

Abstract
Disruption of protein:protein interactions (PPIs) that regulate the function of voltage-gated Na+ (Nav) channels leads to neural circuitry aberrations that have been implicated in numerous channelopathies. One example of this pathophysiology is mediated by dysfunction of the PPI between Nav1.6 and its regulatory protein fibroblast growth factor 14 (FGF14). Thus, peptides derived from FGF14 might exert modulatory actions on the FGF14:Nav1.6 complex that are functionally relevant. The tetrapeptide Glu-Tyr-Tyr-Val (EYYV) mimics surface residues of FGF14 at the β8–β9 loop, a structural region previously implicated in its binding to Nav1.6. Here, peptidomimetics derived from EYYV (6) were designed, synthesized, and pharmacologically evaluated to develop probes with improved potency. Addition of hydrophobic protective groups to 6 and truncation to a tripeptide (12) produced a potent inhibitor of FGF14:Nav1.6 complex assembly. Conversely, addition of hydrophobic protective groups to 6 followed by addition of an N-terminal benzoyl substituent (19) produced a potentiator of FGF14:Nav1.6 complex assembly. Subsequent functional evaluation using whole-cell patch-clamp electrophysiology confirmed their inverse activities, with 12 and 19 reducing and increasing Nav1.6-mediated transient current densities, respectively. Overall, we have identified a negative and positive allosteric modulator of Nav1.6, both of which could serve as scaffolds for the development of target-selective neurotherapeutics.
Keywords: protein:protein interactions (PPIs), voltage-gated Na+ (Nav) channels, fibroblast growth factor 14 (FGF14), peptidomimetics, molecular docking, neurotherapeutics
1. Introduction
In excitable cells, voltage-gated sodium (Nav) channels are the primary molecular determinants of the generation and conduction of action potentials [1]. Of the nine different pore-forming α-subunits that have been described (Nav1.1–1.9), the Nav1.1–1.3 and 1.6 isoforms are the primary Nav channels expressed in the central nervous system (CNS) [2]. Given their primacy in modulating neuronal excitability, it is unsurprising that aberrant activity of these isoforms has been implicated in the etiologies of numerous neurologic and neurodevelopmental disorders, including epilepsy [3,4,5,6], migraines [7,8,9], and autism [10,11,12]. Lending credence to the involvement of Nav channel aberrations in neuropsychiatric disorders, Nav channel blockers are also commonly used as adjunct therapies for the treatment of bipolar disorder [13,14], anxiety [15], and schizophrenia [16,17,18]. Given this primacy of Nav channel dysfunction in the etiology of virtually all CNS disorders, they have been the target of many drug discovery campaigns. Such efforts, however, have been of limited success, as developed lead compounds often fail to demonstrate appreciable isoform selectivity and resultantly confer undesirable off-target side effects [19].
Nav1.6 channels are abundantly expressed in medium spiny neurons (MSNs) of the nucleus accumbens (NAc), a neuroanatomical region that governs mesocorticolimbic circuitry and is involved in motivation [20], reward processing [21], learning [22], and locomotion [23,24,25]. With translational studies increasingly illuminating linkages between dysfunction of MSNs in the NAc and neuropsychiatric and neurological symptomologies [26,27,28,29], the development of chemical probes to modulate these neurons would enable further mechanistic elucidation of these neuropathophysiological processes. Given the abundant expression of Nav1.6 and its regulatory protein fibroblast growth factor 14 (FGF14) in these neurons, a feasible approach for developing such probes is through the rational design of small molecules targeting the FGF14:Nav1.6 protein:protein interaction (PPI) interface [30,31].
To that end, we previously identified two peptides capable of modulating FGF14:Nav1.6 complex assembly [32]. One of these peptides, Phe-Leu-Pro-Lys (FLPK; 1, Figure 1A), was derived from a four amino acid sequence located on the β12 sheet of FGF14 at the FGF14:Nav1.6 PPI interface, whereas the other, Glu-Tyr-Tyr-Val (EYYV; 5, Figure 1B), was derived from an amino acid sequence located on the exposed β8–β9 loop of FGF14 [32]. To improve the potency and drug-like properties of 1, we acetylated and aminated its N-terminus and C-terminus respectively (2, Figure 1A), and subsequently designed, synthesized, and pharmacologically evaluated peptidomimetics derived from the protected FLPK scaffold [30,31]. Briefly, altering the N-terminal substituent to Cbz and adding a Boc protective group to the constituent lysine residue of the protected FLPK scaffold produced ZL181 (4, Figure 1A), which inhibited FGF14:Nav1.6 complex assembly (half maximal inhibitory concentration (IC50) = 63 µM) and decreased maximal and instantaneous firing frequencies in MSNs of the NAc [31]. Subsequent efforts to optimize 2 included altering the C-terminal substituent to methoxyl (OMe) and the addition of a Fmoc protective group to the constituent lysine residue of the protected FLPK scaffold, which produced ZL0177 (3, Figure 1A). Crucially, this analog displayed markedly improved potency, inhibiting FGF14:Nav1.6 complex assembly with an IC50 value of 11 µM. Additionally, mechanism of action (MOA) studies using whole-cell patch-clamp electrophysiology revealed that ZL0177 significantly reduced Nav1.6-mediated peak current density and caused a depolarizing shift in voltage-dependence of Nav1.6 channel activation, suggesting that the peptidomimetic’s activity is conferred by functioning as a partial FGF14 mimic [30].
Figure 1.

Structural modification strategy for development of peptidomimetics based on hot segments of FGF14 at its interface with Nav1.6. (A) The previously reported parental peptide Phe-Leu-Pro-Lys (FLPK) and its analogs, ZL177 (3) and ZL181 (4). (B) Structural modification strategy for development of chemical probes derived from the Glu-Tyr-Tyr-Val (EYYV) scaffold.
To develop additional probes to further elucidate the intricate biophysical changes Nav1.6 channels undergo on account of their PPI with FGF14, we herein report our derivation and pharmacological evaluation of EYYV analogs (6, Figure 1B). To do so, all newly synthesized peptidomimetics were screened using an in-cell assay, which revealed that addition of hydrophobic protective groups to 6 followed by truncation to a tripeptide produced a potent inhibitor of FGF14:Nav1.6 complex assembly. Conversely, addition of hydrophobic protective groups to 6 followed by addition of an N-terminal benzoyl substituent produced a potentiator of the complex’s assembly. After surface plasmon resonance (SPR) studies revealed that both peptidomimetics exhibited great affinity for Nav1.6, subsequent functional evaluation of the two analogs using whole-cell patch-clamp electrophysiology confirmed their inverse activities, with the inhibitor and potentiator of FGF14:Nav1.6 complex assembly reducing and increasing Nav1.6-mediated transient current densities, respectively. Overall, by employing this novel approach that combines in-cell screening with orthogonal validation measures, including SPR and whole-cell patch-clamp electrophysiology, we were able to identify both a negative and positive allosteric modulator of Nav1.6, both of which could serve as ideal scaffolds for the development of targeted pharmacological probes and, with further chemical optimization, potential neurotherapeutic drug leads.
2. Results
2.1. Chemistry
As outlined in Scheme 1, the diverse EYYV analogs were synthesized with modifications to both their C- and N-termini (Figure 1B). The dipeptide analogs (10 and 11) were generated by condensation of commercially available compound 7 with compound 8 and 9, respectively. Deprotecting the Fmoc group of compounds 8 and 9, followed by coupling with compound 7, led to tripeptide compounds 12 and 13, respectively. Following a similar synthetic procedure to that of the preparation of compound 12, compounds 12 and 13 were converted into tetrapeptide compounds 15 and 16 in excellent yields. Compounds 17–24 were prepared by deprotection of compounds 15 and 16 and the subsequent introduction of the corresponding substituents, including acetyl, benzoyl, adamantanyl carbamic, adamantane-1-carbonyl, decanoyl, cyclohexanecarbonyl, and Cbz. Copies of 1H and 13C NMR spectra of all newly synthesized analogs are included in the Supplementary Materials.
Scheme 1.

Synthesis of compound 6 analogs. Reagents and conditions: (a) HBTU, HOBt, DIPEA, DMF, 0 °C to room temperature (rt), overnight, 93% for 10, 91% for 11. (b) (1) HNEt2, MeCN, rt, 1 h, quant; (2) 7, HBTU, HOBt, DIPEA, DMF, 0 °C to rt, overnight, 78% for 12, 73% for 13. (c) (1) HNEt2, MeCN, rt, 1 h, quant; (2) HBTU, HOBt, DIPEA, DMF, 0 °C to rt, overnight, 77% for 15, 74% for 16. (d) for 17–20 and 24, (1) HNEt2, MeCN, rt, 1 h, quant; (2) Et3N, CH2Cl2, 0 °C to rt, overnight, 56–78%; for 21–23, HBTU, HOBt, DIPEA, DMF, 0 °C to rt, overnight, 58–64%. Abbreviations: HBTU = 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOBt = 1-Hydroxybenzotriazole; DIPEA = N,N-Diisopropylethylamine; and DMF = N,N-dimethylformamide.
2.2. Biology
2.2.1. In-Cell Testing of Analogs Using the Split Luciferase Complementation Assay (LCA)
We previously developed, optimized, and reported an in-cell LCA that allows for the reproducible identification of modulators of FGF14:Nav1.6 complex assembly [33]. Briefly, a HEK293 cell line stably expressing both CLuc-FGF14 and CD4-Nav1.6-NLuc recombinant proteins (hereafter referred to as Clone V cells) was developed. In this system, binding of FGF14 to the C-terminal tail of Nav1.6 facilitates the reconstitution of the luciferase enzyme, which in the presence of the substrate luciferin, produces luminescence. This assay was employed in the present investigation by reconstituting all the newly synthesized analogs derived from 6 in dimethyl sulfoxide (DMSO), bringing them to a preliminary screening concentration of 25 µM, administering them to Clone V cells seeded into 96-well plates, and subsequently normalizing the luminescence values observed in treatment wells to per plate controls (0.5% DMSO alone), the results of which are summarized in Figure 2A and Table 1. Before proceeding with further analyses, however, we first ensured that the observed modulatory effects of these peptidomimetics were not merely artifacts stemming from cytotoxicity. To do so, all EYYV analogs were tested using the CellTiter-Blue® Cell Viability (CTB) assay (Figure 2B), which revealed that none of the screened analogs demonstrated cytotoxicity (Figure 2B).
Figure 2.

Screening of peptidomimetics against the FGF14:Nav1.6 complex using the split luciferase complementation assay (LCA). (A) Clone V cells were treated with compounds (25 μM; n = 8 wells per compound) in 96-well plates for 12 h prior to luminescence reading, and maximal luminescence for each well was normalized to per plate 0.5% DMSO controls (n = 32 per plate). (B) To assess effects of compounds on cell viability, the CellTiter Blue reagent was dispensed immediately following luminescence reading in (A). Fluorescence was detected after incubation for 18 h. Tamoxifen, known to be toxic to HEK293 cells [33], was used as a positive control. Individual replicate values with mean ± standard error of the mean (SEM) are shown. Significance was assessed using one-way ANOVA with post hoc Dunnett’s multiple comparisons test. ** p < 0.0005. * p < 0.005.
Table 1.
Representation of Glu-Tyr-Tyr-Val (EYYV) analog modifications and corresponding LCA results.
| Compound | R1 (for 15–24) |
R2 (for 15–24) |
cLogP a | tPSA b | LCA (%Lum) c |
|---|---|---|---|---|---|
| 10 (PW01100) |
|
4.94 | 119.76 | 92.9 ± 2.1 | |
|
12
(PW0201) |
|
6.84 | 158.09 | 31.1 ± 2.0 | |
|
15
(PW0202) |
NH2 | Fmoc | 7.45 | 213.49 | 109.4 ± 2.5 |
|
16
(PW0210) |
OMe | Fmoc | 8.14 | 196.70 | 101.7 ± 2.0 |
|
17
(PW0203) |
NH2 | Ac | 4.05 | 204.26 | 128.4 ± 2.6 |
|
18
(PW0220) |
OMe | Ac | 4.74 | 187.47 | 104.3 ± 1.3 |
|
19
(PW0204) |
NH2 |
|
5.34 | 204.26 | 131.8 ± 1.9 |
|
20
(PW0205) |
NH2 |
|
6.18 | 216.29 | 95.3 ± 3.4 |
|
21
(PW0206) |
NH2 |
|
6.25 | 204.26 | 101.1 ± 2.2 |
|
22
(PW0207) |
NH2 |
|
7.17 | 204.26 | 123.4 ± 2.8 |
|
23
(PW0208) |
NH2 |
|
5.60 | 204.26 | 93.8 ± 3.3 |
|
24
(PW0211) |
NH2 | Cbz | 5.84 | 213.49 | 111.5 ± 2.5 |
a clogP: http://biosig.unimelb.edu.au/pkcsm/prediction. The LogP value of a compound, which is the logarithm of its partition coefficient between n-octanol and water-log (Coctanol/Cwater), is a well-established measurement of a compound′s hydrophilicity. Low hydrophilicities and, therefore, high LogP values cause poor absorption and permeation. b tPSA: https://www.molinspiration.com/cgi-bin/properties. tPSA (topological polar surface area) is defined as the surface sum over all polar atoms or molecules, which is predictive of a drug′s ability to permeate into cells. Exceedingly high tPSA values cause poor cell permeation and poor ability of cross the blood–brain barrier. c LCA results from compound screening (25 µM), as represented in Figure 2A. Percent luminescence values were calculated relative to per plate 0.5% DMSO controls and are shown as mean ± SEM.
As shown in Figure 2A and Table 1, we first investigated how the addition of hydrophobic protective groups to, and truncation of, 6 to a dipeptide (10) and tripeptide (12) modulated in-cell pharmacological activity. Whereas truncation to a dipeptide resulted in a loss of activity, the tripeptide analog (12) markedly inhibited FGF14:Nav1.6 complex assembly (Figure 2A and Table 1) and displayed low micromolar potency (IC50 = 23.7 µM; Figure 3 and Table 2).
Figure 3.

Dose response curves of top FGF14:Nav1.6 complex modulators. To determine potency (IC50 or EC50) and efficacy (percent luminescence at the bottom or top plateau for inhibitors (IMin) or enhances (EMax), respectively) each compound was tested at 14 concentrations (range: 0.25–175 µM; n = 4 replicates per concentration) using the LCA in 96-well plates. Data are shown as mean percent luminescence (relative to per plate 0.5% DMSO controls) values ± SEM. Non-linear regression curve fitting was performed using GraphPad Prism 8 to determine potency and efficacy values for each compound, as shown in Table 2.
Table 2.
Potency and efficacy of top peptidomimetics.
| Compound | IC50/EC50 (µM) a | IMin/EMax (%)a |
|---|---|---|
| 12 | 23.7 ± 0.7 | 18.3 ± 1.2 |
| 17 | 12.5 ± 1.9 b | 153.4 ± 3.5 e |
| 19 | 24.5 ± 1.7 c | 192.4 ± 3.2 f |
| 22 | 47.5 ± 10.6 d | 163.2 ± 6.5 g |
a Potency (estimated IC50/EC50) and efficacy (percent luminescence at the bottom or top plateau for inhibitors (IMin) or enhancers (EMax), respectively) values ± SEM for top peptidomimetics were calculated from the dose response non-linear regression curves represented in Figure 3. Significance was determined via unpaired t-test compared with the indicated group. b p < 0.005 compared to 19. c p = 0.0759 compared to 22. d p < 0.05 compared to 17. e p < 0.0005 compared to 19. f p < 0.05 compared to 22. g p = 0.2326 compared to 17.
After characterizing the in-cell activity of the dipeptide and tripeptide analog, we subsequently sought to design, synthesize, and pharmacologically evaluate tetrapeptide analogs with improved cLogP values and stability compared to the parental EYYV peptide. To do so, hydrophobic protective groups were added to the protected EYYV scaffold, which produced 17. In-cell screening of 17 revealed that it markedly potentiated assembly of the FGF14:Nav1.6 complex (Figure 2A and Table 1) and displayed low micromolar potency (EC50 = 12.5 µM; Figure 3 and Table 2). Having identified a potentiator of FGF14:Nav1.6 complex assembly, further chemical interventions were employed to develop a more efficacious enhancer. To do so, we first replaced the NH2 substituent at the C-terminus of compound 17 with a methoxyl (OMe) group, which produced 18. This R1 modification to OMe resulted in a complete loss of in-cell activity, a recurring finding that was observed in the only other analog with a C-terminal OMe (16), suggesting that a C-terminal NH2 substituent played a crucial role in conferring analogs with in-cell activity. To investigate how altering the hydrophobicity of peptidomimetics modulated their pharmacological activity, the acetyl group at the N-terminus of 17 was modified to various lipophilic groups, which produced 15, 19, 20, and 21. Substituting the Ac group with a Fmoc group (15) resulted in a significant loss of in-cell activity, a phenomenon that is likely attributable to the substituent conferring the analog with a highly steric structure and unfavorable cLogP value (cLogP = 7.45; Table 1). Further efforts to optimize 17 by introducing a N-terminal aryl group produced 19. Despite this analog displaying lessened potency relative to 17 (half maximal effective concentration (EC50) = 24.5 ± 1.7 µM versus 12.5 ± 1.9 µM for 19 and 17, respectively; p < 0.005), its efficacy was significantly improved (Emax = 192.4% ± 3.2% versus 153.4% ± 3.5% for 19 and 17, respectively; p < 0.0005; Figure 3 and Table 2). Whereas replacing the Ac group of 17 with an aryl group improved efficacy, modification of the R2 locale to fused alkyl ring substituents (20 and 21) abrogated in-cell activity. Additional attempts to optimize 17 entailed altering its N-terminal substituent to a long-chain alkyl, cycloalkyl, and Cbz group, which produced 22, 23, and 24, respectively. Although 22 and 24 retained activity, 22 displayed lessened potency (EC50 = 47.5 µM; Figure 3 and Table 2) relative to 17, and 24′s potentiation of FGF14:Nav1.6 complex assembly was comparably mild (Figure 2A and Table 1). Overall, these SAR studies using the LCA identified three potential positive allosteric modulators (PAMs; 17, 19, and 22) and one negative allosteric modulator (NAM; 12) of FGF14:Nav1.6 complex assembly, the four of which were selected for protein:ligand binding studies using SPR.
2.2.2. Characterization of Peptidomimetic Interactions with FGF14 and Nav1.6
After SAR studies using the LCA-identified potential PAMs (17, 19, and 22) and a NAM (12), we next sought to examine their kinetic interactions with FGF14 and Nav1.6 C-terminal tail. SPR-based kinetic analysis of interactions between drug-like compounds and target proteins has become a key method for drug discovery [34,35]. The determined kon and koff rate constants provide important information about interaction mechanisms and compound properties, enabling investigation of SARs and the rational modification of compounds. Therefore, we used SPR to assess and compare the binding of peptidomimetics to FGF14 and Nav1.6 C-terminal tail protein. Proteins were purified as previously described [31,36], immobilized to CM5 sensor chips, and increasing concentrations of compounds (0.195–100 µM) were flown over the chips at 60 µL/min. The results are shown in Figure 4 and Table 3 and Table 4. With the exception of 22, all compounds bound appreciably to FGF14 with affinities ranging from 2.9 to 14.3 μM (Table 3, left). While 12 and 19 bound strongly to Nav1.6, with affinities ranging from 2.3 to 9.7 μM, lower affinities were observed for 17 and 22 (Table 3, right).
Figure 4.

Determination of peptidomimetic binding kinetics to FGF14 and Nav1.6 C-terminal tail by SPR. Increasing concentrations of peptidomimetics (0.195–100 µM) were flown over purified FGF14 or Nav1.6 C-tail protein bound to CM5 chips using a flow rate of 60 µL/min. Kinetic analysis of each ligand/analyte interaction was obtained by fitting the response data to the simplest Langmuir 1:1 interaction model (KD = koff/kon), and binding sensorgrams are shown for FGF14 (left) and Nav1.6 (middle), with fitted curves represented in black. Steady-state saturation plots (right) are shown for comparison of compound binding to FGF14 (red) and Nav1.6 (blue), with response units (RU) normalized to the maximal binding response. The resulting equilibrium dissociation constants (KD), as well as kinetic association (kon) and dissociation (koff) rates, are provided in Table 3.
Table 3.
Biophysical characterization of peptidomimetic binding to FGF14 and Nav1.6 by Surface Plasmon Resonance (SPR).
| FGF14 | Nav1.6 | |||||
|---|---|---|---|---|---|---|
| Compound | KD
a (µM) |
kon (M−1 s−1) |
koff (s−1) |
KD
a (µM) |
kon (M−1 s−1) |
koff (s−1) |
| 12 | 2.98 ± 0.10 b | 6.44 × 103 | 1.86 × 10−2 | 9.69 ± 0.34 e | 4.29 × 103 | 4.23 × 10−2 |
| 17 | 14.3 ± 0.96 c | 6.03 × 103 | 8.80 × 10−2 | 54.4 ± 10.2 f | 9.01 × 103 | 7.83 × 10−2 |
| 19 | 6.49 ± 0.39 d | 2.06 × 103 | 1.30 × 10−2 | 2.20 ± 0.08 g | 6.65 × 103 | 1.50 × 10−2 |
| 22 | >100 | 6.90 × 102 | 1.09 × 10−1 | >100 | 7.30 × 102 | 1.30 × 10−1 |
a The equilibrium dissociation constants (KD) were derived from data shown in Figure 4. The KD shown for each compound is an average of that calculated using the simplest Langmuir 1:1 interaction model (KD = koff/kon, where KD is the dissociation constant, and koff and kon are the first-order rate constants for the dissociation and association, respectively, of the protein:ligand complex) and the steady-state saturation model. b p < 0.005 compared to 17.c p < 0.05 compared to 19. d p < 0.05 compared to 12. e p < 0.05 compared to 17. f p < 0.05 compared to 19. g p < 0.005 compared to 12.
Table 4.
Functional modulation of Nav1.6 by 12 and 19 relative to control a.
| Condition | Peak Current Density (pA/pF) b | Tau of Fast Inactivation (ms) c | V1/2 of Activation (mV) d | V1/2 of Steady-state Inactivation (mV) e | Long Term Inactivation (Depolarization Cycles 2–4) f | ||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | |||||
| DMSO | –55.39 ± 2.16 (10) | 1.17 ± 0.09 (7) | –20.04 ± 0.85 (10) | –58.58 ± 1.90 (5) | 0.89 ± 0.03 | 0.86 ± 0.03 | 0.85 ± 0.04 (6) |
| 12 | –18.30 ± 2.02 ** (10) | 1.30 ± 0.11 (7) | –20.40 ± 0.89 (10) | –57.59 ± 0.56 (6) | 0.88 ± 0.03 | 0.76 ± 0.03 * | 0.70 ± 0.04 * (7) |
| 19 | –79.63 ± 3.55 ** (8) | 1.12 ± 0.07 (7) | –22.49 ± 0.72 * (8) | –58.53 ± 1.69 (8) | 0.83 ± 0.02 | 0.73 ± 0.02 * | 0.73 ± 0.03 * (8) |
a Summary of the electrophysiological evaluation of 12 and 19. Results are expressed as the mean ± SEM. The number of independent experiments is shown in parentheses. Significance was determined via unpaired t-test compared with 0.1% DMSO. * p < 0.05; ** p < 0.005. b Peak current density, which describes the number of channels in a conductive (open) state, is a measure of the maximum influx of Na+ current (pA) into the cell normalized to membrane capacitance (pF) to control for variable cell sizes. c Tau of fast inactivation measures the decay phase of Na+ currents to characterize the time required for channels to transition from the conductive (open) state to a nonconductive state resulting from fast inactivation. d V1/2 of activation is a measure of the voltage at which half of available channels transition from the closed to the conductive (open) state. e V1/2 of steady-state inactivation is a measure of the voltage at which half of channels are available to transition into the conductive (open) state, while the other half are non-conductive due to steady-state (closed-state) inactivation. f Long-term (slow) inactivation is a type of inactivation induced by successive or prolonged depolarization cycles that causes collapse of the S5–S6 pore module of Nav channels resulting in a prolonged nonconductive inactivated state.
Interestingly, the tripeptide (12) demonstrated two-fold greater binding affinity toward FGF14 compared to 19 (KD of 2.98 vs. 6.49 µM, respectively; p < 0.05). As similar dissociation kinetics were observed for the two compounds (koff = 1.86 × 10−2 and 1.30 × 10−2 s−1 for 12 and 19, respectively; p > 0.4), this difference was largely driven by the faster association rate of 12, which can also be observed by the leftward shift of the FGF14 steady-state saturation curve (red) for 12 relative to 19 (Figure 4, right-top and lower middle). The affinities of the other two tetrapeptide analogs, 17 and 22, toward FGF14, were reduced by an even greater extent, collectively indicating that the presence of the glutamic acid residue, regardless of the R2 substituent present, prevents high-affinity FGF14 binding. This may suggest that the binding region of FGF14 is topologically constrained such that it cannot readily accommodate the larger tetrapeptide analogs, or that the presence of the glutamic acid residue masks crucial interactions between the YYV motif and FGF14.
Whereas the presence of the glutamic acid residue reduced the affinity of analogs toward FGF14, its inclusion conferred tighter binding to Nav1.6. This finding was underscored by the tripeptide exhibiting the lowest detectable binding strength toward Nav1.6. This suggests that the region of Nav1.6 bound to by these analogs is less topologically constrained relative to FGF14. Additionally, compared to 12 and 17, 19 exhibited about 4-fold greater binding affinity toward Nav1.6 (p < 0.005 for 12 vs. 19; p < 0.05 for 12 vs. 19), a difference largely driven by its comparatively slow dissociation rate (koff = 1.5 × 10−1 s−1). This difference can similarly be observed via the leftward shift of the Nav1.6 steady-state saturation curve (Figure 4, right, blue curve).
Consistent with the SAR studies using the LCA, which revealed that modification of 17′s R2 substituent from Ac to benzoyl (19) improved efficacy (Table 2), this modification also resulted in an approximately 2-fold increase in binding to FGF14 and a 4-fold increase in binding to Nav1.6. Kinetically, this improved binding affinity was mediated via the benzoyl substitution conferring slower association and dissociation rates toward both proteins (as depicted by greater curvature in the SPR sensorgrams), a change likely attributable to the substituent’s constitutive π bonds enabling additional interactions with both binding partners. Overall, given 19′s heightened efficacy and binding affinities toward both FGF14 and Nav1.6 relative to the other potential PAMs (17 and 22), it, along with the potential NAM (12), were selected for functional evaluation.
While it is potentially somewhat surprising that 12, 17, and 19 bind to both FGF14 and the C-terminal tail of Nav1.6, this finding is consistent with a previous investigation that revealed both overlap and structural divergence between the FGF14:FGF14 homodimer and FGF14:Nav1.6 complex PPI interfaces [36]. That these peptidomimetics bind to both FGF14 and Nav1.6 is desirable in that it affords multiple mechanisms by which they could modulate the PPI between FGF14 and Nav1.6. In the case of the inhibitor (12), its disruption of the PPI between FGF14 and Nav1.6 could be conferred via direct binding to the FGF14 interaction site on the C-terminal tail of Nav1.6 or via binding to FGF14 and causing the protein to undergo a conformational change that makes it inaccessible to its native interaction site on the C-terminal tail of Nav1.6. Similarly, for the potentiator of FGF14:Nav1.6 complex assembly (19), its modulatory effects on the PPI could be conferred by both binding to the C-terminal tail of Nav1.6 that makes the FGF14 interaction site increasingly accessible to the regulatory protein, or, conversely, by binding to FGF14 and causing it to undergo a conformational change that affords it with increased accessibility to its interaction site on the C-terminal tail of Nav1.6. In the native system, it is expected that the protein:ligand interactions of 12 and 19 with FGF14 and the C-terminal tail of Nav1.6 will concomitantly occur and collectively enable functionally relevant modulation of FGF14:Nav1.6 complex assembly.
2.2.3. Electrophysiological Evaluation of Compounds 12 and 19
Heretofore, it had been shown that addition of a N-terminal benzoyl substituent (19) to tetrapeptide analogs produced an efficacious potentiator of FGF14:Nav1.6 complex assembly, whereas truncation to a tripeptide (12) yielded an inhibitor of the complex′s assembly. Additionally, both 12 and 19 demonstrated promising protein:ligand binding interactions. As such, in an effort to further investigate their seemingly inverse activities, 12 and 19 were selected for functional evaluation as modulators of Nav1.6. To do so, HEK293 cells stably expressing Nav1.6 (HEK-Nav1.6) were incubated for 30 min with 0.1% DMSO (control), 20 µM 12, or 20 µM 19. After incubation, the modulatory effects of these peptidomimetics on Nav1.6-mediated currents were assessed using whole-cell patch-clamp electrophysiology (Figure 5 and Table 4).
Figure 5.

Functional evaluation of 12 and 19 as modulators of Nav1.6 using whole-cell patch-clamp electrophysiology. (A) Representative traces of Na+ transient currents recorded from HEK-Nav1.6 cells treated with 0.1% DMSO (black), 20 µM 12 (red), or 20 µM 19 (blue) in response to voltage steps ranging from −100 to +60 mV from a holding potential of −70 mV (inset). (B) Plot comparing current–voltage relationships among HEK-Nav1.6 cells treated as described in (A). (C) Bar graph summary of (B) comparing peak current densities among the indicated experimental groups. (D) Bar graph summary of tau of inactivation at −10 mV among the indicated experimental groups. (E) Normalized conductance plotted as a function of membrane potential (mV) from cells treated with 0.1% DMSO (black), 20 µM 12 (red), or 20 µM 19 (blue) to characterize their respective effects on voltage-dependence of Nav1.6 activation. (F) Bar graph derived from (E) comparing V1/2 of Nav1.6 activation among the indicated experimental groups. V1/2 of Nav1.6 activation was calculated by fitting data plotted in (E) with the Boltzmann equation. (G) Normalized current plotted as a function of membrane potential (mV) from cells treated with 0.1% DMSO (black), 20 µM 12 (red), or 20 µM 19 (blue) to characterize their respective effects on steady-state inactivation of Nav1.6 channels. (H) Bar graph derived from (G) comparing V1/2 of Nav1.6 steady-state inactivation among the indicated experimental groups. V1/2 of Nav1.6 steady-state inactivation was calculated by fitting data plotted in (G) with the Boltzmann equation. (I) Effects of successive depolarization cycles on HEK-Nav1.6 cells treated with 0.1% DMSO (black), 20 µM 12 (red), or 20 µM 19 (blue). Long-term inactivation was characterized by plotting the fraction of Nav1.6 channels available as a function of depolarization cycle. The long-term inactivation voltage steps were as follows: four depolarizations at 0 mV for 16 ms separated by three recovery intervals at −90 mV for 40 ms. (J) Bar graph summary of (I). No leak cancellation protocol was employed in these electrophysiological investigations. Results are expressed as the mean ± SEM. Significance was determined via unpaired t-test compared with 0.1% DMSO. *, p < 0.05; **, p < 0.005.
The first kinetic property of Nav1.6 channels we investigated as potentially being modulated by 12 and 19 was peak current density. Investigation of how this property was modulated by these analogs revealed that treatment with 12 (–18.30 ± 2.02 picoampere per picofarad (pA/pF); n = 10) significantly reduced Nav1.6-mediated peak current density derived from transient Na+ current relative to treatment with 0.1% DMSO (–55.39 ± 2.16 pA/pF; n = 10), whereas treatment with 19 had the inverse effect and potentiated peak current density (–79.63 ± 3.55 pA/pF; n = 8) (Figure 5B,C). The observed phenotypes could be attributable to the compounds altering channel availability by favoring or inhibiting steady-state (closed) inactivation, single channel conductance, or the number of channels at the plasma membrane. Such alterations could be mediated by 12 and 19 binding to the channel and inducing conformational changes that favor the channel adopting non-conductive (closed or inactivated) and conductive (open) states, respectively.
Lending further credence to 19′s potentiation of Nav1.6 channel activity, subsequent investigations revealed that treatment with 19 induced a –20.03 ± 0.85 mV (0.1% DMSO; n = 10) to –22.49 ± 0.72 mV hyperpolarizing shift (n = 8; p < 0.05) of V1/2 of activation (Figure 5E,F), which was not accompanied by a change in the V1/2 of steady-state inactivation (n = 8; p = 0.984) (Figure 5G,H). Conversely, treatment with 12 affected neither the V1/2 of activation (n = 10; p = 0.756) nor the V1/2 of steady-state inactivation (n = 6; p = 0.600) relative to treatment with 0.1% DMSO (Figure 5E–H). Interestingly, both 12 and 19 were shown to significantly increase the long-term inactivation of Nav1.6 channels relative to the control (Figure 5I,J). Overall, these findings suggest that both peptidomimetics have complex mechanisms of action mediated via modulation of multiple kinetic properties of Nav1.6 channels that collectively confer them with inverse activities.
2.2.4. Molecular Docking Studies of Compounds 12 and 19
With compounds 12 and 19 functionally validated as negative and positive allosteric modulators of Nav1.6 respectively, we next docked these compounds to the Nav1.6 C-terminal tail homology model [36] in silico to further elucidate their binding modes using Schrödinger Small-Molecule Drug Discovery Suite. Both compounds 12 and 19 docked well into the Nav1.6 C-terminal tail at a binding site identical to the EYYV motif (within the β8–β9 loop) of FGF14, enabling interactions with the same group of key residues, as shown in Figure 6. For compound 12 (as depicted in Figure 6A,B), the constituent oxygen atom of the backbone carboynl group between Y and V forms hydrogen bonds with Arg1891. The NH2 group at the C-terminus of compound 12 forms two hydrogen bonds with residues Met1832 and Asp1833, whereas the Fmoc protecting group at the N-terminus of compound 12 interacts with Arg1866 via a π-cation interaction.
Figure 6.

Molecular docking studies of compounds 12 and 19. The Nav1.6 C-terminal tail homology model was generated using the crystal structure of the C-terminal tail of the voltage-gated sodium channel 1.5 (Nav1.5) in complex with fibroblast growth factor 13 (FGF13) (Protein Data Bank (PDB) Code: 4DCK) as a template [36]. (A) Docking of compound 12 (green) into the pocket of Nav1.6 C-terminal. Important residues are drawn as sticks. Hydrogen bonds are shown as dashed red lines. (B) Interaction diagram of binding site between compound 12 and Nav1.6 C-terminal tail. Representations: green lines, hydrophobic; light blue lines, polar; red lines, Pi-cation; purple arrows, hydrogen bonds. (C) Docking of compound 19 (magenta) into the pocket of Nav1.6 C-terminal. Important residues and key interactions are presented similarly. (D) Interaction diagram of binding site between compound 19 and Nav1.6 C-terminal tail. (E) Overlay of Nav1.6 C-terminal tail docked poses for compound 12 (green), 17, (blue), and 19 (magenta) with both FGF14 (green ribbons) and Nav1.6 C-terminal (yellow ribbons) shown. The YYV motif of FGF14 (within the β8/9 loop) is highlighted in orange.
The predicted docking model of compound 19 with the Nav1.6 C-terminal tail (Figure 6C,D) is fundamentally different from that of compound 12, which may help elucidate the opposing actions of the two compounds on Nav1.6 activity. Whereas the C-terminus of 12 interacted strongly with Asp1833 and Met1832 via hydrogen bonds, compound 19 was inversely oriented, with its C-terminus engaging with Ser1859 and Asp1863. NH groups between the N-terminal benzoyl substituent and the glutamic acid residue of the backbone and between the two tyrosine residues of the backbone form hydrogen bonds with Ala1831 and Asp1858, respectively. Meanwhile, the constituent oxygen atom of the ester bond at the sidechain of the glutamic acid residue of the tetrapeptide backbone interacts with Arg1891 through hydrogen bonding. Like compound 12, the NH2 group at the C-terminus of compound 19 also forms two hydrogen bonds, thus providing a structural rationale for why replacing the C-terminal NH2 with OMe abrogated the in-cell activity of analogs. The N-terminal benzoyl substituent of compound 19 forms hydrophobic interactions with a small hydrophobic pocket in the Nav1.6 C-terminal tail that is surrounded by Ile1827, Glu1828, Ile1830, Ala1831, and Met1832, a result in agreement with our SAR studies demonstrating that introducing a benzene ring at the N-terminus is essential for heightened efficacy.
Additionally, an overlay of the FGF14:Nav1.6 complex with the highest scoring binding poses for compounds 12, 17, and 19 (Figure 6E) exemplifies the drastic binding differences that result from the chemical changes, with the greatest disparity observed between 12 and 19. Importantly, the docking results for the tripeptide (12) point toward its binding interactions most resembling those of the native surface residues on the β8–β9 loop of FGF14 at its interface with Nav1.6 (shown as orange in Figure 6E). Theses similarities between the binding interactions of 12 and FGF14 with the Nav1.6 C-terminal tail suggest that 12′s truncated size relative to tetrapeptide analogs confers it with heightened mimicry of FGF14, thereby giving rise to its marked suppression of Nav1.6-mediated peak current density. Based upon theses structural analyses, and consistent with current models of Nav channel function [37,38,39], we propose that 12 and 19 exert opposite effects on Nav1.6-mediated currents as a result of differential interactions with the EF hand-like (EFL) and IQ domains of the C-terminal tail of the channel at sites that have established roles in channel trafficking and inactivation.
3. Discussion
Although it has been reported that aberrant firing of striatal neurons is implicated in a multitude of neuropsychiatric and neurological symptomologies [26,27,28,29,40,41], elucidation of how perturbations in their constituent PPIs contribute to these neuropathophysiological processes remains hampered by a lack of target-selective chemical probes. To that end, we sought to identify small molecule modulators of the PPI between FGF14 and Nav1.6, two proteins that are abundantly expressed in MSNs of the NAc and whose PPI, when perturbed, leads to neural circuitry aberrations [42,43]. To do so, we designed peptidomimetics derived from the EYYV parental tetrapeptide by employing a number of chemical interventions, including: (A) truncation of the tetrapeptide to a tripetide (YYV) and dipeptide (YV), (B) protection of the C-terminus of 6, and (C) modification of the N-terminus of 6 with various substituents. After these newly designed peptidomimetics were synthesized, their modulatory effects on FGF14:Nav1.6 complex assembly were assessed using the LCA. In-cell screening of these analogs revealed that a tripeptide (12) displayed low micromolar inhibitory activity against FGF14:Nav1.6 complex assembly (IC50 = 23.7 µM), whereas addition of hydrophobic protective groups to 6 followed by addition of a N-terminal acetyl (17) or benzoyl (19) substituent produced potentiators of the complex’s assembly. Subsequent dose response analyses using the LCA revealed that 19 was the most efficacious analog among the potentiators. Protein:ligand binding studies of the four most promising hits revealed that 12 showed strong binding affinities toward both FGF14 and Nav1.6 C-terminal tail proteins, with KD values of 2.88 and 9.86 µM, respectively. Additionally, 19 displayed appreciable binding affinities toward both of these proteins (KD = 6.3 and 2.25 µM for FGF14 and Nav1.6, respectively). Functional evaluation of these two analogs using whole-cell patch-clamp electrophysiology confirmed their inverse activities, with 12 and 19 reducing and increasing Nav1.6-mediated transient current densities, respectively. Lastly, molecular docking studies of compounds 12 and 19 with the Nav1.6 C-terminal tail homology model were performed, which indicated that these two compounds formed crucial hydrogen bonds and hydrophobic interactions with Nav1.6.
The findings of the present investigation are consistent with previous efforts to develop small molecule modulators of PPIs between Nav channels and their regulatory accessory proteins [30,31], although the results differ in subtle, but crucial ways. For example, we previously presented ZL0177 [30], a novel peptidomimetic derived from FLPK, a four amino acid sequence located on the β12 sheet of FGF14 at its interface with Nav1.6 [32]. Like the herein reported compound 12, ZL0177 suppressed Nav1.6-mediated peak current density; however, it also accelerated the kinetics of current decay and induced a depolarizing shift in the voltage-dependence of Nav1.6 channel activation [30]. That these phenotypes are not also induced by compound 12, despite 12 inducing an even stronger suppression of Nav1.6-mediated peak current density relative to ZL0177 (−18.30 ± 2.02 pA/pF vs. –26.65 ± 6.3 pA/pF for 12 and ZL0177, respectively) likely stems from 12 being derived from EYYV, which, unlike FLPK, is located on the exposed β8–β9 loop of FGF14 at its interface with the C-terminal tail of Nav1.6. As such, it is expected that 12 and ZL0177 will differentially bind to the FGF14 interaction site on the C-terminal tail of Nav1.6 and induce slightly divergent phenotypes.
Currently, these peptidomimetics are being employed as novel pharmacological probes to interrogate the biophysical properties of Nav channels. Given their high molecular weights, as well as their high clogP and tPSA values, chemical optimization of these peptidomimetics by replacing their natural amino acids with non-natural amino acids and substituting functional groups to confer improved water solubility is likely necessary to permit blood-brain permeability and will be the subject of future investigations [44]. By pursuing these chemical optimization strategies, we envision that the negative and positive allosteric modulator of Nav1.6 herein identified could be developed into CNS penetrant lead compounds with promising potential to be advanced into latter stages of preclinical testing as PPI-based neurotherapeutics.
4. Materials and Methods
4.1. Chemistry
4.1.1. General
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical thin-layer chromatography (TLC) was carried out employing silica gel 60 F254 plates (Merck, Darmstadt, Germany). Visualization of the developed chromatograms was performed with detection by ultraviolet (UV) light (254 nm). NMR spectra were recorded on a Brucker-300 (1H, 300 MHz; 13C, 75 MHz; Brucker, Billerica, MA, USA) spectrometer. 1H and 13C NMR spectra were recorded with tetramethylsilane (TMS) as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from a Thermo Fisher LTQ Orbitrap Elite mass spectrometer (Thermo Fischer Scientific, Grand Island, NY, USA) Parameters include the following: Nano electrospray ionization (ESI) spray voltage was 1.8 kV, capillary temperature was 275 °C, and the resolution was 60,000, ionization was achieved by positive mode. Purities of final compounds were established by analytical high-performance liquid chromatography (HPLC), which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm), flow rate 0.5 mL/min, UV detection at 270 and 254 nm, linear gradient from 10% acetonitrile in water to 100% acetonitrile in water. All biologically evaluated compounds are >95% pure.
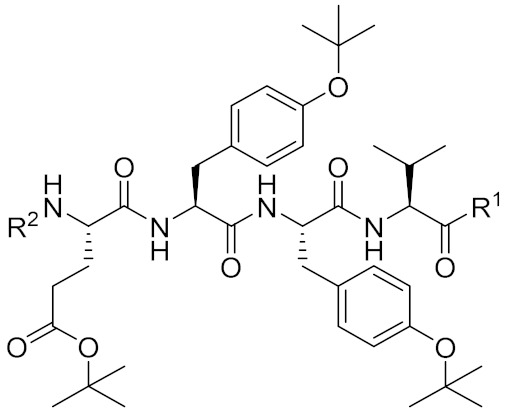
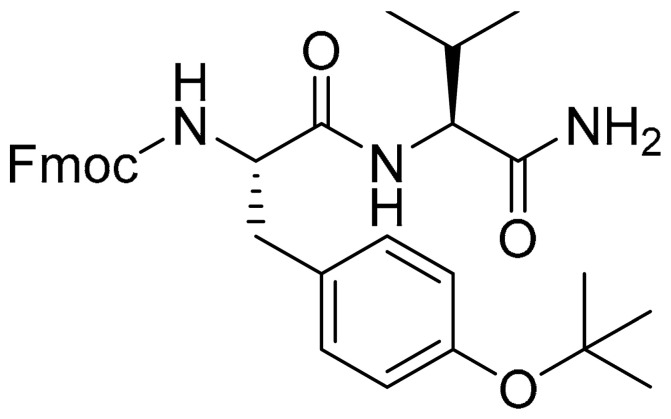
4.1.2. Synthesis of (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)carbamate (10)
(S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(tert-butoxy)phenyl)propanoic acid (7) (1.4 g, 3 mmol) and (S)-2-amino-3-methylbutanamide hydrochloride (8) (459 mg, 3 mmol) were dissolved in 20 mL of CH2Cl2 and the mixture solution was cooled to 0 °C with an ice bath. 1-hydroxybenzotriazole (HOBt) (405 mg, 3 mmol), 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (2.3 mg, 6 mmol), and N,N-Diisopropylethylamine (DIPEA) (2 mL, 12 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (CH2Cl2/MeOH = 50/1) to obtain 10 (1.8 g, 93%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.87 (d, J = 7.5 Hz, 2H), 7.76 (d, J = 8.9 Hz, 1H), 7.68–7.63 (m, 2H), 7.41 (t, J = 7.4 Hz, 3H), 7.34-7.28 (m, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.06 (s, 1H), 6.80 (d, J = 8.3 Hz, 2H), 4.39–4.02 (m, 5H), 3.43–3.36 (m, 1H), 2.97 (dd, J = 13.8, 3.8 Hz, 1H), 2.75 (d, J = 12.3 Hz, 1H), 1.97 (h, J = 6.8 Hz, 1H), 1.18 (s, 9H), 0.85 (t, J = 6.4 Hz, 6H). 13C NMR (75 MHz, DMSO) δ 173.16, 171.76, 156.22, 153.81, 144.15, 141.10, 133.13, 130.16, 128.06, 127.50, 125.73, 123.69, 120.50, 77.99, 66.16, 57.74, 56.71, 47.00, 37.21, 31.17, 28.94, 19.72, 18.34. HRMS (ESI) calcd for C33H40N3O5 558.2962 [M + H]+, found 558.2958.
4.1.3. Synthesis of (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)carbamate (12)
To a solution of HNEt2 (1 mL) and MeCN (4 mL), compound 10 (905 mg, 2.7 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the residue was dissolved in 20 mL of dry CH2Cl2 and Fmoc-Tyr(OtBu)-OH (7) (1.2 g, 2.7 mmol) was added. The mixture solution was cooled to 0 °C with an ice bath. HOBt (364 mg, 2.7 mmol), HBTU (2.0 g, 5.4 mmol), and DIPEA (2 mL, 12 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, and saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/MeOH = 50/1) to obtain compound 12 (1.6 g, 78%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.19 (d, J = 8.1 Hz, 1H), 7.87 (d, J = 7.5 Hz, 2H), 7.77 (d, J = 8.9 Hz, 1H), 7.64 (d, J = 7.4 Hz, 2H), 7.55 (d, J = 8.9 Hz, 1H), 7.46–7.25 (m, 5H), 7.19–7.03 (m, 5H), 6.79 (t, J = 8.1 Hz, 4H), 4.29–3.95 (m, 5H), 3.62 (td, J = 6.6, 4.0 Hz, 1H), 3.19–2.97 (m, 2H), 2.84 (dt, J = 12.6, 5.3 Hz, 2H), 2.04–1.91 (m, 1H), 1.18 (d, J = 6.5 Hz, 18H), 0.84 (t, J = 6.4 Hz, 6H). 13C NMR (75 MHz, DMSO) δ 173.13, 171.88, 171.26, 166.61, 156.08, 153.82, 144.16, 141.11, 133.06, 132.71, 130.15, 128.06, 127.48, 125.72, 123.80, 123.65, 120.49, 77.97, 66.19, 57.83, 56.71, 54.28, 54.07, 46.99, 42.32, 37.04, 31.07, 28.93, 19.72, 18.53, 18.27, 17.16, 12.92. HRMS (ESI) calcd for C46H57N4O7 777.4221 [M + H]+, found 777.4218.
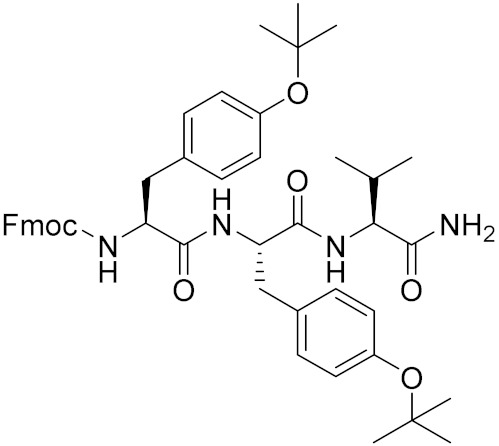
4.1.4. Synthesis of tert-butyl (S)-4-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-5-oxopentanoate (15)
To a solution of HNEt2 (1 mL) and MeCN (4 mL), compound 12 (714 mg, 1.3 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the residue was dissolved in 20 mL of dry CH2Cl2 and Fmoc-Glu(OtBu)-OH (14) (552 mg, 1.3 mmol) was added. The mixture solution was cooled to 0 °C with an ice bath. HOBt (199 mg, 1.3 mmol), HBTU (985 mg, 2.6 mmol), and DIPEA (1 mL, 6 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, an saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/MeOH = 20/1) to obtain compound 15 (975 mg, 77%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 8.24 (d, J = 7.7 Hz, 1H), 8.07–7.54 (m, 7H), 7.51–7.21 (m, 6H), 7.09 (dd, J = 19.4, 8.5 Hz, 5H), 6.78 (dd, J = 25.5, 7.9 Hz, 3H), 4.53 (d, J = 26.4 Hz, 2H), 4.22 (td, J = 30.5, 29.7, 13.2 Hz, 4H), 3.95 (s, 1H), 3.02–2.67 (m, 3H), 2.20–1.63 (m, 4H), 1.37 (s, 9H), 1.28–0.96 (m, 20H), 0.83 (d, J = 7.5 Hz, 6H). 13C NMR (75 MHz, DMSO) δ 174.53, 173.13, 172.63, 171.39, 171.29, 153.83, 132.81, 132.55, 130.26, 130.12, 123.78, 123.63, 79.81, 78.03, 77.96, 57.86, 54.55, 54.40, 53.53, 31.79, 31.05, 30.42, 29.00, 28.20, 19.72, 18.32. HRMS (ESI) calcd for C55H72N5O10 962.5273 [M + H]+, found 962.5270.
4.1.5. Synthesis of methyl (5S,8S,11S,14S)-5-(3-(tert-butoxy)-3-oxopropyl)-8,11-bis(4-(tert-butoxy)benzyl)-1-(9H-fluoren-9-yl)-14-isopropyl-3,6,9,12-tetraoxo-2-oxa-4,7,10,13-tetraazapentadecan-15-oate (16)
(S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(tert-butoxy)phenyl)propanoic acid (7) (1.4 g, 3 mmol) and methyl L-valinate (9) (504 mg, 3 mmol) were dissolved in 20 mL of CH2Cl2 and the mixture solution was cooled to 0 °C with an ice bath. HOBt (405 mg, 3 mmol), HBTU (2.3 mg, 6 mmol), and DIPEA (2 mL, 12 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, and saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (CH2Cl2/MeOH = 100/1) to obtain 11 (1.6 g, 91%) as a white solid.
To a solution of HNEt2 (0.1 mL) and MeCN (0.4 mL), compound 11 (1.7 g, 3 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the residue was dissolved in 20 mL of dry CH2Cl2 and Fmoc-Tyr(OtBu)-OH (7) (1.3 g, 3 mmol) was added. The mixture solution was cooled to 0 °C with an ice bath. HOBt (459 mg, 3 mmol), HBTU (2.3 g, 6 mmol), and DIPEA (2 mL, 12 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, and saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/MeOH = 50/1) to obtain compound 13 (1.7 g, 73%) as a white solid.
To a solution of HNEt2 (0.1 mL) and MeCN (0.4 mL), compound 13 (2.4 g, 3 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the residue was dissolved in 20 mL of dry CH2Cl2 and Fmoc-Glu(OtBu)-OH (14) (1.3 g, 3 mmol) was added. The mixture solution was cooled to 0 °C with an ice bath. HOBt (459 mg, 3 mmol), HBTU (2.3 g, 6 mmol), and DIPEA (2 mL, 12 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, and saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/MeOH = 50/1) to obtain compound 16 (2.2 g, 74%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.82 (d, J = 7.5 Hz, 2H), 7.67 (d, J = 7.4 Hz, 2H), 7.46–7.24 (m, 4H), 7.23–7.03 (m, 5H), 7.00–6.72 (m, 6H), 4.32 (ddt, J = 20.7, 14.7, 7.2 Hz, 4H), 4.05 (dd, J = 8.9, 5.4 Hz, 1H), 3.69 (s, 3H), 3.06 (dd, J = 13.5, 5.5 Hz, 2H), 2.86 (s, 2H), 2. 28–2.09 (m, 3H), 1.86 (ddt, J = 38.6, 14.5, 7.2 Hz, 2H), 1.46 (s, 9H), 1.32–1.30 (m, 9H), 1.25 (s, 9H), 0.94 (dd, J = 7.1, 3.2 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 172.54, 172.49, 171.80, 171.69, 171.51, 166.60, 153.90, 143.93, 143.72, 141.20, 141.18, 131.80, 129.58, 129.51, 129.46, 129.41, 127.41, 126.78, 124.88, 124.85, 124.13, 123.94, 123.79, 123.75, 119.54, 80.43, 78.05, 77.98, 66.76, 57.87, 54.48, 51.06, 36.79, 36.48, 31.21, 30.52, 27.79, 26.99, 26.77, 18.03, 17.30. HRMS (ESI) calcd for C56H73N4O11 977.5270 [M + H]+, found 977.5266.
4.1.6. Synthesis of tert-butyl (S)-4-acetamido-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-5-oxopentanoate (17)
To a solution of HNEt2 (0.1 mL) and MeCN (0.4 mL), compound 15 (50 mg, 0.05 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the compound was dissolved in 5 mL of CH2Cl2 and the solution was cooled to 0 °C with an ice bath. Then, Et3N (11 mg, 0.1 mmol) and acetyl chloride (7.9 mg, 0.1 mmol) were added. The mixture was stirred at room temperature overnight. The solution was diluted with 20 mL of CH2Cl2 and washed with 1 N NaHSO4, saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/CH3OH = 50/1 to 20/1) to obtain compound 17 (26 mg, 66%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 8.14 (s, 1H), 7.92 (dd, J = 21.0, 7.5 Hz, 1H), 7.12 (dd, J = 25.2, 8.0 Hz, 4H), 6.90 (dd, J = 14.9, 8.0 Hz, 4H), 4.60 (d, J = 27.3 Hz, 2H), 4.21 (dd, J = 19.2, 11.7 Hz, 2H), 3.48 (s, 1H), 3.19–2.80 (m, 4H), 2.17 (dt, J = 48.6, 7.0 Hz, 3H), 1.97 (s, 3H), 1.82 (dd, J = 16.5, 9.0 Hz, 1H), 1.38 (d, J = 41.4 Hz, 27H), 0.97 (t, J = 5.9 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 174.33, 172.37, 172.26, 172.09, 171.77, 153.96, 131.95, 131.74, 129.47, 129.42, 123.85, 123.70, 80.40, 78.10, 78.01, 58.54, 54.88, 54.64, 52.90, 36.46, 31.15, 30.42, 29.25, 27.82, 26.97, 26.63, 21.12, 18.35, 17.21. HRMS (ESI) calcd for C42H64N5O9 782.4698 [M + H]+, found 782.4692.
4.1.7. Synthesis of methyl (2S,5S,8S,11S)-11-(3-(tert-butoxy)-3-oxopropyl)-5,8-bis(4-(tert-butoxy)benzyl)-2-isopropyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazatetradecanoate (18)
To a solution of HNEt2 (0.1 mL) and MeCN (0.4 mL), compound 16 (50 mg, 0.05 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the compound was dissolved in 5 mL of CH2Cl2 and the solution was cooled to 0 °C with an ice bath. Then, Et3N (11 mg, 0.1 mmol) and acetyl chloride (7.9 mg, 0.1 mmol) were added. The mixture was stirred at room temperature overnight. The solution was diluted with 20 mL of CH2Cl2 and washed with 1 N NaHSO4, saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/CH3OH = 50/1 to 20/1) to obtain compound 18 (31 mg, 78%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.23–7.04 (m, 4H), 6.96–6.80 (m, 4H), 4.64 (ddd, J = 26.2, 8.4, 5.9 Hz, 2H), 4.31 (dt, J = 5.8, 4.0 Hz, 2H), 3.70 (s, 3H), 3.14–2.80 (m, 4H), 2.26 (t, J = 7.7 Hz, 2H), 2.18–2.07 (m, 1H), 1.96 (s, 3H), 1.89–1.74 (m, 1H), 1.46 (d, J = 2.3 Hz, 9H), 1.32 (d, J = 2.1 Hz, 18H), 0.95 (dd, J = 6.9, 3.7 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 172.39, 172.06, 171.96, 171.82, 171.70, 171.54, 153.94, 131.81, 131.76, 129.73, 129.51, 129.46, 123.91, 123.77, 123.69, 80.37, 78.02, 77.98, 57.85, 54.48, 54.43, 52.67, 51.07, 36.85, 36.63, 31.18, 30.55, 27.83, 27.78, 26.98, 26.93, 26.76, 21.13, 18.05, 17.32. HRMS (ESI) calcd for C43H65N4O10 797.4695 [M + H]+, found 797.4692.
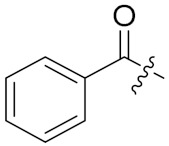
4.1.8. Synthesis of tert-butyl (S)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-4-benzamido-5-oxopentanoate (19)
Compound 19 (42 mg, 70%) was synthesized by a procedure similar to that used to prepare compound 17 as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.87 (d, J = 7.6 Hz, 2H), 7.65–7.43 (m, 3H), 7.10 (dd, J = 23.3, 8.0 Hz, 4H), 6.83 (dd, J = 34.4, 8.1 Hz, 4H), 4.70–4.47 (m, 3H), 4.20 (d, J = 7.3 Hz, 1H), 3.17–2.80 (m, 4H), 2.34 (t, J = 7.2 Hz, 2H), 2.09 (ddt, J = 25.1, 18.1, 8.2 Hz, 3H), 1.44 (s, 9H), 1.28 (d, J = 16.7 Hz, 18H), 0.97 (dd, J = 6.8, 4.0 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 174.37, 172.72, 172.38, 171.85, 171.79, 168.89, 153.92, 133.45, 131.94, 131.70, 131.63, 129.45, 129.38, 128.19, 127.27, 123.85, 123.73, 80.53, 78.10, 77.94, 58.56, 54.87, 54.77, 53.71, 36.56, 36.48, 31.37, 30.42, 27.80, 26.96, 26.26, 18.36, 17.21. HRMS (ESI) calcd for C47H66N5O9 844.4855 [M + H]+, found 844.4855.
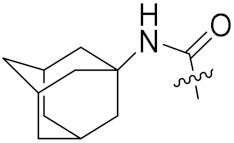
4.1.9. Synthesis of tert-Butyl (S)-4-(3-((3R,5R,7R)-adamantan-1-yl)ureido)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-5-oxopentanoate (20)
Compound 20 (35 mg, 56%) was synthesized by a procedure similar to that used to prepare compound 17 as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.12 (dd, J = 25.8, 8.0 Hz, 4H), 6.90 (dd, J = 13.5, 8.0 Hz, 4H), 4.66 (d, J = 7.8 Hz, 1H), 4.49 (d, J = 7.1 Hz, 1H), 4.20 (d, J = 7.6 Hz, 1H), 4.04 (t, J = 6.9 Hz, 1H), 3.19–2.84 (m, 4H), 2.32–1.82 (m, 13H), 1.70 (s, 7H), 1.46 (s, 9H), 1.32 (s, 18H), 0.97 (dd, J = 6.8, 3.7 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 174.45, 173.73, 172.41, 171.91, 166.61, 157.98, 153.93, 132.11, 131.64, 129.40, 123.85, 80.34, 77.97, 58.74, 55.07, 54.71, 53.35, 50.25, 41.94, 36.61, 36.10, 31.11, 30.37, 29.57, 27.85, 27.79, 27.47, 26.98, 18.37, 17.26. HRMS (ESI) calcd for C51H77N6O9 917.5752 [M + H]+, found 917.5746.
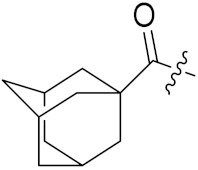
4.1.10. Synthesis of tert-Butyl (4S)-4-((1S,3R,5S)-adamantane-1-carboxamido)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-5-oxopentanoate (21)
To a solution of HNEt2 (1 mL) and MeCN (4 mL), compound 15 (68 mg, 0.07 mmol) was added and the solution was stirred at room temperature for 1 h. After the reaction was completed, the solution was concentrated to remove the solvent. Then, the residue was dissolved in 2 mL of dry CH2Cl2 and 1-adamantanecarboxylic acid (12 mg, 0.07 mmol) was added. The mixture solution was cooled to 0 °C with an ice bath. HOBt (9 mg, 0.07 mmol), HBTU (50 mg, 0.14 mmol), and DIPEA (22 mg, 0.14 mmol) were added to the solution at 0 °C. Then, the ice bath was removed, and the mixture solution was stirred at room temperature overnight. After the reaction was completed (detected by TLC), the mixture was washed with 1 N NaHSO4, and saturated in NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and the residue was purified with silica gel column (CH2Cl2/MeOH = 50/1) to obtain compound 21 (38 mg, 61%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.12 (dd, J = 25.3, 8.2 Hz, 4H), 6.90 (dd, J = 15.6, 8.3 Hz, 4H), 4.66 (dd, J = 8.2, 5.9 Hz, 1H), 4.54 (dd, J = 8.5, 5.2 Hz, 1H), 4.34–4.13 (m, 2H), 3.74 (p, J = 6.6 Hz, 1H), 3.15–2.82 (m, 4H), 2.27 (t, J = 7.2 Hz, 1H), 2.03 (s, 3H), 1.86–1.74 (m, 8H), 1.46 (s, 9H), 1.39 (d, J = 6.6 Hz, 7H), 1.32 (s, 18H), 0.97 (t, J = 6.1 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 179.52, 174.35, 172.84, 172.42, 171.80, 171.70, 154.08, 153.96, 131.91, 131.50, 129.46, 129.44, 123.86, 123.74, 80.54, 78.09, 77.94, 58.57, 54.79, 54.55, 54.45, 52.97, 42.39, 40.47, 38.62, 36.54, 36.10, 31.26, 30.43, 28.18, 27.85, 27.82, 27.00, 26.28, 18.36, 17.24, 15.87, 11.74. HRMS (ESI) calcd for C51H76N5O9 902.5637 [M + H]+, found 902.5632.
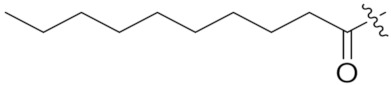
4.1.11. Synthesis of tert-Butyl (S)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-4-decanamido-5-oxopentanoate (22)
Compound 22 (36 mg, 58%) was synthesized by a procedure similar to that used to prepare compound 21 as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.11 (dd, J = 27.5, 8.0 Hz, 4H), 6.89 (dd, J = 16.9, 8.0 Hz, 4H), 4.68–4.50 (m, 2H), 4.24 (dd, J = 28.9, 7.0 Hz, 2H), 3.20–2.76 (m, 5H), 2.36–1.78 (m, 7H), 1.38 (d, J = 41.7 Hz, 40H), 0.94 (dt, J = 19.6, 6.4 Hz, 9H). 13C NMR (75 MHz, MeOD) δ 175.10, 174.35, 172.36, 171.79, 171.74, 153.99, 153.95, 131.96, 131.67, 129.47, 129.41, 123.86, 123.73, 80.40, 78.10, 77.97, 58.55, 54.89, 54.67, 52.80, 36.59, 36.44, 35.42, 31.64, 31.15, 30.42, 29.22, 29.08, 29.02, 28.97, 27.85, 27.82, 26.98, 26.56, 25.46, 22.31, 18.36, 17.23, 13.03. HRMS (ESI) calcd for C50H80N5O9 894.5950 [M + H]+, found 894.5956.
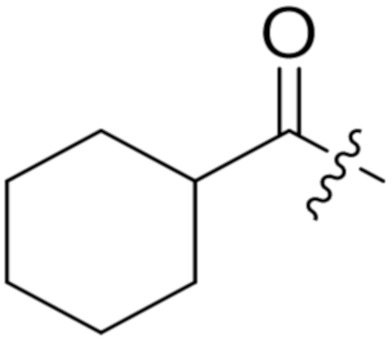
4.1.12. Synthesis of tert-Butyl (S)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-4-(cyclohexanecarboxamido)-5-oxopentanoate (23)
Compound 23 (38 mg, 64%) was synthesized by a procedure similar to that used to prepare compound 21 as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.12 (dd, J = 28.0, 8.1 Hz, 4H), 6.89 (dd, J = 17.8, 8.1 Hz, 4H), 4.70–4.50 (m, 2H), 4.31–4.15 (m, 2H), 3.17–2.81 (m, 4H), 2.39–1.61 (m, 11H), 1.45 (s, 10H), 1.32 (s, 22H), 0.97 (t, J = 6.1 Hz, 6H). 13C NMR (75 MHz, MeOD) δ 177.96, 174.35, 172.44, 172.37, 171.80, 171.70, 154.03, 153.95, 131.93, 131.57, 129.44, 123.85, 123.70, 80.43, 78.10, 77.95, 58.56, 54.84, 54.58, 52.69, 44.62, 36.58, 36.52, 31.19, 30.42, 29.44, 29.13, 27.85, 27.82, 26.98, 26.60, 25.46, 25.38, 25.32, 18.36, 17.23. HRMS (ESI) calcd for C47H72N5O9 850.5330 [M + H]+, found 850.5326.
4.1.13. Synthesis of tert-Butyl (S)-5-(((S)-1-(((S)-1-(((S)-1-amino-3-methyl-1-oxobutan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-3-(4-(tert-butoxy)phenyl)-1-oxopropan-2-yl)amino)-4-(((benzyloxy)carbonyl)amino)-5-oxopentanoate (24)
Compound 24 (36 mg, 61%) was synthesized by a procedure similar to that used to prepare compound 17 as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.49–7.23 (m, 5H), 7.17–7.06 (m, 4H), 6.94–6.84 (m, 4H), 5.21–5.02 (m, 2H), 4.59 (ddd, J = 13.9, 8.5, 3.5 Hz, 2H), 4.08 (dd, J = 8.9, 5.5 Hz, 1H), 3.22–2.78 (m, 5H), 2.25 (t, J = 7.5 Hz, 2H), 2.16–2.05 (m, 1H), 1.98–1.75 (m, 2H), 1.44 (s, 9H), 1.31 (d, J = 3.2 Hz, 18H), 1.00–0.94 (m, 6H). 13C NMR (75 MHz, MeOD) δ 174.33, 172.66, 172.46, 171.74, 153.94, 136.58, 131.92, 131.74, 129.46, 129.43, 128.09, 127.64, 127.45, 123.87, 123.76, 80.42, 78.13, 78.02, 66.50, 58.53, 54.85, 54.60, 36.52, 31.16, 30.43, 27.81, 26.96, 18.35, 17.20. HRMS (ESI) calcd for C48H68N5O10 874.4960 [M + H]+, found 874.4956.
4.2. Cell Culture
HEK293 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) and F-12 (Invitrogen, Carlsbad, CA, USA), supplemented with 0.05% glucose, 0.5 mM pyruvate, 10% fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin (Invitrogen), and incubated at 37 °C with 5% CO2. The double stable HEK293 cell line expressing CD4-Nav1.6C-tail-NLuc and CLuc-FGF14 used for LCA experiments was described in a previous study [33] and was maintained using selective antibiotics (0.5 mg/mL G418 and 5 µg/mL puromycin). HEK293 cells stably expressing the human Nav1.6 channel (hereafter referred to as HEK-Nav1.6 cells) were maintained similarly, except for the addition of 500 μg/mL G418 (Invitrogen) to maintain stable Nav1.6 expression. Cells were transfected at 80–90% confluence with equal amount (1 μg each) of plasmid pairs using Lipofectamine 3000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. HEK-Nav1.6 cells were washed and re-plated at very low density prior to electrophysiological recordings [31,33,36,45].
4.3. Split-Luciferase Complementation Assay (LCA)
Cells were trypsinized (0.25%), triturated in a medium, and seeded in white, clear-bottom CELLSTAR μClear® 96-well tissue culture plates (Greiner Bio-One, Monroe, NC, USA) at ~0.9 × 105 cells per well in 50 μL of serum-free, phenol red-free DMEM/F12 medium (Invitrogen, Waltham, MA, USA). For transiently transfected cells, the trypsinization occurred 48 h post-transfection. The cells were incubated for 2 h, followed by addition of 50 µL of serum-free medium containing either 0.5% DMSO alone (vehicle; n = 32 replicates per plate) or compounds (25 µM for screening and 0.1–175 µM for dose responses; n = 4 replicates per treatment condition per plate) dissolved to a final concentration of 0.5% DMSO, with a minimum of 3 independent experiments per compound. Following 12 h incubation at 37 °C, the reporter reaction was initiated by injection of 100 μL substrate solution containing 1.5 mg/mL of d-luciferin (final concentration = 0.75 mg/mL) by the Synergy™ H1 Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA). Luminescence readings (integration time 0.5 s) were performed at 2-min intervals for 20 min, and the cells were maintained at 37 °C throughout the measurements. Signal intensity for each well was calculated as a mean value of peak luminescence. The calculated values were expressed as percentage of mean signal intensity of the per plate control samples. Detailed methods for LCA can be found in previous studies [31,32,33,36,46,47].
Dose response curves were obtained using GraphPad Prism 8 by fitting the data with a non-linear regression:
| (1) |
where x is log10 of the compound concentration in M, x0 is the inflection point (EC50 or IC50), A is the bottom plateau effect, B is the top plateau effect, and H is the Hill slope. Potency (IC50/EC50) and efficacy (percent luminescence at the bottom or top plateau for inhibitors (IMin) or enhancers (EMax), respectively) were calculated from the dose response non-linear regression.
4.4. Cell Viability Assay
The CellTiter-Blue® Cell Viability (CTB) assay (Promega, Madison, WI, USA) was used to counter-screen top compounds. Immediately following LCA luminescence reading from cells treated with experimental compounds or Tamoxifen (positive control with known toxicity toward HEK293 cells [33]), 30 µL of 1X CTB reagent was dispensed into 96-well plates, plates were incubated overnight (16 h) at 37 °C, and fluorescence was detected using the Synergy H1 reader (excitation λ = 560 nm, emission λ = 590 nm). Cell viability was expressed as percent of the mean fluorescent signal intensity of on-plate negative controls.
4.5. Protein Expression and Purification
The two plasmids (pET28a-FGF14; pET30a-Nav1.6) for protein expression and purification of FGF14 (accession number NP_787125; aa 64–252) and Nav1.6 C-tail (accession number #NP_001171455; aa 1763–1912) have been previously described [36,45] and were transformed into E. coli BL21 (DE3) pLys (Invitrogen). Cells were grown until OD600 = 0.7, and the recombinant proteins were expressed after induction with 0.1 mM isopropyl thio-β-d-galacto-pyranoside (IPTG) for 24 h at 16 °C. Cells were harvested and lysed by sonication at 4 °C in lysis/binding buffer containing the following components (mM): 10 sodium phosphate (prepared from 0.5 M of Na2HPO4 and NaH2PO4), 25 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 NaCl, phenyl methyl sulphonyl fluoride (PMSF) 0.1, 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS) 0.1% pH 7.0 (for FGF14), and with glycerol 10% (for Nav1.6 C-tail) pH 7.5. The respective proteins were centrifuged at 40,000× g for 1 h at 4 °C. For purification of FGF14, the supernatant was applied to pre-equilibrated heparin and the proteins were then eluted with NaCl 0.2–2.0 M (sodium phosphate 10 mM, NaCl 0.2–2.0 M, pH 7.0) buffer. For purification of Nav1.6 C-tail, the supernatant was first applied to a cobalt column (Thermo Fisher Scientific) and eluted with imidazole (200 mM). The Nav1.6 C-tail was further purified using a HiTrap Q Sepharose Fast Flow column (GE Healthcare, Pittsburgh, PA, USA) using a buffer containing Tris-HCl 50 mM and eluted with NaCl (10–500 mM) at pH 7.5. Finally, all concentrated proteins were purified on an AKTATM fast protein liquid chromatography (FPLC) using a Superdex 200 HiLoad 16 × 60 column and equilibrated in Tris-HCl 50 mM + NaCl 150 mM, pH 7.5 (GE Healthcare Bio-Sciences, Chicago, IL, USA). Protein concentrations were determined using UV absorbance with a Thermo NANODROP.
4.6. Surface Plasmon Resonance Spectroscopy
SPR experiments were performed on a Biacore T100 instrument (GE Healthcare, Chicago, IL, USA). Proteins were immobilized on CM5 sensor chips using 10 mM sodium acetate buffer (pH 5.5) with the Amine Coupling Kit (GE Healthcare), as per the manufacturer’s instructions, to a final RU value of 17,896 for FGF14 and 6,800 for Nav1.6 C-tail. No protein was coupled to the control flow channels of the chip (Lanes 1 and 3). The interaction of experimental compounds against FGF14 and Nav1.6 proteins were studied at 25 °C using a flow rate of 60 µL/min. Compounds were serially diluted (0.195–200 µM) in PBS supplemented with Tween-20 0.005% and 2% DMSO. Each sample was injected over the chip for 120 s followed by a dissociation period of 150 s, and finally, chip surface regeneration (600 mM NaCl, 5% DMSO) for 120 s. Each compound was tested over at least two independent experiments with concentrations of 0.195, 0.39, 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 200 µM, including 12.5 and 25 µM, and blanks (buffer alone) in duplicate. A DMSO calibration was performed for each experiment (1.5–2.8% (v/v) DMSO) to correct for bulk refractive index shifts [48]. For each compound injection, nonspecific responses (“0 μM” solution prepared similarly to experimental compound samples) for each corresponding experiment were subtracted from compound sensorgrams/traces prior to data analysis. Kinetic data were analyzed using the Biacore T100 Analysis software. Following visual inspection of the binding curves, the equilibrium constant (KD) was calculated using two methods: (1) maximal responses were plotted against compound concentration, and the steady-state KD was calculated from the fitted saturation binding curve, and (2) a kinetic analysis of each ligand/analyte interaction was obtained by fitting the response data to the simplest Langmuir 1:1 interaction model (KD = koff/kon). The kinetic constants generated from the fitted binding curves were assessed for accuracy based on the distribution of the residuals (even and near zero to baseline). Compounds failing to achieve saturation of binding over the concentration range tested are reported as >100 μM. Graphs were plotted in GraphPad Prism 8 Software (La Jolla, CA, USA).
4.7. Electrophysiology
HEK293 cells stably expressing Nav1.6 (HEK-Nav1.6) were grown in culture as described above. Prior to experimentation, HEK-Nav1.6 cells were dissociated using Gibco TrypLE (Thermo Fisher Scientific, Walthman, MA, USA) and plated at low density onto glass coverslips situated at the base of wells in CELLSTAR® 24-Well Multi-Well Plates (Greiner Bio-One, Monroe, NC, USA). Cells were then incubated on glass cover slips for 2–3 h. After incubation, glass cover slips were transferred to the recording chamber containing 3 mL of extracellular solution comprised of the following salts (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.3. After incubating for thirty minutes in the extracellular solution containing either DMSO or experimental compound, recordings were then performed at room temperature (20–22 °C) using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) and borosilicate glass pipettes (resistance of 3–6 MΩ) containing intracellular solution comprised of the following salts (mM): 130 CH3O3SCs, 1 ethylene glycol tetraacetic acid (EGTA), 10 NaCl, 10 HEPES, pH 7.3. Dial settings on the amplifier were used to estimate membrane capacitance and series resistance and compensated for electronically by 70–80%. Prior to digitization and storage, data were acquired at 20 kHz and filtered at 5 kHz. Clampex 9.2 software (Molecular Devices), interfaced to the electrophysiological equipment using a Digidata 1200 analog-digital interface (Molecular Devices), was used to control all experimental parameters. To assess the relationship of INa and voltage, HEK-Nav1.6 cells were evoked by depolarizations to test potentials ranging from –100 to +60 mV from a holding potential of −70 mV, followed by a voltage pre-step pulse of −120 mV. To assess steady-state inactivation of Nav channels, HEK-Nav1.6 cells were evoked using a paired-pulse protocol in which cells were stepped, from the holding potential, to varying test potentials ranging from −120 to +20 mV (pre-pulse) prior to a test pulse to −20 mV. To assess long-term inactivation, HEK-Nav1.6 cells were subjected to four depolarizations at 0 mV for 16 ms separated by three recovery intervals at –90 mV for 40 ms.
4.8. Electrophysiology Data Analysis
INa was normalized to membrane capacitance to determine current density by dividing INa amplitude by membrane capacitance. Current density was then plotted as a function of the holding potential to characterize current-voltage relationships. Tau of inactivation was calculated by fitting the decay phase of currents at −10 mV with a one-term exponential function. To assess voltage-dependence of Nav1.6 activation, conductance (GNa) was first calculated using the following equation:
| (2) |
where INa is current amplitude at voltage Vm, and Erev is the Na+ reversal potential. Steady-state activation curves were then generated by plotting normalized GNa as a function of test potential. Plotted data was then fitted with the Boltzmann equation to determine V1/2 of Nav1.6 activation values using the following equation:
| (3) |
where GNa,Max is the maximum conductance, Va is the membrane potential of half-maximal activation, Em is the membrane voltage, and k is the slope factor. To assess steady-state inactivation, INa normalized to max INa (INa/INa,Max) at the test potential was plotted as a function of pre-pulse potential. Data was then fitted with the Boltzmann function to determine V1/2 using the following equation:
| (4) |
where Vh is the potential of half-maximal inactivation, Em is the membrane voltage, and k is the slope factor. For long-term inactivation, the peak INa of depolarization cycles 2–4 was normalized to the peak INa of depolarization cycle 1 (INa/INa,Cycle 1) and plotted as a function of depolarization cycle. Data analysis was performed using Clampfit 11.1 (Molecular Devices) and GraphPad Prism 8 (La Jolla, CA, USA) software. Results were expressed as mean ± standard error of the mean (SEM). Statistical significance was determined via unpaired t-tests comparing 0.1% DMSO to an experimental group, with p < 0.05 being considered statistically significant.
4.9. Molecular Docking Method
The molecular docking study was performed using Schrödinger Small-Molecule Drug Discovery Suite (Schrödinger, LLC, New York, NY, USA). FGF14:Nav1.6 homology model [36] was generated using the FGF13:Nav1.5:CaM ternary complex crystal structure (PDB code: 4DCK) as a template. The FGF14:Nav1.6 homology model was prepared with Schrödinger Protein Preparation Wizard using default settings. During this step, hydrogens were added, crystal waters were removed, and partial charges were assigned using the OPLS-2005 force field. The SiteMap (Schrödinger, LLC, New York, NY, USA) calculation was performed and a potential binding site was identified on the PPI of FGF14:Nav1.6. The chain of FGF14 was excluded from the model and the grid center was chosen on Nav1.6 C-terminal tail at the previously identified binding site generated from SiteMap results. The grid box was sized in 24 Å on each side to cover the PPI surface on Nav1.6. The 3D structure of ligands 12, 17, and 19 were created using Schrödinger Maestro and a low-energy conformation was calculated using LigPrep. Docking was employed with Glide using the SP protocol. Docked poses were incorporated into Schrödinger Maestro for a ligand–receptor interactions visualization. Top ranking of Glide GScore, as well as biological and chemical rationales were used for ligand docked pose evaluation and selection. The top docked poses of ligands 12, 17, and 19 were superimposed with FGF14:Nav16 complex homology model for an overlay analysis.
Supplementary Materials
The following are available online, 1H and 13C NMR spectra can be found in supplementary materials.
Author Contributions
Conceptualization, N.M.D., P.A.W., and F.L.; methodology, J.Z. and F.L.; formal analysis, N.M.D., P.A.W., and P.W.; investigation, N.M.D., P.A.W., P.W., and H.C.; resources, J.Z. and F.L.; data curation, N.M.D., P.A.W., P.W., and H.C.; writing—original draft preparation, N.M.D. and P.A.W.; writing—review and editing, N.M.D. and F.L.; visualization, P.A.W., N.M.D., and P.W.; supervision, J.Z. and F.L.; project administration, J.Z. and F.L.; funding acquisition, J.Z. and F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institute of Health (NIH) Grants R01 MH095995 (F.L.), R01 MH111107 (F.L. and J.Z.), R01 MH095995 (F.L.), R01 MH124351 (F.L.), S10 OD023576 (to support instrumentation within the Sealy Center for Structural Biology and Molecular Biophysics), the UTMB Technology Commercialization Program (F.L.), P30 DA028821 (J.Z.), John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.), John Sealy Memorial Endowment Fund (F.L.), NIA T32 Fellowship Grant No. T32 AG051131 (P.A.W.), and the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation Pre-doctoral Fellowship in Pharmacology/Toxicology (P.A.W.).
Conflicts of Interest
The corresponding author, F.L., is the founder and president of IonTx Inc., a start-up company focusing on developing regulators of voltage-gated Na+ channels. However, this activity does not represent a conflict with the present study.
Footnotes
Sample Availability: Samples of all experimental compounds herein reported are available from the authors.
References
- 1.Hille B. Ionic channels in excitable membranes. Current problems and biophysical approaches. Biophys. J. 1978;22:283–294. doi: 10.1016/S0006-3495(78)85489-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldin A.L. Resurgence of Sodium Channel Research. Annu. Rev. Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 3.Mantegazza M., Gambardella A., Rusconi R., Schiavon E., Annesi F., Cassulini R.R., Labate A., Carrideo S., Chifari R., Canevini M.P., et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. USA. 2005;102:18177–18182. doi: 10.1073/pnas.0506818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claes L., Del Favero J., Ceulemans B., Lagae L., Van Broeckhoven C., De Jonghe P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. Am. J. Hum. Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Escayg A., Macdonald B.T., Meisler M.H., Baulac S., Huberfeld G., An-Gourfinkel I., Brice A., LeGuern E., Moulard B., Chaigne D., et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 6.Mullen S.A., Scheffer I.E. Translational Research in Epilepsy Genetics. Arch. Neurol. 2009;66:21–26. doi: 10.1001/archneurol.2008.559. [DOI] [PubMed] [Google Scholar]
- 7.Pellacani S., Sicca F., Di Lorenzo C., Grieco G.S., Valvo G., Cereda C., Rubegni A., Santorelli F.M. The Revolution in Migraine Genetics: From Aching Channels Disorders to a Next-Generation Medicine. Front. Cell. Neurosci. 2016;10:156. doi: 10.3389/fncel.2016.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahlig K.M., Rhodes T.H., Pusch M., Freilinger T., Pereira-Monteiro J.M., Ferrari M.D., Maagdenberg A.M.J.M.V.D., Dichgans M., George A.L.J. Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. USA. 2008;105:9799–9804. doi: 10.1073/pnas.0711717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dichgans M., Freilinger T., Eckstein G., Babini E., Lorenz-Depiereux B., Biskup S., Ferrari M.D., Herzog J., Maagdenberg A.M.V.D., Pusch M., et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- 10.Weiss L.A., Escayg A., Kearney J.A., Trudeau M., Macdonald B.T., Mori M., Reichert J., Buxbaum J.D., Meisler M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry. 2003;8:186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- 11.Sanders S.J., Campbell A.J., Cottrell J.R., Møller R.S., Wagner F.F., Auldridge A.L., Bernier R.A., Catterall W.A., Chung W.K., Empfield J.R., et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018;41:442–456. doi: 10.1016/j.tins.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruth K.A., Grisolano T.M., Ahern C.A., Williams A.J. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: A role for pluripotent stem cells? Mol. Autism. 2020;11:1–11. doi: 10.1186/s13229-020-00330-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Post R.M., Ketter T.A., Uhde T., Ballenger J.C. Thirty Years of Clinical Experience with Carbamazepine in the Treatment of Bipolar Illness. CNS Drugs. 2007;21:47–71. doi: 10.2165/00023210-200721010-00005. [DOI] [PubMed] [Google Scholar]
- 14.Post R., Frye M.A., Denicoff K.D., Leverich G.S., Kimbrell T.A., Dunn R.T. Beyond Lithium in the Treatment of Bipolar Illness. Neuropsychopharmacology. 1998;19:206–219. doi: 10.1016/S0893-133X(98)00020-7. [DOI] [PubMed] [Google Scholar]
- 15.Mula M., Pini S., Cassano G.B. The Role of Anticonvulsant Drugs in Anxiety Disorders. J. Clin. Psychopharmacol. 2007;27:263–272. doi: 10.1097/jcp.0b013e318059361a. [DOI] [PubMed] [Google Scholar]
- 16.Tiihonen L., Hallikainen T., Ryynänen O.-P., Repo-Tiihonen E., Kotilainen I., Eronen M., Toivonen P., Wahlbeck K., Putkonen A. Lamotrigine in treatment-resistant schizophrenia: A randomized placebo-controlled crossover trial. Boil. Psychiatry. 2003;54:1241–1248. doi: 10.1016/S0006-3223(03)00524-9. [DOI] [PubMed] [Google Scholar]
- 17.Dursun S.M., Deakin B. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: A naturalistic caseseries outcome study. J. Psychopharmacol. 2001;15:297–301. doi: 10.1177/026988110101500409. [DOI] [PubMed] [Google Scholar]
- 18.Dursun S.M., McIntosh D., Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch. Gen. Psychiatry. 1999;56:950. doi: 10.1001/archpsyc.56.10.950. [DOI] [PubMed] [Google Scholar]
- 19.Focken T., Burford K., Grimwood M.E., Zenova A., Andrez J.-C., Gong W., Wilson M., Taron M., Decker S., Lofstrand V., et al. Identification of CNS-Penetrant Aryl Sulfonamides as Isoform-Selective NaV1.6 Inhibitors with Efficacy in Mouse Models of Epilepsy. J. Med. Chem. 2019;62:9618–9641. doi: 10.1021/acs.jmedchem.9b01032. [DOI] [PubMed] [Google Scholar]
- 20.Ikemoto S., Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: A unifying interpretation with special reference to reward-seeking. Brain Res. Rev. 1999;31:6–41. doi: 10.1016/S0165-0173(99)00023-5. [DOI] [PubMed] [Google Scholar]
- 21.Day J.J., Carelli R.M. The nucleus accumbens and Pavlovian reward learning. Neuroscientist. 2007;13:148–159. doi: 10.1177/1073858406295854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Setlow B. The nucleus accumbens and learning and memory. J. Neurosci. Res. 1997;49:515–521. doi: 10.1002/(SICI)1097-4547(19970901)49:5<515::AID-JNR1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 23.Gong W., Neill D., Lynn M., Justice J. Dopamine D1/D2 agonists injected into nucleus accumbens and ventral pallidum differentially affect locomotor activity depending on site. Neuroscientist. 1999;93:1349–1358. doi: 10.1016/S0306-4522(99)00235-3. [DOI] [PubMed] [Google Scholar]
- 24.Roberts M.D., Gilpin L., Parker K.E., Childs T.E., Will M., Booth F.W. Dopamine D1 receptor modulation in nucleus accumbens lowers voluntary wheel running in rats bred to run high distances. Physiol. Behav. 2012;105:661–668. doi: 10.1016/j.physbeh.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 25.Dreher J.K., Jackson D.M. Role of D1 and D2 dopamine receptors in mediating locomotor activity elicited from the nucleus accumbens of rats. Brain Res. 1989;487:267–277. doi: 10.1016/0006-8993(89)90831-7. [DOI] [PubMed] [Google Scholar]
- 26.Lim B.K., Huang K.W., Grueter B.A., Rothwell P.E., Malenka R.C. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–189. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bessa J.M., Morais M., Marques F., Pinto L., Palha J.A., Almeida O.F.X., Sousa N. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl. Psychiatry. 2013;3:e266. doi: 10.1038/tp.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francis T.C., Chandra R., Friend D.M., Finkel E., Dayrit G., Miranda J., Brooks J.M., Iñiguez S.D., O’Donnell P., Kravitz A.V., et al. Nucleus Accumbens Medium Spiny Neuron Subtypes Mediate Depression-Related Outcomes to Social Defeat Stress. Boil. Psychiatry. 2015;77:212–222. doi: 10.1016/j.biopsych.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace D.L., Han M.-H., Graham D.L., Green T.A., Vialou V., Iñiguez S.D., Cao J.-L., Kirk A., Chakravarty S., Kumar A., et al. CREB regulation of nucleus accumbens excitability mediates social isolation–induced behavioral deficits. Nat. Neurosci. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Wadsworth P., Singh A.K., Chen H., Wang P., Folorunso O., Scaduto P., Ali S.R., Laezza F., Zhou J. Identification of peptidomimetics as novel chemical probes modulating fibroblast growth factor 14 (FGF14) and voltage-gated sodium channel 1.6 (Nav1.6) protein-protein interactions. Bioorganic Med. Chem. Lett. 2018;29:413–419. doi: 10.1016/j.bmcl.2018.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali S.R., Liu Z., Nenov M., Folorunso O., Singh A.K., Scala F., Chen H., James T.F., Alshammari M., Panova-Elektronova N.I., et al. Functional Modulation of Voltage-Gated Sodium Channels by a FGF14-Based Peptidomimetic. ACS Chem. Neurosci. 2018;9:976–987. doi: 10.1021/acschemneuro.7b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali S., Shavkunov A., Panova N., Stoilova-McPhie S., Laezza F. Modulation of the FGF14:FGF14 homodimer interaction through short peptide fragments. CNS Neurol. Disord.-Drug Targets. 2014;13:1559–1570. doi: 10.2174/1871527313666141126103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wadsworth P.A., Folorunso O., Nguyen N., Singh A.K., D’Amico D., Powell R.T., Brunell D., Allen J., Stephan C., Laezza F. High-throughput screening against protein:protein interaction interfaces reveals anti-cancer therapeutics as potent modulators of the voltage-gated Na+ channel complex. Sci. Rep. 2019;9:1–15. doi: 10.1038/s41598-019-53110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patching S.G. Surface plasmon resonance spectroscopy for characterisation of membrane protein-ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta BBA Biomembr. 2014;1838:43–55. doi: 10.1016/j.bbamem.2013.04.028. [DOI] [PubMed] [Google Scholar]
- 35.Swinney D.C. The role of binding kinetics in therapeutically useful drug action. Curr. Opin. Drug Discov. Dev. 2009;12:31–39. [PubMed] [Google Scholar]
- 36.Ali S.R., Singh A.K., Laezza F. Identification of Amino Acid Residues in Fibroblast Growth Factor 14 (FGF14) Required for Structure-Function Interactions with Voltage-gated Sodium Channel Nav1.6*. J. Boil. Chem. 2016;291:11268–11284. doi: 10.1074/jbc.M115.703868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardill B.R., Rivera-Acevedo R.E., Tung C.-C., Van Petegem F. Crystal structures of Ca2+-calmodulin bound to NaV C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc. Natl. Acad. Sci. USA. 2019;116:10763–10772. doi: 10.1073/pnas.1818618116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardill B.R., Rivera-Acevedo R.E., Tung C.-C., Okon M., McIntosh L.P., Van Petegem F. The voltage-gated sodium channel EF-hands form an interaction with the III-IV linker that is disturbed by disease-causing mutations. Sci. Rep. 2018;8:4483. doi: 10.1038/s41598-018-22713-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C., Chung B.C., Yan H., Lee S.-Y., Pitt G.S. Crystal Structure of the Ternary Complex of a NaV C-Terminal Domain, a Fibroblast Growth Factor Homologous Factor, and Calmodulin. Structure. 2012;20:1167–1176. doi: 10.1016/j.str.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerfen C.R., Surmeier D.J. Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calabresi P., Picconi B., Tozzi A., Ghiglieri V., Di Filippo M. Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat. Neurosci. 2014;17:1022–1030. doi: 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- 42.Laezza F., Gerber B.R., Lou J.-Y., Kozel M.A., Hartman H., Craig A.M., Ornitz D.M., Nerbonne J.M. The FGF14F145S Mutation Disrupts the Interaction of FGF14 with Voltage-Gated Na+ Channels and Impairs Neuronal Excitability. J. Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lou J.-Y., Laezza F., Gerber B.R., Xiao M., Yamada K.A., Hartmann H., Craig A.M., Nerbonne J.M., Ornitz D.M. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelay-Gimeno M., Glas A., Koch O., Grossmann T.N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scala F., Nenov M., Crofton E., Singh A.K., Folorunso O., Zhang Y., Chesson B.C., Wildburger N., James T.F., Alshammari M.A., et al. Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway. Cell Rep. 2018;23:555–567. doi: 10.1016/j.celrep.2018.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shavkunov A., Panova N., Prasai A., Veselenak R., Bourne N., Stoilova-McPhie S., Laezza F. Bioluminescence Methodology for the Detection of Protein–Protein Interactions Within the Voltage-Gated Sodium Channel Macromolecular Complex. ASSAY Drug Dev. Technol. 2012;10:148–160. doi: 10.1089/adt.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu W.-C.J., Nenov M.N., Shavkunov A., Panova N., Zhan M., Laezza F. Identifying a Kinase Network Regulating FGF14:Nav1.6 Complex Assembly Using Split-Luciferase Complementation. PLoS ONE. 2015;10:e0117246. doi: 10.1371/journal.pone.0117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frostell-Karlsson Å., Remaeus A., Roos H., Andersson K., Borg P., Hämäläinen M., Karlsson R. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J. Med. Chem. 2000;43:1986–1992. doi: 10.1021/jm991174y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.