

In order for a pathogen to establish a successful infection, it must undermine the host cell responses inhibitory to the pathogen. As such, herpesviruses encode multiple viral proteins that antagonize each host antiviral response, thereby allowing for efficient viral replication. Human Cytomegalovirus encodes several factors that limit host countermeasures to infection, including pp71. Herein, we identified a previously unreported posttranslational modification of pp71, protein S-nitrosylation. Using site-directed mutagenesis, we mutated the specific sites of this modification thereby blocking this pp71 posttranslational modification. In contexts where pp71 is not protein S-nitrosylated, host antiviral response was inhibited. The net result of this posttranslational modification is to render a viral protein with diminished abilities to block host responses to infection. This novel work supports a model in which protein S-nitrosylation may be an additional mechanism in which a cell inhibits a pathogen during the course of infection.
KEYWORDS: HCMV, protein S-nitrosylation, STING, UL82, cytomegalovirus, human herpesviruses, pp71
ABSTRACT
Human Cytomegalovirus (HCMV) is a ubiquitous pathogen that has coevolved with its host and, in doing so, is highly efficient in undermining antiviral responses that limit successful infections. As a result, HCMV infections are highly problematic in individuals with weakened or underdeveloped immune systems, including transplant recipients and newborns. Understanding how HCMV controls the microenvironment of an infected cell so as to favor productive replication is of critical importance. To this end, we took an unbiased proteomics approach to identify the highly reversible, stress-induced, posttranslational modification (PTM) protein S-nitrosylation on viral proteins to determine the biological impact on viral replication. We identified protein S-nitrosylation of 13 viral proteins during infection of highly permissive fibroblasts. One of these proteins, pp71, is critical for efficient viral replication, as it undermines host antiviral responses, including stimulator of interferon genes (STING) activation. By exploiting site-directed mutagenesis of the specific amino acids we identified in pp71 as protein S-nitrosylated, we found this pp71 PTM diminishes its ability to undermine antiviral responses induced by the STING pathway. Our results suggest a model in which protein S-nitrosylation may function as a host response to viral infection that limits viral spread.
IMPORTANCE In order for a pathogen to establish a successful infection, it must undermine the host cell responses inhibitory to the pathogen. As such, herpesviruses encode multiple viral proteins that antagonize each host antiviral response, thereby allowing for efficient viral replication. Human Cytomegalovirus encodes several factors that limit host countermeasures to infection, including pp71. Herein, we identified a previously unreported posttranslational modification of pp71, protein S-nitrosylation. Using site-directed mutagenesis, we mutated the specific sites of this modification thereby blocking this pp71 posttranslational modification. In contexts where pp71 is not protein S-nitrosylated, host antiviral response was inhibited. The net result of this posttranslational modification is to render a viral protein with diminished abilities to block host responses to infection. This novel work supports a model in which protein S-nitrosylation may be an additional mechanism in which a cell inhibits a pathogen during the course of infection.
INTRODUCTION
Human cytomegalovirus (HCMV) is a widespread betaherpesvirus that has successfully established infections within the majority of the human population. The incidence of HCMV seropositivity increases with age (1), and infections are lifelong, as HCMV undergoes both lytic and latent life cycles (2). The lytic life cycle is highly efficient at undermining the host antiviral responses (3), whereas during the latent stage, the virus remains dormant, thereby ensuring immune evasion and allowing for lifelong infections (4). Primary infections are often resolved by a competent immune system, and maintenance of the latent genome is often clinically inapparent. However, HCMV infections in individuals with weakened or underdeveloped immune systems often result in poor outcomes. For example, HCMV infections of neonates can cause developmental issues, including premature births, blindness, deafness, and learning disabilities, with incidence rates exceeding that of congenital birth defects due to Zika virus infection (5). Equally problematic are viral reactivation events within immunosuppressed or immunodepleted patient populations, such as organ recipients and those undergoing aggressive chemotherapy. Viral reactivations within these individuals can lead to viremia, end-stage organ failure, donor organ rejection, and graft-versus-host disease, all resulting in a poor patient prognosis (6). Consequently, HCMV is a significant economic and medical burden.
HCMV is a large DNA virus that encodes more than 200 open reading frames (ORFs) (7, 8), of which many remain uncharacterized, partly due to the lack of nonhuman model systems. While there are related species of cytomegaloviruses, each has evolved to restrict their infections to tissues derived only from their host species. Even with this restricted tropism, cytomegaloviruses are highly successful pathogens, as these viruses have coevolved with their hosts (9) and have acquired mechanisms to challenge known antiviral responses (10). In many cases, cytomegaloviruses, including HCMV, not only undermine these responses but also utilize them for the virus’ own replication. One such example is the viral tegument protein, pp71, encoded by the UL82 ORF (11). pp71 is critical for efficient HCMV replication, as viruses that lack this ORF have severe growth defects (12, 13). pp71 functions by binding to both retinoblastoma protein (pRb) (14, 15) and the death-associated protein 6 (DAXX) (16, 17), which results in their degradation. pRb interacts with histone deacetylases and leads to the establishment of transcriptionally repressive nucleosome formation on chromatin (18). DAXX is also a potent transcriptional repressor that functions by binding to transcription factors (19). Thus, pp71-mediated binding and degradation of these two proteins permit the virus to establish a subcellular environment conducive for viral lytic replication. pp71 expression also reduces the expression of major histocompatibility complex (MHC) on the cell surface (20) and interacts with components of the innate immune cytoplasmic DNA sensing machinery, including pp71-mediated inhibition of the stimulator of interferon genes (STING) pathway (21). STING, encoded by the transmembrane protein 173 (TMEM173) gene, is a potent mediator of the interferon signaling pathway (22). Activation of cytoplasmic DNA sensors results in the production of cyclic dinucleotides that serve as activators of STING. Cyclic dinucleotide-bound STING results in stimulation of both NF-κB and interferon regulatory factor 3 (IRF3), thereby inducing the type I interferon response to invading intracellular pathogens (23). Thus, pp71’s ability to circumvent STING activation is critical for undermining the host antiviral response and promoting a proviral state.
While viruses can challenge host responses, this is not without countermeasures from the host, including posttranslational modifications (PTMs) of viral proteins. PTMs are responsible for altering functions, localization, and stability of proteins. The most frequently reported PTMs are phosphorylation of serine, threonine, and tyrosine, methylation, SUMOylation and ubiquitination of lysine and arginine, acetylation of lysine, disulfide bond linkage of cysteine, and glycosylation and lipidation of several different amino acids. One modification that is highly prevalent, yet understudied in the field of PTMs, is protein S-nitrosylation. Protein S-nitrosylation is the covalent linkage of a nitric oxide group to the reduced thiol of cysteine, thereby increasing the size and polarity of this amino acid (24). There are multiple consequences of protein S-nitrosylation, including the allosteric regulation of a protein’s enzymatic activity in response to intracellular nitric oxide levels, regulation of protein-protein interactions, and alterations in protein folding by blocking potential disulfide linkages and preventing palmitoylation of cysteine (25), all of which would serve as effective mechanisms to limit the function of viral proteins during productive infections. The reversible nature of the S-nitrosylation linkage to the thiol group makes studying this PTM difficult. S-Nitrosylated reactions are reversed under reducing conditions and are often lost during standard isolation techniques utilized in protein purification and subsequent analyses (26). Thus, this PTM is significantly underrepresented in the literature.
With recent advancements in whole-cell proteomic analyses coupled with innovative stable labeling of S-nitrosylated cysteines, the detection of protein S-nitrosylation can now be applied to complex biological systems (27). As the stress of HCMV infections results in global and profound changes within host cells, including the stimulation of inducible nitric oxide synthase (iNOS) (28), we sought to determine the biological impact of protein S-nitrosylation following HCMV infection. Herein, we evaluated the impact of HCMV infection on the protein S-nitrosylation state of the proteome of infected fibroblasts using unbiased whole-cell proteomics. We identified specific protein S-nitrosylated PTMs for a subset of HCMV proteins, including pp71, a viral protein that limits host antiviral responses, including STING activation. Additionally, mutation of the protein S-nitrosylated cysteines within pp71 to the closely related amino acid serine resulted in increased inhibition of STING antiviral responses, suggesting protein S-nitrosylation may negatively impact the functions of viral proteins. We report that protein S-nitrosylation of pp71 is an important regulatory mechanism that controls this viral protein’s biological function. Importantly, these findings suggest that protein S-nitrosylation of viral proteins by the host cell machinery may function as a universal antiviral defense mechanism that may inhibit multiple types of viruses.
RESULTS
Distinct HCMV proteins are S-nitrosylated during infection of fibroblasts.
Infection with viruses induces significant changes within a cell’s proteome, including PTM alterations. To monitor the changes in host cell protein S-nitrosylation in response to infection, we infected human fibroblasts (multiplicity of infection [MOI] = 3.0) with a bacterial artificial chromosome (BAC)-derived clinical isolate of HCMV that expresses a viral late protein, pp28 (encoded by UL99), containing an in-frame fusion to enhanced green fluorescent protein (eGFP), so as to monitor viral replication. We collected protein lysates at 96 h postinfection (hpi) under nonreducing conditions to preserve the protein S-nitrosylation modification of cysteines, since this modification is highly labile under standard reducing conditions. We then biotinylated the protein S-nitrosylated cysteines using a biotin switch assay (29). The lability of the protein S-nitrosylation modification allows for the chemical removal of the NO− group followed by the efficient substitution of the highly stable biotin group at the same cysteine, thus allowing for enrichment and subsequent peptide identification by tandem mass spectrometry (MS/MS) analysis. MS/MS analysis of fragmented peptides not only allows us to identify peptide amino acid sequences but also affords us the platform to characterize the specific cysteines that are increased in mass due to the conjugated biotin. Thus, we can confirm protein S-nitrosylation of specific cysteines. Using this methodology, we analyzed three independent biological replicates each of mock and infected MRC5 cells and identified more than 2,000 peptides, representing 954 host proteins differentially S-nitrosylated in response to HCMV infection. Importantly, we identified S-nitrosylation of 23 viral peptides originating from 13 different viral ORFs at 96 hpi (Fig. 1A) in which we also confirmed the presence of a modified cysteine in each peptide (represented as a lowercase “c” in the peptide sequence).
FIG 1.

HCMV pp71 is protein S-nitrosylated during infection. (A) NuFF-1 cells were infected at a multiplicity of 3 IU/cell with TB40/E-mCherry-UL99eGFP virus; 96 hpi, total lysate was isolated and subjected to a biotin switch assay. Trypsinized proteins were analyzed by mass spectrometry to identify viral peptides that contain a cysteine conjugated to biotin, thereby reveling the site of protein S-nitrosylation. Peptides from 13 viral proteins are shown with the identified sequences. Lowercase “c” denotes the identified biotinylated cysteine. Xcorr*, cross correlation, a measure of the goodness of fit of experimental peptide fragments to theoretical spectra created from the sequence b and y ions; PSM**, frequency of peptide detection; ***, due to partial trypsin digestion, two of the three UL44 peptides and both of the UL25 peptides represent the same identified S-nitrosylated cysteine. Results are from three independent biological replicates. (B) A representative MS/MS spectrum showing the mass shift for the pp71 peptide containing the LxCxD domain. (C) NuFF-1 cells were infected at a multiplicity of 1.0 IU/cell with TB40/E-mCherry-UL99eGFP, and cells were harvested at 96 hpi. After the biotin switch assay, lysates were affinity purified (AP) with avidin beads (top) or immunoprecipitated by pp71-specific antibody (bottom). Purified lysates were separated by 8% SDS-PAGE and transferred to a nitrocellulose membrane. Proteins were detected using a pp71-specific antibody (top) or antibody against biotin (bottom). n = 3; representative blots are shown. (D) Specificity of the biotin switch reaction was assessed by performing the reaction in the absence of added biotin. NuFF-1 cells were infected at a multiplicity of 3 IU/cell with TB40/E-mCherry-UL99eGFP virus; 96 hpi, total lysate was isolated and subjected to a biotin switch assay in the presence or absence of biotin. Lysates were affinity purified with avidin beads, and then purified lysates were separated by 8% SDS-PAGE and blotted to nitrocellulose membrane (top). A fraction of the lysates (4%) was run in parallel to confirm protein expression (bottom). Proteins were detected by using specific antibodies. A representative blot from three biological repeats is shown.
We were intrigued by S-nitrosylation of the UL82-encoded viral protein pp71, which is important for mediating host antiviral responses involving the interferon pathway, as a central cysteine within the LxCxD domain of that protein is protein S-nitrosylated (Fig. 1B). In order to validate our MS/MS results, we repeated the infections and isolated equal amounts of whole-cell lysates for biotin switch labeling. We then enriched the S-nitrosylated biotin-labeled proteins by coupling them to streptavidin beads and then eluted them for immunoblot analyses. A single protein band of the correct size and antigen specificity for HCMV pp71 was detected from the biotin switch assay-enriched proteins, thus confirming the modifications we identified in the MS/MS analyses (Fig. 1C, top). In addition, we performed the reverse pulldown, in which lysates from the biotin switch reaction were immunoprecipitated with a pp71-specific antibody followed by blotting with an anti-biotin specific antibody. We identified biotinylated pp71 protein (Fig. 1C, bottom), thereby verifying pp71 is specifically S-nitrosylated during infection of fibroblasts. A significant number of proteins are naturally biotinylated (30). Thus, to assess the specificity of the immunoprecipitation and immunodetection efficiency, we repeated the protein purification and biotin labeling protocol in the presence and absence of exogenous biotin. As expected, in the absence of biotin in the reaction mixture, we were not able to detect purification of pp71 from the avidin bead affinity purification (Fig. 1D). These results suggest that pp71 is protein S-nitrosylated within infected fibroblasts and that this modification occurs on specific cysteines within the protein.
Mutation of protein S-nitrosylation identified cysteine residues within pp71 does not inhibit viral production.
To determine the biological function of the S-nitrosylated residue(s), we conducted a mutational analysis of pp71. We introduced mutations within pp71 in the context of the viral BAC genome by changing each of the three identified S-nitrosylated cysteines using site-directed mutagenesis. We elected to introduce serines at these locations, as they are structurally similar to cysteines yet lack the thiol group required for protein S-nitrosylation (C34S, C94S, and C218S). The resulting BAC was used to reconstitute the triple mutant pp71 virus (pp71-TM). To determine if the cysteine-to-serine (Cys-to-Ser) mutations within pp71 inhibit the viability of virus production, we monitored replication kinetics by viral growth analysis. To this end, we compared the single-step growth of the pp71-TM virus to that of the wild-type/parental virus in MRC5 fibroblasts, quantifying cell-free virus over a time course of 7 days (Fig. 2A). We did not observe a significant viral growth defect when all three identified S-nitrosylated cysteines were mutated to serine within pp71 (pp71-TM). In fact, pp71-TM viruses exhibited a slight yet consistent increase in both growth kinetics and cell-free viral yield compared to that of wild-type virus (Fig. 2A). To determine whether the triple mutation described above altered viral protein expression of pp71, we analyzed viral protein accumulation over a course of infection by immunoblotting. We found that pp71-TM-infected fibroblasts displayed similar viral protein expression to that of its wild-type counterpart (Fig. 2B). To determine when the protein S-nitrosylation of cysteines occurs on pp71, we analyzed the kinetics of this PTM on pp71 in relation to de novo pp71 protein synthesis. Fibroblasts were infected with wild-type HCMV, and protein lysates were isolated at various time points. Lysates were either used for Western blot analysis of total pp71 protein expression or used in a biotin switch assay, where S-nitrosylated proteins were captured with streptavidin beads and then analyzed by Western blotting to assess pp71 protein S-nitrosylation (Fig. 3A). We observed similar kinetics between total pp71 expression and the appearance of the protein S-nitrosylation PTM on pp71, suggesting the modification on the protein occurs in a similar time frame as for protein synthesis. Thus, we focused on characterizing the biological effects of pp71 S-nitrosylation.
FIG 2.

S-Nitrosylation-defective viruses have similar growth kinetics to that of WT virus. (A) NuFF-1 cells were infected at a multiplicity of 1 IU/cell with TB40/E-mCherry-UL99eGFP virus (WT) or a recombinant virus containing three-point mutations in pp71 ORF at amino acids C34S, C94S, and C218S (pp71-TM). Medium containing cell-free virus was collected over the indicated time course, and virus yields were determined by TCID50 assay on NuFF-1 cells. Ino, inoculum; LOD, limit of detection. Samples were analyzed in triplicates. Error bars represent the standard deviations of the replicates. (B) Total de novo expression of wild-type pp71 or pp71-TM was monitored by immunoblotting. NuFF-1 cells were infected at a multiplicity of 1 IU/cell with WT or pp71-TM virus. Cells were harvested at the indicated time points after infection, and the proteins were separated by 8% SDS-PAGE and transferred to a nitrocellulose membrane. Cellular tubulin levels served as a control for equal protein loading. n = 3, representative blots are shown.
FIG 3.

Protein S-nitrosylation-defective pp71 is expressed with wild-type kinetics and incorporated into tegument. (A) NuFF-1 cells were infected at a multiplicity of 1 IU/cell with WT virus. Cells were harvested at the indicated time points after infection, and 4% (20 μg) protein lysates were separated by SDS-PAGE and blotted to nitrocellulose membrane (top, total lysate); the remaining 96% (0.5 mg) of protein lysate was used in a biotin switch assay, affinity purified on streptavidin beads (AP), and then separated by SDS-PAGE and transferred to a nitrocellulose membrane (bottom, AP/WB). Cellular tubulin levels served as a control for equal protein loading. n = 3; representative blots are shown. (B) NuFF-1 cells were infected at a multiplicity of 1 IU/cell with WT virus, a recombinant virus containing single point mutation in pp71 ORF at amino acid C218S (pp71-C218S), or a revertant of that mutant (pp71-C218Srev). Medium containing cell-free virus was collected over the indicated time course, and virus yields were determined by TCID50 assay on NuFF-1 cells. Ino, inoculum. Samples were analyzed in triplicates. Error bars represent the standard deviations of the replicates. A representative analysis of three independent biological replicates is shown. (C) MRC5 cells were infected at a multiplicity of 30 IU/cell with the TB40/E-mCherry-UL99eGFP virus, pp71-C218S, or pp71-TM. Cells were harvested 6 hpi to detect pp71 and pp65 delivery into newly infected cells. Total cell lysates were separated by 8% SDS-PAGE and analyzed by Western blotting. Cellular tubulin levels served as a control for equal protein loading. n = 3; representative blots are shown.
pp71 is a multifunctional protein critical for efficient viral infection. Among its many roles, pp71 interacts with pRb, resulting in its degradation, mediated by the consensus pRb binding motif LxCxD in pp71, where the central cysteine is located at amino acid 218 (14). Our proteomic analysis revealed that pp71 cysteine 218 (C218), the central cysteine of the pRb binding motif, is S-nitrosylated (Fig. 1A). To elucidate the biological consequence(s) of C218 S-nitrosylation, we focused our efforts on studying the effects of protein S-nitrosylation of the LxCxD domain. To this end, we introduced a single Cys-to-Ser mutation of C218 into the viral genome, thereby generating pp71-C218S. To ensure any observed phenotype with this virus is specific to our introduced mutation, we then reverted this serine mutant back to the wild-type cysteine, thus generating pp71-C218Srev. A growth curve analysis revealed that, similarly to the triple mutant virus (Fig. 2A), the LxCxD domain mutant exhibited a growth advantage compared to that of either the wild-type or revertant virus (Fig. 3B). We then determined whether C218 S-nitrosylation was critical for tegument delivery of pp71 upon infection. We generated recombinant viruses and assessed their ability to package tegument pp71 as well as the well-characterized tegument protein pp65 as a control. At high MOI of fibroblasts (MOI = 30), we detected the wild type, as well as pp71-TM and pp71-C218S at 6 hpi (Fig. 3C). As pp71 is a late kinetic viral protein, its detection in newly infected cells early postinfection (Fig. 2B) and prior to de novo synthesis is consistent with tegument-delivered pp71 (31). Together, these results suggest the ability to inhibit protein S-nitrosylation of pp71 did not significantly alter the total accumulation of viral progeny but may have expediated growth kinetics compared to that of the parental virus. In addition, mutation of the identified protein S-nitrosylation-modified cysteines did not alter the expression or tegument loading of pp71 to newly infected fibroblasts.
Protein S-nitrosylated pp71 interacts with STING.
We next assessed the importance of protein S-nitrosylation for known biological functions of pp71. Recent studies found pp71 is able to inhibit STING activation (21). Inhibition of potent antiviral responses, such as STING, would aid in successful viral infections. For example, both adenovirus E1a and human papillomavirus E7 also inhibit STING antiviral functions (32). E1a and E7 mediate these interactions through their conserved LxCxE pRb binding domain. As our analysis identified the central cysteine in the pp71 LxCxD domain was S-nitrosylated during infection, we next asked whether this PTM affected pp71-STING interactions. HEK293T cells lacking endogenous STING (33) were cotransfected with either empty vector or a vector expressing hemagglutinin (HA)-tagged STING (STING-HA) (34) in conjunction with either FLAG-tagged wild-type pp71 (FLAG-WT-pp71) or FLAG-tagged pp71 bearing a C218S mutation within the pRb binding domain (FLAG-pp71-C218S). First, we confirmed the expression of each epitope-tagged protein by immunoblot analysis (Fig. 4A and B). Transiently cotransfected cell lysates precipitated with anti-FLAG antibody followed by immunoblotting revealed both FLAG-WT-pp71 and FLAG-pp71-C218S each interact with STING-HA (Fig. 4A). We also found the same interaction between FLAG-pp71-C218S or FLAG-WT-pp71 with STING-HA when we reversed the order of immunoprecipitation, using an HA antibody followed by immunoblotting with an anti-FLAG antibody (Fig. 4B). Collectively, these results suggest STING interacts with pp71, and the interaction may occur with greater affinity when pp71 C218 within its pRb binding domain cannot undergo protein S-nitrosylation.
FIG 4.

pp71 interacts with STING in vitro and colocalizes with STING during infection. (A) HEK293 cells were cotransfected with pCDNA-STING-HA (STING-HA) and pCMSeGFP-FLAG-pp71-WT (FLAG-pp71-WT) or pCMSeGFP-FLAG-pp71-C218S (FLAG-pp71-C218S). After 72 h, total cell lysates were collected. Equal amounts of total cell lysates were incubated with α-HA (A) or α-FLAG affinity beads (B). The bound complexes were washed, eluted, separated by 8% SDS-PAGE, and immunoblotted (IB) with an α-FLAG (A) or an α-HA antibody (B). n = 3; representative blots are shown.
We next wanted to determine if the interaction of pp71 with STING impacts the levels of the cellular protein. To test this, we infected NuFF-1 cells with either wild-type (WT) HCMV or recombinant virus expressing pp71-C218S (MOI = 1.0). We collected total cell lysates at time points relevant for tegument-delivered pp71 (Fig. 3B), as well as at time points consistent with de novo protein synthesis of pp71 (Fig. 3A). Neither tegument-delivered nor de novo protein synthesis of WT or mutant pp71 increased the relative expression of endogenous STING compared to that of α-tubulin (Fig. 5A and B). These results reveal pp71 does not regulate endogenous STING expression levels.
FIG 5.

pp71 does not alter the protein expression levels of STING at early or late times during lytic infection. MRC5 cells were infected at a multiplicity of 1 IU/cell with WT or pp71-C218S virus. Infected cells were harvested over 48 h prior to de novo pp71 expression (A) or after 5 days, when de novo pp71 is expressed (B). Total protein (20 μg) was separated by SDS-PAGE and transferred to a nitrocellulose membrane. Cellular tubulin is shown as a loading control. n = 3; representative blots are shown.
pp71 protein S-nitrosylation limits its ability to undermine the STING antiviral pathway.
Since we observed small but reproducible increases in both growth kinetics and absolute titer following lytic infection with the S-nitrosylation-deficient recombinant virus compared to those of the WT virus, we next investigated the biological impact of pp71 S-nitrosylation on the STING pathway in the context of viral infection. To this end, we generated a repair of the TB40/E-mCherry-pp71-C218S mutant, in which we restored the serine mutation to cysteine, thus reverting the sequence to wild type (C218Srev). The use of this revertant ensures any phenotype we observed with the pp71p-C218S recombinant virus was not caused by off-site mutations due to the recombineering. To determine the effect of STING activation on viral replication, we treated MRC5 cells with 2′,3′-cyclic GMP-AMP (2′,3′-cGAMP) and then infected the cells with WT, pp71-C218S, or pp71-C218Srev virus. 2′,3′-cGAMP is a natural ligand for STING activation (35), and a dose-response curve showed treatment of MRC5 fibroblasts with 10 μM 2′,3′-cGAMP had no adverse effect on cell viability, as determined by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Fig. 6A), and was sufficient to activate STING signaling (Fig. 6B). We then monitored cell-associated HCMV genomes in MRC5s over a time course of viral infection. As expected, activation of the potent antiviral STING pathway yielded low copy numbers of cell-associated viral genomes during WT or pp71-C218Srev infection (Fig. 6C). However, infection with the S-nitrosylation-deficient pp71 mutant virus, pp71-C218S, resulted in a significant increase in cell-associated viral genomes (Fig. 6C), suggesting S-nitrosylation prevents pp71-mediated inactivation of STING.
FIG 6.

Activation of the STING pathway inhibits DNA replication and viral production of pp71-WT and pp71-C218Srev but not pp71-C218S virus. (A) MRC5 cells were treated with indicated concentrations of 2′,3′-cGAMP; 48 h later cell viability was assessed by quantifying the metabolic activity of the treated cells using an NADH-dependent cellular oxidoreductase enzyme assay (MTT assay). MTT conversion results were normalized to the vehicle-treated conditions, and graphed results are shown as percent cell viability. Results are shown as the means from three independent biological replicates. (B) MRC5 cells were treated with indicated concentrations of 2′,3′-cGAMP, and 24 h later, total RNA was isolated. IFN-β1 transcripts were profiled by RT-qPCR using transcript-specific primers and plotted as threshold cycle (ΔΔCT) relative to the control transcript, GAPDH. Results are shown as mean fold change in the mRNA levels. (C) MRC5 cells were infected with either WT, pp71-C218S, or pp71-C218Srev virus (MOI = 1) in the presence of 10 μM 2′,3′-cGAMP. DNA was isolated over the indicated days, and genome copy number was measured by qPCR using eGFP-specific primers to detect viral genomes and normalized to human mdm2. (D) Parallel cultures were treated as for panel C, and medium was collected over the indicated days. Cell-free virus accumulation was quantified by TCID50 analysis. Ino, inoculum. (n = 3). *, P < 0.01 as determined by Student's t test; NS, not significant.
We extended this analysis to monitor the impact of pp71 protein S-nitrosylation on the accumulation of infectious cell-free virus in the presence of activated STING by single-step growth analysis. MRC5 cells were infected with WT, pp71-C218S, or pp71-C218Srev virus (MOI = 1) in the presence of 2′,3′-cGAMP. Cell-free viral supernatants were collected over time for quantification of viral production by 50% tissue culture infective dose (TCID50). Analysis of the initial infectious inoculum confirmed equal amounts of each virus were used in the experiment (Fig. 6D, Ino). We observed markedly diminished viral yield in both the WT- and pp71-C218Srev-infected fibroblasts in the presence of 10 μM 2′,3′-cGAMP up to day 4 of the experiment (Fig. 6D). However, by 6 days postinfection (dpi), both the WT- and pp71-C218Srev-infected cells began to produce infectious progeny from the 2′,3′-cGAMP-treated cells (Fig. 6D). Importantly, infection with pp71-C218S, a mutant virus that is deficient for protein S-nitrosylation within the LxCxD domain, efficiently accumulated infectious progeny even in the presence of 2′,3′-cGAMP across the time course of infection. Together, these results suggest protein S-nitrosylation-deficient pp71 at position C218 undermines potent STING activation compared to that of wild-type pp71.
Activated STING inhibits viral transcription of WT virus but not of pp71-C218S.
To determine the stage of viral replication affected by pp71 S-nitrosylation following STING activation, we monitored the accumulation of a subset of HCMV transcripts within MRC5 cells. To this end, these fibroblasts were infected with WT, pp71-C218S, or pp71-C218Srev virus in the presence or absence of STING activation. Viral transcripts UL122, UL54, and UL32 were selected as representative genes of each infection stage (e.g., immediate early [IE], early [E], and late [L], respectively [36]). In the absence of STING activation, the expression and accumulation of HCMV transcripts were similar to each other compared to those across each virus infection (Fig. 7A). As expected, activation of the STING pathway with 2′,3′-cGAMP treatment prior to infection significantly inhibited the expression and accumulation of HCMV transcripts in the fibroblasts that were infected with either WT or pp71-C218Srev virus. However, 2′,3′-cGAMP treatment did not impair the transcription of these viral genes in the pp71-C218S infected fibroblasts (Fig. 7B). Indeed, the viral transcript levels in the pp71-C218S-infected cells in the presence of 2′,3′-cGAMP (Fig. 7B) mirrored the levels we observed during HCMV infection in the absence of activated STING (Fig. 7A). These data indicate STING activation inhibits HCMV infection, and expression of S-nitrosylation-deficient pp71 effectively undermines this antiviral response.
FIG 7.

STING activation impacts viral RNA accumulation in WT virus- and pp71-C218Srev-infected cells but not pp71-C218S-infected cultures. MRC5 cells infected with either WT, pp71-C218S, or pp71-C218Srev (MOI = 1) in the absence or presence of 10 μM 2′,3′-cGAMP (added at time of infection). Total RNA was isolated from the cultures over the indicated times. Transcripts IE (UL122), E (UL54), and L (UL32) were profiled by RT-qPCR using transcript-specific primers and normalized to cellular GAPDH. Results are shown as mean fold change in the mRNA levels for each of three different kinetic classes of viral transcripts within cells infected by the listed viruses in the presence of vehicle (A) or 10 μM 2′,3′-cGAMP (B). AU, arbitrary units. (n = 3) *, P < 0.01 as determined by Student's t test; NS, not significant.
Protein S-nitrosylation of pp71 impacts STING-induced transcriptional activation.
Activated STING promotes a potent antiviral state within infected cells, leading to the induction of both nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)- and interferon regulatory factor 3 (IRF3)-mediated transcription (23). This leads to the accumulation of cytokines and interferon genes, both of which limit viral replication. To determine whether protein S-nitrosylation of pp71 impacts these transcriptional pathways after STING activation, 2′,3′-cGAMP-treated MRC5 cells were infected with WT, pp71-C218S, or pp71-C218Srev virus, and the expression of NF-κB- and IRF3-responsive genes was quantified by reverse transcriptase quantitative PCR (RT-qPCR). As expected, expression of beta 1 interferon (IFN-β1) was upregulated at both 8 and 24 hpi in WT and pp71-C218Srev virus-infected cells following STING activation; however, induction of IFN-β1 was suppressed in pp71-C218S-infected cells (Fig. 8). Expression of the NF-κB-responsive interleukin 1 beta (IL-1β) transcript was equally suppressed with each of the viruses used, suggesting the inhibition was independent of C218 S-nitrosylation. Previous studies showed HCMV-encoded proteins pUL26 and pp65 inhibit the NF-κB pathway (37, 38). As pUL26 and pp65 were expressed in the three viruses used in this study, we desired to evaluate pp71 protein S-nitrosylation in the absence of other viral proteins. However, our observations strongly suggest the induction of IFN-β1 transcription following the activation of STING is inhibited by pp71 deficient for protein S-nitrosylation at C218.
FIG 8.
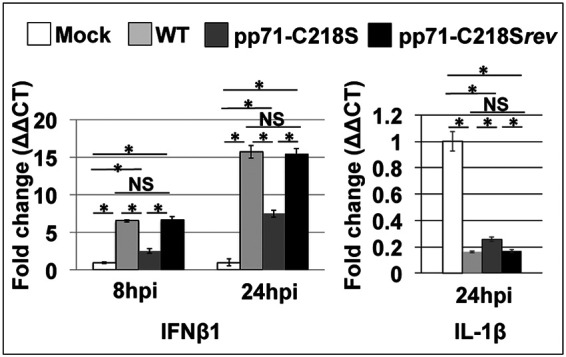
S-nitrosylation of the LxCxD domain of pp71 impacts 2′,3′-cGAMP induced IFN-β1 and IL-1β transcription. MRC-5 cells were mock infected or infected with WT, pp71-C218S, or pp71-C218Srev virus (MOI = 1) in the presence of 10 μM 2′,3′-cGAMP. Total RNA was isolated at 8 or 24 hpi. IFN-β1 and IL-1β transcripts were profiled by RT-qPCR using transcript-specific primers and plotted as ΔΔCT relative to the control transcript, GAPDH. Results are shown as mean fold change in the mRNA levels of IFN-β1 and IL-1β in the cells. (n = 3) *, P < 0.01 as determined by Student's t test; NS, not significant.
pp71 functions to inhibit STING-mediated transcription independent of other viral factors.
We have shown that blocking protein S-nitrosylation of pp71 is necessary for efficient inhibition of STING-induced transcription. Thus, we next determined if blocking this PTM is sufficient by assessing pp71 in the absence of viral infection. To this end, MRC5s stably transduced with lentiviral constructs that allow for the stable expression of either WT-pp71 (M5-pp71WT) or S-nitrosylation-deficient pp71, pp71-C218S (M5-pp71c218S), were generated (Fig. 9A). We next determined whether exogenously expressed pp71 was S-nitrosylated in the absence of HCMV infection. We performed the biotin switch assay on lysates from M5-pp71WT-transduced cells and found pp71 was indeed S-nitrosylated (Fig. 9B), suggesting this PTM occurs independent of any additional viral proteins. We did not perform this analysis on the M5-pp71c218S cells, as this mutant pp71 construct still contains the remaining two cysteines (C34 and C94) that we showed are nitrosylated (Fig. 1A). Thus, this mutant would still display protein S-nitrosylation, as this methodology does not distinguish the location of the PTM.
FIG 9.

S-Nitrosylation of pp71 is independent of viral infection and impacts STING-induced transcription in the absence of additional HCMV factors. (A) Recombinant retroviruses, pLXSN-pp71-WT or pLXSN-pp71-C218S, were used to transduce MRC5 cells that were then selected in G418 for two cell doublings yielding the parental MRC5 cells (M5), M5-pp71WT, and M5-pp71C218S. Cell lysates were isolated, and total protein (20 μg) was separated by SDS-PAGE and then transferred to a nitrocellulose membrane. Immunoblotting was performed with α-pp71 antibody and α-tubulin antibody, which served as a loading control; n = 3; representative blots are shown. (B) Lysates from M5 and M5-pp71WT were harvested and used in a biotin switch assay. Lysates were either affinity purified (AP) with avidin beads or immunoprecipitated (IP) with pp71 antibody. Proteins were separated by SDS-PAGE and then transferred to nitrocellulose membrane. Protein S-nitrosylated pp71 was detected by α-biotin or α-pp71 antibody. n = 3; representative blots are shown. (C) 2′,3′-cGAMP (10 μM) was added to M5-pp71WT and M5-pp71C218S, and 24 h later, total RNA was isolated. IFN-β1 and IL-1β transcripts were profiled by RT-qPCR using transcript-specific primers and plotted as ΔΔCT relative to the control transcript, GAPDH. Results are shown as mean fold change in the mRNA levels of IFN-β1 and IL-1β in M5-pp71WT and M5-pp71C218S cells (n = 3). *, P < 0.01 as determined by Student's t test.
To determine whether stable expression of either WT or mutant pp71 impacts interferon induction in fibroblasts in the absence of infection, we treated the transduced cells with 2′,3′-cGAMP and monitored transcription of STING-responsive transcripts by RT-qPCR. We found the M5-pp71c218S cells displayed a significant reduction of IFN-β1 compared to that of the M5-pp71WT cells (Fig. 9C). As we performed these experiments in the absence of the expression of any other HCMV proteins that may impact NF-κB, we were able to characterize the impact of pp71 S-nitrosylation on the NF-κB-mediated pathway by quantifying IL-1β expression. In support of our hypothesis that pp71 S-nitrosylation affects the inhibition of the STING pathway, our data revealed IL-1β expression was significantly inhibited by overexpression of pp71-C218S compared to that by WT-pp71 (Fig. 9C). Together, these results reveal pp71 inhibited STING-mediated signal transduction, as previously reported, thereby attenuating the STING antiviral response. Importantly, pp71 S-nitrosylation impeded this STING inhibition, and a single amino acid substitution within the pRb binding domain of pp71 was sufficient to relieve this repression.
DISCUSSION
We undertook an unbiased approach to identify and characterize the HCMV viral proteome that undergoes protein S-nitrosylation during lytic infection. As this modification is reversible, we hypothesized this PTM would play distinct roles at different points of the lytic life cycle. To this end, we characterized the S-nitrosylated proteome at a time point during infection in which viral DNA has initiated and assembly of infectious progeny has begun (96 hpi). We chose this time point for two reasons: (i) most viral proteins are expressed at high levels at this time, and (ii) protein S-nitrosylation is a known host response to stress (39). It should be noted this methodology may overlook viral proteins with short half-lives, transient protein S-nitrosylation modifications, and peptides from viral ORFs not present at this time during infection. In three biological replicates, we consistently identified 13 viral proteins from each kinetic class of HCMV proteins that are S-nitrosylated (Fig. 1A), with more than one-half of these peptides originating from tegument proteins.
Of particular interest was the identification of three distinct S-nitrosylation sites within pp71. The tegument viral protein pp71 has multiple critical biological functions which promote efficient viral replication by undermining host antiviral responses. Specifically, tegument-delivered pp71 functions to degrade human DAXX (hDAXX) and pRb, thus relieving repression of viral transcription and promoting entry into the cell cycle of the infected host cell, respectively (14–17, 40). Thus, evaluating the impact of pp71 protein S-nitrosylation was a primary focus. With advancements in bacterial artificial chromosome (BAC) recombineering technology, we are able to mutate and evaluate the requirement of specific protein motifs in viral proteins within the context of an active infection. We mutated each of the S-nitrosylated cysteines we identified in pp71 to the closely related amino acid serine, which is incapable of protein S-nitrosylation. Unexpectedly, mutation of either all three modified cysteines within pp71 or only the single cysteine within the LxCxD domain resulted in a viable virus that replicated with a modest yet consistently better efficiency than wild-type virus (Fig. 2A and 3B). We had hypothesized any deleterious mutation would result in a growth defect in the virus, especially as one of the mutated cysteines was located within the LxCxD domain previously identified in pp71 to mediate the pRb binding functions (14). However, mutations of all three identified protein S-nitrosylated cysteines to serines within pp71 resulted in nearly equal amounts of viral protein accumulation compared to that of the wild type (Fig. 3A) and no apparent defect in tegument loading of pp71 into cell-free virions (Fig. 3C), suggesting that these mutations resulted in no apparent biological defect within highly permissive fibroblasts. It remains to be determined if these targeted mutations within pp71 are critical for efficient viral replication in different cell types.
More recently, pp71 expression has been implicated in undermining the STING-induced response in HCMV-infected cells (21). STING is an adaptor protein involved in mediating the response between activated cytoplasmic DNA sensors and the type I interferon response pathway. STING activation, induced by encountering the secondary signaling mediator 2′,3′-cGAMP, promotes a potent antiviral state resulting in NF-κB-mediated transcriptional activation of cytokines and IRF3-mediated transcriptional activation of interferon genes. Thus, pp71’s ability to undermine STING activation is critical for efficient viral replication. Importantly, proteins encoded by other viruses undermine STING responses. Human papillomavirus E7 and human adenovirus E1a both interact with STING, and these interactions require the LxCxE/D pRb binding motif found in each of these viral proteins (32). Importantly the best-characterized protein S-nitrosylation motif is the I/LxCx2E/D motif, initially characterized in regulating the function of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) within the interferon gamma-activated inhibitor of translation (GAIT) complex (41). Protein S-nitrosylation regulates the function of the GAIT complex, thus restoring translation of specific transcripts during times of interferon activation (42). This motif differs from the canonical pRb binding motif by only the spacing of a single amino acid. Thus, we hypothesized the protein S-nitrosylation of the central cysteine within pp71’s pRB binding domain is critical for the interaction of this viral protein with STING.
We further investigated the biological impact of pp71 S-nitrosylation on the STING pathway. We observed, as previously reported (21), pp71 immunoprecipitated with STING, but we noted a qualitative increase in the precipitation of the two proteins when we mutated the central cysteine in pp71’s pRb binding domain to serine, thus blocking S-nitrosylation (Fig. 4A and B). This suggests that S-nitrosylation of pp71 may be inhibitory to its biological functions. The interaction of pp71 with STING did not result in degradation of this host protein (Fig. 5A and B), suggesting the mechanism of inhibiting the interferon pathway differs from how pp71 inhibits the transcriptional inhibitor, hDAXX.
To evaluate the impact of pp71 S-nitrosylation on the STING pathway, we assessed viral replication in the presence of the natural ligand for STING activation, 2′,3′-cGAMP. In support of our model, we observed inhibition of wild-type HCMV during activation of the STING pathway, but this attenuation was less pronounced when pp71 S-nitrosylation was blocked by the Cys-to-Ser mutation (Fig. 6C and D). The net effect was a decrease in the production of antiviral cytokine and interferon transcripts (Fig. 8). This led us to the novel interpretation that protein S-nitrosylation occurs at a basal level even in the absence of infection, yet increased host-directed S-nitrosylation of virulence factors may function as an additional antiviral measure, thus limiting viral replication. As viral proteins essential for replication of other unrelated viruses (e.g., human papillomavirus and human adenovirus) have conserved motifs compared to those of pp71’s protein S-nitrosylation site through which they interact with STING, we favor a model where host cell-mediated PTMs function to limit additional pathogens beyond HCMV, thus suggesting a more universal function for protein S-nitrosylation.
Why would HCMV pp71 evolve to maintain a motif that limits its capacity to replicate in the face of potent antiviral responses? Data suggest basal and stress-induced S-nitrosylation of cysteines within proteins is not random and that this modification is mediated by specific nitrosyltransferases selective for specific conserved cysteines (43). One could speculate that there would be a benefit to pp71 altering this central cysteine; however, the pp71’s pRb-binding function is important during infection of certain cell types (44). Thus, maintaining this cysteine has a fitness benefit for the virus. It will be interesting to discern the extent of increased protein S-nitrosylation of different viral proteins to determine if this mechanism of host-mediated antiviral responses is more universal and potentially exploitable as an antiviral therapy. In sum, we have identified and characterized a novel pp71 PTM that functions as an additional host-mediated antiviral measure in response to infection. The balance between a successful infection and an abortive infection pivots on the ability of a virus to undermine host countermeasures to infection. We favor a model in which host cells are able to sense the initial shift toward successful viral replication, and in response, the host cell induces pathways resulting in the accumulation of posttranslational modifications that incapacitate or limit potent viral virulence factors, such as pp71. In doing so, host cells can slow viral spread. Identifying the direct factors that dictate these responses will be of high value.
MATERIALS AND METHODS
Cell culture.
Primary newborn human foreskin fibroblasts (NuFF-1; GlobalStem) or primary human embryonic lung fibroblasts (MRC5; ATCC) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml each penicillin and streptomycin. Human embryonic kidney 293 FT cells (293FT; ATCC) and amphotropic phoenix cells (Phoenix-Ampho; ATCC) were maintained in DMEM supplemented with 10% newborn calf serum (NCS) and 100 U/ml each penicillin and streptomycin. Unless indicated, all cells were maintained at 37°C with 5% CO2.
Biotin switch assay and mass spectrometry.
NuFF-1 cells were infected with TB40/E-mCherry-UL99eGFP (45) (herein referred to as wild-type [WT] virus) at a multiplicity of infection (MOI) of 3. Infected cells were harvested 96 h postinfection (hpi), and the biotin switch assay was conducted using an S-Nitrosylated Protein Detection kit (Cayman), according to the manufacturer’s instructions. Proteins were trypsinized, and biotinylated peptides were isolated by Avidin agarose resin (Pierce). The eluates were evaporated using a SpeedVac centrifuge concentrator. Dried samples were resuspended in 10 μl of 0.1% trifluoroacetic acid and then purified by solid-phase extraction (SPE) using C18 Ziptip (stage-tip; EMD Millipore) according to the manufacture’s protocol. Next, 5 μl of the extract in 1% acetic acid was injected, and the peptides were eluted from the column (Dionex 15 cm by 75-μm inside diameter [i.d.] Acclaim Pepmap C18, 2 μm, 100-Å reversed-phase capillary chromatography column) by an acetonitrile-0.1% formic acid gradient at a flow rate of 0.3 μl/min and were introduced into the source of the mass spectrometer (LTQ-Obitrap Elite hybrid mass spectrometer; Finnigan). The microelectrospray ion source was operated at 2.1 kV. The digest was analyzed using the data-dependent multitask capability of the instrument, acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. The data were analyzed by using all collision-induced dissociation (CID) spectra collected in the experiment to search the human and HCMV/HHV5 reference sequence (NCBI:txid295027) with the search program Sequest (46). Biological replicates from three independent infections of both HCMV-infected and mock samples were analyzed by mass spectrometry.
BAC recombineering, virus production, and viral growth analyses.
Based on the mass spectrometry results, the identified S-nitrosylated cysteines (C) were mutated to serine (S). S-Nitrosylation-resistant mutants (pp71 single mutant, C218S, and triple mutant C34S/C94S/C218S) were generated using the BAC-derived parental virus, TB40/E-mCherry-UL99eGFP, using the galK-positive/counterselection scheme of BAC recombineering, described previously (47). In brief, galK was amplified using primers designed with 50-bp homology to the flanking region of each mutation site (Table 1). Purified PCR products were transformed into electrocompetent SW105 Escherichia coli cells harboring the TB40/E-mCherry-UL99eGFP BAC genome. GalK-positive clones were screened by sequence verification. Positive recombinants were made electrocompetent, and double-stranded DNA oligonucleotides in which the codon for cysteine (TGT) was replaced by the codon for serine (TCA) flanked by homology arms were then transformed into electrocompetent SW105 E. coli cells that harbored the galK-positive intermediate BAC genome. Successful recombinants were then counterselected on 2-deoxygalactose-containing plates, and the desired mutation was confirmed by Sanger sequencing.
TABLE 1.
Oligonucleotides used in this study
| Primer use | Sequence (5′→3′) | Targeta |
|---|---|---|
| Oligonucleotides for making targeted galK insertions into pp71 | CGG AAG CGG CCG CGA TCA GCG AGG CCG AAG CCG CCA GCG GCA GCT TTG GTC CTG TTG ACA ATT AAT CAT CGG CA | pp71 C34S GalK FWD |
| CGT AGA CGA CCG GCT TCC AGC GAG CCG CCT TCC ACG TTG GTG ATG AGC CGT CAG CAC TGT CCT GCT CCT T | pp71 C34S GalK REV | |
| AGG AGA ACA AAT CTC CGC ACG ACA CCG TAG ACC TGA CCG ATC TAA ACA TCC CTG TTG ACA ATT AAT CAT CGG CA | pp71 C94S GalK FWD | |
| AGG CGT CGT GGG CCA AAG TTG TTG AGG TCC ACC AGT AGT CGG TCC TGT TCT CAG CAC TGT CCT GCT CCT T | pp71 C94S GalK REV | |
| TCG CCT TTC GCG CCG GCG CTA TCC CGC TGA CGT TGG TAG ACG CCC TGG AGC CTG TTG ACA ATT AAT CAT CGG CA | pp71 C218S GalK FWD | |
| ATG ATC CAT TGG CCT CGC TCG TCC GTC TCC GTT TTG TGG ATG TAC GTA TTT CAG CAC TGT CCT GCT CCT T | pp71 C218S GalK REV | |
| Oligonucleotides for making S-nitrosylation-resistant mutants (galK reversions) | CAG CGA GGC CGA AGC CGC CAG CGG CAG CTT TGG TCG CCT GCA CTC ACA GGT GCT TCG GCT CAT CAC CAA CGT GGA AGG CGG CTC GCT GG | pp71 C34S Mut FWD |
| CCA GCG AGC CGC CTT CCA CGT TGG TGA TGA GCC GAA GCA CCT GTG AGT GCA GGC GAC CAA AGC TGC CGC TGG CGG CTT CGG CCT CGC TG | pp71 C34S Mut REV | |
| GCA CGA CAC CGT AGA CCT GAC CGA TCT AAA CAT CAA GGG CCG CTC AGT GGT GGG CGA ACA GGA CCG ACT ACT GGT GGA CCT CAA CAA CT | pp71 C94S Mut FWD | |
| AGT TGT TGA GGT CCA CCA GTA GTC GGT CCT GTT CGC CCA CCA CTG AGC GGC CCT TGA TGT TTA GAT CGG TCA GGT CTA CGG TGT CGT GC | pp71 C94S Mut REV | |
| CGC TAT CCC GCT GAC GTT GGT AGA CGC CCT GGA GCA GCT GGC CTC ATC GGA CCC TAA TAC GTA CAT CCA CAA AAC GGA GAC GGA CGA GC | pp71 C218S Mut FWD | |
| GCT CGT CCG TCT CCG TTT TGT GGA TGT ACG TAT TAG GGT CCG ATG AGG CCA GCT GCT CCA GGG CGT CTA CCA ACG TCA GCG GGA TAG CG | pp71 C218S Mut REV | |
| Sequencing primers for confirming S-nitrosylation-resistant mutants | CGA CCT CCC ACG AAG ACC C | pp71 C34S Seq FWD |
| GTC TAC GGT GTC GTG CGG AG | pp71 C34S Seq REV | |
| AGG TGC TTC GGC TCA TCA CC | pp71 C94S Seq FWD | |
| CCG CTG GCT CTG AAA GAG GT | pp71 C94S Seq REV | |
| CCG CAC GGC TCA CTA CTG G | pp71 C218S Seq FWD | |
| GGT GCG GGT ACG GAT TGT GT | pp71 C218S Seq REV | |
| Primers for cloning pp71 (WT/C218S) into pCMSeGFP vector | TCG AGA ATT CGC CAC CAT GGA CTA CAA AGA CGA TGA CGA CAA GAT GTC TCA GGC ATC GTC CTC GC | EcoRI-Kozak-Flag-pp71 FWD |
| AGC TGA ATT CCT AGA TGC GGG GTC GAC TGC GTG GGG TGC TGG AAG TGG AAG CGG TG | EcoRI-Kozak-Flag-pp71 REV | |
| Primers for cloning pp71 (WT/C218S) into pLXSN vector | TCG AGA ATT CGC CAC CAT GTC TCA GGC ATC GTC CTC GCC CGG TGA GGG ACC CTC GTC GGA | EcoRI-Kozak-pp71 FWD |
| AGC TGA ATT CCT AGA TGC GGG GTC GAC TGC GTG GGG TGC TGG AAG TGG AAG CGG TG | EcoRI-Kozak-pp71 REV | |
| Real-time PCR primers | CAG CAA TTT TCA GTG TCA GAA GC | IFN-β1 FWD |
| TCA TCC TGT CCT TGA GGC AGT | IFN-β1 REV | |
| GAA TGG GTT TGC TAG AAT GTG ATA | IL-1β FWD | |
| CAG ACT AGG GTT GCC AGA TTT AAC | IL-1β REV | |
| ATG GTT TTG CAG GCT TTG ATG | UL122 FWD | |
| ACC TGC CCT TCA CGA TTC C | UL122 REV | |
| CCC TCG GCT TCT CAC AAC AAT | UL54 FWD | |
| CGA GTT AGT CTT GGC CAT GCA T | UL54 REV | |
| GGT TTC TGG CTC GTG GAT GTC G | UL32 FWD | |
| CAC ACA ACA CCG TCG TCC GAT TAC | UL32 REV | |
| ACC CAC TCC TCC ACC TTT GAC | GAPDH FWD | |
| CTG TTG CTG TAG CCA AAT TCG T | GAPDH REV | |
| ACC ACT ACC TGA GCA CCC AGT C | eGFP FWD | |
| GTCCATGCCGAGAGTGATCC | eGFP REV | |
| CCC CTT CCA TCA CAT TGC A | mdm2 FWD | |
| AGT TTG GCT TTC TCA GAG ATT TCC | mdm2 REV |
FWD, forward primer; REV, reverse primer.
Viral BAC DNA was isolated as described previously (48). Viral BAC DNA and the HCMV pp71-expressing plasmid, pCGN1-pp71 (1 μg) (49), were transfected into low-passage-number MRC5 cells by electroporation, which were transferred to 10-cm dishes and incubated at 37°C with 5% CO2 overnight. At 24 h posttransfection, cells were washed with 1× with phosphate-buffered saline (PBS) to remove dead cells and refed with fresh DMEM supplemented with 10% FBS and 100 U/ml each penicillin and streptomycin. After 7 days, the transfected cells were trypsinized, and one-tenth of the transfection was divided between 12 145-mm dishes containing confluent MRC5 cells, each cultured in 20 ml DMEM, supplemented with 10% new born calf serum (NBCS) and 100 U/ml each penicillin and streptomycin. After 100% cytopathic effect (CPE) was observed, infected cells and media were collected and transferred to centrifuge tubes. Cells were pelleted and supernatant was reserved. Virus was released from the cell pellets by resuspension in 10 ml of medium and disruption by bath sonication. The medium was then cleared of cellular debris, and the supernatant was added to the reserved medium. Virus was then purified by ultracentrifugation through a 20% sorbitol cushion (20% sorbitol in 1× PBS) for 90 min at 72,000 × g at 25°C. The resulting pellet was resuspended in 5 ml of DMEM supplemented with 10% FBS and 1.5% bovine serum albumin (BSA) in a 1:1 ratio, and aliquots were stored at −80°C following snap-freezing in liquid nitrogen. The titers of virus stocks were determined by 50% tissue culture infectious dose (TCID50) on naive NuFF-1 cells.
Newly generated recombinant S-nitrosylation-resistant viruses (pp71 single mutant, C218S, and triple mutant C34S/C94S/C218S) were compared to the parental wild-type TB40/E-mCherry-UL99eGFP (WT) by infecting a confluent monolayer of MRC5 cells at an MOI of 1 infectious unit (IU)/cell as quantified by a TCID50 assay. Supernatants were collected every day for one week, and viral titers quantified by a TCID50 assay on naive NuFF-1 cells.
Generation of plasmids for transfection and transduction.
Primers were generated to amplify the different pp71 ORFs from the WT and recombinant viruses for subsequent cloning into expression constructs using the primer sets (Table 1). FLAG-epitope tagged pp71-WT or pp71-C218S was generated using the primers in Table 1 and the respective BAC constructs as the templates. The PCR products were digested with EcoRI and then cloned into pCMSeGFP (Clontech) using the EcoRI site within the multicloning site. To generate pp71-expressing MRC5 cells, pp71 WT or pp71 C218S was PCR amplified from the above-described BAC constructs and subsequently cloned into the pLXSN retroviral vector (Clontech) using the EcoRI site. pCDNA-STING-HA was a generous gift from Blossom A. Damania (UNC-Chapel Hill) (34).
To generate pp71-expressing retroviruses, Phoenix cells were transfected with 1 μg of either pLXSN-pp71WT or pLXSN-pp71 C218S using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Transfected Phoenix cells were washed, replenished with medium, and maintained in at 32°C with 5% CO2 for 2 days. The retrovirus-containing medium was then clarified by low-speed centrifugation and added to MRC5 cells at 70% confluence. The following day, successfully transduced MRC5 cells were selected by adding 300 μg/ml G418 (Invitrogen) to the medium. This selection treatment was repeated for a total of two cell doublings of the MRC5 cells.
Co-immunoprecipitation and Western blotting.
For Western blot experiments, total protein was extracted in lysis buffer (50) containing 20 mM HEPES (pH 7.4), 50 mM NaCl, 1.5 mM MgCl2, 2 mM dithiothreitol (DTT), 2 mM EGTA, 10 mM NaF, 12.5 mM β-glycerophosphate, 1 mM Na3VO4, 5 mM Na-pyrophosphate, 0.2% (vol/vol) Triton X-100, and Complete protease inhibitors (Roche Applied Science). Equal amounts of protein (20 μg) were separated by 8% SDS-PAGE, transferred to nitrocellulose or polyvinylidene difluoride (PVDF) by semidry transfer, and then probed with the following primary antibodies: anti-pp71 (1:100, 2H10-9 [14]), anti-IE2 (1:100, 3A9 [51]), anti-pp65 (1:100, 8A8 [52]), anti-STING (1:1,000, D2P2F; Cell Signaling), anti-biotin (1:75; Cayman), anti-FLAG (1:2,000; Sigma-Aldrich), anti-HA (1:1,000; Sigma-Aldrich), and anti-α-tubulin (1:5,000; Sigma-Aldrich). Membranes were incubated with a secondary antibody: IRDye 800CW anti-rabbit antibody (1:15,000; LI-COR) or IR-Dye 680RD anti-mouse antibody (1:15,000; LI-COR). Membranes were then washed with 1× tris-buffered saline (TBS), pH 7.6, supplemented with 0.05% Tween 20 (1× TBS-T) and scanned using an Odyssey (LI-COR), and protein bands were visualized by Image Studio software (LI-COR).
For all affinity/immunoprecipitation (IP) experiments, cells were harvested at 4 dpi and lysed in IP lysis buffer containing 20 mM HEPES (pH 7.4), 50 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 2 mM EGTA, 10 mM NaF, 12.5 mM β-glycerophosphate, 1 mM Na3VO4, 5 mM Na-pyrophosphate, 0.2% (vol/vol) Triton X-100, and Complete protease inhibitors (Roche Applied Science). To confirm S-nitrosylation of pp71, NuFF-1 cells were infected at a multiplicity of 1 IU/cell with TB40/E-mCherry-UL99eGFP. After the biotin switch assay, lysates were affinity purified with avidin beads or immunoprecipitated with the designated antibody. Purified lysates were separated by 8% SDS-PAGE and blotted onto a Protran 0.45-μm nitrocellulose membrane (Amersham). S-Nitrosylated proteins were detected by using HCMV-specific antibodies (1:100), as indicated in the text and/or legends, or an antibody against biotin (1:75).
For IP, lysates were precleared for 1 h with mouse immunoglobulin G (IgG) agarose or rabbit immunoglobulin G (IgG) agarose (Sigma), dependent on the species of antibody used in IP, and incubated overnight with IP antibodies and then with protein A/G beads for 1 h with rotation. After incubation, beads were washed with IP lysis buffer and boiled with 4× Laemmli sample buffer. Equal amounts of protein (20 μg) were separated by 8% SDS-PAGE, transferred to nitrocellulose/PVDF, and then probed with the following primary antibodies: anti-m2 FLAG (1:7,500; Sigma-Aldrich) and anti-α-tubulin (1:5,000; Sigma-Aldrich). Both were visualized using a secondary antibody conjugated to horseradish peroxidase (HRP) (goat-anti-mouse HRP, 1:10,000; Jackson ImmunoResearch).
Quantification of DNA and RNA.
Total DNA was extracted by phenol-chloroform-isoamyl purification followed by ethanol precipitation. DNA concentrations were then determined, and equal amounts of DNA were analyzed by quantitative PCR (qPCR) in triplicates using Power SYBR green Master Mix (Applied Biosystems) and an Eppendorf Mastercycler RealPlex2 real-time PCR machine. Copy number was calculated using a standard curve generated from HCMV BAC DNA, containing eGFP and mdm2, a kind gift from Christine O’Connor (Cleveland Clinic). Primers used for qPCR are listed in Table 1.
Total RNA was isolated using TRIzol reagent (Invitrogen) under RNase-free conditions according to the manufacturer’s instructions and then precipitated with isopropanol. DNA-contaminating templates were removed by DNase treatment using the DNA-free DNA Removal kit (Ambion). RNA concentrations were determined, and equal amounts (0.9 μg) were used to generate cDNA using the TaqMan reverse transcription kit with random hexamers (Applied Biosystems) according to the manufacturer’s protocol. Equal amounts of cDNA (10 ng) were then analyzed by qPCR in triplicates using Power SYBR green Master Mix (Applied Biosystems) and an Eppendorf Mastercycler RealPlex2 real-time PCR machine. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA levels were quantified in parallel and used to normalize the RNA isolation efficiency of each sample. Viral RNA copy number was calculated using a standard curve generated from HCMV BAC DNA, as described above.
Data availability.
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD019208.
ACKNOWLEDGMENTS
We thank Belinda Willard of the Cleveland Clinic Proteomics and Metabolomics Core Facility for her assistance with the proteomics reported in the manuscript.
REFERENCES
- 1.Britt WJ, Prichard MN. 2018. New therapies for human cytomegalovirus infections. Antiviral Res 159:153–174. doi: 10.1016/j.antiviral.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. doi: 10.1007/978-3-540-77349-8_23. [DOI] [PubMed] [Google Scholar]
- 3.McCormick AL, Mocarski ES Jr. 2007. Viral modulation of the host response to infection In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 4.Sinclair JH, Reeves MB. 2013. Human cytomegalovirus manipulation of latently infected cells. Viruses 5:2803–2824. doi: 10.3390/v5112803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frenkel LD. 2016. Comparing congenital Zika and cytomegalovirus affliction. Pediatr Infect Dis J 35:1371–1372. doi: 10.1097/INF.0000000000001273. [DOI] [PubMed] [Google Scholar]
- 6.Yang R, Zhang R, Zhang Y, Huang Y, Liang H, Gui G, Gong S, Wang H, Xu M, Fan J. 2020. Risk factors analysis for human cytomegalovirus viremia in donor+/recipient+ hematopoietic stem cell transplantation. Lab Med 51:74–79. doi: 10.1093/labmed/lmz030. [DOI] [PubMed] [Google Scholar]
- 7.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy E, Rigoutsos I, Shibuya T, Shenk TE. 2003. Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci U S A 100:13585–13590. doi: 10.1073/pnas.1735466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murthy S, O'Brien K, Agbor A, Angedakin S, Arandjelovic M, Ayimisin EA, Bailey E, Bergl RA, Brazzola G, Dieguez P, Eno-Nku M, Eshuis H, Fruth B, Gillespie TR, Ginath Y, Gray M, Herbinger I, Jones S, Kehoe L, Kuhl H, Kujirakwinja D, Lee K, Madinda NF, Mitamba G, Muhindo E, Nishuli R, Ormsby LJ, Petrzelkova KJ, Plumptre AJ, Robbins MM, Sommer V, Ter Heegde M, Todd A, Tokunda R, Wessling E, Jarvis MA, Leendertz FH, Ehlers B, Calvignac-Spencer S. 2019. Cytomegalovirus distribution and evolution in hominines. Virus Evol 5:vez015. doi: 10.1093/ve/vez015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engel P, Angulo A. 2012. Viral immunomodulatory proteins: usurping host genes as a survival strategy. Adv Exp Med Biol 738:256–276. doi: 10.1007/978-1-4614-1680-7_15. [DOI] [PubMed] [Google Scholar]
- 11.Hensel GM, Meyer HH, Buchmann I, Pommerehne D, Schmolke S, Plachter B, Radsak K, Kern HF. 1996. Intracellular localization and expression of the human cytomegalovirus matrix phosphoprotein pp71 (ppUL82): evidence for its translocation into the nucleus. J Gen Virol 77:3087–3097. doi: 10.1099/0022-1317-77-12-3087. [DOI] [PubMed] [Google Scholar]
- 12.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A 100:3263–3268. doi: 10.1073/pnas.0538058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalejta RF, Shenk T. 2003. The human cytomegalovirus UL82 gene product (pp71) accelerates progression through the G1 phase of the cell cycle. J Virol 77:3451–3459. doi: 10.1128/jvi.77.6.3451-3459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338. doi: 10.1016/j.virol.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 18.Stiegler P, De Luca A, Bagella L, Giordano A. 1998. The COOH-terminal region of pRb2/p130 binds to histone deacetylase 1 (HDAC1), enhancing transcriptional repression of the E2F-dependent cyclin A promoter. Cancer Res 58:5049–5052. [PubMed] [Google Scholar]
- 19.Yang X, Khosravi-Far R, Chang HY, Baltimore D. 1997. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD. 2006. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J Virol 80:951–963. doi: 10.1128/JVI.80.2.951-963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, Luo WW, Li S, Luo MH, Wang YY, Shu HB. 2017. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21:231–243. doi: 10.1016/j.chom.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. 1992. S-Nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. 2013. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem 288:26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindermayr C, Sell S, Durner J. 2008. Generation and detection of S-nitrosothiols. Methods Mol Biol 476:217–229. doi: 10.1007/978-1-59745-129-1_15. [DOI] [PubMed] [Google Scholar]
- 27.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. 2006. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A 103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dighiero P, Reux I, Hauw JJ, Fillet AM, Courtois Y, Goureau O. 1994. Expression of inducible nitric oxide synthase in cytomegalovirus-infected glial cells of retinas from AIDS patients. Neurosci Lett 166:31–34. doi: 10.1016/0304-3940(94)90833-8. [DOI] [PubMed] [Google Scholar]
- 29.Jaffrey SR, Snyder SH. 2001. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Waye MM, Taricani M, Buckingham K, Sandham HJ. 1993. Biotin-containing protein as a cause of false positive clones in gene probing with streptavidin/biotin. Biotechniques 14:209–212. [PubMed] [Google Scholar]
- 31.Bresnahan WA, Shenk TE. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A 97:14506–14511. doi: 10.1073/pnas.97.26.14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau L, Gray EE, Brunette RL, Stetson DB. 2015. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350:568–571. doi: 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B. 2015. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112:E4306–E4315. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers J, Angulo A, Amaratunga D, Guo H, Jiang Y, Wan JS, Bittner A, Frueh K, Jackson MR, Peterson PA, Erlander MG, Ghazal P. 1999. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J Virol 73:5757–5766. doi: 10.1128/JVI.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A 100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathers C, Schafer X, Martinez-Sobrido L, Munger J. 2014. The human cytomegalovirus UL26 protein antagonizes NF-kappaB activation. J Virol 88:14289–14300. doi: 10.1128/JVI.02552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. 2005. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem 280:14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
- 40.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/mcb.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, Fox PL. 2014. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 159:623–634. doi: 10.1016/j.cell.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia J, Arif A, Willard B, Smith JD, Stuehr DJ, Hazen SL, Fox PL. 2012. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol Cell 47:656–663. doi: 10.1016/j.molcel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guerra D, Ballard K, Truebridge I, Vierling E. 2016. S-Nitrosation of conserved cysteines modulates activity and stability of S-nitrosoglutathione reductase (GSNOR). Biochemistry 55:2452–2464. doi: 10.1021/acs.biochem.5b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalejta RF. 2004. Human cytomegalovirus pp71: a new viral tool to probe the mechanisms of cell cycle progression and oncogenesis controlled by the retinoblastoma family of tumor suppressors. J Cell Biochem 93:37–45. doi: 10.1002/jcb.20177. [DOI] [PubMed] [Google Scholar]
- 45.Nukui M, O’Connor C, Murphy E. 2018. The natural flavonoid compound deguelin inhibits HCMV lytic replication within fibroblasts. Viruses 10:614. doi: 10.3390/v10110614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 47.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishna B, Miller W, O’Connor C. 2018. US28: HCMV's Swiss army knife. Viruses 10:445. doi: 10.3390/v10080445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baldick CJ Jr, Marchini A, Patterson CE, Shenk T. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J Virol 71:4400–4408. doi: 10.1128/JVI.71.6.4400-4408.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, Barik S, Sen GC. 2017. STING Requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe 21:788. doi: 10.1016/j.chom.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 51.Nevels M, Brune W, Shenk T. 2004. SUMOylation of the human cytomegalovirus 72-kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2 protein and promotes viral replication. J Virol 78:7803–7812. doi: 10.1128/JVI.78.14.7803-7812.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bechtel JT, Shenk T. 2002. Human cytomegalovirus UL47 tegument protein functions after entry and before immediate-early gene expression. J Virol 76:1043–1050. doi: 10.1128/jvi.76.3.1043-1050.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD019208.