

Abstract
TNF-related apoptosis-inducing ligand (TRAIL) is known to induce apoptosis in cancer cells, although non-apoptotic functions have also been reported for this cytokine in various cell types. TRAIL and its receptor TRAIL-R2 are expressed in skeletal muscles, but a potential role of muscle-derived TRAIL in myogenesis has not been explored. Here we report that TRAIL is an autocrine regulator of myogenic differentiation. Knockdown of TRAIL or TRAIL-R2 enhanced C2C12 myoblast differentiation, and recombinant TRAIL inhibited expression of the cell cycle inhibitor p21, accompanied by suppression of myoblasts from exiting the cell cycle, a requisite step in the myogenic differentiation process. Blocking cell cycle progression restored differentiation from inhibition by recombinant TRAIL, supporting the notion that TRAIL exerts its effect in myogenesis through modulating cell cycle exit. We also found that TRAIL knockdown led to enhanced muscle regeneration in mice upon injury, recapitulating the in vitro observation. Additionally, inhibition of ERK activation reversed the negative effect of recombinant TRAIL on p21 expression and myoblast differentiation, suggesting that ERK signaling may be a mediator of TRAIL’s function to suppress cell cycle withdrawal and inhibit differentiation. Taken together, our findings uncover a muscle cell-autonomous non-apoptotic function of TRAIL in skeletal myogenesis.
Keywords: TRAIL, myogenic differentiation, muscle regeneration, cell cycle withdrawal, ERK
Introduction
TRAIL, also known as TNFSF10 (tumor necrosis factor superfamily member 10) and Apo2L, was initially identified as a member of the TNF family with a capacity to induce apoptosis in various types of transformed cells (1,2). Since then, TRAIL has been widely studied in oncology because of its antitumor properties. For example, suppression of tumor growth by treatment with TRAIL has been reported from mouse xenograft models (3,4). Conversely, TRAIL knockout mice were more susceptible to transplanted lymphoma, as indicated by the knockout animals’ lower survival rates than the wild-type counterparts due to tumor burden (5).
TRAIL is expressed as a type II transmembrane protein, which can be subsequently cleaved to release a soluble form that retains biological activities of full-length TRAIL. Both membrane-bound TRAIL and soluble TRAIL are present as homotrimers. In humans, five TRAIL receptors have been identified (6,7). TRAIL receptors 1–4 (TRAIL-R1–4) are type I transmembrane proteins. TRAIL-R1 (also known as DR4 and TNFRSF10A) and TRAIL-R2 (also known as DR5 and TNFRSF10B) can induce apoptosis through their cytoplasmic death domain (8). Thus, binding of TRAIL by TRAIL-R1 or TRAIL-R2 leads to formation of the death-inducing signaling complex (DISC) and triggers a caspase cascade, resulting in apoptosis (6). TRAIL can also bind TRAIL-R3 (also known as DCR1 and TNFRSF10C) and TRAIL-R4 (also known as DCR2 and TNFRSF10D). However, due to the lack of a cytoplasmic death domain, these two receptors cannot further transduce apoptotic signals and they act as decoy receptors by competing for ligand binding (9). Lastly, TRAIL can bind to a soluble decoy receptor, osteoprotegerin (OPG, also known as TNFRSF11B), which also functions as a decoy receptor for receptor activator of NF-kappaB ligand (RANKL) (10,11). In mice, TRAIL-R2 is the only apoptosis-related receptor displaying marked sequence homology with human TRAIL-R1 (43%) and TRAIL-R2 (49%) (12). Two decoy receptors without signaling capacity, mDcTRAIL-R1 (or TNFRSF23) and mDcTRAIL-R2 (or TNFRSF22), have been identified in mice (13,14). In addition, mouse OPG is believed to also function as a soluble decoy receptor (6).
Aside from the pro-apoptotic signaling through DISC, TRAIL and its receptors can induce apoptosis-independent signaling (15). It has been reported that TRAIL can activate NF-κB, leading to proliferation of Jurkat cells (16) or migration and invasion of human cholangiocarcinoma (CCA) cells (17). TRAIL also activates the Jun N-terminal kinase (JNK) and p38 Mitogen-Activated Protein Kinase (MAPK) pathways in HT1080 cells (15). Moreover, it has been shown that another member of the MAPK family, ERK1/2 is activated by TRAIL, leading to human endothelial cell proliferation (18) or proliferation and migration of vascular smooth muscle cells (19).
Adult skeletal muscles in mammals have the capacity to undergo robust regeneration upon damage or injury, owing to the activation of quiescent satellite cells (20). Activated satellite cells proliferate and then withdraw from the cell cycle, followed by differentiation and fusion to form new or repaired myofibers. This sequential process of proliferation, cell cycle withdrawal, differentiation and fusion can be recapitulated in vitro by cultured myoblasts, such as the mouse satellite cell-derived C2C12 cells (21,22). TRAIL and TRAIL-R2 were reported to express in cultured mouse myoblasts, in which a recombinant TRAIL induced apoptosis through TRAIL-R2 (23). TRAIL is also expressed in human myositis skeletal muscles, and it is implicated in apoptotic or autophagic cell death of the diseased muscles (24,25). Whether muscle-derived TRAIL plays a non-cell death role in myogenic differentiation had not been explored. In the current study, we find that TRAIL functions as a negative regulator of myogenic differentiation, and that TRAIL signals through TRAIL-R2 and ERK to suppress cell cycle withdrawal of myoblasts.
Materials and Methods
Antibodies and other reagents
Anti-MHC (MF20, used at 1:500), anti-myogenin (F5D, 1:500) and Ad5-luciferase adenovirus were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD, National Institutes of Health and maintained by the University of Iowa, Department of Biological Sciences. Anti-TRAIL (sc-7877, 1:500) for detection of human TRAIL was from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-TRAIL (PA5–47073, 1:500) for detection of mouse TRAIL, ELISA kit for detection of mouse TRAIL (EMTNFSF10), peroxidase-conjugated anti-goat IgG antibody (A27014, 1:1000), Alexa Fluor 594-conjugated goat anti rabbit secondary antibody (A-11072, 1:500) and U0126 were from Thermo Fisher Scientific (Waltham, MA, USA). Anti-tubulin (ab11304, 1:10000) and anti-p21 (ab109199, 1:500) were from Abcam (Cambridge, MA, USA). Anti-TRAIL-R2 (AF721, 1:1000) and human TRAIL recombinant protein (375-TEC) were from R&D Systems (Minneapolis, MN, USA). Anti-pERK (4370, 1:500), anti-ERK (4696, 1:1000) and anti-Ki-67 (12202, 1:400) were from Cell Signaling Technology (Danvers, MA, USA). Peroxidase-conjugated anti-rabbit (115-036-003, 1:1000) and anti-mouse (111-036-003, 1:1000) IgG antibodies and FITC-conjugated anti-mouse IgG (115-096-003, 1:100) were from Jackson Immuno Research Laboratories (West Grove, PA, USA). 5-ethynyl-2’-deoxyuridine (EdU) was from Carbosynth Limited (Compton, UK). Fluorescein azide 5-isomer was from Lumiprobe (Hunt Valley, MD, USA). Cytosine β-D-arabinofuranoside hydrochloride (Ara-C), gelatin and collagen type I from calf skin were from Sigma-Aldrich (St. Louis, MO, USA). Adenovirus expressing human full-length TRAIL (140841A) was from Applied Biological Materials (Richmond, BC, Canada). pCMV-untagged empty vector (CV011) and pCMV-untagged mouse full-length TRAIL (MG50166-UT) were from Sino Biological (Wayne, PA, USA).
Cell Culture
C2C12 myoblasts were a gift from S. Kaufman at the University of Illinois and originally obtained from ATCC. Cells were maintained in DMEM containing 4.5 g/L glucose, 10% fetal bovine serum, and 1% penicillin-streptomycin at 37°C with 7.5% CO2. To induce differentiation, cells were plated on tissue culture plates coated with 0.2% gelatin, grown to 100% confluence, and then cultured in differentiation medium (DMEM containing 2% horse serum). The cells were replenished with fresh differentiation medium daily for 3 days. Myoblasts were transfected by using TransIT-LT1 (Mirus, Madison, WI) according to the manufacturer’s recommendations. Cells were selected in 0.5 mg/mL Hygromycin.
Mouse primary myoblast isolation and differentiation
Primary myoblasts were isolated from 2 to 5-day-old FVB mice as described previously (26), and cultured on 0.1 mg/mL collagen-coated tissue culture plates. Differentiation was induced at 50–70% cell density in differentiation medium for 2 days.
Immunofluorescence microscopy and quantitative analysis of myocytes in vitro
C2C12 cells and primary myoblasts differentiated in 12-well plates were fixed with 3.7% formaldehyde and incubated with anti-MHC, -Ki-67 or -myogenin antibody, followed by incubation with DAPI and FITC- or Alexa-conjugated secondary antibody. Cells were observed with a Leica DMI 4000B fluorescence microscope (Leica, Wetzlar, Germany). The fluorescence images were captured using a RETIGA EXi camera (QImaging, Surry, BC, Canada) and Image Pro Express software (Media Cybernetics, Rockville, MD, USA). Images were analyzed using ImageJ software (NIH, Bethesda, MD, USA). The differentiation index or fusion index was calculated as the percentage of nuclei in MHC-positive cells or in MHC-positive cells with more than one nucleus, respectively. The percentage of Ki-67- or myogenin-positive cells was calculated by taking DAPI signals as the total number of cells. Each data point was generated from quantifying five randomly chosen microscopic fields.
Lentivirus-delivered RNA interference
shRNAs in the pLKO.1-puro vector were purchased from Sigma-Aldrich (MISSION TRC). Clone IDs are: shTRAIL#1, TRCN0000066235; shTRAIL#2, TRCN0000066236; shTRAIL-R2#1, TRCN0000012323; shTRAIL-R2#2, TRCN0000012327. A hairpin of scrambled sequence (shScramble) used for a negative control and lentivirus packaging were previously described (27). C2C12 cells were transduced with lentiviruses in growth medium with 8 μg/mL polybrene and selected in 3 μg/mL puromycin for 2 days, followed by plating into 12-well plates to induce differentiation. For transduction of primary myoblasts, polybrene and puromycin selection were omitted to preserve cell viability.
Quantitative RT-PCR
C2C12 cells or isolated muscle tissues were lysed in Trizol (Invitrogen, Carlsbad, CA, USA), and RNA was isolated following the manufacturer’s protocol. cDNA was synthesized from 1 μg RNA using the qScript™ cDNA Synthesis kit (Quantabio, Beverly, MA, USA) following the manufacturer’s protocol. Quantitative PCR was performed with a StepOne Plus (Applied Biosystems, Foster City, CA, USA) using gene-specific primers. β-actin or Hprt was used as a reference to obtain the relative fold change for target samples using the comparative CT method. Mouse β-actin primers: forward 5’-ttgctgacaggatgcagaag-3’; reverse 5’-atccacatctgctggaaggt-3’. Mouse Hprt primers: forward 5’-atggactgattatggacaggactg-3’; reverse 5’-tccagcaggtcagcaaagaac-3’. Mouse TRAIL primers: forward 5’-tccaatctccaaggatggaaaga-3’; reverse 5’-gatgtaatacaggccctcctgctc-3’.
Western blotting
Cells were lysed in SDS sample buffer with 5% β-mercaptoethanol. Proteins were resolved on SDS-PAGE, transferred onto polyvinylidene fluoride membrane (EMD Millipore, Darmstadt, Germany) and incubated with various antibodies in accordance with manufacturers’ recommendations. Detection of horseradish peroxidase-conjugated secondary antibodies was performed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA), and images were captured on an iBright CL1000 imaging system (Thermo Fisher Scientific, Waltham, MA, USA). Quantification of western blot band intensities was performed with densitometry of images using ImageJ software (NIH, Bethesda, MD, USA).
Cell proliferation and apoptosis assays
To measure proliferation of C2C12 cells, EdU labelling was performed as previously described (27). To examine apoptosis, TUNEL assays were performed according to manufacturer’s protocol (Promega, Madison, WI, USA).
Injury-induced muscle regeneration and intramuscular TRAIL knockdown in mice
Male (D7AI and D14AI) or female (D3AI) C57BL/6 wild-type mice aged 10–12 weeks were used in all the regeneration experiments. Animals were randomly allocated to the different experimental groups. Muscle injury was induced by injection of 50 μL of 1.2% (w/v) BaCl2 dissolved in saline into mouse TA muscles as previously described (28). To knockdown TRAIL, lentivirus expressing shTRAIL (and shScramble as negative control) was concentrated to 1× 107 – 1×108 IU/mL via ultracentrifugation and co-injected with BaCl2 into mouse TA muscles. The injured muscles were collected at 3, 7 or 14 days after injury and subjected to RNA isolation or cryosection and staining.
Muscle tissue cryosection, hematoxylin and eosin staining, and immunohistochemistry
TA muscles were isolated and frozen in liquid nitrogen-cooled 2-methylbutane and embedded in TBS tissue freezing medium (Thermo Fisher Scientific, Waltham, MA, USA). Sections of 10 μm thickness were obtained with a Microm HM550 (Thermo Fisher Scientific, Waltham, MA, USA) at −20°C, placed on uncoated slides, and stained with hematoxylin and eosin (H&E). Separately, the sections from D3AI were fixed by 1.5% paraformaldehyde, incubated with anti-Ki-67 antibody, followed by incubation with DAPI and Alexa-conjugated secondary antibody. Images were obtained by DMI 4000B fluorescence microscope (Leica, Wetzlar, Germany) with a 20X dry objective (Fluotar, numerical aperture 0.4; Leica). Five bright field or fluorescence images were randomly captured using RETIGA EXi camera (QImaging, Surry, BC, Canada) and Image Pro Express software (Media Cybernetics, Rockville, MD, USA). The images were analyzed for cross-sectional area of all centrally nucleated regenerating myofibers (H&E staining) or counting Ki-67-positive nuclei using ImageJ. All procedures were performed by an investigator that were blinded to sample identification.
Statistical analysis
All the data shown are representative results of at least three independent experiments with cells, or 5–8 mice for each data point in animal experiments. The exact sample size for each experiment is described in figure legends. All the quantitative values are presented as mean ± SEM. Whenever necessary, statistical significance of the data was analyzed by performing two-tailed paired Student’s t-tests. The specific types of test and the P-values, when applicable, are indicated in the figure legends. Statistical comparison was considered significant when P < 0.05. All statistical analysis was performed using Excel.
Study approval
All the animal experiments in this study followed protocols approved by the Animal Care and Use Committee at the University of Illinois at Urbana-Champaign.
Data availability:
All data are contained within the manuscript.
Results
TRAIL is a negative regulator of myoblast differentiation
Since the expression of TRAIL had been reported in myoblasts (23), we wondered whether its expression level changed during myogenesis. C2C12 mouse myoblasts were induced to differentiate by serum deprivation for up to 72 hrs. We observed that the level of TRAIL mRNA increased by ~3-fold at 24 hrs of differentiation and returned to pre-differentiation level by 72 hrs (Fig. 1A). The secreted TRAIL protein level increased by ~2-fold at 24 hrs and this level was largely maintained throughout differentiation (Fig. 1B).
Figure 1. Depletion of TRAIL promotes myoblast differentiation.
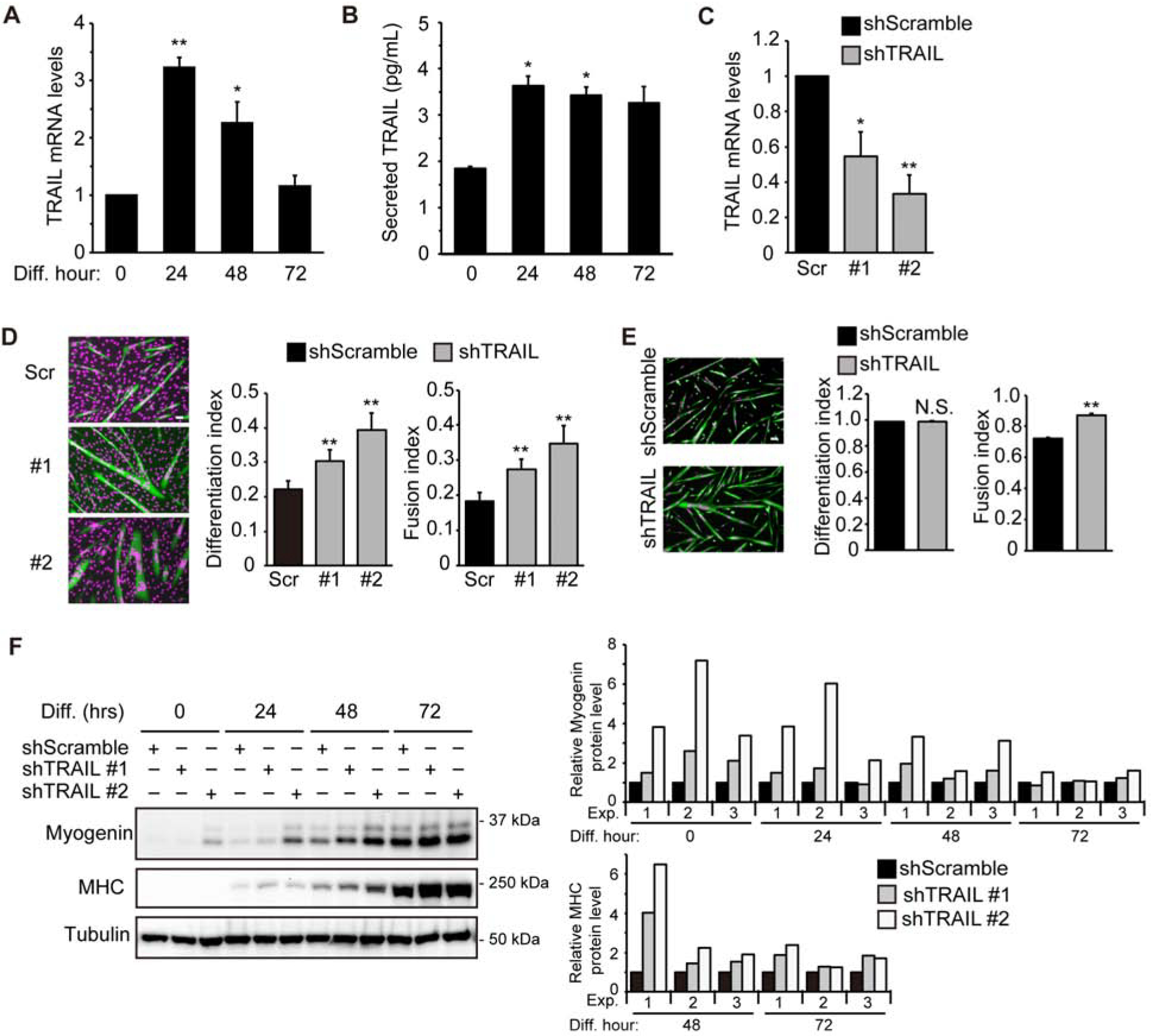
(A) RNA was isolated from differentiating C2C12 cells at different time points followed by qRT-PCR analysis (n=4). (B) Culture medium during C2C12 differentiation was collected at different time points and ELISA assay was performed to determine secreted TRAIL levels (n=3). (C) C2C12 cells were transduced with lentiviruses expressing shTRAIL or shScramble (Scr) followed by 2 days of puromycin selection. Cell lysates were then collected and subjected to qRT-PCR analysis (n=5). (D) Cells treated as in (C) were induced to differentiate for 72 hrs, followed by staining for MHC (green) and DAPI (magenta), and differentiation and fusion indices were quantified (n=7). (E) Mouse primary myoblasts were transduced with lentiviruses expressing shRNAs overnight, cultured in growth medium for 1 day, and then induced to differentiate for 48 hrs, followed by staining for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=3). (F) C2C12 cells treated as in (C) were induced to differentiate, and at different time points, cells were lysed and subjected to western analysis. Representative blots are shown. Results of quantification by densitometry are shown for 3 independent experiments. The MHC and myogenin protein levels were determined relative to the level of tubulin. Data were normalized to shScramble at each differentiation time point. When applicable, data are presented as mean with error bars representing SEM. Two-tailed t-test was performed to compare each data to control. *P < 0.05; **P < 0.01. NS, not significant. Scale bars: 50 μm.
To examine a potential role of endogenous TRAIL in myogenesis, we delivered shRNAs by lentiviral transduction into C2C12 myoblasts. Two independent shRNAs (shTRAIL) knocked down TRAIL mRNA by ~50% and ~70% compared to a negative control shRNA with a scrambled hairpin sequence (shScramble) (Fig. 1C). Increased differentiation was observed in correlation with the knockdown, as quantified by differentiation index (% of myosin heavy chain (MHC)-positive cells) and fusion index (% of MHC-positive cells with two or more nuclei) (Fig. 1D). Knockdown of TRAIL in mouse primary myoblasts also resulted in statistically significant increase in fusion index (Fig. 1E). However, unlike C2C12 myoblasts, which mimic satellite cells in vivo and maintain their stemness by limiting the degree of differentiation (29), primary myoblasts achieved a differentiation index of near 100%, precluding any further increase by TRAIL knockdown (Fig. 1E).
Expression of the early myogenic marker myogenin was found to be up-regulated upon TRAIL knockdown in C2C12 cells from 0 to 48 hrs of differentiation, and the late myogenic marker MHC was elevated by the knockdown at 48 and 72 hrs (Fig. 1F). These data suggest that TRAIL may have a negative role in myogenic differentiation.
Consistent with a negative function of TRAIL, overexpression of TRAIL from a cDNA construct inhibited differentiation of C2C12 cells treated with the shScramble virus (Fig. 2A). In addition, the effect of TRAIL knockdown was reversed by TRAIL overexpression (Fig. 2A), validating the shRNA to be on-target. Endogenous TRAIL was not detected by western blotting of the cell lysates (Fig. 2A) although it was detected in the medium by ELISA (Fig. 1B). Similar to overexpression of TRAIL, a soluble recombinant TRAIL (sTRAIL) added to the cell medium also inhibited myoblast differentiation and countered the effect of TRAIL knockdown (Fig. 2B). Taken together, our observations suggest that TRAIL is likely a muscle cell-derived negative regulator of myogenic differentiation.
Figure 2. Exogeneous TRAIL inhibits myoblast differentiation.
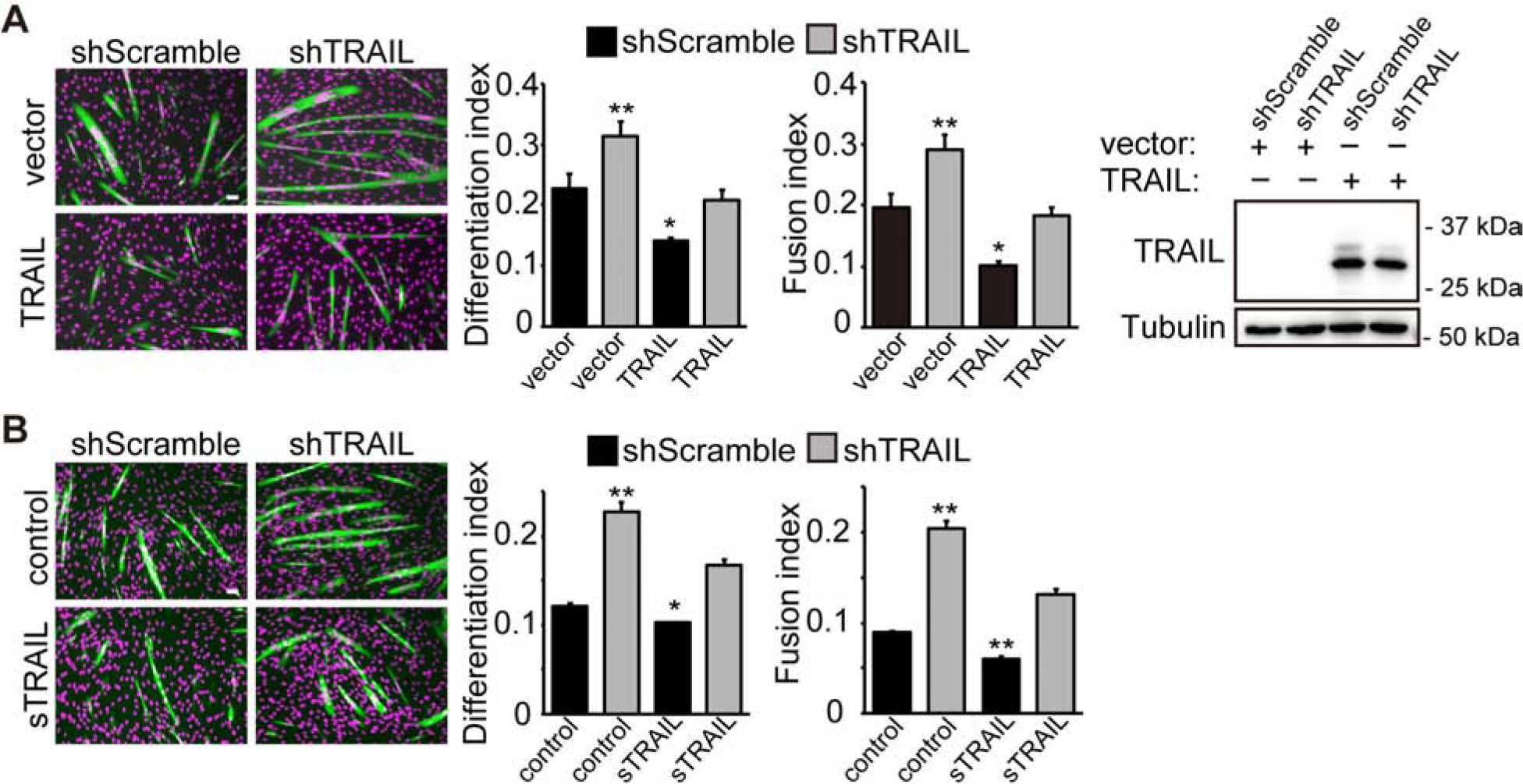
(A) C2C12 cells were transduced with lentiviruses expressing shTRAIL or shScramble followed by 2 days of puromycin selection. Cells were then transfected with a mouse full-length TRAIL expression construct (TRAIL) or the corresponding empty vector (vector), selected with hygromycin, and differentiated for 72 hrs, followed by staining for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=4). Overexpression of TRAIL was confirmed by western blotting. (B) Cells treated as in (A) were cultured with 100 ng/mL sTRAIL or vehicle (control) for 6 to 9 hrs and then induced to differentiate for 72 hrs with sTRAIL present for the first 24 hrs, followed by staining for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=5). All error bars represent SEM. Two-tailed t-test was performed to compare each data to control (first bar in each graph). *P < 0.05; **P < 0.01.
TRAIL-R2, the receptor of TRAIL, negatively regulates myoblast differentiation
Next, we wished to test whether the myogenic function of TRAIL was mediated by TRAIL-R2, the only known functional receptor for TRAIL in mice (12). With lentivirus-delivered shRNAs we knocked down the TRAIL-R2 protein in C2C12 cells by 50–70% (Fig. 3A) and observed that, similar to TRAIL knockdown, depletion of TRAIL-R2 increased both differentiation and fusion indices (Fig. 3B). Myogenin expression was upregulated by TRAIL-R2 knockdown during the early phase of differentiation (0–24 hrs), whereas elevated MHC expression was observed later through 72 hrs (Fig. 3C). Interestingly, the protein level of TRAIL-R2 decreased during differentiation (see shScramble in Fig. 3C), in contrast to the increase of TRAIL (Fig. 1A&1B). Importantly, upon TRAIL-R2 knockdown, TRAIL overexpression no longer had an effect on myoblast differentiation (Fig. 3D). These results are consistent with the notion that TRAIL functions through TRAIL-R2 to negatively regulate myogenic differentiation.
Figure 3. Depletion of TRAIL-R2 promotes myoblast differentiation.
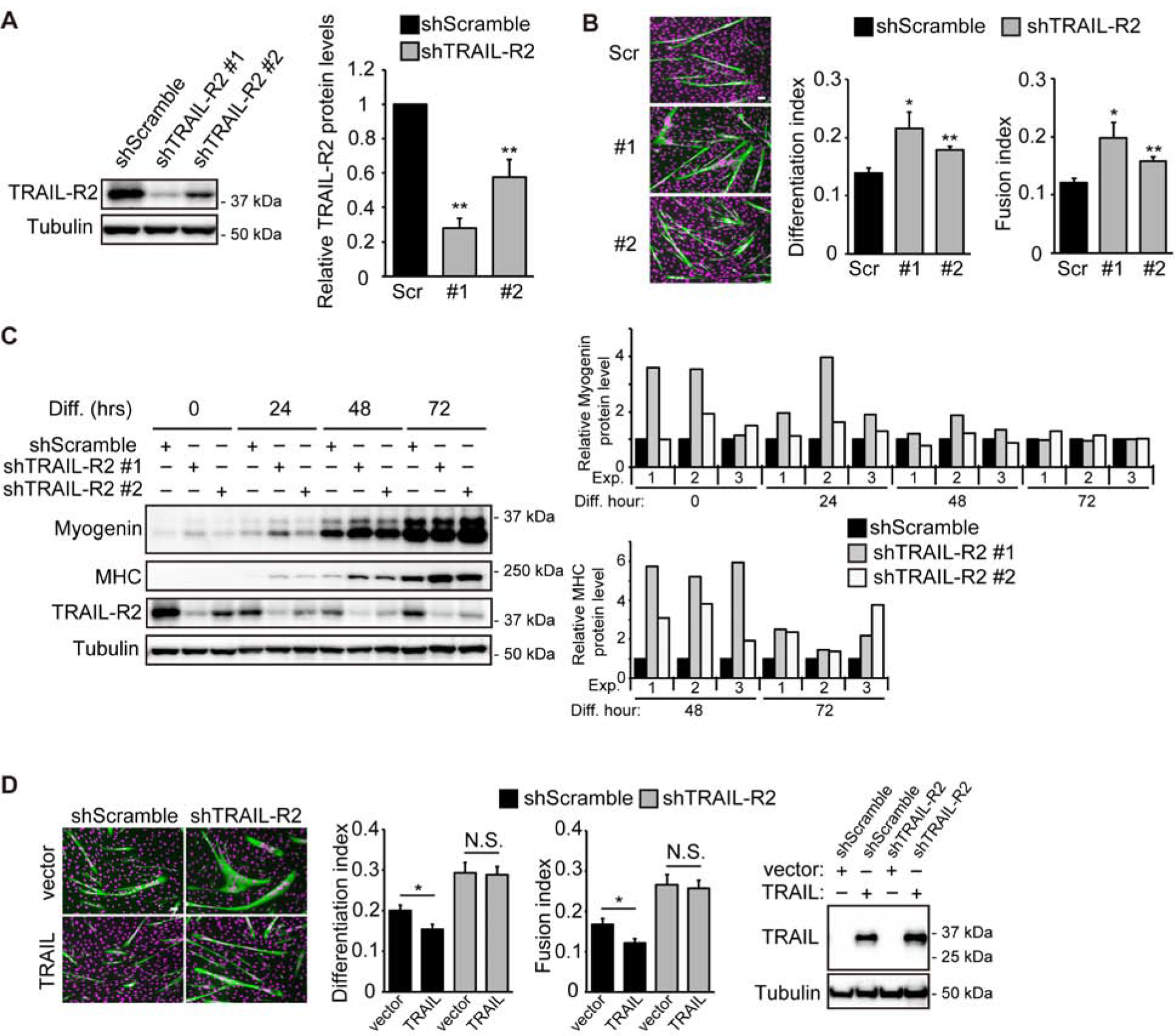
(A) C2C12 cells were transduced with lentiviruses expressing shTRAIL-R2 or shScramble followed by 2 days of puromycin selection. Cell lysates were then collected and subjected to western analysis. Representative blots and results of quantification are shown for 5 independent experiments. (B) Cells treated as in (A) were induced to differentiate for 72 hrs, followed by staining for MHC (green) and DAPI (magenta), and differentiation and fusion indices were quantified (n=5). (C) Cells treated as in (A) were induced to differentiate, and at different time points, cells were lysed and subjected to western analysis. Representative blots and results of quantification are shown for 3 independent experiments. (D) Cells treated as in (A) were transfected with TRAIL or empty vector, selected with hygromycin, and differentiated for 72 hrs, followed by staining for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=4). Expression of TRAIL was confirmed by western blotting. For (A) and (C), quantification of western blots was performed by densitometry. The level of each specific protein was determined relative to the level of tubulin, and the data were normalized to shScramble at each time point. When applicable, data are presented as mean with error bars representing SEM. Two-tailed t-test was performed to compare each data to control. *P < 0.05; **P < 0.01. NS, not significant. Scale bars: 50 μm.
TRAIL inhibits myoblast differentiation through impeding cell cycle withdrawal
We set out to investigate the mechanism by which TRAIL/TRAIL-R2 regulates myogenic differentiation. In the early stage of myogenesis, proliferating myoblasts exit the cell cycle, which is a requisite step prior to myoblast fusion into myotubes. Since TRAIL had been reported to promote proliferation of other types of cells (18,19), we wondered whether it might impact cell cycle withdrawal of myoblasts. Knockdown of TRAIL decreased the number of EdU-labeled C2C12 cells at the onset of differentiation (Fig. 4A), indicating a reduced rate of proliferation. TRAIL knockdown in primary myoblasts at the time of differentiation also led to a modest but statistically significant decrease in EdU-labeled cells (Fig. 4B). Conversely, overexpression of TRAIL (Fig. 4C), as well as addition of sTRAIL in cell medium (Fig. 4D), increased the number of EdU-labeled C2C12 cells. Interestingly, neither the effect of TRAIL knockdown (Fig. 4E) nor that of sTRAIL addition (Fig. 4F) on EdU-labeling was observed when C2C12 cells were actively proliferating, suggesting that TRAIL may impact cell cycle specifically at the time of differentiation.
Figure 4. TRAIL and TRAIL-R2 promote myoblast proliferation at the onset of differentiation.
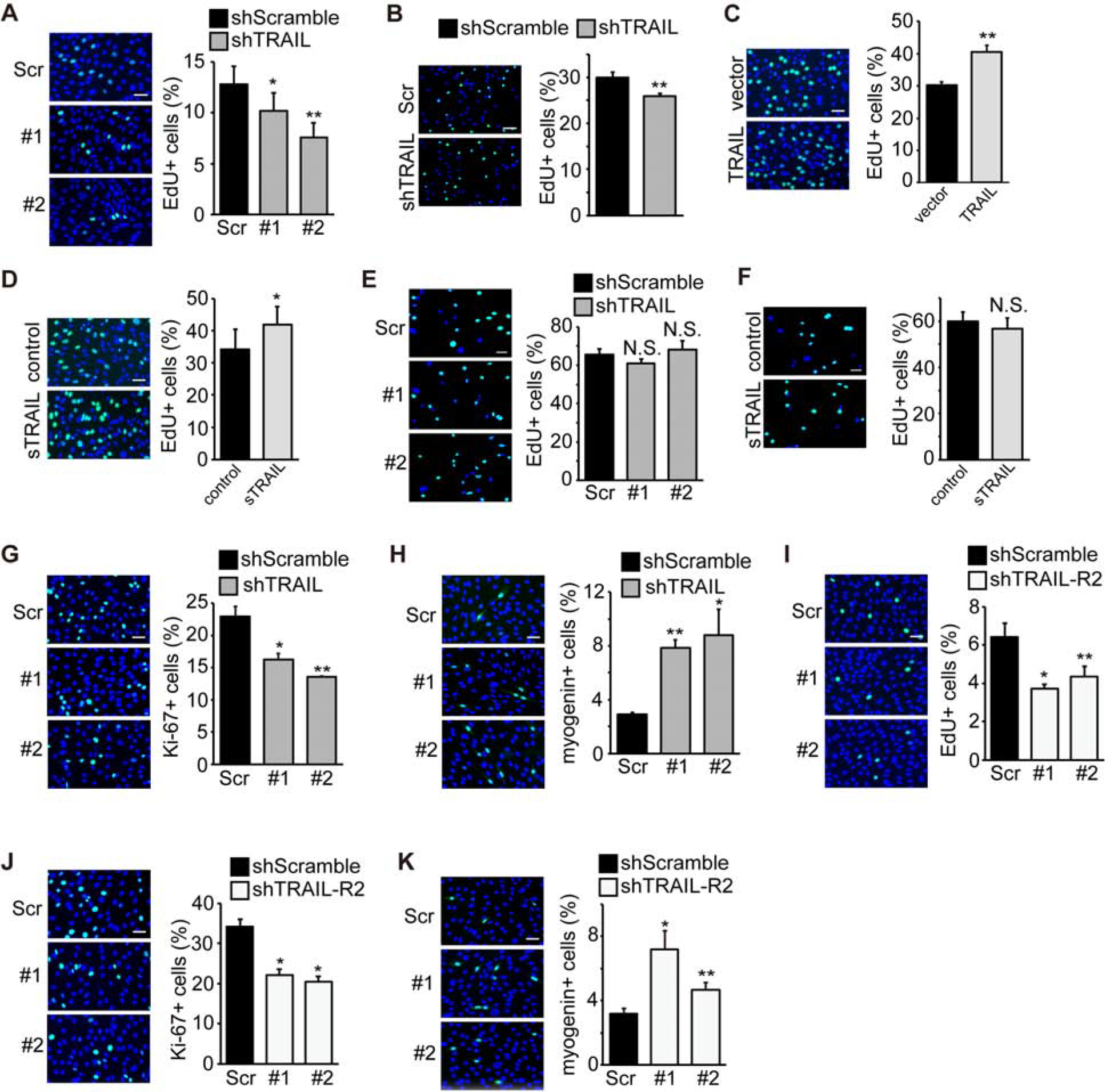
(A) C2C12 cells were transduced with lentiviruses expressing shTRAIL or shScramble followed by 2 days of puromycin selection. At 24 hr of differentiation, cells were labeled with EdU for 2 hrs in differentiation medium, stained for EdU (green) and DAPI (blue), and the percentage of cells positive for EdU staining was quantified (n=6). (B) Mouse primary myoblasts were transduced with lentiviruses expressing shRNAs overnight, cultured in growth medium for 1 day, and then labeled with EdU for 2 hrs in differentiation medium at 0 hr of differentiation and processed as in (A) (n=5). (C) C2C12 cells were transfected with TRAIL or empty vector and selected with hygromycin for 2 days. At 0 hr of differentiation, cells were labeled with EdU for 2 hrs in differentiation medium, and processed as in (A) (n=5). (D) C2C12 cells were grown in the presence of vehicle (Control) or 100 ng/mL sTRAIL for 24 hrs. At 0 hr of differentiation, cells were labeled with EdU for 2 hrs in differentiation medium, and processed as in (A) (n=6). (E) C2C12 cells were transduced with lentiviruses expressing shTRAIL or shScramble followed by 2 days of puromycin selection. Cells were then grown to ~20% confluence, labeled with EdU for 2 hrs in growth medium, and processed as in (A) (n=5). (F) C2C12 cells were grown to ~20% in the presence of vehicle (Control) or 100 ng/mL sTRAIL for 24 hrs, labeled with EdU for 2 hrs in growth medium, and processed as in (A) (n=4). (G) C2C12 cells treated as in (A) were stained for Ki-67 (green) and DAPI (blue), and the percentage of cells positive for Ki-67 staining was quantified (n=4). (H) C2C12 cells treated as in (A) were stained for myogenin (green) and DAPI (blue), and the percentage of cells positive for myogenin staining was quantified (n=4). (I) C2C12 cells were transduced with lentiviruses expressing shTRAIL-R2 or shScramble followed by 2 days of puromycin selection. At 24 hr of differentiation, cells were labeled with EdU for 2 hrs in differentiation medium and processed as in (A) (n=5). (J) C2C12 cells treated as in (I) were stained for Ki-67 (green) and DAPI (blue), and the percentage of cells positive for Ki-67 staining was quantified (n=4). (K) C2C12 cells treated as in (I) were stained for myogenin (green) and DAPI (blue), and the percentage of cells positive for myogenin staining was quantified (n=4). All error bars represent SEM. Two-tailed t-test was performed to compare each data to control. *P < 0.05; **P < 0.01. NS, not significant. Scale bars: 50 μm.
As a marker of cell proliferation, Ki-67 was also examined in C2C12 cells by immunostaining. We found a reduced number of Ki-67-positive cells upon TRAIL knockdown at the onset of differentiation (Fig. 4G), which was accompanied by an increased number of C2C12 cells expressing the early myogenic marker myogenin (Fig. 4H).
Knockdown of the receptor TRAIL-R2 in C2C12 had an effect mirroring that of TRAIL knockdown: decreased numbers of EdU-positive (Fig. 4I) and Ki-67-positive cells (Fig. 4J), and an increased number of myogenin-positive cells (Fig. 4K). Taken together, the observations thus far suggest that muscle-derived TRAIL may signal to suppress cell cycle withdrawal that is necessary for myogenic differentiation.
It has been well established that p21CIP, a cell cycle inhibitor, plays a critical role in myogenic differentiation by promoting cell cycle withdrawal (30,31). We observed that p21 expression was upregulated when TRAIL (Fig. 5A) or TRAIL-R2 (Fig. 5B) was knocked down in C2C12 cells. TRAIL knockdown in primary myoblasts also resulted in a modest but statistically significant increase of p21 expression (Fig. 5C). Conversely, TRAIL overexpression in C2C12 cells decreased p21 expression, and reversed the effect of TRAIL knockdown on p21 upregulation (Fig. 5D). Adding sTRAIL to C2C12 cells revealed an acute (and thus potentially direct) effect – p21 expression was reduced as early as 30 min after sTRAIL addition (Fig. 5E). These observations are consistent with p21, and subsequently cell cycle withdrawal, being a mediator of the myogenic effects of TRAIL.
Figure 5. TRAIL inhibits p21 expression.

(A) C2C12 cells were transduced with lentiviruses expressing shTRAIL or shScramble followed by 2 days of puromycin selection. Cells were then lysed at 0 hr of differentiation followed by western analysis (n=6). (B) C2C12 cells were transduced with lentiviruses expressing shTRAIL-R2 or shScramble followed by 2 days of puromycin selection. Cells were then lysed at 0 hr of differentiation and subjected to western analysis (n=7). (C) Mouse primary myoblasts were transduced with lentiviruses expressing shRNAs overnight, cultured in growth medium for 1 day, and then lysed at 0 hr of differentiation followed by western analysis (n=4). (D) C2C12 cells treated as in (A) were transfected with TRAIL or empty vector, selected with hygromycin and lysed at 0 hr of differentiation, followed by western analysis (n=4). (E) C2C12 cells were induced to differentiate for 6 to 9 hrs, and then treated with vehicle or 100 ng/mL sTRAIL for 15, 30 or 60 min, followed by cell lysis and western analysis (n=5). (F) C2C12 cells were transduced with adenoviruses expressing human full-length TRAIL or luciferase (Control), followed by differentiation for 72 hrs with or without 2 μg/mL Ara-C during the first 24 hr of differentiation. Cells were stained for MHC (green) and DAPI (magenta), and differentiation and fusion indices were quantified (n=3). Expression of TRAIL was confirmed by western blotting. Quantification of p21 protein was performed by densitometry analysis of western blots. The level of p21 protein was determined relative to the level of tubulin, and the data were always normalized to control at each time point. All error bars represent SEM. For (F), one-tailed t-test was performed to compare each data point to control (without Ara-C). For all other panels, two-tailed t-test was performed to compare each data to control (first bar in graph unless otherwise noted). *P < 0.05; **P < 0.01. NS, not significant. Scale bar: 50 μm.
To directly probe a causal role of cell cycle withdrawal in the regulation of myogenesis by TRAIL, we treated C2C12 cells with the cell cycle inhibitor Ara-C. As shown in Fig. 5F, Ara-C treatment rescued differentiation from the inhibition by overexpression of TRAIL, as reflected in both differentiation and fusion indices. Collectively, our data suggest that TRAIL inhibits myogenic differentiation by preventing myoblasts from exiting the cell cycle.
TRAIL depletion enhances muscle regeneration in vivo
To probe a physiological relevance of the myogenic function of TRAIL discovered above in cell culture, we turned to a well-established mouse model of injury-induced muscle regeneration. Barium chloride (BaCl2) injection into the tibialis anterior (TA) muscle of the hindlimb leads to confined muscle necrosis, which is followed by de novo regeneration (28,32). Here, we co-injected lentivirus expressing TRAIL shRNA with BaCl2 into TA muscles and isolated the injected muscles for analysis on various days after injury (AI). Knockdown of TRAIL was confirmed by analyzing the mRNA isolated from injected muscles on Day 3 AI. Because lentivirus delivery was only expected in the injured part, which was typically 1/3 to 1/2 of the TA muscle, the observed knockdown efficiency of 20% in the entire TA was fully expected (Fig. 6A). Importantly, we observed statistically significant, ~25% increase in the average regenerating myofiber size (cross sectional area) upon TRAIL knockdown on both Day 7 and Day 14 AI (Fig. 6B), suggesting that endogenous TRAIL has a negative role in muscle regeneration.
Figure 6. TRAIL knockdown enhances muscle regeneration post-injury.
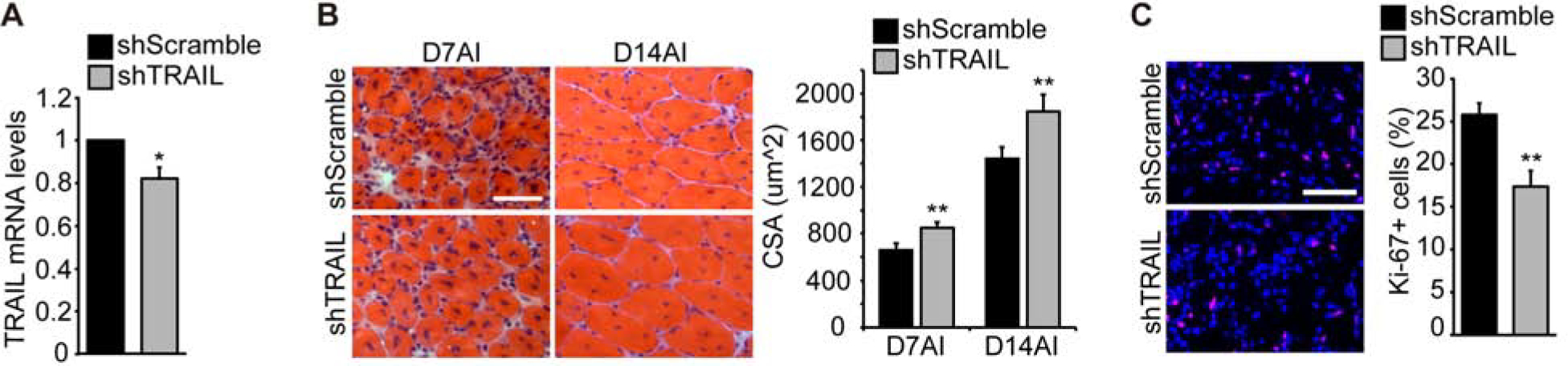
(A) Mouse TA muscles were co-injected with BaCl2 and lentiviruses expressing shTRAIL or shScramble, isolated on Day 3 AI and subjected to RNA isolation and qRT-PCR (n=5). (B) TA muscles injected as in (A) were isolated on Day 7 or Day 14 AI and subjected to cryosection. Upon H&E staining, cross-sectional area (CSA) was quantified for regenerating myofibers (n=6 for D7AI, n=8 for D14AI). (C) TA muscles injected as in (A) were isolated on Day 3 AI, cryosectioned and immunostained for Ki-67 (red) along with DAPI (blue). The percentage of Ki-67 positive cells was quantified (n=5). All error bars represent SEM. Two-tailed t-test was performed to compare shTRAIL to shScramble. *P < 0.05; **P < 0.01. Scale bars: 50 μm.
To further test the possibility that the upregulated muscle regeneration upon TRAIL knockdown in vivo was due to enhanced cell cycle withdrawal as we observed in vitro, we probed Day 3 AI TA muscle sections for the proliferation marker Ki-67. As shown in Fig. 6C, TRAIL knockdown muscles had fewer Ki-67-positive cells, indicating a lower level of proliferation at the early stage of muscle regeneration. This observation is consistent with TRAIL preventing proliferating cells from exiting the cell cycle.
ERK mediates myogenic function of TRAIL
We next searched for a potential signaling mechanism that mediates the myogenic function of TRAIL. The MAP kinase ERK1/2, a well-known regulator of cell proliferation, suppresses differentiation, likely by preventing cell cycle withdrawal (33). In addition, TRAIL has been reported to activate ERK in non-muscle cells (18,34). Therefore, we decided to test a potential role of ERK1/2 signaling in the regulation of myogenic differentiation by TRAIL. First, we treated myoblasts with recombinant TRAIL and examined phosphorylation of ERK1/2 at various time points. As shown in Fig. 7A, after 2, 4 and 6 hrs of TRAIL treatment, phosphorylation of ERK1/2 was found to increase modestly. Consistently, TRAIL knockdown in primary myoblasts led to a decrease in pERK (Fig. 7B). The specificity of the anti-pERK antibody was confirmed with lysates of serum-starved and serum-stimulated C2C12 cells (Fig. S1).
Figure 7. TRAIL inhibits differentiation through ERK signaling.
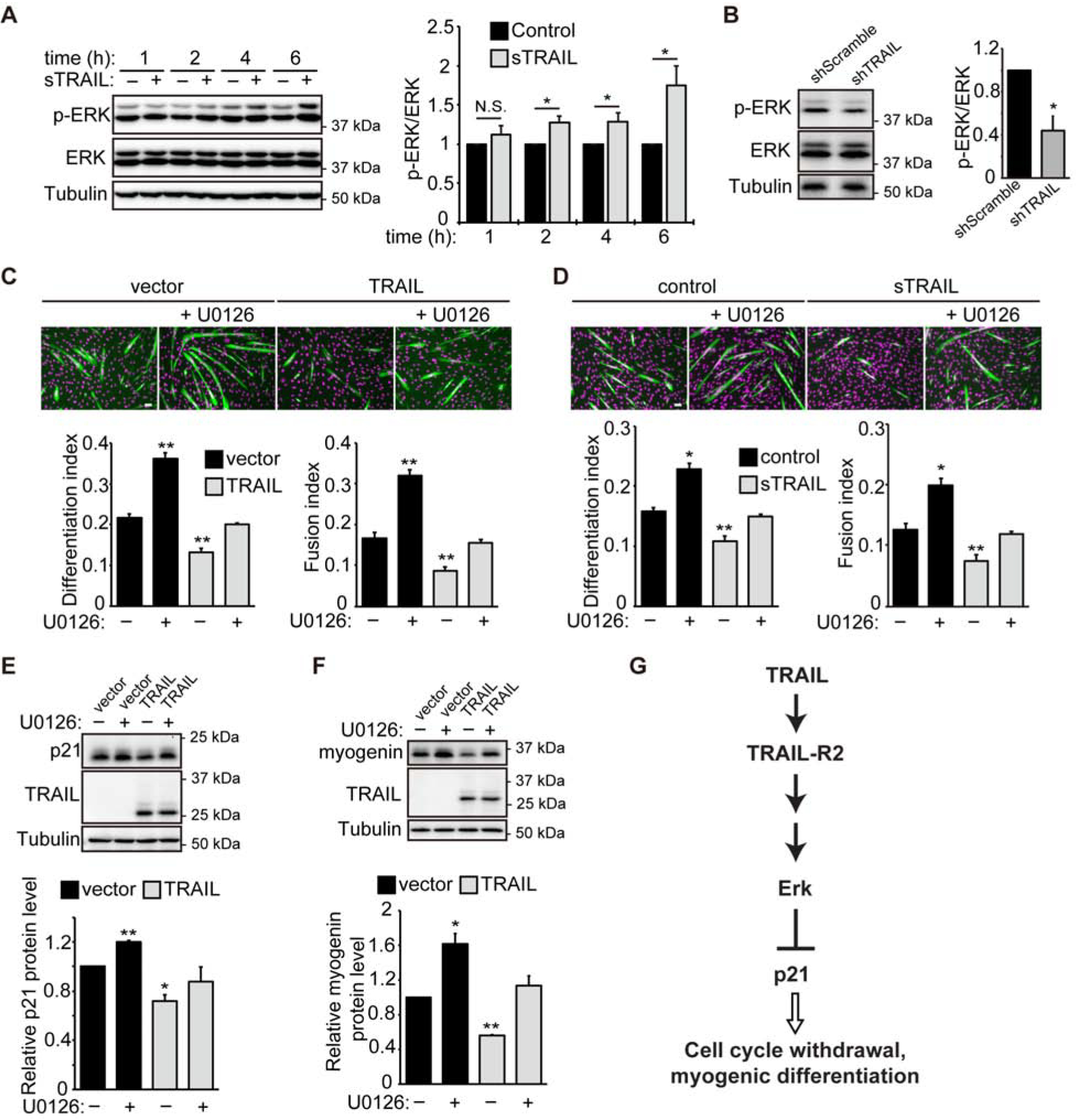
(A) C2C12 cells were induced to differentiation for 6 to 9 hrs, and then treated with 100 ng/mL sTRAIL or vehicle (control). Cells were lysed at 1, 2, 4 or 6 hrs, followed by western analysis (n=5). (B) Mouse primary myoblasts were transduced with lentiviruses expressing shRNAs overnight, cultured in growth medium for 1 day, and then induced to differentiate for 24 hrs, followed by lysis and western analysis (n=4). (C) C2C12 cells were transfected with TRAIL or empty vector, selected with hygromycin, and induced to differentiate for 72 hrs in the presence or absence of U0126 (3 μM) for the first 24 hrs of differentiation. Cells were then stained for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=4). (D) C2C12 cells were grown with 100 ng/mL sTRAIL or vehicle (Control) for 6 to 9 hrs and induced to differentiate for 72 hrs in the presence or absence of sTRAIL for the first 24 hrs of differentiation. U0126 (3 μM) was also added for the first 24 hrs of differentiation. Cells were than stained for MHC (green) and DAPI (magenta). Differentiation and fusion indices were quantified (n=3). (E) Cells treated as in (C) were induced to differentiate for 6 hrs in the presence or absence of U0126 (3 μM). Cells were then lysed and subjected to western analysis (n=4). (F) Cells treated as in (C) were induced to differentiate for 48 hrs in the presence or absence of U0126 (3 μM) for the first 24 hrs of differentiation. Cells were then lysed and subjected to western analysis (n=4). Quantification of western blots was performed by densitometry. The level of each specific protein was determined relative to the level of tubulin, and the data were normalized to control. All error bars represent SEM. Two-tailed t-test was performed to compare each data to control. *P < 0.05; **P < 0.01. Scale bars: 50 μm. (G) A proposed model: TRAIL/TRAIL-R2 negatively regulates myogenesis by inhibiting cell cycle withdrawal through activation of ERK signaling and inhibition of p21 expression.
To directly probe a functional relevance of ERK1/2 signaling in mediating TRAIL’s effect, we utilized the highly specific MEK inhibitor U0126. Treatment by U0126 reversed the inhibitory effect of overexpressed TRAIL (Fig. 7C) and sTRAIL (Fig. 7D) on myoblast differentiation such that differentiation and fusion indices were restored to the control levels (Fig. 7E). In cells overexpressing TRAIL, the reduced p21 expression was restored by U0126 treatment to a level no longer significantly different from the control (without TRAIL overexpression), consistent with p21 being a mediator of the effect of ERK signaling on cell cycle withdrawal. Finally, expression of the differentiation marker myogenin was also restored in TRAIL-overexpressing cells by U0126 (Fig. 7F). Taken together, our observations support the notion that TRAIL negatively regulates myogenic differentiation in an ERK1/2-dependent manner.
Discussion
In this report we have provided evidence for a role of TRAIL in negatively regulating skeletal myogenesis both in vitro and in vivo, a non-canonical function distinct from the widely studied pro-apoptotic signaling of TRAIL. Our data suggest that TRAIL exerts this myogenic function by suppressing cell cycle withdrawal necessary for myoblast differentiation. We further show that TRAIL-R2, the only known functional TRAIL receptor in mice, ERK signaling, and the cell cycle inhibitor p21 likely mediate TRAIL’s myogenic function (see Fig. 7G for a proposed model). Previously TRAIL-R2 (DR5) was reported to have a positive role in myoblast differentiation through the FADD/Caspase pathway that regulates MyoD expression (35). This discrepancy with our findings could be due to the different myoblast cell lines utilized – 23A2 in that study and C2C12 in ours. It is important to note that our in vitro findings have been recapitulated in vivo by our observations in a muscle regeneration model.
It was reported that a soluble TRAIL induced apoptosis in murine myoblasts (23). However, we did not observe any change in apoptosis of C2C12 cells upon either TRAIL knockdown (Fig. S2A) or recombinant sTRAIL addition at the concentration (100 ng/mL) found to inhibit differentiation (Fig. S2B). In the aforementioned study induction of apoptosis was observed in 23A2 myoblasts treated with a soluble TRAIL at 0.5–2 μg/mL, and indeed we found that apoptosis was induced in C2C12 cells by TRAIL at those high concentrations (Fig. S2B). Therefore, it is likely that TRAIL has dose-dependent dual effects on skeletal muscle cells – at low doses it suppresses myogenic differentiation and it can induce apoptosis at high concentrations. Dose-dependent pleiotropic functions have been reported for other TNF superfamily members. For instance, in primary culture of Schwann cells, treatment with low dosages (1–2 ng/mL) of TNF-α stimulated cell proliferation, whereas higher dosages (10–40 ng/mL) induced apoptosis (36). Similarly, low dosages (0.156–1.56 ng/mL) of Fas ligand (FasL, also known as CD95 and Apo-1L) stimulated proliferation of brain endothelial cells, and higher dosages (100–1000 ng/mL) induced apoptosis (37). Hence, it appears to be a common theme that TNF factors promote cell proliferation and induce cell death at low and high dosages, respectively. However, our data show that TRAIL does not affect the proliferation of myoblasts per se; only when myoblasts are poised for differentiation does TRAIL prevent their withdrawal from the cell cycle.
We observed enhanced muscle regeneration in mice that received intramuscular delivery of TRAIL shRNA. However, neither TRAIL−/− mice (38) nor TRAIL-R2−/− mice (39) displayed any developmental phenotype, although they have not been studied in any context of adult skeletal muscle functions. Compensatory effects could take place in the knockout mice, given the known redundancy of functions of TNF superfamily members. Alternatively, it is possible that TRAIL has a specific function in adult muscle regeneration that is distinct from embryonic muscle development. It also cannot be ruled out that the knockdown phenotype is dependent on the residual TRAIL, which is absent upon knockout.
It is important to acknowledge that our intramuscular RNAi delivery method would result in the knockdown of TRAIL in both muscle cells and infiltrating immune cells (macrophages, in particular). Hence, we are not able to conclude that it is the muscle-derived TRAIL that acts as a brake on muscle regeneration. In fact, there is a general paucity of experimental evidence in the literature that definitively proves the function of muscle-secreted cytokines (or myokines) on myogenesis in vivo. Studies with muscle-specific manipulation of endogenous gene expression would be necessary to establish the physiological relevance of those muscle-derived cytokines, including TRAIL.
TRAIL has long been studied as a candidate anti-cancer agent (6). Our current findings implicate TRAIL in a potentially new therapeutic strategy in muscle disease, regardless of the relevant source of TRAIL (muscle versus infiltrating immune cells). In a recent study, injection of an anti-TRAIL antibody resulted in improved strength of dystrophic EDL muscles in mdx mice, a model for Duchenne muscular dystrophy (DMD) (40). Although this effect was postulated to be due to suppressed apoptosis, in light of our results a role of the anti-TRAIL antibody on myogenic differentiation in the regeneration of dystrophic muscles is also possible. Indeed, DMD muscles contain elevated numbers of satellite cells, which fail to enter the myogenic program, possibly due to prolonged cell division (41). TRAIL may contribute to preventing the satellite cells from exiting the cell cycle.
Our previous RNAi screen identified a number of cytokines as candidates of autocrine regulators of C2C12 myoblast differentiation (42). Two of them, the chemokine C-X-C motif ligand 14 (Cxcl14) and Fms-like tyrosine kinase 3 ligand (Flt3L) have been found to regulate myogenic differentiation via suppressing and promoting cell cycle withdrawal, respectively, both mediated by ERK signaling and expression of the cell cycle inhibitor p21 (43,44). Additional muscle-derived cytokines with a similar function and mechanism are likely to continue to emerge. It is reasonable to speculate that nature has devised a cytokine network to ensure fail-proof and/or finely-tuned control over a point of no return in the myogenic process such as exiting the cell cycle. Future investigation of these cytokines at the systems level is warranted.
Supplementary Material
TRAIL, as well as its receptor TRAIL-R2, has a non-apoptotic function in skeletal muscle as a negative regulator of myogenic differentiation.
TRAIL signaling inhibits cell cycle withdrawal, a requisite step for myogenic differentiation.
Depletion of TRAIL enhances skeletal muscle regeneration in vivo.
The negative myogenic function of TRAIL is mediated by ERK signaling.
Funding information
Funding for this work was provided by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR048914 to J.C.).
Abbreviations:
- Ara-C
Cytosine β-D-arabinofuranoside hydrochloride
- EdU
5-ethynyl-2’-deoxyuridine
- MHC
myosin heavy chain
- TA
tibialis anterior
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflicts of interest in regards to this manuscript.
References
- 1.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, and Ashkenazi A (1996) Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 271, 12687–12690 [DOI] [PubMed] [Google Scholar]
- 2.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, and et al. (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 [DOI] [PubMed] [Google Scholar]
- 3.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, and Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5, 157–163 [DOI] [PubMed] [Google Scholar]
- 4.Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM, and Repasky EA (2002) Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients’ colon tumors grown in SCID mice. Cancer Res 62, 5800–5806 [PubMed] [Google Scholar]
- 5.Sedger LM, Glaccum MB, Schuh JC, Kanaly ST, Williamson E, Kayagaki N, Yun T, Smolak P, Le T, Goodwin R, and Gliniak B (2002) Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur J Immunol 32, 2246–2254 [DOI] [PubMed] [Google Scholar]
- 6.von Karstedt S, Montinaro A, and Walczak H (2017) Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer 17, 352–366 [DOI] [PubMed] [Google Scholar]
- 7.Kimberley FC, and Screaton GR (2004) Following a TRAIL: update on a ligand and its five receptors. Cell Res 14, 359–372 [DOI] [PubMed] [Google Scholar]
- 8.Pan G, Ni J, Wei YF, Yu G, Gentz R, and Dixit VM (1997) An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 [DOI] [PubMed] [Google Scholar]
- 9.LeBlanc HN, and Ashkenazi A (2003) Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 10, 66–75 [DOI] [PubMed] [Google Scholar]
- 10.Vitovski S, Phillips JS, Sayers J, and Croucher PI (2007) Investigating the interaction between osteoprotegerin and receptor activator of NF-kappa B or tumor necrosis factor-related apoptosis-inducing ligand. Journal of Biological Chemistry 282, 31601–31609 [DOI] [PubMed] [Google Scholar]
- 11.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, and Young PR (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273, 14363–14367 [DOI] [PubMed] [Google Scholar]
- 12.Wu GS, Burns TF, Zhan Y, Alnemri ES, and El-Deiry WS (1999) Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 59, 2770–2775 [PubMed] [Google Scholar]
- 13.Schneider P, Olson D, Tardivel A, Browning B, Lugovskoy A, Gong D, Dobles M, Hertig S, Hofmann K, Van Vlijmen H, Hsu YM, Burkly LC, Tschopp J, and Zheng TS (2003) Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem 278, 5444–5454 [DOI] [PubMed] [Google Scholar]
- 14.Esperon ED, Cordier G, and Engel N (2012) A genomic reservoir for Tnfrsf genes is developmentally regulated and imprinted in the mouse. Epigenetics-Us 7, 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, and Ashkenazi A (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem 280, 40599–40608 [DOI] [PubMed] [Google Scholar]
- 16.Ehrhardt H, Fulda S, Schmid I, Hiscott J, Debatin KM, and Jeremias I (2003) TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene 22, 3842–3852 [DOI] [PubMed] [Google Scholar]
- 17.Ishimura N, Isomoto H, Bronk SF, and Gores GJ (2006) Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am J Physiol Gastrointest Liver Physiol 290, G129–136 [DOI] [PubMed] [Google Scholar]
- 18.Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D, and Zauli G (2003) TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 107, 2250–2256 [DOI] [PubMed] [Google Scholar]
- 19.Secchiero P, Zerbinati C, Rimondi E, Corallini F, Milani D, Grill V, Forti G, Capitani S, and Zauli G (2004) TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell Mol Life Sci 61, 1965–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, and Buckingham M (2005) Direct isolation of satellite cells for skeletal muscle regeneration. Science 309, 2064–2067. Epub 2005 Sep 2061. [DOI] [PubMed] [Google Scholar]
- 21.Yaffe D, and Saxel O (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727. [DOI] [PubMed] [Google Scholar]
- 22.Blau HM, Chiu CP, and Webster C (1983) Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell 32, 1171–1180. [DOI] [PubMed] [Google Scholar]
- 23.O’Flaherty J, Mei Y, Freer M, and Weyman CM (2006) Signaling through the TRAIL receptor DR5/FADD pathway plays a role in the apoptosis associated with skeletal myoblast differentiation. Apoptosis 11, 2103–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y, Fedczyna TO, McVicker V, Caliendo J, Li H, and Pachman LM (2007) Apoptosis in the skeletal muscle of untreated children with juvenile dermatomyositis: impact of duration of untreated disease. Clin Immunol 125, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alger HM, Raben N, Pistilli E, Francia DL, Rawat R, Getnet D, Ghimbovschi S, Chen YW, Lundberg IE, and Nagaraju K (2011) The role of TRAIL in mediating autophagy in myositis skeletal muscle: a potential nonimmune mechanism of muscle damage. Arthritis Rheum 63, 3448–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge Y, Sun Y, and Chen J (2011) IGF-II is regulated by microRNA-125b in skeletal myogenesis. J Cell Biol 192, 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Son K, You JS, Yoon MS, Dai C, Kim JH, Khanna N, Banerjee A, Martinis SA, Han G, Han JM, Kim S, and Chen J (2019) Nontranslational function of leucyl-tRNA synthetase regulates myogenic differentiation and skeletal muscle regeneration. Journal of Clinical Investigation 129, 2088–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge Y, Wu AL, Warnes C, Liu J, Zhang C, Kawasome H, Terada N, Boppart MD, Schoenherr CJ, and Chen J (2009) mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am J Physiol Cell Physiol 297, C1434–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida N, Yoshida S, Koishi K, Masuda K, and Nabeshima Y (1998) Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J Cell Sci 111, 769–779. [DOI] [PubMed] [Google Scholar]
- 30.Guo K, Wang J, Andres V, Smith RC, and Walsh K (1995) MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol 15, 3823–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, and Lassar AB (1995) Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 267, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 32.Caldwell CJ, Mattey DL, and Weller RO (1990) Role of the basement membrane in the regeneration of skeletal muscle. Neuropathol Appl Neurobiol 16, 225–238 [DOI] [PubMed] [Google Scholar]
- 33.Bennett AM, and Tonks NK (1997) Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278, 1288–1291 [DOI] [PubMed] [Google Scholar]
- 34.Zoller V, Funcke JB, Roos J, Dahlhaus M, Abd El Hay M, Holzmann K, Marienfeld R, Kietzmann T, Debatin KM, Wabitsch M, and Fischer-Posovszky P (2017) Trail (TNF-related apoptosis-inducing ligand) induces an inflammatory response in human adipocytes. Sci Rep 7, 5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freer-Prokop M, O’Flaherty J, Ross JA, and Weyman CM (2009) Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation. 78, 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang X, Wang Y, Zhou S, Qian T, and Gu X (2013) Signaling pathways regulating dose-dependent dual effects of TNF-alpha on primary cultured Schwann cells. Mol Cell Biochem 378, 237–246 [DOI] [PubMed] [Google Scholar]
- 37.Zhang C, Gao F, Teng F, and Zhang M (2015) Fas/FasL Complex Promotes Proliferation and Migration of Brain Endothelial Cells Via FADD-FLIP-TRAF-NF-kappaB Pathway. Cell Biochem Biophys 71, 1319–1323 [DOI] [PubMed] [Google Scholar]
- 38.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, and Smyth MJ (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168, 1356–1361 [DOI] [PubMed] [Google Scholar]
- 39.Finnberg N, Gruber JJ, Fei P, Rudolph D, Bric A, Kim SH, Burns TF, Ajuha H, Page R, Wu GS, Chen Y, McKenna WG, Bernhard E, Lowe S, Mak T, and El-Deiry WS (2005) DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol 25, 2000–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dufresne SS, Boulanger-Piette A, Bosse S, Argaw A, Hamoudi D, Marcadet L, Gamu D, Fajardo VA, Yagita H, Penninger JM, Russell Tupling A, and Frenette J (2018) Genetic deletion of muscle RANK or selective inhibition of RANKL is not as effective as full-length OPG-fc in mitigating muscular dystrophy. Acta Neuropathol Commun 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang NC, Chevalier FP, and Rudnicki MA (2016) Satellite Cells in Muscular Dystrophy - Lot in Polarity. Trends Mol Med 22, 479–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge Y, Waldemer RJ, Nalluri R, Nuzzi PD, and Chen J (2013) RNAi screen reveals potentially novel roles of cytokines in myoblast differentiation. PLoS One 8, e68068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waldemer-Streyer RJ, Reyes-Ordonez A, Kim D, Zhang R, Singh N, and Chen J (2017) Cxcl14 depletion accelerates skeletal myogenesis by promoting cell cycle withdrawal. NPJ Regen Med 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ge Y, Waldemer RJ, Nalluri R, Nuzzi PD, and Chen J (2013) Flt3L is a novel regulator of skeletal myogenesis. J Cell Sci 126, 3370–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.