

Abstract
The fibroblast is a key mediator of wound healing in the heart and other organs, yet how it integrates multiple time-dependent paracrine signals to control extracellular matrix synthesis has been difficult to study in vivo. Here, we extended a computational model to simulate the dynamics of fibroblast signaling and fibrosis after myocardial infarction (MI) in response to time-dependent data for nine paracrine stimuli. This computational model was validated against dynamic collagen expression and collagen area fraction data from post-infarction rat hearts. The model predicted that while many features of the fibroblast phenotype at inflammatory or maturation phases of healing could be recapitulated by single static paracrine stimuli (interleukin-1 and angiotensin-II, respectively), mimicking the reparative phase required paired stimuli (e.g. TGFβ and endothelin-1). Virtual overexpression screens simulated with either static cytokine pairs or post-MI paracrine dynamic predicted phase-specific regulators of collagen expression. Several regulators increased (Smad3) or decreased (Smad7, protein kinase G) collagen expression specifically in the reparative phase. NADPH oxidase (NOX) overexpression sustained collagen expression from reparative to maturation phases, driven by TGFβ and endothelin positive feedback loops. Interleukin-1 overexpression had mixed effects, both enhancing collagen via the TGFβ positive feedback loop and suppressing collagen via NFκB and BAMBI (BMP and activin membrane-bound inhibitor) incoherent feedforward loops. These model-based predictions reveal network mechanisms by which the dynamics of paracrine stimuli and interacting signaling pathways drive the progression of fibroblast phenotypes and fibrosis after myocardial infarction.
Introduction
Wound healing is a complex process that involves a dynamic interplay between inflammatory and reparative signaling. This process is especially important following injury to the heart, where cardiomyocytes are unable to regenerate. Scar formation and the preservation of viable heart muscle are important for continued cardiac function[1]. After myocardial infarction (MI), ACE inhibitors and beta blockers are prescribed to prevent adverse cardiac remodeling and heart failure [2], but the risk of heart failure and cardiac-related death post-MI remains high[3–5]. This is partly because wound healing is a balancing act between clearance of debris and formation of new scar, and the regulators of this dynamic process are not fully understood. Therefore, attempts to find therapeutic targets that allow for adequate collagen expression while avoiding excessive fibrosis have been largely unsuccessful. For example, although increased levels of interleukin-1 (IL1) have been linked to fibrosis and diminished cardiac index post-MI[6], blocking IL1 post-MI does not consistently improve healing and is actually associated with an increased risk of secondary MI [7, 8].
Myocardial infarcts follow a healing process that is similar to that in other organs [9, 10]. There is first an inflammatory phase characterized by extracellular matrix (ECM) breakdown and myocyte necrosis, which lasts around 2 days in rats and 5 days in larger mammals [11]. Then, the reparative phase lasts around 2–5 days in rats (2 weeks in large mammals), during which fibroblasts proliferate, migrate into the wound, differentiate into myofibroblasts, and generate large amounts of collagen I and III and other ECM proteins [11, 12]. Ultimately, the wound matures into a stable scar with balanced ECM production and degradation. In the adult heart, cardiomyocytes do not proliferate sufficiently to re-populate the wound, so the ultimate fate of cardiac tissue depends on the behavior of cardiac fibroblasts. Excessive degradation can lead to ventricular dilation and wall rupture due to the loss of structural integrity in the heart wall [13]. Conversely, excessive ECM deposition, particularly in myocardium remote from the infarct, can lead to diastolic dysfunction [14, 15]. Many patients with heart failure post-MI have both dilation and fibrosis [1].
A beneficial infarct healing process likely involves a transient burst of collagen deposition that replaces lost cardiomyocytes with strong ECM without a sustained increase in ECM synthesis that leads to adverse remodeling [16]. This “transient fibrosis” is likely facilitated by many different factors including inflammatory cell phenotype and number, the pre-infarct signaling state, the size of the infarct, and the health of the remaining cardiac vessels [17, 18]. Fibroblasts play a prominent role throughout the entire wound healing process, and therefore present a good system for studying how cells respond to the dynamic signaling environment of wound healing[19]. Additionally, understanding how fibroblasts respond during the different phases of wound healing could identify mechanisms by which fibrosis develops in other organs [“Cardiac Fibroblast Diversity in Health and Disease” Rossi, Matrix Biology Special Issue on Fibroblasts].
Myocardial infarct healing is notoriously difficult to investigate because it involves many dynamic and interacting signaling processes [Cardiac Fibroblast Heterogeneity after Myocardial Infarction” Lindsey, Matrix Biology Special Issue on Fibroblasts]. Fibroblasts are particularly difficult to study in situ during wound healing because they can differentiate from many different cell types and there is no clear consensus on fibroblast markers [20]. Computational modeling has been useful for investigating complex dynamic processes in many areas of biology. Although models have been constructed to study the wound healing process post-MI [21], no such model has yet been applied to study fibroblast intracellular signaling and phenotypic changes during myocardial wound healing [20, 22]. This study integrates a large-scale computational model of cardiac fibroblast signaling [23] together with a tissue-level model that can predict collagen accumulation post-MI. To simulate post-MI wound healing, we used post-MI experimental data for nine time-dependent paracrine stimuli to identify key paracrine and intracellular drivers of fibroblast phenotype after MI. The simulation of post-MI fibroblast dynamics was validated against experimental in vivo time courses of collagen mRNA and collagen area fraction throughout infarct healing. Next, we applied the computational model to identify simpler conditions with static single or paired paracrine stimuli that induce fibroblast phenotypes that mimic specific phases of post-MI healing as predicted by the dynamic simulations. Virtual overexpression screens predicted both context-independent drivers of collagen synthesis as well as regulators that differentially affect collagen expression in the context of specific paracrine stimuli or phases of post-MI wound healing. Further model simulations generated experimentally testable mechanistic hypotheses for the signaling underlying such context-dependent responses, highlighting the utility of this computational model for interrogating the complex roles of fibroblasts in the post-MI wound healing environment.
Methods
Updates to Network Model of Fibroblast Signaling
We extended a large-scale computational model of fibroblast signaling [23] to be more relevant for subsequent experimental study of fibroblast phenotype dynamics in vivo. As described in previous studies [22, 24], the signaling model was implemented as a system of logic-based differential equations with default normalized reaction and node parameters. Here, the input nodes were separated from their associated ligand, and outputs associated with fibroblast phenotype collagen maturation (e.g. LOX) and myofibroblast differentiation (e.g. contraction) were added [25–32]. See Supplementary Methods for representative equations and additional details. A schematic of the network model highlighting the added interactions and nodes is shown in Figure S1.
Modeling the Dynamics of Post-MI Signaling and Collagen Expression
To simulate a post-MI setting, we used experimental time course data for the nine paracrine signals that act as inputs to the signaling model: IL1, IL6, tumor necrosis factor α (TNFα), angiotensin II (AngII), endothelin-1 (ET1), TGFβ, norepinephrine (NE), platelet-derived growth factor (PDGF), brain natriuretic peptide (BNP; corresponding node in the model is natriuretic peptide or NP). Given the known differences in the timing of infarct healing between species [33], all experimental data used to define paracrine inputs were obtained from studies of myocardial infarction in rats [34–41]. We used data on protein levels from the infarct tissue when available (AngII, PDGF), but otherwise we included data such as mRNA levels from the infarct tissue (IL1β, TNFα, NP, ET1), mRNA levels from whole heart samples (TGFβ), or protein levels in plasma (AngII, NE). Data used for Figure 1 and additional studies used to inform the general shape of the paracrine input timecourses are tabulated in Supplementary File 1.
Figure 1: Computational model of post-MI fibroblast dynamics.
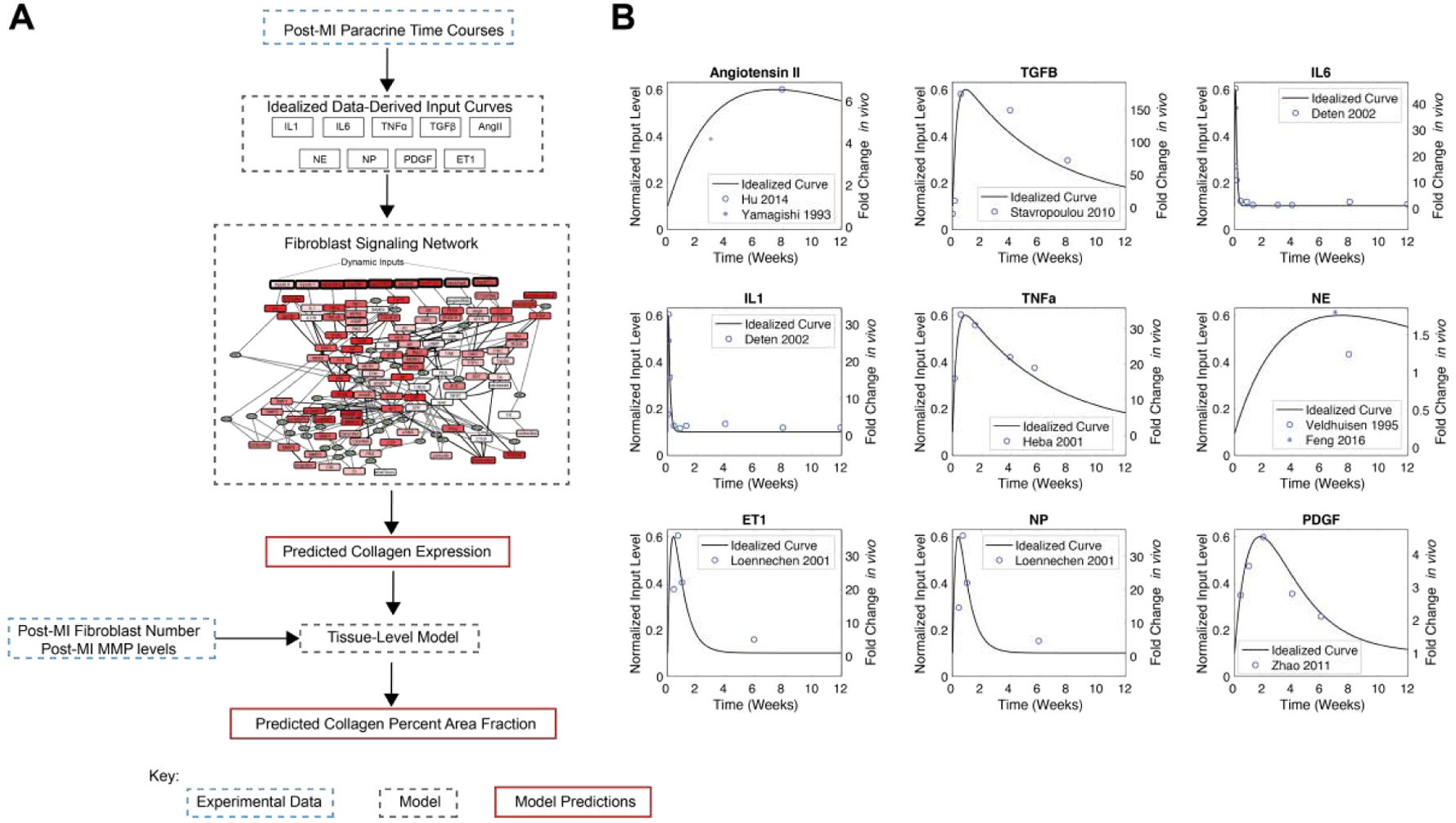
A) Schematic of coupled model of post-MI fibroblast dynamics, incorporating dynamic paracrine stimuli, a fibroblast signaling network, and tissue-level collagen metabolism. B) Dynamic paracrine stimuli were modeled by fitting idealized bi-exponential curves to post-MI timecourse data from the literature[40, 52–59]. These time-dependent signals provide inputs to the signaling network model. Experimental data were digitized from the indicated sources.
In the logic-based modeling formalism, all inputs and signaling nodes are normalized to a continuous range from 0 to 1. Idealized paracrine input curves were derived by fitting time-dependent bi-exponential curves to the paracrine experimental data and then normalizing these curves from 0.1 to 0.6 (see Supplementary Methods). Similar to our past logic-based model of cardiomyocytes in vivo [66], we chose a low uniform baseline paracrine input of 0.1 and a peak height of 0.6 based on the default EC50 of 0.6 used in the fibroblast model [1]. We reasoned this would lead to approximately half-maximal activation of these pathways after MI, resulting in a moderate level of collagen expression that would enable simulations perturbations that are known to either up-regulate (e.g. β1 integrin, ET1) or down-regulate (e.g. Smad7) collagen. In dynamic post-MI simulations, all nine paracrine input curves were applied together in the same simulation (Figures 2, 3, 5, 6, S2, S3, S4, S7, S8). The mechanical input was maintained at a level of 0.6 throughout the post-MI simulation, because mechanical stretch has been shown to be approximately constant throughout the infarct healing time course in rats, likely due to the fact that increasing infarct stiffness is offset by decreasing infarct thickness [42, 43].
Figure 2: Modeling of post-MI fibroblast signaling reproduces dynamics of post-MI collagen expression and deposition.
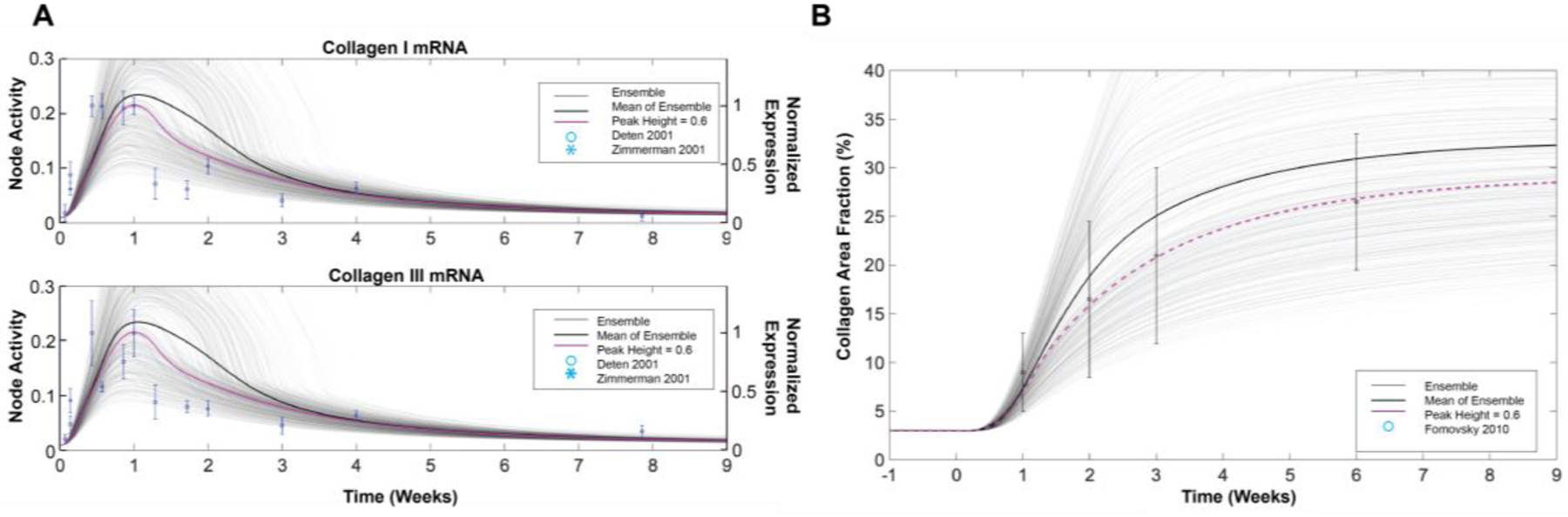
A) Validation of the predicted timing of collagen expression post-MI against experimental data from rat infarcts. B) Validation of predicted collagen accumulation (area fraction) post-MI from the tissue-level model. Experimental data were digitized from [37, 51].Model predictions are shown for the default model which has a uniform paracrine input peak height, along with an ensemble of 500 simulations in which the paracrine peak heights were randomly varied.
To test the impact of biological variation and uncertainty in paracrine inputs on post-MI fibrosis, we further performed population ensemble simulations in which the peak height of all paracrine input curves was randomly sampled independently from a normal distribution with a mean of 0.6 and a standard deviation of 0.05. Ensemble predictions from 500 such simulations is shown as light gray lines in Figure 2.
To predict how single-cell changes in collagen expression lead to tissue-level changes in percent collagen area fraction, we adapted a previously published model of tissue-level collagen accumulation dynamics [44] and coupled it to the signaling model described above. The tissue-level model integrates time-dependent MMP activity and fibroblast numbers derived from previous studies of rat infarcts. Additionally, the collagen I and III mRNA levels predicted by the signaling model are used to drive collagen protein expression in the tissue-level model (Figure S2). MMP activity was modeled as a time-dependent function based on data from rat or mouse infarcts [45–48] rather than being defined by predictions from the signaling model, because MMPs are expressed by many cell types post-MI and fibroblast-specific expression does not fully account for cardiac MMP levels [49]. The time course of fibroblast numbers was also based on experimental data from rat infarcts [11]. The degradation and accumulation rate constants for mature collagen in the ECM were optimized such that the predicted collagen mRNA levels from a standard dynamic simulation with uniform peak height of 0.6 would match the experimentally measured magnitude of collagen accumulation (see Figure 2B)[50]. This model is described in more detail in the Supplementary Methods. MATLAB code of the computational model is freely available on GitHub at: https://github.com/saucermanlab/Zeigler_MIdynamics.
We validated the network model predictions of post-MI collagen I and III mRNA dynamics against independent experimental data collected in rat infarcts [37, 51], in order to be consistent with the species used to develop the model and input curves. The collagen accumulation predicted by the tissue-level model was validated against the collagen area fraction measured in rat infarcts [50].
Modeling the Response to Static Paracrine Stimuli and Comparison with post-MI Dynamics
In contrast to modeling post-MI dynamics where all nine dynamic paracrine inputs are applied in the same simulation, some simulations were performed with static level of single or double-paracrine stimuli (Figures 3, 4, S6). A constant stimulus of each single paracrine input and each input pair was set to a normalized level of 0.6, and the simulation was run to steady state. Steady-state predictions in response to static paracrine stimuli were compared to predictions at specific time points from the dynamic post-MI simulations (0 days, 0.5 days, 7 days, and 42 days) that correspond to different phases of infarct healing (pre-infarct, inflammatory, reparative, and maturing, respectively). We used principal component analysis (pca function in MATLAB) on mean-centered columns to compare static simulations and specific time points from the dynamic simulations based on output profiles that characterize the fibroblast phenotype (MMPs, collagens, other ECM proteins, proliferation, contraction, and αSMA). The single- and paired-paracrine static simulations that most closely matched the time points of interest from the dynamic post-MI simulation were identified by calculating their Euclidean distance based on activity levels of all outputs (see Figure S5).
Figure 3: Simplified paracrine stimuli that mimic distinct phases of the post-MI fibroblast phenotype.
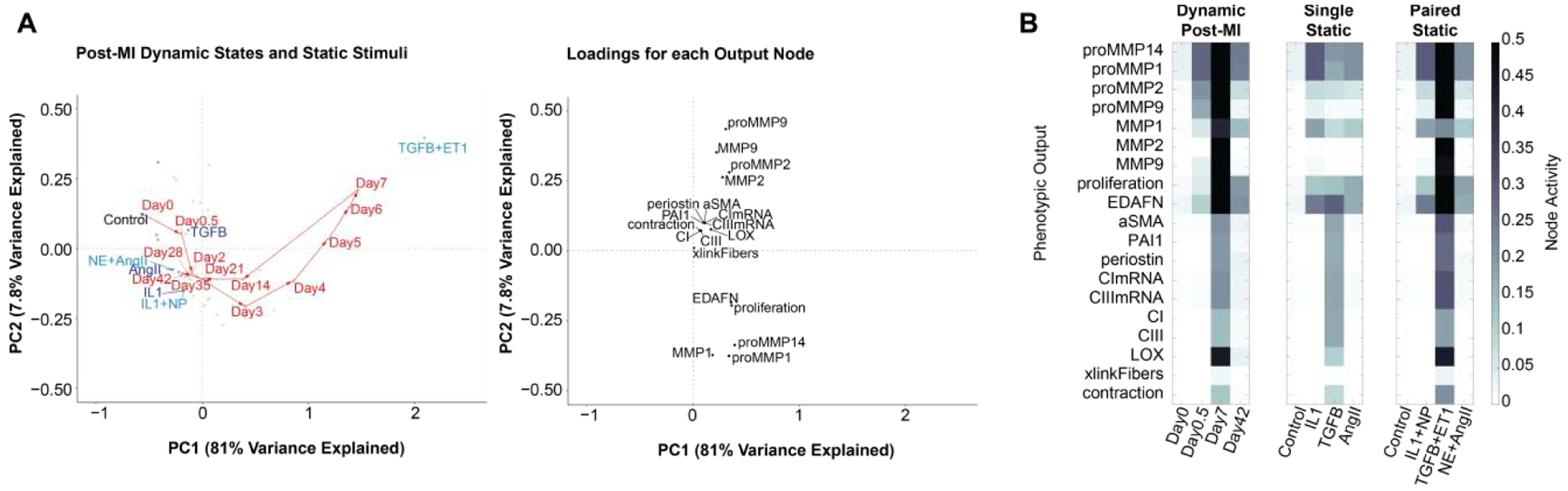
A) Principal component analysis (PCA) to visualize the fibroblast phenotype at specific times post-MI (red circles) or at steady state with 45 static paracrine conditions (representative singles in dark blue, pairs in light blue). B) PCA node loadings show the contribution of each node towards the overall predicted fibroblast phenotype in the first two principal components. C) Activity profile of fibroblast phenotype nodes at selected timepoints from dynamic post-MI simulations (left), or at steady-state with the best matching single (center) or paired (right) paracrine stimuli.
Screen for Intracellular Modulators of Collagen Expression
To identify modulators of collagen expression, we individually simulated overexpression of each node in the network and quantified its effect on the collagen expression nodes. These comprehensive screens were performed under conditions with pairs of static paracrine inputs representative of the different phases of post-MI healing, as well as with the time-dependent post-MI simulation that include nine dynamic paracrine stimuli. Overexpression of a given network node was simulated by increasing its normalized expression parameter (ymax) 10-fold. Change in collagen expression was quantified as the difference between the sum of collagen I mRNA and collagen III mRNA with (ymax 10) or without (ymax = 1) overexpression of that node, at the same timepoint and with the same paracrine stimuli.
Results
Expanding a fibroblast signaling network model to predict post-MI fibroblast phenotype dynamics
A published computational model of cardiac fibroblast signaling network [23] was extended to make it more suited to predict the dynamics of fibroblast phenotype in vivo (see Methods and Figure S1). While the signaling model was originally developed and validated using a wealth of in vitro experimental data [23], we hypothesized that this model could be extended to predict post-MI fibroblast dynamics because it is capable of predicting semi-quantitative time-dependent behavior and it incorporates many of the pathways involved in infarct healing. Thus, we coupled the fibroblast network model with experimentally-based dynamic post-MI paracrine stimuli and a tissue-level model of collagen accumulation (Figure 1A). Timecourses for the nine paracrine model inputs (AngII, TGFβ, IL6, IL1, TNFα, NE, ET1, NP, PDGF) were fit with bi-exponential curves based on published experimental data from rats following myocardial infarction (Figure 1B).
When driven by dynamic post-MI paracrine stimuli, the model predicts a wide range of distinct time-dependent network responses (Figure S3) due to the distinct behaviors of individual stimuli as well as pathway crosstalk and intra-network dynamics (Supplementary File S2). The model predicts transient collagen I and III mRNA expression dynamics that are similar to those observed in independent experimental data from rat infarcts (Figure 2A)[37, 51], and the coupled network and tissue models predicted changes in collagen area fraction consistent with those measured in rat infarcts (Figure 2B)[42]. Further, the model accurately predicts post-MI increases in MMP2, MMP9, EDA-FN, AP1, NFκB, p38, JNK, and ERK (Figure S3), which have been measured in rat infarcts post-MI [60–63]. These results support the model’s ability to predict fibroblast phenotype in these complex signaling environments.
Given the uncertainty and biological variability in the level of paracrine inputs post-MI, we examined how variation in paracrine inputs affected the predicted post-MI fibrosis. Predictions of collagen dynamics were robust to uniform variation in baseline paracrine levels, but more sensitive to variation in peak height (Figure S4). Thus to simulate individual variability, we generated an ensemble of paracrine input curves where each peak height was individually randomly sampled. Individual simulations predicted variable collagen expression and fibrosis, although the mean ensemble predictions were qualitatively similar to those resulting from using a uniform peak height (Figure 2). Final predicted collagen area fraction correlated with TGFβ levels across ensemble simulations, but was not sensitive to small variations in other inputs (Figure S4B). Thus, idealized input curves from 0.1 to 0.6 were used in subsequent Figures.
Relationship between fibroblast phenotypes induced by dynamic post-MI vs. static paracrine stimuli
In vitro experiments can provide precise control of simplified environmental conditions, but it is unclear to what extent such conditions can reproduce the phenotype of fibroblasts during the more dynamic and complex process of infarct healing [64, 65]. Therefore, we used the model to identify individual or pairs of static paracrine stimuli that drive phenotypes that best mimic distinct phases of the post-MI fibroblast phenotype. In contrast to the dynamic simulations described above, the fibroblast model was run to steady-state under stimulation with static levels of the nine paracrine stimuli as well as all pairwise combinations (Figure S5). Using principal component analysis, we visualized the phenotypic relationship between fibroblasts stimulated by these 45 static paracrine conditions and representative time points in the post-MI fibroblast phenotype time course (Figure 3 and Figure S5). TGFβ stimulation had a very distinct effect on fibroblast phenotype, correlating with node loadings of pro-fibrotic outputs such as periostin, collagens, and αSMA near the positive PC2 axis of Figure 3 and positive PC3 axis of Figure S5. In contrast, the node loadings for MMP1/MMP14/proliferation and MMP2/MMP9 groups did not strongly associate with any single stimulus and had distinct node loadings on PC2 and PC3.
We next examined the extent to which stimulation with one or two static paracrine factors mimicked fibroblast phenotypes at specific times post-MI. Such analysis can identify paracrine signals that drive specific phases of the post-MI fibroblast phenotype. The inflammatory phase (0.5 days post-MI) was fairly well mimicked by static IL1 (Figure 3A), which predicted proliferation, EDAFN, MMP1, and MMP14 expression but insufficient proMMP2 or proMMP9 (Figure 3C). Day 0.5 could also be mimicked fairly well by AngII or ET1, but pairs of cytokines did not noticeably further improve predictions of MMP2/MMP9 activity. In contrast, the period we defined as reparative signaling at 7 days post-MI was not well mimicked by any single stimulus (Figure 3A), highlighting the signaling complexity of this phase (Figure S3 and Figure S4). TGFβ mimicked aspects of the 7 day post-MI fibroblast phenotype (e.g. expression of collagens I/III, αSMA, periostin), consistent with previous studies showing that mouse fibroblasts at intermediate stages of post-MI healing are most similar to fibroblasts stimulated by TGFβ [64]. However, TGFβ was predicted to be insufficient to fully mimic the post-MI expression of multiple MMPs, proliferation, LOX, and collagen fiber cross-linking. Combination of TGFβ with ET1 significantly improved predictions of MMP activity and fibroblast proliferation (Figure 3C). The late (42 day) time point was mimicked well with AngII or ET1, and there was no significant improvement with paired stimuli such as NE+AngII. The paracrine pairs that induced phenotypes most similar to post-MI day 0.5, 7, and 42 fibroblasts are consistent with their elevation at those times post-MI as shown in Figure 1. Overall, these model predictions indicate that early (day 0.5) and late (day 42) stages of the post-MI fibroblast phenotype are more readily mimicked using single static stimuli, but intermediate post-MI time points (day 7) involve many interacting stimuli and a complex network state that is more challenging to mimic in vitro.
Predicting post-MI phase-specific regulators of fibroblast phenotype
Identification of regulators that drive distinct fibroblast phenotypes would provide a better understanding of the complex transitions that occur in vivo and may lead to potential phase-targeted therapeutics with reduced side effects. We simulated a virtual screen for regulators of collagen I/III mRNA expression by overexpressing each node in the context of paired paracrine static stimuli that best mimic the inflammatory (IL1+NP), reparative (TGFβ+ET1), and maturation (NE+AngII) post-MI phases, as well as the control condition representative of the pre-infarct environment (Figure 4). For each node, overexpression was simulated by increasing that node’s normalized expression parameter 10-fold. Several nodes such as β1 integrin pathway members, ET1 pathway members, AP1, and JNK were predicted to drive collagen expression across multiple paracrine contexts.
Figure 4: Modulators of collagen mRNA in the context of static paracrine stimuli that mimic inflammatory (IL1+NP), reparative (TGFβ+ET1) and maturation (NE+AngII) phases.
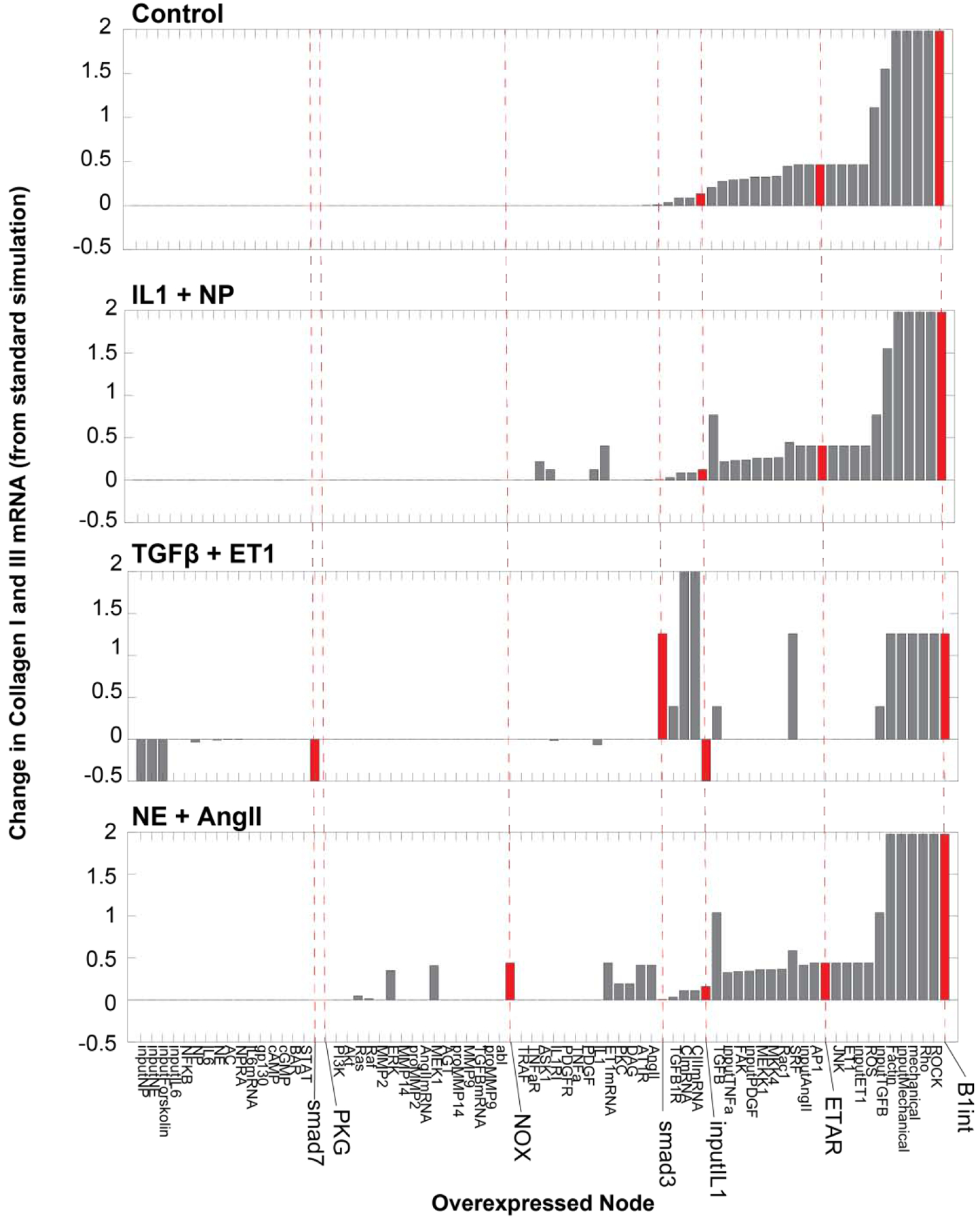
Network nodes were each overexpressed 10-fold (normalized expression parameter ymax = 10) in the context of the indicated static paracrine stimuli (set to 0.6 normalized activity), predicting the change in collagen I and III mRNA compared to no overexpression. Nodes were rank-ordered by their predicted effect on collagen I and III mRNA expression with no paracrine stimulus (Control). Smad3, PKG, NOX, IL1, ETAR and B1int are emphasized for comparison with subsequent simulations of post-MI dynamics. Overexpressed nodes that did not affect collagen mRNA in any condition are not shown.
The overexpression screen also identified a number of context-specific regulators of collagen expression. In the TGFβ+ET1 stimulated condition, overexpression of TGFβ pathway members such as Smad3 enhanced collagen mRNA expression, while Smad7 overexpression suppressed collagen mRNA. However, overexpression of these nodes did not significantly affect collagen mRNA expression in control or IL1+NP contexts. Overexpression of the “input IL1” node moderately increased collagen expression under control or AngII+NP stimulation, but it decreased collagen expression in the TGFβ+ET1 context. This effect was dose-dependent, as overexpression of the “IL1” node itself had smaller effects in the IL1+NP and TGFβ+ET1 conditions.
As static paracrine stimuli only partially mimic the history-dependent effect of the dynamic post-MI paracrine environment (Figure 3), it is unknown to what extent intracellular regulators of fibroblast phenotype identified under simplified conditions in vitro can explain their role in vivo. To identify post-MI phase-specific regulators, we simulated post-MI paracrine dynamics using the idealized paracrine input curves described above, and performed a virtual overexpression screen of the 106 network nodes. The activity time courses for each of the overexpressed nodes in each of these 106 simulations are shown in Figure S6. Node overexpression induced a diverse range of phase-specific effects on the post-MI time courses of collagen I/III mRNA expression (Figure 5). Overexpression of some nodes was predicted to induce sustained collagen mRNA expression even before MI, such as TGFβ, ETAR, JNK, and β1-integrin pathway members. These context-independent nodes had an effect on collagen expression similar to that identified under static control conditions (Figure 4). Some regulators (e.g PKG) had the strongest effect on collagen expression during the reparative phase when collagen mRNA expression peaks. Many regulators, such as NOX, not only increased collagen mRNA in the reparative phase but sustained it into many weeks post-MI. Other regulators had dichotomous effects, such as input IL1, which enhanced collagen mRNA during the inflammatory and late phases but decreased collagen during the reparative phase. Conversely, NFκB, IL6 and Akt overexpression modestly suppressed early and late collagen mRNA expression but enhanced collagen mRNA expression at intermediate timepoints.
Figure 5: Post-MI overexpression screen to identify phase-specific regulators of collagen mRNA expression.
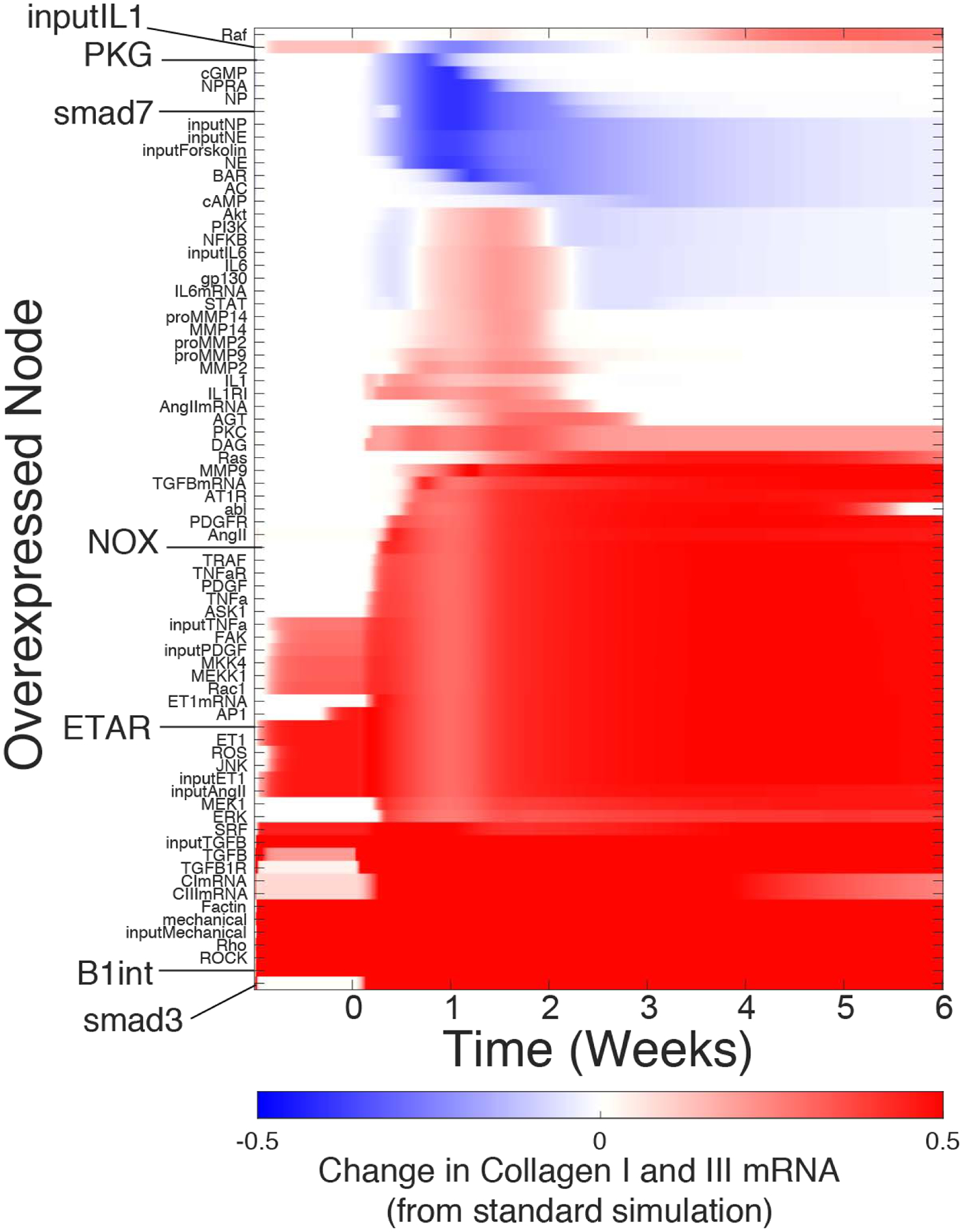
Each row shows the effect of 10-fold overexpression of the indicated node. The predicted change in collagen expression is calculated as [Collagen I mRNA + Collagen III mRNA]Overexpressed.-[Collagen I RNA + Collagen III mRNA]control. overexpressed nodes that did not affect collagen mRNA in any condition are not shown.
Mechanisms contributing to post-MI phase-specific regulation of collagen
The screen above identified pro-fibrotic pathways where the mechanism by which they up-regulate collagen expression has been well-studied. Both moderate and strong overexpression of β1-integrin induced substantial fibrosis even before MI, consistent with its effect across all simplified paracrine contexts (Figure 4, Figure S8) and the continual mechanical stress in the beating heart. Moderate ET1AR overexpression induced strong and sustained collagen expression beginning in the inflammatory phase post-MI, while strong ET1AR overexpression induced fibrosis even in baseline conditions (Figure S8).
However, this in silico screen also identified regulators with more complex relationships to collagen expression. The mechanistic nature of the computational model enables identification of the specific conditions and network mechanisms that underlie phase-specific regulation of collagen expression. For specific influential nodes of interest, we performed additional simulations representing different levels of overexpression or knockdown using the dynamic post-MI model. We used the model’s mechanistic network structure along with sensitivity analysis as described previously [23] (data not shown) to identify simplified network mechanisms that mediate the effects of these notable regulators of collagen expression. For example, the model predicted that overexpression of Smad7 amplifies its activity and decreases collagen mRNA expression only in the reparative phase (1–2 weeks), resulting in decreased collagen area fraction in the infarct throughout post-MI remodeling (Figure 6A). This prediction is consistent with in vitro studies showing Smad7 overexpression decreases collagen expression in vitro[66], as well as our simulations of the simplified TGFβ+ET1 context (Figure 4).
Figure 6: Mechanisms contributing to context-dependent regulators of collagen expression post-MI.
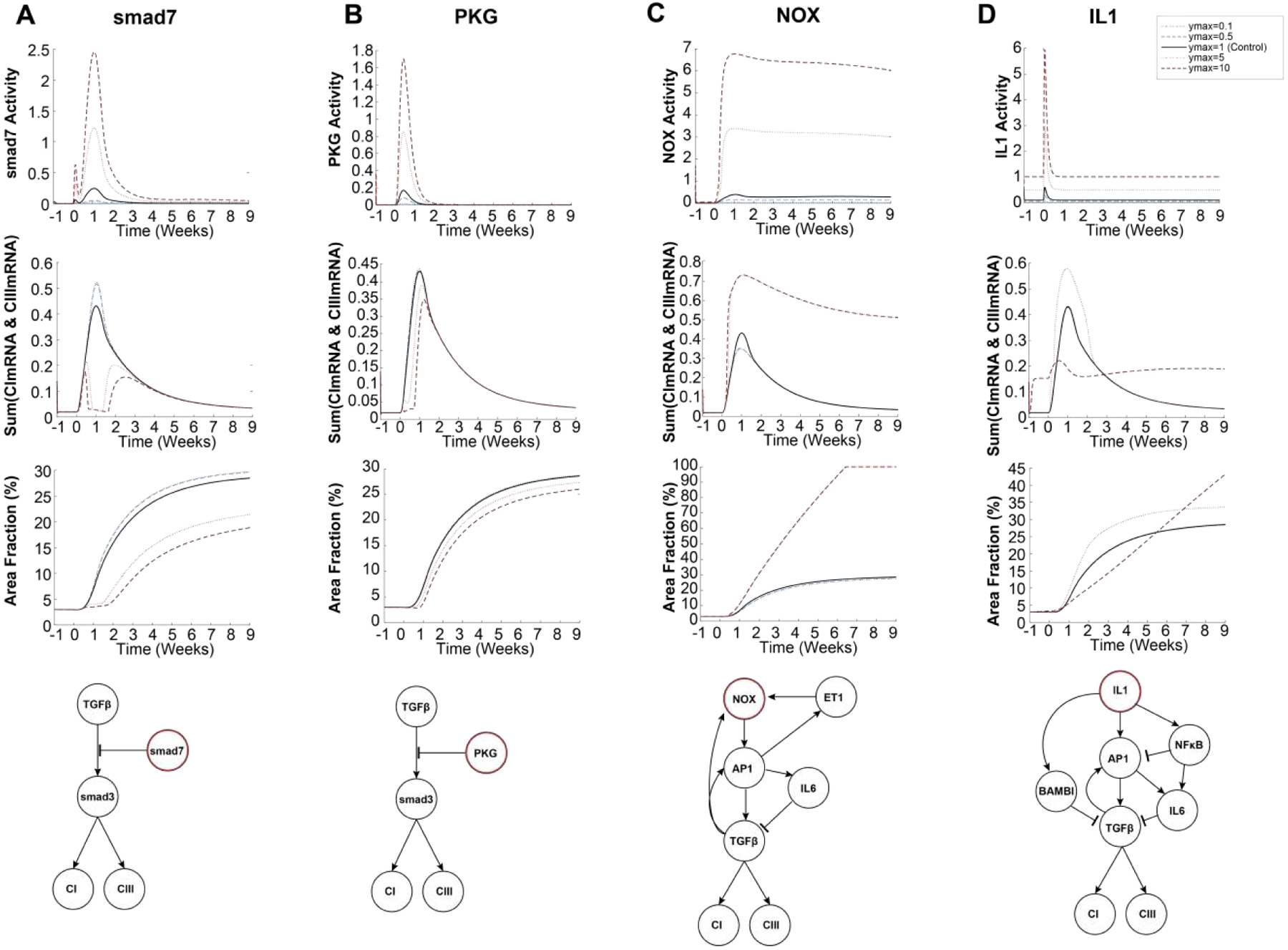
Overexpression or knockdown were simulated by increasing or decreasing the normalized expression parameter (ymax) for Smad7 (panel A), PKG (B), NOX (C), IL1 (D). The resulting post-MI dynamics of that node’s activity, collagen mRNA expression, and collagen area fraction are shown. Simplified schematics indicate the network mechanisms by which these nodes regulate collagen expression.
Like Smad7, PKG was predicted to reduce collagen expression by inhibiting Smad3 activity. However, its inhibitory effect on collagen mRNA was more limited to the first week post-MI due to the transient stimulation by the upstream paracrine NP (Figure 6B). Interestingly, PKG was not predicted as a negative regulator in the static TGFβ+ET1 context (Figure 4), as that context lacked the NP stimulation of PKG activity (Figure S5). In contrast to Smad7 and PKG, Smad3 overexpression strongly enhanced collagen mRNA in the reparative phase with subsequent and sustained increase in collagen area fraction (Figure S7).
NADPH oxidase (NOX) overexpression was predicted to induce sustained NOX activity beginning in the reparative phase post-MI, primarily due to activation of positive feedbacks by AP1-TGFβ and AP1-ET1. The predicted AP1-TGFβ feedback involves both AP1-dependent transcription and MMP-induced activation of latent TGFβ. Together, these feedbacks induced sustained collagen I/III mRNA expression (Figure 6C). NOX also induced partial negative regulation of collagen mRNA via an incoherent feedforward circuit involving AP1-driven expression of IL6 and activation of Smad7. Overall, NOX overexpression induced a strong increase in collagen area fraction after MI.
IL1 exhibited a complex post-MI regulation of collagen expression that was phase- and dose-dependent. Moderate IL1 overexpression amplified collagen mRNA in the reparative phase, driven primarily by the AP1-TGFβ positive feedback through JNK (Figure 6D). In contrast, strong IL1 overexpression negatively regulated collagen mRNA during the reparative phase via 1) inhibition of the TGFβ receptor through the decoy receptor BAMBI (BMP and activin membrane-bound inhibitor; 2) NFκB inhibition of JNK-AP1 signaling; and 3) NFκB stimulation of the IL6/Smad7 pathway. In later phases, sustained collagen mRNA expression was primarily driven by continued activation of the AP1-TFGβ positive feedback loop, resulting in delayed but continually elevating fibrosis post-MI (Figure 6D). IL1 knockdown was not predicted to significantly alter post-MI collagen expression. These predictions are in line with studies that have shown IL1 inhibition is most effective in limiting collagen expression post-MI when levels of inflammatory cytokines are high[8].
Both moderate and strong overexpression of β1-integrin induced substantial fibrosis even before MI, consistent with its effect across all simplified paracrine contexts (Figure 4, Figure S6) and the continual mechanical stress in the beating heart. Moderate ET1AR overexpression induced strong and sustained collagen expression beginning in the inflammatory phase post-MI, while strong ET1AR overexpression induced fibrosis even in baseline conditions (Figure S7).
3.5. Discussion
Cardiac fibroblasts are central mediators of wound healing and cardiac function after myocardial infarction [16, 50, 67], yet the complexity of the dynamic in vivo paracrine environment and the fibroblast intracellular signaling network hinders therapeutic targeting [68]. Here, we extended a large-scale computational model of the fibroblast signaling network to identify paracrine and intracellular drivers of extracellular matrix synthesis in specific phases of post-infarct healing. By integrating experimentally-based post-MI dynamics of nine paracrine stimuli, the model accurately predicted the dynamics of collagen expression as validated against experimental data from rat infarcts. The model was applied to screen for drivers of collagen expression in the context of static paracrine stimuli representative of specific post-MI phases, as well as the more complex dynamic post-MI paracrine environment. These virtual overexpression screens identified post-MI phase-specific regulators such as PKG and Smad7, which suppress collagen expression in the reparative phase via inhibition of Smad3; NOX, which sustained fibrosis in reparative and late phases via AP1-TGFβ feedback; and IL1, which induced mixed enhancement and suppression of collagen expression. This study highlights how fibroblast signaling pathways coordinate to dynamically control matrix synthesis during wound healing, and invites further investigation into therapeutics that target fibroblasts at specific post-MI phases.
Loci of control in the fibroblast signaling network
The complex, concomitant dynamics of many signaling pathways and cell types during infarct healing has made it difficult to experimentally identify and therapeutically target the drivers of collagen production[52, “Master Regulators of Fibroblast Cell State” Davis][Matrix Biology Special Issue on Fibroblasts]. In particular, studies that have performed fibroblast-specific genetic perturbations in the heart have emerged only recently[69]. Computational modeling enabled a detailed mechanistic investigation into the determinants of collagen production by integrating many sources of experimental data into a single framework. TGFβ is well recognized as a primary driver of fibrosis, as seen in both model predictions and experiments of fibroblast-specific TGFβ receptor [70]. However targeting TGFβ itself with treatments such as TGFβ-blocking antibodies decreased the collagen necessary for post-MI wound healing, resulting in increased cardiac dilation and mortality [71]. The model predicted that Smad7 overexpression decreases collagen expression during the reparative phase, which is consistent with previous studies that found decreased collagen expression in fibroblasts after overexpressing Smad7 in vitro [71] or using ultrasound-mediated cardiac gene transfer after AngII-induced cardiac remodeling in mice [72]. The prediction that PKG overexpression reduces collagen expression is consistent with a previous study showing that protein delivery of its upstream regulator NP decreases post-MI fibrosis [73]. The literature-based fibroblast network model predicts that both Smad7 and PKG affect collagen expression via mechanisms that inhibit Smad3 [66, 74]. Further, Smad3 knockdown was predicted to decrease post-MI fibrosis, consistent with previous studies with global [75] or fibroblast-specific Smad3 deletion [76]. Overall, these simulations indicate that Smad3 is a critical target for negative regulators of collagen expression in cardiac fibroblasts, which could direct future studies into other inhibitors of Smad3 activation as a way to modulate excessive fibrosis.
Inflammation is strongly associated with post-MI fibrosis, but therapeutic targeting has had mixed effects that indicate context-dependent roles. Higher pre-infarct levels of inflammation, high post-infarct levels of inflammatory mediators, and longer duration of inflammation are associated with an increase in fibrosis and a decrease in cardiac function post-MI[6, 8, 68, 77]. However, the use of broad anti-inflammatory treatments such as NSAIDs or corticosteroids as well as therapies targeted to specific inflammatory mediators worsened outcomes post-MI[7, 13, 17, 68, 78–81]. While IL1 receptor inhibitors (e.g. Anakinra or canakinumab) consistently decrease post-MI inflammatory markers, they have been shown to have more modest or subgroup-dependent effects on long-term incidence of cardiovascular events [7, 79, 82]. Sano et al recently showed that mice with TET2 deletion in hematopoietic cells exhibited enhanced IL1 secretion and post-MI fibrosis, along with greater responsiveness to inhibition of IL1 secretion compared to wildtype mice [8]. These studies are consistent with our model predictions that perturbations to IL1 have dose- and time-dependent effects on post-MI fibrosis.
Network analysis predicted mechanisms that may mediate context-dependent roles of IL1. Moderate IL1 overexpression increased collagen expression in the reparative phase, while high IL1 overexpression increased longer-term collagen expression- both via enhanced AP1 signaling. In contrast, the model predicted that high IL1 attenuated TGFβ-induced collagen expression in the reparative phase due to a combination of NFκB stimulation of IL6, NFκB inhibition of AP1, and inhibition of TGFβR through upregulation of BAMBI. IL1 has previously been shown to up-regulate fibroblast expression of IL6, which has been shown to decrease collagen synthesis[83]. Examination of both bulk RNA-seq from fibroblasts post-MI and single-cell RNA-seq from Wnt-expressing fibroblasts post-MI shows increased expression of BAMBI post-MI [19, 84]. In isolated cardiac fibroblasts, IL1 attenuated TGFβ-induced collagen and αSMA expression, which was associated with increased BAMBI expression [85]. Further, global knockout of BAMBI enhanced pro-fibrotic responses of isolated cardiac fibroblasts stimulated with TGFβ, or in vivo after transverse aortic constriction [86]. Additional experiments are needed to validate whether fibroblast-specific perturbations to NFκB or BAMBI modulate post-MI fibrosis.
NOX is downstream of TGFβ, ET1, and AngII, which both our model predictions and previous experiments demonstrate to enhance cardiac fibrosis[87, 88]. Previous studies found that overexpression of NOX in cardiomyocytes induced cardiac fibrosis [89], and inhibiting NOX with hydrogen sulfide attenuated collagen expression post-MI [90]. Cardiac fibroblast-specific inhibition or overexpression of NOX in vivo has not been reported.
MAP kinases are well appreciated as mediators of cytokine signaling in fibroblasts [91], but the specific roles for different MAPK classes and isoforms have not been fully elucidated. The model predicted that overexpression of several nodes including IL1β, NOX, and ET1 can induce fibrosis via AP1-TGFβ positive feedback. Consistent with this prediction, the AP1 component c-Jun is expressed in a wide range of human fibrotic diseases (including heart), c-Jun overexpression induces multi-organ fibrosis in mice, and TGFβR inhibition blocks c-Jun induced fibroblast migration [92]. AP1 has been shown to be a major mechanism by which both inflammatory cytokines such as TNFα or pro-fibrotic hormones such as AngII induce TGFβ expression in cultured mesenchymal stem cells[93, 94], lung fibroblasts, and our previous modeling and experiments with mechano-activated cardiac fibroblasts [23, 92]. The model predicted that, post-MI, AP1 activity was primarily driven by c-Jun N-terminal kinase (JNK), although ERK can also regulate c-Jun. This prediction is consistent with experiments where pharmacologic inhibition of JNK or ERK mitigated fibrosis and dilated cardiomyopathy associated with a lamin gene mutation [95, 96]. Thus, the role of AP1-TGFβ feedback, JNK, and ERK could be the focus of further studies that use fibroblast-specific perturbations or test potential therapies post-MI.
Several studies have characterized an important role for p38 in cardiac fibrosis. Some studies in isolated cardiac fibroblasts found p38 inhibition to increase collagen expression in response to AngII [97] or coxsackivirus [98], potentially through p38 inhibition of ERK signaling as reported in human dermal and synovial fibroblasts [99, 100]. However a number of others have shown that pharmacologic p38 inhibition decreases collagen expression in TGFβ-stimulated cardiac fibroblasts [101][102] or pressure overload in vivo [102]. Strikingly, fibroblast-specific knockout of p38 attenuated fibrosis due to AngII or MI, while activation of p38 via MKK6 overexpression induced multi-organ fibrosis and amplified post-MI fibrosis [103]. These distinct effects of p38 may reflect differences between stimuli, species, p38 isoforms, or in vitro/in vivo experimental systems. While the literature-curated fibroblast signaling network contains mechanisms linking p38 to both pro- and anti-fibrotic pathways, the model did not predict p38 overexpression to strongly modulate post-MI fibrosis. Experiments that clarify the context-dependent roles for MAPK pathways could be used to revise and improve predictions of the fibroblast signaling network model [89].
The dynamic model as a tool for investigating wound healing
There is a need for computational tools for investigation of wound healing in order to more fully understand the development of fibrosis and identify potential therapeutic strategies against it [20]. In the heart, early collagen production is important for generating a strong scar [104], but continued production and fibrosis contribute to diastolic dysfunction [1, 16, 105]. It has been shown that the timing of collagen production is important in determining the ultimate health of the heart[80].
The dynamic modeling approach outlined in this study allowed for a direct comparison between specific signaling pathways and the phases of fibroblast phenotype, and this could be a tool for further investigation into cardiac fibrosis. We identified representative stimuli that mimic fibroblast activity in the pre-infarct (control), inflammatory (IL1+NP), reparative (TGFβ+ET1), and mature (AngII+NE) phases of wound healing. The timing of these phases is variable and depends on the species and individual [33, 106]. Therefore, we used model-predicted fibroblast phenotypes to define the phases of healing and identify static stimuli that can induce those phenotypes. These stimuli can be used to design in vitro experiments mimic in vivo fibroblast phenotypes characteristic of specific time points during infarct healing. Future studies could investigate how changing the timing of these signals affect collagen expression. The model could be used to test the effect of potential novel therapeutics on fibroblast phenotype. The modeling of differential expression of nodes in the network invites the possibility of integrating gene expression data to investigate potential genetic causes of adverse wound healing that lead to heart failure, patient-specific variability, or single-cell heterogeneity. Indeed, our simulations of variable paracrine input magnitudes predicted amplified variability in fibrosis for individual simulations, although population means were similar (Figure 2; ensemble versions of Figures 3, 5, and 6 are similar and not shown). Furthermore, the model includes explicit network mechanisms, which guide in vitro experiments to validate promising collagen regulators identified in this study.
Limitations and future directions
The main limitation of this study is that the model predicts post-MI contributions of fibroblasts but not other cell types such as macrophage and cardiomyocytes that can alter cardiac remodeling. The tissue level model only indirectly accounts for the activity of these cells in collagen degradation and expression. Of note, the tissue level model does not account for changes in MMP expression by fibroblasts, as the tissue-level MMP activity that drives collagen degradation is assumed to be largely due to inflammatory cells. Furthermore, processes such as re-vascularization (which can improve infarct healing) or further cardiomyocyte injury (which can re-start or prolong wound healing and induce pathologic remodeling) are not captured by this model. On the molecular level, the model also does not capture how changes in ECM composition affect fibroblast signaling. For example, changes in cross-linking, protease-mediated cleavage, and local binding of proteins to the ECM can all feed back to regulate fibroblast phenotype, often through incompletely understood mechanisms [106, 107]. Despite these limitations, this model provides a framework with which to begin to investigate these additional features of fibroblast signaling and intercellular cross-talk with systems models.
In this study, we demonstrate that by integrating multiple time-dependent paracrine stimuli, the fibroblast network model can predict many features and regulators of post-MI fibroblast signaling and matrix remodeling. These predictions should be validated in vivo, but initial validations may be most practical in vitro with the static paracrine stimuli that we identified to mimic post-MI phases. Further studies could test how variations in the timing of input stimuli affect fibroblast activity or incorporate this model into a multi-scale model that can predict the effect of cell-cell interactions between macrophage and cardiomyocytes. As in many experimental screens and our past modeling of in vivo hypertrophy, here we performed virtual overexpression screens[65]. This approach has advantages of testing whether overexpression is sufficient to modulate fibroblast phenotype and is more readily validated experimentally with transgenic mice or viral vectors. Future studies may more systematically explore simulations of genetic knockouts or small molecule inhibitors. Future in vivo studies will also be necessary to determine how therapeutic modulation of collagen dynamics can be leveraged to optimally improve cardiac remodeling and contractility after myocardial infarction.
Supplementary Material
Highlights.
Computational model predicts in vivo dynamics of fibroblast signaling network and collagen accumulation after myocardial infarction
Many features of fibroblast phenotype after myocardial infarction may be recapitulated by static stimulation with single or pairs of paracrine factors
After myocardial infarction, Smad3, Smad7, protein kinase G, NADPH oxidase, and IL1 induce phase-specific regulation of collagen due to feedforwards and feedbacks
Acknowledgements
We thank Dr. William Richardson for valuable discussion of this work. This work was supported by the National Institutes of Health [grant numbers HL127944, HL137755, HL116449, HL007284]; the National Science Foundation [grant numbers 1560282, 1252854] and the University of Virginia Center for Engineering in Medicine. The funding sources had no involvement in the conduct of the research or decision to publish.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict
The authors have declared that no conflict of interest exists.
References:
- [1].Beltrami CA, Finato N, Rocco M, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation; 89: 151–163. [DOI] [PubMed] [Google Scholar]
- [2].O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of st-elevation myocardial infarction: Executive summary: A report of the American college of cardiology foundation/american heart association task force on practice guidelines. J Am Coll Cardiol 2013. Epub ahead of print 2013. DOI: 10.1016/j.jacc.2012.11.018. [DOI] [PubMed] [Google Scholar]
- [3].Schocken DD, Benjamin EJ, Fonarow GC, et al. Prevention of heart failure: A scientific statement from the American Heart Association Councils on epidemiology and prevention, clinical cardiology, cardiovascular nursing, and high blood pressure research; Quality of Care and Outcomes Research Interdisc. Circulation 2008. Epub ahead of print 2008. DOI: 10.1161/CIRCULATIONAHA.107.188965. [DOI] [PubMed] [Google Scholar]
- [4].Gottdiener JS, Arnold a M, Aurigemma GP, et al. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol; 35: 1628–1637. [DOI] [PubMed] [Google Scholar]
- [5].He J, Ogden LG, Bazzano LA, et al. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med; 161: 996–1002. [DOI] [PubMed] [Google Scholar]
- [6].Orn S, Ueland T, Manhenke C, et al. Increased interleukin-1β levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J Intern Med; 272: 267–276. [DOI] [PubMed] [Google Scholar]
- [7].Morton AC, Rothman AMK, Greenwood JP, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur Heart J 2015. Epub ahead of print 2015. DOI: 10.1093/eurheartj/ehu272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sano S, Oshima K, Wang Y, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J Am Coll Cardiol 2018. Epub ahead of print 2018. DOI: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fishbein MC, Maclean D, Maroko PR. The histopathologic evolution of myocardial infarction. Chest; 73: 843–849. [DOI] [PubMed] [Google Scholar]
- [10].Palatinus JA, Rhett JM, Gourdie RG. Translational lessons from scarless healing of cutaneous wounds and regenerative repair of the myocardium. Journal of Molecular and Cellular Cardiology; 48: 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Virag JI, Murry CE. Myofibroblast and Endothelial Cell Proliferation during Murine Myocardial Infarct Repair. Am J Pathol; 163: 2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chistiakov DA, Orekhov AN, Bobryshev YV. The role of cardiac fibroblasts in post-myocardial heart tissue repair. Experimental and Molecular Pathology; 101: 231–240. [DOI] [PubMed] [Google Scholar]
- [13].Hwang MW, Matsumori A, Furukawa Y, et al. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol; 38: 1546–53. [DOI] [PubMed] [Google Scholar]
- [14].Volders PG, Willems IE, Cleutjens JP, et al. Interstitial collagen is increased in the noninfarcted human myocardium after myocardial infarction. J Mol Cell Cardiol; 25: 1317–23. [DOI] [PubMed] [Google Scholar]
- [15].Litwin SE, Litwin CM, Raya TE, et al. Contractility and stiffness of noninfarcted myocardium after coronary ligation in rats. Effects of chronic angiotensin converting enzyme inhibition. Circulation; 83: 1028–1037. [DOI] [PubMed] [Google Scholar]
- [16].Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair; 6: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation; 81: 1161–1172. [DOI] [PubMed] [Google Scholar]
- [18].Sun Y, Weber KT. Infarct scar: A dynamic tissue. Cardiovascular Research; 46: 250–256. [DOI] [PubMed] [Google Scholar]
- [19].Mouton AJ, Ma Y, Rivera Gonzalez OJ, et al. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol 2019. Epub ahead of print 2019. DOI: 10.1007/s00395-019-0715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ma Y, Iyer RP, Jung M, et al. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends in Pharmacological Sciences 2017. Epub ahead of print 2017. DOI: 10.1016/j.tips.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jin Y-F, Han H-C, Berger J, et al. Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricular remodeling. BMC Syst Biol; 5: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zeigler AC, Richardson WJ, Holmes JW, et al. Computational modeling of cardiac fibroblasts and fibrosis. J Mol Cell Cardiol. Epub ahead of print 19 November 2015. DOI: 10.1016/j.yjmcc.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zeigler AC, Richardson WJ, Holmes JW, et al. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol; 94: 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kraeutler MJ, Soltis AR, Saucerman JJ. Modeling cardiac β-adrenergic signaling with normalized-Hill differential equations: comparison with a biochemical model. BMC Syst Biol; 4: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].López B, Querejeta R, González A, et al. Impact of treatment on myocardial lysyl oxidase expression and collagen Cross-linking in patients with heart failure. Hypertension; 53: 236–242. [DOI] [PubMed] [Google Scholar]
- [26].Voloshenyuk TG, Hart AD, Khoutorova E, et al. TNF-?? increases cardiac fibroblast lysyl oxidase expression through TGF-?? and PI3Kinase signaling pathways. Biochem Biophys Res Commun; 413: 370–375. [DOI] [PubMed] [Google Scholar]
- [27].Voloshenyuk TG, Landesman ES, Khoutorova E, et al. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine; 55: 90–97. [DOI] [PubMed] [Google Scholar]
- [28].Cox TR, Bird D, Baker AM, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res; 73: 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yuan W, Varga J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem; 276: 38502–10. [DOI] [PubMed] [Google Scholar]
- [30].Hall MC, Young DA, Waters JG, et al. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-β1. J Biol Chem; 278: 10304–10313. [DOI] [PubMed] [Google Scholar]
- [31].Hinz B, Celetta G, Tomasek JJ, et al. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell 2001. Epub ahead of print 2001. DOI: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huet E, Vallée B, Szul D, et al. Extracellular matrix metalloproteinase inducer/CD147 promotes myofibroblast differentiation by inducing α-smooth muscle actin expression and collagen gel contraction: Implications in tissue remodeling. FASEB J 2008. Epub ahead of print 2008. DOI: 10.1096/fj.07-8748com. [DOI] [PubMed] [Google Scholar]
- [33].Dewald O, Ren G, Duerr GD, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol; 164: 665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].White M, Rouleau JL, Hall C, et al. Changes in vasoconstrictive hormones, natriuretic peptides, and left ventricular remodeling soon after anterior myocardial infarction. Am Heart J; 142: 1056–1064. [DOI] [PubMed] [Google Scholar]
- [35].Schieffer B, Wirger A, Meybrunn M, et al. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994. Epub ahead of print 1994. DOI: 10.1161/01.CIR.89.5.2273. [DOI] [PubMed] [Google Scholar]
- [36].Herskowitz a, Choi S, Ansari a a, et al. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol; 146: 419–428. [PMC free article] [PubMed] [Google Scholar]
- [37].Deten A, Hölzl A, Leicht M, et al. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J Mol Cell Cardiol; 33: 1191–207. [DOI] [PubMed] [Google Scholar]
- [38].Hao J, Ju H, Zhao S, et al. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-β in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol 1999. Epub ahead of print 1999. DOI: 10.1006/jmcc.1998.0902. [DOI] [PubMed] [Google Scholar]
- [39].Deten A, Volz HC, Briest W, et al. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res; 55: 329–340. [DOI] [PubMed] [Google Scholar]
- [40].Heba G, Krzemiński T, Porc M, et al. The time course of tumor necrosis factor-α, inducible nitric oxide synthase and vascular endothelial growth factor expression in an experimental model of chronic myocardial infarction in rats. J Vasc Res 2001. Epub ahead of print 2001. DOI: 10.1159/000051057. [DOI] [PubMed] [Google Scholar]
- [41].Zhao W, Zhao T, Huang V, et al. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol 2011. Epub ahead of print 2011. DOI: 10.1016/j.yjmcc.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fomovsky GM, Rouillard AD, Holmes JW. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol; 52: 1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Richardson WJ, Clarke SA, Alexander Quinn T, et al. Physiological implications of myocardial scar structure. Compr Physiol 2015. Epub ahead of print 2015. DOI: 10.1002/cphy.c140067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Richardson WJ, Clarke SA HJ. Modifying the Mechanics of Healing Infarcts: Better the Enemy of Good? J Mol Cell Cardiol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Carlyle WC, Jacobson AW, Judd DL, et al. Delayed reperfusion alters matrix metalloproteinase activity and fibronectin mRNA expression in the infarct zone of the ligated rat heart. J Mol Cell Cardiol 1997. Epub ahead of print 1997. DOI: 10.1006/jmcc.1997.0482. [DOI] [PubMed] [Google Scholar]
- [46].Cleutjens JPM, Kandala JC, Guarda E, et al. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol 1995. Epub ahead of print 1995. DOI: 10.1016/S0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- [47].Peterson JT, Li H, Dillon L, et al. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res 2000. Epub ahead of print 2000. DOI: 10.1016/S0008-6363(00)00029-8. [DOI] [PubMed] [Google Scholar]
- [48].Tao Z-Y, Cavasin M a, Yang F, et al. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci; 74: 1561–1572. [DOI] [PubMed] [Google Scholar]
- [49].Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nature Reviews Cardiology 2018. Epub ahead of print 2018. DOI: 10.1038/s41569-018-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fomovsky GM, Holmes JW. Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol; 298: H221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zimmerman SD, Thomas DP, Velleman SG, et al. Time course of collagen and decorin changes in rat cardiac and skeletal muscle post-MI. Am J Physiol Heart Circ Physiol; 281: H1816–22. [DOI] [PubMed] [Google Scholar]
- [52].Hu J, Li Y, Cheng W, et al. A comparison of the efficacy of surgical renal denervation and pharmacologic therapies in post-myocardial infarction heart failure. PLoS One 2014. Epub ahead of print 2014. DOI: 10.1371/journal.pone.0096996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yamagishi H, Kim S, Nishikimi T, et al. Contribution of Cardiac Renin-Angiotensin System to Ventricular Remodelling in Myocardial-Infarcted Rats. Journal of Molecular and Cellular Cardiology 1993. Epub ahead of print 1993. DOI: 10.1006/jmcc.1993.1149. [DOI] [PubMed] [Google Scholar]
- [54].Stavropoulou A, Philippou A, Halapas A, et al. uPA, uPAR and TGFβ1 expression during early and late post myocardial infarction period in rat myocardium. In Vivo (Brooklyn) 2010. [PubMed] [Google Scholar]
- [55].Deten A, Volz HC, Driest W, et al. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res 2002. Epub ahead of print 2002. DOI: 10.1016/S0008-6363(02)00413-3. [DOI] [PubMed] [Google Scholar]
- [56].van Veldhuisen DJ, Brodde OE, van Gilst WH, et al. Relation between myocardial β-adrenoceptor density and hemodynamic and neurohumoral changes in a rat model of chronic myocardial infarction: Effects of ibopamine and captopril. Cardiovasc Res 1995. Epub ahead of print 1995. DOI: 10.1016/S0008-6363(95)00058-5. [DOI] [PubMed] [Google Scholar]
- [57].Feng G, Yang Y, Chen J, et al. Ranolazine attenuated heightened plasma norepinephrine and B-type natriuretic peptide-45 in improving cardiac function in rats with chronic ischemic heart failure. Am J Transl Res 2016. [PMC free article] [PubMed] [Google Scholar]
- [58].Loennechen JP, Støylen A, Beisvag V, et al. Regional expression of endothelin-1, ANP, IGF-1, and LV wall stress in the infarcted rat heart. Am J Physiol - Hear Circ Physiol 2001. Epub ahead of print 2001. DOI: 10.1152/ajpheart.2001.280.6.h2902. [DOI] [PubMed] [Google Scholar]
- [59].Zhao T, Zhao W, Chen Y, et al. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am J Physiol Heart Circ Physiol; 304: H1719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang NP, Wang ZF, Tootle S, et al. Curcumin promotes cardiac repair and ameliorates cardiac dysfunction following myocardial infarction. Br J Pharmacol 2012. Epub ahead of print 2012. DOI: 10.1111/j.1476-5381.2012.02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Magda MW, Anique MH, Daemen MJAP, et al. Increased expression of fibronectin isoforms after myocardial infarction in rats. J Mol Cell Cardiol 1997. Epub ahead of print 1997. DOI: 10.1006/jmcc.1997.0486. [DOI] [PubMed] [Google Scholar]
- [62].Shimizu N, Yoshiyama M, Omura T, et al. Activation of mitogen-activated protein kinases and activator protein- 1 in myocardial infarction in rats. Cardiovasc Res 1998. Epub ahead of print 1998. DOI: 10.1016/S0008-6363(97)00327-1. [DOI] [PubMed] [Google Scholar]
- [63].Wang X, Lv H, Gu Y, et al. Protective effect of lycopene on cardiac function and myocardial fibrosis after acute myocardial infarction in rats via the modulation of p38 and MMP-9. J Mol Histol 2014. Epub ahead of print 2014. DOI: 10.1007/s10735-013-9535-2. [DOI] [PubMed] [Google Scholar]
- [64].Squires CE, Escobar GP, Payne JF, et al. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol; 39: 699–707. [DOI] [PubMed] [Google Scholar]
- [65].Frank DU, Sutcliffe MD, Saucerman JJ. Network-based predictions of in vivo cardiac hypertrophy. J Mol Cell Cardiol 2018. Epub ahead of print 2018. DOI: 10.1016/j.yjmcc.2018.07.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang B, Hao J, Jones SC, et al. Decreased Smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol - Hear Circ Physiol 2002. [DOI] [PubMed] [Google Scholar]
- [67].Sun Y Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovascular Research; 81: 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacology and Therapeutics 2018. Epub ahead of print 2018. DOI: 10.1016/j.pharmthera.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nature Reviews Cardiology 2017. Epub ahead of print 2017. DOI: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. In: Journal of Clinical Investigation. 2017. Epub ahead of print 2017. DOI: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Frantz S, Hu K, Adamek A, et al. Transforming growth factor beta inhibition increases 40 mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol; 103: 485–492. [DOI] [PubMed] [Google Scholar]
- [72].Wei LH, Huang XR, Zhang Y, et al. Smad7 inhibits angiotensin II-induced hypertensive cardiac remodelling. Cardiovasc Res 2013. Epub ahead of print 2013. DOI: 10.1093/cvr/cvt151. [DOI] [PubMed] [Google Scholar]
- [73].Soeki T, Kishimoto I, Okumura H, et al. C-type natriuretic peptide, a novel antifibrotic and antihypertrophic agent, prevents cardiac remodeling after myocardial infarction. J Am Coll Cardiol 2005. Epub ahead of print 2005. DOI: 10.1016/j.jacc.2004.10.067. [DOI] [PubMed] [Google Scholar]
- [74].Ikeuchi M, Tsutsui H, Shiomi T, et al. Inhibition of TGF-?? signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res; 64: 526–535. [DOI] [PubMed] [Google Scholar]
- [75].Bujak M, Ren G, Kweon HJ, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007. Epub ahead of print 2007. DOI: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- [76].Kong P, Shinde AV., Su Y, et al. Opposing actions of fibroblast and cardiomyocyte smad3 signaling in the infarcted myocardium. Circulation 2018. Epub ahead of print 2018. DOI: 10.1161/CIRCULATIONAHA.117.029622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ono K, Matsumori a, Shioi T, et al. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Circulation; 98: 149–156. [DOI] [PubMed] [Google Scholar]
- [78].Abbate A, Kontos MC, Grizzard JD, et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am J Cardiol 2010. Epub ahead of print 2010. DOI: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- [79].Abbate A, Van Tassell BW, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the virginia commonwealth university-anakinra remodeling trial (2) (vcu-art2) pilot study]. Am J Cardiol 2013. Epub ahead of print 2013. DOI: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Garcia RA, Go KV., Villarreal FJ. Effects of timed administration of doxycycline or methylprednisolone on post-myocardial infarction inflammation and left ventricular remodeling in the rat heart. Mol Cell Biochem 2007. Epub ahead of print 2007. DOI: 10.1007/s11010-006-9379-0. [DOI] [PubMed] [Google Scholar]
- [81].Kirlin PC, Romson JL, Pitt B, et al. Ibuprofen-mediated infarct size reduction: Effects on regional myocardial function in canine myocardial infarction. Am J Cardiol 1982. Epub ahead of print 1982. DOI: 10.1016/0002-9149(82)91244-9. [DOI] [PubMed] [Google Scholar]
- [82].Ridker PM, MacFadyen JG, Everett BM, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 2018. Epub ahead of print 2018. DOI: 10.1016/S0140-6736(17)32814-3. [DOI] [PubMed] [Google Scholar]
- [83].Siwik D a Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res; 86: 1259–1265. [DOI] [PubMed] [Google Scholar]
- [84].Farbehi N, Patrick R, Dorison A, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019. Epub ahead of print 2019. DOI: 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Saxena A, Chen W, Su Y, et al. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J Immunol 2013. Epub ahead of print 2013. DOI: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Villar AV., García R, Llano M, et al. BAMBI (BMP and activin membrane-bound inhibitor) protects the murine heart from pressure-overload biomechanical stress by restraining TGF-β signaling. Biochim Biophys Acta - Mol Basis Dis 2013. Epub ahead of print 2013. DOI: 10.1016/j.bbadis.2012.11.007. [DOI] [PubMed] [Google Scholar]
- [87].Xu J, Carretero OA, Lin CX, et al. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. Am J Physiol-Hear Circ Physiol 2007. Epub ahead of print 2007. DOI: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ammarguellat F, Larouche I, Schiffrin EL. Myocardial fibrosis in DOCA-salt hypertensive rats: Effect of endothelin ETA receptor antagonism. Circulation 2001. Epub ahead of print 2001. DOI: 10.1161/01.CIR.103.2.319. [DOI] [PubMed] [Google Scholar]
- [89].Zhao QD, Viswanadhapalli S, Williams P, et al. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 2015. Epub ahead of print 2015. DOI: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Pan LL, Liu XH, Shen YQ, et al. Inhibition of NADPH oxidase 4-related signaling by sodium hydrosulfide attenuates myocardial fibrotic response. Int J Cardiol 2013. Epub ahead of print 2013. DOI: 10.1016/j.ijcard.2013.06.007. [DOI] [PubMed] [Google Scholar]
- [91].Mitchell MD, Laird RE, Brown RD, et al. IL-1β stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol - Hear Circ Physiol 2007. Epub ahead of print 2007. DOI: 10.1152/ajpheart.00881.2005. [DOI] [PubMed] [Google Scholar]
- [92].Wernig G, Chen SY, Cui L, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A 2017. Epub ahead of print 2017. DOI: 10.1073/pnas.1621375114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sullivan DE, Ferris M, Nguyen H, et al. TNF-α induces TGF-β1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J Cell Mol Med 2009. Epub ahead of print 2009. DOI: 10.1111/j.1582-4934.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].González-Ramos M, Mora I, De Frutos S, et al. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: Studies on TGF-β1 regulation. Int J Biochem Cell Biol 2012. Epub ahead of print 2012. DOI: 10.1016/j.biocel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- [95].Wu W, Muchir A, Shan J, et al. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in Lamin A/C gene. Circulation 2011. Epub ahead of print 2011. DOI: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wu W, Shan J, Bonne G, et al. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta - Mol Basis Dis 2010. Epub ahead of print 2010. DOI: 10.1016/j.bbadis.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chichger H, Vang A, O’Connell KA, et al. PKC δ and βII regulate angiotensin II-mediated fibrosis through p38: A mechanism of RV fibrosis in pulmonary hypertension. Am J Physiol - Lung Cell Mol Physiol 2015. Epub ahead of print 2015. DOI: 10.1152/ajplung.00184.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jiang S, Jiang D, Zhao P, et al. Activation of AMP-activated protein kinase reduces collagen production via p38 MAPK in cardiac fibroblasts induced by coxsackievirus B3. Mol Med Rep 2016. Epub ahead of print 2016. DOI: 10.3892/mmr.2016.5319. [DOI] [PubMed] [Google Scholar]
- [99].Westermarck J, Li S-P, Kallunki T, et al. p38 Mitogen-Activated Protein Kinase-Dependent Activation of Protein Phosphatases 1 and 2A Inhibits MEK1 and MEK2 Activity and Collagenase 1 (MMP-1) Gene Expression. Mol Cell Biol 2001. Epub ahead of print 2001. DOI: 10.1128/mcb.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Jones DS, Jenney AP, Joughin BA, et al. Inflammatory but not mitogenic contexts prime synovial fibroblasts for compensatory signaling responses to p38 inhibition. Sci Signal 2018. Epub ahead of print 2018. DOI: 10.1126/scisignal.aal1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Liu H, Liu A, Shi C, et al. Curcumin suppresses transforming growth factor-β1-inducedcardiac fibroblast differentiation via inhibition of smad-2 and p38 MAPK signaling pathways. Exp Ther Med 2016. Epub ahead of print 2016. DOI: 10.3892/etm.2016.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kojonazarov B, Novoyatleva T, Boehm M, et al. P38 mapk inhibition improves heart function in pressure-loaded right ventricular hypertrophy. Am J Respir Cell Mol Biol 2017. Epub ahead of print 2017. DOI: 10.1165/rcmb.2016-0374OC. [DOI] [PubMed] [Google Scholar]
- [103].Molkentin JD, Bugg D, Ghearing N, et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase in Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017. Epub ahead of print 2017. DOI: 10.1161/CIRCULATIONAHA.116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Jugdutt BI, Joljart MJ, Khan MI. Rate of Collagen Deposition during Healing and Ventricular Remodeling after Myocardial Infarction in Rat and Dog Models. Circulation 1996. Epub ahead of print 1996. DOI: 10.1161/01.CIR.94.1.94. [DOI] [PubMed] [Google Scholar]
- [105].Weber KT, Sun Y, Bhattacharya SK, et al. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol; 10: 15–26. [DOI] [PubMed] [Google Scholar]
- [106].Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. Journal of Molecular and Cellular Cardiology 2010. Epub ahead of print 2010. DOI: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction. Circulation Research 2016. Epub ahead of print 2016. DOI: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.