

Supplemental Digital Content is Available in the Text.
Placebo and tramadol-controlled study assessing long-term safety and efficacy of tanezumab in patients with chronic low back pain and inadequate response to standard analgesics.
Keywords: Tanezumab, Chronic low back pain, Clinical study, Nerve growth factor
Abstract
This randomized, double-blind, phase 3 study (56-week treatment; 24-week follow-up) assessed tanezumab in patients with chronic low back pain and history of inadequate response to standard-of-care analgesics (NCT02528253). Patients received placebo, subcutaneous tanezumab (5 or 10 mg every 8 weeks), or oral tramadol prolonged-release (100-300 mg/day). Primary endpoint was change in low back pain intensity (LBPI) at week 16 for tanezumab vs placebo. Key secondary endpoints were proportion of patients with ≥50% decrease in LBPI at week 16, change in Roland Morris Disability Questionnaire at week 16, and change in LBPI at week 2 for tanezumab vs placebo. Adverse events and joint safety were assessed through weeks 56 and 80, respectively. Tanezumab 10 mg met the primary endpoint by significantly improving LBPI at week 16 vs placebo; least squares (LS) mean (95% CI) difference = −0.40 (−0.76 to −0.04; P = 0.0281). Tanezumab 10 mg significantly improved all key secondary endpoints. Tanezumab 5 mg did not meet the primary endpoint (LS mean [95% CI] treatment difference vs placebo = −0.30 [−0.66 to 0.07; P = 0.1117]), preventing formal testing of key secondary endpoints for this dose. The proportion of patients with ≥50% improvement in LBPI at week 16 was 37.4% in the placebo group, 43.3% in the tanezumab 5 mg group (Odds ratio [95% CI] vs placebo = 1.28 [0.97 to 1.70; P = 0.0846]), and 46.3% in the tanezumab 10 mg group (Odds ratio [95% CI] vs placebo = 1.45 [1.09 to 1.91; P = 0.0101]). Prespecified joint safety events were more frequent with tanezumab 10 mg (2.6%) than tanezumab 5 mg (1.0%), tramadol (0.2%), or placebo (0%). Seven patients, all in the tanezumab 10 mg group (1.4%), underwent total joint replacement. In conclusion, tanezumab 10 mg significantly improved pain and function vs placebo in patients with difficult-to-treat chronic low back pain. Tanezumab was associated with a low rate of joint safety events, some requiring joint replacement.
1. Introduction
Chronic low back pain (CLBP) is a leading contributor to global disability.14 A multidisciplinary approach is often used to manage CLBP.1,29 Among pharmacologic options, nonsteroidal anti-inflammatory drugs (NSAIDs) are recommended as first-line therapy, and duloxetine or tramadol as second-line.29 Opioids are also commonly prescribed for CLBP in North America despite being recommended only as a last resort due to safety concerns, lack of functional improvement, and scarcity of long-term data.3,4,7,29,32
Recognition of the role nociceptor sensitization plays in the pathogenesis of CLBP has spurred characterization of novel drug targets that modulate hyperalgesia, and accumulating evidence indicates that nerve growth factor (NGF) is a promising target in CLBP syndromes.2,6,24 Preclinical models demonstrate that a majority of fibers innervating lumbar intervertebral disks express NGF receptors.2 Administration of exogenous NGF triggers prolonged myofascial pain in the lumbar paraspinal muscles of healthy adults.34 Studies have also detected increased nerve density accompanied by higher levels of NGF in disks resected from spinal segments associated with painful symptoms as compared with disk material sampled from asymptomatic segments.12 A recent systematic review identified the need for further study of anti-NGF therapies in CLBP populations refractory to commonly used therapies and the need for long-term follow-up to assess adverse events (AEs).22
Tanezumab is a monoclonal antibody against NGF.15 A subcutaneous (SC) formulation of tanezumab has recently demonstrated efficacy in a pivotal 16-week dose-titration phase 3 study for osteoarthritis (OA) pain.31 Previous phase 2 studies in patients with nonradicular CLBP have demonstrated efficacy for intravenous (IV) tanezumab, up to 16 weeks, vs placebo and naproxen.19,21 Here, we report findings from the first phase 3 study to assess SC tanezumab (5 and 10 mg) in patients with CLBP and a history of inadequate response (insufficient pain relief at maximum tolerated dose or intolerance/contraindication) to standard-of-care analgesics. Patients with more than mild radiographic and clinical evidence of OA were excluded from the study because advanced evidence of OA seems to increase the risk of joint safety events in patients exposed to higher doses of tanezumab.17 Thus, the study design aimed to assess tanezumab efficacy and safety in a treatment refractory population at potentially lower risk for joint safety events. The primary objective was to evaluate efficacy of SC tanezumab vs placebo after 16 weeks of treatment. Secondary objectives were to evaluate long-term (56 week) safety and efficacy of tanezumab, including comparisons to oral tramadol.
2. Methods
2.1. Study oversight
This randomized, double-blind, placebo- and tramadol-controlled, parallel-group, phase 3 study (ClinicalStudys.gov: NCT02528253) was conducted from August 2015 to December 2018 at 191 sites in the United States, Canada, France, Hungary, Japan, Spain, Sweden, and South Korea. The protocol was approved by an institutional review board or independent ethics committee for each participating investigational center, and all patients provided written informed consent. This study was conducted in compliance with ethical principles of the Declaration of Helsinki and Good Clinical Practice Guidelines.
2.2. Patients
Investigators recruited patients from their own practices and institutions. On receiving informed consent, the site investigator evaluated the patient based on inclusion/exclusion criteria described in the study protocol. The screening period (maximum duration of 37 days) included a washout period for prohibited medications (minimum 2 days for all medications that have an elimination half-life of less than 10 hours or at least 5 times the elimination half-life for medications with longer half-lives) and an initial pain assessment period (the 5 days immediately preceding randomization). During the screening period, patients were trained in the use of the sponsor's Interactive Response Technology to assess pain, NSAID use, and rescue medication use throughout the trial.
Patients aged 18 years and older with axial predominant CLBP (primary location between the 12th thoracic vertebra and lower gluteal folds, with or without radiation into the posterior thigh) of ≥3 months' duration were eligible. Key inclusion criteria included an average low back pain intensity (LBPI) score ≥5 (on an 11-point numeric rating scale; patients must have completed at least 4 daily pain diaries during the 5 day initial pain assessment period), Patient's Global Assessment of Low Back Pain score of fair, poor, or very poor (on a 5-point scale from very good to very poor), and history of inadequate response (insufficient pain relief at maximum tolerated dose or intolerance/contraindication) to ≥3 different categories of standard-of-care analgesics. These included acetaminophen/paracetamol, nonprescription NSAIDs, prescription NSAIDs, opioids (excluding tramadol), tapentadol, tricyclic antidepressants, benzodiazepines or muscle relaxants, lidocaine patch, and serotonin–norepinephrine reuptake inhibitors. Key exclusion criteria included history of lumbosacral radiculopathy and a diagnosis of OA of the knee or hip based on American College of Rheumatology combined clinical and radiographic criteria. Patients with Kellgren–Lawrence (KL) radiographic evidence of hip (grade ≥ 2) or knee (grade ≥ 3) OA were excluded (asymptomatic knee KL grade ≤2 was allowed). Patients with symptoms and radiographic evidence of shoulder OA were excluded. Other exclusionary criteria included history or evidence of spinal disease (eg, ankylosing spondylitis, rheumatoid arthritis, tumor, and Paget disease), clinically significant cardiac disease, neurological disease, or psychiatric disorder, and other conditions that could confound assessment of CLBP (eg, fibromyalgia and back pain referred from visceral sources).
2.3. Randomization and masking
The study consisted of 56-week double-blind treatment followed by 24-week safety follow-up. A computer-generated (Oracle Clinical, Redwood Shores, CA), blocked, static randomization scheme assigned patients in a 1:1:2:2:3 ratio to placebo/SC tanezumab 5 mg (switch from placebo to tanezumab at week 16), placebo/SC tanezumab 10 mg (switch from placebo to tanezumab at week 16), SC tanezumab 5 mg, SC tanezumab 10 mg, or oral tramadol prolonged-release (Fig. 1). All patients, investigators, and staff (sponsor and site) involved in the study were blinded to treatment allocation.
Figure 1.

Study design. Subcutaneous (SC) treatment was administered every 8 weeks and oral treatment was administered daily. At week 16, patients in the placebo arm were transitioned in a blinded 1:1 ratio to tanezumab 5 or 10 mg. Oral treatment was initiated at 100 mg/day and could be adjusted in 100 mg increments at weeks 1, 2, 3, and 4 to a maximum of 300 mg/day. The dose of oral medication remained stable from weeks 5 through 56 (titration of oral medication was allowed in Europe after week 16). Scheduled in-clinic visits occurred at baseline, and weeks 2, 4, 8, 16, 24, 32, 40, 48, 56, 64, and 80. Scheduled phone contact with the subject occurred at weeks 1, 3, 12, 20, 28, 36, 44, 52, 60, 68, 72, and 76. *Before receiving treatment at week 16, patients must have had a ≥30% reduction from baseline in average low back pain intensity (LBPI) score at week 16 and a ≥15% reduction from baseline in mean weekly LBPI score at any week from weeks 1 through 15 to continue the study. †Before receiving treatment at week 32, patients must have had a ≥30% reduction from baseline in LBPI score to continue the study. Numbers based on the safety population.
2.4. Interventions
Oral tramadol prolonged-release (tablet) was administered once daily with titration from 100 mg/day up to a maximum of 300 mg/day, based on efficacy and tolerability, over 4 weeks. Tanezumab was provided in a prefilled syringe of 1 mL volume and was administered subcutaneously in the abdomen or thigh by site staff every 8 weeks. Subcutaneous and oral placebo were provided and administered in an identical manner (minus active ingredient) to tanezumab and tramadol.
Before third SC administration at week 16, patients were required to demonstrate i) a ≥30% reduction from baseline in average LBPI at week 16 and ii) a ≥15% reduction from baseline in mean weekly LBPI at any week from weeks 1 through 15 to continue the study. Before fifth SC administration at week 32, patients were required to demonstrate a ≥30% reduction from baseline in LBPI to continue. These requirements were included in the study design with safety in mind, to ensure only patients with consistent clinical pain response across the initial 16-week placebo-controlled period continued onto the non–placebo-controlled later stages of the trial.
Rescue medication (acetaminophen/paracetamol; max daily dose of 3000 mg/day) was allowed daily during screening (except 24 hours before baseline), ≤3 days/week from baseline through week 16, and daily thereafter. Adjunctive analgesics including pregabalin, gabapentin, skeletal muscle relaxants, benzodiazepines, sedative/hypnotics, anxiolytics, antidepressants (except monoamine oxidase inhibitors), and topical analgesics were allowed after week 16; NSAIDs and opioids remained prohibited. During the study, patients were permitted to continue with preexisting nonpharmacologic treatment regimens, but it was required that such regimens remained stable and that new treatments were not initiated until after week 16.
2.5. Assessments and endpoints
Efficacy was characterized by LBPI and Roland Morris Disability Questionnaire (RMDQ) scores. Patients used a handheld device (daily) to record average LBPI over the past 24 hours on a scale from 0 = no pain to 10 = worst possible pain. The 24-item RMDQ assessed current functional status at baseline and weeks 2, 4, 8, 16, 24, 32, 40, 48, 56, 64, and 80 with scores ranging from 0 = no disability to 24 = severe disability.
Primary endpoint was change in LBPI from baseline to week 16 (tanezumab vs placebo). Key secondary endpoints were the percentage of patients with ≥50% reduction from baseline in LBPI at week 16, change in RMDQ from baseline to week 16, and change in LBPI from baseline to week 2 (tanezumab vs placebo). Other secondary endpoints included weekly changes from baseline in LBPI and RMDQ through week 16 (tramadol vs placebo and vs tanezumab) and through week 56 (tanezumab vs tramadol), and the proportion of patients with ≥30% reduction from baseline in LBPI at week 16 (tanezumab vs placebo and vs tramadol, tramadol vs placebo). Low back pain intensity for a particular week was defined as the mean of all daily scores for that week.
Safety assessments included treatment-emergent AE reporting, physical examinations, laboratory tests, vital signs, 12-lead electrocardiograms, and tanezumab antibody assessments. Adverse events were coded using Medical Dictionary for Regulatory Activities version 21.1 with severity and causality assessed by investigators. Radiographs of the shoulders, knees, and hips were required for all patients at screening, week 24, week 56, and week 80 or early termination, and were reviewed by a central reader for identification of potential joint safety events. Potential joint safety events, including total joint replacement (TJR), were adjudicated by a blinded external interdisciplinary adjudication committee. Events adjudicated as rapidly progressive OA (RPOA) type 1 or 2, primary osteonecrosis, subchondral insufficiency fracture, or pathologic fracture were included in a composite joint safety endpoint. Rapidly progressive OA type 1 was defined as a significant loss of joint space width ≥2 mm (predicated on optimal joint positioning) within approximately 1 year, without gross structural failure.26 Rapidly progressive OA type 2 was defined as abnormal bone loss or destruction, including limited or total collapse of at least 1 subchondral surface, which is not normally present in conventional end-stage OA.26
2.6. Statistical analyses
A sample size of 400 patients per group provides ≥80% power to achieve significance (2-sided 5% level) for comparisons of tanezumab vs placebo for the primary LBPI endpoint, based on expected placebo-adjusted decreases of 0.5, 0.9, and 0.4 points from baseline in LBPI at week 16 in the tanezumab 5 mg, tanezumab 10 mg, and tramadol groups, respectively, and a within-group standard deviation of 2.4. These differences were based on actual observed differences in a phase 2b dose ranging study of tanezumab in patients with CLBP and in dose-response modeling studies.21 To balance groups after week 16, the planned number of patients randomized at baseline was increased to 1800 (tramadol N = 600; other groups N = 400 each).
Testing of primary and key secondary endpoints followed a graphical approach of gatekeeping strategy to control the family-wise type 1 error rate of 5% (2-sided) across multiple comparisons.5 First, tanezumab 10 mg was tested vs placebo for LBPI at week 16 at α = 0.05. Pending the outcome of this analysis, remaining primary and key secondary endpoints were analyzed in the prespecified hierarchy presented in Figure S1 (available at http://links.lww.com/PAIN/B46) until all endpoints were tested or no remaining hypotheses could be rejected at their corresponding α level. No control for multiplicity was performed for analyses beyond the primary and key secondary endpoints.
Continuous endpoints were assessed using an analysis of covariance model with treatment as a fixed effect; baseline score (and baseline LBPI score for RMDQ endpoints) as covariate; and study site as a random effect. A multiple imputation approach was used for missing data, with the method for imputation dependent on the reason for missing data. Data missing due to treatment discontinuation due to death, AEs, insufficient clinical response, or not meeting prespecified efficacy criteria used a multiple imputation version of a baseline-observation-carried-forward single imputation method. Data missing due to other reasons used a multiple imputation version of a last-observation-carried-forward single imputation method. The 30% and 50% LBPI responder endpoints were assessed using a logistic regression model with treatment as a fixed effect, baseline LBPI score as a covariate, and a mixed baseline-observation-carried-forward/last-observation-carried-forward approach for missing data. Statistical analyses were performed using SAS version 9.4 (Cary, NC). Adverse events and adjudication of joint safety events, including the composite joint safety endpoint, were summarized descriptively. Adverse events were summarized through the initial 16- and full 56-week treatment periods. Joint safety was summarized through 80 weeks, recognizing that joint events may have a delayed presentation.
Efficacy analyses included all patients who received ≥1 dose of SC study medication grouped according to their assigned treatment (intention-to-treat). Safety analyses included all patients who received ≥1 dose of SC study medication grouped according to treatment received (3 patients randomized to tramadol did not receive treatment were grouped to placebo). Figure 2 shows the number of patients included in efficacy and safety analyses for each group.
Figure 2.

Patient disposition. Efficacy analyses included all patients who received ≥1 dose of SC study medication grouped according to their assigned treatment (intention-to-treat). Safety analyses included all patients who received ≥1 dose of SC study medication grouped according to treatment received. Three patients randomized to tramadol received SC placebo but did not receive oral tramadol treatment. These patients, therefore, were assigned to the “placebo to SC tanezumab 5 mg” group for the safety population. Thus, in the “placebo to SC tanezumab 5 mg” group, there were 3 more patients in the safety population (205) than were randomized (202), leading to >100% for the safety population (all % are based on N patients randomized). *Other screened but not randomized indicates patients who were screened but not randomized for a reason not related to a specific eligibility criterion. †At week 16, patients receiving placebo were switched in a blinded 1:1 manner to 5 mg or 10 mg subcutaneous (SC) tanezumab. Patients in the placebo to SC tanezumab 5 mg and placebo to SC tanezumab 10 mg were analyzed as a single placebo group up to week 16. ‡Percentages based on the number of patients randomized. §Percentages based on the safety population. Before receiving treatment at week 16, patients must have had a ≥30% reduction in average low back pain intensity (LBPI) score at week 16 and a ≥15% reduction in mean weekly LBPI score at any week from weeks 1 through 15 to continue the study. ‖Before receiving treatment at week 32, patients must have had a ≥30% reduction in LBPI score to continue the study. If a patient withdrew due to efficacy reasons other than the specific criteria at weeks 16 and 32, the reason was classified as “insufficient clinical response.
Key secondary endpoints in the study protocol were specified as change from baseline to week 16 in RMDQ (tanezumab vs placebo) and change from baseline to week 16 in LBPI (tanezumab vs tramadol). Before unblinding, database lock, and finalization of the analysis plan, key secondary endpoints were amended to those reported in the manuscript.
2.7. Role of the funding source
The study was sponsored by Pfizer Inc. (manufacturer of tanezumab) and Eli Lilly and Company. Authors from Pfizer Inc. and Eli Lilly and Company contributed to study design; data collection (Pfizer), management (Pfizer), and interpretation of data; and preparation, review, and approval of the manuscript. All authors had full access to study data and final responsibility for submission.
3. Results
3.1. Patients
A total of 6518 patients were screened, 1832 were randomized, and 1825 received ≥1 dose of SC medication. Approximately 65% of patients discontinued treatment by week 56, commonly for reasons related to inadequate efficacy (Fig. 2). Patient demographics and baseline characteristics were similar across groups (Table 1). Overall, 57.0% of patients were female and 72.4% were white. Mean age was approximately 49 years. Mean baseline LBPI and RMDQ scores were approximately 7.2 and 15.0, respectively. Common prior inadequate treatments for CLBP were acetaminophen or low-dose NSAIDs (91.7%), prescription NSAIDs (85.8%), and opioids (70.2%) (Table S1, available at http://links.lww.com/PAIN/B46). Mean daily dose of tramadol was 203 mg and 209 mg at weeks 16 and 56, respectively.
Table 1.
Subject demographics and baseline characteristics.

There was little use of non-NSAID concomitant drug treatments for CLBP during the first 16 weeks because they were prohibited (placebo = 2.9%, tanezumab 5 mg = 3.2%, tanezumab 10 mg = 2.7%, and tramadol = 1.8%) and no individual concomitant treatment was markedly more frequent than others. The use of non-NSAID concomitant drug treatments through week 64 was 9.3%, 13.4%, 13.3%, and 11.8% in the placebo, tanezumab 5 mg, tanezumab 10 mg, and tramadol groups, respectively. The only individual concomitant treatment taken by more than 2% of patient in any group was tramadol (placebo = 1.4%, tanezumab 5 mg = 3.6%, tanezumab 10 mg = 3.6%, and tramadol = 3.3%).
3.2. Primary endpoint
Tanezumab 10 mg met the primary endpoint by providing significantly greater improvement in LBPI at week 16 vs placebo; least squares (LS) mean (95% confidence interval [CI]) difference = −0.40 (−0.76 to −0.04; P = 0.0281) (Fig. 3A). Improvements in LBPI with tanezumab 5 mg were not significantly different from placebo at week 16; LS mean (95% CI) difference = −0.30 (−0.66 to 0.07; P = 0.1117).
Figure 3.
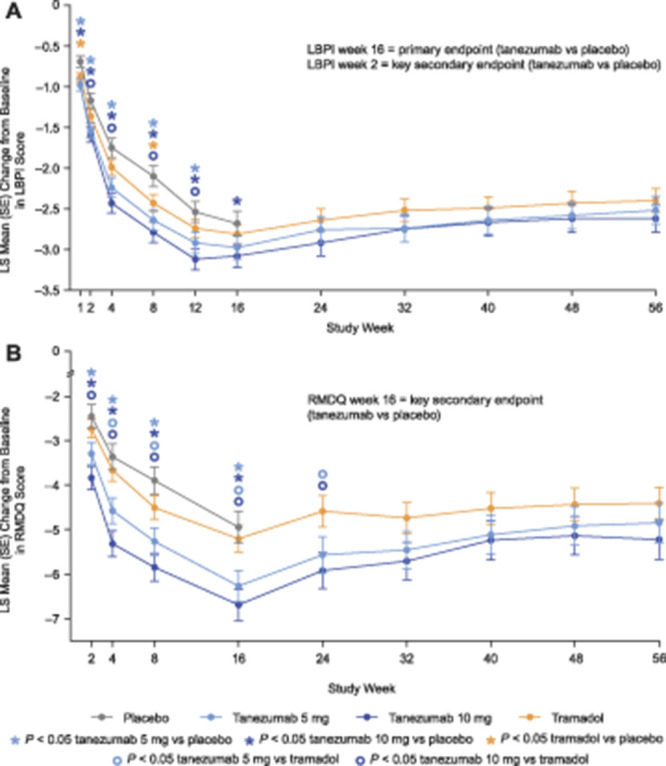
Change in LBPI (A) and RMDQ (B) scores from baseline to week 56. Change in low back pain intensity (LBPI) score at week 16 (tanezumab vs placebo) was the primary efficacy endpoint. Change in LBPI score at week 2 (tanezumab vs placebo) was a key secondary endpoint. Change in Roland Morris Disability Questionnaire (RMDQ) score at week 16 (tanezumab vs placebo) was a key secondary endpoint. Comparisons of tanezumab to placebo at other time points, and comparisons of tramadol to other treatment groups at any time point, were secondary endpoints. The primary and key secondary endpoint analyses were adjusted for multiple comparisons; other secondary analyses were not adjusted for multiplicity. See text for details. LS, least squares; SE, standard error.
3.3. Key secondary endpoints
Tanezumab 10 mg also met all key secondary endpoints. Significantly more patients achieved ≥50% improvement in LBPI at week 16 (46.3%) vs placebo (37.4%); odds ratio [OR] (95% CI) = 1.45 (1.09 to 1.91; P = 0.0101). Improvement in RMDQ was significantly greater at week 16 vs placebo; LS mean (95% CI) difference = −1.74 (−2.64 to −0.83; P = 0.0002) (Fig. 3B). Improvement in LBPI was significantly greater at week 2 compared with placebo (Fig. 3A); LS mean (95% CI) difference = −0.42 (−0.65 to −0.19; P = 0.0004).
Per the predefined analysis plan, the primary endpoint result prevented formal testing of the key secondary endpoints for tanezumab 5 mg. Therefore, although improvements in RMDQ at week 16 and LBPI at week 2 were greater with tanezumab 5 mg compared to placebo, a conclusion of superiority could not be made for the 5 mg dose for these endpoints. The percentage of patients achieving ≥50% improvement in LBPI at week 16 was 43.3% in the tanezumab 5 mg group relative to 37.4% for placebo; OR (95% CI) = 1.28 (0.97 to 1.70; P = 0.0846). LS mean (95% CI) difference in RMDQ at week 16 was −1.32 (−2.21 to −0.43; P = 0.0035) relative to placebo. LS mean (95% CI) difference in LBPI at week 2 was −0.37 (−0.60 to −0.14; P = 0.0015) relative to placebo.
3.4. Other secondary endpoints
3.4.1. Tanezumab vs placebo
Both doses of tanezumab improved LBPI and RMDQ vs placebo (P < 0.05) at every time point assessed (through week 16) with the exception of the 5 mg dose for LBPI at week 16 (Fig. 3A). The percentage of patients achieving ≥30% improvement in LBPI at week 16 was greater in the tanezumab 5 mg (64.8%) and tanezumab 10 mg (65.5%) groups compared to placebo (55.9%). Odds ratio (95% CI) vs placebo was 1.45 (1.09 to 1.92; P = 0.0101) for tanezumab 5 mg and 1.50 (1.13 to 1.99; P = 0.0054) for tanezumab 10 mg. As shown in Figure S2 (available at http://links.lww.com/PAIN/B46), the proportion of patients with a >0% to ≥90% improvement in LBPI at week 16 was larger in both tanezumab groups than in the placebo group, although the treatment difference incrementally reduced with increasing level of response threshold. The number needed to treat for an additional beneficial (NNTB) outcome, based on a decrease of ≥30% in LBPI from baseline at week 16, was 10 for tanezumab 10 mg.
3.4.2. Tramadol vs placebo
Tramadol improved LBPI vs placebo (P < 0.05) only at weeks 1 and 8, whereas changes in RMDQ were not different between tramadol and placebo at any time point through week 16. At week 16, LS mean (95% CI) differences, vs placebo, in LBPI and RMDQ were −0.12 (−0.46 to 0.21; P = 0.4620) and −0.26 (−1.09 to 0.57; P = 0.5412), respectively, for tramadol. The percentage of patients achieving ≥30% improvement in LBPI at week 16 was not different between the placebo (55.9%) and tramadol (57.9%) groups; odds ratio (95% CI) vs placebo was 1.08 (0.84 to 1.39; P = 0.5493).
3.4.3. Tanezumab vs tramadol
Tanezumab 10 mg improved LBPI compared to tramadol (P < 0.05) at weeks 2 through 12. At week 16, LS mean (95% CI) difference, vs tramadol, in LBPI was −0.28 (−0.60 to 0.05; P = 0.0958) for tanezumab 10 mg and −0.17 (−0.50 to 0.16; P = 0.3118) for tanezumab 5 mg. The percentage of patients achieving ≥30% improvement in LBPI at week 16 was greater in the tanezumab 5 mg (64.8%) and tanezumab 10 mg (65.5%) groups compared to tramadol (57.9%). Odds ratio (95% CI) vs tramadol was 1.34 (1.03 to 1.74; P = 0.0269) for tanezumab 5 mg and 1.38 (1.07 to 1.80; P = 0.0144) for tanezumab 10 mg. At week 56, LS mean (95% CI) difference, vs tramadol, in LBPI was −0.21 (−0.61 to 0.18; P = 0.2887) for tanezumab 10 mg and −0.11 (−0.51 to 0.28; P = 0.5763) for tanezumab 5 mg.
Both doses of tanezumab improved RMDQ compared with tramadol (P < 0.05) over the first 24 weeks of treatment with exception of the 5 mg dose at week 2 (first time point assessed). Numerical improvements over tramadol were evident for both doses of tanezumab at later time points. LS mean (95% CI) difference, vs tramadol, in RMDQ at week 16 was −1.48 (−2.29 to −0.66; P = 0.0004) for tanezumab 10 mg and −1.06 (−1.87 to −0.25; P = 0.0107) for tanezumab 5 mg. LS mean (95% CI) difference, vs tramadol, in RMDQ at week 56 was −0.83 (−1.84 to 0.18; P = 0.1089) for tanezumab 10 mg and −0.44 (−1.47 to 0.58; P = 0.3981) for tanezumab 5 mg.
3.5. Adverse events
Relative to other groups over the 56-week treatment period, back pain, fall, and sinusitis were more frequent (≥1% difference) in the tanezumab 5 mg group; arthralgia, nasopharyngitis, upper respiratory tract infection, pain in extremity, paresthesia, and hypoesthesia were more frequent in the tanezumab 10 mg group; and nausea, constipation, dizziness, somnolence, musculoskeletal pain, vomiting, dry mouth, and fatigue were more frequent in the tramadol group (Table 2). An increased rate of serious AEs through week 56 in the tanezumab 10 mg group (4.6%), relative to tramadol (3.2%), was largely a result of musculoskeletal and connective tissue events including intervertebral disk compression (n = 1), intervertebral disk protrusion (n = 1), OA (n = 1), RPOA (n = 4), and rotator cuff syndrome (n = 1), none of which occurred with tramadol. Seven deaths were reported during the study; none were deemed by investigators to be treatment-related (Table S2, available at http://links.lww.com/PAIN/B46).
Table 2.
Summary of treatment-emergent adverse events (all causalities) and joint safety.

3.6. Joint safety events
Overall, 30 patients had joint safety events meeting criteria for adjudication: tanezumab 5 mg n = 9 (1.8%), tanezumab 10 mg n = 17 (3.4%), tramadol n = 4 (0.7%), and placebo n = 0. Seven patients had a TJR, all in the tanezumab 10 mg group (knee n = 4, hip n = 1, shoulder n = 2). Of these 7 patients, 2 were adjudicated as RPOA type 1, 2 as RPOA type 2, 1 as subchondral insufficiency fracture, and 2 as “other” (meniscal tear and trauma). Three TJRs occurred during the treatment period, 2 after patients completed the treatment period but discontinued the study during the follow-up period, and 2 during the safety follow-up period after the patient discontinued the treatment period.
Incidence of the composite joint safety endpoint was higher in the tanezumab 10 mg group (n = 13; 2.6%) than in the tanezumab 5 mg (n = 5; 1.0%), and tramadol (n = 1; 0.2%) groups (Table 2). The most common event was RPOA; tanezumab 5 mg (n = 5; 1.0%), tanezumab 10 mg (n = 9; 1.8%), and tramadol (n = 1; 0.2%). Rapidly progressive OA type 1 occurred in 13 patients (15 joints: knee n = 14 and hip n = 1); 5 with tanezumab 5 mg (1.0%), 7 with tanezumab 10 mg (1.4%), and 1 with tramadol (0.2%) groups. Eleven of the 15 joints with RPOA type 1 had possible (KL grade 1 n = 4) or mild (KL grade 2 n = 7) radiographic evidence of OA at screening, and 4 had no evidence of OA (tanezumab 5 mg n = 1; tanezumab 10 mg n = 2; tramadol n = 1). Rapidly progressive OA type 2 occurred in 2 (0.4%) patients treated with tanezumab 10 mg (hip n = 1; both shoulders n = 1). There was no radiographic evidence of OA at screening for any of the 3 joints (2 patients) adjudicated to RPOA type 2. Subchondral insufficiency fracture occurred in 4 (0.8%) patients in the tanezumab 10 mg group (knee n = 4), 2 of which had possible (KL grade 1) radiologic evidence of OA at screening.
4. Discussion
In this large, multinational, phase 3, double-blind, randomized, placebo-controlled study, SC tanezumab 10 mg significantly reduced pain after 16 weeks of treatment in patients with difficult-to-treat CLBP. Tanezumab 10 mg was also superior to placebo for all key secondary efficacy endpoints: back-specific function (RMDQ at week 16), clinically meaningful pain relief (50% LBPI responder rate at week 16), and onset of action (LBPI at week 2). Together, these primary and key secondary endpoints provide evidence of clinically meaningful analgesia.11 The magnitude of pain relief with tanezumab 10 mg was 0.4 (on a standard 0-10 numeric scale) better than placebo, a modest but clinically significant difference consistent with other nonopioid treatments commonly prescribed for CLBP.7,30 Tanezumab 10 mg was associated with the highest rate of joint safety events, even in joints without evidence of OA. Although tanezumab 5 mg did not meet the primary study endpoint, this dose exhibited a more favorable joint safety profile than tanezumab 10 mg, particularly in patients without evidence of OA.
The findings of this study replicate and confirm efficacy observed in a smaller phase 2b/3 CLBP study in which tanezumab 10 mg IV significantly improved pain and function compared with placebo and naproxen after 16 weeks.21 Several factors may account for the lower LS mean difference in LBPI for tanezumab 10 mg, relative to placebo, in the current SC study (−0.4) compared to the previous IV study (−0.8).21 In contrast to the previous study, the current study was enriched for a treatment-refractory population and enrolled a more severe CLBP population based on baseline LBPI (approximately 7.2 vs 6.7) and RMDQ (approximately 15.0 vs 12.8) scores.21 Despite including patients with axial syndromes (Quebec classification 1 and 2) in both studies, it is possible the current study had fewer patients with osteoarthritic pain of the lumbar facet joints. This study included screening procedures specifically designed to exclude patients with osteoarthritic joints of the appendicular skeleton; patients with OA at large joints often have similar radiographic features in small joints of the axial spine.13 Response to placebo was also greater in the current study, possibly due to methodologic factors including greater expectation for relief among patients based on prior knowledge of favorable results with tanezumab.
The NNTB, based on a decrease of ≥ 30% in LBPI from baseline at week 16, was 10 for tanezumab 10 mg. The threshold of 30% response was chosen because it represents at least a moderately clinically important improvement in chronic pain conditions.10 With the exception of duloxetine, there are few NNTB values reported in the literature for the management of CLBP.28 The current study of tanezumab enrolled a CLBP population that is substantially different from previous trials of other agents for CLBP (patients were required to have a history of inadequate response to standard-of-care analgesics). Thus, the NNTB observed here for tanezumab should not be directly compared to NNTBs observed previously for other agents used to treat CLBP. This is based on that rationale that calculation of NNTB is highly dependent on the characteristics of the study population, the outcome measure being assessed, the response level chosen as the dichotomous cutoff point, and the timeframe of assessment.20,25 Thus, comparison of NNTBs across different treatments is inappropriate unless these treatments were tested in similar populations, with similar disease status, using similar comparators, periods, and outcome measures.25
The AE profile of tanezumab was consistent with previous studies. The most common AEs in the tanezumab 10 mg group, over the 56-week treatment period, were arthralgia, nasopharyngitis, upper respiratory tract infection, pain in extremity, paresthesia, and hypoesthesia. Tanezumab seemed to be better tolerated than tramadol, which had a higher rate of discontinuation due to AEs and a higher proportion of AEs over the 56-week treatment period.
The joint safety profile of tanezumab over 80 weeks was also similar to previous studies. All NGF antibody programs were paused by regulatory authorities from 2010 to 2012 due to joint safety concerns and these concerns spurred risk mitigation strategies evident in the current study's design.16 Prespecified joint safety events (most commonly RPOA type 1) occurred more frequently with tanezumab than with placebo or tramadol. Tanezumab 5 mg demonstrated a more favorable joint safety profile than tanezumab 10 mg because the incidence of prespecified joint safety events (1.0% vs 2.6%), RPOA type 2 (0% vs 0.4%), subchondral insufficiency fracture (0% vs 0.8%), and number of TJRs (0 vs 7) was lower with 5 mg relative to 10 mg. The only joint-related event in the tanezumab 5 mg group was RPOA type 1, and 4 of the 5 instances had some (possible to mild) radiographic evidence of OA at screening. The overall rate of RPOA observed in this 80-week study (1.4%) was similar to a recent 40-week study of tanezumab for OA (1.3%).31 The rate of RPOA in the current study, despite exclusion of patients with more than mild radiographic and clinical evidence of OA at baseline, may be due to the use of a higher doses of tanezumab (10 mg) than what is used for OA patients and the enrollment of patients with radiographic evidence of mild (KL Grade 2) or possible (KL grade 1) OA.
Despite increased use of surgical, pharmacological, and nonpharmacological interventions in recent decades, the public health burden of CLBP continues to increase and there remains an unmet need for safe and effective treatments.8 Chronic low back pain is a leading indication in some regions for instrumented lumbar fusion and other interventions associated with variable efficacy compared with sham intervention.9,18,23 Tanezumab differs in important ways from existing treatments. First, it requires administration only once every 8 weeks. Second, tanezumab alleviates chronic pain through a reduction in sensitization via inhibition of NGF, a novel mechanism of action.6,15,24 The replication of tanezumab efficacy in this study reinforces the role of sensitization and hyperalgesia in the potentiation and persistence of CLBP. Third, patients treated with tanezumab are not exposed to increased risk for respiratory depression, gastrointestinal bleeding, cardiovascular events, or addiction commonly associated with NSAIDs or opioids.27,33
Risk of joint safety events with tanezumab must be considered with respect to the mortality and serious AEs of the most commonly prescribed CLBP therapies such as opioids and NSAIDs. In this study, over 85% of patients had previous inadequate response to prescription NSAIDs and over 70% had previous inadequate response to opioids. A paucity of long-term data and safety concerns for these analgesics likely contribute to wide variation in care of CLBP. The results of this randomized controlled study of a pharmacologic agent (ie, tramadol) for CLBP with duration of ≥1 year further underscores the need for controlled studies examining long-term efficacy, tolerability, and safety of drugs commonly recommended for CLBP. Serious risks of opioid treatment include abuse, overdose, and death.33 Nonsteroidal anti-inflammatory drugs pose potentially serious risks of gastrointestinal ulceration/bleeding and cardiovascular thrombotic events.27 By contrast, potentially serious AEs associated with tanezumab are predominantly joint-related events, based on rigorous monitoring (eg, baseline radiographic studies and surveillance for NSAID coadministration) in controlled clinical study settings.
Secondary objectives of this study included assessment of short- and long-term efficacy of tanezumab vs tramadol. Changes in LBPI and RMDQ with tramadol were not different from placebo at week 16. Tanezumab 10 mg improved LBPI compared with tramadol from weeks 2 to 12, whereas both the 5 mg and 10 mg doses improved RMDQ compared with tramadol over the first 24 weeks of treatment (except 5 mg at week 2). Such durable improvements in function are not evident in clinical studies of opioids (and to a lesser extent for NSAIDs) for CLBP.3 Notably, the proportion of patients with ≥30% improvement in LBPI from baseline to week 16 was greater in both tanezumab groups than in the tramadol group. Benefits over tramadol may have persisted at later time points, but study design after week 16 was optimized for safety evaluation rather than long-term efficacy comparisons. Thus, conclusions on longer-term efficacy are limited. First, the study was not powered to compare long-term efficacy because nonresponders were excluded at weeks 16 and 32 as part of the risk minimization strategy. Second, adjunctive analgesics (excluding NSAIDs and opioids) were allowed after week 16 and may confound comparisons of tanezumab to tramadol. Likewise, the permitted use of rescue medication increased from ≤3 days per week (before week 16) to daily after week 16 in all groups. The observed efficacy for tramadol may have also been affected by study design, which enriched for patients with inadequate response or intolerant to opioids; opioids having a mechanism of action overlapping that of tramadol. Patients with a history of inadequate response or intolerance to tramadol were excluded from the trial. The mean dose of tramadol, after titration for efficacy and tolerability, was ∼200 mg/day with 38% (119 of 310) and 41% (85 of 208) of patients receiving the maximum of 300 mg/day at weeks 16 and 56, respectively.
Although this study was rigorous in design and substantial in sample size, generalizing conclusions regarding tanezumab efficacy and safety to broader CLBP populations may be limited by characteristics of the study population (treatment-refractory with no-to-mild evidence of OA). As noted, conclusions on long-term tanezumab efficacy are limited by study design, although a 56-week duration was essential to assess long-term safety of both tanezumab and tramadol. Another limitation is the lack of analysis of psychosocial prognostic factors (eg, catastrophizing) known to amplify CLBP intensity. The magnitude of placebo response was also high and future studies should consider measures to minimize placebo response. Finally, although NGF levels are elevated in several painful conditions, this study was not powered to assess the utility of serum NGF biomarkers in CLBP.6,24 Development of reliable biomarkers and sampling methods to identify subgroups of patients in whom these levels correlate with pain intensity would help optimize the therapeutic index of tanezumab.
5. Conclusions
Tanezumab 10 mg provided long-term pain relief and functional improvement in patients with difficult-to-treat CLBP but was associated with more joint safety events than placebo or tramadol, even in patients with no or possible radiologic evidence of OA. Tanezumab 5 mg did not meet the primary efficacy endpoint, but was associated with improvements in secondary analyses of pain and function and was associated with fewer joint safety events than tanezumab 10 mg.
Conflict of interest statement
The study was sponsored by Pfizer and Eli Lilly and Company. J.D. Markman has served on an advisory board for Clexio Biosciences, Flexion Therapeutics, Quark Pharmaceuticals, QuartetMedicine, Collegium Pharmaceutical, Purdue Pharma, Biogen, Novartis, Aptinyx, Nektar, Allergan, Grü nenthal, Eli Lilly and Company, Depomed, Janssen Pharmaceuticals, Teva Pharmaceutical Industries, KemPharm, Abbott Laboratories, Plasma Surgical, Chromocell, Convergence Pharmaceuticals, Inspirion, Pfizer, Sanofi, Daiichi Sankyo, Braeburn, Sophren, Swanbio, SK Lifescience and Trevena; has served as a consultant to Trigemina, Editas Medicine, Averitas, and Plasma Surgical; and has served on data safety monitoring boards for Novartis and Allergan. Equity interest in YellowBlackcorp. R.B. Bolash has served on advisory boards for AcelRx Pharmaceuticals, Pfizer, Premier, Salix Pharmaceuticals, and TerSera Therapeutics; has served as a consultant to Jazz Pharmaceuticals, Medtronic, Nuvectra, and Stimwave Technologies; and has participated in sponsored research with Abbott Laboratories, Boston Scientific, Neurana Pharmaceuticals, Nuvectra, Pfizer, and Stimwave Technologies. T.E. McAlindon has served on an advisory board for Pfizer, Samumed, Seikagaku, Flexion Therapeutics, Sanofi, Roche, Astellas, and Regeneron. A.J. Kivitz has served on an advisory board for Janssen Pharmaceuticals, Pfizer, UCB, Genzyme, Sanofi, Regeneron, Boehringer Ingelheim, and Genentech; has served as a speaker for Celgene, Horizon Pharma, Merck & Company, Novartis, Pfizer, Genzyme, Sanofi, Regeneron, and Flexion Therapeutics; has served as a consultant to Pfizer, Sanofi, Regeneron, and SunPharma Advanced Research; and owns stock in Novartis. M. Pombo-Suarez has received consultant and/or speaker fees from AbbVie, Janssen Pharmaceuticals, Eli Lilly and Company, Merck Sharp & Dohme, and Sanofi. F.W. Roemer is Chief Medical Officer and a shareholder of Boston Imaging Core Lab, LLC. L. Viktrup is a full-time employee of Eli Lilly & Company. C. Bramson, K. Verburg, and C. R. West are full-time employees of, and own stock and/or options in, Pfizer. D. J. Li was a full-time employee of, and owned stock and/or options in, Pfizer Inc at the time of the study and is now a full-time employee of Eisai Inc. The remaining authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B46.
Supplementary Material
Acknowledgements
Editorial support was provided by Matt Soulsby, PhD, CMPP, of Engage Scientific Solutions and was funded by Pfizer and Eli Lilly and Company.
Data sharing: Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-studys/study-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the United States and/or EU or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer and Lilly will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer studies 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, through a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Allegri M, Montella S, Salici F, Valente A, Marchesini M, Compagnone C, Baciarello M, Manferdini ME, Fanelli G. Mechanisms of low back pain: a guide for diagnosis and therapy. F1000Research 2016;5:F1000 Faculty Rev-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Aoki Y, Ohtori S, Takahashi K, Ino H, Takahashi Y, Chiba T, Moriya H. Innervation of the lumbar intervertebral disc by nerve growth factor-dependent neurons related to inflammatory pain. Spine 2004;29:1077–81. [DOI] [PubMed] [Google Scholar]
- [3].Ashworth J, Green DJ, Dunn KM, Jordan KP. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? PAIN 2013;154:1038–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baldini A, Von Korff M, Lin EH. A review of potential adverse effects of long-term opioid therapy: a practitioner's guide. Prim Care companion CNS Disord 2012;14:11m01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bretz F, Posch M, Glimm E, Klinglmueller F, Maurer W, Rohmeyer K. Graphical approaches for multiple comparison procedures using weighted Bonferroni, Simes, or parametric tests. Biometrical J Biometrische Z 2011;53:894–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang DS, Hsu E, Hottinger DG, Cohen SP. Anti-nerve growth factor in pain management: current evidence. J Pain Res 2016;9:373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chou R, Deyo R, Friedly J, Skelly A, Weimer M, Fu R, Dana T, Kraegel P, Griffin J, Grusing S. Systemic pharmacologic therapies for low back pain: a systematic review for an American college of physicians clinical practice guideline. Ann Intern Med 2017;166:480–92. [DOI] [PubMed] [Google Scholar]
- [8].Deyo RA, Dworkin SF, Amtmann D, Andersson G, Borenstein D, Carragee E, Carrino J, Chou R, Cook K, DeLitto A, Goertz C, Khalsa P, Loeser J, Mackey S, Panagis J, Rainville J, Tosteson T, Turk D, Von Korff M, Weiner DK. Report of the NIH Task Force on research standards for chronic low back pain. J Pain 2014;15:569–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deyo RA, Hallvik SE, Hildebran C, Marino M, O'Kane N, Carson J, Van Otterloo J, Wright DA, Millet LM, Wakeland W. Use of prescription opioids before and after an operation for chronic pain (lumbar fusion surgery). PAIN 2018;159:1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, Haythornthwaite JA, Jensen MP, Kerns RD, Ader DN, Brandenburg N, Burke LB, Cella D, Chandler J, Cowan P, Dimitrova R, Dionne R, Hertz S, Jadad AR, Katz NP, Kehlet H, Kramer LD, Manning DC, McCormick C, McDermott MP, McQuay HJ, Patel S, Porter L, Quessy S, Rappaport BA, Rauschkolb C, Revicki DA, Rothman M, Schmader KE, Stacey BR, Stauffer JW, von Stein T, White RE, Witter J, Zavisic S. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain 2008;9:105–21. [DOI] [PubMed] [Google Scholar]
- [11].Dworkin RH, Turk DC, McDermott MP, Peirce-Sandner S, Burke LB, Cowan P, Farrar JT, Hertz S, Raja SN, Rappaport BA, Rauschkolb C, Sampaio C. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. PAIN 2009;146:238–44. [DOI] [PubMed] [Google Scholar]
- [12].Freemont AJ, Watkins A, Le Maitre C, Baird P, Jeziorska M, Knight MT, Ross ER, O'Brien JP, Hoyland JA. Nerve growth factor expression and innervation of the painful intervertebral disc. J Pathol 2002;197:286–92. [DOI] [PubMed] [Google Scholar]
- [13].Gellhorn AC, Katz JN, Suri P. Osteoarthritis of the spine: the facet joints. Nat Rev Rheumatol 2013;9:216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017;390:1211–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hefti F. Pharmacology of nerve growth factor and discovery of tanezumab, an anti-nerve growth factor antibody and pain therapeutic. Pharmacol Res 2020;154:104240. [DOI] [PubMed] [Google Scholar]
- [16].Hochberg MC, Tive LA, Abramson SB, Vignon E, Verburg KM, West CR, Smith MD, Hungerford DS. When is osteonecrosis not osteonecrosis?: adjudication of reported serious adverse joint events in the tanezumab clinical development program. Arthritis Rheumatol 2016;68:382–91. [DOI] [PubMed] [Google Scholar]
- [17].Holmes D. Anti-NGF painkillers back on track? Nat Rev Drug Discov 2012;11:337–8. [DOI] [PubMed] [Google Scholar]
- [18].Juch JNS, Maas ET, Ostelo RWJG, Groeneweg JG, Kallewaard J-W, Koes BW, Verhagen AP, van Dongen JM, Huygen FJPM, van Tulder MW. Effect of radiofrequency denervation on pain intensity among patients with chronic low back pain: the mint randomized clinical trials. JAMA 2017;318:68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Katz N, Borenstein DG, Birbara C, Bramson C, Nemeth MA, Smith MD, Brown MT. Efficacy and safety of tanezumab in the treatment of chronic low back pain. PAIN 2011;152:2248–58. [DOI] [PubMed] [Google Scholar]
- [20].Katz N, Paillard FC, Van Inwegen R. A review of the use of the number needed to treat to evaluate the efficacy of analgesics. J Pain 2015;16:116–23. [DOI] [PubMed] [Google Scholar]
- [21].Kivitz AJ, Gimbel JS, Bramson C, Nemeth MA, Keller DS, Brown MT, West CR, Verburg KM. Efficacy and safety of tanezumab versus naproxen in the treatment of chronic low back pain. PAIN 2013;154:1009–21. [DOI] [PubMed] [Google Scholar]
- [22].Leite VF, Buehler AM, El Abd O, Benyamin RM, Pimentel DC, Chen J, Hsing WT, Mazloomdoost D, Amadera JE. Anti-nerve growth factor in the treatment of low back pain and radiculopathy: a systematic review and a meta-analysis. Pain Physician 2014;17:E45–60. [PubMed] [Google Scholar]
- [23].Mafi JN, McCarthy EP, Davis RB, Landon BE. Worsening trends in the management and treatment of back pain. JAMA Intern Med 2013;173:1573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology 2011;115:189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McAlister FA. The “number needed to treat” turns 20-and continues to be used and misused. Can Med Assoc J 2008;179:549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miller CG, Guermazi A, Roemer F. The current status of imaging in anti-NGF clinical trials. Osteoarthritis Cartilage 2015;23(suppl 1):S3–7. [DOI] [PubMed] [Google Scholar]
- [27].Moore RA, Derry S, McQuay HJ. Cyclo-oxygenase-2 selective inhibitors and nonsteroidal anti-inflammatory drugs: balancing gastrointestinal and cardiovascular risk. BMC Musculoskelet Disord 2007;8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Moore RA, Cai N, Skljarevski V, Tolle TR. Duloxetine use in chronic painful conditions-individual patient data responder analysis. Eur J Pain 2014;18:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qaseem A, Wilt TJ, McLean RM, Forciea MA. Noninvasive treatments for acute, subacute, and chronic low back pain: a clinical practice guideline from the American college of physicians. Ann Intern Med 2017;166:514–30. [DOI] [PubMed] [Google Scholar]
- [30].Roelofs PDDM, Deyo RA, Koes BW, Scholten RJPM, van Tulder MW. Nonsteroidal anti-inflammatory drugs for low back pain: an updated cochrane review. Spine 2008;33:1766–74. [DOI] [PubMed] [Google Scholar]
- [31].Schnitzer TJ, Easton R, Pang S, Levinson DJ, Pixton G, Viktrup L, Davignon I, Brown MT, West CR, Verburg KM. Effect of tanezumab on joint pain, physical function, and patient global assessment of osteoarthritis among patients with osteoarthritis of the hip or knee: a randomized clinical trial. JAMA 2019;322:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shmagel A, Ngo L, Ensrud K, Foley R. Prescription medication use among community-based U.S. adults with chronic low back pain: a cross-sectional population based study. J Pain 2018;19:1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].US Department of Health and Human Services. What is the U.S. opioid epidemic? Vol. 2018 Washington: US Department of Health and Human Services, 2019. [Google Scholar]
- [34].Weinkauf B, Deising S, Obreja O, Hoheisel U, Mense S, Schmelz M, Rukwied R. Comparison of nerve growth factor-induced sensitization pattern in lumbar and tibial muscle and fascia. Muscle Nerve 2015;52:265–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B46.