

Abstract
Background
Fanconi anemia (FA) is phenotypically diverse, hereditary condition associated with bone marrow failure, multiple physical abnormalities, and an increased susceptibility to the development of malignancies. Less recognized manifestations of FA include endocrine abnormalities. International discourse has highlighted that these abnormalities are widespread among children and adults with FA. To date there has been no systematic study that has evaluated the endocrine abnormalities in a cohort of patients with FA, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG. The objectives of the study were to evaluate endocrine gland function in patients with FA of a single FA genotype, and to determine the frequency and nature of endocrine abnormalities in this group.
Methods
Cross‐sectional, descriptive study of 24 South African patients of African ancestry with FA (homozygous for a FANCG founder mutation). Outcomes measured included growth, pubertal status, growth hormone axis screening, thyroid gland function, glucose and insulin metabolism and bone age (BA).
Results
Endocrine dysfunction was present in 70.8% (17 of 24), including abnormal insulin‐like growth factor 1 (IGF‐1)/insulin‐like growth factor‐binding protein 3 (IGFBP‐3) in 25.0% (6 of 24), insulin resistance in 41.7% (10 of 24), abnormal thyroid function in 16.7% (4 of 24) and short stature in 45.8% (11 of 24). No abnormalities of glucose metabolism were identified. Abnormal pubertal status was seen in three males (12.5%). Abnormal BAs were present in 34.8% (8 of 23).
Conclusion
Endocrine abnormalities occur at a high frequency in patients with FA, homozygous for a FANCG founder mutation, similar to other FA cohorts. Our data are specific to FA patients with a single genotype, and therefore provide the first genotype‐phenotype information on endocrine abnormalities in South African patients, homozygous for a FANCG founder mutation. Recommendations regarding endocrine screening in this patient subgroup are made, including, but not limited to, baseline testing of thyroid function, fasted insulin and glucose, and IGF‐1 and IGFBP‐3.
Keywords: endocrine abnormalities, FANCG, founder mutation, short stature, thyroid function
Endocrine abnormalities are a lesser known complication of Fanconi anemia. Endocrine abnormalities occur at a high frequency in South African patients with Fanconi anemia, homozygous for a FANCG founder mutation.

1. INTRODUCTION
Fanconi anemia (FA) is an uncommon, phenotypically diverse, hereditary chromosome breakage disorder characterized by deoxyribonucleic acid (DNA) hypersensitivity to cross‐linking agents at a molecular level, with resultant chromosome instability (Mehta & Tolar, 2018). To date, 22 FA‐associated genes have been identified, designated FANCA (OMIM: 607139)—W (OMIM: 617784) (RFWD3 (OMIM: 614151)), demonstrating the marked genetic heterogeneity that FA exhibits (The Rockefellar University Fanconi anemia mutation database, 2019). These FANC genes encode FA proteins, which operate together in a shared FA pathway, considered a DNA repair pathway that regulates the cells’ resilience to harmful DNA interstrand cross‐linking agents (Mehta & Tolar, 2018; Taniguchi & D’Andrea, 2006). If this pathway becomes disrupted, by a pathogenic variant in a FA‐related gene, the cellular and clinical abnormalities suggestive of FA manifest (Garcia‐Higuera et al., 2001). The FA subtypes are inherited predominantly in an autosomal recessive manner; however, heterozygous dominant‐negative mutations in the RAD51 gene (OMIM: 179617) (also known as FANCR (OMIM: 617244)) and hemizygous mutations in the FANCB gene (OMIM: 300515) result in the less common autosomal dominant and X‐linked forms of FA, respectively (Meetei et al., 2004; Mehta & Tolar, 2018; Vaz et al., 2010).
Although FA is thought to be a rare disorder, the prevalence in certain South African population groups, such as the Afrikaner and Black populations, has been found to be much higher (Tipping et al., 2001). The term “Black” has been used to describe individuals deriving from sub‐Saharan Bantu‐speaking indigenous ancestry groups (Feben, Wainstein, Kromberg, Essop, & Krause, 2018). Morgan et al. (2005) proposed that the birth incidence of FA in the Black South African population is higher than 1 in 40,000 based on carrier frequency data obtained from gene frequency studies. The likely reason for this higher incidence is a genetic founder mutation in the FANCG gene (OMIM: 602956) (Morgan et al., 2005). In the Black South African FA population studied, a deletion mutation (c.637_643del (p.Tyr213Lysfs*6)) was identified in 82.5% of individuals tested (present in a homozygous state in 77.5%) (Morgan et al., 2005). These patients with FA thus represent a unique patient cohort from a genetic homogeneity perspective. When compared to other FA cohorts, individuals with FA, specifically homozygous for the FANCG founder mutation, have been found to have significant growth restriction and a higher incidence of renal abnormalities, abnormal skin pigmentary lesions and present with severe cytopenia (Feben et al., 2014, 2015). Given this predominantly genetically homogeneous group, and the limited availability of chromosome breakage testing in the state healthcare sector in South Africa, molecular genetic testing for the FANCG founder mutation is now the favored first‐line diagnostic test for South African patients, with African ancestry, suspected to have FA (Wainstein et al., 2013).
Clinically, FA is associated most commonly with bone marrow failure, multiple congenital physical abnormalities, and an increased susceptibility to the development of hematological and solid tissue malignancies (Mehta & Tolar, 2018). Less recognized manifestations of FA include a wide range of abnormalities of endocrine gland function, which are influenced to a certain extent by the various treatments used in the management of patients with FA, such as androgen therapy or hematopoietic stem cell transplantation (HSCT) (Giri, Batista, Alter, & Stratakis, 2007). More recent discourse by Giri et al. (2007) and Rose et al. (2012) have highlighted that endocrine abnormalities are widespread (79%) among both children and adults with FA. Of the endocrine abnormalities identified, the most notable were short stature and/or growth hormone (GH) deficiency (GHD) (51%), abnormal gonadal function (65%), hypothyroidism (37%), and dysfunctional glucose/insulin metabolism (39%) (Giri et al., 2007). Strikingly, a more recent study identified at least one endocrine abnormality in 79% of the overall study group (Rose et al., 2012). Under‐nutrition, low body mass index (BMI), raised BMI, reduced bone mineral density, and pituitary gland abnormalities are other endocrine abnormalities that have been documented in patients with FA (Petryk et al., 2015).
While numerous previous research studies have documented the major endocrine abnormalities in patients with FA, these studies have assessed individuals with FA of various genotypes to give general frequencies of these disorders. Very little genotype‐specific information has yet been documented in the literature. Our study aimed to evaluate the need for routine screening of endocrine status in patients with FA, with a specific focus on patients homozygous for the FANCG founder mutation.
2. MATERIALS AND METHODS
2.1. Ethical compliance
Ethics clearances were obtained from The University of the Witwatersrand Human Research Ethics Committee (certificates M160220 and M1703108), the Faculty of Health Sciences Research Ethics Committee of the University of Pretoria (certificate 547/2017) and the Health Sciences Research Ethics Committee of the University of the Free State (certificate UFS‐HSD2017/1406/3107).
2.2. Subjects and methods
Patients were recruited from the Paediatric Haematology/Oncology Units at four tertiary academic hospitals in South Africa. Recruitment took place over 19 months (January 2017 to August 2018).
The present study included 24 South African patients, confirmed to be homozygous for the seven base‐pair deletion (c.637_643del (p.Tyr213Lysfs*6)) in FANCG (NM_004629.1). Targeted mutation analysis had been performed by the Molecular Genetics Laboratory of the National Health Laboratory Service (NHLS) in Braamfontein, South Africa. Patients who met the study inclusion criteria and their parent/guardian were required to read the information document, available in English, Afrikaans or Sesothu, and read and sign informed consent/assent indicating their wish to participate in the study.
Patients underwent a clinical examination, which included anthropometric measurements and Tanner pubertal staging. Anthropometric measurements included weight (to the nearest 0.1 kg) using a Seca© electronic scale, height (to the nearest mm) using a floor‐standing Seca© stadiometer, and head circumference (to the nearest mm) using a tape measure. Measurements were expressed as standard deviation (SD) scores (SDS), based on age‐ and sex‐matched growth charts from the Handbook of Physical Measurements (Hall, Allanson, Gripp, & Slavotinek, 2007). BMI was calculated for each patient using the standard BMI formula (weight (kg)/height (m)2). BMI was expressed as a SDS calculated using the World Health Organization (WHO)‐AnthroPlus software (WHO, 2009).
Tanner staging was assessed using a Tanner staging chart and a Prader orchidometer to assess testicular volume in the male patients (Marshall & Tanner, 1969, 1970). The normal age range for the onset of puberty for females was considered as eight years to 13 years, and for males was between nine years six months and 13 years six months (Carel & Léger, 2008; Marshall & Tanner, 1969, 1970). These are the ages at which 95% of children attain Tanner stage two pubertal development (Carel & Léger, 2008; Marshall & Tanner, 1969, 1970). Pubarche is the development of pubic and axillary hair, thelarche the development of breast buds, and gonadarche is testicular volume equal to or greater than 4 milliliters (ml) (Emmaneul & Bokor, 2019).
Short stature is defined as height‐for‐age more than 2 SD below the WHO growth reference mean; underweight‐for‐age is defined as weight more than 2 SD below the mean on a WHO weight‐for‐age growth chart; overweight‐for‐age is defined as weight‐for‐height greater than 2 SD above WHO growth reference mean (for children under 5 years) and BMI greater than 1 SD above WHO growth reference mean (for children aged 5–19 years) (WHO, 2008); microcephaly is considered as head circumference more than 2 SD below the growth reference mean (Pang, Atefy, & Sheen, 2008).
Endocrine hormone testing included glucose and insulin metabolism (by measuring fasting plasma glucose and insulin levels); thyroid gland function (by measuring thyroid stimulating hormone [TSH] and free thyroxine [FT4]); and screening of the GH axis (consisting of insulin‐like growth factor 1 [IGF‐1] and insulin‐like growth factor‐binding protein 3 [IGFBP‐3] measurements). GH stimulation testing was not performed. Measurements were assessed from a single, overnight fasted venepuncture sample, and were evaluated in the same private laboratory, in Johannesburg and Bloemfontein. Blood measurements were evaluated against the laboratory age‐ and sex‐matched control reference ranges.
Fasting insulin and glucose levels were used to calculate the homeostasis model assessment of insulin resistance (HOMA‐IR) index. This model uses the following formula: fasting insulin concentration (milliunits per litre) multiplied by fasting glucose concentration (millimoles per liter) divided by 22.5 (Wallace, Levy, & Matthews, 2004). A HOMA‐IR value of greater than two was considered as insulin resistant, in order to standardize with the cut off value used by Giri et al.’s (2007) FA cohort and to allow direct comparison.
Subclinical hypothyroidism is defined as serum TSH above the upper limit of normal age‐matched ranges with a normal level of FT4, and overt hypothyroidism is characterized by a raised TSH in combination with a lower than normal FT4 (Garber et al., 2012).
The diagnosis of GHD is not obtained from a single test but rather is a process involving clinical evaluation, radiological assessment (bone age [BA] and central nervous system imaging), and biochemical testing (including provocative (stimulation) GH testing and IGF‐1 and IGFBP‐3 testing) (GH Research Society, 2000).
Left wrist and hand X‐rays were performed on the day of the study visit and reviewed by a Paediatric Endocrinologist, to assess BA using the published standards of Greulich and Pyle (Greulich & Pyle, 1950). Bone age was expressed as being normal, advanced (more than 2 SD above the mean for age and sex) or delayed (more than 2 SD below the mean for age and sex) (European Society of Paediatric Radiology, 2019). The X‐rays were also examined for bony abnormalities. It is important to note that the BA standards published by Greulich and Pyle in 1959 are based on European population data (Greulich & Pyle, 1950). These standards have low accuracy when estimating skeletal age of African individuals, and skeletal age standards specific to African populations still need to be developed (Govender & Goodier, 2018). For this reason, the reporting of BA in the present cohort may be inaccurate.
Data were captured and analyzed statistically using Microsoft Excel (2013). The frequency of growth disturbances and endocrine abnormalities were documented, and comparisons were made, where possible, with the Giri et al. (2007) and Rose et al. (2012) cohorts. These cohorts included individuals with FA with varying genotypes. Continuous variables (such as weight, height and head circumference) were compared to the Giri et al. and Rose et al. FA cohorts using an unpaired t test. Fisher's exact test was used to calculate the p value from a 2 × 2 contingency table of categorical variables (such as number of individuals with an endocrine abnormality). Differences in means were considered statistically significant with p values < .05.
3. RESULTS
3.1. Demographic data
The total study group consisted of 24 patients. Thirteen (54%) of the patients were male and 11 (46%) female. The median age was 9.5 years (range 3–19 years), with the majority of patients (23 (96%)) under 18 years of age at the time of data collection.
Giri et al.’s (2007) FA cohort consisted of 45 patients (19 males and 26 females) between the ages of two to 49 years (28 were 18 years or younger, and 17 were over 18 years old). Giri et al.’s (2007) study analyzed retrospective endocrine data, whereas the present study is a cross‐sectional study. Rose et al.’s (2012) FA cohort consisted of 78 children (43 females and 35 males) between the ages of 0.3 and 15.9 years, and 42 adults (19 females and 23 males) between the ages of 13.5 and 31 years. Twenty‐six patients of the Rose et al. (2012) FA cohort were 18 years or older (14 females and 12 males). Our study makes comparisons to the entire Giri et al. (2007) cohort, and where possible to the pediatric Giri et al. (2007) cohort specifically, and to the pediatric Rose et al. (2012) cohort specifically.
3.2. Growth measurements
Growth measurements (weight, height, and head circumference) are depicted in Figure 1. The growth measurements of the FA FANCG founder mutation cohort were compared with those of the patients in Giri et al.’s (2007) cohort (Table 1). The median weight SDS for the study cohort was −1.6 (range 0.6 to <−3.0; mean −1.7 ± 0.9). The median BMI for the study cohort was −0.7 (range 1.2 to −2.3; mean −0.7 ± 1.0). Only 8.3% (2 of 24) were overweight‐for‐age, compared to almost 27.0% overweight in Giri et al.’s (2007) cohort, although this was not statistically significant (p value = .12), and 11.0% of children (p value = 1.00) with FA in Rose et al.’s (2012) cohort. The median height SDS for the study cohort was −1.9 (range 0.32 to <−3; mean −1.8 ± 1.0). The median head circumference SDS for the study cohort was −1.7 (range 0.0 to <−3; mean −1.7 ± 0.8). Of the eight patients with microcephaly, three had a normal weight and height, one was underweight, one was short, and three were both underweight and short.
FIGURE 1.

Growth measurement distribution of patients with Fanconi anemia, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG (N = 24)
TABLE 1.
Comparison of the growth measurements between the present FA cohort, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG, and Giri et al.’s (2007) FA cohort
| Growth measurement | Present FA cohort (N = 24), n/N (%) | Giri et al.’s FA cohort (N = 45), n/N (%) | p value |
|---|---|---|---|
| Underweight | 10/24 (41.7) | 10/45 (22.2) | .10 |
| Overweight | 2/24 (8.3) | 12/45 (26.7) | .12 |
| Short stature | 11/24 (45.8) | 23/45 (51.1) | .80 |
| Microcephaly | 8/24 (33.3) | NA | NA |
Abbreviations: FA, Fanconi anemia; NA, not available.
3.3. Growth measurements and their relationship to pubertal status and BA
Eighteen percent (2 of 11) of the FA individuals with short stature had abnormal pubertal development (one precocious and one delayed gonadarche). Of the FA individuals with short stature, 45.5% (5 of 11) had abnormal (delayed) BAs.
3.4. Pubertal assessment
As nine of the 11 females in the present study were receiving androgen therapy (which can cause virilization as a side effect) only thelarche, and not pubarche, was used to assess pubertal stage. All 11 females had appropriate pubertal development, according to breast development.
As 11 of the 13 males in the present study were receiving androgen therapy, only gonadarche, and not pubarche, was used to assess pubertal stage. Seventy‐seven percent (10 of 13) of males had age‐appropriate pubertal development. Two of the males had delayed gonadarche (15.4%) and one had precocious gonadarche (7.7%). All of the males with delayed and precocious gonadarche had normal BAs.
3.5. Endocrine hormone testing
Fifty‐eight percent (14 of 24) of the study cohort had at least one endocrine abnormality (including abnormal screening of the GH axis, insulin resistance and abnormal thyroid functions (hypothyroidism and subclinical hypothyroidism). The majority (37.5% (9 of 24)) of these patients had one endocrine abnormality, 16.7% (4 of 24) had two endocrine abnormalities and one patient (4.2%) had three endocrine abnormalities. Figure 2 shows the percentage of endocrine abnormalities identified in the present study cohort. Frequency of endocrine abnormalities was compared to those in Giri et al.’s (2007) FA cohort (Table 2). Of the patients with low IGF‐1/IGFBP‐3, 33.3% (2 of 6) also had abnormal thyroid function (one subclinical hypothyroidism and one hypothyroidism). Fifty percent (3 of 6) of the patients with low IGF‐1/IGFBP‐3 had short stature.
FIGURE 2.

Endocrine abnormalities in patients with Fanconi anemia, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG (N = 24). Overt hypothyroidism refers to increased TSH and decreased FT4 (Garber et al., 2012); subclinical hypothyroidism refers to Increased TSH and normal FT4 (Garber et al., 2012); impaired fasting glucose refers to a laboratory fasting blood glucose reference greater than 6.0 mmol/L; insulin resistance was determined by a HOMA index greater than two. FT4, free thyroxine; GH, growth hormone; HOMA, homeostasis model assessment; TSH, thyroid stimulating hormone
TABLE 2.
Comparison of endocrine abnormalities between the present FA cohort, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG, and Giri et al.’s (2007) FA cohort
| Endocrine abnormality | Present FA cohort (N = 24), n/N (%) | Giri et al.’s FA cohort (N = 35), n/N (%) | p value (Fisher's exact test) |
|---|---|---|---|
| Thyroid function abnormality | 4/24 (16.7) | 18/35 (51.4) | .01 |
| Hypothyroidism a | 1/24 (4.2) | 13/35 (37.1) | .00 |
| Subclinical hypothyroidism b | 3/24 (12.5) | 5/35 (14.3) | 1.00 |
| Impaired fasting glucose | 0/24 (0.0) c | 10/41 (24.4) d | .01 |
| Insulin resistance e | 10/24 (41.7) | 10/24 (41.7) | 1.00 |
| Low IGF−1 f | 2/24 (8.3) | 6/9 (66.7) | .00 |
| Low IGFBP−3 f | 5/24 (20.8) | 3/9 (33.3) | .65 |
Abbreviations: FA, Fanconi anemia; FT4, free thyroxine; HOMA, homeostasis model assessment; IGF‐1, insulin‐like growth factor 1; IGFBP‐3, insulin‐like growth factor‐binding protein 3; TSH, thyroid stimulating hormone.
Increased TSH and decreased FT4 (Garber et al., 2012).
Increased TSH and normal FT4 (Garber et al., 2012).
Laboratory fasting blood glucose reference greater than 6.0 mmol/L.
Laboratory fasting blood glucose range greater than 5.6–6.9 mmol/L (Giri et al., 2007).
Insulin resistance was determined by a HOMA index greater than two.
Age and sex‐matched reference ranges; Statistically significant p values are highlighted in italics.
The laboratory‐provided normal TSH reference range for the individuals in the present study cohort was 0.35–4.94 uIU/ml, and the normal FT4 range was 9.0–19.0 pmol/L. Overt hypothyroidism was seen in one individual (4.2%) in the present study, compared to 37.1% of Giri et al.’s (2007) entire FA cohort (p value = .00) (and 20.0% (5 of 20) of Giri et al.’s (2007) patients aged 18 years or less (p value = .08)), and 61% of Rose et al.’s (2012) FA cohort (p value = .00). Of the 10 individuals with insulin resistance, two (20.0%) were also overweight‐for‐age.
3.6. Radiographic studies
Hand and wrist X‐rays were available in 23 of the 24 patients. BA was abnormal in 34.8% (8 of 23) of the present study cohort. Advanced BA was seen in 8.7% (2 of 23), a similar frequency to that of Giri et al.’s (2007) cohort (7.7%) (p value = 1.00). Delayed BA was seen in 26.1% (6 of 23) of the present cohort, a higher, although not statistically significant, frequency compared to Giri et al.’s (2007) cohort (15.4%) (p value = .68). Only one of the patients with advanced BA was receiving androgen therapy, and all but one of the patients with delayed BA were receiving androgen therapy. Although not an endocrine abnormality, it was incidentally noted that three male individuals (13.0% of the study cohort) had abnormal fusion of the same two carpal (triquetral and lunate) bones (Figure 3).
FIGURE 3.
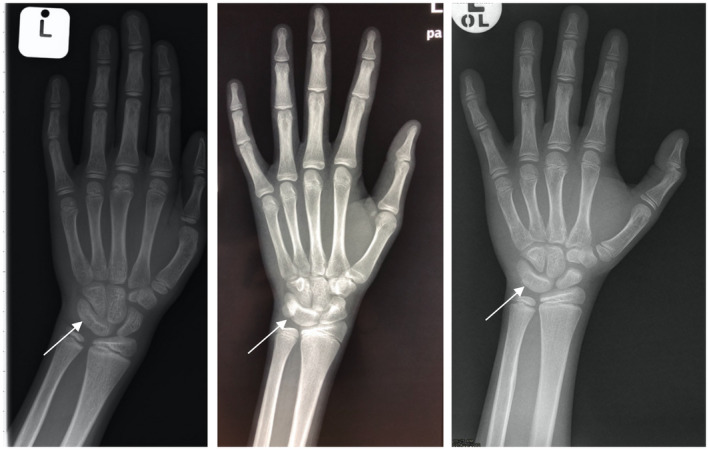
Left hand and wrist anteroposterior X‐rays of three male patients with Fanconi anemia, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG, demonstrating fusion of the triquetral and lunate carpal bones. Fusion of the carpal bones is indicated by the arrows
3.7. Endocrine abnormalities and their relationship to growth, pubertal status, and BA
Table 3 details the endocrine abnormalities associated with the various abnormalities in growth measurements. In the present study, 70.0% (7 of 10) of the underweight individuals were also of short stature. Of the two overweight individuals, one had short stature.
TABLE 3.
Association of abnormal growth measurements and endocrine abnormalities a in patients with Fanconi anemia, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG (N = 24)
| Abnormal growth measurement | |||||
|---|---|---|---|---|---|
| UWFA | OWFA | SS | UWFA + SS | MC | |
| Percentage of study cohort (%) (n/N) | 41.7 (10/24) | 8.3 (2/24) | 45.8 (11/24) | 29.2 (7/24) | 33.3 (8/24) |
| Percentage with at least one endocrine abnormality a (%) (n/N) | 50.0 (5/10) | 100.0 (2/2) | 72.7 (8/11) | 57.1 (4/7) | 50.0 (4/8) |
| Endocrine abnormality (%) | |||||
| Subclinical hypothyroidism | 10.0 (1/10) | 50.0 (1/2) | 0.0 (0/11) | 0.0 (0/7) | 0.0 (0/8) |
| Hypothyroidism | 0.0 (0/10) | 0.0 (0/2) | 9.1 (1/11) | 0.0 (0/7) | 0.0 (0/8) |
| Insulin resistance | 30.0 (3/10) | 100.0 (2/2) | 63.6 (7/11) | 42.9 (3/7) | 37.5 (3/8) |
| Abnormal IGF‐1/IGFBP‐3 | 50.0 (2/10) | 50.0 (1/2) | 27.3 (3/11) | 14.3 (1/7) | 12.5 (1/8) |
Abbreviations: IGF‐1, insulin‐like growth factor 1; IGFBP‐3, insulin‐like growth factor‐binding protein 3; MC, microcephaly; OWFA, overweight‐for‐age; SS, short stature; UWFA, underweight‐for‐age.
Excluding short stature as an endocrine abnormality.
Of the individuals with at least one endocrine abnormality (insulin resistance, abnormal thyroid function, abnormal testing of the GH axis), 57.1% (8 of 14) had short stature. Of the individuals with no endocrine abnormality, 30.0% (3 of 10) had short stature. However, patients with one or more endocrine abnormality were not significantly shorter than those with no endocrine abnormality; mean height SDS −2.0 ± 0.9 compared to −1.6 ± 1.1, respectively (p value = .35). This differs to Giri et al.’s (2007) cohort whose patients with one or more endocrine abnormality were statistically significantly shorter than those without (mean height SDS −2.7 ± 2.0 compared to −1.3 ± 1.4, respectively (p value = .01).
3.8. Pubertal assessment and endocrine abnormalities
Of the three individuals with abnormal pubertal development, one had insulin resistance and two had abnormal testing of the GH axis (both individuals had delayed gonadarche). Thirty‐three percent (2 of 6) of patients with abnormal testing of the GH axis had abnormal (delayed) gonadarche. In view of the small sample size, statistical conclusions about pubertal status and endocrine abnormalities cannot be reliably made.
3.9. BA and endocrine abnormalities
Two thirds (4 of 6) of the individuals with delayed BA had at least one endocrine abnormality (including abnormal thyroid functions, abnormality of GH axis testing and insulin resistance). Fifty percent (2 of 4) had abnormal IGF‐1/IGFBP‐3 testing, 50.0% (2 of 4) had abnormal thyroid functions (one subclinical hypothyroidism and overt hypothyroidism), and 75.0% (3 of 4) had insulin resistance. Neither of the two individuals with advanced age had an endocrine abnormality.
4. DISCUSSION
The Fanconi Anemia Research Foundation (FARF) recommends standard endocrine screening and testing in all patients with FA based on the predicted prevalence of the endocrine dysfunction summarized in a 2015 comprehensive literature review conducted by Petryk et al. (2015).
In South Africa, there are currently no similar guidelines for the investigation and treatment of endocrine disorders in patients with FA. Although patients with FA are managed in tertiary care units, protocols for endocrine profiling differ between centres. Most patients who attend these hematology/oncology units reside in poor communities and have limited resources and access to healthcare services, apart from their hematology/oncology visits. This, together with the constrained public healthcare sector, prescribes that endocrine screening programs be financially prudent and designed to detect significant endocrine abnormalities, for which management is available.
We report for the first time the nature and frequency of endocrine abnormalities in a cohort of patients with FA, homozygous for the same FANCG founder mutation (c.637_643del (p.Tyr213Lysfs*6)). These observations provide important genotype‐phenotype correlations, as our study cohort was comprised solely of FA patients with the same FANCG mutation, which occurs in >78% of local patients with African ancestry (Morgan et al., 2005). Additionally, these observations may serve as the basis for the development of standardized guidelines for endocrine profiling in patients with FA due to this FANCG founder variant.
Nutrition and weight gain of an individual with FA is governed by both endocrine and non‐endocrine factors. International studies have found the BMI‐for‐age to be normal in only approximately 50% of individuals with FA (Giri et al., 2007; Rose et al., 2012). The present study found a similarly high frequency of underweight‐for‐age patients with FA to the study cohorts of Giri et al. (2007) and Rose et al. (2012). Food security has improved in South Africa over the past decade; however, many households still do not have access to nutritionally valuable foods in sufficient quantities, which may be adding to this high frequency of underweight‐for‐age patients in the present study (Labadarios et al., 2011).
Two patients in our study cohort were found to be overweight‐for‐age. Both of these patients had insulin resistance, and one had the additional finding of subclinical hypothyroidism. Increased weight gain can occur with both of these endocrine derangements. The frequencies of overweight or obese patients in previous cohorts are higher than that of the FANCG FA cohort, 26.6% versus 8.3% respectively (Giri et al., 2007). Future studies could evaluate the nutritional statuses of the South African patients with FA, to determine the extent of food insecurity as a confounding factor when evaluating growth parameters.
Short stature is one of the most well recognized physical abnormalities associated with FA. The etiology of short stature in patients with FA is manifold, including complications from FA treatment (Petryk et al., 2015). Commonly observed endocrine causes of short stature in FA consist of GHD, hypothyroidism, and hypogonadism. International studies have documented the presence of short stature at high frequencies of between 51.0% and 60.0% (Giri et al., 2007; Rose et al., 2012). In the present study, 45.8% of the patients were of short stature. When compared to Giri et al.’s (2007) FA cohort, these findings are slightly lower and not statistically significant, but nonetheless remain a high frequency.
International studies have shown that individuals with FA who have hormone deficiencies are markedly shorter compared to their counterparts without hormone deficiencies, suggesting that superimposed endocrine dysfunction further influences the short stature that is inherently associated with FA (Giri et al., 2007; Rose et al., 2012; Wajnrajch et al., 2001). In the 2012 study by Rose et al. (2012), short stature was associated with hormone abnormalities in 42.3% of child patients and 30% of short statured adults. A higher frequency (72.7%) of patients with short stature (of whom 10 were ≤18 years and one was >18 years) had one or more endocrine abnormalities in the present cohort, although this finding was not statistically significant. Interestingly, our study showed that patients with at least one endocrine abnormality were not significantly shorter than their counterparts without endocrine dysfunction.
Puberty, gonadal function, and fertility can all be affected in patients with FA (Petryk et al., 2015). Children with FA can suffer from peripheral precocious puberty or they can have delayed onset of puberty (Petryk et al., 2015). Abnormal pubertal development was present in a minority (12.5%) (3 of 24) of FA patients in the present study (all of whom were males), based on clinical Tanner staging assessing only thelarche and gonadarche. Direct assessment of gonadal function (by biochemical stimulation testing) was not included in our study due to financial constraints. A larger sample size is required to make any meaningful correlations between pubertal stage and endocrine dysfunction. In addition, the prospective assessment of participants in the prepubertal age range would ideally need to be performed, in order to more accurately comment on the frequency of pubertal abnormalities in this FA genotype.
GHD is a recognized abnormality observed in patients with FA, with half of the patients evaluated by Giri et al. (2007) having documented GHD. These patients were noted to be significantly shorter when compared to those individuals with FA who had normal GH levels (Giri et al., 2007). Presently, in South Africa the cost of performing a GH stimulation test is approximately two and a half times greater than the cost of IGF‐1 and IGFBP‐3 combined (personal communication with Lancet Laboratory, 29 January 2019). Funding constraints limited the testing of the GH axis in the present study cohort to IGF‐1 and IGFBP‐3 testing.
As IGF‐1 and IGFBP‐3 are screening tests, we are unable to comment on confirmed GHD in the present cohort, and thus unable to directly compare it to other international FA cohorts. Despite this limitation, we observed a statistically significantly lower frequency of patients with low IGF‐1 levels compared to Giri et al.’s (2007) cohort (8.3% vs. 66.7% respectively), an unexpected finding as IGF‐1 is negatively influenced by poor nutrition (Petryk et al., 2015). We also observed a lower frequency of patients with low IGFBP‐3 levels compared to that of Giri et al.’s (2007) cohort; 20.8% versus 33.3%, respectively. These findings suggest that GHD may be a less common finding in patients with FA, homozygous for the FANCG founder mutation. Poor growth and growth velocity should be used as a means of screening to guide which patients should undergo GH stimulation testing in the FANCG founder mutation FA cohort.
International recommendations suggest initiating GH treatment in patients with FA only if GHD has been compellingly documented, as there is currently no consensus on the long‐term safety of GH treatment in patients with FA (Petryk et al., 2015). In the state healthcare sector in Johannesburg, South Africa, GH therapy is provided to only a limited number of patients and even those with documented GH deficiency may not be able to access the medication even when a documented clinical need exists (personal communication with Professor David Segal, 29 January 2019). If GHD were to be diagnosed in patients with FA in South Africa in the state healthcare sector, there is concern as to whether the patient would have access to GH therapy.
Thyroid function is affected in individuals with FA; however, the pathophysiological explanation for thyroid disturbances in FA is not well understood. Hypothyroidism was identified in a (statistically significant) lower frequency (4.2%) in our study cohort than in international studies, which may be related to the cross‐sectional design of the present study or due to the non‐HSCT status of the present cohort (Giri et al., 2007; Rose et al., 2012). The presence of thyroid antibodies, as a potential cause of hypothyroidism, was not tested for.
Various disturbances in glucose metabolism occur frequently (39.0%) in children with FA; including impaired fasting glucose, insulin resistance, and overt type two diabetes mellitus (DM) (Elder et al., 2008; Giri et al., 2007). Patients with FA are at a higher risk of developing DM when compared to the general population (Morrell, Chase, Kupper, & Swift, 1986). Previous FA studies have documented an increased frequency of glucose homeostatic abnormalities among individuals with FA; Giri et al. (2007) documented impaired fasting glucose (fasting blood glucose greater than 5.6–6.9 mmol/L) and/or glucose intolerance in 24% and overt DM (fasting blood glucose equal to or greater than 7 mmol/L) in 10% of their study cohort; Rose et al. (2012) documented higher levels of glucose intolerance with 30% of their adult cohort and 68% of their pediatric cohort affected. Factors extrinsic to the presence of FA (such as androgen treatment) and factors intrinsic to FA (such as the presence of reactive oxygen species) play a role in an individual's susceptibility to a disturbance in glucose homeostasis (Giri et al., 2007; Li et al., 2012; Morrell et al., 1986).
Interestingly, in the present study, none of the patients with FA were found to have impaired fasting glucose or overt type two DM, despite the majority of these patients having received long‐standing androgen therapy and multiple blood transfusions. Blood ferritin levels (used to assess total body iron stores) were not measured in the present study, thereby comments regarding the influence of multiple blood transfusions on the endocrine status of this cohort cannot be made. Both fasting plasma glucose testing and the two hour plasma glucose value obtained during an oral glucose tolerance test (OGTT) (performed with 75 grams of oral glucose) are deemed acceptable diagnostic modalities for DM and prediabetes mellitus; however, according to the American Diabetes Association’s, 2019 position statement, the two hour plasma glucose value identifies more individuals with DM and prediabetes than the fasting plasma glucose testing (American Diabetes Association, 2019). An OGTT was not performed on the present study patients due to cost limitations, and thus some individuals with impaired glucose homeostasis may not have been detected. Although at present, in South Africa, the cost of an OGTT is approximately two and a half times more expensive than that of a fasting plasma glucose test (personal communication with Lancet Laboratory, 29 January 2019) it would be the preferred diagnostic test for DM.
Insulin resistance was identified in 41.7% of our study population, which corresponded to the incidence of insulin resistance in Giri et al.’s (2007) cohort and contrasted with the minimal numbers of insulin resistance in Rose et al.’s (2012) cohort.
Abnormal BA was found at a moderate (34.8%) frequency in the present study cohort. Twenty‐six percent of the cohort had delayed BA, contrasting (not statistically significantly) with the lower frequency (15.4%) of delayed BA identified in Giri et al.’s (2007) cohort. Advanced BA was noted at a lower frequency (8.7%) than delayed BA in the present cohort, and at a similar frequency to that reported by Giri et al. (2007) (7.7%). Endocrine abnormalities were not seen in the individuals with advanced BA but were noted at a high frequency (66.7%) in the delayed BA group. Androgen therapy, used to enhance red cell formation and improve platelet counts in patients with FA, accelerates bone maturation and thereby increases BA (Allen, 1996; Lindberg et al., 2005). Only one of the two individuals with advanced BA in the present study was taking androgen therapy (danazol). Interestingly, almost all of the individuals with delayed BA in our cohort were also receiving danazol, suggesting the possible presence of a yet unidentified strong intrinsic or extrinsic factor delaying BA.
Various factors cause a delay in BA, including reduced sex hormones, hypothyroidism, malnutrition, chronic illness, GHD, and treatment with corticosteroids (Martin et al., 2011; Petryk et al., 2015). Half of the delayed BA group in the present cohort also had abnormal testing of the GH axis, and the same two individuals also had abnormal thyroid functions (subclinical and overt hypothyroidism) suggesting these may be contributing factors to the delay in bone maturation seen in these patients.
Of interest it was noted that three male individuals (13.0% of the study cohort) had abnormal fusion of the same two carpal (triquetral and lunate) bones. Whether or not this lunotriquetral coalition is merely an incidental finding or a finding associated with FA requires further investigation, as lunotriquetral coalition has been documented in the general population but its true population incidence is unknown (Gottschalk, Danilevich, & Gottschalk, 2016). No documented association between carpal coalition and FA could be found in the literature.
We considered the cross‐sectional study design and small sample size of 24 patients with FA, homozygous for the c.637_643del (p.Tyr213Lysfs*6) mutation in FANCG, to be the main identified limitations of our study. A more accurate assessment of the frequency and nature of endocrine abnormalities in patients with FA of this genotype could be achieved by assessing a larger study size, in conjunction with a prospective study design; however, this may prove to be difficult due to the rarity of this disease. Despite study limitations, the present study is the first endocrine profiling study on patients with FA, who are homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG. In addition, the present study provides insight into specific FA genotype‐endocrine phenotype correlations, which provides useful information tailored to the South African population.
Our study confirms that endocrine abnormalities (including abnormal IGF‐1/IGFBP‐3, insulin resistance, abnormal thyroid functions, and short stature) occur at a high frequency (70.8%) in patients with FA, homozygous for a founder mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG. Short stature (45.8%), abnormal IGF‐1/IGFBP‐3 (25.0%), insulin resistance (41.7%), and abnormal thyroid functions (hypothyroidism and subclinical hypothyroidism) (16.7%) were all documented. The total frequency of endocrine abnormalities in the present cohort closely resembled the frequency identified in Giri et al.’s (2007) cohort (73%), although the observed frequencies of the individual endocrine abnormalities differ somewhat, suggesting possible genotype‐phenotype correlations. Genotype‐phenotype correlations for the seven base‐pair founder deletion mutation (c.637_643del (p.Tyr213Lysfs*6)) in FANCG possibly include lower frequencies of GHD, overt hypothyroidism and impaired fasting glucose. Based on the frequencies of endocrine abnormalities observed in the present cohort, and given the resource limitations in the South Africa state healthcare sector, it would be a pragmatic recommendation that baseline fasted thyroid function (FT4 and TSH), glucose and insulin levels, and IGF‐1 and IGFBP‐3 levels be performed at the time of diagnosis. However, where financially possible, it would be more appropriate to base screening guidelines for endocrine abnormalities (even for this FANCG cohort of patients) on the recommendations provided by FARF. Due to the cross‐sectional nature of the present study, timing intervals for follow‐up testing cannot be commented on. Pubertal assessment (through Tanner staging), and growth measurements (including growth velocity) should be assessed at baseline and at regular intervals (at least six monthly) thereafter. BA should be monitored in response to growth velocity concerns, at an interval advised by a pediatric endocrinologist. Patients shown to have abnormalities would require review by a pediatric endocrinologist such, that appropriate therapy can be instituted.
CONFLICT OF INTEREST
The authors declare no conflicts of interest in this work.
AUTHORS’ CONTRIBUTION
CF conceived the study topic. BD developed and wrote the study protocol, was involved in participant recruitment, data collection, analysis of data and research write‐up, under the supervision of CF, AK and DS. JDP and DR assisted with participant identification and recruitment. RW and JP assisted with participant identification.
ACKNOWLEDGMENTS
The authors thank Lindiwe Lamola and Elzette Nienaber for translation of study documents; Professor Jennifer Kromberg for reviewing the study protocol; Engela Honey for assistance in initiating collaboration; staff from the Epidemiological Data Centre of The University of the Witwatersrand for statistical assistance; and the project funders (AstraZeneca Pharmaceuticals (Pty) Ltd (Research Trust Grant), The Colleges of Medicine of South Africa Phyllis Knocker/Bradlow Award (2016), NHLS Research Trust Grant, and The University of the Witwatersrand Faculty Research Committee Individual Research Grant (2016)). Lastly, the authors thank the patients and their families for their willingness to participate, without whom this research would not be possible.
Dillon B, Feben C, Segal D, et al. Endocrine profiling in patients with Fanconi anemia, homozygous for a FANCG founder mutation. Mol Genet Genomic Med. 2020;8:e1351 10.1002/mgg3.1351
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon reasonable request from the corresponding author.
REFERENCES
- Allen, D. B. (1996). Growth suppression by glucocorticoid therapy. Endocrinology and Metabolism Clinics of North America, 25(3), 699–717. 10.1016/S0889-8529(05)70348-0 [DOI] [PubMed] [Google Scholar]
- American Diabetes Association . (2019). Standards of medical care in diabetes – 2019 position statement. Diabetes Care, 37(1), 11–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carel, J. C. , & Léger, J. (2008). Precocious puberty. NEJM., 358(22), 2366–2377. 10.1056/NEJMcp0800459 [DOI] [PubMed] [Google Scholar]
- Elder, D. A. , D’Alessio, D. A. , Eyal, O. , Mueller, R. , Smith, F. O. , Kansra, A. R. , & Rose, S. R. (2008). Abnormalities in glucose tolerance are more common in children with Fanconi anemia and associated with impaired insulin secretion. Pediatric Blood & Cancer, 51(2), 256–260. 10.1002/pbc.21589 [DOI] [PubMed] [Google Scholar]
- Emmaneul, M. , & Bokor, B. (2019).Tanner stages. NCBI StatPearls. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK470280/ [PubMed]
- European Society of Paediatric Radiology . (2019). Bone age for chronological age determination.Recommendation from the ESPR musculoskeletal task force group. Vienna. Retrieved from http://www.espr.org/ [DOI] [PubMed]
- Feben, C. , Kromberg, J. , Wainwright, R. , Stones, D. , Poole, J. , Haw, T. , & Krause, A. (2015). Hematological consequences of a FANCG founder mutation in Black South African patients with Fanconi anemia. Blood Cells, Molecules, & Diseases, 54(3), 270–274. 10.1016/j.bcmd.2014.11.011 [DOI] [PubMed] [Google Scholar]
- Feben, C. , Kromberg, J. , Wainwright, R. , Stones, D. , Sutton, C. , Poole, J. , … Krause, A. (2014). Phenotypic consequences in black South African Fanconi anemia patients homozygous for a founder mutation. Genetics in Medicine, 16(5), 400–406. 10.1038/gim.2013.159 [DOI] [PubMed] [Google Scholar]
- Feben, C. , Wainstein, T. , Kromberg, J. , Essop, F. , & Krause, A. (2018). Fanconi anaemia in South Africa: Past, present and future. South African Medical Journal, 108(5), 393–398. 10.7196/SAMJ.2018.v108i5.13004 [DOI] [PubMed] [Google Scholar]
- Garber, J. , Cobin, R. , Gharib, H. , Hennessey, J. , Klein, I. , Mechanick, J. , … Woeber, K. (2012). Clinical practice guidelines for hypothyroidism in adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid, 22(12), 1200–1234. 10.1089/thy.2012.0205 [DOI] [PubMed] [Google Scholar]
- Garcia‐Higuera, I. , Taniguchi, T. , Ganesa, S. , Meyn, M. S. , Timmers, C. , Hejna, J. , … D’Andrea, A. D. (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular Cell, 7(2), 249–262. 10.1016/S1097-2765(01)00173-3 [DOI] [PubMed] [Google Scholar]
- GH Research Society . (2000). Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: Summary statement of the GH Research Society. Journal of Clinical Endocrinology and Metabolism, 85(11), 3990–3993. 10.1210/jcem.85.11.6984 [DOI] [PubMed] [Google Scholar]
- Giri, N. , Batista, D. L. , Alter, B. P. , & Stratakis, C. A. (2007). Endocrine abnormalities in patients with Fanconi anemia. Journal of Clinical Endocrinology and Metabolism, 92(7), 2624–2631. 10.1210/jc.2007-0135 [DOI] [PubMed] [Google Scholar]
- Gottschalk, M. B. , Danilevich, M. , & Gottschalk, H. P. (2016). Carpal coalitions and metacarpal synostoses: A review. HAND, 11(3), 271–277. 10.1177/1558944715614860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govender, D. , & Goodier, M. (2018). Bone of contention: The applicability of the Greulich‐Pyle method for skeletal age assessment in South Africa. South African Journal of Radiology, 22(1), a1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich, W. W. , & Pyle, S. I. (1950). Radiographic atlas of skeletal development of the hand and wrist (pp. 30–33). London, UK: Oxford University Press. [Google Scholar]
- Hall, J. G. , Allanson, J. E. , Gripp, K. W. , & Slavotinek, A. M. (2007). Handbook of physical measurements (5th ed., pp. 24–81). New York, NY: Oxford University Press. [Google Scholar]
- Labadarios, D. , Mchiza, Z. J. R. , Steyn, N. P. , Gericke, G. , Maunder, E. M. W. , Davids, Y. D. , & Parker, W. (2011). Food security in South Africa: A review of national surveys. Bulletin of the World Health Organization., 89, 891–899. 10.2471/BLT.11.089243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Sipple, J. , Maynard, S. , Mehta, P. A. , Rose, S. R. , Davies, S. M. , & Pang, Q. (2012). Fanconi anemia links reactive oxygen species to insulin resistance and diabetes. Antioxidants & Redox Signaling, 17(8), 1083–1098. 10.1089/ars.2011.4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg, M. K. , Vandenput, L. , Movèrare Skrtic, S. , Vanderschueren, D. , Boonen, S. , Bouillon, R. , & Ohlsson, C. (2005). Androgens and the skeleton. Minerva Endocrinologica, 30(1), 15–25. [PubMed] [Google Scholar]
- Marshall, W. A. , & Tanner, J. M. (1969). Variations in the patterns of pubertal changes in girls. Archives of Disease in Childhood, 44(235), 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, W. A. , & Tanner, J. M. (1970). Variations in the patterns of pubertal changes in boys. Archives of Disease in Childhood, 45(239), 13–23. 10.1136/adc.45.239.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. , Wit, J. , Hochberg, Z. , Sävendahl, L. , van Rijn, R. , Fricke, O. , … Ranke, M. (2011). The use of bone age in clinical practice – Part 1. Hormone Research in Paediatrics, 76, 1–9. 10.1159/000329372 [DOI] [PubMed] [Google Scholar]
- Meetei, A. R. , Levitus, M. , Xue, Y. , Medhurst, A. L. , Zwaan, M. , Ling, C. , … Joenje, H. (2004). X‐linked inheritance of Fanconi anemia complementation group B. Nature Genetics, 36(11), 1219–1224. 10.1038/ng1458 [DOI] [PubMed] [Google Scholar]
- Mehta, P. A. , & Tolar, J. (2018).Fanconi anemia. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1401/
- Morgan, N. V. , Essop, F. , Demuth, I. , de Ravel, T. , Jansen, S. , Tischkowitz, M. , … Mathew, C. G. (2005). A common Fanconi anemia mutation in black populations of sub‐Saharan Africa. Blood, 105(9), 3542–3544. 10.1182/blood-2004-10-3968 [DOI] [PubMed] [Google Scholar]
- Morrell, D. , Chase, C. L. , Kupper, L. L. , & Swift, M. (1986). Diabetes mellitus in ataxia‐telangiectasia, Fanconi anemia, xeroderma pigmentosum, common variable immune deficiency, and severe combined immune deficiency families. Diabetes, 35, 143–148. 10.2337/diab.35.2.143 [DOI] [PubMed] [Google Scholar]
- Pang, T. , Atefy, R. , & Sheen, V. (2008). Malformations of cortical development. Neurologist., 14(3), 181–191. 10.1097/NRL.0b013e31816606b9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk, A. , Kanakatti Shankar, R. , Giri, N. , Hollenberg, A. N. , Rutter, M. M. , Nathan, B. , … Rose, S. R. (2015). Endocrine disorders in Fanconi anemia: Recommendations for screening and treatment. Journal of Clinical Endocrinology and Metabolism, 100(3), 803–811. 10.1210/jc.2014-4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, S. R. , Myers, K. C. , Rutter, M. M. , Mueller, R. , Khoury, J. C. , Mehta, P. A. , … Davies, S. M. (2012). Endocrine phenotype of children and adults with Fanconi anemia. Pediatric Blood & Cancer, 59, 690–696. 10.1002/pbc.24095 [DOI] [PubMed] [Google Scholar]
- Taniguchi, T. , & D’Andrea, A. D. (2006). Molecular pathogenesis of Fanconi anemia: Recent progress. Blood, 107(11), 4223–4233. 10.1182/blood-2005-10-4240 [DOI] [PubMed] [Google Scholar]
- The Rockefellar University Fanconi Anemia Mutation Database. (2019). Retrieved from http://www2.rockefeller.edu/fanconi/
- Tipping, A. J. , Pearson, T. , Morgan, N. V. , Gibson, R. A. , Kuyt, L. P. , Havenga, C. , … Mathew, C. G. (2001). Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population in South Africa. Proceedings of the National Academy of Sciences of the United States of America, 98(10), 5734–5739. 10.1073/pnas.091402398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz, F. , Hanenberg, H. , Schuster, B. , Barker, K. , Wiek, C. , Erven, V. , … Mathew, C. G. (2010). Mutation of the RAD51C gene in a Fanconi anemia‐like disorder. Nature Genetics, 42(5), 406–409. 10.1038/ng.570 [DOI] [PubMed] [Google Scholar]
- Wainstein, T. , Kerr, R. , Mitchell, C. L. , Madaree, S. , Essop, F. B. , Vorster, E. , … Krause, A. (2013). Fanconi anaemia in black South African patients heterozygous for the FANCG c.637‐643delTACCGCC founder mutation. South African Medical Journal, 103(12), 970–973. 10.7196/SAMJ.7215 [DOI] [PubMed] [Google Scholar]
- Wajnrajch, M. P. , Gertner, J. M. , Huma, Z. , Popovic, J. , Lin, K. , Verlander, P. C. , … Auerback, A. D. (2001). Evaluation of growth and hormonal status in patients referred to the international Fanconi anemia registry. Pediatrics, 107(4), 744–754. 10.1542/peds.107.4.744 [DOI] [PubMed] [Google Scholar]
- Wallace, T. M. , Levy, J. C. , & Matthews, D. R. (2004). Use and abuse of HOMA modeling. Diabetes Care, 27(6), 1487–1495. 10.2337/diacare.27.6.1487 [DOI] [PubMed] [Google Scholar]
- World Health Organization . (2008). Training course on child growth assessment. Geneva, Switzerland: World Health Organization. [Google Scholar]
- World Health Organization . (2009). WHO AntroPlus for personal computers manual: Software for assessing growth of the world’s children and adolescents. Geneva, Switzerland: WHO; Retrieved from https://www.who.int/growthref/tools/en/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available upon reasonable request from the corresponding author.