

Abstract
Isoflavones isolated from members of the Fabaceae (primarily Leguminosae) family have been characterized for their phytoestrogenic properties, but certain derivatives have also shown potential as possible cancer therapeutic agents. ME-344, related to phenoxodiol (Figure 1), is a second generation isoflavone with a recent history of both preclinical and early clinical testing. The drug has unusual cytotoxicity profiles, where cancer cell lines can be categorized as either intrinsically sensitive or resistant to the drug. Evolving studies show that the cytotoxic properties of the drug are enacted through targeting mitochondrial bioenergetics. While the drug has undergone early Phase I/II trials in solid tumors with confined dose limiting effects and some evidence of disease response, there is a continuing need to define specific cellular targets that determine sensitivity, with the long-term goal of applying such information to individualized therapy. This review article details some of the existing and ongoing studies that are assisting in the continued drug development processes that may lead to new drug application (NDA) status.
Keywords: Clinical trials, Drug resistance, Energy metabolism, Heme oxygenase, Isoflavones, Mitochondria, ME-344, Reactive oxygen species
1. Introduction
Among many adaptive changes, altered responses to caspase-dependent programmed cell death have been identified as a contributory cause of resistance to anticancer drugs (Igney and Krammer, 2002). Various isoflavones have been recognized in both cancer prevention (Sarkar and Li, 2003) and as inducers of cell death via caspase-independent pathways in drug resistant cancer cell lines. As such, there has been an emphasis on identifying those structures that may possess cytotoxic properties that can translate into a beneficial therapeutic index in cancer patients (Mor et al., 2006). Most isoflavones have been purified as natural plant compounds produced by members of the Leguminous family and can act as phytoestrogens with structures that bear resemblance to 17-β-estradiol (Yu et al., 2000). They share the same biosynthetic pathways used for flavonoid biosynthesis (Barnes, 2010), which produce active isoflavones such as daidzein and genistein. Glycoside conjugated forms of isoflavones frequently embody highly polar, water-soluble properties. Aglycone forms can be produced by intestinal bacteria, and these can have better absorption and biological activities than glycosidic forms (Vitale et al., 2013). Soybeans are the most significant dietary sources of isoflavones, with food processing conditions impacting their chemical structures. Genistein and daidzein have been extensively studied in clinical chemoprevention trials, particularly breast and prostate. Other therapeutic activities have been identified in cardiovascular disease, diabetes and post-menopausal disorders (Prasad et al., 2010; Setchell, 1998). A number of chemical activities have been ascribed to isoflavones, but in general, common to their mechanisms are effects upon redox homeostasis. These can then impact downstream events including protein tyrosine kinase (PTK) inhibition and deactivation of oncogenic proteins by catalysis of tyrosine phosphorylation. Inhibition of PTKs suppresses downstream oncogenic signaling pathways. Genistein and daidzein can also down-regulate the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2), upregulate the pro-apoptotic protein Bcl-2 associated X protein (Bax) and induce cell cycle G2/M arrest (Piontek et al., 1993). In addition, genistein inhibits DNA topoisomerase activity and acts as a gene regulator (Kurzer and Xu, 1997).
NV-128 is a synthetic derivative of natural isoflavone compounds and has been investigated for its pre-clinical anti-cancer activities (Alvero et al., 2009; Alvero et al., 2011). This agent is an analogue of phenoxodiol, a derivative of genistein. In contrast to phenoxodiol, NV-128 has been shown to induce caspase-independent cell death. Extrapolating from these compounds, a second generation isoflavone, ME-344, structurally similar to NV-128, but with greater anticancer potency in resistant cancer cells and stem cells has been developed. ME-344 has been evaluated in clinical situations (Bendell et al., 2015; Diamond et al., 2017), but there are numerous ongoing efforts to define how ME-344 mediates its cytotoxic properties. In this review, we provide an update of the pre-clinical and clinical data for ME-344 and provide perspectives on how this agent may move through development towards eventual potential FDA approval.
2. Mitochondria as drug target organelles in cancer
Mitochondria, the double-membrane-bound organelles in most eukaryotic cells, participate in bioenergetic metabolism producing ATP through electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). Their relevance to ME-344 pharmacology resides in shared properties of overlapping ROS production, Ca2+ homeostasis and apoptosis initiation (Cheng and Ristow, 2013). Under physiological conditions, mitochondria are essential for cellular homeostasis, but during stress response, death pathways may be induced. Mitochondrial dysfunction has been implicated in many human diseases, including cancer (Fulda et al., 2010) and they have been targeted by different approaches in therapy, including: (i) Induction of mitochondrial permeability transition (MPT) leading to destruction of mitochondrial integrity. For example, Bcl-2 family members are critical regulators of MPT. Bcl-2 and Bcl-xL proteins protect and stabilize mitochondrial membranes and prevent pro-apoptotic protein release into the cytosol. Conversely, Bax and Bcl-2 antagonist/killer (Bak) can create channels on outer mitochondrial membranes, inducing MPT causing cell death (Shamas-Din et al., 2013). (ii) Multiple copies of the mitochondrial genome (mtDNA), some of which code for OXPHOS complexes, can cause instability and can be targets. Mutations or aberrant copy numbers within the mtDNA creates possible targets that may determine drug sensitivities (Singh et al., 1999). Either increased or decreased mtDNA copy number can occur in various solid tumors. In general, mtDNA loss in mammalian cells can be a precursor to cell death and the antibiotic ciprofloxacin can cause a disruption of mitochondrial function by cleaving mtDNA (Lawrence et al., 1996). *(iii) Enzymes in the OXPHOS pathway are also potential targets. OXPHOS is localized within inner mitochondrial membranes, composed of five mitochondrial enzyme complexes (complexes I-V) (Signes and Fernandez-Vizarra, 2018). The ETC is built with membrane channel pumps, which include complexes I-IV, involved in promoting redox reactions and pumping H+ ions into mitochondrial intermembrane spaces. NADH and FADH from the Krebs Cycle act as electron carriers and fuel of the ETC. H+ ions flow into the inter-membrane space of mitochondria to be channeled by electron acceptors and O2, localized in complex IV, to produce water. These stepwise transfers lead to the serial pumping of protons into the mitochondrial inter-membrane space. These protons are used for ATP production by ATP synthase (complex V). Mitochondrial OXPHOS is important in macromolecular biosynthesis, bioenergetics, but particularly in redox signaling and homeostasis (Chandel, 2015). By attenuating mitochondrial functions, deficiencies in OXPHOS have been implicated in drug resistance in cancer. Importantly in terms of cancer therapy, cancer cells, because of their high proliferative indexes, require and produce high levels of ATP and possess more active OXPHOS. Moreover, cancer stem cells also exhibit high levels of mitochondrial OXPHOS and generate more mitochondrial ROS (Viale et al., 2014). Thus, endogenously high levels of mitochondrial OXPHOS are seen in a variety of tumors (Roesch et al., 2013), creating a scenario where mitochondrial OXPHOS components may be considered as conceivable targets for anti-cancer drug development. Mitochondrial respiratory chain dysfunction, particularly involvement of complex I, is a contributory component in the pathogenesis of a variety of complex diseases including psychiatric disorders, neurodegenerative or cardiovascular diseases and cancer (Archer, 2013; Bansal and Kuhad, 2016; Bergman and Ben-Shachar, 2016; Kato, 2017; Looney and Childs, 1934). Mitochondrial complexes I and III have also been considered to be the two major enzyme complexes to generate ROS. Therefore, inhibition of mitochondrial complexes provides another perspective in treating human diseases. It is within this setting that ME-344 has gained credence as a cytotoxic anticancer drug.
3. Cell components targeted by ME-344
The biological effects of ME-344 have been reported to be quite broad, perhaps reflecting promiscuity of drug targeting. For example, ME-344 can activate caspase-dependent and independent cell death pathways, with implications to a number of plausible mechanisms of action. An analysis of the present literature allows us to deduce that these can include: inhibition of the respiratory complexes, i.e. complex I-V (Alvero, et al., 2011; Lim et al., 2015; Manevich et al., 2016; Navarro et al., 2016) and translocation of HO-1 from rough endoplasmic reticulum to mitochondria (Manevich, et al., 2016), accompanied by the downstream signaling events, e.g., (a) reduction in ATP production, activation of AMPK leading to mTOR-mediated autophagy, (b) increase in mitochondrial ROS production, activation of ERK leading to Bax-mediated loss of mitochondrial membrane potential (MMP, ΔΨm), endonuclease G (EndoG) translocation into nucleus which cleaves DNA, resulting in chromatin condensation. Possible inhibition of tubulin polymerization (Jeyaraju et al., 2016) has also been shown, however, it is not uncommon for estrogen-like molecules to have such effects (Speicher et al., 1992; Speicher et al., 1994). Figure 2 summarizes such information and the following sections provide further explanatory details.
Figure 2.
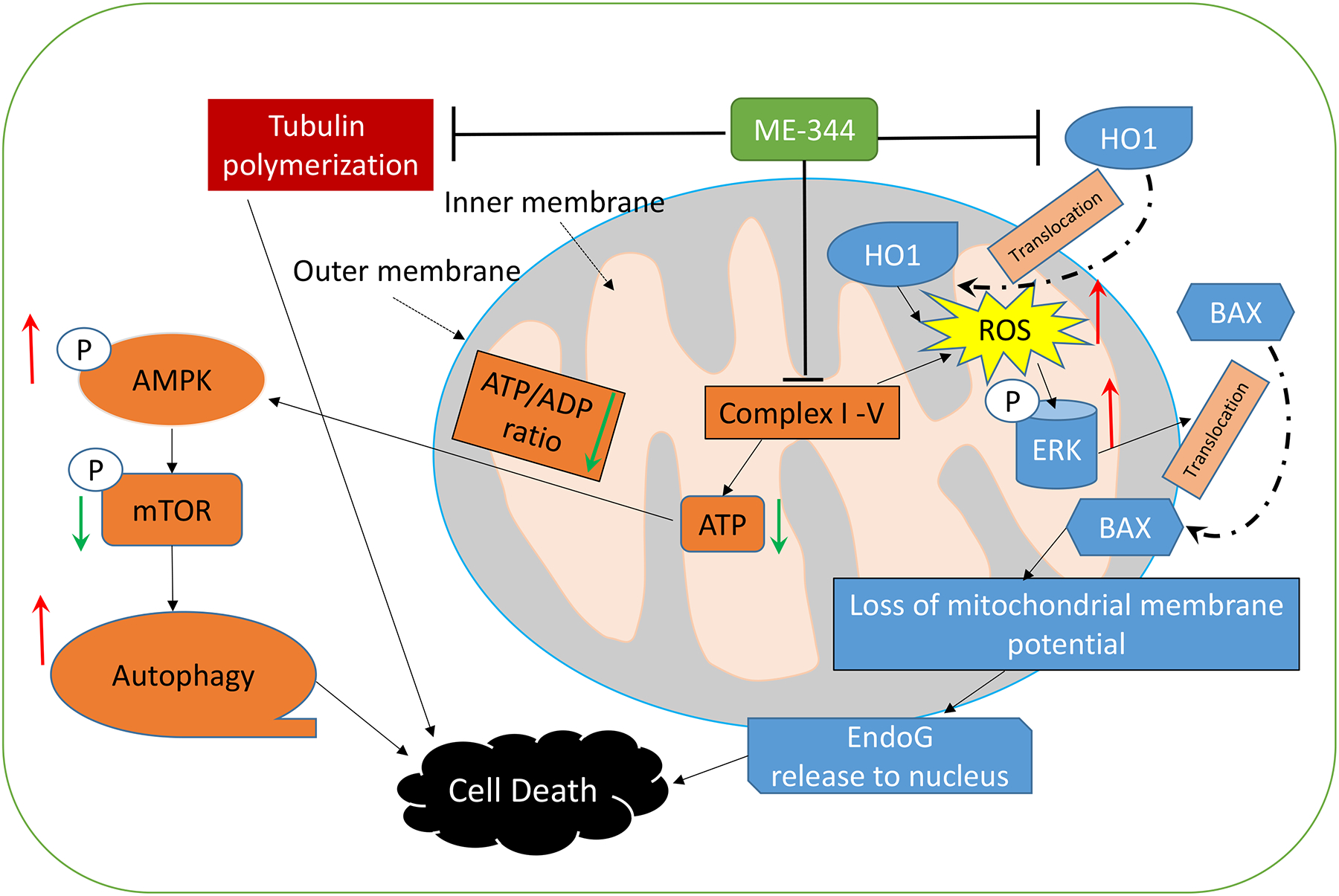
Mechanisms of ME-344 action include (1) inhibition of the respiratory complexes, such as complex I-V; (2) inhibition of HO-1 and induction of its translocation to mitochondria, accompanied by downstream signaling events exemplified by, (a) reduction in ATP production, activation of AMPK leading to mTOR mediated autophagy, (b) increase in mitochondrial ROS production, activation of ERK leading to Bax-mediated loss of mitochondrial membrane potential, Endo G nuclease translocation into nucleus which cleaves DNA, resulting in chromatin condensation. Possible inhibition of tubulin polymerization has also been shown.
3.1. Mitochondrial enzyme complexes
Complex I (NADH-coenzyme Q oxidoreductase) is the first enzyme complex of the ETC, reducing ubiquinone to ubiquinol by using the electrons donated from the oxidation of NADH from the Krebs cycle (Lenaz et al., 2004). It is also the largest multi-subunit enzyme of the ETC to generate an electrochemical proton gradient by pumping hydrogen ions across the inner mitochondrial membrane, driving ATP production and consequently the efficiency of the OXPHOS process (Holper et al., 2018; Yano, 2002). The impact of ME-344 on mitochondria was investigated by Lim et al., who showed that ME-344 suppresses mitochondrial respiration and complex I activity (and to some extent complex III). These effects were associated with a reduction in mitochondrial oxygen consumption and maximum ADP stimulated respiration rates. This inhibition has been compared to some of the characteristics of rotenone (Lim, et al., 2015), a known complex I inhibitor, interfering with the ETC in mitochondria by preventing electron transfer from iron-sulfur centers in complex I to ubiquinone, subsequently inhibiting energy production and creating ROS byproducts.
ME-344 also induced the dissipation of ΔΨm, comparable in extent to FCCP, a mitochondrial uncoupling agent that can depolarize mitochondrial membranes. This outcome was found in ovarian cancer stem cells treated with a related isoflavone, NV-128 (Figure 1; (Alvero, et al., 2011; Lim, et al., 2015)). A normal ΔΨm maintains steady-state levels of OXPHOS complexes, but in a variety of cancer cell lines and in ovarian cancer stem cells, ME-344 treatment was found to degrade the complex I subunit NDUFA9 and the complex IV subunits, COXI and IV. While such results support the principle that ME-344 interferes with mitochondrial respiration and dissipates ΔΨm, specific drug targets were not defined (Alvero, et al., 2011; Lim, et al., 2015).
Figure 1.
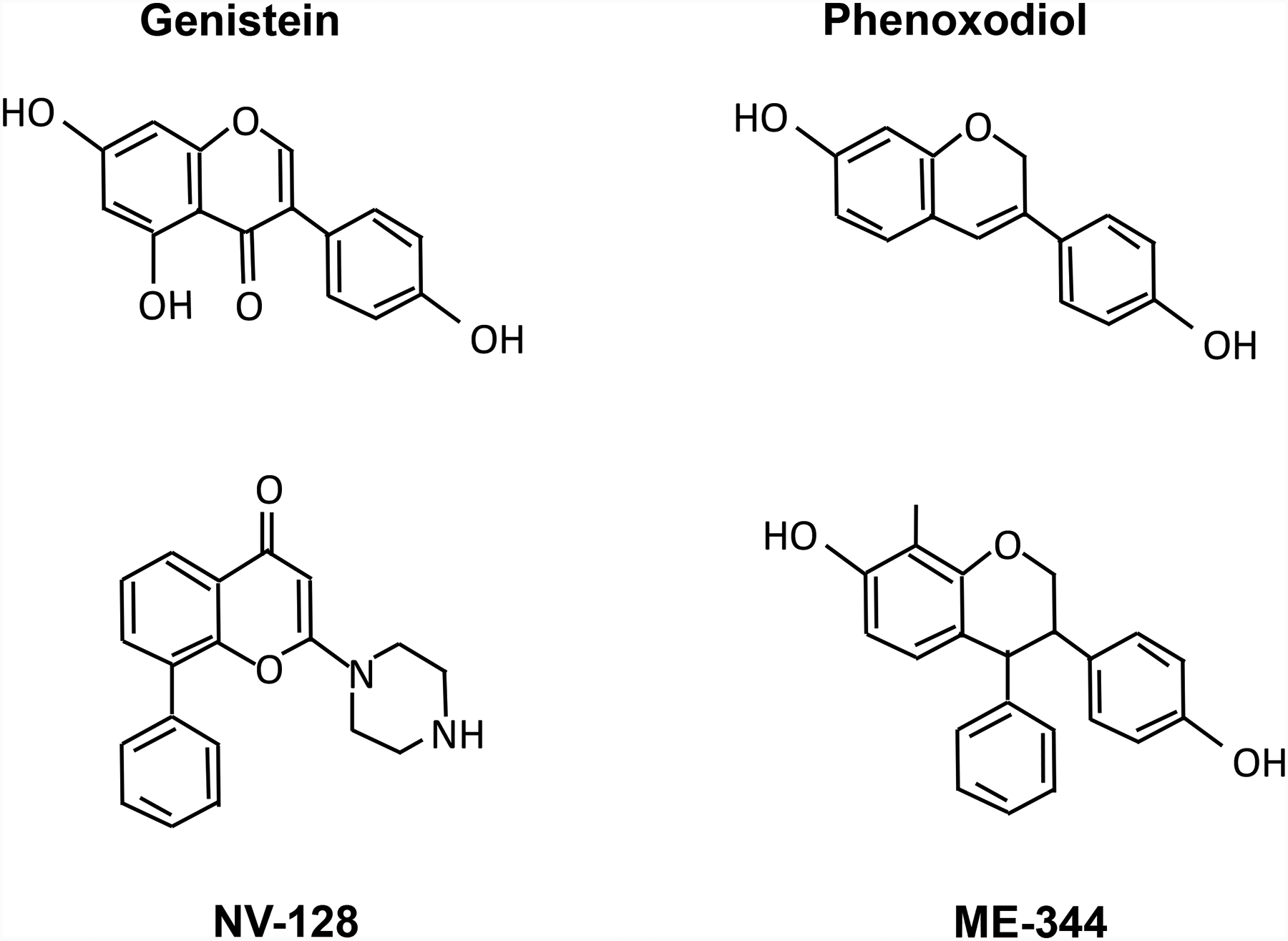
Structures of common isoflavone molecules.
In a cytotoxicity screen of 240 human tumor cell lines, 20 were shown to be intrinsically resistant to ME-344, while the remainder were sensitive. For our own studies with ME-344, we compared representative examples of both sensitive (SHP-77 and H460) and resistant (H596 and SW900) lung cancer cell lines, with the goal of identifying cellular traits that might be determinants of the mechanism of action of ME-344 (Manevich, et al., 2016). Needless to say, such information should have relevance to how the drug is clinically applied. Consistent with other results which demonstrated ME-344 interference with mitochondrial energy production, we found that ME-344 caused substantial reduction in cellular oxygen consumption rates (OCR), rapid hyperpolarization (instead of depolarization) in inner mitochondrial membranes, and oxidation of the protein thiols, with the effect more pronounced in sensitive lung cancer cell lines. In addition, ME-344 dose-dependently reduced ATP levels, ATP/ADP ratios, and increased the generation of H2O2 and ●OH (not O2●-) only in sensitive lung cancer cell lines, not in resistant lung cancer cell lines or in normal lung fibroblasts (MRC-5), implying a certain degree of specificity of ME-344 toward the sensitive cell lines. Furthermore, ME-344 induced a rapid increase and subsequent slow decrease of NADPH to below normal resting levels in sensitive, but not in resistant cancer or normal cell lines. Taken together, these results implicated several proteins and pathways that may be associated with the drug’s mechanism of action. (1) ME-344 may target the oligomycin-sensitive F0 component (proton pump) of ATP synthase (complex V), as evidenced by its ability to inhibit OCR and ATP production, inducing inner mitochondrial membrane hyperpolarization, and minimizing the effects of subsequent oligomycin in sensitive lung cancer cells. (2) ME-344 may have a direct impact on glycolysis. Most cancer cell lines depend on aerobic glycolysis for ATP synthesis and produce large amounts of lactate. As expected, initial extracellular acidification rate (ECAR) values of lung cancer cells were higher than those of normal cells. ME-344 has little effect on ECAR under glucose starvation. However, after addition of glucose, ME-344 significantly increased ECAR and glycolytic stress in sensitive lung cancer cells. (3) ME-344 influences heme oxygenase-1 (HO-1) expression, activity and subcellular localization. HO-1 is the rate-limiting enzyme involved in the oxidative degradation of free heme to produce biliverdin, carbon monoxide, and ferrous iron (Fe2+) (Ryter et al., 2006). HO-1 is generally regarded as cytoprotective, however, studies have shown that certain isoflavones can induce HO-1 expression leading either to protective events or to cause cell death (Park et al., 2011; Podkalicka et al., 2018), functional paradoxes perhaps linked with the mitochondrial translocation of HO-1 (Bansal et al., 2014). The cytotoxicity of ME-344 in sensitive lung cancer cells might be the consequence of increased ●OH production via the Fenton reaction, where the intermediate product of HO-1, Fe2+, reacts with H2O2. ME-344 generated ROS imbues activities associated with a major redox transcription factor, nuclear factor, erythroid 2-like 2 (Nrf2), which regulates a series of antioxidant response genes (Tebay et al., 2015). In addition, the BTB and CNC Homology 1 (Bach1) protein can compete with Nrf2 for binding to antioxidant response elements (ARE), thereby suppressing some Nrf2 target genes, including heme oxygenase-1 (HO-1) (Reichard et al., 2007; Shan et al., 2006). Under basal conditions, Bach1 binds to ARE’s that regulate HO-1 expression (Sun et al., 2002), but ROS can disrupt this and enable activation of HO-1 by Nrf2. These regulatory switches can influence the scope and extent of response to ME-344. (4) ME-344 may have an indirect (resulting from F0-ATP synthase inhibition, inner mitochondrial membrane hyperpolarization and the compensatory proton fluxes) or direct impact on nicotinamide nucleotide transhydrogenase, an enzyme that uses energy from the mitochondrial proton gradient to inter-convert NADH to NADPH, as implied by the immediate effects of ME-344 on NADPH in sensitive lung cancer cells.
Each of these proteins or pathway targets is presently under continuing investigation, but our own ongoing studies (Zhang, 2019) have confirmed that ME-344 can bind to HO-1, inhibit its activity and initiate a robust Nrf2 signaling response to induce HO-1 which is accompanied by mitochondrial translocation of HO-1 in drug sensitive lung cancer cells. Similar effects were not seen in either drug resistant or normal lung fibroblasts cells, where basal levels of HO-1 were lower. This differential would be consistent with the reported ROS events and provide a rationale for an advantageous therapeutic index.
3.2. mTOR inhibition through AMPK activation
In a variety of sensitive cancer cell lines, ME-344 effects on mitochondrial complex subunits leads to significant loss of ATP, enhanced phosphorylation of AMPKα1, decreased phosphorylation of the mammalian target of rapamycin (mTOR; consisting of two distinct complexes, mTOR complex I (mTORC1) and mTOR complex II (mTORC2)) and decreased phosphorylation of p70 S6 kinase (Alvero, et al., 2009; Alvero, et al., 2011; Bendell, et al., 2015; Lim, et al., 2015; Manevich, et al., 2016). With low cellular ATP levels, AMP is more likely to activate AMPK by direct binding to its regulatory subunit (Oakhill et al., 2011; Xiao et al., 2011). One mechanism by which AMPK interacts with mTOR to regulate cell growth is by phosphorylating tumor suppressor tuberous sclerosis complex 2 (TSC2) (Mihaylova and Shaw, 2011). TSC1/2 gene mutation product TSC1/2 regulates cell growth negatively by acting upstream of mTOR and downstream of AMPK (Gwinn et al., 2008; Inoki et al., 2006). TSC2 accepts the signals and then regulates mTORC1 by interacting with the GTP-binding protein Ras homolog enriched in the brain (Rheb). GTPase-activating proteins (GAP) possessed by TSC2 increase the activity of Rheb to convert Rheb-GTP to Rheb-GDP which then inhibits the activation of mTORC1, with the end result that TSC2 suppresses the mTORC1 pathway (Gao and Pan, 2001; Kalender et al., 2010). ME-344 also decreases p-Akt, considered to have a role in induction of autophagy in cancer cells by interacting with mTOR. In addition, ME-344 increased the expression of mitochondrial beclin-1, which has been identified as a pro-autophagic molecule. Taken together, under energy starvation conditions, activation of AMPKα1 by ME-344 suppressed mTOR activity and since the mTOR pathway plays an integral role in regulating autophagy in eukaryotic cells (Yang et al., 2005), increasing levels of LC-3 II caused by ME-344 treatment implied some cause:effect relationship with autophagy pathways in cancer cells (Alvero, et al., 2009; Alvero, et al., 2011; Bendell, et al., 2015). Nevertheless, these sorts of studies although informative did not identify direct drug binding targets.
3.3. ERK activation through increased ROS.
Whenever drugs impact mitochondria and energy metabolism, there is a likelihood that cellular levels of ROS will be adversely impacted, resulting in excessive damage to a variety of biomolecules in the inner mitochondrial membrane and matrix (Adam-Vizi and Chinopoulos, 2006). ME-344 was shown to increase the levels of mitochondrial superoxide and cellular hydroxyl radicals and hydrogen peroxide (H2O2), but primarily (although not exclusively) in drug-sensitive cancer cells and cancer stem cells (Alvero, et al., 2011; Bendell, et al., 2015). ME-344 induced inhibition of OCR may also be responsible for oxidative stress generated by impairment of mitochondrial ETC. Characteristic of most cancer cells is aberrations and inefficiencies in OXPHOS and accompanying antioxidant systems and even under cellular homeostasis, cancer cells produce high levels of ROS and have lower levels of antioxidant enzymes (Mates and Sanchez-Jimenez, 1999). Since intermediate signaling events can be regulated by ROS, instead of inducing cell death, these molecules can alter tumor growth. Alvero et al. (date) showed that ME-344, by inhibiting ETC can increase mitochondrial ROS, leading to the activation of mitochondrial ERK1/2 in cancer stem cells. Consistently, pre-treatment with MnTBAP, a cell permeable superoxide dismutase mimetic, diminishes ME-344-induced activation of ERK1/2, indicating that ERK activation in cancer stem cells is linked with increasing mitochondrial ROS (Alvero, et al., 2011; Bendell, et al., 2015). Different sub-cellular localization of MEK/ERK signaling pathways can regulate either cell death or survival (Alavi et al., 2003; Chu et al., 2004). For example, activation can cause ERK1/2 nuclear translocation and subsequent activation of transcription factors leading to cell survival (Seger and Krebs, 1995). In contrast, mitochondrial ERK can participate in cell death pathways by regulating various mitochondrial functions. In Parkinson’s disease, ERK activity has been linked with oxidative injury through initiation of mitochondrial ROS (Kulich et al., 2007). ME-344 can induce a pro-apoptotic effect in cancer stem cells through up-regulation of the Bcl-2 family member, Bax. Pre-treatment with the ERK inhibitor U0126 can abrogate the effects of ME-344 on Bax mitochondrial translocation (Alvero, et al., 2011; Bendell, et al., 2015). Bcl-2 family members play a role in mitochondrial targeting of MEK/ERK signaling pathways, where for example, Bax as a pro-apoptotic member, is expressed in the cytosol of healthy cells. Under stress conditions, Bcl-2 is phosphorylated by ERK signaling leading to Bax translocation from cytosol to mitochondria by integrating into the outer mitochondrial membrane via its carboxyl terminus (Mikhailov et al., 2001; Nechushtan et al., 1999). ME-344 induces MPT, caused when Bax forms channels in the outer mitochondrial membrane in conjunction with ME-344 induced dissipation of ΔΨm. ME-344 initiated beclin-1 mitochondrial translocation might also contribute to the MPT, since mitochondrial integrity is regulated to some degree by Bcl-2 family members. Under normal conditions, Bcl-2 and Bcl-xL reside in mitochondria and stabilize membrane structure. ME-344 induced mitochondrial translocation of beclin-1 allows it to bind to Bcl-2 and Bcl-xL through its BH domain and abrogates the inhibition of Bax by Bcl-2, leading to Bax induced MPT. Therefore, ME-344-induced ERK signaling activation can drive cellular loss of ΔΨm via the interaction between Bax and mitochondrial components (Alvero, et al., 2011; Bendell, et al., 2015). ME-344 also induces EndoG nuclear translocation, resulting in chromatin condensation. ME-344 induced the loss of ΔΨm and MPT caused release of some mitochondrial proteins, including the mitochondrial nucleases EndoG and AIF. Nuclear translocation of EndoG, not AIF, can contribute to the cleavage of DNA and chromatin condensation effects initiated by ME-344 (Alvero, et al., 2009; Bendell, et al., 2015).
*3.4. Tubulin
ME-344 was shown to cause morphological changes in the plasma membranes of acute myeloid leukemia (AML) cells, effects that were considered to be similar to vinblastine, a characterized tubulin inhibitor (Jeyaraju, et al., 2016). Indeed, ME-344 was shown to inhibit tubulin polymerization in AML cells by interacting at the colchicine binding site. Consequently, ME-344 caused specific G2/M cell cycle arrest by suppression of microtubule dynamics and demonstrated synergistic cell killing effects with vinblastine. Because estrogenic molecules are known to have antimicrotubule effects (Speicher, et al., 1992; Speicher, et al., 1994), this result may reflect the phytoestrogenic properties (Leclercq and Jacquot, 2014) of the isoflavone backbone. Interestingly, combinations of such molecules have been previously tested in prostate cancer patients (Hudes et al., 1992), although the actual specificities of this tubulin binding and its importance, at low concentrations, to the in vivo or clinical effects remains to be established.
4. Preclinical investigational new drug (IND) enabling studies for ME-344
Characteristics of many tumors are high glycolytic rates, abnormal tumor angiogenesis and hypoxia. Some antiangiogenic agents can correct pathologic tumor angiogenesis and normalize tumor hypoxia (Jain, 2013; Kerbel, 2006). Antiangiogenics are widely used in the treatment of tumors such as kidney, liver, breast, ovarian, colorectal and lung. However, all patients receiving these biological agents eventually experience acquired resistance to the drugs and subsequent progressive disease, frequently as a consequence of up-regulation of aerobic glycolysis mediated by mutations in MAPK and PI3K/AKT pathways (Mastri et al., 2016; Quintela-Fandino, 2016; Sennino and McDonald, 2012). Recent studies suggested that ME-344 could overcome acquired resistance to antiangiogenics such as multi-tyrosine-kinase inhibitor (TKIs) (Navarro, et al., 2016). TKI’s (nintedanib or dovitinib) caused 60% tumor growth inhibition (TGI), accompanied by a 2-fold decrease in glucose uptake and reduced glycolysis through decreased HIF1α and AKT signaling, as evidenced by diminished glycolysis metabolites and end products of the pentose-phosphate shunt. How tumors were able to continue growing under such nutritional stress conditions was the subject of this study by Quintela-Fandino’s group (Navarro, et al., 2016). This suggested that tumor metabolism can be reprogrammed in response to TKI or decreased glycolysis, with mitochondrial metabolism increasing under the control of PPAR-α, AMPK and PKA activation. Evidence of increased mitochondrial metabolism in TKI- versus vehicle-treated tumors included: (1) an ~2-fold increase in ketones and fatty acids, substrates for the tricarboxylic acid cycle, was detected in tumors treated with TKI. (2) A >2.5-fold up-regulation of the ketone-body transporters MCT1 and 4 was detected in TKI-treated tumors, coupled with a >4-fold increase in the levels of ACAT1, the enzyme that allows re-use of ketone bodies as mitochondrial substrates. (3) Adipose deposits almost disappeared with TKI treatment, coupled with increased levels of the fatty acid transporters FABP3, CAV1, and SLC27A1/A2 and the lipases PNPLA2 and 3, and an almost 4-fold up-regulation of CPT1B, which shuttles the carnitine-bound species committed for mitochondrial degradation into the mitochondrial matrix. (4) Succinate dehydrogenase (SDH) or complex II activity increased from week one of TKI treatment and was maintained for more than 2 months. (5) OCR was ~2-fold higher in tumors treated with TKI. These adaptive changes in energy metabolism were mechanistic adaptations that permitted resistance to chronic TKI treatment and indicated that pharmacologic modulation of mitochondrial respiration might not be efficient if used as monotherapy, but may enhance the effects of the TKIs. When one energy source (glycolysis) is pharmacologically restricted, tumors become vulnerable to inhibition of the other (mitochondrial metabolism). Indeed, while neither ME-344 nor phenformine was effective when administered as monotherapy, the combination demonstrated synergistic effects with TKI (nintedanib, dovitinib or regorafenib) through inhibition of both mitochondrial and glycolytic metabolisms. The therapeutic benefit of such a drug combination has been successfully extended to two lung cancer models: Pulm24 lung-cancer-patient-derived xenograft and spontaneous Lewis lung carcinoma cancer model. This approach certainly appears to hold future promise and partly as a consequence of the broad spectrum of cytotoxic activity of ME-344 in human tumor cell lines (including a number of drug resistant variants) led to its application in reversing TKI resistance in a spontaneous HER-2 negative (estrogen receptor positive, and progesterone receptor negative) breast cancer model (Navarro, et al., 2016). After priming with TKI antiangiogenics, mitochondrial metabolism, as compensation for reduced glycolysis, becomes essential for tumor survival and can lead to acquired resistance against these drugs. For example, nintedanib in combination with ME-344, demonstrated synergistic effects, increasing the TGI from 64% to 92%. Effective therapeutic synergy was also observed with ME-344 and regorafenib (another TKI), increasing the TGI to 88% (Navarro, et al., 2016). On the basis of these findings, an early-stage clinical trial that combines antiangiogenic treatment (bevacizumab, 15 mg/kg at day 1) with weekly doses of ME-344 (10 mg/kg on days 8, 15 and 22) has been launched for patients with early HER-2 negative breast cancer. So far, the authors claim that interim data from 19 patients are encouraging and suggest that completion of enrollment of the clinical study of ME-344 in combination with bevacizumab is warranted (Miguel Quintela-Fandino, 2018).
AML accounts for 10 to 15 percent of all leukemias diagnosed in children and is the most common acute leukemia in adults. With AML, the incidence escalates with age and those >60 have poorer prognosis due to toxicity and low response to standard chemotherapies. AML is not only a highly aggressive disease, but also develops resistance to further chemotherapy (Estey and Dohner, 2006; Siegel et al., 2013). Studies have been carried out to investigate the cytotoxic effect of ME-344 on leukemia cell lines, primary AML patient samples and normal hematopoietic cells (Jeyaraju, et al., 2016). ME-344 treatment resulted in inhibition of cell growth and viability in all the leukemia cell lines tested, e.g., OCI-AML2, TEX, HL60, K562, KG1a, U937 and NB4, with IC50 values in the range of 70–260 nM. In addition, ME-344 was toxic to AML patient samples rather than normal hematopoietic samples. Furthermore, in leukemia xenograft models using OCI-AML2 or MDAY-D2, ME-344 reduced tumor growth by up to 95% of control without evidence of toxicity. Taken together, these results implied that ME-344 has anti-leukemia efficacy both in vitro and in vivo, which supports its clinical evaluation in patients with AML and other hematological malignancies.
5. Clinical studies
Results from Phase I and Ib trials have been published. ME-344 has been employed to treat patients with locally advanced or metastatic refractory solid tumors by single agent or combination therapies. The original rationale for such trials was the preclinical data showing that activation of caspase-independent cell death pathways in malignant cells may create a beneficial therapeutic index. Two clinical trials with ME-344 as a single agent or in combination with topotecan (Table 1) have been completed in patients with refractory solid tumors and were designed to show safety, adverse events, pharmacokinetics and disease response (Bendell, et al., 2015; Diamond, et al., 2017).
Table 1:
Summary of clinical trials with ME-344
| Clinical trials | Patients / type of cancer, no. (%) | Median no. of prior therapies, no. (%) | Pharmacokinetics | Adverse events, no. (%) | Response, no. (%) |
|---|---|---|---|---|---|
| Phase I | 30 patients: 5 (16.7) NSCLC 5 (16.7) colorectal 3 (10.0) endometrial 2 (6.7) ovarian 2 (6.7) squamous cell carcinoma of vagina 2 (6.7) urothelial carcinoma 11 (36.7) other* |
3 (10.0) 0–1 prior therapies 5 (16.7) 2 prior therapies 8 (26.7) 3 prior therapies 14 (46.7) 4 prior therapies |
Cmax/dose (ug/mL/mg) : 1.99 t1/2 (hr) : 6 AUC/dose (hr*ug/mL/mg) : 0.008 |
6 (20.0) neuropathy 6 (20.0) nausea 6 (20.0) dizziness 5 (16.7) fatigue 4 (13.3) vomiting 3 (10.0) diarrhea 3 (10.0) asthenia |
1 (3.3) partial response 10 (33.3) stable disease 10 (33.3) progressive disease 9 (30.0) not evaluable |
| Phase Ib | 46 patients: 28 (60.9) ovarian 13 (28.3) SCLC 5 (10.9) cervical |
28 (60.9) 1–4 prior therapies in ovarian cancer 13 (28.3) 1–3 prior therapies in SCLC 5 (10.9) 1–4 prior therapies in cervical cancer |
Cmax (ng/mL) : 20,880 Cmin (ng/mL) : 25.30 tmax (hr) : 0.500 AUC0-t (hr*ng/mL) : 21,830 AUC0-inf (hr*ng/mL) : 22,040 AUC% extrap (%) : 1.03 t1/2 (hr) : 5.30 kel (1/h) : 0.14 CL,ss (L/h) : 42.59 Vd,ss (L) : 104.65 CL,ss/kg (L/h) : 0.50 Vd,ss/kg (L) : 1.20 |
30 (65.2) fatigue 26 (56.5) neutropenia 23 (50.0) thrombocytopenia 22 (47.8) nausea 21 (45.7) diarrhea 19 (41.3) decreased appetite 19 (41.3) hypertension 18 (39.1) vomiting 16 (34.8) anemia 15 (32.6) constipation 9 (19.6) weight decreased 8 (17.4) arthralgia 8 (17.4) back pain 8 (17.4) dyspnea 7 (15.2) abdominal pain 7 (15.2) asthenia 7 (15.2) cough |
1 (2.4) partial response 21 (51.2) stable disease 9 (22.0) progressive disease 10 (24.4) not evaluable |
Abbreviations: NSCLC, Non-small cell lung carcinoma; SCLC, Small cell lung carcinoma.
Other includes bladder, breast, carcinoid of the ileum, cervical, cervical leiomyosarcoma, small cell lung, melanoma, pancreatic, peritoneal, sarcoma, and unknown primary (one patient each).
5.1. Phase I: ME-344 as a single agent
The first-in-human phase 1 study of ME-344 identified safety and adverse events (AEs) that might provide dose-limiting toxicities (DLTs) and maximum tolerated dose (MTD). Thirty patients with refractory solid tumors received intravenous ME-344 at six dose levels (1.25 – 20 mg/kg) on days 1, 8, 15, and 22 of each 28-day treatment cycle until disease progression. DLTs developing at doses 15 and 20 mg/kg included grade 3 neuropathy, vomiting, nausea and hypertension, but each reverted to normal with discontinuation of treatment. Therefore, in subsequent studies 10 mg/kg was selected to be the MTD. Common treatment-related AEs included neuropathy (20.0%), nausea (20.0%), dizziness (20.0%), fatigue (16.7%), vomiting (13.3%), diarrhea (10.0%) and asthenia (10.0%) (Table 1). 6 of 30 patients discontinued treatments as a consequence of serious AEs (SAE). Pharmacokinetic parameters were assessed on day 1 and day 15. The mean terminal half-life of ME-344 was 6 hours; maximal plasma concentration Cmax/dose was 1.99 ug/mL/mg; mean area under the concentration curve AUC/dose was 0.008 hour* ug/mL/mg. There were no significant differences in volume of distribution at steady state (Vdss) and drug clearance (CI). Efficacy was assessed in 30 patients. 21 of 30 received at least 3 cycles of treatment. One (3.3%) patient with small cell lung cancer (SCLC) experienced a partial response (PR) at the 11th cycle of treatment at a dose of 5 mg/kg. Laminography images showed decreased tumor bulk at week 52 compared to baseline. Within the cohort, 4 of 10 patients were reported to have stable disease (SD), including: urothelial carcinoma for 47 weeks; carcinoid of the ileum for 40 weeks; cervical leiomyosarcoma for 39 weeks; cervical cancer for 31 weeks. 10 patients (33.3%) developed progressive disease (PD) within the treatment duration. Overall, ME-344 was judged to have demonstrated promising efficacy as a monotherapy in this phase I study and provided a baseline for future combination studies.
5.2. Phase Ib: ME-344 in combination with topotecan.
A subsequent phase Ib study was implemented to evaluate ME-344 in combination with topotecan in patients with SCLC, ovarian or cervical cancer. Topotecan is an established drug in the management of these diseases (Hirte et al., 2015; Riemsma et al., 2010). 46 patients (32 of whom had been previously treated) received ME-344 intravenously at 10 mg/kg on days 1, 8, 15, and 22, with topotecan at 4 mg/m2 on days 1, 8, and 15 of each 28-day treatment cycle. Discontinuation was enacted if disease progression occurred (Diamond, et al., 2017; Safra et al., 2013; Sehouli et al., 2011). Common treatment-related AEs were similar to the other trial, including fatigue (65.2%), neutropenia (56.5%), thrombocytopenia (50.0%), nausea (47.8%), diarrhea (45.7%), decreased appetite (41.3%) and hypertension (41.3%) (Table 1). Only two patients (4.3%) experienced neuropathy with no grade 3/4 AEs. 3 patients (6.5%) who developed ME-344-related AEs discontinued treatment. One patient (2.1%) experienced 1 ME-344-related grade 3 diarrhea; 7 patients (15.2%) experienced 6 topotecan-related SAEs, including grade 4 thrombocytopenia, grade 3 fatigue, grade 3 febrile neutropenia, grade 3 blood-stream infection and neutropenic sepsis. 5 patients (10.9%) died within 30 days of discontinuing treatment. Pharmacokinetic parameters were similar to the earlier trial and decreasing plasma levels of ME-344 suggested little drug accumulation. One patient (2.4%) with ovarian cancer obtained a PR and continued for 14 cycles. 21 patients (51.2%) experienced SD, translating into a clinical benefit of 53.7% and an overall response rate of 2.4%. These results were unspectacular, but did not preclude the possibility that different combinations should be explored (Diamond, et al., 2017). As discussed in a previous section, the combination of ME-344 with antiangiogenic agents has become the next-line clinical approach.
6. Conclusions and future perspectives
The current literature contains significant preclinical data and more limited clinical results that support the continued development of ME-344 as a cytotoxic anticancer drug. Isoflavones have been the subject of numerous interrogations as cancer preventative agents. However, the phenoxodiol scaffold of ME-344 has cytotoxic properties that have been transmitted into the general pharmacological characteristics of ME-344. Screening of numerous cancer cell lines showed that cells were either intrinsically sensitive or resistant to the drug. Evidence so far is that energy metabolism and mitochondria constitute targets for the drug, although precisely which mitochondrial proteins are specifically inhibited remains under study. Delineation of such (a) target(s) will assist significantly in moving drug development forward, perhaps predicting which tumor characteristics may predetermine drug response. In the future, such information may be applied to a precision medicine or individualized therapy setting, where biomarkers of drug sensitivity may be predicted.
Acknowledgments
This work was supported by grants from the National Institutes of Health (5P20GM103542, COBRE in Oxidants, Redox Balance and Stress Signaling), support from the South Carolina Centers of Excellence program and was conducted in a facility constructed with the support from the National Institutes of Health, Grant Number C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. Financial support from MEI Pharma is also acknowledged.
Footnotes
Conflict of Interest
KDT has been the recipient of research support from MEI Pharma.
References
- Adam-Vizi V, and Chinopoulos C (2006). Trends Pharmacol Sci 27, 639–45. [DOI] [PubMed] [Google Scholar]
- Alavi A, Hood JD, Frausto R, Stupack DG, and Cheresh DA (2003). Science 301, 94–6. [DOI] [PubMed] [Google Scholar]
- Alvero AB, Montagna MK, Chen R, Kim KH, Kyungjin K, Visintin I, Fu HH, Brown D, and Mor G (2009). Cancer 115, 3204–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, and Mor G (2011). Mol Cancer Ther 10, 1385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL (2013). N Engl J Med 369, 2236–51. [DOI] [PubMed] [Google Scholar]
- Bansal S, Biswas G, and Avadhani NG (2014). Redox Biol 2, 273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal Y, and Kuhad A (2016). Curr Neuropharmacol 14, 610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes S (2010). Lymphat Res Biol 8, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendell JC, Patel MR, Infante JR, Kurkjian CD, Jones SF, Pant S, Burris HA 3rd, Moreno O, Esquibel V, Levin W, and Moore KN (2015). Cancer 121, 1056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman O, and Ben-Shachar D (2016). Can J Psychiatry 61, 457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2015). Cell Metab 22, 204–6. [DOI] [PubMed] [Google Scholar]
- Cheng Z, and Ristow M (2013). Antioxid Redox Signal 19, 240–2. [DOI] [PubMed] [Google Scholar]
- Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, and DeFranco DB (2004). Eur J Biochem 271, 2060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JR, Goff B, Forster MD, Bendell JC, Britten CD, Gordon MS, Gabra H, Waterhouse DM, Poole M, Ross Camidge D, Hamilton E, and Moore KM (2017). Invest New Drugs 35, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estey E, and Dohner H (2006). Lancet 368, 1894–907. [DOI] [PubMed] [Google Scholar]
- Fulda S, Galluzzi L, and Kroemer G (2010). Nat Rev Drug Discov 9, 447–64. [DOI] [PubMed] [Google Scholar]
- Gao X, and Pan D (2001). Genes Dev 15, 1383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). Mol Cell 30, 214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirte H, Kennedy EB, Elit L, and Fung Kee Fung M (2015). Curr Oncol 22, 211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holper L, Ben-Shachar D, and Mann JJ (2018). Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudes GR, Greenberg R, Krigel RL, Fox S, Scher R, Litwin S, Watts P, Speicher L, Tew K, and Comis R (1992). J Clin Oncol 10, 1754–61. [DOI] [PubMed] [Google Scholar]
- Igney FH, and Krammer PH (2002). Nat Rev Cancer 2, 277–88. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, and Guan KL (2006). Cell 126, 955–68. [DOI] [PubMed] [Google Scholar]
- Jain RK (2013). J Clin Oncol 31, 2205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaraju DV, Hurren R, Wang X, MacLean N, Gronda M, Shamas-Din A, Minden MD, Giaever G, and Schimmer AD (2016). Oncotarget 7, 49777–49785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, and Thomas G (2010). Cell Metab 11, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T (2017). Schizophr Res 187, 62–66. [DOI] [PubMed] [Google Scholar]
- Kerbel RS (2006). Science 312, 1171–5. [DOI] [PubMed] [Google Scholar]
- Kulich SM, Horbinski C, Patel M, and Chu CT (2007). Free Radic Biol Med 43, 372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzer MS, and Xu X (1997). Annu Rev Nutr 17, 353–81. [DOI] [PubMed] [Google Scholar]
- Lawrence JW, Claire DC, Weissig V, and Rowe TC (1996). Mol Pharmacol 50, 1178–88. [PubMed] [Google Scholar]
- Leclercq G, and Jacquot Y (2014). The Journal of steroid biochemistry and molecular biology 139, 237–44. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Fato R, Baracca A, and Genova ML (2004). Methods Enzymol 382, 3–20. [DOI] [PubMed] [Google Scholar]
- Lim SC, Carey KT, and McKenzie M (2015). Am J Cancer Res 5, 689–701. [PMC free article] [PubMed] [Google Scholar]
- Looney JM, and Childs HM (1934). J Clin Invest 13, 963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Reyes L, Britten CD, Townsend DM, and Tew KD (2016). J Pharmacol Exp Ther 358, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastri M, Rosario S, Tracz A, Frink RE, Brekken RA, and Ebos JM (2016). Curr Drug Targets 17, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mates JM, and Sanchez-Jimenez F (1999). Front Biosci 4, D339–45. [DOI] [PubMed] [Google Scholar]
- Miguel Quintela-Fandino, J. VA, Alfonso Cortes Salgado, Silvana Andrea Mouron, Juan Antonio Guerra, Mana Gion Cortes, Manuel Morente, Luis Manso. (2018). J Clinical Oncology 36, abstract 2552. [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). Nat Cell Biol 13, 1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, and Saikumar P (2001). J Biol Chem 276, 18361–74. [DOI] [PubMed] [Google Scholar]
- Mor G, Fu HH, and Alvero AB (2006). Curr Opin Investig Drugs 7, 542–8. [PubMed] [Google Scholar]
- Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mouron S, Munoz J, Gomez-Lopez G, Jimenez-Renard V, Mulero F, Chandel NS, and Quintela-Fandino M (2016). Cell Rep 15, 2705–18. [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, and Youle RJ (1999). EMBO J 18, 2330–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, and Kemp BE (2011). Science 332, 1433–5. [DOI] [PubMed] [Google Scholar]
- Park JS, Jung JS, Jeong YH, Hyun JW, Le TK, Kim DH, Choi EC, and Kim HS (2011). J Neurochem 119, 909–19. [DOI] [PubMed] [Google Scholar]
- Piontek M, Hengels KJ, Porschen R, and Strohmeyer G (1993). Anticancer Res 13, 2119–23. [PubMed] [Google Scholar]
- Podkalicka P, Mucha O, Jozkowicz A, Dulak J, and Loboda A (2018). Contemp Oncol (Pozn) 22, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Phromnoi K, Yadav VR, Chaturvedi MM, and Aggarwal BB (2010). Planta Med 76, 1044–63. [DOI] [PubMed] [Google Scholar]
- Quintela-Fandino M (2016). Mol Cell Oncol 3, e1217368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard JF, Motz GT, and Puga A (2007). Nucleic Acids Res 35, 7074–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemsma R, Simons JP, Bashir Z, Gooch CL, and Kleijnen J (2010). BMC Cancer 10, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, and Herlyn M (2013). Cancer Cell 23, 811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Alam J, and Choi AM (2006). Physiol Rev 86, 583–650. [DOI] [PubMed] [Google Scholar]
- Safra T, Berman T, Yachnin A, Bruchim I, Meirovitz M, Barak F, Atlas I, Levy T, and Rosengarten OS (2013). Int J Gynecol Cancer 23, 475–80. [DOI] [PubMed] [Google Scholar]
- Sarkar FH, and Li Y (2003). Cancer Invest 21, 744–57. [DOI] [PubMed] [Google Scholar]
- Seger R, and Krebs EG (1995). FASEB J 9, 726–35. [PubMed] [Google Scholar]
- Sehouli J, Stengel D, Harter P, Kurzeder C, Belau A, Bogenrieder T, Markmann S, Mahner S, Mueller L, Lorenz R, Nugent A, Wilke J, Kuznik A, Doering G, Wischnik A, Sommer H, Meerpohl HG, Schroeder W, Lichtenegger W, and Oskay-Oezcelik G (2011). J Clin Oncol 29, 242–8. [DOI] [PubMed] [Google Scholar]
- Sennino B, and McDonald DM (2012). Nat Rev Cancer 12, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setchell KD (1998). Am J Clin Nutr 68, 1333S–1346S. [DOI] [PubMed] [Google Scholar]
- Shamas-Din A, Kale J, Leber B, and Andrews DW (2013). Cold Spring Harb Perspect Biol 5, a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Lambrecht RW, Donohue SE, and Bonkovsky HL (2006). FASEB J 20, 2651–3. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, and Jemal A (2013). CA Cancer J Clin 63, 11–30. [DOI] [PubMed] [Google Scholar]
- Signes A, and Fernandez-Vizarra E (2018). Essays Biochem 62, 255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Russell J, Sigala B, Zhang Y, Williams J, and Keshav KF (1999). Oncogene 18, 6641–6. [DOI] [PubMed] [Google Scholar]
- Speicher LA, Barone L, and Tew KD (1992). Cancer Res 52, 4433–40. [PubMed] [Google Scholar]
- Speicher LA, Laing N, Barone LR, Robbins JD, Seamon KB, and Tew KD (1994). Mol Pharmacol 46, 866–72. [PubMed] [Google Scholar]
- Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, and Igarashi K (2002). EMBO J 21, 5216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, and Hayes JD (2015). Free Radic Biol Med 88, 108–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, and Draetta GF (2014). Nature 514, 628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale DC, Piazza C, Melilli B, Drago F, and Salomone S (2013). Eur J Drug Metab Pharmacokinet 38, 15–25. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, and Gamblin SJ (2011). Nature 472, 230–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YP, Liang ZQ, Gu ZL, and Qin ZH (2005). Acta Pharmacol Sin 26, 1421–34. [DOI] [PubMed] [Google Scholar]
- Yano T (2002). Mol Aspects Med 23, 345–68. [DOI] [PubMed] [Google Scholar]
- Yu O, Jung W, Shi J, Croes RA, Fader GM, McGonigle B, and Odell JT (2000). Plant Physiol 124, 781–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LZ, Ye J, Manevich Z, Ball Y, Bethard LE, Jiang JR, Broome Y, Wang AM, Townsend DM G. Y and Tew KD (2019). Can Res In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]