

Abstract
Background
Duchenne muscular dystrophy (DMD) is a progressive muscle‐wasting disease caused by mutations in the dystrophin gene, which leads to structural instability of the dystrophin–glycoprotein‐complex with subsequent muscle degeneration. In addition, muscle inflammation has been implicated in disease progression and therapeutically addressed with glucocorticosteroids. These have numerous adverse effects. Treatment with human immunoglobulin G (IgG) improved clinical and para‐clinical parameters in the early disease phase in the well‐established mdx mouse model. The aim of the present study was to confirm the efficacy of IgG in a long‐term pre‐clinical study in mdx mice.
Methods
IgG (2 g/kg body weight) or NaCl solution as control was administered monthly over 18 months by intraperitoneal injection in mdx mice beginning at 3 weeks of age. Several clinical outcome measures including endurance, muscle strength, and echocardiography were assessed. After 18 months, the animals were sacrificed, blood was collected for analysis, and muscle samples were obtained for ex vivo muscle contraction tests, quantitative PCR, and histology.
Results
IgG significantly improved the daily voluntary running performance (1.9 m more total daily running distance, P < 0.0001) and slowed the decrease in grip strength by 0.1 mN, (P = 0.018). IgG reduced fatigability of the diaphragm (improved ratio to maximum force by 0.09 ± 0.04, P = 0.044), but specific tetanic force remained unchanged in the ex vivo muscle contraction test. Cardiac function was significantly better after IgG, especially fractional area shortening (P = 0.012). These results were accompanied by a reduction in cardiac fibrosis and the infiltration of T cells (P = 0.0002) and macrophages (P = 0.0027). In addition, treatment with IgG resulted in a significant reduction of the infiltration of T cells (P ≤ 0.036) in the diaphragm, gastrocnemius, quadriceps, and a similar trend in tibialis anterior and macrophages (P ≤ 0.045) in gastrocnemius, quadriceps, tibialis anterior, and a similar trend in the diaphragm, as well as a decrease in myopathic changes as reflected by a reduced central nuclear index in the diaphragm, tibialis anterior, and quadriceps (P ≤ 0.002 in all).
Conclusions
The present study underscores the importance of an inflammatory contribution to the disease progression of DMD. The data demonstrate the long‐term efficacy of IgG in the mdx mouse. IgG is well tolerated by humans and could preferentially complement gene therapy in DMD. The data call for a clinical trial with IgG in DMD.
Keywords: Inflammation, Immunmodulation, Immunoglobulin G, Voluntary wheel running, mdx mouse model, Muscular dystrophy
1. Introduction
Duchenne muscular dystrophy (DMD) is the most common myopathy in childhood with an incidence of 1 per 3500 newborn boys.1 The X‐linked inherited disease is characterized by progressive skeletal muscle weakness starting between 3 and 5 years of age and leading to wheelchair dependency in youth. The affected patients often die before the age of 30, mainly due to cardiac or respiratory failure.
DMD is mainly caused by frame shift deletion, duplication, or nonsense mutations in the DMD gene on the X chromosome (Xp21.2), which result in an absent or faulty production of dystrophin.2, 3 Dystrophin is a subsarcolemmal structure protein that links actin to the dystrophin–glycoprotein‐complex.4, 5 The disrupted dystrophin–glycoprotein‐complex results in sarcolemmal instability, an increased vulnerability to mechanical stress, changes in calcium homeostasis,6 and the replacement of muscle tissue by adipose and connective tissue. The cycles of muscle de‐ and regeneration are closely linked to inflammatory mechanisms.7 The characteristic immune cells that invade the dystrophic muscle are CD4 and CD8 positive T cells, macrophages, eosinophils, and natural killer T cells.8 The skeletal muscle itself enhances the inflammatory response by releasing certain cyto‐ or chemokines, also known as myokines.9 The inflammatory milieu is more than a coincidental reaction as shown by various mouse studies, in which specific depletion of either myeloid or lymphocyte populations reduced muscle necrosis.10, 11 In addition, non‐steroidal anti‐inflammatory drugs or inhibitors of the NF‐κB‐ and tumour necrosis factor‐pathways modulated muscle morphology and inflammatory cell infiltration, supporting a role of inflammation in the progression of the dystrophinopathy.12, 13, 14
Different treatment strategies for DMD have evolved over the years. The most promising therapeutic candidates, which include myoblast transfer and gene correcting treatments,15, 16, 17 aim at restoring dystrophin function. In 2015 and 2016, the first dystrophin‐restoring therapies were approved: ataluren (PTC Therapeutics, South Plainfield, New Jersey, USA) and eteplirsen (Sarepta Therapeutics, Cambridge, Massachusetts, USA). These promote readthrough of nonsense mutation18 or exon skipping.19 Due to the specific mutations, each of these therapies can benefit only approximately 15% of DMD patients.
With respect to muscle inflammation in the DMD muscle, pre‐clinical studies have shown that gene therapeutic approaches must be accompanied by immunosuppression.15 So far, the current international treatment guidelines20 recommend glucocorticosteroids (GS) as the standard treatment for DMD. GS reduce sarcolemmal damage and muscle degeneration, suppress inflammation, and increase calcium concentrations in skeletal muscle.21, 22 GS also improve pulmonary und cardiac function in DMD patients and lead to a longer life expectancy.23, 24 The disadvantages of long‐term treatment with GS are numerous adverse effects such as osteoporosis and Cushing syndrome.25 For this reason, several studies aim to find alternative immunosuppressive agents. Such an agent could be human immunoglobulin G (IgG). IgG is a well‐established treatment for a number of neurological disorders such as chronic inflammatory polyneuropathy. In addition, children with autoimmune diseases and immunodeficiencies tolerate IgG very well.26, 27 Although IgG is widely used, its mode of action is not fully understood. Thus far, IgG is known to neutralize autoantibodies by anti‐idiotypic binding, inhibit complement deposition, increase catabolism of pathological antibodies by saturating FcRn, modulate cyto‐ and chemokines, and reduce inflammatory T cells, macrophages, or eosinophils.28, 29, 30, 31
The efficacy of IgG treatment was recently demonstrated in the early disease stage of the well‐established mdx mouse model for DMD32. The clinical phenotype of mdx mice is milder than that of DMD patients. The pathological and clinical disease course is biphasic33. In the first phase, muscle damage appears in week 3 to 4 with an ongoing progress of de‐ and regeneration until week 8 followed by a chronic stable disease period during the subsequent 10 months33. After 12 months, muscular damage is accompanied by fibrotic replacement of necrotic muscles, which reflects the human pathology more closely than the early disease phase34.
This study demonstrates the efficacy of monthly IgG treatment over a period of 18 months in the mdx mouse model.
2. Materials and methods
2.1. Animals
The animals were bred in the central animal facility of the University Medical Center Göttingen. The mdx mice (C57BL/10ScSn mdx) used for breeding were kindly provided by Ralf Herrmann (University of Essen, Germany); male mice were heterozygous and female mice homozygous for the mdx gene. The experiments were approved by the responsible ethics committee of the state of Lower Saxony, Germany, and performed in accordance with the ethical guidelines of the national animal protection law. All animals received the usual rodent feed, as well as drinking water ad libitum. After a successful weaning phase, mdx female and male mice were block randomized into two groups and placed in individual cages equipped with a running wheel at post‐natal day 21. Two days after adaptation to the individual cage and running wheel (day 23), the animals were injected i.p., either with human IgG (2 g/kg body weight, liquid solution containing 10% IgG) (Privigen®, CSL Behring, King of Prussia, PA, USA) or the same volume of a 0.9 % NaCl solution. The i.p. injections were repeated monthly during the 18 month observation period.
2.2. Grip strength and weight assessment
Body weight and forelimb grip strength (Grip strength meter TSE‐Systems, Bad Homburg) were measured weekly. Grip strength was determined as the maximum force attained while the mouse gripped a horizontal metal bar with its forelimbs and the examiner pulled it away from the bar by its tail. The force was measured in triplicate, and the mean value was recorded.
2.3. Voluntary running in running wheel
The mice had free access to a fully computerized running wheel (circumference 38 cm), which was equipped with an aligned rotation sensor (resolution of a one‐sixteenth turn). The MATLAB software (MathWorks, Natick, MA, USA) recorded continuously the daily number of runs, maximum velocity, and cumulative running time and distance.32, 35, 36 The daily data were used for statistical analysis.
2.4. Echocardiography
Echocardiography was performed by the same, blinded examiner at three months of age, that is 2 months after the first IgG injection and then at 12 and 18 months of age. The mice were anaesthetized with isoflurane, and the heart was examined with the Vevo 1000 system (Visual Sonics) under continuous monitoring of the heart rate. Anterior and posterior wall thickness as well as systolic and diastolic left ventricular diameter and area were measured in the short axis by B‐ and M‐mode, while the distance from the aortic valve to the apex was measured in the long axis by B‐mode. The obtained left ventricular dimensions were used to calculate fractional area shortening (FAS) and ejection fraction (EF) using the equations: FAS = [(Area d − Area s)/Area d] × 100 and EF =[(Vol d − Vol s) /Vol d]× 100.
2.5. Tissue sampling, cyrosectioning, histology, and microphotography
After 18 months, the mice were euthanized by CO2 inhalation for blood and tissue collection. Blood was collected via cardiac puncture and centrifuged at 16 000 g for 10 min to obtain serum. Using routine protocols, the serum concentration of creatinine kinase (CK) was determined by the Department of Clinical Chemistry of the University Medical Center Göttingen. Right or left tibialis anterior, gastrocnemius, and quadriceps muscles, as well as the diaphragm and heart were removed for histology and the diaphragm for the ex vivo muscle contraction test. The muscle samples were placed on a cork disc coated with Tissue Tek® (Sakura, The Netherlands) and snap frozen in liquid nitrogen. The samples were then stored at −80° C until histological sections were prepared. Cross sections with a thickness of 10 μm were cut with a Leica CM3050 S cryomicrotome (Leica Mikrosysteme, Wetzler) and stained with haematoxilin and eosin stain (Mayer's Hämalaun solution, Merck, Darmstadt/Eosin G, Merck, Darmstadt). Dried slides were examined under an upright light microscope (10× objective), and 300 to 500 fibres were imaged with a digital microscope camera (ColorView, Soft Imaging Systems, Olympus, Hamburg). Using the Analysis B‐1045 software (Olympus, Hamburg), the following parameters were assessed by a blinded observer: percentage of central nucleated muscle fibers and muscle fiber diameters.
Immunohistochemical staining was performed according to a previously established protocol. The sections were air‐dried and incubated overnight with the primary antibodies for macrophages (rat monoclonal anti‐mouse F4/80 antibody, clone mCA 497, diluted 1/500 in 1% BSA; Serotec, Düsseldorf, Germany) or T cells (rat monoclonal antibody against CD3, clone KT3, diluted 1/200 1% BSA; Serotec). The sections were then rinsed and incubated for 1 hour at 20° with a second anti‐rat IgG (H + L) antibody (BA‐4001, diluted 1/200; Vector Labratories, Burlingame, CA, USA). Extravidin peroxidase (Sigma‐Aldrich, Steinheim) was blocked and antibody binding detected using the ABC system (Dako, Hamburg, Germany) and 3,3‐diaminobenzedine tetrahydrocloride (Merck) as chromogenic substrate. Finally, the slides were counterstained with haematoxylin and covered with Entellan® (Merck, Darmstadt). For quantification, five non‐overlapping representative areas were analysed under a light microscope (40× objective) by a blinded observer.
To detect myocardial fibrosis, sections were stained with the Elastica van Giesson staining method:
First, the sections were dehydrated in an ascending alcohol series and incubated with Resorcin Fuch's solution for 15 min. The stains were then incubated with van Giesson picrofuchsin solution, washed with an ascending alcohol series (80, 96, and 99%) and finally fixed with xylene. The proportion of fibrosis was analysed on the entire cardiac section (40× objective) by a blinded observer.
2.6. Ex vivo muscle contraction test
Contractile properties were measured using diaphragm strips from mdx mice as described previously.32, 37 In short, the tendons were affixed with surgical twine, and the strips were mounted in a recording chamber. The proximal tendon was connected to the transducer hook (force transducer type KG4, Scientific Instruments GmbH, Heidelberg Germany). The chamber was continuously perfused with Krebs–Henseleit solution (pH 7.4) that was bubbled with 95% O2 and 5% CO2, and the pH was continuously monitored. All experiments were performed at 25°C. The muscle strips were stimulated electrically with single or repetitive 0.1 ms stimuli. The optimum muscle length was determined before the experiments started. Responses to single stimuli and to 500 ms pulse trains at 10, 50, and 120 Hz were recorded. This was followed by a fatigue protocol consisting of 50 Hz tetanic stimulations lasting 500 ms that were repeated every 2 seconds for a total of 440 seconds. After the force recordings, the muscle strips were weighed, and their length and weight were used to calculate muscle cross‐sectional area.38 Specific muscle force was calculated as force/cross‐sectional area.
2.7. Statistical analysis
The data were statistically evaluated by one‐way analysis of variance for several groups or by t‐test for two group comparisons (GraphPad Software, Inc.). Grubbs test (QuickCal, GraphPad Software, Inc.) was used to exclude outliers. The analyses of muscle performance were adjusted for sex (Table 1). Two types of longitudinal analyses were performed (R software, version 3.2.2): The time‐course of voluntary running performance, grip strength, and body weight were analysed by longitudinal linear regression with the covariates treatment and time, adjustment for sex, and a random subject intercept that accounted for repeated measurements. Cardiac function was analysed with the longitudinal rank–sum test39 with adjustment for sex, testing an overall (main) effect of IgG treatment across time. All reported P values are two‐sided.
Table 1.
Benefits of long‐term IgG treatment in mdx mice compared with NaCl placebo
| Phenotype | Descriptive statisticsa | IgG treatment | NaCl placebo | P valueb | |||
|---|---|---|---|---|---|---|---|
| n | Descriptive statistics | n | Descriptive statistics | ||||
| Voluntary running (average of months 13 to 16) | |||||||
| Maximum velocity (m/s) | mean ± SD | 12 | 0.73 ± 0.08 | 17 | 0.66 ± 0.09 | 0.0348 | |
| Number of daily runs | mean ± SD | 12 | 120 ± 64 | 17 | 78 ± 40 | 0.0416 | |
| Total time per day (s) | mean ± SD | 12 | 4333 ± 2102 | 17 | 2450 ± 1012 | 0.0030 | |
| Total daily distance (m) | mean ± SD | 12 | 1445 ± 756 | 17 | 699 ± 298 | 0.0012 | |
| Grip strength (mN, average of months 13 to 16) | mean ± SD | 16 | 109 ± 10 | 18 | 104 ± 9 | 0.0397 | |
| Cardiac function | |||||||
| Fractional area shortening (%) | 3rd month | mean ± SD | 10 | 39.0 ± 4.3 | 10 | 33.4 ± 7.2 | 0.0119 c |
| 12th month | mean ± SD | 9 | 41.0 ± 4.6 | 11 | 38.3 ± 5.1 | ||
| 18th month | mean ± SD | 8 | 39.4 ± 4.5 | 9 | 33.2 ± 3.8 | ||
| Ejection fraction (%) | 3rd month | mean ± SD | 10 | 45.0 ± 4.2 | 8 | 47.3 ± 9.1 | 0.0269 c |
| 12th month | mean ± SD | 9 | 48.3 ± 3.9 | 11 | 46.1 ± 5.9 | ||
| 18th month | mean ± SD | 8 | 48.0 ± 4.3 | 9 | 41.6 ± 3.9 | ||
| CK concentration (U/l) | median | 9 | 829 | 10 | 853 | 0.5429 | |
| IQ | (560, 1043) | (546, 1287) | |||||
| range | 144–1756 | 329–2014 | |||||
| Diaphragm | |||||||
| Fatigability (normalized force after 300 s) | mean ± SD | 9 | 0.59 ± 0.06 | 6 | 0.50 ± 0.08 | 0.0439 | |
| 50 Hz specific tetanic force (mN/mm2) | median | 10 | 50.4 | 8 | 56.9 | 0.1964 | |
| IQ | (40.0, 57.5) | (37.8, 105.5) | |||||
| range | 28.6–70.4 | 34.1–129.8 | |||||
Long‐term IgG treatment improved voluntary wheel running and grip strength, reduced fatigability of diaphragm, and had a beneficial effect on cardiac function. Displayed are descriptive statistics of the cohort and sex‐adjusted inferential statistics that tested group differences between IgG‐treated and control animals. P values < 0.05 are highlighted in bold. Fatigability is characterized by the normalized force after 300 s of diaphragm muscle fatigue protocol. For voluntary wheel running (daily measurement) and grip strength (weekly measurement), the table gives the individual averages over the 4 month period after completion of the 12th month until completion of the 16th month of life. This period corresponds most closely to the human DMD phenotype and lies well within the lifespan of mdx mice, avoiding possible non‐random dropouts.
Displayed descriptive statistics are mean and standard deviation (SD) (for normally distributed or longitudinal cardiac phenotypes), or median, interquartile (IQ), and range (for non‐normally distributed phenotypes).
Non‐normally distributed data (creatinine kinase levels, 50 Hz specific tetanic force) were standardized to normal distribution by Blom transformation before statistical testing. All statistical tests were adjusted for sex.
Comparing IgG with NaCl‐treated control animals, longitudinal rank‐sum tests jointly analysed measurements of cardiac function across the 3rd, 12th, and 18th month. Tested was an overall (main) effect of IgG treatment across time with adjustment for sex.
3. Results
3.1. IgG improved voluntary running
Over the initial 3 to 4 weeks of the experiment, that is until reaching the age of 6 to 7 weeks, all mice displayed a continuous increase in the use of the running wheel and an improvement in all running parameters, which can be attributed to weight gain and increasing muscle strength (Figure 1). This initial growth period was followed by a 6‐ to 7‐week‐long plateau phase. These findings are in line with previous studies 32, 36.
Figure 1.
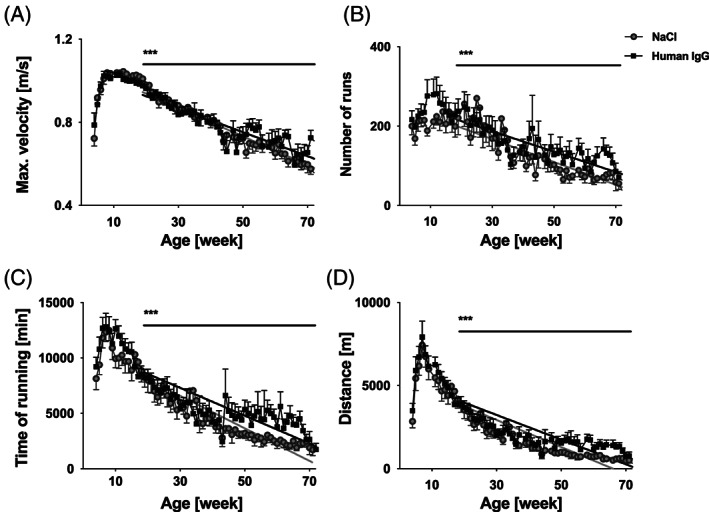
Running performance during the 18 month IgG treatment period. Running performance is displayed as daily average per group (mean ± SE), IgG‐treated mice (black squares, n = 12), NaCl control animals (grey filled circles, n = 17). (A) Maximum running speed, (B) number of daily runs, (C) total daily time in running wheel, and (D) total daily running distance decreased significantly more slowly in the IgG group (*** P < 0.0001, group differences: (A) 0.0003 m/(s·day), (B) 0.2 runs per day, (C) 5.2 s/day, and (D) 1.9 m/day, longitudinal linear regression with adjustment for sex). IgG, immunoglobulin G.
From 12 to 14 weeks of age, all mdx mice displayed a linear decline in wheel running performance (Figure 1). This time‐course was analysed by longitudinal linear regression with adjustment for sex. The decline in running performance over time was slower in the IgG group, indicating a better preservation of physical performance in this group.
Maximum running speed, number of daily runs, total daily time spent in the running wheel, and total daily running distance all decreased at a significantly slower rate in the IgG‐treated mice. The mean differences over time in the rates of decrease were: maximum running speed 0.0003 m/s per day (Figure 1A, P < 0.0001), number of daily runs 0.2 runs per day (Figure 1B, P < 0.0001), total daily time in the running wheel 5.2 s per day (Figure 1C, P < 0.0001), and total daily running distance 1.9 m per day (Figure 1D, P < 0.0001).
The longitudinal regression analysis of daily voluntary running (Figure 1, P < 0.0001) included more than 300 data points per animal, with adjustment for sex. In a secondary analysis (Table 1), a single average of each running parameter was obtained per animal, averaging over more than 100 daily data during the months 13 to 16. The course of the disease during this period corresponds most closely to the human DMD phenotype and lies well within the lifespan of mdx mice, avoiding possible non‐random dropouts (Table 1). During this 4 month period, the preservation of physical performance under IgG administration became particularly apparent. All running parameters were substantially positively affected under IgG, particularly total daily running time (IgG was 64% better than control with a sex‐adjusted group difference 1562 s ± 476 s, P = 0.0030) and total daily running distance (IgG was 94% better than control with a sex‐adjusted group difference 657 m ± 181 m, P = 0.0012).
3.2. IgG slowed the decrease of grip strength
Body weight and grip strength were determined weekly (Figure 2). No significant change in body weight was observed (Figure 2A). Grip strength decreased from week 12 to 14 onward, but the rate of decrease was 0.099 mN per week less in the IgG‐treated animals (P = 0.018, Figure 2B).
Figure 2.
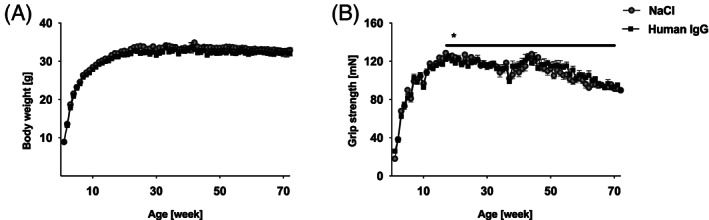
Course of body weight and grip strength over the 18 month period. (A) No significant differences in body weight were noted between the two groups. (B) Grip strength decreased significantly more slowly in the IgG group (difference 0.099 mN/week, P = 0.018, longitudinal linear regression with adjustment for sex). Data are presented as mean ± standard error. IgG, immunoglobulin G.
3.3. IgG improved cardiac function
Systolic cardiac function was measured by high‐resolution echocardiography, acquiring multiple parameters at several time points. Descriptive statistics are presented in Table 1. The longitudinal analyses jointly analysed echocardiography across the 3rd, 12th, and 18th month. Compared with control, left ventricular FAS was consistently and significantly greater in IgG‐treated animals across all three time‐points (P = 0.0119, Figure 3C).
Furthermore, IgG treatment stabilized EF more effectively over time (P = 0.0269 across all three time‐points, Figure 3D). Of note, IgG particularly improved EF during the 12th to the 18th month of life (P = 0.0050) at which time mdx and human DMD phenotypes most closely resemble each other. Other parameters, such as wall thickness or left ventricle diameter did not show relevant changes in either treatment group.
Figure 3.
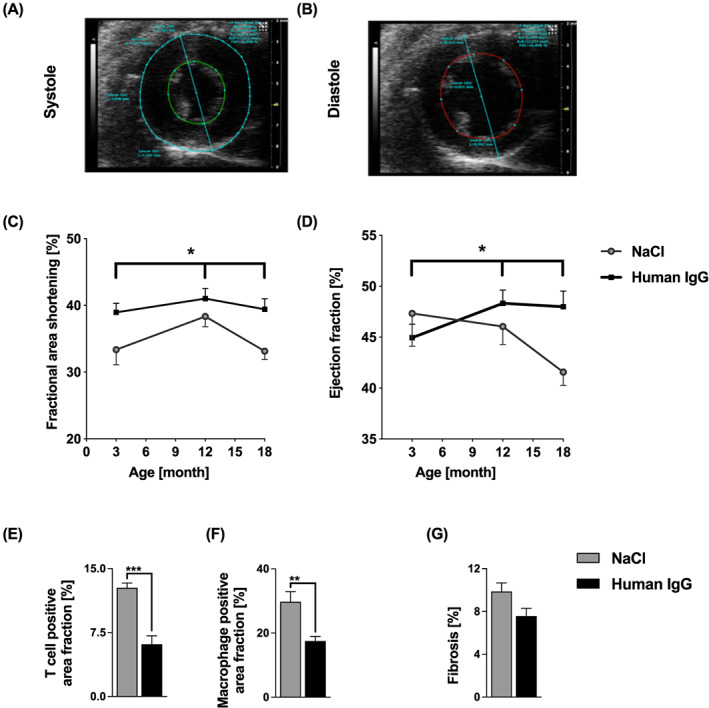
High‐resolution echocardiography and myocardial histology. (A, B) Representative echocardiograms in (A) systole and (B) diastole (three‐dimensional B‐mode, short axis), (C) Left ventricular FAS was greater in IgG‐treated animals at 3rd, 12th and 18th months (P = 0.0119). (D) IgG stabilized ejection fraction (EF) better over time (P = 0.0269, at 3rd, 12th, and 18th months), and gave a larger EF, especially at months 12 and 18 (P = 0.0050, longitudinal rank–sum test with adjustment for sex). Data are presented as mean ± standard error (SE) (* P < 0.05). (E, F) Myocardial infiltration by T cells and macrophages. IgG significantly reduced the staining signal for (E) T cells (P = 0.0002) and (F) macrophages (P = 0.0027) with a tendency towards reduced (G) cardiac fibrosis (P = 0.0574). Sample sizes were n = 6 to 8 for each analysed cardiac muscle in each treatment group (E–F) after outlier elimination with Grubbs test. Unpaired t‐test (** P < 0.01, *** P < 0.001). Data are presented as mean ± SE. See Methods Subsection Tissue sampling for detailed description. IgG, immunoglobulin G.
IgG treatment significantly reduced T cell (P = 0.0002) and macrophage (P = 0.0027) infiltration of the heart muscle. Fibrotic replacement was also reduced after treatment with IgG, but the difference did not quite reach statistical significance (P = 0.0574) (Figure 3 E–G).
3.4. Ex vivo muscle contraction test and serum CK levels
Diaphragm segments of IgG‐treated animals were less susceptible to fatigue. Normalized force after 300 s of the diaphragm muscle fatigue protocol was significantly higher than in control animals (P = 0.044, see Table 1 and Figure 4F). No significant difference in specific tetanic force was observed (P = 0.196, Table 1 and Figure 4E). Serum CK levels did not differ significantly (P = 0.543, Table 1).
Figure 4.
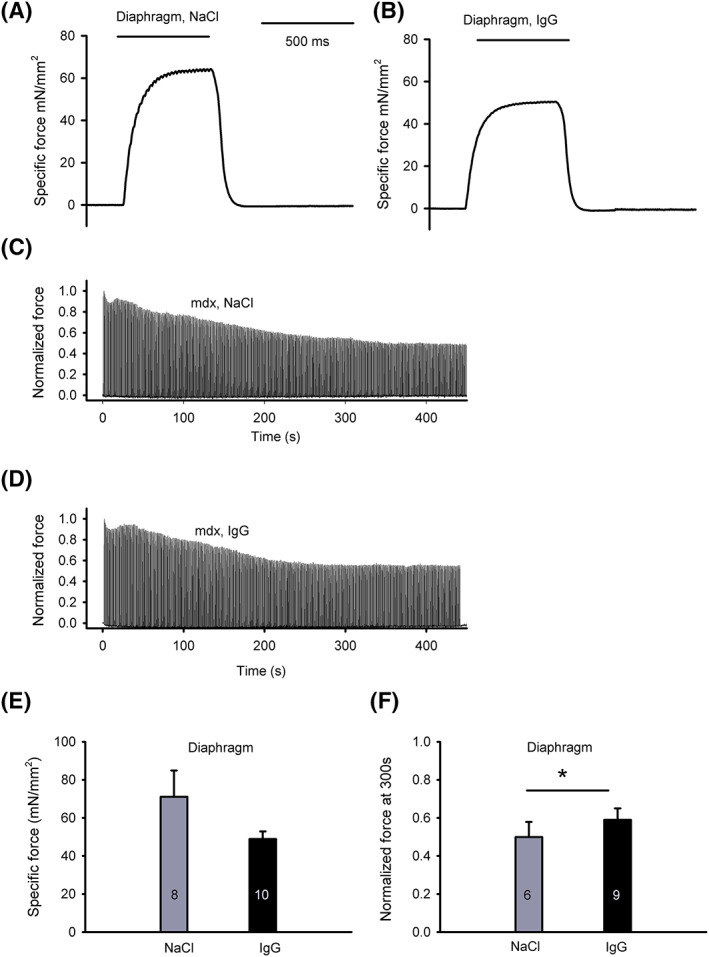
Specific tetanic force and force decrease during sustained repetitive stimulation of diaphragm strips. (A, B) Representative recordings of 50 Hz tetanic stimulation of mdx diaphragm strips at 18 months of age after monthly NaCl (A) or IgG (B) injections. (C, D) Representative recordings of the course of contractile force of diaphragm strips from NaCl controls (C) and IgG‐treated mice (D) from t = 0 to t = 440 s. 500 ms tetanic stimulation was applied every 2 seconds. (E) Peak amplitude of diaphragm specific force with 50 Hz tetanic stimulation. Mean values ± standard error (SE) are given for the indicated number of tested muscles; not significant (P = 0.196, adjusted for sex). (F) Normalized tetanic force at 300 s during the diaphragm muscle fatigue protocol. Fatigability was significantly reduced under IgG (P = 0.044, adjusted for sex). The numbers of tested muscle samples are shown in columns, data given as mean ± SE. See Methods Subsection Ex vivo muscle contraction test for detailed description. IgG, immunoglobulin G.
3.5. IgG reduced macrophage and T cell infiltration and improved myopathic changes
Macrophages and T cells are the dominant inflammatory cells involved in dystrophin‐deficient muscles. A reduction in macrophages and T cells was found in all examined muscle groups upon IgG therapy (Figure 5 and 6). The staining signal for T cells was significantly diminished after treatment with IgG in the diaphragm (P = 0.0032), gastrocnemius (P = 0.0016), and quadriceps muscles (P = 0.036) compared with controls; however, the difference was not significant in the tibialis anterior muscle (P = 0.0829). Macrophages were significantly reduced by IgG treatment in the gastrocnemius (P = 0.0097), tibialis anterior (P = 0.0014), and quadriceps muscles (P = 0.0446), while the reduction in the diaphragm was not significant (P = 0.1663; Figure 6A).
Figure 5.
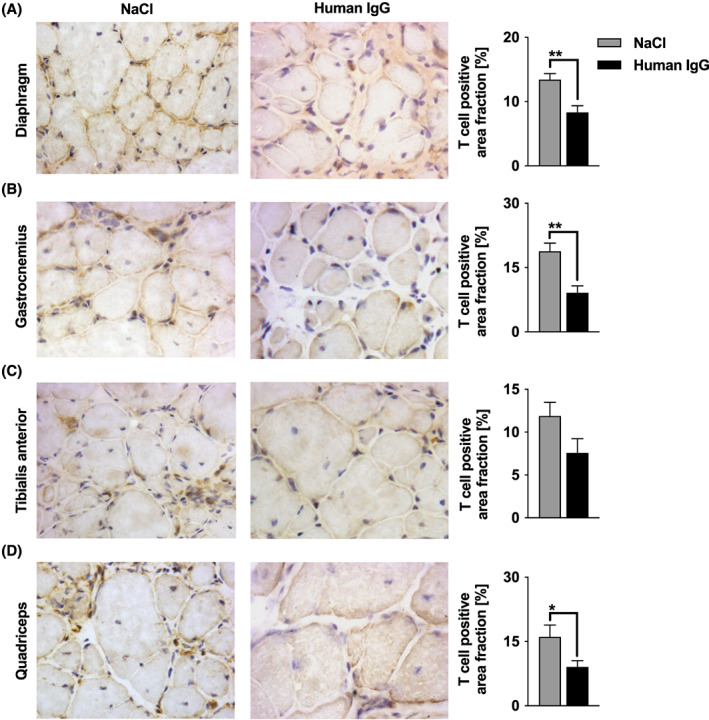
Infiltration of T cells in diaphragm, M. gastrocnemius, M. tibilialis anterior, and M. quadriceps femoris. Muscle cross sections were incubated with antibody against CD3 for T cells. (A–D) Cells stained dark brown are 3,3‐diaminobenzidine tetrahydrochloride positive T cells. In five non‐overlapping areas, T cells were quantified using a 40× objective. Sample sizes were n = 7 to 9 for each analysed muscle in each treatment group after outlier elimination with Grubbs test. IgG treatment led to a significantly reduced staining signal for T cells in (A) diaphragm (P = 0.0032), (B) gastrocnemius (P = 0.0016), and (D) quadriceps (P = 0.036). (C) M. tibialis anterior displayed a trend towards diminished T cell infiltration (P = 0.083). Unpaired t‐test (* P < 0.05, ** P < 0.01). Data are presented as mean ± SE. Photomicrographs were obtained using a 40× objective. See Methods Subsection Tissue sampling for details on staining. IgG, immunoglobulin G.
Figure 6.
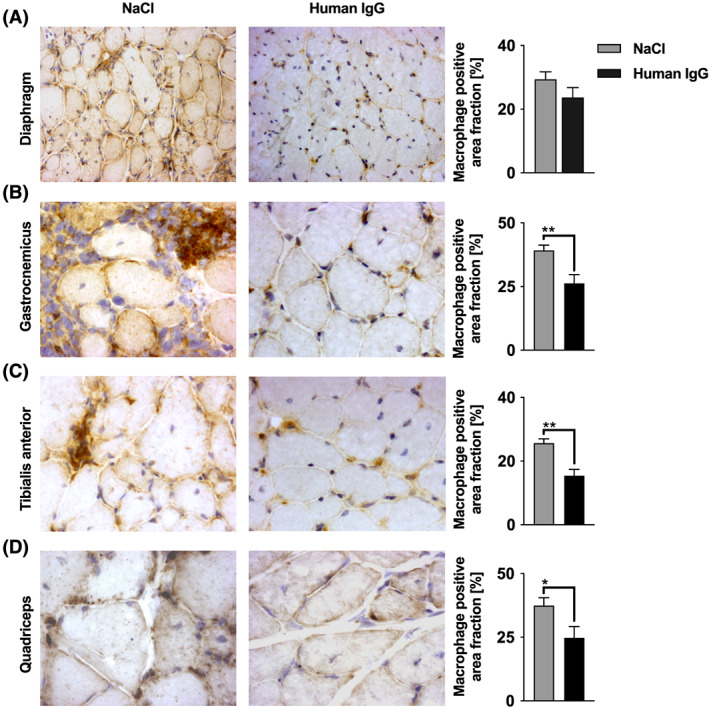
Staining signal for macrophages. Muscle cross sections were incubated with an antibody against F4/80 for macrophages. Cells stained dark brown are 3,3‐diaminobenzidine tetrahydrochloride positive macrophages. Five non‐overlapping areas were quantified using a 40× objective. Sample sizes were n = 7 to 9 for each analysed muscle in each treatment group after outlier elimination with Grubbs test. IgG treatment led to a significantly reduced staining signal for macrophages in (B) gastrocnemicus (P = 0.0097), (C) tibialis anterior (P = 0.0014), and (D) quadriceps (P = 0.0446). Unpaired t‐test (* P < 0.05, ** P < 0.01). No significant difference was seen in the (A) diaphragm (P = 0.1663). Data are presented as mean ± standard error. Photomicrographs were obtained using a 40× objective. See Methods Subsection Tissue sampling for details on staining. IgG, immunoglobulin G.
Myopathic changes, such as number of centralized cell nuclei and fiber size were assessed manually in haematoxylin‐ and eosin‐stained sections. The fiber diameter varied between 16.5 and 46.8 μm. The central nuclear index (percentage of fibers with centralized nuclei) was significantly reduced in the diaphragm (P = 0.002), as well as the tibialis anterior (P = 0.002) and quadriceps (p < 0.001) muscles after IgG treatment. A reduction of the central nuclear index indicates a therapeutic effect on de‐ and regenerative mechanisms in dystrophin‐deficient muscles (Figure 7).
Figure 7.
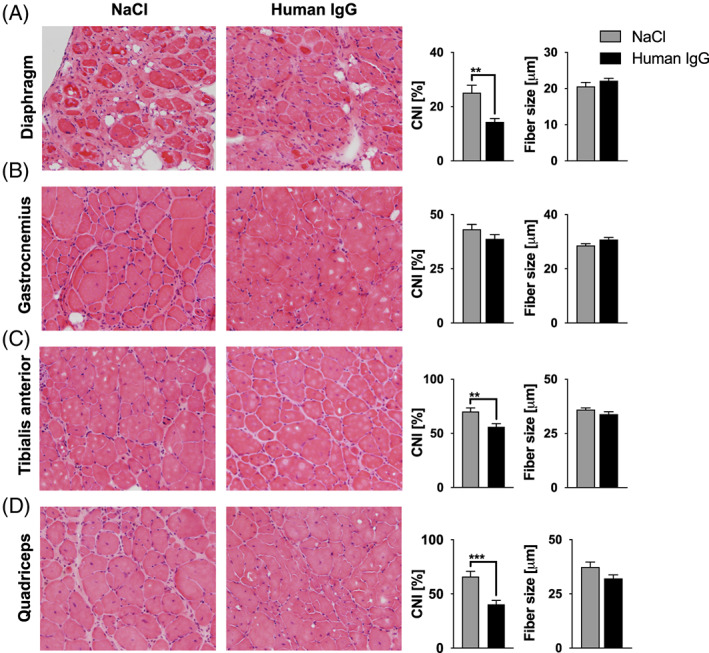
Muscle morphology. Central nuclear index (CNI) and mean fiber size were assessed in (A) diaphragm, (B) M. gastrocnemicus, (C) M tibialis anterior and (D) M. quadriceps femoris stained with haematoxylin and eosin. A lower percentage of muscle fibers with central nuclei was found after treatment with IgG, resulting in a significantly lower CNI in diaphragm (P = 0.002), tibialis anterior (P = 0.002), and quadriceps (P < 0.001). No treatment effect on CNI was detected in the gastrocnemius nor on fiber size in all analysed muscles. Statistical analysis was performed with unpaired t‐test (** P < 0.01, *** P < 0.001). Values are presented as mean ± standard error. Sample sizes were n = 7 to 9 for each analysed skeletal muscle in each treatment group after outlier elimination with Grubbs test. Photomicrographs were obtained using a 20× objective. See Methods Subsection Tissue sampling for details on staining. IgG, immunoglobulin G.
4. Discussion
IgG treatment had been proven to be effective during the early disease phase in the mdx mouse model with respect to clinical and paraclinical outcome measures.32 We hypothesized that immunomodulation with IgG would also be beneficial over a 18 month period in this model. The same treatment regimen as in the 3 month trial was used with monthly i.p. injections of human IgG (2 g/kg body weight).
The present study shows that the i.p. application of human IgG over an 18 month period was effective as it improved in vivo motor performance significantly with reduced fatigability in ex vivo muscle tests, maintained a stable cardiac function, reduced inflammatory cell infiltration in skeletal muscles, and ameliorated myopathic changes. The data also demonstrate tolerability of IgG in mdx mice over a period of 18 months. The results suggest that intravenous human IgG could be a promising treatment option for DMD patients in addition to mutation‐based therapeutic efforts.
Strategies aimed at correcting the underlying genetic mutation include antisense therapy such as eteplirsen, stop codon readthrough such as ataluren, and gene therapies such as CRISPR gene editing or micro and mini dystrophin gene delivery to express a truncated but functional dystrophin.16 While promising, these strategies are still not sufficient to cure or markedly improve DMD. Although dystrophin‐restoring therapies have already been studied in several clinical trials,40, 41, 42, 43 one can expect that years will pass before gene therapeutics are routinely used in a wide range of DMD patients.
There are at least two good reasons for supplementing the gene‐based therapies with immunosuppression or immunomodulation. First of all, when DMD is diagnosed, inflammation is predominant. And second, gene therapy does not reduce inflammation44 or fibrosis.15 In fact, an additional acute or chronic inflammatory response was observed.45, 46, 47
Several non‐steroidal immunosuppressive agents have already been successfully tested in the mdx mouse model,48, 49, 50 but, so far, the transfer to clinical trials has been rather disappointing for DMD patients.51 GS are still the recommended first line therapy, and the search for an alternative immunosuppressant is ongoing. Human IgG could be a suitable immunomodulator with this respect.
It is known from the regular use of human IgG in neurological and paediatric patients that it has fewer side effects and a more acceptable side‐effect profile than GS and other immunosuppressants. In the present study, IgG was well tolerated by mdx mice over a period of 18 months. This is in line with an 8 month treatment with human IgG in a mouse model of Alzheimer's disease.52 The rationale for our long‐term approach results from the clinical disease course in mdx mice, which develop a chronic dystrophic phase and an accumulation of muscular pathological changes after the first year of life that closely resemble the human disease.53 The rare long‐term treatment studies in mdx mice have been performed with a morpholino antisense oligomer over 12 months,54 naproxcinod over 9 months,55 and NF‐kappaB inhibitor pyrrolidine dithiocarbamate56 and prednisolone over up to 50 weeks.57
To compensate for the milder phenotype of mdx mice compared with DMD patients, a sensitive, multi‐parameter evaluation running wheel system32, 58 was used to detect therapeutic effects as recommended by the international network Treat NMD.59 In earlier studies, mdx mice displayed poorer running wheel performance with greater interindividual fluctuations compared with wild type mice.36, 60 In the present study, the human IgG‐treated animals displayed a significantly higher voluntary wheel running activity than control animals over the entire study period. Linear regression analysis demonstrated a significantly reduced long‐term decrease in all recorded running parameters, that is, maximum velocity, total time, number of runs, and total distance in the IgG group.
In mdx mice, physical training effects on muscles depend on the onset, duration, and type of training. 61 Acute and high‐intensity exercise protocols, like downhill running and treadmill training, are used to induce myofiber damage in mdx mice.62, 63 Voluntary running wheel exercise counts as low intensity training. Different data exist on how beneficial or harmful this intervention is on muscles in mdx mice.61 In our previous experiments, we demonstrated a negligible effect of running wheel exercise on muscle pathology.36 Voluntary running wheel exercise assesses the progression of the dystrophic state and enables a reliable judgement of the effectiveness of experimental drugs.
In line with stabilized running performance after IgG treatment, our mdx mice displayed a significantly slower decrease in grip strength over time compared with controls from week 30 onwards. Grip strength is influenced by many variables, such as fatigue, cognition, and learning ability, which are independent of muscle function. A possible explanation for the observed stabilized long‐term effect of IgG on grip strength is the reduction of inflammation in the forelimbs as described earlier for other treatment regimens.55
Specific muscle strength in aged mdx mice was reported to be decreased.64 Therefore, it was reasonable to examine the therapeutic effects of IgG in adult mdx animals. No beneficial effect of IgG on muscle isometric force in the ex vivo force measurements was detected in the present study. However, resistance to muscle fatigue was increased under IgG, which was in line with our previous observations32 and the present grip strength results.
CK levels in this study were not significantly changed in either group, although IgG‐treated animals displayed better running performance. Overall, both groups displayed the previously described interindividual CK level variations.55 The results are in accordance with the findings of the short‐term study.32
Progressive heart failure is one of the ultimately life‐limiting factors in DMD. The characteristic cardiomyopathic alterations include systolic dysfunction and cardiac arrhythmias.65 Cardiac function was examined with high‐resolution echocardiography at regular intervals. FAS and EF were significantly better in the IgG‐treated animals than in controls after 3, 12, and 18 months of treatment. Most studies describe first echocardiographic evidence of cardiac symptoms around an age of 9 to 12 months.66, 67, 68, 69 In this study, improved cardiac function upon IgG treatment was associated with less pathological remodelling and a significant reduction in macrophage and T cell infiltration and fibrosis in the hearts of IgG‐treated mdx mice. Similar results have been achieved with anti‐inflammatory drugs such as the cyclooxygenase‐inhibiting compound naproxcinod.55 By contrast, other tested immunosuppressants, such as tumour necrosis factor‐α antibodies70 and GS,57 were associated with decreased cardiac function. There is even evidence that tumour necrosis factor‐α antibodies can induce dilated cardiomyopathy.71, 72 Although the precise mechanisms underlying the myocardial remodelling triggered by anti‐inflammatory drugs are still not sufficiently elucidated, evidence for a crosslink between myocardial inflammation and the regenerative capacity of the heart is growing.73, 74 Non‐cardiomyocyte cells of the heart particularly contribute to this regeneration process.75
In DMD and mdx mice, inflammation is an important component of skeletal muscle pathology. Especially CD4 and CD8 T cells, macrophages, eosinophils, and natural killer T cells invade the dystrophic muscle and contribute to muscle wasting.76 We found that immunomodulatory therapy with IgG significantly reduced macrophage and T cell infiltration and ameliorated myopathic changes. The reversal of the pathological changes might be explained by an interaction between the immune system and the regenerative capacity of the muscle.7 The highly specialized immune response is guided by subpopulations of immune cells. Macrophages and T cells in the muscle display a broad continuum of activation in vivo. In the early immune response, M1 phagocytic macrophages and TH1 T cells are activated by proinflammatory cytokines and the latter promote the release of appropriate soluble agents such as interferon gamma and nitric oxide to initiate the removal of muscle debris. In this phase of inflammation and myogenesis, satellite cells are activated and begin to proliferate.77 As the process continues, macrophages and T cells change their polarization, and the so‐called M2 macrophages are induced by Th2 cytokines such as interleukin‐4 and interleukin‐10,7 hereby inhibiting the proinflammatory components and regulating myoblast fusion and myofiber differentiation78. If one of these pathways is inhibited or impaired, for example by immunosuppressants such as ciclosporin,51 muscle regeneration is insufficient or even fails. We assume that immunomodulation with IgG, as opposed to classical immunosuppression, may modify the cytokine‐driven shift from early immune M1/Th1 to M2/Th2 phenotypes. This point has to be clarified in subsequent studies.
In conclusion, long‐term treatment of mdx mice with IgG improved all clinical outcome measures for skeletal muscle strength and cardiac function as well as paraclinical parameters. The results underline the importance of the inflammatory response in dystrophin‐deficient muscles and point towards IgG as an immunmodulatory and myoregulatory agent, which is presumed to cause fewer side effects than GS. Human IgG is expensive and cannot cure DMD, yet it has a highly acceptable risk/benefit ratio compared with several immunosuppressive therapies. This is particularly important for long‐term treatment and treatment of children and adolescents. Given the positive results of two pre‐clinical studies using human IgG in the mdx mouse model, future clinical studies should explore the use of human IgG in DMD.
Author contributions
J.Z. and J.S. conceived, designed, and executed the study. J.Z., P.V.J., and Y.Z. carried out the experiments. D.M., J.Z., P.V.J., and Y.Z. analysed the data. J.Z., P.V.J., and Y.Z. drafted the manuscript, which was critically revised by J.S., D.M., F.K., D.L., M.T., and H.B.
Conflict of interest
J.Z. has received reimbursements for travel from Bayer, CSL Behring, Temmler and Kedplasma. J.S. has received honoraria, research grants, or reimbursements for travel from Bayer, Biogen, Biotest, CSL Behring, Grifols, Novartis, and Octapharma. All other authors (Y.Z., D.M., P.J.V., H.B., F.K., D.L., and M.T.) declare no conflicting financial interests.
Funding
This work was supported by research grants from CSL Behring including an Interlaken Leadership Award to J.S. Parts of the study were supported by the Aktion Benni & Co and the Else‐Kröner‐Fresenius‐Stiftung (2016_A183).
Ethical approval
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. All procedures involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted (Nds. Landesamt für Verbraucherschutz und Lebensmittelsicherheit 33.9‐42502‐04‐13/1048).
Acknowledgements
We thank Fatima Agdas, Marit Strotbek, and Iris Iben for technical assistance. The CRC/Sonderforschungsbereich (SFB) 1002 service unit (S01) is acknowledged for the echocardiographic measurements. J.S., J.Z., and D.L. are members of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO‐NMD).
Zschüntzsch J., Jouvenal P. V., Zhang Y., Klinker F., Tiburcy M., Liebetanz D., Malzahn D., Brinkmeier H., and Schmidt J. (2020) Long‐term human IgG treatment improves heart and muscle function in a mouse model of Duchenne muscular dystrophy, Journal of Cachexia, Sarcopenia and Muscle, 11, 1018–1031. 10.1002/jcsm.12569.
Contributor Information
Jana Zschüntzsch, Email: j.zschuentzsch@med.uni-goettingen.de.
Jens Schmidt, Email: j.schmidt@gmx.org.
References
- 1. Emery AE. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscul Disord 1991;1:19–29. [DOI] [PubMed] [Google Scholar]
- 2. Bonilla E, Samitt CE, Miranda AF, Hays AP, Salviati G, DiMauro S, et al. Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell 1988;54:447–452. [DOI] [PubMed] [Google Scholar]
- 3. Ryder‐Cook AS, Sicinski P, Thomas K, Davies KE, Worton RG, Barnard EA, et al. Localization of the mdx mutation within the mouse dystrophin gene. EMBO J 1988;7:3017–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772;2007:108–117. [DOI] [PubMed] [Google Scholar]
- 5. Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 2000;150:1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol 2006;33:657–662. [DOI] [PubMed] [Google Scholar]
- 7. Villalta SA, Rosenberg AS, Bluestone JA. The immune system in Duchenne muscular dystrophy: friend or foe. Rare Dis (Austin, Tex) 2015;3:e1010966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr Neurol 2007;36:1–7. [DOI] [PubMed] [Google Scholar]
- 9. De Paepe B, Creus KK, Martin J‐J, De Bleecker JL. Upregulation of chemokines and their receptors in Duchenne muscular dystrophy: potential for attenuation of myofiber necrosis. Muscle Nerve 2012;46:917–925. [DOI] [PubMed] [Google Scholar]
- 10. Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol 2001;155:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spencer MJ, Montecino‐Rodriguez E, Dorshkind K, Tidball JG. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin‐deficient muscle. Clin Immunol 2001;98:235–243. [DOI] [PubMed] [Google Scholar]
- 12. Serra F, Quarta M, Canato M, Toniolo L, De Arcangelis V, Trotta A, et al. Inflammation in muscular dystrophy and the beneficial effects of non‐steroidal anti‐inflammatory drugs. Muscle Nerve 2012;46:773–784. [DOI] [PubMed] [Google Scholar]
- 13. Acharyya S, Villalta SA, Bakkar N, Bupha‐Intr T, Janssen PML, Carathers M, et al. Interplay of IKK/NF‐kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest 2007;117:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Radley HG, Davies MJ, Grounds MD. Reduced muscle necrosis and long‐term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul Disord 2008;18:227–238. [DOI] [PubMed] [Google Scholar]
- 15. Cordova G, Negroni E, Cabello‐Verrugio C, Mouly V, Trollet C. Combined therapies for Duchenne muscular dystrophy to optimize treatment efficacy. Front Genet 2018;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nelson CE, Robinson‐Hamm JN, Gersbach CA. Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat Rev Neurol 2017;nrneurol.2017;126. [DOI] [PubMed] [Google Scholar]
- 17. Min Y‐L, Bassel‐Duby R, Olson EN. CRISPR Correction of Duchenne Muscular Dystrophy. Annu Rev Med 2019;70:239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet (London, England) 2017;390:1489–1498. [DOI] [PubMed] [Google Scholar]
- 19. Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- 21. Fisher I, Abraham D, Bouri K, Hoffmann EP, Hoffman EP, Muntoni F, et al. Prednisolone‐induced changes in dystrophic skeletal muscle. FASEB J 2005;19:834–836. [DOI] [PubMed] [Google Scholar]
- 22. Wehling‐Henricks M, Lee JJ, Tidball JG. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin‐deficient skeletal muscle. Neuromuscul Disord 2004;14:483–490. [DOI] [PubMed] [Google Scholar]
- 23. Meyers TA, Townsend D. Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int J Mol Sci 2019;20(17):4098 10.3390/ijms20174098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sawnani H, Horn PS, Wong B, Darmahkasih A, Rybalsky I, Shellenbarger KC, et al. Comparison of pulmonary function decline in steroid‐treated and steroid‐naïve patients with Duchenne muscular dystrophy. J Pediatr 2019;210:194–200.e2. [DOI] [PubMed] [Google Scholar]
- 25. Angelini C, Peterle E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol myopathies cardiomyopathies Off J Mediterr Soc Myol 2012;31:9–15. [PMC free article] [PubMed] [Google Scholar]
- 26. Oates‐Whitehead RM, Baumer JH, Haines L, Love S, Maconochie IK, Gupta A, et al. Intravenous immunoglobulin for the treatment of Kawasaki disease in children. Cochrane Database Syst Rev 2003;2003(4):CD004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singhi SC, Jayshree M, Singhi P, Banerjee S, Prabhakar S. Intravenous immunoglobulin in very severe childhood Guillain‐Barre syndrome. Ann Trop Paediatr 1999;19:167–174. [DOI] [PubMed] [Google Scholar]
- 28. Berger M, McCallus DE, Lin CS‐Y. Rapid and reversible responses to IVIG in autoimmune neuromuscular diseases suggest mechanisms of action involving competition with functionally important autoantibodies. J Peripher Nerv Syst 2013;18:275–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA 2004;291:2367–2375. [DOI] [PubMed] [Google Scholar]
- 30. Klehmet J, Goehler J, Ulm L, Kohler S, Meisel C, Meisel A, et al. Effective treatment with intravenous immunoglobulins reduces autoreactive T‐cell response in patients with CIDP. J Neurol Neurosurg Psychiatry 2015;86:686–691. [DOI] [PubMed] [Google Scholar]
- 31. Samuelsson A, Towers TL, Ravetch JV. Anti‐inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 2001;291:484–486. [DOI] [PubMed] [Google Scholar]
- 32. Zschüntzsch J, Zhang Y, Klinker F, Makosch G, Klinge L, Malzahn D, et al. Treatment with human immunoglobulin G improves the early disease course in a mouse model of Duchenne muscular dystrophy. J Neurochem 2016;136:351–362. [DOI] [PubMed] [Google Scholar]
- 33. McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 2015;8:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGeachie JK, Grounds MD, Partridge TA, Morgan JE. Age‐related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J Neurol Sci 1993;119:169–179. [DOI] [PubMed] [Google Scholar]
- 35. Kutschenko A, Reinert M‐C, Klinker F, Paulus W, Hesse S, Liebetanz D. Accurate quantification of tetanus neurotoxin‐induced focal spasticity in mice using complex running wheels. J Neurosci Methods 2012;205:45–48. [DOI] [PubMed] [Google Scholar]
- 36. Weller C, Zschüntzsch J, Makosch G, Metselaar JM, Klinker F, Klinge L, et al. Motor performance of young dystrophic mdx mice treated with long‐circulating prednisolone liposomes. J Neurosci Res 2012;90:1067–1077. [DOI] [PubMed] [Google Scholar]
- 37. Steinberger M, Foller M, Vogelgesang S, Krautwald M, Landsberger M, Winkler CK, et al. Lack of the serum‐ and glucocorticoid‐inducible kinase SGK1 improves muscle force characteristics and attenuates fibrosis in dystrophic mdx mouse muscle. Pflugers Arch 2015;467:1965–1974. [DOI] [PubMed] [Google Scholar]
- 38. Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 1988;404:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Malzahn D, Schillert A, Müller M, Bickeböller H. The longitudinal nonparametric test as a new tool to explore gene‐gene and gene‐time effects in cohorts. Genet Epidemiol 2010;34:469–478. [DOI] [PubMed] [Google Scholar]
- 40. Bushby K, Finkel R, Wong B, Barohn R, Campbell C, Comi GP, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014;50:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Havens MA, Hastings ML. Splice‐switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res 2016; gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kenji RQL, Rika M, Toshifumi Y. Eteplirsen in the treatment of duchenne muscular dystrophy. Drug Des Devel Ther 2017;II:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mendell JR, Rodino‐Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 44. Farini A, Sitzia C, Erratico S, Meregalli M, Torrente Y. Influence of immune responses in gene/stem cell therapies for muscular dystrophies. Biomed Res Int 2014;2014:818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vila MC, Novak JS, Benny Klimek M, Li N, Morales M, Fritz AG, et al. Morpholino‐induced exon skipping stimulates cell‐mediated and humoral responses to dystrophin in mdx mice. J Pathol 2019;248:339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hareendran S, Balakrishnan B, Sen D, Kumar S, Srivastava A, Jayandharan GR. Adeno‐associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev Med Virol 2013;23:399–413. [DOI] [PubMed] [Google Scholar]
- 47. Mendell JR, Campbell K, Rodino‐Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med 2010;363:1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Luca A. Pre‐clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol 2012;31:40–47. [PMC free article] [PubMed] [Google Scholar]
- 49. Grounds MD, Torrisi J. Anti‐TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J Off Publ Fed Am Soc Exp Biol 2004;18:676–682. [DOI] [PubMed] [Google Scholar]
- 50. Miyatake S, Shimizu‐Motohashi Y, Takeda S, Aoki Y. Anti‐inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle‐releasing factors. Drug Des Devel Ther 2016;10:2745–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kirschner J, Schessl J, Schara U, Reitter B, Stettner GM, Hobbiebrunken E, et al. Treatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double‐blind, placebo‐controlled multicentre trial. Lancet Neurol 2010;9:1053–1059. [DOI] [PubMed] [Google Scholar]
- 52. Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H. Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer's disease. J Neuroinflammation 2012;9:618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Muntoni F, Mateddu A, Marchei F, Clerk A, Serra G. Muscular weakness in the mdx mouse. J Neurol Sci 1993;120:71–77. [DOI] [PubMed] [Google Scholar]
- 54. Wu B, Xiao B, Cloer C, Shaban M, Sali A, Lu P, et al. One‐year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol Ther 2011;19:576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uaesoontrachoon K, Quinn JL, Tatem KS, Van Der Meulen JH, Yu Q, Phadke A, et al. Long‐term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum Mol Genet 2014;23:3239–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Siegel AL, Bledsoe C, Lavin J, Gatti F, Berge J, Millman G, et al. Treatment with inhibitors of the NF‐kappaB pathway improves whole body tension development in the mdx mouse. Neuromuscul Disord 2009;19:131–139. [DOI] [PubMed] [Google Scholar]
- 57. Sali A, Guerron AD, Gordish‐Dressman H, Spurney CF, Iantorno M, Hoffman EP, et al. Glucocorticoid‐treated mice are an inappropriate positive control for long‐term preclinical studies in the mdx mouse. PLoS One 2012;7(4):e34204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liebetanz D, Merkler D. Effects of commissural de‐ and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp Neurol 2006;202:217–224. [DOI] [PubMed] [Google Scholar]
- 59. Grounds MD, Radley HG, Lynch GS, Nagaraju K, De Luca A. Towards developing standard operating procedures for pre‐clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol Dis 2008;31:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA, Lowe DA. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 2008;38:1290–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hyzewicz J, Ruegg UT, Takeda S. Comparison of experimental protocols of physical exercise for mdx mice and Duchenne muscular dystrophy patients. J Neuromuscul Dis 2015;2:325–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Terrill JR, Radley‐Crabb HG, Grounds MD, Arthur PG. N‐Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul Disord 2012;22:427–434. [DOI] [PubMed] [Google Scholar]
- 63. Mathur S, Vohra RS, Germain SA, Forbes S, Bryant ND, Vandenborne K, et al. Changes in muscle T2 and tissue damage after downhill running in mdx mice. Muscle Nerve 2011;43:878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol 2001;535:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fayssoil A, Nardi O, Orlikowski D, Annane D. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail Rev 2010;15:103–107. [DOI] [PubMed] [Google Scholar]
- 66. Fayssoil A, Renault G, Guerchet N, Marchiol‐Fournigault C, Fougerousse F, Richard I. Cardiac characterization of mdx mice using high‐resolution doppler echocardiography. J Ultrasound Med 2013;32:757–761. [DOI] [PubMed] [Google Scholar]
- 67. Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord 2004;14:491–496. [DOI] [PubMed] [Google Scholar]
- 68. Van Erp C, Loch D, Laws N, Trebbin A, Hoey AJ. Timeline of cardiac dystrophy in 3‐18‐month‐old MDX mice. Muscle Nerve 2010;42:504–513. [DOI] [PubMed] [Google Scholar]
- 69. Spurney CF, Cha H‐J, Sali A, Pandey GS, Pistilli E, Guerron AD, et al. Evaluation of skeletal and cardiac muscle function after chronic administration of thymosin beta‐4 in the dystrophin deficient mouse. PLoS One 2010;5:e8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ermolova NV, Martinez L, Vetrone SA, Jordan MC, Roos KP, Sweeney HL, et al. Long‐term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul Disord 2014;24:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cush JJ. Unusual toxicities with TNF inhibition: heart failure and drug‐induced lupus. Clin Exp Rheumatol 2004;22:S141–S147. [PubMed] [Google Scholar]
- 72. Synetos A, Toutouzas K, Aznaouridis K, Lerakis S, Stefanadis C. Dilated cardiomyopathy induced by anti‐tumor necrosis factor. Int. J. Cardiol. 2009;132:e26–e27. [DOI] [PubMed] [Google Scholar]
- 73. Gentek R, Hoeffel G. The Innate Immune Response in Myocardial Infarction, Repair, and Regeneration. Adv Exp Med Biol 2017;1003:251–272. [DOI] [PubMed] [Google Scholar]
- 74. Zlatanova I, Pinto C, Silvestre J‐S. Immune modulation of cardiac repair and regeneration: the art of mending broken hearts. Front Cardiovasc Med 2016;3:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Varga I, Kyselovič J, Galfiova P, Danisovic L. The Non‐cardiomyocyte Cells of the Heart. Their Possible Roles in Exercise‐Induced Cardiac Regeneration and Remodeling. Adv Exp Med Biol 2017;999:117–136. [DOI] [PubMed] [Google Scholar]
- 76. Evans NP, Misyak SA, Robertson JL, Bassaganya‐Riera J, Grange RW. Immune‐mediated mechanisms potentially regulate the disease time‐course of duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R 2009;1:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tidball JG. Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol 2017;17:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Duchesne E, Dufresne SS, Dumont NA. Impact of inflammation and anti‐inflammatory Modalities on skeletal muscle healing: from fundamental research to the clinic. Phys Ther 2017;97:807–817. [DOI] [PubMed] [Google Scholar]