusion under elevated copy number conditions, all downstream experiments were performed using single-copy chromosomal fusions.
Two observations are noteworthy. First, the extent to which the activity of the deaD-lacZ fusion was increased, when expressed at low or single-copy, was similar to the increase observed for the full-length deaD transcript (Fig. 2A), suggesting that the deaD-lacZ fusion contains all determinants that are required for DeaD autoregulation. Second, the similar increase in deaD mRNA levels and deaD-lacZ translation fusion activity suggested that their regulation can be fully explained by changes in mRNA levels and is not likely to involve any significant regulation at the translational level.
Autoregulation of DeaD is jointly mediated by mRNA stability and transcription termination
Although DeaD is implicated in the regulation of several genes, only cat and uvrY have been studied in some detail (Butland et al. 2007; Vakulskas et al. 2014). In each case, it has been proposed that DeaD regulates these genes through a translational mechanism, possibly by unwinding secondary structures near the translational initiation codon. In theory, DeaD could utilize a similar mechanism for autoregulation, for example, by using its RNA remodeling activity to create secondary structures that impair ribosome binding. However, given that DeaD regulates deaD mRNA levels to the same extent as deaD-lacZ translation fusions, it seemed more likely that its autoregulation is effected via mRNA stability or transcription.
To test whether DeaD negatively regulates its expression via mRNA stability, deaD mRNA half-life was measured in WT and DM strains. Exponentially growing strains were treated with the RNA polymerase inhibitor, rifampicin, and cells were harvested at various times thereafter for RNA preparation, with an initial time of one min to allow the completion of deaD mRNA transcription. The RNA samples were analyzed by Northern blot using a deaD-specific probe and the amount of mRNA remaining at different times after rifampicin addition was quantified to determine deaD mRNA half-life in the WT and mutant strains. Initial efforts to measure half-lives at 37°C did not yield reliable data due to low deaD mRNA expression in the WT strain, therefore, the strains were analyzed at 30°C, a condition under which the mRNA levels were found to be increased. These analyses showed that the half-life of the deaD mRNA in the WT strain was 2.7 min, but it increased to 7.7 min in the DM strain (Fig. 3A,B). Thus, in the absence of DeaD activity, the deaD mRNA is stabilized by nearly threefold. To identify the enzyme responsible for deaD mRNA degradation, the levels of deaD mRNA were determined in a WT strain and a derivative containing the temperature-sensitive rne-3071 mutation. RNase E is implicated in the degradation of most of the E. coli mRNAs (Mackie 2013; Mohanty and Kushner 2016), and was thus a likely candidate to be the enzyme that initiates deaD mRNA degradation. As expected, the levels of deaD mRNA were found to be increased by 14-fold in the rne-3071 strain when cells were harvested after a temperature shift to 42°C (Fig. 3C), implicating RNase E in deaD mRNA degradation. In contrast, a smaller difference was seen between the DM and the DM rne-3071 strains, consistent with the increased stability of the deaD mRNA in the former strain.
FIGURE 3.

Autoregulation of deaD is mediated via mRNA stability and transcription termination. (A) Northern-blot analysis. WT or DM strains were treated with rifampicin and aliquots of the cell cultures were harvested at different times after rifampicin addition, as indicated at the top. Total RNA was prepared from these cultures and analyzed by Northern blot using a probe for deaD mRNA. (B) Half-life analysis. The levels of deaD mRNA from (A) were normalized and plotted as a function of time after rifampicin addition on a semi-logarithmic scale. Each data point represents an average value from three experiments. The calculated half-life of the deaD mRNA in each strain is indicated. (C) Inactivation of RNase E increases deaD mRNA levels. WT and DM strains, as well as their rne-3071 derivatives, were transferred to 42°C for 30 min after growth to mid-log phase at 30°C, followed by harvesting of cultures, RNA preparation and Northern blot analysis. (D) Effects of Rho inactivation on deaD mRNA. WT or DM strains containing either wild-type rho or a rhots allele were grown to mid-log phase at 30°C and then transferred to 42°C for 30 min before harvesting. RNA was isolated from the strains and analyzed by Northern blotting. (E) Effect of BCM addition on deaD mRNA. Cultures of WT or DM strains were grown at 37°C and were either untreated or treated with 20 or 40 µg/mL of BCM for 20 min before harvesting. RNA was isolated from the strains and analyzed by Northern blotting. (F) RNA was isolated from WT, rhots or DM strains after growth at 42°C for 30 min and the levels of RNA transcription from 538 to 638 bp downstream from the guaC coding region were quantified by qRT-PCR. The convergently oriented guaC and hofC coding regions are shown in blue and yellow, respectively. The arrow depicts transcription from the guaC promoter with dotted lines indicating readthrough antisense transcription into the hofC coding region. The region analyzed by qRT-PCR is indicated by a red line.
Despite its effects on deaD mRNA half-life, the observed threefold increase in the DM strain was insufficient to explain the ∼25-fold increase in steady state levels of the deaD transcript (Fig. 2A), suggesting that changes in deaD mRNA stability can only partially explain the latter effect. Among the possible explanations for the residual differences were that transcription initiation or elongation might be different between the two strains. However, as DBPs generally act on RNA rather than on DNA, any effects of DeaD on the concentration of its own mRNA would be expected to be effected after the transcription initiation stage.
In E. coli transcription regulation following initiation commonly occurs either via a factor-independent mode, in which intrinsic RNA sequences cause termination, or by a factor-dependent mode, whereby extrinsic factors, most commonly Rho protein, actively dissociate RNA polymerase during transcription (Nudler and Gottesman 2002; Kriner et al. 2016; Ray-Soni et al. 2016). Two approaches were taken to determine whether Rho has a role in deaD mRNA regulation. First, we used a strain that contains a temperature sensitive mutation in Rho, an essential protein. Wild-type and rhots derivatives of the WT and DM strains were initially grown at a semipermissive temperature (30°C), then transferred to 42°C, and cells were harvested 30 min later for RNA analysis. Northern-blot analysis of the RNA showed that the amount of full-length deaD mRNA was significantly increased in the rhots strain, suggesting that Rho regulates deaD mRNA synthesis (Fig. 3D). Additionally, the nearly 20-fold difference in mRNA levels between the WT and the DM strains was reduced to less than threefold between the rhots derivatives of these strains, indicating that regulation of deaD mRNA occurs primarily through a Rho-dependent mechanism, with the residual difference attributable to effects on mRNA stability. To further test the role of Rho in deaD autoregulation, bicyclomycin (BCM), a Rho inhibitor, was added to growing cultures, and RNA was harvested 20 min after addition of this antibiotic. As with the effects observed with the rhots strain, BCM addition was also found to increase deaD mRNA levels in the WT strain (Fig. 3E), albeit to a lower degree, possibly because of incomplete Rho inactivation at the BCM concentrations used. To rule out a possibility that DeaD might be affecting Rho activity indirectly, we used quantitative reverse transcription-PCR (qRT-PCR) to measure the abundance of a validated Rho target. One of the RNA regions whose transcription is sensitive to Rho activity lies downstream from the guaC coding region (Dar and Sorek 2018). Transcription over this region is antisense to the convergently oriented downstream hofC coding region, and Rho has been shown to be important for suppression of pervasive antisense transcription (Peters et al. 2012). As expected, the abundance of the guaC mRNA downstream region was dramatically increased upon Rho inactivation (Fig. 3F). In contrast, no such increase was observed in the DM strain, suggesting that the DM mutation does not affect Rho function, and thereby, the effects of the DM mutation on deaD mRNA levels (Fig. 2A) are not due to indirect effects on Rho activity. Collectively, these results suggest that DeaD regulates its own mRNA directly through both mRNA degradation and Rho-dependent transcription termination mechanisms.
Rho-dependent transcription termination in vitro
To further define the action of Rho on deaD mRNA, in vitro transcription reactions were performed. A DNA template that contains the deaD promoter and 1.0 kb of the downstream region was incubated with E. coli RNA polymerase in the presence of varying amounts of purified Rho protein. In the absence of added Rho, most of the transcripts synthesized during multiround transcription were found to correspond to the full-length runoff transcript (Fig. 4A, lane 1). The addition of Rho, however, resulted in multiple shorter transcripts primarily ranging in size from 400 to 900 nt, consistent with a role of Rho in promoting termination within the deaD mRNA (lanes 2–4), whereas the further addition of BCM abrogated Rho activity (lane 5). To compare the efficacy of Rho-dependent termination on deaD mRNA, transcription reactions were also performed on a template containing the rho gene, since Rho also autoregulates its own synthesis through transcription termination within the 5′ UTR of its own mRNA (Matsumoto et al. 1986). A comparison between the deaD and rho templates revealed that Rho-dependent termination in vitro was more efficient on the deaD template than on the rho template (Fig. 4B,C), consistent with the significant degree of regulation seen in vivo (Fig. 2).
FIGURE 4.

Rho promotes deaD mRNA transcription termination in vitro. (A) E. coli RNA polymerase and different amounts of purified Rho protein, as indicated, were added to a DNA template containing the deaD promoter and 955 bp of DNA downstream from the transcription initiation site. The radiolabeled transcription products were resolved by denaturing gel electrophoresis. The full-length runoff product is indicated by an arrow, whereas prematurely terminated transcripts are indicated by asterisks. Radiolabeled DNA molecular weight markers, run alongside the samples, are shown on the left, along with their sizes in nucleotides. (B) Transcription reactions were performed as in (A), except that a rho template was used. (C) Quantitation of transcription termination efficiency using deaD and rho templates. The relative amounts of full-length mRNA synthesis observed is plotted as a function of Rho concentration. Each data point represents an average from three to four replicates.
Given that multiple deaD mRNA termination products were observed in vitro, we investigated whether comparable products could also be seen in vivo by Northern blot analysis of RNA isolated from WT and DM strains using radiolabeled probes complementary to the 5′ UTR. However, no significant accumulation of short deaD mRNA transcripts that would have been consistent with Rho-dependent termination could be seen (not shown), suggesting that if such products arise, they might be getting rapidly degraded. Recent transcriptome-wide studies have shown that Rho-dependent termination products often undergo extensive degradation or 3′-end trimming (Dar and Sorek 2018). We also noted that although the results from Figure 3 indicate that Rho action on the deaD mRNA is stimulated by DeaD, prematurely terminated transcripts were observed in vitro even without DeaD addition, suggesting a basal level of transcription termination. Efforts to recapitulate enhanced transcription termination by adding purified DeaD to the in vitro reactions did not lead to increased termination, either because the conditions used for deaD mRNA transcription in vitro do not allow DeaD to stimulate Rho termination activity or because DeaD requires other cellular factors to do so in vivo.
Multiple regions in the deaD 5′ UTR contribute to feedback regulation
Given the results implicating DeaD in regulating its own mRNA, we hypothesized that the 5′ UTR plays a key role in mediating this response. To test whether the removal of the 5′ UTR affects the regulation of the deaD-lacZ fusion, a deletion from 101 to 820 nt upstream of the start codon was made, which upon testing in the WT and ΔdeaD strains was found to exhibit only a 1.4-fold difference between the two strains, in contrast with a 25-fold difference for the wild-type fusion (Fig. 5A). Thus, the 5′ UTR is critical for regulation of the deaD-lacZ fusions. Moreover, the loss of regulation as a consequence of an internal deletion downstream from the TSS suggests that changes in transcription initiation play little, if any, role in deaD regulation.
FIGURE 5.
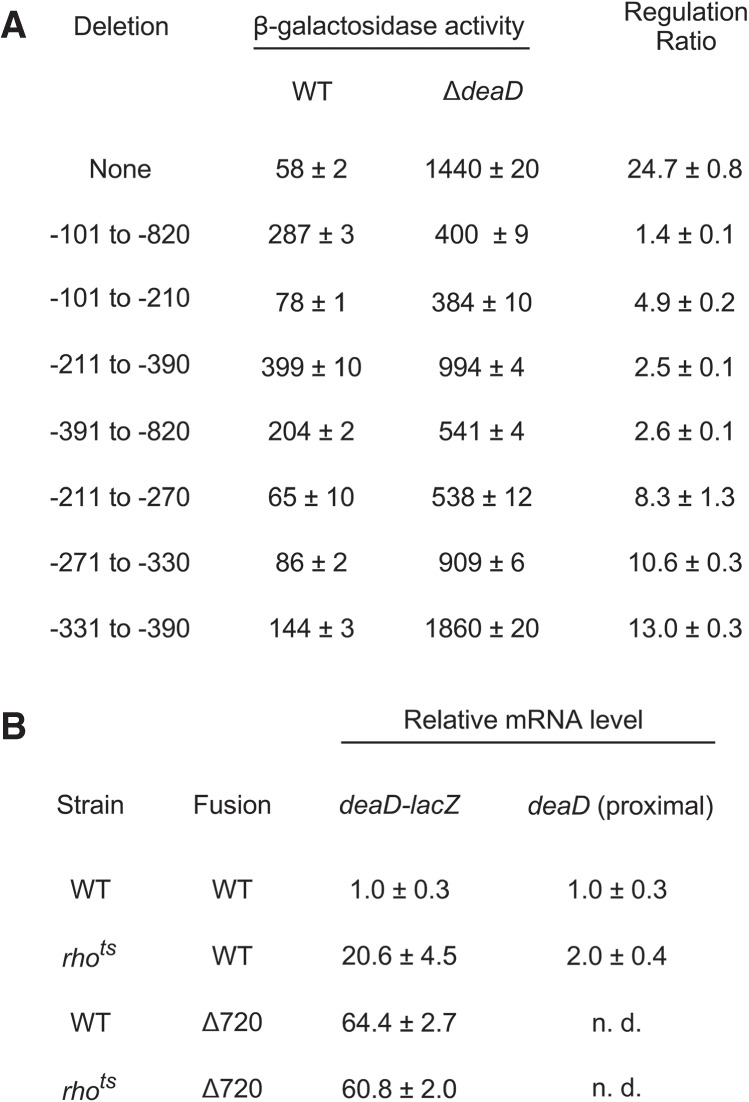
(A) Deletion mapping of regions important for autoregulation. A wild-type single-copy deaD-lacZ fusion or variants containing the indicated deletions within the deaD 5′ UTR were incorporated into ΔlacZ derivatives of the WT or ΔdeaD strains and the β-galactosidase activity of the resulting strains was measured. (B) RNA was isolated from WT or rhots strains containing a WT deaD-lacZ fusion or a fusion containing a 720 nt deletion in the deaD 5′ UTR after growth at 42°C for 30 min. The RNA samples were analyzed by qRT-PCR, with amplification performed either over the junction region of the deaD-lacZ transcript (100 nt of deaD and 24 nt of lacZ sequence) or a 123 nt region near the 5′ end of the transcript. n. d., not determined.
We next tested whether the effect of the 720 nt 5′ UTR deletion can be delimited to a smaller region. Accordingly, we made three smaller deletions that remove 110, 180, or 430 nt of the deaD 5′ UTR and tested them in the WT and ΔdeaD strain backgrounds. Each derivative deletion construct reduced the extent of feedback regulation by fivefold or more relative to the WT fusion, suggesting that they all contribute to the regulation of this transcript, although none of these deletions had as pronounced an effect on regulation as the larger 720 nt deletion.
We further investigated whether the reduced regulatory effect of the 180 nt deletion can be localized to a smaller region, and therefore, we tested three 60 nt deletions spanning this region. Each of the smaller deletions reduced the extent of regulation of the deaD-lacZ fusion, though none was as defective as the parental 180 nt deletion. Overall, these results suggest that multiple regions within its large 5′ UTR contribute to deaD autoregulation.
As the earlier results had indicated that deaD autoregulation is primarily regulated by Rho (Fig. 3D), based on the observation that regulation is lost when the 5′ UTR is deleted, we hypothesized that removal of the 5′ UTR within the context of a deaD-lacZ fusion should abolish Rho-dependence. To test that hypothesis, we first measured deaD-lacZ mRNA levels in WT and rhots strains that contained a WT deaD-lacZ fusion by using qRT-PCR and primers that amplify sequences at the deaD-lacZ junction. We found that the RNA levels within this region of the transcript were increased by 20-fold in the rhots strain, consistent with the effects of Rho-mediated negative regulation (Fig. 5B). In contrast, when the same analysis was performed on strains containing a deaD-lacZ fusion with a 720 nt 5′ UTR deletion, both the WT and rhots strains were found to display an equivalent increase in mRNA levels. These results suggest that deletion of the 5′ UTR abrogates Rho-dependent regulation of the deaD mRNA. As a control, to verify that the changes observed are not caused by differences in transcription initiation frequency, we also performed qRT-PCR over an initial part of the deaD mRNA, 16–138 nt downstream from the TSS. Since Rho generally requires a minimal region to load before it can confer termination (Kriner et al. 2016), it would be expected that any effects of Rho inactivation within the initial regions of a regulated transcript should be reduced. Consistent with this expectation, a relatively smaller twofold increase in the level of the deaD mRNA was observed in the rhots strain over the proximal region of this transcript.
DISCUSSION
DBPs represent a prominent class of RNA-remodeling factors whose functions have been of significant interest to understand how they remodel RNA structure to influence gene expression and RNA biology. However, the regulatory aspects of DBP expression have not been investigated in any significant detail. In this work, we have focused on the regulation of DeaD, one of the first DBPs to be identified. An analysis of the deaD transcript, as well as the mechanisms by which it is regulated, has led to several novel findings. First, we have found that the deaD mRNA contains an 838 nt 5′ UTR, which is longer than the longest primary 5′ UTR (300 nt) annotated in a genome-wide study (Thomason et al. 2015). The presence of such an unusually long 5′ UTR suggested that DeaD could be subject to multiple levels of regulation. Indeed, we show that DeaD regulates its own expression, arguably providing the first example of a DBP that is subject to autoregulatory control. Moreover, we have found that deaD mRNA is regulated at the level of both mRNA stability and transcription termination. To the best of our knowledge, such a dual level of regulation by a DBP on any transcript has not been reported before. However, examples of dual-level regulation at the both the mRNA stability and transcription termination level by metabolites that bind to riboswitches have been reported for the lysC riboswitch in E. coli and for the FMN riboswitch that regulates ribM expression in Corynebacterium glutamicum (Caron et al. 2012; Takemoto et al. 2015; Bastet et al. 2017).
In regard to regulation by transcription termination, we found that the Rho termination factor is important for this process, as Rho mutations or the addition of BCM significantly increased full-length transcript expression (Fig. 3D,E). In contrast, strains containing DeaD-inactivating mutations were found to express high levels of deaD mRNA even when Rho was functional. The latter observation suggests that DeaD activity is required to remodel deaD mRNA so that it becomes accessible to Rho function, leading to transcription termination. This example is reminiscent of control of gene expression by certain riboswitches where ligand-dependent changes in the 5′ UTR region enable Rho access (Hollands et al. 2012).
So far, prokaryotic DBPs have been found to regulate gene expression either by regulating translation or RNA stability (Khemici et al. 2005; Resch et al. 2010; Vakulskas et al. 2014). The finding that Rho plays a role in deaD regulation thus provides a new mechanism of gene expression regulation by an RNA helicase. We speculate that this mode of regulation is not unique to the deaD mRNA and anticipate that future studies will yield additional examples of DeaD, and possibly other DBPs, regulating gene expression through a transcription termination mechanism, rather than via translation or RNA degradation.
To identify the regions that could be important for DeaD autoregulation, we constructed a deaD-lacZ fusion construct and found that its activity was regulated by DeaD. However, a large 720 nt deletion within the deaD mRNA 5′ UTR resulted in an almost complete loss of regulation, indicating that deaD regulation is primarily mediated through its 5′ UTR (Fig. 5). We next attempted to determine whether the cis-acting elements that control regulation can be localized to a smaller region. Each of three smaller deletions of 110 to 430 nt reduced the extent of regulation even though none of these smaller deletions quantitatively captured the effect of the larger deletion, suggesting that the determinants for deaD autoregulation are spread out over the 5′ UTR. The contribution of multiple regions to autoregulation is also suggested by transcription termination assays, which showed Rho can terminate transcription at multiple sites within the deaD 5′ UTR in vitro (Fig. 4A).
Based on the results obtained, we suggest that the deaD mRNA initially adopts a conformation that contains multiple structured regulatory regions, which upon unwinding by DeaD, become sensitive to Rho and RNase E action. Thus, in the presence of DeaD, deaD mRNA undergoes enhanced levels of premature termination, whereas the small fraction of transcripts that escape premature termination get degraded by RNase E. The collective influence of these two processes is a reduction of deaD mRNA levels by ∼25-fold. This first example of multiple mechanisms used by a DBP to negatively regulate the production of its own transcript underscores the importance of tightly controlling any fluctuations in its expression inside the cell. We expect that future studies will reveal additional examples of autoregulatory control not only for DBPs but also for other RNA-remodeling factors that perform important regulatory functions inside the cell.
MATERIALS AND METHODS
Strains and plasmids
Strains were derived from MG1655*, a derivative of MG1655 that contains a point mutation in rph that restores the reading frame of its prematurely terminated gene product (Gutgsell and Jain 2012). ΔdeaD, ΔlacZ, and ΔpcnB mutations were derived from the Keio collection of strains containing the respective deletion alleles marked with a kanamycin-resistant (kanR) element, which were transferred to MG1655* by P1 transduction (Baba et al. 2006). For the ΔdeaD::kan allele, the kanR marker was removed by using FLP recombinase. Point mutations in deaD were introduced via two steps. First, deaD codons 48–339 were replaced by a tetA-sacB cassette by using recombineering (Li et al. 2013). Second, PCR products spanning the deleted deaD region and containing the desired mutations were recombined into the derivative strain and grown on plates containing fusaric acid and sucrose, which provide a counter-selection against the tetA-sacB cassette. Colonies growing under the selection conditions were isolated and sequenced to confirm the presence of the desired mutations. A strain containing a rhots allele was obtained from Dr. Sankar Adhya, and the rhots allele was transduced into MG1655* by P1 transduction using a linked ilvC::kan marker (Gulletta et al. 1983). An rnets allele (rne-3071) was derived from strain CJ1826 (Jain and Belasco 1995) and transduced to recipient strains using a linked plsX::chloramphenicol-resistant marker.
The deaD-lacZ plasmid was constructed by cloning PCR amplified deaD DNA spanning from 973 bp upstream of the coding region to 100 bp downstream between the BamHI and XhoI sites of pLACZY1A, a ColE1-based plasmid with expected copy number of ∼20/cell (Jain 1993). Derivatives of this plasmid were made using a Q5 site-directed mutagenesis kit (NEB). Single copy deaD-lacZ fusions were made by recombining the plasmid-borne fusions with modified λ phage, integrating the recombinant phage at the λ att site on the chromosome, and identifying monolysogens by PCR-based screening (Simons et al. 1987; Powell et al. 1994).
Northern blotting
Total RNA was isolated using the hot-phenol method. Equal amounts of total RNA samples were fractionated on 1.2% agarose gels in MOPS buffer containing 0.66 M formaldehyde. After electrophoresis, the RNA was transferred to positively charged membrane (Nytran), cross-linked using 254 nM ultraviolet light, and hybridized overnight with radiolabeled riboprobes at 65°C. The DNA template for riboprobe synthesis was made by PCR using E. coli DNA as a template and primers with the sequences 5′-AATTTAATACGACTCACTATAGGTTACGCATCACCACCGAAAC-3′ and 5′-GTTCGTCATATCGTTGGTGC-3′. Riboprobe synthesis was performed using the HiScribe kit (NEB) with γ-32P UTP added. For mRNA half-life measurements, samples of total cellular RNA were isolated from cultures after rifampicin addition to a final concentration of 200 µg/mL. All experiments were performed a minimum of three times.
In vitro transcription termination assays
Transcription reactions were performed on a 1.1 kb DNA template synthesized by using PCR on E. coli DNA. The sequences for the primers used to synthesize the deaD template were 5′-CGGTACCATTGCAACCGACTTTAC-3′ and 5′-ATACACTCTGCCTGAATTGG-3′. The primer sequences for synthesis of a 0.4 kb rho template were 5′-TGGACGCCCGGCGTGAGTCA-3′ and 5′-GCAAAAATAATGTCCTGCTT-3′. Transcription reactions, performed in 10 µL, contained 10 ng of template in 10% v/v glycerol, 40 mM Tris–HCl (pH 7.9), 50 mM KCl, 5 mM MgCl2, 0.1 mM dithiothreitol, 1 mM ATP, GTP and CTP, 20 µM UTP, 1 µCi γ-32P UTP (3000 Ci/mmol), and 1 unit E. coli RNA polymerase holoenzyme (NEB). The reactions were supplemented with Rho and BCM (100 µM), as indicated. After incubation for 20 min at 37°C, the reactions were terminated by adding 2 µL of acidic phenol, and the samples were vortexed and centrifuged. 7 µL of the aqueous phase was removed and added to 5 µL of loading dye. The samples were heated at 95°C for 5 min and the transcription products were resolved on denaturing 4% polyacrylamide gel and visualized using a Personal Molecular Imager (BioRad).
5′ RACE
Primer extension was performed on RNA from a WT strain as previously described (Diwa et al. 2000), using a primer with the sequence 5′-CAGAATCAAACGCTTCATAG-3′. The cDNA product was precipitated and 1 µL of the cDNA product was added to 1 µL of a 5′ phosphorylated, 3′ blocked oligonucleotide (5′-AGATCGGAAGAGCACACGTCT-NH2-3′) at 1 µM and 1 µL of dimethyl sulfoxide. The mix was heated to 75°C for three min and placed on ice. The reaction was supplemented with 3 µL of T4 RNA ligase I (NEB), 1.5 µL of 10× reaction buffer, 6.5 µL of 50% PEG-8000, and 1 µL of 10 mM ATP. The reaction was incubated at room temperature for 1 h and an aliquot was used to amplify the ligation product using primers with the sequences 5′-CCTAAGTAATTGAATACTTC-3′ and 5′-AGACGTGTGCTCTTCCGATCT-3′. The PCR product was cloned into pGEM-T easy (Promega), amplified by using primers with the sequences 5′-AGTCACGACGTTGTAAAACG-3′ and 5′-GAGCGGATAACAATTTCACACAGG-3′, and the resulting product was sequenced using Sanger sequencing (Genewiz).
qRT-PCR
One microgram of total RNA was treated with 0.4 units of DNase I (Promega) and 0.2 units of Turbo-DNase (Ambion) in 1× DNase buffer for 45 min at 37°C in a volume of 10 µL. An amount of 90 µL of water was added to these reactions and 2 µL of each mix was used for qRT-PCR using the Luna qRT-PCR kit from New England Biolabs (cat # E3005) and 200 nM of primers in a total volume of 5 µL using a BioRad CFX 96 instrument. The sequences of the primers used for quantification of RNA downstream from guaC mRNA were 5′-GCAACTTATTGGCTATCGCC-3′ and 5′-GGCACTAACGCAGGGGATC-3′; for quantification of deaD mRNA were 5′-CGCGTAACGATTTTTCGCAAGCG-3′ and 5′-CAGAATCAAACGCTTCATAG-3′; and for quantification of the deaD-lacZ fusion RNA were 5′-GATGGCTGAATTCGAAA-3′ and 5′-AGTCACGACGTTGTAAAACG-3′. ΔCt measurements were converted into fold-differences assuming an amplification efficiency of two per PCR cycle. All RNA quantitation measurements reported are based on three to four replicates, with standard errors indicated.
Protein analysis
Cell extracts were prepared by heating cells resuspended in sample buffer at 100°C for 10 min. The cells were fractionated on a 4%–15% gel (BioRad) and the gels were stained using PageBlue protein staining solution (Fermentas).
β-galactosidase assays
β-galactosidase assays were performed as previously described (Jain and Belasco 2000). The β-galactosidase activities from deaD-lacZ plasmids or single-copy fusions were measured in ΔlacZ derivatives of the WT and ΔdeaD strains. All β-galactosidase values reported are based on three to seven replicates, with standard errors indicated.
ACKNOWLEDGMENTS
We thank Dr. Evgeny Nudler (New York University) for the gift of purified Rho protein. This work was supported by grant GM114540 from the National Institutes of Health.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.074112.119.
REFERENCES
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006 0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastet L, Chauvier A, Singh N, Lussier A, Lamontagne AM, Prevost K, Masse E, Wade JT, Lafontaine DA. 2017. Translational control and Rho-dependent transcription termination are intimately linked in riboswitch regulation. Nucleic Acids Res 45: 7474–7486. 10.1093/nar/gkx434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Krogan NJ, Xu J, Yang WH, Aoki H, Li JS, Krogan N, Menendez J, Cagney G, Kiani GC, et al. 2007. Investigating the in vivo activity of the DeaD protein using protein-protein interactions and the translational activity of structured chloramphenicol acetyltransferase mRNAs. J Cell Biochem 100: 642–652. 10.1002/jcb.21016 [DOI] [PubMed] [Google Scholar]
- Caron MP, Bastet L, Lussier A, Simoneau-Roy M, Masse E, Lafontaine DA. 2012. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc Natl Acad Sci 109: E3444–E3453. 10.1073/pnas.1214024109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charollais J, Pflieger D, Vinh J, Dreyfus M, Iost I. 2003. The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli. Mol Microbiol 48: 1253–1265. 10.1046/j.1365-2958.2003.03513.x [DOI] [PubMed] [Google Scholar]
- Charollais J, Dreyfus M, Iost I. 2004. CsdA, a cold-shock RNA helicase from Escherichia coli, is involved in the biogenesis of 50S ribosomal subunit. Nucleic Acids Res 32: 2751–2759. 10.1093/nar/gkh603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar D, Sorek R. 2018. High-resolution RNA 3′-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res 46: 6797–6805. 10.1093/nar/gky274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwa A, Bricker AL, Jain C, Belasco JG. 2000. An evolutionarily conserved RNA stem-loop functions as a sensor that directs feedback regulation of RNase E gene expression. Genes Dev 14: 1249–1260. [PMC free article] [PubMed] [Google Scholar]
- Ettwiller L, Buswell J, Yigit E, Schildkraut I. 2016. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genomics 17: 199 10.1186/s12864-016-2539-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulletta E, Das A, Adhya S. 1983. The pleiotropic ts15 mutation of E. coli is an IS1 insertion in the rho structural gene. Genetics 105: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell NS, Jain C. 2012. Gateway role for rRNA precursors in ribosome assembly. J Bacteriol 194: 6875–6882. 10.1128/JB.01467-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollands K, Proshkin S, Sklyarova S, Epshtein V, Mironov A, Nudler E, Groisman EA. 2012. Riboswitch control of Rho-dependent transcription termination. Proc Natl Acad Sci 109: 5376–5381. 10.1073/pnas.1112211109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iost I, Bizebard T, Dreyfus M. 2013. Functions of DEAD-box proteins in bacteria: current knowledge and pending questions. Biochim Biophys Acta 1829: 866–877. 10.1016/j.bbagrm.2013.01.012 [DOI] [PubMed] [Google Scholar]
- Jagessar KL, Jain C. 2010. Functional and molecular analysis of Escherichia coli strains lacking multiple DEAD-box helicases. RNA (New York, NY) 16: 1386–1392. 10.1261/rna.2015610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C. 1993. New improved lacZ gene fusion vectors. Gene 133: 99–102. 10.1016/0378-1119(93)90231-Q [DOI] [PubMed] [Google Scholar]
- Jain C, Belasco JG. 1995. RNase E autoregulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: unusual sensitivity of the rne transcript to RNase E activity. Genes Dev 9: 84–96. 10.1101/gad.9.1.84 [DOI] [PubMed] [Google Scholar]
- Jain C, Belasco JG. 2000. Rapid genetic analysis of RNA-protein interactions by translational repression in Escherichia coli. Methods Enzymol 318: 309–332. 10.1016/S0076-6879(00)18060-7 [DOI] [PubMed] [Google Scholar]
- Jones PG, Mitta M, Kim Y, Jiang W, Inouye M. 1996. Cold shock induces a major ribosomal-associated protein that unwinds double-stranded RNA in Escherichia coli. Proc Natl Acad Sci 93: 76–80. 10.1073/pnas.93.1.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khemici V, Poljak L, Toesca I, Carpousis AJ. 2005. Evidence in vivo that the DEAD-box RNA helicase RhlB facilitates the degradation of ribosome-free mRNA by RNase E. Proc Natl Acad Sci 102: 6913–6918. 10.1073/pnas.0501129102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriner MA, Sevostyanova A, Groisman EA. 2016. Learning from the leaders: gene regulation by the transcription termination factor Rho. Trends Biochem Sci 41: 690–699. 10.1016/j.tibs.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XT, Thomason LC, Sawitzke JA, Costantino N, Court DL. 2013. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res 41: e204 10.1093/nar/gkt1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder P, Jankowsky E. 2011. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12: 505–516. 10.1038/nrm3154 [DOI] [PubMed] [Google Scholar]
- Mackie GA. 2013. RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol 11: 45–57. 10.1038/nrmicro2930 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Shigesada K, Hirano M, Imai M. 1986. Autogenous regulation of the gene for transcription termination factor rho in Escherichia coli: localization and function of its attenuators. J Bacteriol 166: 945–958. 10.1128/JB.166.3.945-958.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR. 2016. Regulation of mRNA decay in bacteria. Annu Rev Microbiol 70: 25–44. 10.1146/annurev-micro-091014-104515 [DOI] [PubMed] [Google Scholar]
- Nudler E, Gottesman ME. 2002. Transcription termination and anti-termination in E. coli. Genes Cells 7: 755–768. 10.1046/j.1365-2443.2002.00563.x [DOI] [PubMed] [Google Scholar]
- Peters JM, Mooney RA, Grass JA, Jessen ED, Tran F, Landick R. 2012. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev 26: 2621–2633. 10.1101/gad.196741.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell BS, Rivas MP, Court DL, Nakamura Y, Turnbough CL Jr. 1994. Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res 22: 5765–5766. 10.1093/nar/22.25.5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme-Genereux A, Beran RK, Iost I, Ramey CS, Mackie GA, Simons RW. 2004. Physical and functional interactions among RNase E, polynucleotide phosphorylase and the cold-shock protein, CsdA: evidence for a ‘cold shock degradosome.’ Mol Microbiol 54: 1409–1421. 10.1111/j.1365-2958.2004.04360.x [DOI] [PubMed] [Google Scholar]
- Ray-Soni A, Bellecourt MJ, Landick R. 2016. Mechanisms of bacterial transcription termination: all good things must end. Annu Rev Biochem 85: 319–347. 10.1146/annurev-biochem-060815-014844 [DOI] [PubMed] [Google Scholar]
- Redder P, Hausmann S, Khemici V, Yasrebi H, Linder P. 2015. Bacterial versatility requires DEAD-box RNA helicases. FEMS Microbiol Rev 39: 392–412. 10.1093/femsre/fuv011 [DOI] [PubMed] [Google Scholar]
- Resch A, Vecerek B, Palavra K, Blasi U. 2010. Requirement of the CsdA DEAD-box helicase for low temperature riboregulation of rpoS mRNA. RNA Biol 7: 796–802. 10.4161/rna.7.6.13768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocak S, Linder P. 2004. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5: 232–241. 10.1038/nrm1335 [DOI] [PubMed] [Google Scholar]
- Simons RW, Houman F, Kleckner N. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53: 85–96. 10.1016/0378-1119(87)90095-3 [DOI] [PubMed] [Google Scholar]
- Takemoto N, Tanaka Y, Inui M. 2015. Rho and RNase play a central role in FMN riboswitch regulation in Corynebacterium glutamicum. Nucleic Acids Res 43: 520–529. 10.1093/nar/gku1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason MK, Bischler T, Eisenbart SK, Forstner KU, Zhang A, Herbig A, Nieselt K, Sharma CM, Storz G. 2015. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197: 18–28. 10.1128/JB.02096-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toone WM, Rudd KE, Friesen JD. 1991. deaD, a new Escherichia coli gene encoding a presumed ATP-dependent RNA helicase, can suppress a mutation in rpsB, the gene encoding ribosomal protein S2. J Bacteriol 173: 3291–3302. 10.1128/JB.173.11.3291-3302.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulskas CA, Pannuri A, Cortes-Selva D, Zere TR, Ahmer BM, Babitzke P, Romeo T. 2014. Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92: 945–958. 10.1111/mmi.12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu FF, Gaggero C, Cohen SN. 2002. Polyadenylation can regulate ColE1 type plasmid copy number independently of any effect on RNAI decay by decreasing the interaction of antisense RNAI with its RNAII target. Plasmid 48: 49–58. 10.1016/S0147-619X(02)00023-9 [DOI] [PubMed] [Google Scholar]