

Abstract
Due to their prominent roles in development, cancer, and HIV, the chemokine receptor CXCR4 and its ligand CXCL12 have been the subject of numerous structural and functional studies, but the determinants of ligand binding, selectivity, and signaling are still poorly understood. Here, building upon our latest structural model, we used a systematic mutagenesis strategy to dissect the functional anatomy of the CXCR4-CXCL12 complex. Key charge swap mutagenesis experiments provided evidence for pairwise interactions between oppositely charged residues in the receptor and chemokine, confirming the accuracy of the predicted orientation of the chemokine relative to the receptor, and providing insight into ligand selectivity. Progressive deletion of N-terminal residues revealed an unexpected contribution of the receptor N terminus to chemokine signaling. This finding challenges a longstanding “two-site” hypothesis about the essential features of the receptor-chemokine interaction in which the N terminus contributes only to binding affinity. Our results suggest that although the interaction of the chemokine N terminus with the receptor binding pocket is the key driver of signaling, the signaling amplitude depends on the extent to which the receptor N terminus binds the chemokine. Together with systematic characterization of other epitopes, these data enable us to propose an experimentally consistent structural model for how CXCL12 binds CXCR4 and initiates signal transmission through the receptor transmembrane domain.
Introduction
Chemokine receptors are members of the class A family of G protein-coupled receptors (GPCRs), best known for their role in controlling cell migration, particularly in the context of immune system function. They are activated by small 8- to 10-kD secreted proteins (chemokines) that are classified into four subfamilies (CC, CXC, CX3C, and XC) according to the pattern of conserved cysteine residues in their proximal N termini. The mechanism by which chemokines activate receptors has long been described as involving “two sites” and “two steps” (1–5). According to this mechanism, the globular domain of the chemokine binds to the N-terminus (NT) of its receptor (an interface referred to as chemokine recognition site 1, CRS1) and contributes primarily to the affinity of the complex, whereas the N-terminus of the chemokine binds in the transmembrane (TM) domain extracellular-facing pocket of the receptor (chemokine recognition site 2, CRS2) to activate signaling (6). The distinction between these two sites arose from the general observation that mutations in chemokine N-termini produce a disproportionately large effect on receptor signaling efficacy compared to mutations in the chemokine globular domains (7, 8), with similar trends observed for chimeric rearrangements (1) or mutations (9) of the corresponding CRS2 and CRS1 regions of the receptors. Indeed, single point mutations or modifications of chemokine N-termini can completely alter ligand pharmacology, producing antagonists and even superagonists in many cases (2, 7, 10–13).
In 2015, our group solved the structure of the human CXC chemokine receptor 4 (CXCR4) in complex with vMIP-II, a CC subfamily chemokine antagonist from human herpesvirus 8 (14). The CXCR4–vMIP-II structure confirmed the presence of CRS1 and CRS2 interactions as expected from the two-site model, but also revealed an intermediate region, CRS1.5, that bridges CRS1 and CRS2 and contributes to a contiguous interaction interface between the chemokine and receptor. Structures of three other complexes have also been determined: those of the virally encoded receptor US28 in complex with the human CX3C chemokine, CX3CL1, and an engineered variant (15, 16), and that of the human chemokine receptor CCR5 bound to [5P7]CCL5, an engineered antagonist variant of human CCL5 (17). All of these crystallized complexes feature a similar contiguous interaction interface involving CRS1, CRS1.5, and CRS2, suggesting that these epitopes constitute an interaction architecture that is conserved in the chemokine receptor family. The structures also suggest that CRS1.5 acts as a pivot point that enables the relative orientations of the chemokine and receptor to differ between complexes, thereby contributing to ligand recognition and signaling specificity (17).
Despite being one of the most intensely studied chemokine receptors, initially because of its role as a cofactor for HIV infection (18–20) and subsequently because of its widespread role in cancer (21–23), a structure of CXCR4 in complex with its endogenous chemokine ligand, CXCL12, has not yet been determined. Several computational models (24–29), along with our own (14, 30, 31) have been put forward, but important geometrical differences between them (31) highlight the need for experimental validation and refinement. Additionally, experimental data are required to understand how the structure of the complex translates into receptor activation, which is poorly understood, even for this well-studied receptor. There are several likely reasons for this. Previous mutational studies, although valuable, have often been focused on limited sets of mutations and originated from different laboratories using different techniques. Moreover, reports of the effects of mutations have often been based on single-point assays rather than full concentration response curves, and typically without accounting for changes in receptor expression that can critically influence the results. Finally, the most frequently used readout of CXCR4 activation, intracellular calcium (Ca2+) mobilization, is subject to signal amplification that can mask the effect of mutations.
Here, we functionally dissect the signaling role of various features proposed in our latest computational model of CXCR4-CXCL12 (31). In addition to single- and multiple-point loss-of-function mutagenesis, our approach features reciprocal charge reversal (“charge swap”), rescue-of-function mutagenesis to provide evidence for the proposed orientation of the chemokine relative to the receptor. Furthermore, our proposed model includes an extensive interface between the full receptor N-terminus and the chemokine, which is not resolved in any chemokine receptor crystal structure determined to date. Here, we demonstrated that mutations and progressive truncations of the receptor N-terminus caused diminished β-arrestin and G protein recruitment, which was unexpected given that the N-terminus has been purported to be primarily an affinity determinant. Building on our previous studies (14, 30–32), the current data enable us to propose a comprehensive, experimentally consistent structural framework explaining how the chemokine binds to CXCR4 and initiates signal transmission through the receptor TM domain. The data also add to accumulating evidence suggesting that receptor-chemokine interactions are more complex than is implied by the two-site mechanism and that residues outside of CRS2 can play an important role in receptor activation.
Results
Full-length model of the CXCR4-CXCL12 signaling complex
A model of the complex between full-length CXCR4 and mature CXCL12 (Fig. 1A) was generated using an integrated approach that combined homology modeling and flexible molecular docking with experimentally derived restraints from disulfide crosslinking (31). The architecture of the complex is consistent with that of all three crystallized receptor-chemokine complexes (14, 15, 17). It features the CRS1 interaction where the N-terminal residues 21-sYDSMKE-26 of the receptor bind in the groove formed by the “N-loop” and “40s loop” of CXCL12, and the CRS2 interaction where the flexible N-terminus of CXCL12 (NH3+−1-KPVSLSYR-8) reaches into the TM domain pocket of CXCR4, making contacts with critical residues from the so-called “engagement layer” [Asp97(2.63), Asp187(ECL2), and Asp262(6.58); Ballesteros and Weinstein numbering in parentheses] and “signal initiation layer” [Trp94(2.60), Tyr116(3.32), and Glu288(7.39)] (30). These two epitopes are joined by the CRS1.5 region (14), in which Pro27(NT) and Cys28(NT) of CXCR4 pack against the first disulfide (Cys9-Cys50) of CXCL12 and its β3-strand in a conserved manner that has been observed not only in all three crystallized receptor-chemokine complexes (17), but also across multiple chemokine-binding proteins that are unrelated to receptors (33, 34). This suggests that CRS1.5 is an anchor point for various proteins that interact with chemokines. The conformations and interactions in these epitopes originate from our previously published partial model that featured CXCR4 residues 21 to 304 (14), and were further refined in another study (31).
Fig. 1. The computational model of the CXCR4-CXCL12 complex used to guide and interpret the experiments in this work.
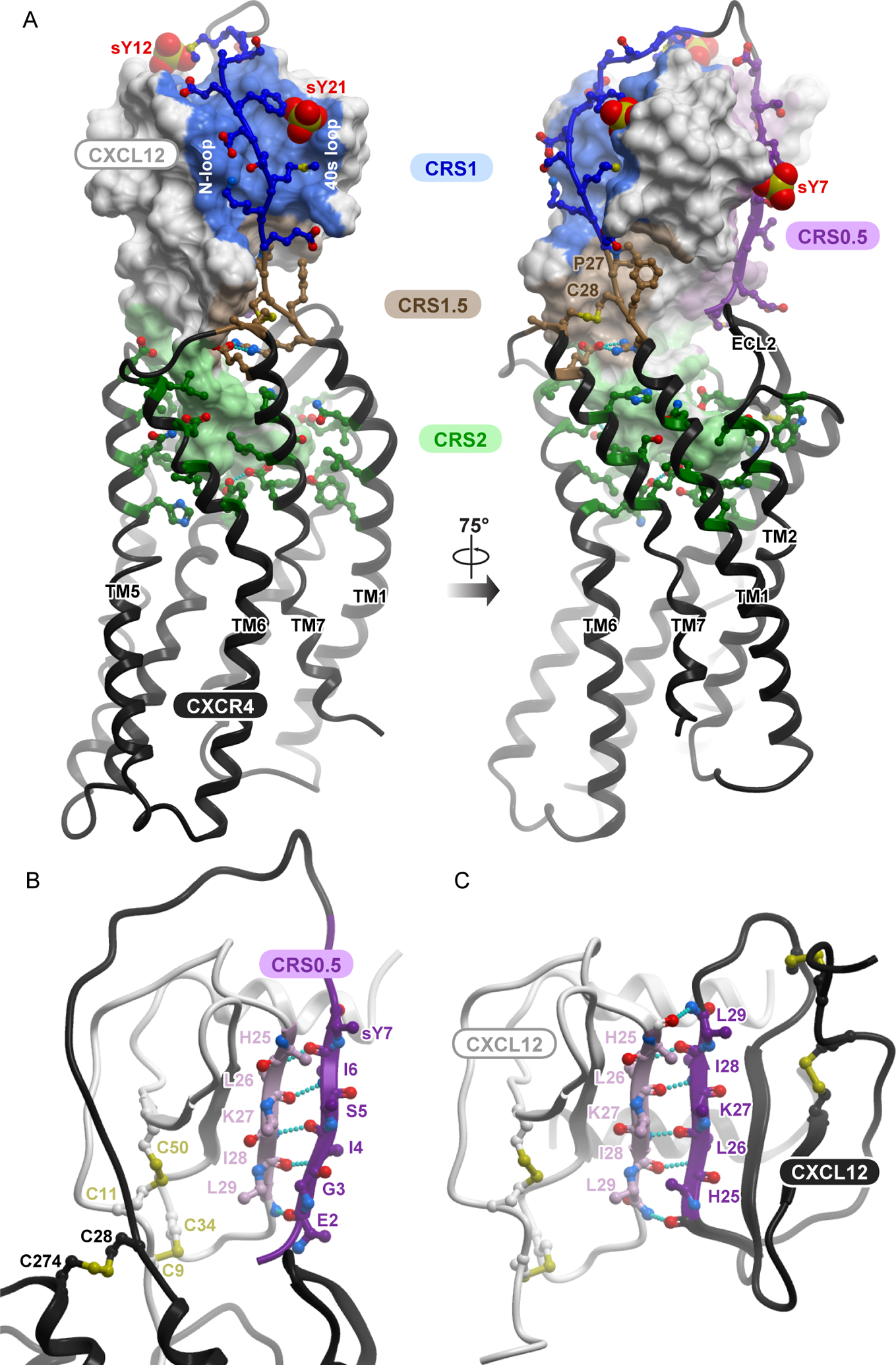
(A) The entire model is viewed along the plane of the membrane. The chemokine is shown as a surface mesh, the receptor as a black ribbon. The distinct interaction epitopes discussed in the paper are labeled. (B) The proposed CRS0.5 interaction involves an antiparallel β-sheet between the distal N terminus of the receptor and the β1 strand of the chemokine. (C) The proposed CRS0.5 interaction between the receptor and the chemokine closely mimics the interaction between CXCL12 monomers in the dimer (PDB ID: 3gv3).
In the current model, we present the complete N-terminus of CXCR4, including residues 1 to 20, a region that has not been resolved in any of the receptor-chemokine complex crystal structures (14, 15, 17). Previous mutagenesis studies suggested that the entire N-terminus of the receptor interacts with the chemokine (30, 35) and highlighted the roles of three sulfated tyrosine residues (sTyr7, sTyr12, and sTyr21) that affect the binding affinity of the chemokine for the receptor (36–38). Our model (Fig. 1A) suggests that the N-terminus of CXCR4 continues on from its position in the CXCR4 crystal structure and “wraps around” the chemokine, engaging residues from the chemokine 310 helix and the C-terminal helix. It also suggests that the distal N-terminal residues 1-MEGISIsY-7 form an anti-parallel β-sheet with the β1-strand of the chemokine (Fig. 1B) in a manner that largely mimics the intermolecular packing in CXCL12 homodimers (Fig. 1C) (39). These residues belong to an interaction epitope referred to as CRS0.5 (40).
Consistent with the abundance of charged residues in both CXCR4 and CXCL12, the model reveals that intermolecular interactions between the receptor and chemokine are mediated by numerous electrostatic contacts. Salt bridges between the N-terminal amine of the chemokine and CXCR4 Asp97(2.63), the side chains of CXCL12 Lys1 and CXCR4 Glu288(7.39), and of CXCL12 Arg8 and CXCR4 Asp262(6.58), form key electrostatic anchors in CRS2 (table S1). The salt bridge between CXCR4 Glu277(7.28) and CXCL12 Arg12 (N-loop) supports the orientation of CXCL12 relative to CXCR4 in the CRS1.5 interaction epitope. The model also predicts several additional pairwise interactions between oppositely charged residues in CRS1 (table S1).
Altogether, the model suggests that the interface between the receptor and the chemokine is compositionally complex. At a minimum, four constituent epitopes can be clearly identified: CRS0.5, CRS1, CRS1.5, and CRS2. Compared to the broadly defined roles of the best-known epitopes, CRS1 and CRS2, almost nothing is known about the role of CRS1.5 contacts in binding and signaling, and the proposed CRS0.5 epitope has never been studied. Moreover, the contributions of the individual residue contacts in these four epitopes (as well as other regions) to the affinity and pharmacology of the CXCR4-CXCL12 complex are unclear. Guided by the model, we set out to quantitatively dissect the anatomy of the CXCR4-CXCL12 interface and the roles of its constituent epitopes in triggering downstream signaling.
Mutagenesis strategy to quantitatively assess the signaling capacity of CXCR4 mutants
Previous mutagenesis studies that investigated CXCR4-CXCL12 interactions almost exclusively focused on G protein signaling, with little attention given to the involvement of β-arrestins, which are also important in CXCR4 function (41–43). Moreover, many of these studies relied on single-point CXCL12 concentration responses, did not account for varying mutant receptor expression, and were based on second messenger assays that are subject to signal amplification (44–46). Although valuable, these studies (most of which are summarized in table S2) do not provide a consistent, uniform, and quantitative assessment of mutants, making it difficult to integrate them into a cohesive model of CXCR4 signaling. Here, we undertook a quantitative and systematic approach. Initially, CXCL12-induced β-arrestin2 recruitment to the receptor mutants was monitored with a bioluminescence resonance energy transfer (BRET)-based assay (fig. S1A) (40, 47). Selected mutants with substantial effects on β-arrestin2 recruitment were then characterized in a BRET-based mini-Gαi association assay (fig. S1B). For each mutant tested in either the β-arrestin2 or Gαi experiments, a full chemokine concentration response curve was generated, and both EC50 (potency) and Emax (efficacy) signaling parameters were obtained (table S3). These direct BRET-based interaction assays are not subject to second messenger signal amplification, in contrast with the commonly used Ca2+ mobilization and inositol monophosphate (IP) accumulation experiments (fig. S1C).
To enable quantitative comparisons between the wild-type (WT) and mutant receptors, we designed the BRET experiments so that the observed potency, efficacy, and fraction of receptor on the cell surface did not vary within a substantial range of total WT CXCR4 abundance (fig. S2, A to E). Total expression of receptor variants was monitored in all experiments, and cell transfections were adjusted as needed to maintain mutant receptor expression within WT range; however, we note that only one mutant required adjustment (fig. S2F, fig. S3A). To account for possible mutation-induced changes in receptor expression, specifically at the cell surface, we independently determined both the total and cell-surface expression of all but two CXCR4 mutants that demonstrated substantial effects in BRET experiments (any statistically significant efficacy impairment of >15% and/or potency impairment >0.1 log10 units) (fig. S3, A to F). Throughout the results, we note all cases in which mutations impaired the fractional surface expression of CXCR4 (fig. S3, C and F). For these mutants, no simple method was available to ensure that their cell surface expression was comparable to that of the WT receptor, because the amount of CXCR4 found at the cell surface did not vary in correspondence with total expression (fig. S2E). Hereafter, we systematically characterized selected mutants across the various CXCR4-CXCL12 interaction epitopes in a model-guided manner. Even though most of the residues mutated here were investigated in previous studies (table S2), taken together, the published data are incomplete and heterogeneous. Here, we attempted to systematically characterize the CXCL12 interface residues of CXCR4 in a uniform and quantitative manner.
Charge swap mutagenesis validates the predicted geometry of the CXCR4-CXCL12 complex
To validate the overall architecture and investigate key polar interactions in the model, we used a strategy of “charge swap” mutagenesis. This strategy is superior to traditional single-sided loss-of-function mutagenesis because it can provide information about direct pairwise residue contacts between the receptor and the chemokine through functional rescue. Our CXCR4-CXCL12 model suggests that Asp262(6.58) of CXCR4 forms a CRS2-anchoring ionic interaction with Arg8 of CXCL12 (Fig. 2A). Consistent with the model, the D262A, D262K, and D262R mutations all nearly abolished CXCL12-mediated β-arrestin2 recruitment, whereas the D262N mutation had a lesser but still substantial effect (Fig. 2B). The D262K mutation also markedly impaired Gαi recruitment to CXCR4 (Fig. 2C). Another important intermolecular salt bridge predicted by the model is between CXCR4 Glu277(7.28) and Arg12 of CXCL12 (Fig. 2A). Mutation of Glu277(7.28) to alanine or glutamine had no negative effect on signaling, whereas the E277K and E277R mutations both showed statistically significant potency deficits and an approximately 30% decrease in β-arrestin2 recruitment efficacy (Fig. 2D, table S3). E277R also impaired both the potency and efficacy of the CXCR4 response to CXCL12 in the Gαi assay (Fig. 2C), to an extent comparable to β-arrestin2 recruitment deficits. By contrast, mutations of neighboring acidic residues Glu268(ECL3) and Glu275(7.26) (fig. S4A) to glutamine, lysine, and arginine resulted in almost no negative effects (fig. S4, B and C). Indeed, we observed slight improvements in β-arrestin2 recruitment efficacy for CXCR4(E268Q/K) and in potency for CXCR4(E275R) (fig. S4, B and C, table S3).
Fig. 2. Reciprocal charge reversal experiments validate the model geometry and establish the roles and the interaction partners of CXCL12 residues Arg8 and Arg12.
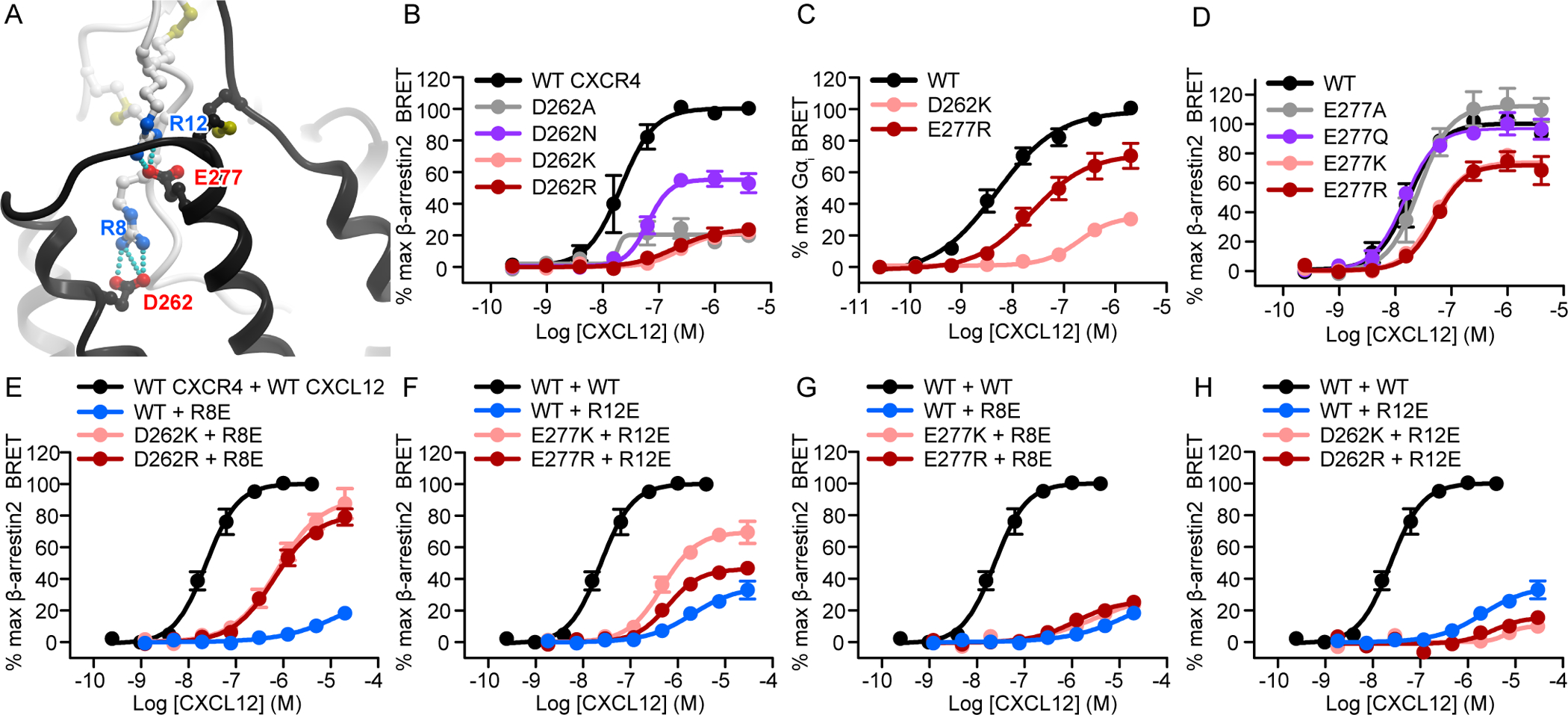
(A) The two salt bridges proposed by the model to determine the orientation of CXCL12 relative to CXCR4 involve CXCL12 Arg8 and Arg12 paired with CXCR4 Asp262(6.58) and Glu277(7.28), respectively. The chemokine and the receptor are shown in white and black ribbons, respectively. (B) β-arrestin2 association BRET ratio data for a series of CXCR4 Asp262(6.58) mutants (D262A/N/K/R) after stimulation with the indicated concentrations of CXCL12 for 20 min. (C) Mini-Gɑi association BRET ratio data for CXCR4(D262K) and CXCR4(E277R) after stimulation with the indicated concentrations of CXCL12 for 1 min. (D) β-arrestin2 association BRET ratio data for a series of CXCR4 Glu277(7.28) mutants (E277A/Q/K/R) after stimulation for 20 min with the indicated concentrations of CXCL12. (E) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 after the stimulation of WT CXCR4 with WT CXCL12, WT CXCR4 with CXCL12(R8E), CXCR4(D262K) with CXCL12(R8E), and CXCR4(D262R) with CXCL12(R8E) for 20 min. (F) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 for WT CXCR4 stimulated with WT CXCL12, WT CXCR4 with CXCL12(R12E), CXCR4(E277K) with CXCL12(R12E), and CXCR4(E277R) with CXCL12(R12E) for 20 min. (G) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 obtained by stimulating WT CXCR4 with WT CXCL12 or by stimulating WT CXCR4, CXCR4(E277K), or CXCR4(E277R) with CXCL12(R8E) for 20 min. (H) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 obtained by stimulating WT CXCR4 with WT CXCL12 or by stimulating WT CXCR4, CXCR4(D262K), or CXCR4(D262R) with CXCL12(R12E) for 20 min. Data in (B) to (H) are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 (WT CXCR4 + WT CXCL12 for the charge swap experiments) tested in the same experiments. The same pooled WT CXCR4 + WT CXCL12 dataset is shown in panels (E) to (H). The same WT CXCR4 + CXCL12(R8E) dataset is shown in panels (E) and (G). The same WT CXCR4 + CXCL12(R12E) dataset is shown in panels (F) and (H). For this and all subsequent figures, error bars smaller than the circle visualizing the mean are not shown.
On the chemokine side, the R8E mutation of CXCL12 practically eliminated its ability to activate CXCR4 (Fig. 2E). However, applying CXCL12(R8E) to cells expressing CXCR4(D262K/R) led to robust activation, with an efficacy greater than those of the same mutants tested individually (Fig. 2E). Indeed, the efficacy of this mutant combination approached that observed for WT CXCR4-CXCL12 signaling. The R12E mutation also greatly decreased CXCL12 potency and efficacy towards CXCR4 activation (Fig. 2F), although not to the same extent as the R8E mutation (Fig. 2E). But again, the E277K mutation in CXCR4 rescued the signaling of CXCL12(R12E) substantially, as did E277R (although with a reduced efficacy) (Fig. 2F). The functional rescue effects were specific, because no rescue was observed when chemokine mutants from each of the two predicted salt bridges were combined with receptor mutants from the other salt bridge [namely CXCL12(R12E) with CXCR4(D262K/R) or CXCL12(R8E) with CXCR4(E277K/R)] (Fig. 2, G and H).
We noted that the potency of either rescuing combination did not reach that of the WT receptor–WT chemokine combination. Furthermore, in the case of CXCR4(E277K/R)-CXCL12(R12E), the rescue was not reciprocal, because the efficacy of this combination exceeded that of WT CXCR4-CXCL12(R12E) but not that of CXCR4(E277K/R)-WT CXCL12. Nevertheless, the fact that receptor mutations restored the signaling deficits of the chemokine mutations suggests that the corresponding residues are in direct contact in the complex. The inability to completely restore signaling to that of the WT receptor can be attributed to the complexity of the interface where other residue interactions play a role, or to the requirement of a precise spatial arrangement of residues for full signaling capacity. Indeed, the salt bridge between CXCR4 Glu277(7.28) and CXCL12 Arg12 is part of a larger, interconnected network of hydrogen bonding interactions also involving CXCR4 Arg30(NT) (Fig. 3A), a residue at the junction of the receptor N-terminus and TM1 that is predicted to be involved in coordination of CRS1.5 interactions. Even a conservative substitution of Arg30(NT) with glutamine resulted in an efficacy loss of approximately 40% in the β-arrestin2 recruitment experiments, whereas an arginine-to-alanine mutant CXCR4(R30A) showed a statistically significant impairment in potency as well (table S3). In the Gαi experiments, CXCR4(R30Q) exhibited a 30% loss in efficacy, as well as a statistically significant reduction in potency (Fig. 3, B and C, table S3).
Fig. 3. CXCR4 residue Arg30(NT) is an important mediator of CXCL12 signaling toward both Gɑi and β-arrestin2.
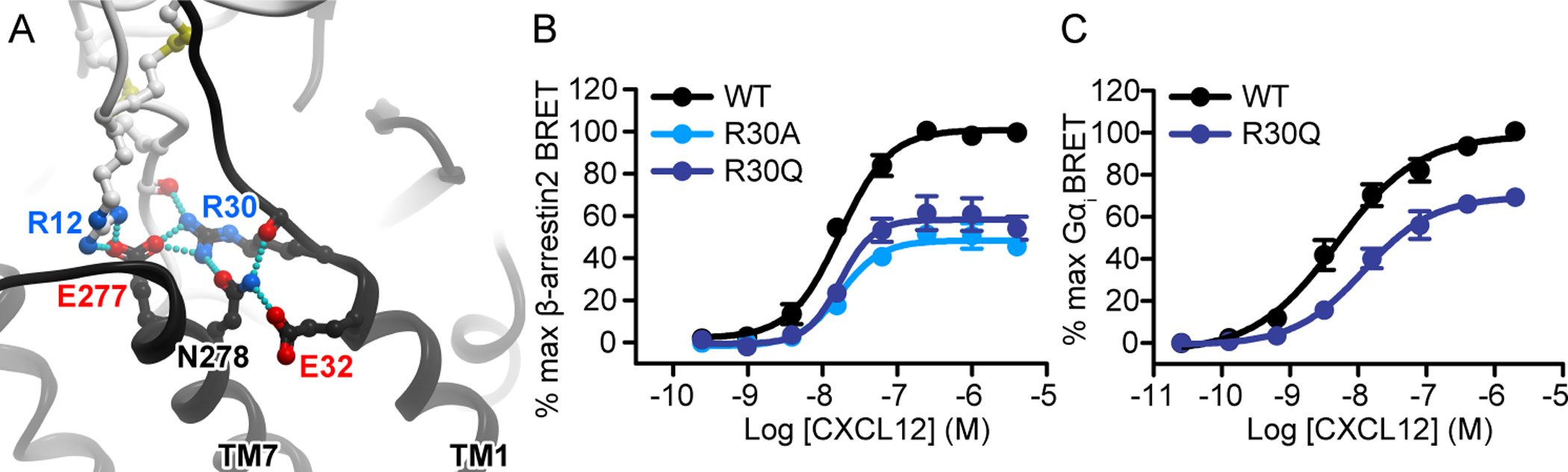
(A) The model predicts CXCR4 Arg30(NT) to be the central residue in the CRS1.5 hydrogen-bonding network between CXCR4 and CXCL12. (B) β-arrestin2 association BRET ratio data for the CXCR4(R30A) and CXCR4(R30Q) mutants after stimulation with the indicated concentrations of CXCL12 for 20 min. (C) Mini-Gɑi association BRET ratio data for CXCR4(R30Q) after stimulation with the indicated concentrations of CXCL12 for 1 min. Data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments.
We also applied charge swap mutagenesis to investigate an alternative geometry of the CXCR4-CXCL12 complex developed by Ziarek et al. (28). In that model, Arg8 of CXCL12 is purported to interact directly with CXCR4 Glu32(NT) rather than with Asp262(6.58), and CXCL12 Arg12 is predicted to interact with CXCR4 Asp181(ECL2) rather than with Glu277(7.28). When tested in the β-arrestin2 recruitment assay, mutations of CXCR4 Glu32(NT) to glutamine, lysine, or arginine resulted in modest impairments in efficacy (8, 15, and 19% reductions, respectively), and CXCR4(E32R) exhibited a statistically significant reduction in potency as well (Fig. 4A, table S3). However, we observed no rescue of the markedly impaired signaling of CXCL12(R8E) when combined with CXCR4(E32K/R) (Fig. 4B). Similarly, we observed no rescue of function when either CXCR4(D181K) or CXCR4(D181R) were combined with CXCL12(R12E) (Fig. 4C). These findings argue against the geometry of the complex proposed by Ziarek et al. (28), but are consistent with our model, in which CXCR4 Glu32(NT) is 15.6 Å away from CXCL12 Arg8 (Cα atom distance), and CXCR4 Asp181(ECL2) is on the opposite side of the CXCR4-binding pocket from Glu277(7.28) and 16.5 Å from CXCL12 Arg12 (Fig. 4D). The discrepancy is due to a different rotational position of the chemokine relative to the receptor in the two models.
Fig. 4. Charge swap experiments do not support the pairing of CXCR4 residues Glu32(NT) and Asp181(ECL2) with CXCL12 Arg8 and Arg12, respectively.
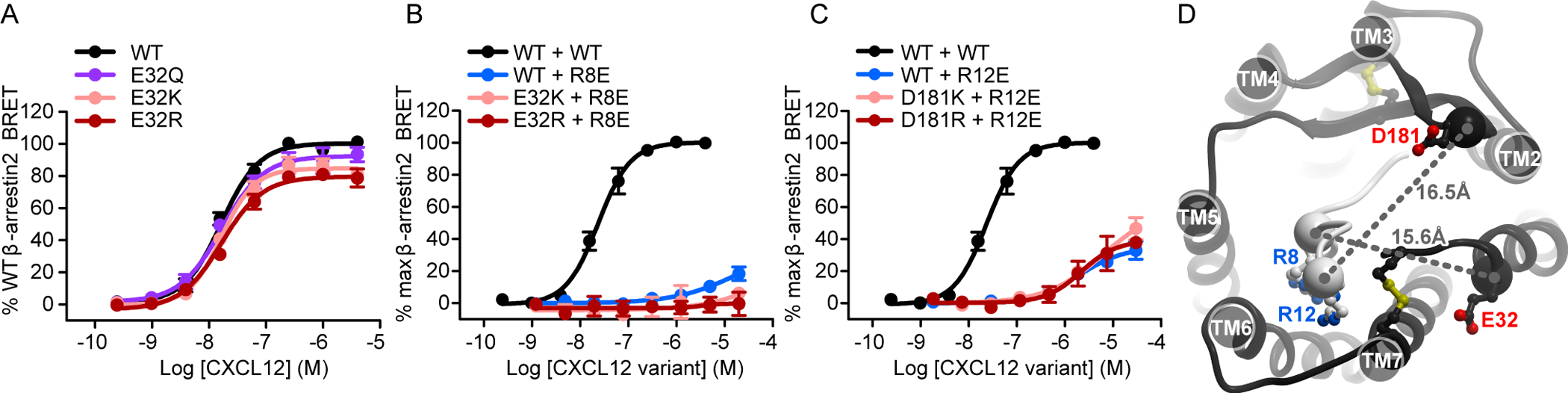
(A) β-arrestin2 association BRET ratio data for a series of CXCR4 Glu32(NT) mutants (E32Q/K/R) after stimulation with the indicated concentrations of CXCL12 for 20 min. (B) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 obtained by stimulating WT CXCR4 with WT CXCL12 or by stimulating WT CXCR4, CXCR4(E32K), or CXCR4(E32R) with CXCL12(R8E). (C) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 obtained by stimulating WT CXCR4 with WT CXCL12 or by stimulating WT CXCR4, CXCR4(D181K), or CXCR4(D181R) with CXCL12(R12E). Data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments. The same pooled datasets for WT CXCR4 + WT CXCL12, WT CXCR4 + CXCL12(R8E), and WT CXCR4 + CXCL12(R12E) are shown in (B) and (C) as were shown in Fig. 2, E to H. (D) The relative location of the two control residue pairs, CXCR4 Asp181(ECL2) and CXCL12 Arg12, and CXCR4 Glu32(NT) and CXCL12 Arg8, in the model of the CXCR4-CXCL12 complex.
The N-terminus of CXCR4, including CRS0.5, contributes to CXCR4 signaling efficacy
Although all three receptor-chemokine complexes crystallized thus far involve full-length receptors, the solved structures lack density for a large stretch of residues in the receptor N-termini (residues 1 to 22 in CXCR4). For the vMIP-II–bound structure of CXCR4 (14), the visible density contains only the proximal N-terminus (residues 23 to 27) interacting with the N-loop and 40s loop groove of the chemokine, and provides no information for the role of any of the putative sulfotyrosines (sTyr7, sTyr12, and sTyr21). As described earlier, our model suggests that the entire receptor N-terminus engages CXCL12 by wrapping around the globular domain of the chemokine, with the distal N-terminus (CRS0.5 residues 3-GISIsY-7) forming an anti-parallel β-sheet with the β1-strand of the chemokine (25-HLKIL-29) (31). To globally test the functional role of the CRS0.5-CRS1 interaction, we generated several CXCR4 constructs with truncations of 7, 10, 15, 19, and 25 residues in the N terminus (Fig. 5A). Successively longer deletions produced progressively larger reductions in β-arrestin2 recruitment efficacy, with CXCR4(Δ1–19) and CXCR4(Δ1–25) displaying efficacies <25% of that of the WT receptor (Fig. 5B, Table S3). Moreover, the efficacy impairment caused by the removal of the first 15 residues (Δ1–15, 30% of WT efficacy remaining) was almost the same as that caused by the truncation of 10 further residues, Δ1–25, suggesting that it is the distal and not the proximal N-terminus that plays a dominant role in the signaling efficacy of the receptor. The truncations did not lead to statistically significant changes in β-arrestin-2 recruitment potency (Fig. 5B, table S3), although CXCR4(Δ1–19) and CXCR4(Δ1–25) yielded such poor responses that their EC50 values could not be accurately fitted. Note that despite the size and location of the truncations, only CXCR4(Δ1–19) showed a notable reduction in cell surface expression (down to 65% of that of the WT receptor), but not to any extent that would explain its impaired signaling efficacy (<15% of that of WT CXCR4; fig. S3, B and C).
Fig. 5. The N terminus of CXCR4 is important for both the efficacy and potency of CXCR4-CXCL12 signaling.
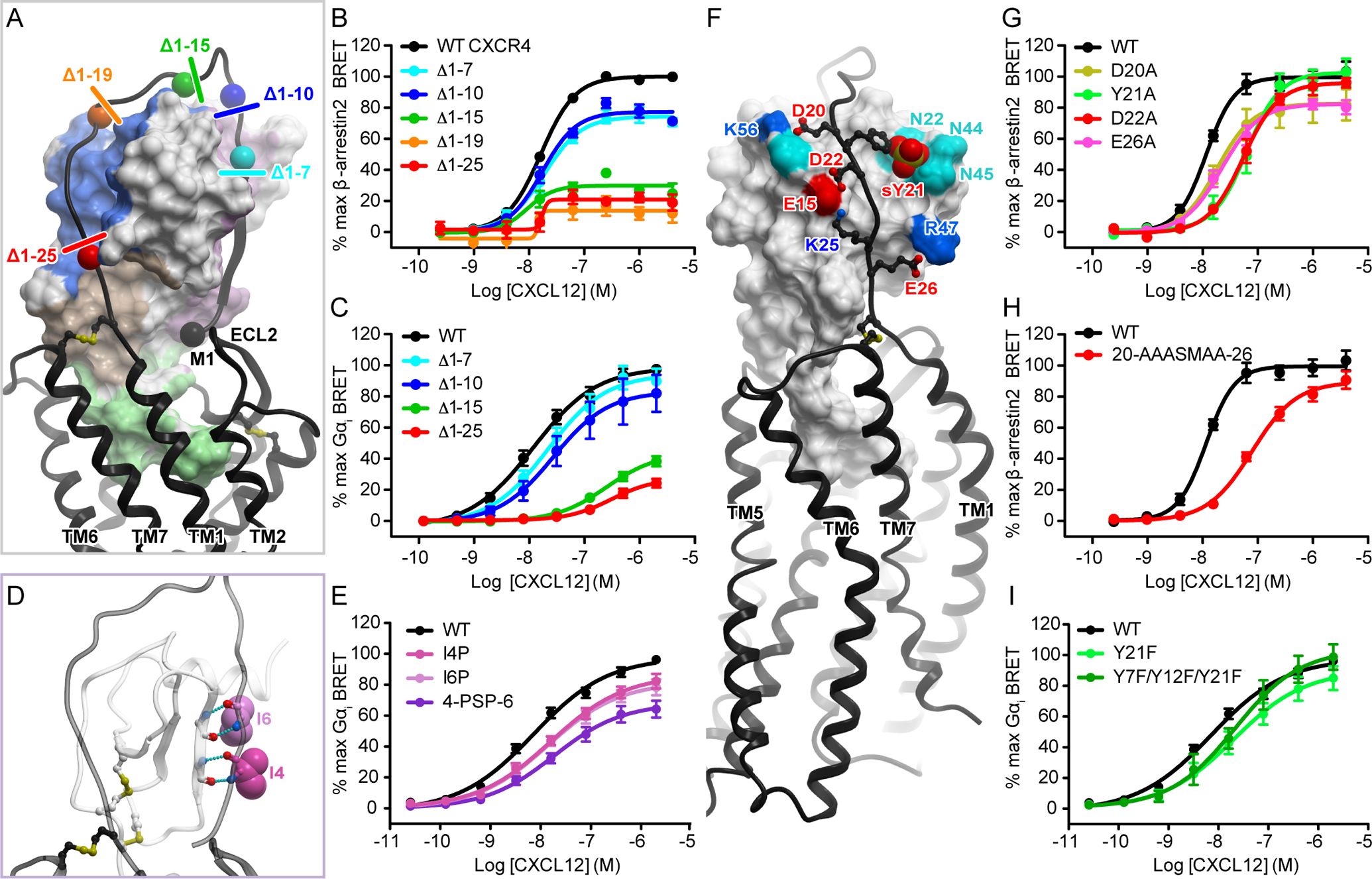
(A) Progressive N-terminal truncations of CXCR4 illustrated in the context of the model of the CXCR4-CXCL12 complex. (B) β-arrestin2 association BRET ratio data for a series of CXCR4 N-terminal truncations (Δ1–7, Δ1–10, Δ1–15, Δ1–19, and Δ1–25) after stimulation with the indicated concentrations of CXCL12 for 20 min. (C) Mini-Gαi association BRET ratio data for a series of CXCR4 N-terminal truncations (Δ1–7, Δ1–10, Δ1–15, and Δ1–25) after stimulation with the indicated concentrations of CXCL12 for 1 min. (D) Predicted interaction of Ile4(NT) and Ile4(NT) (shown as spheres) in the CRS0.5 region of the model, including the hydrogen bonds that would be disrupted by mutation of isoleucine to proline. (E) Mini-Gαi association BRET ratio data for CXCR4(I4P), CXCR4(I6P), and CXCR4(4-PSP-6) after stimulation with the indicated concentrations of CXCL12 for 1 min. (F) The predicted polar interactions between the proximal N terminus of CXCR4 and the N-loop/40s loop groove of CXCL12. (G and H) β-arrestin2 association BRET ratio data for the indicated concentrations of CXCL12 for CXCR4(D20A), CXCR4(Y21A), CXCR4(D22A), and CXCR4(E26A) (G) or for CXCR4(20-AAASMAA-26) (H), in which all four CXCR4 residues tested in (G) together with Lys25(NT) were mutated to alanines. (I) Mini-Gαi association BRET ratio data for the stimulation of CXCR4(Y21F) and CXCR4(Y7F/Y12F/Y21F) with the indicated concentrations of CXCL12. Data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments.
When examined in the Gαi BRET assay, the same overall pattern of reduced efficacy with increasing truncation length was observed (Fig. 5C), although in this case the efficacy impairment caused by the seven-residue truncation was not statistically significant compared to WT CXCR4 (Table S3). In contrast to what we saw with the β-arrestin2 recruitment assay, receptor truncations also led to statistically significant impairments in the potency of Gαi association (Fig. 5C, table S3). Because in the Gαi recruitment assays, the CXCR4(Δ1–15) and CXCR4(Δ1–25) truncation mutants showed cell surface abundances equivalent to 40 and 33% of that of the WT receptor, respectively (fig. S3F), we exercise caution in interpreting the apparent efficacy impairments (33 and 23% of WT efficacy remaining). Therefore, we also decided to investigate the role of the CRS0.5 interaction in a less disruptive, more targeted way: we mutated two CXCR4 isoleucine residues at positions 4 and 6 within CRS0.5 to prolines to exclusively perturb the predicted β-sheet interaction. Introduction of a proline instead of Ile4(NT) reduced the potency of Gαi association, substituting Ile6(NT) with proline reduced the efficacy of Gαi association, and mutating both isoleucines to prolines (4-PSP-6) impaired both potency and efficacy (Fig. 5, D and E, Table S3). Both the total and cell surface expression of CXCR4(I4P), CXCR4(I6P), and CXCR4(4-PSP-6) were similar to that of the WT receptor (fig. S3, D to F), enabling us to attribute the observed signaling defects to the disrupted CRS0.5 interaction.
Charge complementarity, as suggested by the model, likely defines the position of the receptor N-terminus as it makes contact with residues from the N-loop, the 40s loop, and the C-terminal helix of the chemokine, culminating with CRS0.5. Specifically, Glu26(NT), Lys25(NT), Asp22(NT), and Asp20(NT) in the proximal N-terminus of CXCR4 interact with CXCL12 Arg47 (40s loop), Glu15 and His17 (N-loop), and Lys56, whereas the sulfated CXCR4 Tyr21(NT) interacts with CXCL12 Asn22 (310 helix), Asn44, and Asn45 (40s loop) (Fig. 5F). Accordingly, in addition to studies of the distal N-terminus of CXCR4, we also examined alanine mutants of these charged residues in the proximal N-terminus. CXCR4(D20A) exhibited a small (<20%) but statistically significant efficacy impairment, whereas CXCR4(Y21A) and CXCR4(D22A) both showed potency impairments, and CXCR4(E26A) was impaired in both efficacy and potency (Fig. 5G, Table S3). When all of the alanine mutations were combined, the potency defect was not much greater than that of either CXCR4(Y21A) or CXCR4(D22A) alone, and a modest 10% decrease in efficacy was observed (Fig. 5H, fig. S5, table S3), consistent with the traditional view of CRS1 as being a region that principally contributes binding affinity to the CXCR4-CXCL12 complex. However, it appears that multiple N-terminal residues function in unison rather than any individual residue dominating the contribution. Together with the results from experiments with the N-terminal truncation and secondary-structure-disrupting proline mutants, these data challenge the established view of the receptor N-terminus as only an affinity determinant and suggest that it also plays a role in signaling efficacy, but without any apparent “hotspot residues.”
To specifically test the role of sulfation of the three putative sulfotyrosine residues in the receptor N-terminus, we also generated tyrosine-to-phenylalanine mutants and tested them in the Gαi recruitment assay. Mutating CXCR4 Tyr21 to phenylalanine resulted in a reduction in potency, which was not quite statistically significant (P = 0.055, Fig. 5I, table S3). A variant in which all three tyrosine residues were mutated to phenylalanine [CXCR4(Y7F/Y12F/Y21F)] showed the same CXCL12 response as that of CXCR4(Y21F), suggesting that Tyr21 plays the most important role. However, these results may underestimate the importance of CXCR4 N-terminal sulfation. This is because the mutants were compared to the WT receptor, which, due to being overexpressed, may overwhelm the capacity of the endogenous tyrosyl sulfotransferases and cofactor regeneration process, and as a result, be incompletely sulfated. Indeed, previous publications concluded that although Tyr21(NT) plays a dominant role, the binding affinity of CXCL12 to the receptor increases when Tyr12(NT) and Tyr7(NT) are also sulfated (36–38). To quantitatively test the effects of a fully sulfated receptor on signaling, the relevant enzymes will need to be overexpressed as well, which is outside the scope of this study. In any case, the data suggest that sulfation re-enforces the interaction of the receptor N-terminus with the chemokine in a manner consistent with the N-terminus functioning as a whole.
Negatively charged residues in the CXCR4 ECL2 hairpin are important for both β-arrestin2 and Gɑi recruitment efficacy
Extracellular loop 2 (ECL2) of CXCR4 forms a β-hairpin whose tip contains three negatively charged residues: Glu179(ECL2), Asp181(ECL2), and Asp182(ECL2). In our model, these residues are in proximity to a positively charged patch of the chemokine involving CXCL12 residues His25, Lys27, and Arg41 (Fig. 6, A and B). To investigate the role of this charge cluster in CXCR4, we mutated the constituent residues, both separately and together. The mutations D181A/N and D182K/R led to modest (20% or less) but statistically significant reductions in efficacy, whereas D181K and D181R resulted in 23 and 31% reductions, respectively, in maximal β-arrestin2 recruitment, and D182A caused a slight but statistically significant increase in efficacy. However, when Glu179(ECL2), Asp181(ECL2), and Asp182(ECL2) were all mutated to either lysine (179-KAKK-182) or arginine (179-RARR-182), β-arrestin2 recruitment to the mutant receptor was reduced to only 39 and 25% of WT efficacy, respectively (Fig. 6F, Table S3). In the Gαi recruitment assay, CXCR4(179-KAKK-182) also displayed a large reduction in efficacy, together with a much larger decrease in potency than was observed in the β-arrestin2 recruitment assay (Fig. 6G, table S3). This likely indicates the lack of specific intermolecular contacts involving these residues, whereas the overall negative charge of the ECL2 hairpin is important for favorable electrostatic attraction to the major basic surface of the chemokine.
Fig. 6. The residues in the ECL2 hairpin loop are cumulatively important for CXCR4 activation.
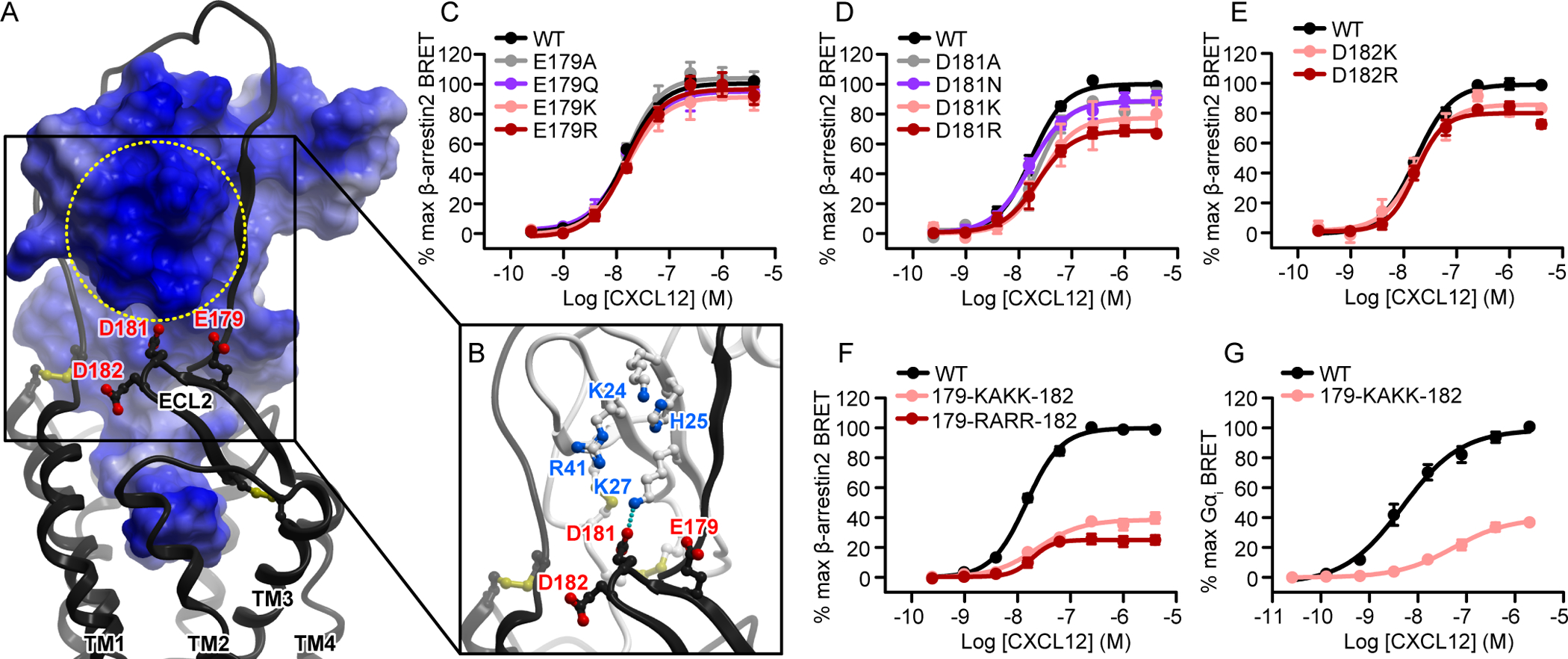
(A and B) The negatively charged β-hairpin of CXCR4 ECL2 (residues 179-EADD-182) is predicted to be proximal to the basic patch on the three-stranded β-sheet of CXCL12. In (A), the receptor is shown as a ribbon and the chemokine as a surface mesh colored blue-to-red by electrostatic potential. The entire chemokine surface is basic, whereas the indicated patch is distinct because of a greater-than-average concentration of positively charged residues. In (B), the chemokine is shown as a ribbon and the basic residues forming the patch are indicated. (C to F) β-arrestin2 association BRET ratio data for (C) a series of Glu179(ECL2) mutations (E179A/Q/K/R), (D) a series of Asp181(ECL2) mutations (D181A/N/K/R), (E) a pair of Asp182(ECL2) mutations (E182K/R), and (F) CXCR4(179-KAKK-182) and CXCR4(179-RARR-182), in which all of the CXCR4 residues tested in (C) to (E) were mutated to lysines or arginines, after stimulation with the indicated concentrations of CXCL12 for 20 min. (G) Mini-Gαi association BRET ratio data for CXCR4(179-KAKK-182) after stimulation with the indicated concentrations of CXCL12 for 1 min. With the exception of CXCR4(D182K) in (E), data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments. In (E), the data for CXCR4(D182K) are means from two independent experiments.
CXCR4 residues directly in contact with the distal N terminus of CXCL12 are critical for activation
CRS2 is the best characterized interaction epitope of the CXCR4-CXCL12 complex (9, 30, 32, 48–52), with numerous studies reporting deleterious effects of mutations of charged residues in the receptor-binding pocket (table S2). This is likely because interactions between these residues and the flexible N-terminus of the chemokine are crucial for CXCL12-mediated CXCR4 activation. Here, we sought to revisit these findings in the context of our 3D model, using the amplification-free assays, quantitative approaches, and cell surface expression monitoring.
In our model, CXCL12 Lys1 interacts with CXCR4 Asp97(2.63) through the N-terminal amine group, and with CXCR4 Glu288(7.39) through the Lys1 side chain (Fig. 7A) (14). Individual mutations of CXCR4 D97N and E288Q eliminated CXCL12-mediated β-arrestin2 recruitment despite being conservative substitutions (Fig. 7B). CXCR4 residue Tyr116(3.32) sits just underneath CXCL12 Pro2 in the model and is thought to couple CXCR4-CXCL12 engagement to intracellular conformational changes within the receptor. In our hands, the Y116A mutation of CXCR4 completely abrogated β-arrestin2 recruitment (Fig. 7B). Finally, in the model, CXCR4 residue Asp187(ECL2), near the base of the CXCR4 ECL2 hairpin, is positioned to interact with CXCL12 Tyr7 and the backbone amide of Val3. Consistent with this, CXCR4(D187A) was almost completely inactive (<15% WT efficacy) in the β-arrestin2 recruitment assay (Fig. 7B, table S3), whereas it retained approximately 30% of the activity of the WT receptor in the Gαi association experiments (Fig. 7C, table S3). These data are consistent with the exceptional sensitivity of CXCR4 activation to residue substitutions in the binding pocket, which also mirrors the sensitivity of receptor activation to modification in the N-terminus of CXCL12 (2). Combined with the structural model, these data provide insight into the initial steps of CXCR4 activation by CXCL12.
Fig. 7. CRS2 receptor binding pocket mutations abrogate β-arrestin2 recruitment.
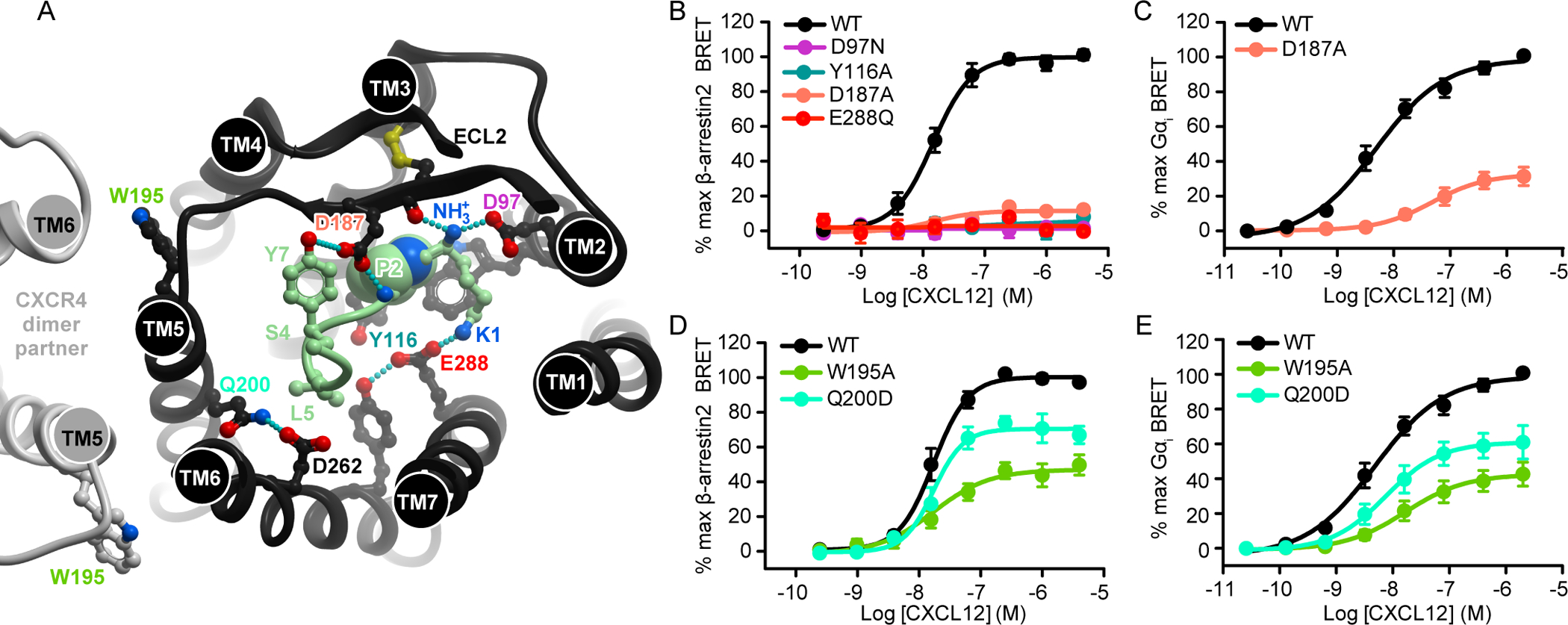
(A) The N terminus of CXCL12 (light green) in the binding pocket of CXCR4. The receptor is viewed “top-down,” or across the plane of the membrane from the extracellular side. Residues mentioned in the text are indicated. The hypothetical position of the CXCR4 dimer (based on the crystallographic dimer identified in PDB ID: 3ODU) is shown on the left in light gray. (B) β-arrestin2 association BRET ratio data for CXCR4(D97N), CXCR4(Y116A), CXCR4(D187A), and CXCR4(E288Q) after stimulation with the indicated concentrations of CXCL12 for 20 min. (C) Mini-Gαi association BRET ratio data for CXCR4(D187A) after stimulation with the indicated concentrations of CXCL12 for 1 min. (D and E) β-arrestin2 association (D) and mini-Gαi association (E) BRET ratio data for the stimulation of CXCR4(W195A) and CXCR4(Q200D) with the indicated concentrations of CXCL12. Data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments.
TM5 mutations cause impaired β-arrestin2 recruitment through unknown mechanisms
In previous mutagenesis studies of CXCR4-CXCL12 signaling, little attention was paid to the role of residues in TM5 and the so-called major subpocket of CXCR4 (part of the binding pocket delineated by TM helices 4 to 6). This is likely because of the established predominant role of residues in the minor subpocket (CXCR4 TM helices 1, 2, and 7), and the sensitivity of the CXCL12 N-terminus to mutations. This view is challenged by our refined model of the complex, which features the CXCL12 N-terminal residues Ser4 and Leu5 in the major subpocket of CXCR4. Additionally, in our studies of the atypical chemokine receptor 3 (ACKR3), a homologous receptor that also binds to CXCL12, we observed that the major subpocket residues Trp208(5.34) and Glu213(5.39) were important for CXCL12-mediated β-arrestin recruitment (40). We therefore tested the effects of mutating the corresponding CXCR4 residues, Trp195(5.34) and Gln200(5.39), on effector recruitment to CXCR4. The CXCR4 Q200D and W195A mutations both led to statistically significantly reductions in the efficacy of β-arrestin2 and Gαi recruitment to the receptor, and W195A also impaired potency in both assays (Fig. 7, D and E). In our model, Gln200(5.39) mediates the TM5 interaction with TM6 by hydrogen bonding to Asp262(6.58), which itself is a key chemokine-coordinating residue (Fig. 7A). Moreover, in many GPCRs, activation is associated with a counter-clockwise rotation of TM5, which helps to shape the G protein–binding pocket at the intracellular side (53–56). The proximity of Gln200(5.39) to CXCL12 Ser4 suggests that these two residues may engage in a direct interaction, facilitating the TM5 rotation and an active-like conformation of the CXCR4 TM bundle. By contrast, Trp195(5.34) points away from the receptor core and is unlikely to engage in direct interaction with the chemokine; therefore, the effect of the W195A mutation is likely indirect or allosteric. This residue has also been proposed to mediate receptor dimerization (57), which may be a possible alternative explanation for the effects of its mutation.
Quantifying the G protein vs β-arrestin pathway bias in mutation-induced signaling impairments
Having collected data on numerous CXCR4 mutants in two amplification-free, direct association assays, we next asked whether the effect of the mutations on the two respective pathways were “balanced,” that is, similarly affecting both G protein and β-arrestin association, or if instead they preferentially affected one effector over the other. Given the quantitative nature of our data, the apparent bias of the mutants could be evaluated using the equiactive comparison method (46). Mutants with unbalanced signaling profiles would be especially intriguing because of the general interest in biased signaling by GPCRs as a strategy for obtaining improved therapeutics and the desire to explain bias from a structural perspective. To calibrate our bias calculations, we first examined residues in the intracellular effector-coupling interface that are known or expected to produce bias, specifically the conserved DRY motif in TM3 that participates directly in G protein coupling (58–60). Mutation of the central Arg(3.50) residue of the DRY motif in other GPCRs, including the homologous CCR5, results in receptor bias wherein G protein signaling is reduced but β-arrestin interactions are preserved or enhanced (61–63). Consistent with these findings, we found that the CXCR4 R134A mutation almost completely abrogated Gαi recruitment (Fig. 8A). However, in the β-arrestin2 recruitment assays, CXCR4(R134A) showed not only increased constitutive association (Fig. 8B), but also increased efficacy and potency of the CXCL12 response (Fig. 8C, table S3). When subjected to the equiactive comparison model, this mutant demonstrated a bias factor of −1.13 ± 0.19 (Fig. 8D, table S3). Although CXCR4(R134A) suffered from a slight decrease in cell surface expression (fig. S3, C and F), this decrease was not great enough to explain the observed bias toward β-arrestin2 vs Gαi recruitment. Indeed, the decreased cell surface expression may reflect increased internalization resulting from its constitutive association with β-arrestin2. Another DRY box mutant, CXCR4(D133N), showed an approximately 50% reduction in β-arrestin2 recruitment efficacy together with a statistically significant improvement in potency (Fig. 8C, Table S3); however, similar to CXCR4(R134A), it displayed essentially no Gαi recruitment (Fig. 8A) and yielded a bias factor of −2.13 ± 0.41 (Fig. 8D, Table S3). This mutant showed particularly reduced cell surface expression in the β-arrestin2 recruitment assay (fig. S3C), so its efficacy may be artificially impaired, although in this case, the true bias for this mutant toward β-arrestin2 would be even greater than is apparent from the data.
Fig. 8. Mutations of CRS0.5 and ECL2 appear to cause biased signaling.
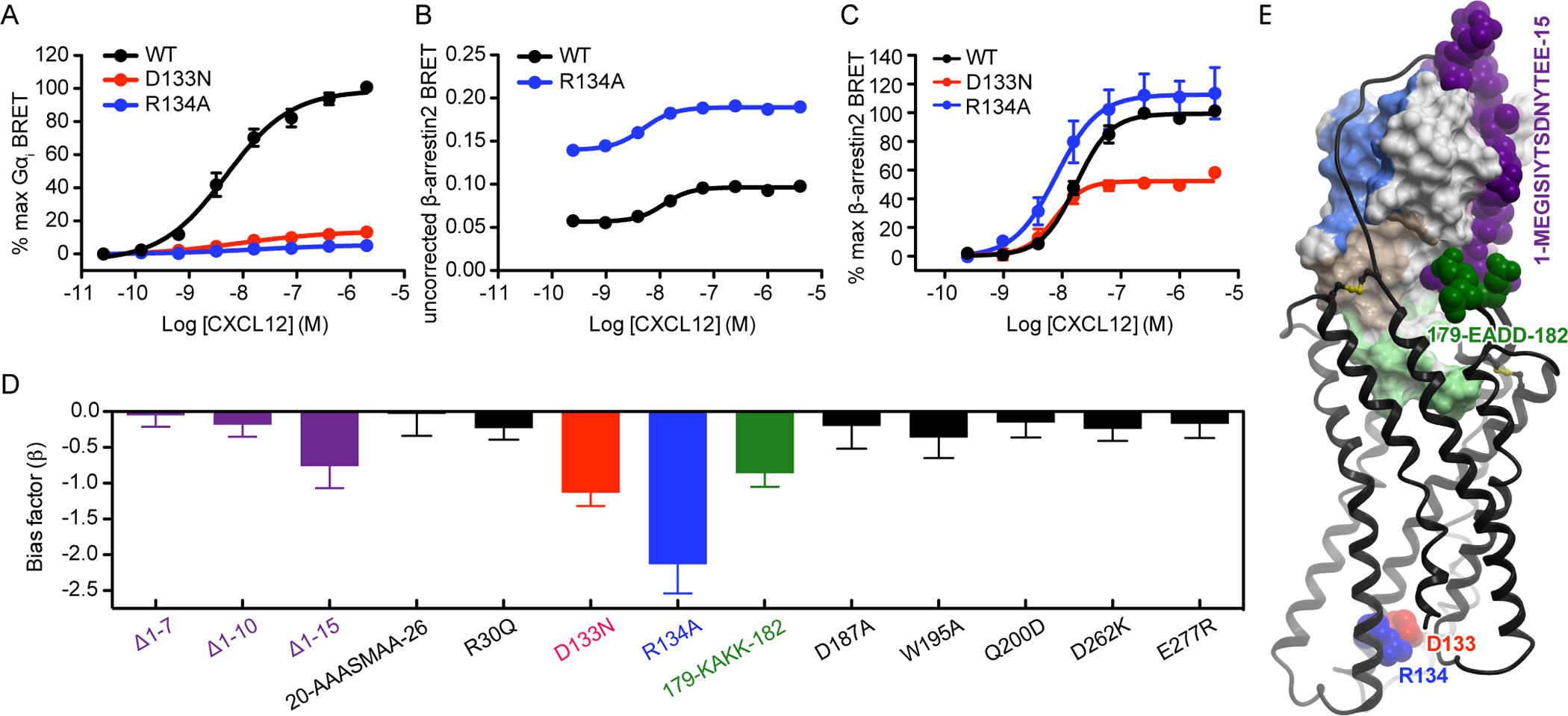
(A) Mini-Gαi association BRET ratio data for CXCR4(D133N) and CXCR4(R134A) after stimulation with the indicated concentrations of CXCL12 for 1 min. (B) Uncorrected β-arrestin2 association BRET ratio data (without background signal subtraction or normalization) for CXCR4(R134A) after stimulation with the indicated concentrations of CXCL12 for 20 min. Data are from a single experiment, performed in duplicate, and are representative of three independent experiments. (C) β-arrestin2 association BRET ratio data for CXCR4(D133N) and CXCR4(R134A) after stimulation with the indicated concentrations of CXCL12. In (A) and (C), data are means ± SEM of at least three independent experiments, each performed in duplicate. Data were normalized to the Emax of WT CXCR4 tested in the same experiments. (D) Equiactive bias factor (β) for all CXCR4 mutants that were tested in both the β-arrestin2 and mini-Gαi association BRET experiments. Error bars indicate the combined errors of the EC50 and Emax values used to calculate β, and colored bars indicate that the absolute value of β > 2 times the combined error. A negative bias factor indicates bias towards β-arrestin2. (E) When mapped onto the model of the CXCR4-CXCL12 complex, bias-associated residues at the chemokine interface cluster on the same side. The intracellular residues Asp133(3.49) and Arg134(3.50) are not in direct contact with the chemokine but are shown for reference.
Next, we calculated bias factors for the chemokine interface mutants in this study and found that most of them were balanced in their effects on Gαi and β-arrestin2 recruitment (Fig. 8D, table S3). However, two variants should be noted: the triple mutant of the ECL2 hairpin, CXCR4(E179K/D181K/D182K), and the 15-residue N-terminal truncation, CXCR4(Δ1–15). As reflected by bias factors of −0.86 ± 0.19 and −0.76 ± 0.31, respectively, these mutants displayed greater reductions in potency in Gαi recruitment experiments than in β-arrestin2 recruitment experiments. The bias was lower than for the control “maximally biased” DRY motif mutants; however, given the location of the mutations at the extracellular, chemokine-binding interface, it is surprising that the bias was present at all. Note also that both of these bias-associated perturbations affect residues interacting with the same general region of the chemokine globular core (Fig. 8E). This finding recalls a previous report of signaling bias by dimeric CXCL12 (42, 64), which effectively prevents receptor interactions with the same residues in the CXCL12 β1 strand (fig. S6) (39). The mechanistic basis for the biased signaling of these mutants remains unclear; however, the results suggest that bias may be introduced by mutations that preferentially disfavor a particular orientation of the chemokine with respect to the receptor. The finding of bias in the CXCR4(Δ1–15) N-terminal truncation mutant again challenges the notion that the N-terminus is important exclusively for binding affinity; our data suggest that it not only affects signaling efficacy but may also do so in a pathway-selective manner.
Quantitative dissection of mutation effects benefits from the use of amplification-free assays
A key distinction between this study and previous mutagenesis studies is the use of direct, BRET-based interaction methods between CXCR4 and its effectors. As previously established (44–46, 65–67), for assays in which measurements ultimately depend on the production of downstream second messengers, the magnitude of the measured signal is amplified as a result. The second messenger amplification concept is best understood for IP3 and Ca2+ (Fig. S1C), both of which are commonly measured in chemokine receptor mutagenesis studies. Moreover, such measurements may reach their experimental maximum, either by saturation of the observation method (such as a fluorescent dye used to measure intracellular Ca2+) or by transient exhaustion of the intracellular second messenger stores or precursors themselves, long before full receptor occupancy, activation, or both are reached. In such cases, differences in the ability of WT vs mutant receptors to activate the associated signaling pathways may be obscured.
To investigate the utility of an amplification-based assay for quantitative dissection of mutation effects on CXCR4 signaling, we selected several CXCR4 mutations and truncations that showed pronounced defects in mini-Gαi BRET experiments, and tested them in Ca2+ mobilization experiments by generating a CXCL12 concentration response curve for each (fig. S7A, C–H, Table S4). The cell surface expression of each mutant was manually adjusted to closely match that of WT CXCR4 (fig. S7B). As expected, the Ca2+ mobilization experiments consistently showed less pronounced impairment of CXCR4 signaling by mutations (fig. S7, C to J, Tables S3 and S4), indicating that these experiments are indeed subject to amplification, and that such amplification masks all but the most pronounced mutant signaling defects. Moreover, if taken at face value, the Ca2+ mobilization data would suggest a large degree of bias in many of the mutants, which, according to our BRET-based recruitment assays, is not the case. This result demonstrates that the quantitative nature of the findings in our study was aided by the use of amplification-free, stoichiometric molecular association assays.
Discussion
With CXCR4 being a prototypical CXC family receptor, CXCL12 being its only known endogenous agonist, and both of them playing pivotal roles in immune system homeostasis and in numerous cancers, CXCR4-CXCL12 is one of the most studied receptor-chemokine complexes biochemically. Extensive mutagenesis efforts directed at both the receptor and the chemokine have generated insight into the roles of a large number of residues; nevertheless, numerous uncertainties remain. First, there are many inconsistent reports on the relative contributions of individual residue interactions to the affinity or signaling capacity of the complex. This is likely because previous mutagenesis efforts largely relied on amplification-based second messenger assays conducted in a single-point rather than concentration-response format, an approach that often fails to detect all but the most severe mutant defects. In combination with mutation-induced variations in receptor expression and trafficking, this has precluded a systematic and quantitative dissection of the receptor-chemokine interface residues (table S2). Second, even when data are consistent, the molecular basis for the effect of the mutations is unclear, because a structure of the complex has not been determined. Thus, the structural role and quantifiable functional effect of the various interaction epitopes, including the sulfotyrosinated extracellular N terminus of the receptor, have remained cryptic.
Here, we addressed these methodological concerns by using BRET-based methods for detecting the stoichiometric association of CXCR4 with Gαi and β-arrestin2, which are not subject to amplification. Moreover, we designed the assays in a way that enabled quantitative interpretation of the results despite mutation-induced variations in receptor expression. Our studies were guided and interpreted with a computationally constructed, high-resolution 3D model of the CXCR4-CXCL12 complex. Built by a hybrid approach combining homology modeling, ab initio structural optimization, and experimentally derived disulfide crosslinking restraints between chemokine and receptor (31), the model features the engagement of the full N-terminus of the receptor with CXCL12, and elucidates other key intermolecular interaction epitopes. Together with the experimental results of this study, it fills major gaps in the structural understanding of CXCR4-CXCL12 signaling.
Key charge swap rescue-of-function mutagenesis experiments established the accuracy of the overall model architecture. In these experiments, loss of function caused by mutation of a charged residue on one of the interacting proteins was rescued by a complementary mutation on the second protein, indicating a pairwise interaction. In this respect, charge swap rescue-of-function approaches provide information that cannot be obtained from traditional single-sided loss-of-function mutagenesis, which only reveal whether specific residues are important for the function of the complex. Using this method, we confirmed the predicted salt bridge between CXCR4 Asp262(6.58) and CXCL12 Arg8, as well as that between CXCR4 Glu277(7.28) and CXCL12 Arg12. Not all of the interacting residue pairs in the CXCR4-CXCL12 complex are amenable to this strategy. For example, residues in CRS2 render CXCR4 completely inactive when mutated, and most mutations of single residues in CRS1 do not result in signaling deficits that are substantial enough to rescue (31). Therefore, the data on the two interacting pairs Asp262(6.58)-Arg8 and Glu277(7.28)-Arg12 provide the strongest possible charge swap support for the model, particularly for the CRS1.5 and CRS2 regions. Concurrently, disulfide crosslinking and charge swap results obtained in a separate study established the geometry of CRS1 and CRS0.5 (31).
The importance of the two intermolecular salt bridges validated by the charge swaps extends beyond simply supporting the predicted geometry of the CXCR4-CXCL12 complex. Indeed, sequence alignments (14, 68) demonstrate conservation of the residues forming the first of the two salt bridges [CXCR4 Asp262(6.58) with CXCL12 Arg8] in the CXC receptors and chemokines, respectively, but not at all in other subfamilies (CC, CX3C, and XC). This suggests that the identified salt bridge may be a universal CXC recognition anchor and a determinant of inter-subfamily selectivity. However, note that the virally encoded chemokine vMIP-II, a rare example of a CC chemokine that binds to a CXC receptor, has an arginine in the proximal N terminus, which, as demonstrated by the crystal structure (14), is engaged with CXCR4 Asp262(6.58) in a salt bridge, largely mimicking the one predicted and validated here. Therefore, the presence of an arginine in the proximal N-terminus and the resulting ability to form a salt bridge with the acidic residue in position 6.58 of the receptor may confer a cross-subfamily activity to CC chemokines. By contrast with Asp262(6.58) and Arg8, residues forming the second salt bridge are not conserved. CXCL12 Arg12 is unique among the CXC chemokines, whereas a glutamate at position 7.28 is only found in CXCR3 and ACKR3 (the latter of which also binds to CXCL12). Therefore, this second salt bridge likely contributes to the intra-CXC-subfamily selectivity for the CXCR4-CXCL12 complex.
Our experiments also established an important role of CXCR4 Arg30(NT), a key predicted feature of CRS1.5. A basic residue in the corresponding position is found in almost every CC (but not CXC or other subfamily) receptor, where it has been proposed to be important for coordination of the uniquely shaped CC-motif backbone of the respective CC chemokines (68). It is thus possible that a basic amino acid residue in this position confers CXCR4 with a CC-like recognition determinant and facilitates its interaction with the CC chemokine, vMIP-II.
Mutations in CXCR4 CRS2 often have large effects, and in some cases completely ablate CXCL12-mediated signaling. This is true not only for G protein signaling, consistent with previous studies (9, 30, 32, 48–52), but also, as demonstrated here, for β-arrestin recruitment. Our findings are consistent with the receptor-binding pocket residues and the N terminus of CXCL12 being the key drivers of signaling (2, 6). By contrast, single-point mutations in other epitopes, and particularly the receptor N terminus, generally have less substantial effects. Accordingly, our discovery of impairments in the efficacy of recruitment of β-arrestin2, Gαi, or both to CXCR4 variants with N-terminal deletions and the secondary structure-perturbing single and double mutations in CRS0.5, was unexpected. This finding challenges the long-standing paradigm in which the receptor N terminus serves only as a docking domain for the chemokine (2, 6). However, even with truncations, the signaling defects were not as substantial as those caused by single-point mutations within CRS2. Nevertheless, the fact that they were observed at all suggests a previously uncharacterized and potentially important role for the N terminus beyond the two-site hypothesis; specifically, that signaling amplitude depends on the extent to which the receptor N terminus binds to the chemokine. Note that the efficacy variations resulting from truncations of the N terminus were only detectable in an amplification-free, molecular association-based assay; this emphasizes the importance of using adequate tools and readouts when characterizing mutants.
There are two possible mechanisms for the observed signaling effects from truncating the receptor N terminus. On the one hand, the N terminus may be directly involved in conformational changes underlying receptor activation. Consistent with this hypothesis, there is a direct covalent (disulfide) bond between the receptor N terminus and ECL3 that effectively links the N terminus to the two activation-related helices, TM6 and TM7. This N terminus-to-ECL3 disulfide is highly conserved across chemokine receptors, suggesting a common underlying mechanism. In addition, on the opposite side of the binding pocket, the packing of CXCL12 against ECL2 and the ability of the receptor to close down around the chemokine, akin to other GPCRs and their ligands (69–71), may both depend on the receptor N terminus locking down around the globular core of the chemokine. This agrees with our observation of the detrimental effects of charge reversal of the CXCR4 ECL2 tip, which produced results similar to those caused by truncating the N terminus. Finally, the intramolecular association between the N terminus and ECL2 of the receptor, stabilized by the bound chemokine, may play a role in establishing the correct signaling geometry.
As an alternative to the conformational mechanism, the receptor N terminus may affect signaling efficacy indirectly by prolonging the residence time of CXCL12 on the receptor. This hypothesis is inspired by our previous study of ACKR3 (which also binds CXCL12), where impairment in β-arrestin2 recruitment efficacy caused by N-terminal truncations in the receptor correlated with an increase in chemokine dissociation rate (72). A mechanistic link between ligand dissociation rates and signaling efficacy has also been established for other GPCRs (44, 73, 74). Specifically, efficacy differences between agonists have been explained by receptor occupancy relative to the kinetics of the signaling process under study. In the case of ACKR3, one can argue that more rapid chemokine dissociation (that is, shorter receptor residence time), prevents the receptor from being phosphorylated by GPCR kinases and coupling to β-arrestin2. Although we have not yet established similar kinetic off-rate assays for CXCR4, it is possible that the observed signaling differences between WT CXCR4 and the truncated receptor are at least partially due to changes in chemokine dissociation rate. Such a role for the N-terminus in slowing the off-rate of the chemokine would also provide a partial explanation for the increased affinity of CXCL12 for CXCR4 in the presence of G protein (75), similar to the role of a G protein-mediated “closed conformation” of β2AR in preventing egress of ligands (69). The geometry of the interaction, wherein the N terminus has an extended structure that wraps around the chemokine, would facilitate a scenario in which alterations, such as tyrosine sulfation, together with allosteric effects from G protein coupling could readily modulate the chemokine interaction with the receptor N terminus, thereby influencing signaling responses.
The involvement of the distal N terminus in signaling may also explain why a disulfide-locked dimer of CXCL12 has reduced signaling efficacy in β-arrestin2 recruitment (42, 64). As described earlier, the distal N terminus of the receptor forms an anti-parallel β-sheet with the β1-strand of the chemokine in a manner mimicking the chemokine dimer interface; thus, binding of the chemokine dimer would displace the receptor N-terminus (fig. S6), likely producing similar conformational or ligand off-rate differences (relative to the complex with monomeric CXCL12) as we suspect for the N-terminal truncations tested herein.
In summary, our study presents insights into the functional anatomy of the CXCR4-CXCL12 complex and the role of various epitopes in regulating the structure, ligand specificity, and signaling responses of the receptor. Some of these findings, such as the conserved CXC-specific salt bridge and the importance of the N terminus, are likely to be broadly applicable in the chemokine receptor family, and provide structural explanations for the previously observed effects of mutations and N-terminal truncations in other receptors (76).
Materials and Methods
DNA constructs and cloning
The cDNA encoding human CXCR4 fused to Renilla luciferase 3 (rluc3, otherwise known as rlucII) and that of GFP fused to human β-arrestin2 (GFP-β-arrestin2), both contained within a pcDNA vector, were kindly donated by Nicolaus Heveker, Université de Montréal, Montreal, Quebec, Canada. An N-terminal HA tag was added to the CXCR4-rluc3 vector, followed by the production of our mutant library. All mutations and truncations (as well as the N-terminal HA tag) were introduced into the CXCR4-coding region of the CXCR4-rluc3 vector using the QuikChange site-directed mutagenesis kit (Stratagene). The plasmid encoding renilla GFP (rGFP) fused to rGFP-mini-Gαsi for the Gαi association BRET assay was a kind gift from Nevin Lambert, Augusta University, Augusta, Georgia, USA. To ensure that the effects of the N-terminal truncations were not confounded by the presence of the N-terminally fused HA tag, mini-Gαi BRET experiments with CXCR4 N-terminal truncations were performed with HA-free (tagless) receptor constructs. In contrast, for β-arrestin2 recruitment experiments with CXCR4 N-terminal truncation mutants, the N-terminal HA tag was retained. Although the signaling results in both sets of experiments assays were similar, we noted a greater negative effect of the truncations on cell surface receptor abundance when the HA tag was absent (fig. S3, E and F).
Mammalian cell culture
HEK293T cells were obtained directly from the American Type Culture Collection (ATCC) and certified mycoplasma-free by ATCC. HEK293T cells were cultured in sterile tissue culture treated T75 flasks in Dulbecco’s Modified Eagle Media (DMEM) with 10% fetal bovine serum (FBS) and were passaged 1:10 every 2 to 3 days. CHO-K1 cells stably expressing the promiscuously coupling Gα15 were described previously (32) and originated from an ATCC-derived, mycoplasma-free certified CHO-K1 cell line. These cells were cultured in sterile tissue culture treated T75 flasks in 1:1 DMEM/F12 nutrient mixture supplemented with 10% FBS and 700 μg/ml active G418 mammalian antibiotic and were passaged 1:15 every 2 to 3 days. Both cell lines were exclusively used for reporter assays (rather than studies of underlying biology). Experiments were restricted to cells at early passages.
BRET-based β-arrestin2 and mini-Gαi association assays
Recruitment of β-arrestin2 and mini-Gαi to CXCR4-rluc3 was measured with the bioluminescence resonance energy transfer 2 (BRET2) assay (77). Four days before each assay, HEK293T cells were passaged and plated at 4.25 × 105 cells per well in 6-well tissue culture plates in DMEM with 10% FBS. For the β-arrestin2 association experiments, the cells were transfected two days later with 0.1 μg DNA/well HA-CXCR4-rluc3 (WT and mutants, with 0.075 ug DNA used for the highly expressing Δ1–10 truncation) and 2.4 μg DNA/well of GFP-β-arrestin2. Transfections were performed with TransIT-LT1 transfection reagent (MirusBio) according to the manufacturer’s recommended protocol. For the Gαi association experiments, the procedure was basically identical except that rGFP-mini-Gαsi was used in place of GFP-β-arrestin2. On the day of the assay, the cells were washed with PBS while still adherent, then resuspended through manual pipetting in BRET buffer consisting of PBS with 0.1% D-glucose, and diluted to a final concentration of 1.5 × 106 cells/ml. The cell suspension was then dispensed at 90 μl/well into a white, clear bottom, tissue culture treated 96-well plate (Corning). The plate was placed into a conditioned incubator (37°C, 5% CO2) for 30 min before GFP-β-arrestin2 or rGFP-mini-Gαsi fluorescence intensity was measured with a SpectraMax M5 fluorescence plate reader (Molecular Devices). The β-arrestin2 recruitment experiment was then performed in four steps. First, 10 μl of WT or mutant CXCL12 in BRET buffer at 10 times the final concentration was added to each well. Second, the plate was incubated for 10 min at 37°C. Third, coelenterazine-400a (also known as DeepBlueC) was added to each well to obtain a final concentration of 5 μM. Fourth, repeated luminescence reads at both 410 and 515 nm were initiated immediately and continued for 20 min at 37°C using the VictorX Light luminescence plate reader (PerkinElmer Life Sciences). In the case of the Gαi association experiments, three steps were performed. First, coelenterazine-400a was added to the plate at the final concentration of 5 μM. Second, CXCL12 was added immediately after. Third, repeated luminescence reads were initiated immediately and continued for 10 min. The difference procedure between the β-arrestin2 and Gαi association experiments reflects the different kinetics of recruitment of these two effectors. For concentration-response curves, BRET ratios (515 nm luminescence/410 nm luminescence) measured 20 min after CXCL12 addition for the β-arrestin2 experiments and 1 min for the Gαi experiments were then calculated using MS Excel, and initial four-parameter agonist concentration response curve fitting was performed with GraphPad Prism version 5.0b for Mac OS X (GraphPad Software) to enable normalization to the WT Emax within each experiment. Plasmid DNA amounts used in the BRET experiment transfections (0.1 and 2.4 μg for HA-CXCR4-rluc3 and GFP-β-arrestin-2/rGFP-mini-Gαsi, respectively), were selected to meet three criteria. First, GFP-effector (BRET acceptor) had to be in sufficient excess relative to that of CXCR4-rluc3 (BRET donor) to ensure donor saturation. Donor saturation ensures that “BRETmax” [the upper limit of the hyperbolic donor:acceptor titration curve (77)] is achieved and prevents misinterpretation of expression-based BRET deviations as apparent efficacy changes. The second criterion was that CXCR4-rluc3 had to be expressed at a sufficient amount to yield an analyzable luminescence signal for both wavelengths measured in the BRET assay. The third criterion stemmed from the limitations on the maximum recommended amount of the DNA for the scale used (2.5 μg/well). The orientation of our BRET assays, with the receptor linked to the energy-donating luminescent enzyme and saturated by a signaling effector fused to the accepting fluorescent protein, provides the advantage of rendering the experiments insensitive to moderate variations in receptor abundance. This is due to the ratiometric BRET signal representing the proportion of donor in close proximity to acceptor rather than the absolute quantity of engaged complexes. As long as there is ample GFP-effector available (enabled by saturation), the proportional BRET signal should represent the per molecule average receptor-effector engagement or complex rearrangement within a sample. To confirm this, we titrated WT CXCR4-rluc3 in both the β-arrestin2 and mini-Gαi BRET experiments, and indeed found no changes in signaling parameters using the Extra sum-of-squares F test (fig. S2). The range of total WT CXCR4-rluc3 expression that produced functionally equivalent activation curves encompassed the range of expression of all of the mutants tested herein, except for CXCR4(Δ1–10) (fig. S3, A and D), which was more abundant than the WT receptor. Note that the surface:total expression ratio was altered to varying degrees for a number of mutants (fig. S3, C and F), and, in extreme cases, may have affected the BRET measurements, because a smaller proportion of the donor-fused receptor available to CXCL12 at baseline would be expected to produce a lower proportional saturation of donor by acceptor upon stimulation (57). We therefore note every such case in the Results (for β-arrestin2 BRET: Δ1–19, D133N, and R134A; for mini-Gαi: Δ1–15, Δ1–25, D133N, and R134A), and interpret these data with caution to the degree warranted by the deficit in cell surface expression.
Ca2+ mobilization G protein signaling assay
Ca2+ mobilization experiments were performed with the FLIPR calcium 4 assay kit (Molecular Devices). As detailed previously (32), for these experiments, we used a modified CHO-K1 cell line that is devoid of endogenous CXCR4 and stably expresses the promiscuously coupling Gα15. Three days before each assay, the cells were passaged and plated at 2×105 cells per dish in 10-cm diameter tissue culture dishes in 1:1 DMEM/F12 nutrient mixture supplemented with 10% FBS and further supplemented with 0.25% DMSO to aid in transfection efficiency (78). The next day, the medium was removed and replaced with a 1:1 DMEM/F12 nutrient mixture supplemented with 10% FBS immediately before transfection of the cells with Trans-IT CHO transfection kit (Mirus Bio), which was used according to the manufacturer’s recommended protocol except that the ratio of reagent (in μl) to DNA (in μg) was adjusted to 4:1. For the current study, the cells in each dish were transfected with 24 μg of either WT or mutant CXCR4-rluc3 DNA. We used the same WT and mutant CXCR4-rluc3 constructs as in the BRET experiments, because we found that CXCR4-rluc3 retained the ability to activate Ca2+ mobilization in the CHO-K1-Gα15 cell line and seemed to improve the data quality (fig. S7A). In the case of Ca2+ mobilization experiments, receptor expression at the cell surface did affect signaling measurements substantially, so the amount of DNA encoding WT CXCR4 used in the transfections was adjusted to match those of mutants as closely as possible using flow cytometry-based monitoring of anti-HA-PE or anti-HA-APC binding to the N-terminal HA tag on the receptor (fig. S7B). On the day after transfection, the cells were washed with PBS before being resuspended in PBS, 5 mM EDTA, and then centrifuged and resuspended in 1:1 DMEM/F12 nutrient mixture supplemented with 10% FBS before being re-plated at 90,000 cells/well in black, clear bottom, poly-D-lysine coated 96-well plates (Corning). On day four, the medium was carefully removed from the adherent cells and 200 μl of a 1:1 mixture of HBSS, 20 mM HEPES, 0.1% BSA (Ca2+ flux buffer) and FLIPR4 dye were added to each well. After a 75-min incubation at 37°C, the assay was performed in a FlexStation-3 multi-mode plate reader (Molecular Devices) using the automated injection function to add 22 μl of CXCL12 at 10x the final indicated concentrations (in Ca2+ flux buffer) while reading fluorescence (excitation at 485 nm, emission at 525 nm) repeatedly (with 1.52-s intervals) over the course of 150 s. Reduced (baseline-corrected) fluorescence values were calculated by subtracting the baseline fluorescence from all measured values. Reduced peak fluorescence four-parameter CXCL12 concentration response curve fitting was performed with GraphPad PRISM.
Flow cytometry-based surface expression testing
The cell surface expression of WT and mutant versions of CXCR4-rluc3 was monitored by flow cytometry as described previously (32). Briefly, cells were resuspended in PBS, 5 mM EDTA, centrifuged, and resuspended in PBS, 0.5% PBS (FACS buffer) to a final concentration of 1 × 105 to 1 × 106 cells/ml. For anti-HA staining, fluorophore-conjugated anti-HA antibody (either anti-HA-APC, catalogue number 130-098-404, or anti-HA-PE, catalogue number 130-092-257, both from Miltenyi Biotec) was added to obtain an 11X dilution, and cells were stained on ice in the dark for 10 min, according to the manufacturer’s recommended procedure. For anti-CXCR4 staining, fluorophore-conjugated anti-CXCR4 antibody, either 12G5 anti-CXCR4-APC (catalogue number 560936, BD Biosciences, La Jolla, California, USA) for N-terminal truncations and CRS1 mutations or 1D9 anti-CXCR4-PE (catalogue number 551510, BD Biosciences for all other mutations), was added to obtain a 50X dilution, and cells were stained on ice in the dark for 45 min, as recommended by the manufacturer. Cells were then washed three times with FACS buffer and fixed with a final concentration of 0.8% PFA. Flow cytometric analysis of the fixed antibody-stained cells was performed with a GUAVA benchtop flow cytometer (EMD Millipore). Flow cytometry data was analyzed with FlowJo version 10 (FlowJo LLC), and geometric mean fluorescence intensity (GMFI) values were normalized to that of WT CXCR4 after subtraction of the low GMFI obtained for cells transfected with pcDNA and stained with the same antibody.
Statistical comparison of WT and mutant signaling parameters
To compare WT and mutant signaling assay results, as well as the results of different combinations of CXCL12 and CXCR4 mutants, the results on each day were normalized to 100% WT efficacy, and mean values from independent experiments (each performed in duplicate) for each CXCL12 concentration were plotted together. Statistical comparisons between WT and mutant CXCR4 concentration response parameters were performed on the same combined dataset in GraphPad Prism version 5.0b for Mac OS X (GraphPad Software) using the Extra sum-of-squares F test with a P value cutoff of 0.05, the results of which are reported in Tables S3 (BRET-based assays) and S4 (Ca2+ mobilization). We applied a secondary P value cutoff of 0.1 (without referring to the changes as being statistically significant) to note the trend of several mutants for which signaling was clearly altered but did not reach the P < 0.05 threshold. It should be noted that the Extra sum-of-squares F test was in some cases inadequate for identifying differences in efficacy despite clearly decreased efficacy (<35% WT efficacy remaining). This is due to near, but not complete, elimination of receptor activation upon mutation, which enables the data to be erroneously fit to a wide range of Emax values. These cases include CXCR4(Δ1–15), CXCR4(Δ1–25), CXCR4(D262K), CXCR4(D133N), and CXCR4(R134A) in the mini-Gαi association experiments, and CXCR4(Δ1–25), CXCR4(D262A), CXCR4(D262R), and CXCR4(D187A) in the β-arrestin2 experiments.
Bias calculations
Bias was calculated according to the equiactive comparison method for bias calculation (46), which requires only two pairs of EC50 and Emax parameters from assays of two different GPCR-initiated signaling pathways. Although this equation is typically used to assess bias between different agonists, it was perfectly suited to our case of seemingly differential mutational effects on β-arrestin2 and Gαi association. The adapted equation is as follows:
where 1 and 2 correspond to pathways 1 and 2, designated arbitrarily. Because G protein signaling is usually considered to be the primary function of GPCRs, we designated Gαi association as pathway 1 and β-arrestin2 association as pathway 2, so that negative results indicate a bias towards β-arrestin2 association. The signaling parameters were applied to the adapted equiactive bias equation, and the error of the parameters was combined in MS Excel.
Supplementary Material
Fig. S1. Schematics of the assays used to characterize WT and mutant CXCR4 in this study.
Fig. S2. A wide range of CXCR4 expression yields identical β-arrestin2 and Gαi signaling parameters.
Fig. S3. Total and cell surface expression of receptors with mutations of residues important for signaling as suggested by BRET experiments.
Fig. S4. CXCR4 residues Glu268(ECL3) and Glu275(7.26) are not critical for CXCL12-mediated CXCR4 activation.
Fig. S5. CRS1 is important for the potency but not the efficacy of CXCL12-mediated G protein engagement by CXCR4.
Fig. S6. Predicted consequence of the binding of dimeric CXCL12 to CXCR4.
Fig. S7. Signal amplification in Ca2+ mobilization experiments obscures mutation-induced defects.
Table S1. Key predicted charge interactions in CRS1, CRS1.5, and CRS2 of the CXCR4-CXCL12 complex.
Table S2. Previously published effects of mutations on the CXCR4 residues tested in this study.
Table S3. Signaling parameters obtained from the β-arrestin2 and mini-Gαi association BRET experiments.
Table S4. Signaling parameters obtained from the Ca2+ mobilization experiments.
Acknowledgments:
We gratefully acknowledge N. Heveker (Université de Montréal), N. Lambert (Augusta University), M. Bouvier (Université de Montréal), and M. Gustavsson (UC San Diego, currently University of Copenhagen) for the BRET reagents used in our studies. We thank M. Pu, of the UCSD Moores Cancer Center Biostatistics Shared Resource for critical review of and assistance with the statistical analyses in the paper. We thank J. J. Ziarek (Indiana University) and B. F. Volkman (Medical College of Wisconsin) for providing the coordinates of their previously published CXCR4-CXCL12 model.
Funding: This work was supported by NIH R01 grants AI118985 and R01 GM117424 to I.K. and T.M.H. B.S.S was supported by Cellular and Molecular Pharmacology Training Grant T32 GM007752 and Molecular Biophysics Training Grant T32 GM008326. T.N. was supported by NHMRC C. J. Martin Early Career Fellowship 1145746.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The coordinates of the CXCR4-CXCL12 complex used in this study are available as supplementary material from Ngo et al. (31). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Monteclaro FS, Charo IF, The Amino-+terminal Extracellular Domain of the MCP-1 Receptor, but Not the RANTES/MIP-1a Receptor, Confers Chemokine Selectivity: Evidence for a Two-Step Mechanism fir MCP-1 Receptor Activation. J Biol Chem 271, 19084–19092 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I, Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. Embo J 16, 6996–7007 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarnagin K, Grunberger D, Mulkins M, Wong B, Hemmerich S, Paavola C, Bloom A, Bhakta S, Diehl F, Freedman R, McCarley D, Polsky I, Ping-Tsou A, Kosaka A, Handel TM, Identification of Surface Residues of the Monocyte Chemotactic Protein 1 That Affect Signaling through the Receptor CCR2b Biochemistry 38, 16167–16177 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Wells TN, Power CA, Lusti-Narasimhan M, Hoogewerf AJ, Cooke RM, Chung CW, Peitsch MC, Proudfoot AE, Selectivity and antagonism of chemokine receptors. Journal of Leukocyte Biology 59, 53–60 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Blanpain C, Doranz BJ, Bondue A, Govaerts C, De Leener A, Vassart G, Doms RW, Proudfoot A, Parmentier M, The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J Biol Chem 278, 5179–5187 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Scholten DJ, Canals M, Maussang D, Roumen L, Smit MJ, Wijtmans M, de Graaf C, Vischer HF, Leurs R, Pharmacological modulation of chemokine receptor function. Br J Pharmacol 165, 1617–1643 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong JH, Clark-Lewis I, Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal residues. The Journal of Experimental Medicine 181, 631–640 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, Sykes BD, Structure-activity relationships of chemokines. Journal of Leukocyte Biology 57, 703–711 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Brelot A, Heveker N, Montes M, Alizon M, Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem 275, 23736–23744 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Hanes MS, Salanga CL, Chowdry AB, Comerford I, McColl SR, Kufareva I, Handel TM, Dual Targeting of the Chemokine Receptors CXCR4 and ACKR3 with Novel Engineered Chemokines. Journal of Biological Chemistry 290, 22385–22397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaertner H, Cerini F, Escola J-M, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I. n., Nedellec R, Salkowitz J, Gorochov G, Mosier D, Hartley O, Highly potent, fully recombinant anti-HIV chemokines: Reengineering a low-cost microbicide. Proceedings of the National Academy of Sciences of the United States of America 105, 17706–17711 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorenzen E, Ceraudo E, Berchiche YA, Rico CA, Fürstenberg A, Sakmar TP, Huber T, G protein subtype–specific signaling bias in a series of CCR5 chemokine analogs. Science Signaling 11, (2018). [DOI] [PubMed] [Google Scholar]
- 13.Chevigne A, Fievez V, Schmit J-C, Deroo S, Engineering and screening the N-terminus of chemokines for drug discovery. Biochemical Pharmacology 82, 1438–1456 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, Abagyan R, Stevens RC, Handel TM, Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, Garcia KC, Structural basis for chemokine recognition and activation of a viral G protein–coupled receptor. Science 347, 1113–1117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miles TF, Spiess K, Jude KM, Tsutsumi N, Burg JS, Ingram JR, Waghray D, Hjorto GM, Larsen O, Ploegh HL, Rosenkilde MM, Garcia KC, Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. eLife 7, e35850 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Y, Han GW, Abagyan R, Wu B, Stevens RC, Cherezov V, Kufareva I, Handel TM, Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46, 1005–1017.e1005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Broder CC, Kennedy PE, Berger EA, HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA, The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B, The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382, 833–835 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Zlotnik A, Burkhardt AM, Homey B, Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol 11, 597–606 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Balkwill F, Cancer and the chemokine network. Nat Rev Cancer 4, 540–550 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Balkwill F, The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol 14, 171–179 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Huang X, Shen J, Cui M, Shen L, Luo X, Ling K, Pei G, Jiang H, Chen K, Molecular Dynamics Simulations on SDF-1a Binding with CXCR4 Receptor. Biophysical Journal 84, 171–184 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mona CE, Besserer-Offroy É, Cabana J, Lefrançois M, Boulais PE, Lefebvre M-R, Leduc R, Lavigne P, Heveker N, Marsault E, Escher E, Structure Activity Relationship and Signaling of New Chimeric CXCR4 Agonists. Journal of Medicinal Chemistry, (2016). [DOI] [PubMed] [Google Scholar]
- 26.Cutolo P, Basdevant N, Bernadat G, Bachelerie F, Ha-Duong T, Interaction of chemokine receptor CXCR4 in monomeric and dimeric state with its endogenous ligand CXCL12: Coarse-grained simulations identify differences. J. Biomol. Struct. Dyn, 1–56 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Xu L, Li Y, Sun H, Li D, Hou T, Structural basis of the interactions between CXCR4 and CXCL12/SDF-1 revealed by theoretical approaches. Molecular BioSystems 9, 2107–2117 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Ziarek JJ, Kleist AB, London N, Raveh B, Montpas N, Bonneterre J, St-Onge G, DiCosmo-Ponticello CJ, Koplinski CA, Roy I, Stephens B, Thelen S, Veldkamp CT, Coffman FD, Cohen MC, Dwinell MB, Thelen M, Peterson FC, Heveker N, Volkman BF, Structural basis for chemokine recognition by a G protein–coupled receptor and implications for receptor activation. Science Signaling 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamamis P, Floudas CA, Elucidating a Key Component of Cancer Metastasis: CXCL12 (SDF-1α) Binding to CXCR4. Journal of Chemical Information and Modeling 54, 1174–1188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, Berdougo E, Rucker JB, Handel TM, Doranz BJ, Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proceedings of the National Academy of Sciences of the United States of America 113, 9928–9933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngo T, Stephens BS, Gustavsson M, Holden LG, Abagyan R, Handel TM, Kufareva I, Crosslinking-guided geometry of a complete CXC receptor-chemokine complex and the basis of chemokine subfamily selectivity. PLOS Biology 18, e3000656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kufareva I, Stephens BS, Holden LG, Qin L, Zhao C, Kawamura T, Abagyan R, Handel TM, Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: Molecular modeling and experimental validation. Proc Natl Acad Sci USA 111, E5363–E5372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kufareva I, Gustavsson M, Zheng Y, Stephens BS, Handel TM, What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annual Review of Biophysics 46, 175–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubman OY, Fremont DH, Parallel Evolution of Chemokine Binding by Structurally Related Herpesvirus Decoy Receptors. Structure 24, 57–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prado GN, Suetomi K, Shumate D, Maxwell C, Ravindran A, Rajarathnam K, Navarro J, Chemokine Signaling Specificity: Essential Role for the N-Terminal Domain of Chemokine Receptors. Biochemistry 46, 8961–8968 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H, The Role of Post-translational Modifications of the CXCR4 Amino Terminus in Stromal-derived Factor 1α Association and HIV-1 Entry. J Biol Chem 277, 29484–29489 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Seibert C, Veldkamp CT, Peterson FC, Chait BT, Volkman BF, Sakmar TP, Sequential Tyrosine Sulfation of CXCR4 by Tyrosylprotein Sulfotransferases†. Biochemistry 47, 11251–11262 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziarek JJ, Getschman AE, Butler SJ, Taleski D, Stephens B, Kufareva I, Handel TM, Payne RJ, Volkman BF, Sulfopeptide Probes of the CXCR4/CXCL12 Interface Reveal Oligomer-Specific Contacts and Chemokine Allostery. ACS Chemical Biology 8, 1955–1963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dealwis C, Fernandez EJ, Thompson DA, Simon RJ, Siani MA, Lolis E, Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusin” coreceptor. Proc Natl Acad Sci USA 95, 6941–6946 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gustavsson M, Wang L, van Gils N, Stephens BS, Zhang P, Schall TJ, Yang S, Abagyan R, Chance MR, Kufareva I, Handel TM, Structural basis of ligand interaction with atypical chemokine receptor 3. Nature Communications 8, 14135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orsini MJ, Parent JL, Mundell SJ, Marchese A, Benovic JL, Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem 274, 31076–31086 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Drury LJ, Ziarek JJ, Gravel S. p., Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB, Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA 108, 17655–17660 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alekhina O, Marchese A, beta-Arrestin1 and Signal-transducing Adaptor Molecule 1 (STAM1) Cooperate to Promote Focal Adhesion Kinase Autophosphorylation and Chemotaxis via the Chemokine Receptor CXCR4. J Biol Chem 291, 26083–26097 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sykes DA, Dowling MR, Charlton SJ, Exploring the mechanism of agonist efficacy: a relationship between efficacy and agonist dissociation rate at the muscarinic M3 receptor. Mol Pharmacol 76, 543–551 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Ehlert FJ, Griffin MT, Sawyer GW, Bailon R, A simple method for estimation of agonist activity at receptor subtypes: comparison of native and cloned M3 muscarinic receptors in guinea pig ileum and transfected cells. J Pharmacol Exp Ther 289, 981–992 (1999). [PubMed] [Google Scholar]
- 46.Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ, Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80, 367–377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benredjem B, Girard M, Rhainds D, St-Onge G, Heveker N, Mutational Analysis of Atypical Chemokine Receptor 3 (ACKR3/CXCR7) Interaction with Its Chemokine Ligands CXCL11 and CXCL12. J Biol Chem 292, 31–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian S, Choi W-T, Liu D, Pesavento J, Wang Y, An J, Sodroski JG, Huang Z, Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1alpha on CXCR4 transmembrane helical domains. J Virol 79, 12667–12673 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong RSY, Bodart V, Metz M, Labrecque J, Bridger G, Fricker SP, Comparison of the Potential Multiple Binding Modes of Bicyclam, Monocylam, and Noncyclam Small-Molecule CXC Chemokine Receptor 4 Inhibitors. Mol Pharmacol 74, 1485–1495 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Thiele S, Mungalpara J, Steen A, Rosenkilde MM, Vabeno J, Determination of the binding mode for the cyclopentapeptide CXCR4 antagonist FC131 using a dual approach of ligand modifications and receptor mutagenesis. Br J Pharmacol 171, 5313–5329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heredia JD, Park J, Brubaker RJ, Szymanski SK, Gill KS, Procko E, Mapping Interaction Sites on Human Chemokine Receptors by Deep Mutational Scanning. J Immunol 200, 3825–3839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenkilde MM, Gerlach L-O, Jakobsen JS, Skerlj RT, Bridger GJ, Schwartz TW, Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: transfer of binding site to the CXCR3 receptor. J Biol Chem 279, 3033–3041 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Manglik A, Kruse AC, Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 56, 5628–5634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R, Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit 22, 307–318 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AGW, Schertler GFX, Tate CG, The structural basis for agonist and partial agonist action on a b1-adrenergic receptor. Nature 469, 241–244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Katritch V, Abagyan R, GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol Sci 32, 637–643 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kufareva I, Stephens B, Gilliland CT, Wu B, Fenalti G, Hamel DJ, Stevens RC, Abagyan R, Handel TM, in Chemokines: Methods and Protocols, Cardona AE, Ubogu EE, Eds. (Springer Science+Business Media, New York, 2013), vol. 1013, chap. 7, pp. 93–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK, Crystal structure of the b2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rovati GE, Capra V, Neubig RR, The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol 71, 959–964 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Rovati GE, Capra V, Shaw VS, Malik RU, Sivaramakrishnan S, Neubig RR, The DRY motif and the four corners of the cubic ternary complex model. Cell Signal 35, 16–23 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Gyombolai P, Toth AD, Timar D, Turu G, Hunyady L, Mutations in the ‘DRY’ motif of the CB1 cannabinoid receptor result in biased receptor variants. J Mol Endocrinol 54, 75–89 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ, Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A 100, 10782–10787 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lagane B, Ballet S, Planchenault T, Balabanian K, Le Poul E, Blanpain C, Percherancier Y, Staropoli I, Vassart G, Oppermann M, Parmentier M, Bachelerie F, Mutation of the DRY motif reveals different structural requirements for the CC chemokine receptor 5-mediated signaling and receptor endocytosis. Mol Pharmacol 67, 1966–1976 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC III, Basnet H, Sakmar TP, Volkman BF, Structural Basis of CXCR4 Sulfotyrosine Recognition by the Chemokine SDF-1/CXCL12. Sci. Signal 1, ra4- (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffin MT, Figueroa KW, Liller S, Ehlert FJ, Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther 321, 1193–1207 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Ehlert FJ, On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol 377, 549–577 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Buchwald P, A three-parameter two-state model of receptor function that incorporates affinity, efficacy, and signal amplification. Pharmacol Res Perspect 5, e00311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kufareva I, Chemokines and their receptors: insights from molecular modeling and crystallography. Current Opinion in Pharmacology 30, 27–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, Fung J-J, Manglik A, Masureel M, Du Y, Matt RA, Pardon E, Steyaert J, Kobilka BK, Sunahara RK, Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK, Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abdul-Ridha A, Lane JR, Mistry SN, Lopez L, Sexton PM, Scammells PJ, Christopoulos A, Canals M, Mechanistic insights into allosteric structure-function relationships at the M1 muscarinic acetylcholine receptor. J Biol Chem 289, 33701–33711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gustavsson M, Dyer DP, Zhao C, Handel TM, Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor N terminus in chemokine binding. Science Signaling 12, eaaw3657 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR, The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7, 10842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao P, Furness SGB, The nature of efficacy at G protein-coupled receptors. Biochemical Pharmacology 170, 113647 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Nijmeijer S, Leurs R, Smit MJ, Vischer HF, The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges G{alpha}i proteins, and constitutively impairs CXCR4 functioning. J Biol Chem 285, 29632–29641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katancik JA, Sharma A, de Nardin E, Interleukin 8, neutrophil-activating peptide-2 and GRO-alpha bind to and elicit cell activation via specific and different amino acid residues of CXCR2. Cytokine 12, 1480–1488 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Bonneterre J, Montpas N, Boularan C, Gales C, Heveker N, Analysis of Arrestin Recruitment to Chemokine Receptors by Bioluminescence Resonance Energy Transfer. Methods Enzymol 570, 131–153 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Ye J, Kober V, Tellers M, Naji Z, Salmon P, Markusen JF, High-level protein expression in scalable CHO transient transfection. Biotechnol Bioeng 103, 542–551 (2009). [DOI] [PubMed] [Google Scholar]
- 79.Zhou N, Luo Z, Luo J, Liu D, Hall JW, Pomerantz RJ, Huang Z, Structural and Functional Characterization of Human CXCR4 as a Chemokine Receptor and HIV-1 Co-receptor by Mutagenesis and Molecular Modeling Studies. J Biol Chem 276, 42826–42833 (2001). [DOI] [PubMed] [Google Scholar]
- 80.Berchiche YA, Chow KY, Lagane B, Leduc M, Percherancier Y, Fujii N, Tamamura H, Bachelerie F, Heveker N, Direct Assessment of CXCR4 Mutant Conformations Reveals Complex Link between Receptor Structure and Gαi Activation. Journal of Biological Chemistry 282, 5111–5115 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW, Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem 276, 14153–14160 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Doranz BJ, Orsini MJ, Turner JD, Hoffman TL, Berson JF, Hoxie JA, Peiper SC, Brass LF, Doms RW, Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J Virol 73, 2752–2761 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou H, Tai HH, Expression and functional characterization of mutant human CXCR4 in insect cells: role of cysteinyl and negatively charged residues in ligand binding. Arch Biochem Biophys 373, 211–217 (2000). [DOI] [PubMed] [Google Scholar]
- 84.Rosenkilde MM, Gerlach L-O, Hatse S, Skerlj RT, Schols D, Bridger GJ, Schwartz TW, Molecular mechanism of action of monocyclam versus bicyclam non-peptide antagonists in the CXCR4 chemokine receptor. J Biol Chem 282, 27354–27365 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Schematics of the assays used to characterize WT and mutant CXCR4 in this study.
Fig. S2. A wide range of CXCR4 expression yields identical β-arrestin2 and Gαi signaling parameters.
Fig. S3. Total and cell surface expression of receptors with mutations of residues important for signaling as suggested by BRET experiments.
Fig. S4. CXCR4 residues Glu268(ECL3) and Glu275(7.26) are not critical for CXCL12-mediated CXCR4 activation.
Fig. S5. CRS1 is important for the potency but not the efficacy of CXCL12-mediated G protein engagement by CXCR4.
Fig. S6. Predicted consequence of the binding of dimeric CXCL12 to CXCR4.
Fig. S7. Signal amplification in Ca2+ mobilization experiments obscures mutation-induced defects.
Table S1. Key predicted charge interactions in CRS1, CRS1.5, and CRS2 of the CXCR4-CXCL12 complex.
Table S2. Previously published effects of mutations on the CXCR4 residues tested in this study.
Table S3. Signaling parameters obtained from the β-arrestin2 and mini-Gαi association BRET experiments.
Table S4. Signaling parameters obtained from the Ca2+ mobilization experiments.