

Abstract
Hepatic glucose metabolism signaling downstream of insulin can diverge to multiple pathways including AKT. Genetic studies suggest that AKT is necessary for insulin to suppress gluconeogenesis. To specifically address the role of AKT2, the dominant liver isoform of AKT in the regulation of gluconeogenesis genes, we generated hepatocytes lacking AKT2 (Akt2−/−). We found that, in the absence of insulin signal, AKT2 is required for maintaining the basal level expression of phosphoenolpyruvate carboxyl kinase (PEPCK) and to a lesser extent G6Pase, two key rate-limiting enzymes for gluconeogenesis that support glucose excursion due to pyruvate loading. We further showed that this function of AKT2 is mediated by the phosphorylation of cyclic AMP response element binding (CREB). Phosphorylation of CREB by AKT2 is needed for CREB to induce the expression of PEPCK and likely represents a priming event for unstimulated cells to poise to receive glucagon and other signals. The inhibition of gluconeogenesis by insulin is also dependent on the reduced FOXO1 transcriptional activity at the promoter of PEPCK. When insulin signal is absent, this activity appears to be inhibited by AKT2 in manner that is independent of its phosphorylation by AKT. Together, this action of AKT2 on FOXO1 and CREB to maintain basal gluconeogenesis activity may provide fine-tuning for insulin and glucocorticoid/glucagon to regulate gluconeogenesis in a timely manner to meet metabolic needs.
Introduction
The primary function of the liver in glucose homeostasis is breaking down glycogen storage to produce glucose through gluconeogenesis during starvation. During fasting when the insulin signal is at minimum, the two rate-limiting enzymes involved in gluconeogenesis, glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxyl kinase (PEPCK) are induced to facilitate the production of glucose from other precursors [1]. This process is regulated by glucagon or glucocorticoid involving the activation of protein kinase A and transcriptional activity of cyclic AMP response element binding factor (CREB) [1]. The expression of PEPCK and G6Pase is also inhibited in fed condition when the hormone levels of insulin are elevated among others [2]. The signaling events downstream of insulin have been investigated and were found to lead to the activation of a serine/threonine kinase AKT [3]. AKT regulates a broad spectrum of cellular processes including cell growth and survival, lipid and glucose metabolism, etc. by phosphorylating a variety of downstream targets [4–6]. One such target is the forkhead transcriptional factor FOXO1 [7–9]. When phosphorylated by AKT, FOXO1 is prevented from translocating to the nucleus to perform its function as a transcriptional factor. This action of AKT is thought to be the primary mechanism for insulin to suppress gluconeogenesis as FOXO1 controls the transcription of both G6Pase and PEPCK [10–12]. Consistently, FOXO1 activity has been found to be increased in the insulin resistant (IR) state and down-regulation of FOXO1 is found to reverse hyperglycemia observed in IR models [13,14]. However, the physiological role of this signaling node under normal unstimulated conditions is not clear.
In this study, we investigated the regulation of PEPCK and G6Pase expression by AKT2, the dominant AKT isoform in the liver [15], when insulin signal is absent. In hepatocytes lacking AKT2, the ability of insulin to inhibit the expression of PEPCK and G6Pase is compromised, consistent with the in vivo hyperglycemia phenotype observed in the Akt2−/− mice [16]. This observation is consistent with a negative regulatory role of insulin/AKT signal on gluconeogenesis and the expression of PEPCK and G6Pase. Surprisingly, in unstimulated cells, the deletion of Akt2 by itself led to down-regulation of PEPCK and G6Pase, suggesting a positive regulatory function of AKT2 on the expression of PEPCK and G6Pase when insulin signal is not present. This observation is supported by analysis showing that loss of AKT2 inhibited the excursion of glucose in a pyruvate loading test in vivo and suppressed the expression of PEPCK and G6Pase in the livers of fasting mice. To further explore the mechanisms responsible for this novel observation, we explored the two key transcriptional factors involved in the regulation of PEPCK and G6Pase, FOXO1 and CREB [17]. Our data suggest that AKT2-dependent CREB phosphorylation is required to sustain the basal expression of PEPCK in the absence of insulin. Additionally, our data suggest that AKT2 may inhibit the FOXO1 regulated transcription of PEPCK independent of its role on phosphorylation of FOXO1. This regulation of AKT2 on CREB and FOXO1 likely primes the promoter of PEPCK to respond to either insulin or glucagon in a timely manner.
Experimental procedures
Tissue culture
Primary hepatocytes were isolated from livers of Akt2−/− (C−;A−/−, Akt2 null) mice [16] and Akt2+/+ (Con) mice. Hepatocytes were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM with 4.5 g/L glucose; Mediatech, Inc) supplemented with 10% heat-inactivated fetal bovine serum FBS (Atlas Biologicals), 5 μg/ml insulin (Sigma), and 10 ng/ml epidermal growth factor (EGF; Invitrogen). Cells were plated in six-well plates at 0.5 × 106 per well density. For insulin stimulation experiments, the medium was changed to free serum DMEM (no glucose) medium when the culture reached 80% confluence. On the second morning, 100 nM Insulin or vehicle was added and incubated for 6 h. prior to lysing the cells for protein and RNA analysis. For forskolin treatment, cells were maintained in low serum (0.2% FBS) media for 16 h at 37°C in 5% CO2. The medium was replaced with fresh medium with 100 μM Forskolin (Alomone Labs Cat# F-500) or vehicle and incubated for 24 h before harvesting. To ectopically express AKT, FOXO-AAA, wtCREB or CREB-S133, 4 μg of plasmid DNA containing the corresponding cDNA sequence were mixed with 10 μl of Lipofectamine and incubated with the cells for 48 h. Protein and RNA were collected for analysis as described [18].
Animals
The Akt2−/− (AM) mice [16] were crossed with PtenloxP/loxP; Alb-Cre+ (PM) mice to generate the double mutant PtenloxP/loxP; Akt2−/−; Alb-Cre+ (DM) as previously described [19–23]. Control animals are PtenloxP/loxP; Alb-Cre− mice. All mice were kept on a standard chow diet and allowed free access to food. All animals were kept in 12 h dark/light cycled facilities. All experiments are performed at USC following USC IACUC approved protocols in accordance with IACUC guidelines.
Mice at 1 month of age were used for experiments. For fasting glucose, animals were fasted overnight (16 h). Blood glucose levels were determined using a handheld Therasense glucometer device. For tissue sections, mice were fasted overnight for 16 h. Blood was collected from cardiac puncture. Livers were then perfused with cold PBS and collected in formalin for histology, collected in OTC for frozen sectioning, and fresh frozen in liquid nitrogen for protein, RNA and lipid analysis. Tissue sections were stained with H&E and Oil Red O to visualize the morphology and lipid accumulation.
Insulin tolerant test (ITT)
An ITT was performed on mice fasted for 6 h. For glucose measurement, tail veins were punctured and a small amount of blood was released and applied onto a handheld Therasense glucometer. For ITT, mice were given a single dose (0.5 unit/kg body weight) of Humulin (Eli Lily) by i.p. injection after a baseline glucose check. Circulating glucose levels were then measured at indicated time points after insulin injection.
Pyruvate tolerant test (PTT)
A PTT was performed on mice fasted for 16 h. For glucose measurement, tail veins were punctured and a small amount of blood was released and applied onto a handheld Therasense glucometer. For PTT, mice were given a single dose (2 g/kg body weight) of l-pyruvate (Sigma) by i.p. injection after a baseline glucose check. Circulating glucose levels were then measured at indicated time points after pyruvate injection.
Quantitative PCR
Total RNA was extracted from liver tissues or cells using Trizol reagent according to the protocol provided by the manufacturer and quantitative PCR performed as previously reported [18].
Protein electrophoresis
Frozen liver tissues were lysed in three times the volume of cold RIPA buffer containing protease inhibitors as previously reported [24]. Seventy-five micrograms of protein lysates were loaded for each sample for electrophoresis using polyacrylamide gel. After protein transfer, membranes were probed with antibodies for PTEN, p-AKT, p-GSK and p-CREB from Cell Signaling. The membranes were also probed with β-actin (Sigma) for loading control.
Statistics
All data are presented as means ± standard errors of the means (SEM). All data were subjected to Student’s t-test for statistical comparison between the two groups. For comparisons among groups of three or more, multivariance ANOVA is applied to determine between group differences followed by TUKEY as post-hoc analysis. A P value less than or equal to 0.05 is considered to be statistically significant.
Results
Regulation of basal expression of PEPCK and G6Pase by AKT2
To genetically dissect the role of AKT signaling in hepatic regulation of gluconeogenesis by insulin, we established hepatocyte cell lines from mice carrying Akt2 deletion (Akt2−/−) [16]. The control hepatocyte lines are established from the Akt2+/+ mice (Con). The two cell lines are morphologically similar and both express albumin (Figure 1A, bottom left panel). In the control hepatocytes, insulin treatment led to the expected induction of AKT and GSK-3 phosphorylation (Figure 1A, bottom right panel). In cells lacking AKT2, AKT2 is completely undetectable. The phosphorylated form of AKT is still detectable, presumably contributed by other AKT isoforms. In hepatocytes lacking AKT2, insulin was incapable of inducing phosphorylation of GSK-3 nor AKT.
Figure 1. AKT2 regulates basal expression of PEPCK And G6Pase.
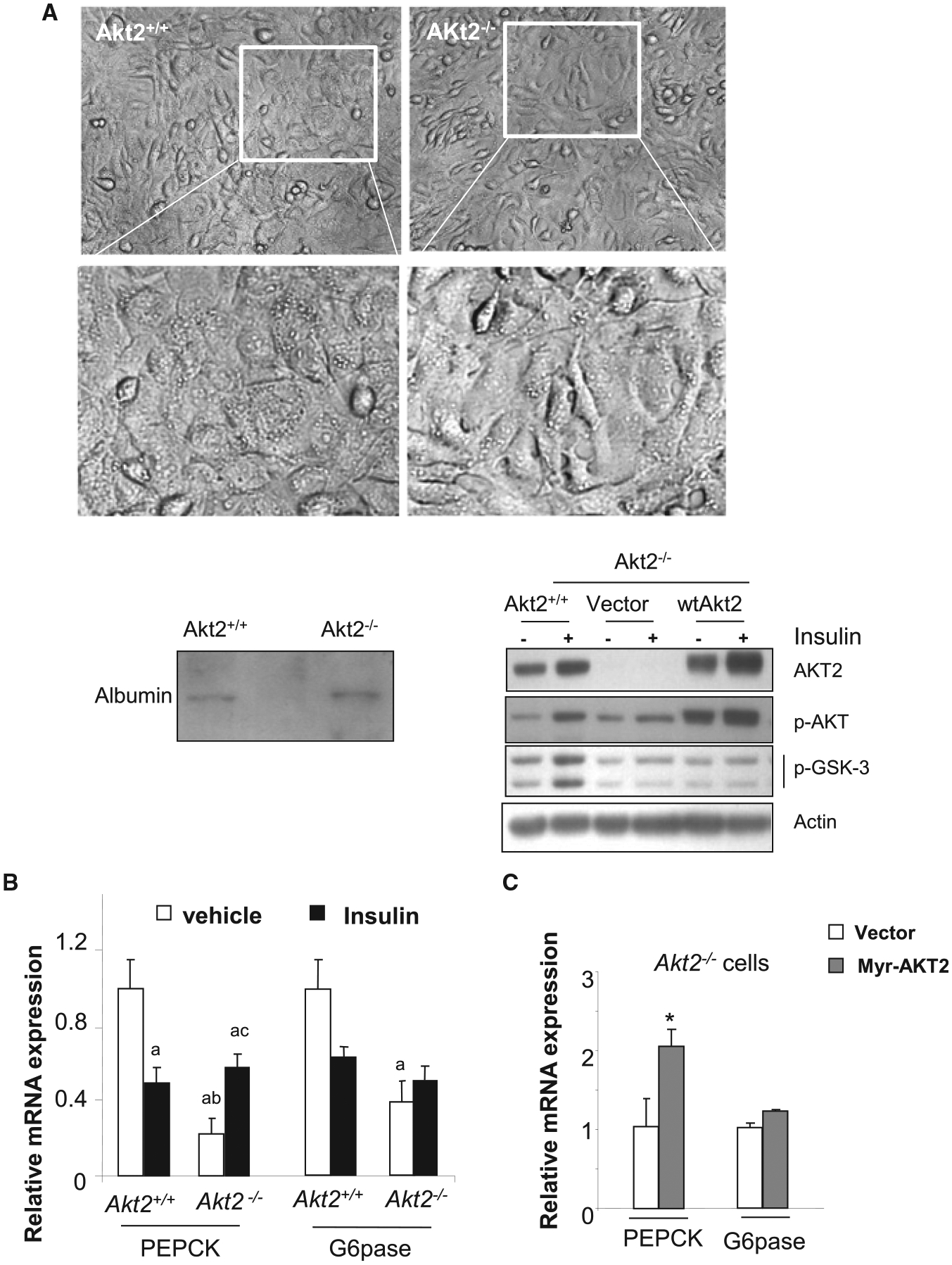
(A) Hepatocyte cell lines established from Akt2−/− mouse liver. Top, representative image of hepatocytes established from control (akt2+/+) and Akt2−/− mouse liver. Bottom left, expression of albumin in hepatocyte cell lines established from the mouse livers. Bottom right, expression of AKT2, phospho-AKT, and p-GSK3 in hepatocytes with or without ectopic expression of AKT2. (B) Akt2+/+ and Akt2−/− hepatocytes are treated with vehicle (open bar) or 100 nM insulin (solid bar) for 6 h. Expression of PEPCK and G6Pase were determined using quantitative PCR analysis. n = 3. Data presented as mean ± SEM. P ≤ 0.05. a, different from Vehicle control treated Akt2+/+ cells; b, different from insulin treated Akt2+/+ cells; c. different from vehicle treated Akt2−/− cells. Experiments repeated at least three times. (C) Akt2−/− hepatocytes were transiently transfected with Myr-AKT2. Expression of PEPCK and G6Pase were determined using quantitative PCR analysis. n = 3. Data presented as mean ± SEM. * P ≤ 0.05. Different from vector transfected Akt2−/− cells. Experiments repeated at least three times.
In Akt2+/+ hepatocytes, insulin treatment led to 50% suppression of PEPCK and 30% suppression of G6Pase as expected (comparing solid vs. open bars in Akt2+/+ hepatocytes) (Figure 1B). Consistent with the requirement of AKT signal in insulin suppression of PEPCK and G6Pase, insulin is incapable of suppressing the expression of PEPCK and G6Pase in the Akt2−/− hepatocytes (Figure 1B). Indeed, expression of both PEPCK and G6Pase increased as a result of insulin stimulation in Akt2−/− cells (comparing solid vs. open bars in Akt−/− hepatocytes). Surprisingly, deletion of Akt2 also significantly attenuated the expressions of PEPCK and G6Pase in the non-stimulated cells (comparing the open bars in Akt2−/− vs. Akt2+/+ cells), suggesting a stimulatory role of AKT2 on the expression of PEPCK and G6Pase when insulin signal is not present (Figure 1B).
Analysis of Akt2−/− hepatocytes with the expression of a constitutively active form of AKT2, myr-AKT2 confirmed this stimulatory role of AKT2 on PEPCK expression (Figure 1C). Reintroduction of myr-AKT2 led to robust up-regulation of p-AKT in the Akt2−/− cells (Figure 1A). Phosphorylation of GSK-3, however, is not restored. As GSK-3 is subjected to regulation by multiple signals [25], other signals interacting with the regulation of GSK-3 may also be at play. Instead of inhibiting the expression of PEPCK, the reintroduction of myr-AKT2 led to almost 2-fold increase in PEPCK expression whereas G6Pase is less affected (Figure 1C). Other enzymes involved in gluconeogenesis and glycolysis such as fructose 1,6-bisphosphatase, glucokinase pyruvate kinases 1 and 2 were not similarly affected (data not shown). Thus, this effect of AKT2 appears to be specific to PEPCK, and to a lesser extend G6Pase.
The effect of AKT2 in pyruvate induced glucose excursion in vivo
To validate this observation in vivo, we examined the expression of PEPCK and G6Pase in livers of the Akt2−/− mice. In these mice, both fasting and random fed glucose levels are increases compared with that observed in the wild type (Akt2+/+) mice, consistent with the proposed role of AKT2 in regulating insulin signal and glucose metabolism (Figure 2A,B). The ability of insulin to suppress plasma glucose is also impaired in the intraperitoneal insulin tolerance (ITT) tests when Akt2 is deleted (Figure 2C). We observed that AKT2 loss resulted in ~50% decreased expression of both PEPCK and G6Pase in these livers of the fasting Akt2−/− vs. Akt2+/+ mice (Figure 2D). These observations confirms the in vitro data shown in Figure 1B,C suggesting that the presence of AKT2 is required to sustain the expression of PEPCK (and G6Pase) when insulin signal is absent.
Figure 2. AKT2 loss suppresses glucose excursion induced by pyruvate in the absence of insulin/nutrient signal.
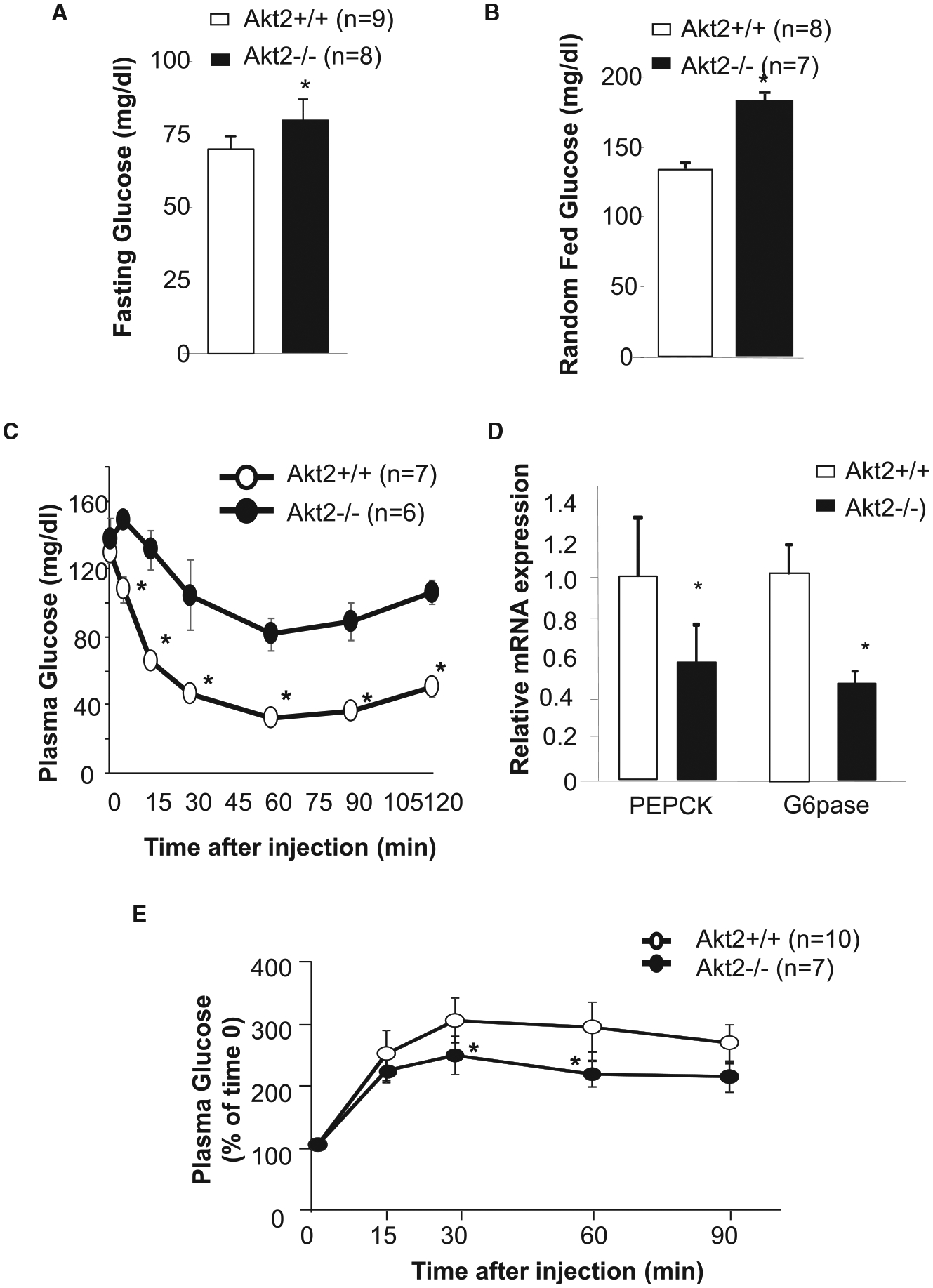
(A) Fasting plasma glucose levels determined in Akt2+/+ (A+/+) and Akt2−/− (A−/−) mice fasted for 16 h. (B) Random fed plasma glucose levels. (C) Insulin tolerance test (ITT) performed in Akt2+/+ (A+/+) and Akt2−/− (A−/−) mice fasted for 6 h. (D) Expression of PEPCK and G6Pase in Akt2+/+ and Akt2−/− mice fasted for 16 h. n = 3. (E) Pyruvate tolerant test (PTT) in Akt2+/+ (A+/+) and Akt2−/− (A−/−) mice fasted for 16 h. Data presented as mean ± SEM. * P ≤ 0.05.
We also subjected the mice to a pyruvate tolerant (or loading) test (PTT) to assess the role of AKT2 loss on the effects of pyruvate overload on glucose excursion. Here, the injection of pyruvate led to increases in plasma glucose levels in both control and Akt2−/− mice as expected (Figure 2E). In the absence of AKT2, however, the glycemic excursion induced by pyruvate loading is significantly lower in Akt2−/− mice (solid circle) compared with that of the controls with intact AKT2 (open circle). These data are consistent with our in vitro observations showing that AKT2 is needed to maintain the expression of PEPCK, and supports a role of AKT2 in sustaining gluconeogenesis (Figure 1).
AKT2 regulates basal gluconeogenic gene expression via CREB phosphorylation
The forkhead transcriptional factor FOXO1 is a well-characterized substrate for AKT and has been reported to mediate the insulin regulated suppression of PEPCK and G6Pase [10–12]. FOXO1 binding element has been reported for both PEPCK and G6Pase promoters [26,27]. To address whether AKT2 regulates PEPCK under the insulin-free condition via FOXO1, we expressed a mutant form of FOXO1, FOXO1-AAA in hepatocytes without insulin stimulation. FOXO1-AAA cannot be phosphorylated by AKT2 and thus is not excluded from the nucleus upon AKT phosphorylation by insulin. The nuclear exclusion of FOXO1 is credited for how insulin suppresses the expression of PEPCK and G6Pase [10–12]. Under the insulin-free conditions, our result shows that FOXO1-AAA stimulated the expression of PEPCK when Akt2 is deleted (Figure 3). This data is consistent with a positive regulatory role of FOXO1 on the expression of PEPCK in the absence of insulin. Interestingly, the introduction of FOXO1-AAA led to moderate but insignificant inhibition of PEPCK in hepatocytes with intact AKT2 (Figure 3). Thus, while FOXO1 binding to the promoter of PEPCK is capable of inducing its expression at basal levels, the presence of AKT2 may negate this effect regardless of FOXO1 phosphorylation by AKT2.
Figure 3. FOXO1 does not mediate AKT2-regulated induction of PEPCK.
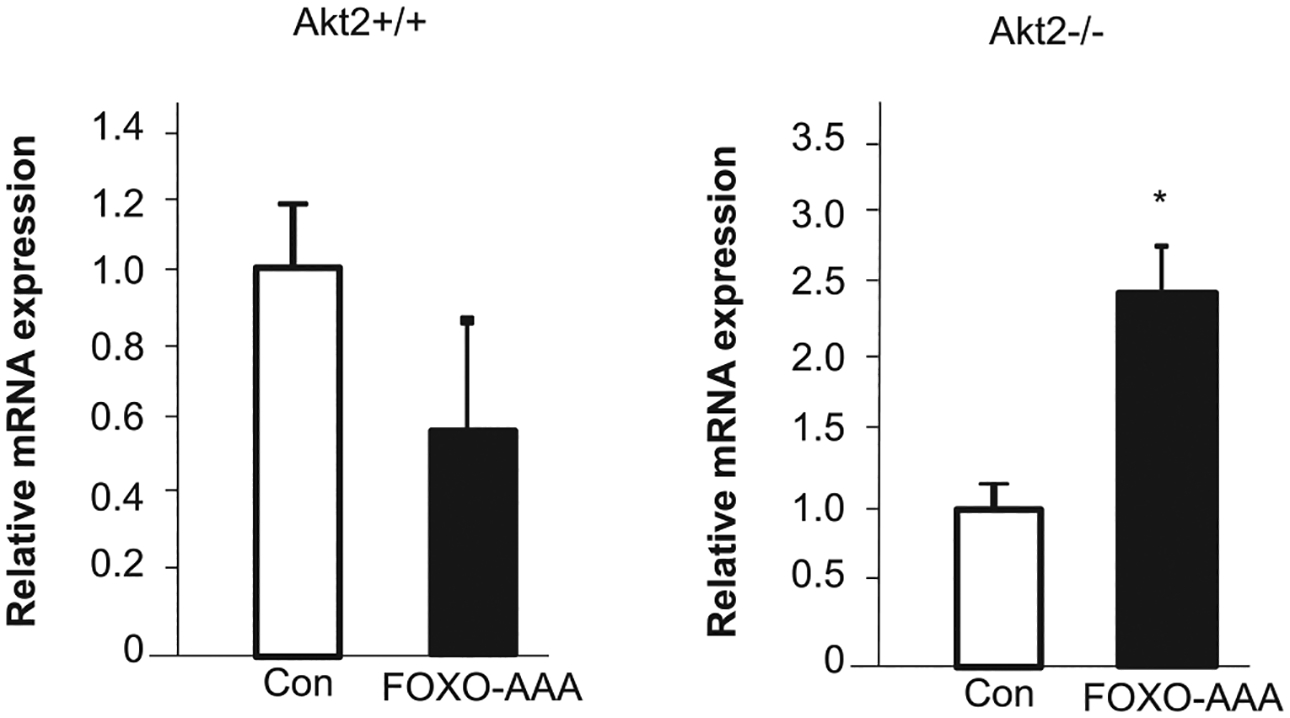
FOXO-AAA is transiently transfected to Akt2+/+ and Akt2−/−. PEPCK expression is determined using quantitative PCR analysis. n = 3. Data presented as mean ± SEM. * P ≤ 0.05. Different from vector transfected cells of the same genotype. Experiments repeated at least three times.
To further explore the mechanisms by which AKT2 may sustain the basal expression of PEPCK, we explored the phosphorylation of CREB. We showed previously that PTEN (phosphatase and tensin homolog deleted on chromosome 10) regulates the phosphorylation of CREB through PI3K/AKT signaling, independent of PKA [28]. PTEN is the phosphatase that negatively regulates PI3K/AKT and loss of PTEN leads to ubiquitous phosphorylation of AKT [29]. Using hepatocytes lacking PTEN, we have identified CREB as a potential substrate for AKT kinase activity [28]. Here, we show that AKT2 is responsible for the regulation of CREB phosphorylation. While PTEN loss led to induction of CREB phosphorylation, deletion of Akt2 in the livers of mice lacking PTEN reversed this phosphorylation (Figure 4A). This observation is confirmed using hepatocytes where AKT2 is deleted (Figure 4B), even though forskolin, which increases cellular levels of cyclic AMP (cAMP) is still capable of inducing CREB phosphorylation in these cells. CREB is the key transcriptional factor utilized by glucagon and glucocorticoid to induce the transcription of PEPCK and G6Pase [30]. To investigate the role of CREB on the basal expression of gluconeogenic genes regulated by AKT2, a dominant-negative mutant CREB carrying S133A mutation was introduced to hepatocytes. CREB-S133A cannot be phosphorylated by AKT and acts as a dominant negative form of CREB to inhibit its activity as a transcriptional factor [28]. Expression of CREB-S133A in control hepatocytes led to reduced expression of PEPCK (Figure 4C, left panel), whereas the introduction of wtCREB to Akt2−/− cells is unable to elicit a response in PEPCK expression (Figure 4C, middle panel). This observation suggests that CREB activation is needed for AKT2 to maintain the expression of PEPCK. In addition, intact AKT2 appears to be needed for p-CREB to perform its transcriptional activity. wtCREB is only able to induce PEPCK when AKT2 signal is present in cells with intact AKT2, i.e. Con cells, but not in Akt2−/− cells. Such manipulation led to robust induction of PEPCK by over 15-folds (Figure 4C, left panel). Thus, the presence of AKT2 granted the induction of PEPCK by CREB and is necessary to maintain the basal levels of PEPCK expression in hepatocytes.
Figure 4. CREB phosphorylation mediates AKT2-regulated induction of PEPCK.
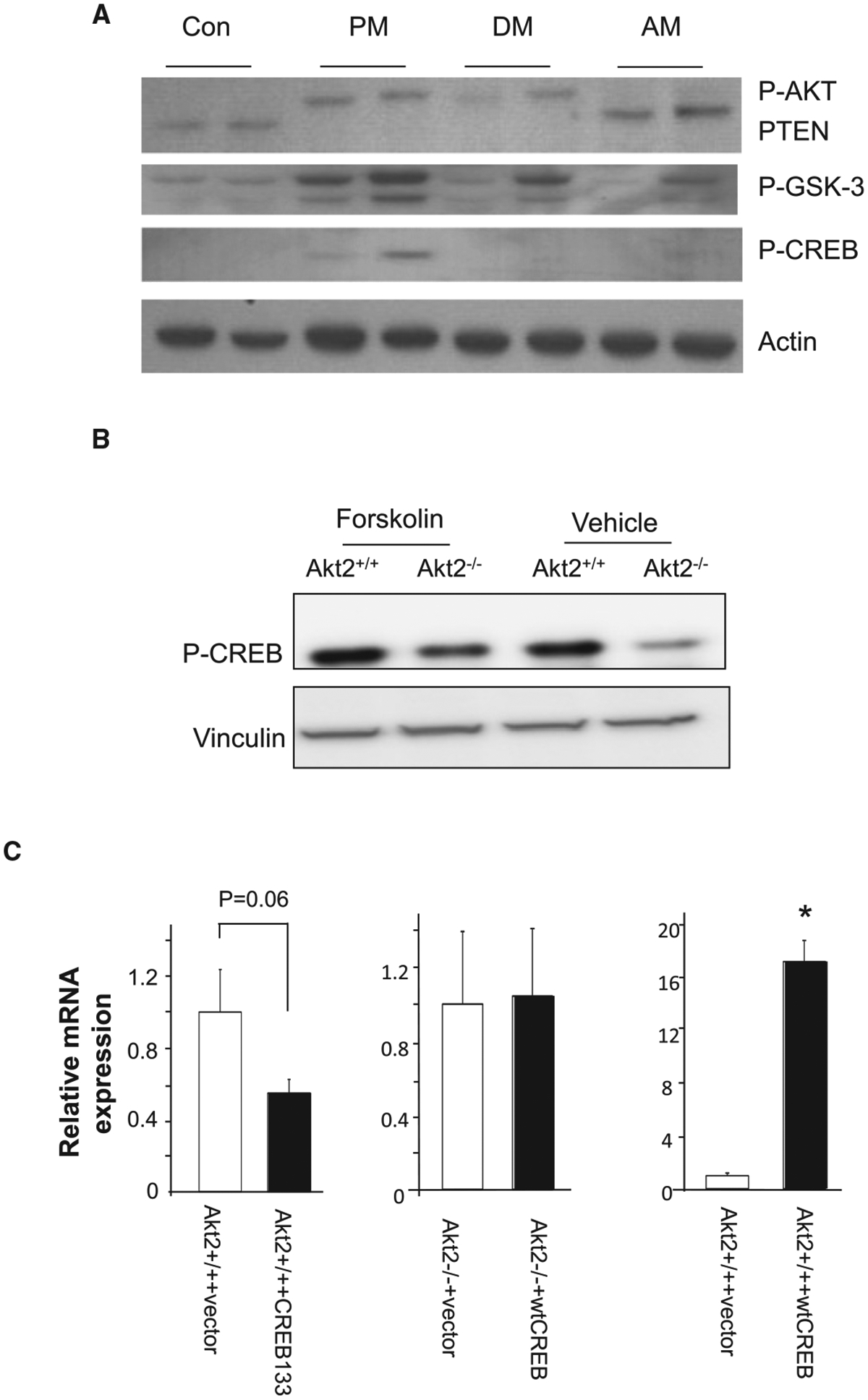
(A) CREB phosphorylation induced by PTEN loss is regulated by AKT2. Liver lysates from mice with different genotypes were analysis for p-CREB. PM, Pten deleted; DM, Pten and Akt2 deleted; AM, Akt2 deleted. Two biological replicas are included for each genotype group. Protein levels of p-AKT, PTEN and p-GSK3 are analyzed and showed up-regulation of AKT and GSK-3 with PTEN loss and inhibition when AKT2 is lost. Phosphorylation of CREB is only observed when PTEN is lost. AKT2 loss together with PTEN blocked this phosphorylation in the DM livers. (B) Akt2+/+ and Akt2−/− cell were treated with forskolin (100 μM) for 24 h to induce cAMP and CREB phosphorylation or vehicle as controls. Phosphorylation of CREB is detected and showed to be lower when AKT2 is lost. When forskolin is added to induce cAMP, protein expression of p-CREB is evaluated in both Akt2+/+ and Akt2−/− cells even though its levels remains lower in the Akt2−/− cells. Vinculin is probed as loading control. (C) Introduction of CREB-133A and wtCREB to Akt2+/+ and Akt2−/− hepatocytes. Expression of PEPCK is analyzed using quantitative PCR analysis. Left panel, CREB-S133A is introduced to Akt2+/+ cells. Middle panel, wtCREB is introduced to Akt2−/− cells; Right panel, wtCREB is introduced to Akt2+/+ cells. n = 3. Data presented as mean ± SEM. * P ≤ 0.05. Different from vector transfected cells of the same genotype. Experiments repeated at least three times.
Discussion
The liver is the major organ involved in the regulation of glucose homeostasis and is the first organ to see insulin produced from the islets. In the liver, insulin inhibits gluconeogenesis via both canonical signals that require AKT and FOXO1 as well as AKT independent and FOXO1 dependent pathways [31]. In this paper, we investigated the role of AKT2 in the regulation of the key rate-limiting enzymes for gluconeogenesis, i.e. PEPCK and G6Pase. We found that in addition to playing a role in the suppression of these genes regulated by insulin, AKT2 positively regulates the expression of PEPCK and G6Pase when insulin signal is not present. We showed that phosphorylation of CREB appears to be necessary for this AKT2 regulated expression of PEPCK and G6Pase. In addition, the induction effect of PEPCK expression by exogenous expression of CREB also depends on the presence of AKT2. On the other hand, while FOXO1 positively regulate the promoter of PEPCK, this ability may be blocked by AKT2 via other means than phosphorylation of FOXO1. Together, our data suggest (1) AKT2 is necessary to sustain the basal expression of PEPCK (and to a lesser extent G6Pase); (2) phosphorylation of CREB by AKT2 is necessary for AKT2 to maintain the expression of these two genes; (3) AKT2 may suppress FOXO1 induced expression of PEPCK via means independent of AKT-regulated phosphorylation of FOXO1. Examining these three points together, we propose that AKT2 primes the promoters of these two gluconeogenic genes, particularly PEPCK, by inducing the phosphorylation of CREB. This primed promoter can then either be stimulated by glucagon/glucocorticoid to promote gluconeogenesis or inhibited by insulin to suppress gluconeogenesis (Figure 5).
Figure 5. Schematic for AKT2 regulated PEPCK expression mediated by CREB phosphorylation and FOXO1 activity.
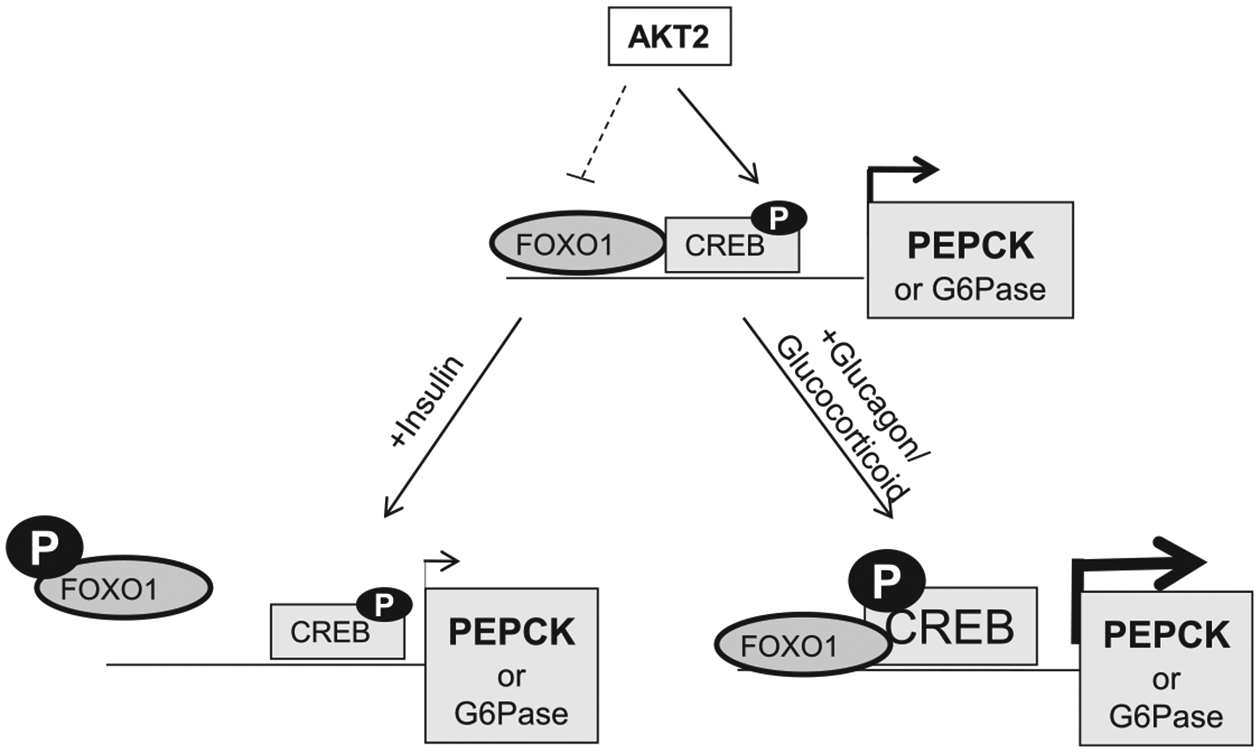
In the absence of insulin signal, AKT2 sustains the expression of PEPCK (and to a lesser extent G6Pase) by inducing the phosphorylation of CREB. This action likely primes the PEPCK promoter for further response to glucagon and glucocorticoid induced phosphorylation of CREB mediated by cAMP-PKA. AKT2 also keeps the FOXO1 transcriptional activity at an inhibitory state. This, however is independent of its phosphorylation on FOXO1 in the absence of insulin.
In the fed state, insulin suppresses the output of glucose from the liver by inhibiting its de novo synthesis [32]. AKT as the kinase downstream of insulin signal has been characterized to play important roles in this process [33]. In particular, the loss of AKT2 has resulted in conditions that are similar to insulin resistance and diabetes [16]. This effect is largely due to higher hepatic glucose output and reduced uptake of glucose in the muscle in Akt2−/− mice [16,34,35]. The inability of insulin to suppress hepatic output of glucose during feeding in Akt2−/− mice leads to glucose intolerance in these mice whereas glucagon plays a minimum role in this process as levels of glucagon only elevated slightly later in aged when mice are already progressing to diabetes [16,34–36]. In contrast, AKT1 loss affected primarily the growth of pancreatic β-cells but is dispensable for maintenance of glucose homeostasis [37]. However, transient loss of AKT2 is not sufficient to suppress refeeding induced inhibition of gluconeogenesis [38]. Loss of both AKT1 and AKT2 are necessary in this immediate response. Our data using isolated hepatocytes suggest that AKT2 indeed mediates the insulin’s ability to suppress the expression of PEPCK and G6Pase, even though AKT1 may also play a role. More importantly, we report the novel finding that AKT2 is necessary to sustain the basal level of expression of PEPCK and G6Pase.
The kinase activity of AKT phosphorylates forkhead transcriptional factor FOXO1. This action results in the exclusion of FOXO1 from the nucleus where it binds to the promoter of PEPCK and G6Pase, leading to suppression of these genes by insulin [39]. In mice where insulin signal is blocked by deletion of irs-1 and −2 and FOXO1 activity is high, deletion of FOXO1 restores inhibition of gluconeogenesis and normalizes euglycemia controls [40]. Together, the canonical signals suggest that AKT-FOXO1 signal is the mediators for insulin to suppress gluconeogenesis. However, a recent report has questioned the role of AKT as a cell-autonomous intermediate in insulin signaling [16,34,35,41]. AKT2 independent but FOXO1 dependent signals have been suggested to exist [38]. Our data here showed that the nuclear-located FOXO1 (FOXO1-AAA) is capable to induce the expression of PEPCK when AKT2 is not present. The presence of AKT2 suppresses this effect, suggesting that AKT2 and FOXO1 mediated PEPCK expression are not linear and other signals are also present for AKT2 to inhibit PEPCK transcription that is likely independent of FOXO1.
The primary signal for inducing the expression of gluconeogenic genes is orchestrated by CREB. At the fasting state when glucagon is the primary metabolic hormone released by the pancreas rather than insulin, mediated by a G-protein coupled receptor, glucagon stimulates the accumulation of cAMP [30]. The cAMP-PKA mediated phosphorylation of CREB at Ser133 is needed for glucagon to induce the transcription of PEPCK and G6Pase. We showed previously that CREB could also be phosphorylated by AKT at Ser133 [28]. This phosphorylation is independent of cAMP-PKA signal. Here, using hepatocyte lacking AKT2, we showed that AKT2 mediates the phosphorylation of CREB. Previously, it had been reported that AKT1 rather than AKT2 is the AKT kinase that mediates phosphorylation and the transcriptional activity of CREB [42]. The study, using HEK293T cells that overexpress Gal4-CREB and different mutant of AKT suggests the regulatory domain of AKT1 is needed for the phosphorylation of CREB. Our work here in hepatocytes where the dominant form of AKT is AKT2 suggest that AKT2 also mediates the phosphorylation and transcriptional activity of CREB. Furthermore, phosphorylation of CREB by AKT2 is needed for CREB to induce the transcription of PEPCK. In addition, our data indicates that phosphorylation of CREB mediates the novel basal regulation of PEPCK expression by AKT2 that we observed.
There are two major consequences of insulin action in the liver: suppression of gluconeogenesis and stimulation of lipogenesis. Distinct signals downstream of insulin that regulates these two processes has been credited to be responsible for ‘selective hepatic insulin resistance’ observed in patients [31]. In these IR patients, elevated insulin becomes incapable of suppressing hepatic gluconeogenesis (resistance), but continues to induce lipogenesis (non-resistance). Others and we have shown previously that AKT2-FOXO1 is required for hepatic lipogenesis [22,43]. Our finding here that AKT2 sustains gluconeogenesis through regulation of p-CREB but not FOXO1 demonstrates how these two signals may diverge. Such discovery elucidates the complexity of AKT regulated hepatic insulin signal and sheds light for future studies aimed at unveiling the mechanism for selective insulin resistance.
Acknowledgements
Dr. Zeng would like to acknowledge a fellowship from the California Institute of Regenerative Medicine.
Funding
Part of this work is supported by support from USC center for Liver Disease (P30DK48522).
Abbreviations
- CREB
cyclic AMP response element binding
- DMEM
Dulbecco’s Modified Eagle’s Medium
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- IR
insulin resistant
- ITT
insulin tolerant test
- PEPCK
phosphoenolpyruvate carboxyl kinase
- PTT
pyruvate tolerant test
Footnotes
Competing Interests
The authors declare that there are no competing interest s associated with the manuscript.
References
- 1.Yabaluri N and Bashyam MD (2010) Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci 35, 473–484 [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, Yang S, Chen J and Su Z (2018) Unraveling the regulation of hepatic gluconeogenesis. Front. Endocrinol 9, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatting M, Tavares CDJ, Sharabi K, Rines AK and Puigserver P (2018) Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci 1411, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manning BD and Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stiles BL (2009) PI-3-K and AKT: onto the mitochondria. Adv. Drug Deliv. Rev 61, 1276–1282 [DOI] [PubMed] [Google Scholar]
- 6.Whiteman EL, Cho H and Birnbaum MJ (2002) Role of Akt/protein kinase B in metabolism. Trends Endocrinol. Metab 13, 444–451 [DOI] [PubMed] [Google Scholar]
- 7.Biggs WH III, Meisenhelder J, Hunter T, Cavenee WK and Arden KC (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl Acad. Sci. U.S.A 96, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo S, Rena G, Cichy S, He X, Cohen P and Unterman T (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem 274, 17184–17192 [DOI] [PubMed] [Google Scholar]
- 9.Rena G, Guo S, Cichy SC, Unterman TG and Cohen P (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem 274, 17179–17183 [DOI] [PubMed] [Google Scholar]
- 10.Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA et al. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F et al. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 12.Schmoll D, Walker KS, Alessi DR, Grempler R, Burchell A, Guo S et al. (2000) Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J. Biol. Chem 275, 36324–36333 [DOI] [PubMed] [Google Scholar]
- 13.Cook JR, Langlet F, Kido Y and Accili D (2015) Pathogenesis of selective insulin resistance in isolated hepatocytes. J. Biol. Chem 290, 13972–13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakae J, Kitamura T, Silver DL and Accili D (2001) The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest 108, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS et al. (2005) Role for Akt3/protein kinase Bγ in attainment of normal brain size. Mol. Cell. Biol 25, 1869–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB III et al. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292, 1728–1731 [DOI] [PubMed] [Google Scholar]
- 17.Oh KJ, Han HS, Kim MJ and Koo SH (2013) CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 46, 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng N, Li Y, He L, Xu X, Galicia V, Deng C et al. (2011) Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Mol. Cancer Res 9, 1708–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, Fu D et al. (2017) Wnt/beta-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene 36, 6020–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA et al. (2010) Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 139, 2170–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He L, Gubbins J, Peng Z, Medina V, Fei F, Asahina K et al. (2016) Activation of hepatic stellate cell in Pten null liver injury model. Fibrogenesis Tissue Repair 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L, Hou X, Kanel G, Zeng N, Galicia V, Wang Y et al. (2010) The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am. J. Pathol 176, 2302–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia C, Medina V, Liu C, He L, Qian D, Taojian T et al. (2017) Crosstalk of LKB1- and PTEN-regulated signals in liver morphogenesis and tumor development. Hepatol. Commun 1, 153–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW et al. (2013) PTEN controls beta-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell 12, 1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hur EM and Zhou FQ (2010) GSK3 signalling in neural development. Nat. Rev. Neurosci 11, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barthel A, Schmoll D, Kruger KD, Bahrenberg G, Walther R, Roth RA et al. (2001) Differential regulation of endogenous glucose-6-phosphatase and phosphoenolpyruvate carboxykinase gene expression by the forkhead transcription factor FKHR in H4IIE-hepatoma cells. Biochem. Biophys. Res. Commun 285, 897–902 [DOI] [PubMed] [Google Scholar]
- 27.Durham SK, Suwanichkul A, Scheimann AO, Yee D, Jackson JG, Barr FG et al. (1999) FKHR binds the insulin response element in the insulin-like growth factor binding protein-1 promoter. Endocrinology 140, 3140–3146 [DOI] [PubMed] [Google Scholar]
- 28.Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C et al. (2013) Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor alpha (ERRalpha). J. Biol. Chem 288, 25007–25024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CY, Chen J, He L and Stiles BL (2018) PTEN: tumor suppressor and metabolic regulator. Front. Endocrinol 9, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altarejos JY and Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol 12, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown MS and Goldstein JL (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96 [DOI] [PubMed] [Google Scholar]
- 32.Saltiel AR (2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104, 517–529 [DOI] [PubMed] [Google Scholar]
- 33.Liao J, Barthel A, Nakatani K and Roth RA (1998) Activation of protein kinase B/Akt is sufficient to repress the glucocorticoid and cAMP induction of phosphoenolpyruvate carboxykinase gene. J. Biol. Chem 273, 27320–27324 [DOI] [PubMed] [Google Scholar]
- 34.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL et al. (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Invest 112, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross DN, van den Heuvel AP and Birnbaum MJ (2008) The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 36.Buzzi F, Xu L, Zuellig RA, Boller SB, Spinas GA, Hynx D et al. (2010) Differential effects of protein kinase B/Akt isoforms on glucose homeostasis and islet mass. Mol. Cell. Biol 30, 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho H, Thorvaldsen JL, Chu Q, Feng F and Birnbaum MJ (2001) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem 276, 38349–38352 [DOI] [PubMed] [Google Scholar]
- 38.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S et al. (2012) Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med 18, 388–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Monks B, Ge Q and Birnbaum MJ (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 447, 1012–1016 [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto M, Pocai A, Rossetti L, Depinho RA and Accili D (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 41.Chen WS, Peng XD, Wang Y, Xu PZ, Chen ML, Luo Y et al. (2009) Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol. Cell. Biol 29, 3151–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato S, Ding J and Du K (2007) Differential activation of CREB by Akt1 and Akt2. Biochem. Biophys. Res. Commun 354, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 43.Leavens KF, Easton RM, Shulman GI, Previs SF and Birnbaum MJ (2009) Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 10, 405–418 [DOI] [PMC free article] [PubMed] [Google Scholar]