

Joint analysis of genomic and transcriptomic data reveals diverse noncoding mutation landscapes in five types of pediatric cancers.
Abstract
Interpreting the function of noncoding mutations in cancer genomes remains a major challenge. Here, we developed a computational framework to identify putative causal noncoding mutations of all classes by joint analysis of mutation and gene expression data. We identified thousands of SNVs/small indels and structural variants as putative causal mutations in five major pediatric cancers. We experimentally validated the oncogenic role of CHD4 overexpression via enhancer hijacking in B-ALL. We observed a general exclusivity of coding and noncoding mutations affecting the same genes and pathways. We showed that integrated mutation profiles can help define novel patient subtypes with different clinical outcomes. Our study introduces a general strategy to systematically identify and characterize the full spectrum of noncoding mutations in cancers.
INTRODUCTION
Extensive research efforts have identified recurrent mutated genes in multiple types of childhood cancer, including two recent pan-cancer studies that have defined landscapes of coding mutations in common pediatric cancers (1, 2). However, current cancer mutation landscapes are far from complete without systematic analysis of the noncoding portion of the genome. For instance, recurrent leukemia-associated genetic alterations cannot be identified in 10 to 20% of children with acute leukemias (1), thereby making it challenging to design targeted therapies for such patients.
The vast majority of somatic mutations in cancer genomes occur in noncoding regions because 98% of the human genome is made up of noncoding sequences, and the somatic mutation rate of noncoding regions is similar to that of coding regions (1). The Therapeutically Applicable Research To Generate Effective Treatments (TARGET) project has sequenced more than 1000 genomes from five common pediatric cancers. Similarly, The Cancer Genome Atlas (TCGA) project has molecularly characterized more than 20,000 primary cancer genomes spanning 33 types of adult cancers. Despite this rapid accumulation of whole-genome sequences (WGS) for both pediatric and adult cancers, identification and interpretation of the functional impact of noncoding mutations remain challenging.
Noncoding regulatory sequences, particularly enhancers and promoters, are key determinants of tissue-specific gene expression. Multiple mutation types have been reported to disrupt enhancers and promoters and expression of their target genes, including single-nucleotide variants (SNVs), small insertion and deletions (indels), and large structural variants (SVs), including deletions, insertions, duplications, inversions, and translocations. Most previous studies of noncoding mutations have focused on SNVs and small indels and revealed a number of noncoding causal mutations. Examples include mutations in the TERT promoter in a number of cancer types (3), a single-nucleotide polymorphism (SNP) located in the enhancer of LMO1 in patients with neuroblastoma (NBL) (4), and heterozygous indels [2 to 18 base pairs (bp)] in a super-enhancer located −7.5 kb from the TAL1 promoter in patients with T-acute lymphoblastic leukemia (T-ALL) (5).
Only about 30% of SVs result in in-frame gene fusions that join the protein-coding regions of two genes (6). Most of the SV break points are located in noncoding regions and do not change gene structure. Although less well studied compared to SNVs and small indels, several seminal studies have revealed oncogenic roles of noncoding SVs by redirecting enhancers/promoters to oncogenes or from tumor suppressor genes (7, 8). Such enhancer rearrangement events have been identified in diverse cancer types, suggesting that such events could be a common mechanism of oncogenesis. For instance, interstitial deletion in the pseudoautosomal region 1 of the X/Y chromosomes places the transcription of CRLF2 (cytokine receptor-like factor 2) under the control of the P2RY8 (purinergic 2 receptor Y 8) enhancer in Philadelphia chromosome–like and Down syndrome–associated B cell acute lymphoblastic leukemia (B-ALL) (9). Other examples include complex SVs in childhood medulloblastoma, which rearranges the DDX31 (DEAD-box helicase 31) enhancer to GFI1B (growth factor independent 1B) (7), and the t(3;8) translocation in B cell lymphoma that rearranges the BCL6 enhancer to MYC (8).
Given the prevalence of noncoding mutations and the drastic increase of whole-genome sequencing data, novel computational methods are critically needed to systematically identify putative causal noncoding mutations. Here, we introduce PANGEA (predictive analysis of noncoding genomic enhancer/promoter alterations), a general computational framework for systematic analysis of noncoding mutations and their impact on gene expression. PANGEA simultaneously identifies all classes of somatic mutations that are associated with gene expression changes, including SNVs, small indels, copy number variations (CNVs), and SVs. Using PANGEA, we have conducted a pan-cancer analysis of noncoding mutations in 501 pediatric cancer patients of five histotypes with matched WGS and RNA-sequencing (RNA-seq) data generated by the TARGET project. We identified a comprehensive list of recurrent noncoding mutations as putative causal mutations in these cancers. An integrated analysis of both coding and noncoding mutations revealed distinct pathways affected by either coding or noncoding mutations. We also show that integrated mutation profiles can help define novel patient subtypes with different clinical outcomes.
RESULTS
A comprehensive catalog of recurrent noncoding mutations
The TARGET data portal (https://ocg.cancer.gov/programs/target/data-matrix) contains matched WGS and RNA-seq data for 501 patients, including 163 patients with B-ALL, 153 patients with acute myeloid leukemia (AML), 100 patients with NBL, 53 patients with Wilms tumor (WT), and 32 patients with osteosarcoma (OS). All patients have WGS data for both tumor and germline or remission samples, which can be used to define alterations as germline or somatic.
We identified somatic SNVs and small indels (<60 bp) for the five cancer types using tumor and remission samples (fig. S1A). We evaluated the accuracy of our mutation calling pipeline using two approaches. First, we used a benchmark set generated by Zook et al. (10). It consists of a set of high-confidence SNVs and indels in subjects NA12878 and NA24631 from the 1000 Genomes Project, identified by integrating 14 datasets using five sequencing technologies, seven read mappers, and three variant callers. Using this benchmark set, we estimated the precision of our pipeline to be ~95%. Second, the TARGET project provides a list of high-confidence SNV calls in B-ALL, AML, NBL, and WT that were validated using multiple experimental protocols including whole-exome sequencing, RNA-seq, and targeted sequencing. Of the 735 high-confidence calls, our variant calling pipeline identified 681 (92%) of them, suggesting high sensitivity of our pipeline (fig. S1B).
Across the five cancer types, the average mutation rate ranges from 0.16 SNV/indel per million bases (Mb) in AML to 0.55 per Mb in OS (fig. S1C). The higher somatic mutation rate in OS is consistent with a previous WGS study, in which regional clusters of hypermutation, termed kataegis, were identified in 85% of patients with pediatric OS (11). As expected, more than 98% of the identified mutations are located in noncoding regions, while less than 1% of the mutations are located in coding regions (fig. S1D). There is no significant difference in the mutation rate between the noncoding region and coding region across all five cancer types (fig. S1E).
Next, we used Delly2 and Lumpy to identify SVs, including large deletions (DELs), tandem duplications (DUPs), inversions (INVs), and translocations (TRANs). We only kept SVs that were called by both methods and passed additional filtering criteria (fig. S1F) as the final set of identified SVs. In total, we identified 26,757 SVs across 501 patients. The average number of identified SVs per patient ranges from 18 in WT to 71 in OS (fig. S1G). These numbers are comparable with published data by the International Cancer Genome Consortium (ICGC) and TCGA consortium. Previously, Roberts et al. (12) identified and experimentally validated 21 SVs that joined exons of two genes in frame in patients with B-ALL. We identified 20 of those SVs, suggesting high sensitivity of our pipeline. Among the identified SVs, 9831 are in-frame changes and potentially generate fusion genes. We evaluated the fusion gene predictions by comparing to matched RNA-seq data from the patients. Of the 9831 fusion genes predicted by our pipeline, 2323 of them were supported by RNA-seq data from the same patients. The TARGET consortium experimentally tested 12 known translocations in 212 patients with leukemia. On the basis of this set of validated SVs, our SV calling pipeline has an accuracy of 98% (fig. S1H). Together, using multiple benchmarking datasets, we show a higher sensitivity of our SV calling pipeline compared to the previous publication.
Noncoding mutations disrupting enhancer/promoter sequences or enhancer-promoter interactions
The functional consequence of noncoding mutations is difficult to interpret without comprehensive annotation of noncoding regulatory DNA sequences in cancer genomes. To this end, we used publicly available epigenomic data of disease-relevant cell/tissue types to construct an enhancer catalog for the five cancer types in this study (see Materials and Methods). Specifically, we used chromatin immunoprecipitation sequencing (ChIP-seq) data of histone modification marks (H3K4Me1, H3K4Me3, and H3K27Ac) to predict transcription enhancers using the chromatin signature identification by artificial neural network (CSI-ANN) algorithm (table S1) (13). In total, we identified 282,021 enhancers for the five cancer types at the false discovery rate (FDR) of 0.05. Overall, 91% of predicted enhancers are supported either by assay for transposase-accessible chromatin sequencing (ATAC-seq) data in cancer-relevant cell types or by sequence conservation across 20 mammalian genomes (fig. S1I) or both, suggesting high quality of the predicted enhancers. Next, we predicted target gene(s) of each enhancer using the integrated method for predicting enhancer targets (IM-PET) algorithm (14), using public histone modification ChIP-seq and RNA-seq data in disease-relevant cell types. In total, we predicted 635,096 enhancer-promoter (EP) pairs across the five cancer types. We compared the predicted EP pairs with published high-resolution Hi-C/ChIA-PET data in human B cells, myeloid cells, and kidney cells (table S1; Materials and Methods). Seventy-four percent (317,698) of our EP predictions are supported by either Hi-C or ChIA-PET data (fig. S1J), suggesting high quality of our predictions.
To identify recurrent mutations that disrupt either enhancer/promoter sequences or EP interactions, we intersected the catalog of somatic mutations with the catalogs of enhancers/promoters and EP interactions. Across the five cancer types, 16% of the identified SNVs or indels overlap with enhancers/promoters in cell types relevant to a given pediatric cancer, 15% of the identified CNVs overlap with the enhancers in relevant cell type, and 45% of the break points of inversions and translocations are located between predicted EP pairs in the disease-relevant cell types. On the other hand, 23.1% (146,707) of the 635,096 EP pairs are affected by SVs, among which 4.4% (27,944) are recurrently affected in multiple patients. On average, the distance between an SV break point and an affected gene is 0.3 million base pairs (Mbp) (largest distance up to 1.4 Mbp), suggesting the potential of SVs to disrupt gene expression across large genomic distance.
Systematic identification of putative causal noncoding mutations
To help prioritize noncoding mutations, we developed the PANGEA method to systematically identify recurrent noncoding mutations that disrupt the transcriptional regulation of a gene. Unlike previous methods, PANGEA considers all types of noncoding mutations, including SNVs, small indels, CNVs, and SVs. These mutation types can either disrupt enhancer/promoter sequences or disrupt the interactions between enhancers and promoters that are critical for transcription activation. After tabulating all such mutations, we use weighted elastic net to perform a regression analysis of gene expression on the sets of noncoding mutations across the patient cohort (Fig. 1A). Because the EP interactions are predicted computationally, we use EP prediction scores as the weights for each predictor (mutation) to include a confidence measure of EP interactions in our regression model. Putative causal mutations are predicted on the basis of the statistical significance of the corresponding regression coefficients (see Materials and Methods for details). The PANGEA software package is available at https://github.com/tanlabcode/PANGEA.
Fig. 1. The PANGEA algorithm for simultaneous identification of putative causal noncoding mutations of all classes.

(A) Overview of the PANGEA algorithm. (B) Fraction of patients stratified by different classes of noncoding mutations affecting enhancers/promoters. CNA, Copy Number Alteration. (C) Proportion of genes whose expression is disrupted by different classes of putative causal noncoding mutations. SNV, single-nucleotide variant; DEL, deletion; DUP, duplication; TRAN, translocation. (D) Predicted mutations affect enhancers that have higher tumor type specificity. Enhancer specificity is defined as the number of cell types in which the enhancer is observed to be active. P value of one-sided t test is shown (n = 282,021). (E) Predicted mutations affect higher percentage of super-enhancers. P value of hypergeometric test is shown (n = 282,021). (F) Predicted mutations affect enhancers with higher sequence conservation across 20 mammalian species. Enhancer conservation is defined as the average PhastCons score of the enhancer sequence. P value of one-sided t test is shown (n = 282,021).
Using multiple-testing adjusted P value of <0.05 as the cutoff, we identified 1405 genes whose expression changes can be predicted by SNVs/small indels in their enhancers/promoters, 55 genes whose expression changes can be predicted by CNVs in their enhancers, and 1082 genes whose expression changes can be predicted by SVs that disrupt their EP interactions (table S2). In total, disruption of enhancer function by recurrent noncoding mutations was found in 477 of 501 patients with pediatric cancer (95%) (Fig. 1B). The quantile-quantile plot shows large deviation of the observed P values for the putative causal noncoding mutations compared to those of random expectations using an independent ICGC cohort (n = 2715 donors), suggesting low false prediction rate (fig. S1K; Supplementary Materials). More than half of the putative causal noncoding mutations are SNVs and small indels (66%), followed by translocations (27%) and other types of SVs (Fig. 1C). However, when adjusted by the overall frequency of each type of mutation, SVs become the most frequent type of putative causal noncoding mutations (fig. S1L).
The enhancers that are affected by putative causal noncoding mutations are more cell type–specific compared to all enhancers in our enhancer catalog (Fig. 1D). In addition, these enhancers have more overlap with published super-enhancers in cell types that are relevant to the given cancer type (Fig. 1E) (15). Last, these enhancers have high levels of sequence conservation across 20 mammalian species (Fig. 1F). Together, these results provide additional support to the putative causal noncoding mutations.
Risk enhancer rearrangements
Several seminal studies have reported “enhancer hijacking,” also known as oncogenic rearrangement of enhancers due to translocation/inversion (7, 8, 16, 17). In this study, we performed a systematic analysis of enhancer hijacking events across the five cancer types. Among 501 total patients, 405 patients (81%) have at least one predicted enhancer hijacking event in their genomes. We identified several known enhancer hijacking events. For example, we found the t(14;X)(q32;p22) translocation in 12 patients with B-ALL, which hijacks multiple enhancers of IGHV to the vicinity of CRLF2, resulting in significant overexpression of CRLF2 in these patients (fig. S2A). We also found enhancer hijacking from three different genomic loci to the common TERT gene locus [t(10;5)(p22;p15), t(5;5)(q34;p15), and t(5;5)(q12;p15)] (fig. S2B). These TERT-related enhancer hijacking events were observed in 15 patients with NBL, and the resultant translocations led to significant overexpression of TERT in these patients (fig. S2B).
Most of our predicted enhancer hijacking events have not been previously reported. One such event involves hijacking of enhancers to up-regulate CHD4, which has three translocation partners [t(12;22)(p13;q13), t(12;19)(p13;p13), and t(9;12)(p24;p13)] (Fig. 2A). In total, 12 patients with B-ALL had this translocation. Most translocations in this region result in fusion genes involving a nearby gene ZNF384. A previous study has focused on the function of ZNF384 fusion genes (18). However, in the TARGET cohort, we observed two patients in which the translocation break point is actually located downstream of ZNF384 and did not generate an ZNF384 fusion (Fig. 2A). Furthermore, ZNF384 fusion is not correlated with patient prognosis. Together, these data suggest a driver of oncogenesis that is independent of ZNF384. In all patients with the translocation, we found that enhancers such as that of EP300, TCF3, and SMARCA2 are hijacked to chr12.p13. The expression level of CHD4, a gene adjacent to ZNF384, is significantly increased in these patients (Fig. 2B). Similarly, in a recently published mixed phenotype acute leukemia (MPAL) cohort, we also observed the CHD4 expression increase in the patients with ZNF384-involved rearrangement (fig. S2C) (19). Moreover, B-ALL patients with the enhancer hijacking event have significantly shorter time to relapse (Fig. 2C). We thus hypothesized that enhancer hijacking translocation events that bring potent enhancers to the promoter of CHD4 may be an independent oncogenic driver in B-ALL.
Fig. 2. Enhancer hijacking to CHD4 by translocation in patients with B-ALL.

(A) Translocations result in enhancer hijacking to CHD4 in B-ALL. Three different translocation partners are identified in the cohort. Histone modification ChIP-seq data of human CD19+ B cells (identifying enhancers), SNVs, and SV break points (BPs; each vertical line represents a patient) are shown. The hijacked enhancers are highlighted in brown. (B) Expression levels of CHD4 in patients with and without translocations. P value of one-sided t test is shown (n = 163). (C) Patients with CHD4 translocation have shorter time to relapse. P value of log-rank test is shown (n = 163). (D) CHD4 knockout impairs growth of B-ALL cell lines, NAML-6 and REH. (E) Luciferase reporter assay of hijacked enhancer. BG, no DNA vector control; EV, vector containing no enhancer; NC, negative control; Enh, test EP300 enhancer. (F) Western blots of CHD4 protein in Ba/F3 cells with and without translocation. P value of one-sided t test is shown (n = 2). (G) mRNA levels of CHD4, NOP2, ZNF384, and EP300 in Ba/F3 cells with and without translocations. P values were calculated using one-sided t test (n = 2 for all tests). TBP, TATA-box Binding Protein. (H) Ba/F3 cells with induced translocation undergo oncogenic transformation. Enhancer hijacking translocation was induced using CRISPR-Cas9. Error bars represent SD.
CHD4 (chromodomain helicase DNA binding protein 4) is a component of the nucleosome remodeling and deacetylase complex, which plays an important role in B cell development by regulating B cell–specific transcription (20). In addition, CHD4 is known to function as a repressor of several tumor suppressor genes, and inhibition of CHD4 reduces the growth of AML and colon cancer cells (21). These data suggest a role of CHD4 in the oncogenesis of B-ALL. To investigate the potential role of CHD4, we first identified differentially expressed genes in patients with CHD4 overexpression. In total, there are 666 up-regulated and 922 down-regulated genes in patients with CHD4 rearrangements. Using published transcription factor (TF) ChIP-seq data in human GM12878 cells, we found that CHD4 binding sites are significantly enriched at enhancers or promoters of the down-regulated genes (fig. S2E). Among the 157 down-regulated genes with CHD4 binding sites, several of them encode well-known regulators of B cell development including PAX5, IRF4, TCF3, and EBF1 (fig. S2, F and G). These results suggest a role of CHD4 in B-ALL by regulating the expression of key TFs in B cell development.
To test the potential oncogenic role of CHD4 in B-ALL, we knocked out CHD4 in the NALM-6 and REH B-ALL cell lines. We performed growth competition assays to compare the growth phenotypes of CHD4 knockout with non-knockout leukemic cells. Both NALM-6 and REH cells showed impaired growth with CHD4 knockout (Fig. 2D). Next, we introduced the translocation t(6;15)(qF2;qE1) in murine Ba/F3 cells using CRISPR-Cas9. The translocation break points were designed to be located in the intergenic regions near CHD4 and EP300, therefore placing the EP300 enhancer (Fig. 2E) upstream of the CHD4 promoter without creating a fusion gene (fig. S2H). In Ba/F3 cells with the translocation, we observed increased expression of CHD4 at both mRNA and protein levels (Fig. 2, F and G). In contrast, expression of nearby genes was not increased in cells with the translocation (Fig. 2G). The introduced translocation enables Ba/F3 cells to proliferate in the absence of interleukin-3 (IL-3) (Fig. 2H), suggesting oncogenic transformation. In addition, the expression levels of TCF3, PAX5, and EBF1 were significantly decreased in the cells with CHD4 rearrangement, suggesting the role of CHD4 in regulating those key regulators (fig. S2I). In summary, these results strongly support an oncogenic role of CHD4 in B-ALL due to enhancer hijacking.
Risk enhancer copy number alterations
We found that 309 patients in the TARGET cohort (62%) had enhancer amplification/deletion and associated target gene expression change. MYCN is frequently amplified in patients with NBL (22). In the NBL cohort, we observed that the body of the MYCN gene is amplified in 34 patients. However, we also observed 11 patients with amplification of the MYCN enhancer rather than the gene body (Fig. 3A). Hi-C data in the NBL cell line, SK-N-SH, support the EP interaction predicted using IM-PET. MYCN expression level is significantly higher (2.8-fold change) in those patients compared to the patients without MYCN amplification (Fig. 3B). Moreover, patients with only MYCN enhancer amplification have shorter time to relapse compared to other patients (Fig. 3C), including patients with MYCN gene body amplification. This result suggests that enhancer amplification alone is sufficient to up-regulate MYCN and drive aggressive NBL. Another example involves deletion of the ATG3 enhancer in 15 patients with B-ALL (fig. S3A), resulting in decreased ATG3 expression in those patients (fig. S3B). Autophagy related 3 (ATG3) is a ubiquitin-like–conjugating enzyme and plays a role in the regulation of autophagy; down-regulation of ATG3 has been reported in patients with myelodysplastic syndrome (MDS) progressing to leukemia (23).
Fig. 3. Examples of disruption of enhancer function by copy number alteration and point mutation.

(A) Segmental duplications result in amplification of MYCN enhancer in NBL. Shown tracks are histone modification ChIP-seq data in human neurocrest cells and identified CNVs. Color bars in CNV track indicate MYCN amplification types. Orange, amplification of MYCN enhancers; green, amplification of both MYCN enhancers and gene body; blue, amplification of MYCN gene body only. (B) Expression level of MYC in patients with and without copy number change. (C) Kaplan-Meier plots of time to relapse for NBL patients with MYCN gene amplification, MYCN enhancer amplification, and without any copy number change in either gene or enhancer. P value of one-sided log-rank test is shown (n = 100). (D) Point mutations result in disruption of GATA2 binding sites that regulate GFI1B expression in AML. Shown tracks are histone modification ChIP-seq data in human neurocrest cells and identified SNVs. (E) Expression level of GFI1B in patients with and without enhancer mutations. P value of one-sided t test is shown (n = 153). (F) Kaplan-Meier plots of time to relapse for AML patients with and without GFI1B enhancer point mutations. P value of one-sided log-rank test is shown (n = 153).
Risk enhancer/promoter SNVs and small indels
We found that 346 patients in the TARGET cohort (69%) had SNVs/small indels located in enhancers/promoters that caused target gene expression change. For instance, we found six patients with AML who have SNVs in the GFI1B +11-kbp enhancer (24). The GATA2 binding sites in the enhancer were disrupted, and GFI1B expression decreased correspondingly (Fig. 3, D and E). Growth factor independence 1b (GFI1B) encodes a key TF regulating dormancy and proliferation of hematopoietic stem cells and development of erythroid and megakaryocytic cells (25). Recent studies had revealed its critical role as a tumor suppressor in AML, as low GFI1B expression is associated with poor patient survival (26). Consistent with previous studies, the six patients with GFI1B enhancer mutation have significantly shorter time to relapse (Fig. 3F). Another example involves nine patients with AML who have mutations in the IDH2 +56-kbp enhancer (fig. S3B). This enhancer is part of a known super-enhancer of IDH2 (27) and is constitutively active in myeloid cells. Previously, nonsynonymous mutations of IDH2 have been reported in 9 to 19% of adult patients with AML but relatively rare in childhood AML (<4%) (28). Mutations in IDH2, as well as in several other genes involved in the regulation of DNA methylation (such as DNMT3, TET2, and IDH1), were found to have higher variant allele frequencies in adult patients with AML, suggesting that they are early mutational events in AML (29). We did not identify nonsynonymous IDH2 mutations but did find recurrent mutations in the IDH2 enhancer. The mutations are predicted to disrupt ERG/FLI1 and E2F4 binding sites in the enhancer. The expression level of IDH2 is significantly lower in patients with these mutations. Together, these results suggest a different mechanism of IDH2 disruption in pediatric AML that was previously unappreciated. We also found four patients who have SNVs in the GATA2 +126-kbp enhancer and two patients who have SNVs in the GATA2 promoter (fig. S3C). The enhancer was previously reported to be rearranged to the vicinity of EVI1 in adult patients with AML (30). The mutations were predicted to disrupt a FUBP1 binding site, and the expression of GATA2 was significantly lower in the patients with mutations in enhancer/promoter regions.
Coding and noncoding mutations affect distinct sets of genes and pathways
In total, our analysis of five pediatric cancer types identified 1175 genes recurrently altered in their coding regions and 2162 genes recurrently altered in their noncoding regions. Unexpectedly, the overlap (62 genes; 2%) between the two groups of genes is smaller than expected by chance (P = 4.1 × 10−12, one-sided Fisher’s exact test), suggesting general exclusivity of coding and noncoding mutations affecting a given gene (Fig. 4A).
Fig. 4. Coding and noncoding mutations affect distinct sets of genes.
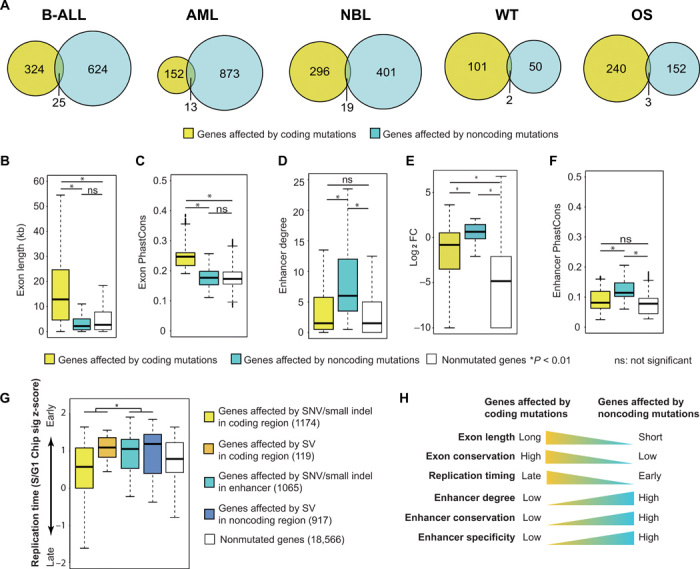
(A) Venn diagrams of genes affected by recurrent coding and noncoding mutations in five cancer types. (B to G) Features of genes affected by coding and noncoding mutations and genes without any mutation: gene exon length (B), exon conservation level measured by PhastCons score (C), enhancer degree (number of enhancers regulating a promoter) (D), gene expression specificity measured by fold change (FC) of average expression in a given cancer type compared to that of all five pediatric cancer types (E), enhancer conservation level measured by PhastCons score (F), and replication timing (G). (H) Summary of different genomic features for the genes affected by coding and noncoding mutations. P values of one-sided t test are shown (n = 21,841).
We investigated the genomic features of the genes affected by coding versus noncoding mutations. We found that genes affected by coding mutations are longer and have higher level of sequence conservation (Fig. 4, B and C). On the other hand, genes affected by noncoding mutations have more enhancers regulating them (Fig. 4D), and their expression is more tumor type specific (Fig. 4E). In addition, the regulating enhancers of those genes are more conserved (Fig. 4F). Previous studies have suggested that SNVs and small indels occur more frequently in genomic regions with late replication timing, while translocations and inversions occur more frequently in genomic regions with early replication timing (31). Consistent with this observation, we found that genes affected by SNVs and small indels in coding regions are located in the relatively late replicating regions, while the genes in other groups have relatively earlier replication timing (Fig. 4G). Pathway analysis reveals that signaling pathways, including Janus kinase (JAK)/signal transducer and activator of transcription (STAT), mitogen-activated protein kinase (MAPK), and Wnt signaling pathways, are mostly affected by coding mutations (fig. S4A). In stark contrast, genes in metabolic pathways are mostly affected by noncoding mutations (fig. S4A). Correspondingly, we found that metabolic genes tend to be located in early replicating regions (fig. S4B). Together, these data suggest that the exclusivity of genes affected by coding versus noncoding mutations is likely due to the different genomic locations and features of those genes (Fig. 4H).
Level of TF regulon disruption correlates with patient prognosis
Genes encoding lineage-specific TFs are frequently mutated in pediatric cancers. For example, TFs that regulate B cell development including IKZF1, EBF1, PAX5, and TCF3 are frequently altered in patients with B-ALL (32). In addition, MYCN and ZNF281 are known prognostic markers for NBLs (33, 34), while RUNX1 and CBFB are also frequently altered in pediatric AML and are linked to unfavorable clinical outcomes (35).
In contrast to coding mutations of TFs, regulatory output of TFs can also be disrupted by mutations affecting individual enhancers/promoters/EP interactions regulating the target genes. Therefore, we compared the effects of coding and noncoding mutations on TF regulons (defined as the set of genes regulated by a TF) and patient outcome. For each cancer type, we report the top 10 most frequently affected regulons by combined coding and noncoding mutations (Fig. 5A and table S3). Most of the identified TFs have a known role in the specific cancer type. Furthermore, we found that regulon disruption by noncoding mutations occurs more frequently than regulon disruption by coding mutations of the TF genes (Fig. 5B). This is probably because mutations of a TF gene lead to a bigger disruption of the TF regulon compared to mutations disrupting regulation of individual TF targets.
Fig. 5. Degree of regulon disruption of key TFs is correlated with clinical outcome.

(A) Top 10 most frequently disrupted regulons in each cancer type. Bar plots show the percentage of patients with regulon disruption. TFs with known role in the given cancer are highlighted in red. (B) Number of patients with regulon disruption (either TF coding mutations or target gene noncoding mutations). Asterisks indicate TFs whose regulon disruptions are correlated with patient time to relapse (log-rank test, P < 0.05; n = 501). (C) Kaplan-Meier plots of time to relapse for B-ALL patients with PAX5 deletion, EP disruption involving PAX5, and with no mutation of the PAX5 regulon. (D) AML patients with RUNX1 fusion, EP disruption involving RUNX1, and without any mutation of the RUNX1 regulon. (E) WT patients with WT1 mutation, EP disruption involving WT1, and with no mutation of the WT1 regulon. (F) B-ALL patients with TCF3 fusion, EP disruption involving TCF3, and with no mutation of the TCF3 regulon. (G) NBL patients with MYC amplification, EP disruption involving MYC, and with no mutation of the MYC regulon. P values of one-sided log-rank test are shown. The numbers of samples are indicated in parentheses.
We hypothesized that the level of regulon disruption is correlated with patient disease outcome. To test this hypothesis, we plotted time to relapse stratified by the degree of regulon disruption (i.e., TF coding mutations versus disruption of individual TF targets by noncoding mutations). We found a strong correlation between time to relapse and the degree of regulon disruption. For instance, in B-ALL, patients with PAX5 deletion have the shortest time to relapse, followed by patients with disruption of PAX5 EP interactions. Last, patients without any PAX5 regulon mutation have the longest time to relapse (Fig. 5C). We found the same correlations for RUNX1 regulon in AML (Fig. 5D) and WT1 regulon in WT (Fig. 5E). For PAX5, RUNX1, and WT1, the mutations are all loss of function of the TFs. We also found correlations involving gain-of-function mutations of the TFs, including TCF3 in B-ALL and MYCN in NBL. For TCF3, because the fusion event leads to gain of function of TCF3, patients with disruption of TCF3 EP interactions have longer time to relapse compared to the patients without any TCF3 regulon mutation (Fig. 5F). Presumably, the gain of function of TCF3 partially mitigates the effect of disruption of T cell factor (TCF) target genes due to noncoding mutations. Similarly, NBL patients with disruption of MYCN EP interactions have longer time to relapse compared to the patients without any MYCN regulon mutation (Fig. 5G). In summary, our data suggest that there is typically a wide spectrum of regulon disruption for key TFs in pediatric cancers. The level of TF regulon disruption is associated with patient prognosis.
Integrated mutation profiles suggest novel disease subtypes
Mutational profiles of coding regions have been widely used for patient stratification and prognosis purposes. This is not the case for noncoding mutational profiles. We therefore constructed mutational profiles of the five pediatric cancers using both coding and noncoding mutations. Clustering analysis of these combined mutational profiles enabled us to discover novel patient subtypes.
For B-ALL, seven patient clusters are identified (Fig. 6A). Of the seven clusters, four are characterized by known translocation events in B-ALL including ZNF384 rearrangement, ETV6-RUNX1, TCF3-PBX1, and IGHV translocation. All these are previously reported to be recurrent SVs in B-ALL (32). However, the mechanism by which these fusion proteins contribute to leukemogenesis has not been fully elucidated. Here, we found that in addition to creating fusion genes, the translocations can alter the expression of nearby genes by rearranging locations of distal enhancers (Fig. 6A; affected genes are listed below the heat map).
Fig. 6. Integrative mutation profiles of pediatric cancers.

(A to E) Mutation profiles of patients with B-ALL, AML, NBL, WT, and OS. Heat maps were generated using recurrently mutated genes (due to either coding or noncoding mutations) in at least five patients. Patients are clustered according to the combined coding and noncoding mutation profiles. The color of each cell indicates the class of mutations. Rows, genes; columns, patients. For presentation purposes, the heat maps are divided into submaps based on noncoding (top) or coding mutations (bottom). The tables below the heat maps list up- or down-regulated genes due to noncoding mutations in all patients in a given cluster. Clusters discussed in the text are highlighted in orange boxes. Mutations and affected genes discussed in the text are indicated on the right edge of each heat map.
Two other clusters (C5 and C6) are characterized by novel inversion or translocation. Patients in C5 have Inv(2) (n = 14). The inversion is predicted to hijack an enhancer to near SUPT7L and ATRAID and up-regulate the expression of these two genes (fig. S5, A and B). SUPT7L is a subunit of the SPT3-TAFII31-GCN5L acetylase complex, which is known to regulate the stability of the TCF3-PBX1 oncoprotein in ALL (36). We found that Inv(2) significantly co-occurs with TCF3-PBX1 in six patients (fig. S5C). Both TCF3-PBX1 and Inv(2) are associated with aggressive clinical outcome in B-ALL. Patients who have both mutations show even shorter time to relapse compared to patients with either type of mutation (fig. S5D), suggesting synergy of these two pathways contributing to disease outcome.
Patient cluster C6 is characterized by interchromosomal translocations at chr2 [t(2;7)(q21;q11) and t(2;11)(q21;q11)]. The translocations occur in 15 patients and are predicted to disrupt the EP interaction involving the ANKDR30BL gene (fig. S5E), resulting in significant decrease of ANKDR30BL expression in patients with this translocation. MIR663B, a microRNA located in the intron of ANKDR30BL, is also down-regulated in those patients (fig. S5F). MIR663B is known to regulate the expression of CCL17, CD40, and PIK3CD in chronic lymphocytic leukemia (37). Consistent with the previous finding, we observed significant expression increase of those genes in patients with MIR663B down-regulation (fig. S5F).
We also identified two potential novel cancer subtypes in NBL (C6 and C7; Fig. 6C). Cluster 7 consists of nine NBL patients with translocation on chr17 that results in enhancer rearrangement and down-regulation of ERBB2 [t(1;17)(p33;q12), t(9;17)(p21;q12), and t(11;17)(q13;q12)] (fig. S6, A and B). ERBB2 is essential for normal embryonic development and has a critical function in oncogenesis and progression of several cancer types including breast cancer, lung cancer, and leukemias. In addition, low ERBB2 expression is associated with poor patient survival in NBL (38). Consistently, patients in cluster 7 have shorter time to relapse (fig. S6C). The other novel NBL subtype (C6) is characterized by amplification of the TGM6 enhancer in 11 patients with NBL and consequently increased TGM6 expression in these patients (fig. S6, D and E). Transglutaminase 6 (TGM6) is a protein associated with nervous system development (39). Transglutaminases, particularly TGM2, is known to play important roles in neurite outgrowth and modulation of neuronal cell survival (40). Our result suggests a potential oncogenic role of TGM6 in NBL.
A novel AML subtype (cluster C5) is characterized by enhancer deletion of ZNF37A in 16 patients (fig. S6G). ZNF37A is involved in fusion events in breast cancer (41) and adult AML (42). Notably, AML patients with ZNF37A enhancer deletion have lower ZNF37A expression and shorter time to relapse (fig. S6, H and I), suggesting the prognosis significance of this noncoding mutation.
A novel WT subtype (cluster C4) is characterized by enhancer rearrangement of GAS6 in six patients (fig. S6J). Growth arrest–specific 6 (GAS6) is a ligand for receptor tyrosine kinases AXL, TYRO3, and MER whose signaling is implicated in cell growth and survival (43). Patients with the translocation have lower GAS6 expression and shorter time to relapse (fig. S6, K and L).
DISCUSSION
The landscape of noncoding mutations in pediatric cancers has not been comprehensively characterized. Here, we developed PANGEA, a general computational method to identify all classes of putative causal noncoding mutations by joint analysis of patients’ mutations and gene expression profiles. Application of PANGEA led to a comprehensive and prioritized list of putative causal noncoding mutations in five major pediatric cancers.
Previous studies on noncoding mutations have been focused on SNVs and small indels. In contrast, systemic analysis of SVs has been lacking. Because of their much larger sizes, SVs may have a bigger impact on shaping the cis-regulatory landscape than SNVs. In support of this notion, we found that SVs are the most frequent class of putative causal noncoding mutations when adjusted for background occurrence frequency (fig. S1L). In total, our analysis has revealed 1137 putative causal SVs affecting the expression of more than 2000 genes across five pediatric cancer types.
Previous studies have also been focused on fusion genes generated by SVs because they are intuitive candidates of driver events. However, only ~30% of SVs generate fusion genes according to the current deposited SVs at ICGC. Moreover, ~35% of the gene fusions in the TARGET cohort are generated via microhomology-mediated end jointing (MMEJ). Because microhomology tends to be located at the break points of nonpathogenic SVs (44), these data suggest that the fusion genes may not be oncogenic drivers in cases of MMEJ. Instead, in our analysis, we found that 55% SVs altered regulatory landscape and expression of nearby genes of the break points. These genes warrant careful investigation for a role in oncogenesis in future studies.
We found that coding and noncoding mutations affect distinct sets of genes and pathways. This mutual exclusivity is likely due to the different genomic locations of these two classes of genes. We found that genes affected by noncoding mutations tend to be located in regions with early replication timing. This trend is consistent with previous reports that the occurrence of genomic rearrangements tends to occur in regions of early replication (45). Whether this correlation indicates a novel oncogenic mechanism needs to be further investigated. For instance, we found that metabolic genes tend to be located in early replicating regions and are more frequently affected by noncoding mutations. Rewiring of metabolism is a hallmark of cancer. Recent systematic analysis of TCGA data for eight cancer types has reported that more than 75% of metabolic genes are differentially expressed in each cancer type (46). However, it is unclear to what degree metabolism rewiring is mediated by noncoding mutations in those cancer types. Here, our analysis suggests that metabolic genes may be preferentially affected by noncoding mutation. In summary, our results highlight the need for comparative analysis of both coding and noncoding because novel cancer-related genes and pathways may be unveiled with comprehensive noncoding mutation analysis.
Many lineage-specific TFs are frequently perturbed in various cancers. However, the underlying oncogenic mechanism is challenging to understand. With comprehensive noncoding analysis, our approach provides a means to understand the detailed molecular mechanism underlying TF perturbation in cancers. For instance, recurrently deregulated target genes of a TF suggest that they can be important mediators of the disrupted TFs. Clinically, we found that the level of disruption of a TF regulon can be used to stratify patient survival. More sophisticated computational models can be developed to prioritize target genes of perturbed TFs that contribute to oncogenesis.
MATERIALS AND METHODS
Identification of SNVs and small indels
We used GATK (version 3.8) and Freebayes (version 1.0.2) to call SNVs and small indels. We first generated a set of initial SNV and indel calls with the default parameters of each software. Several filters were applied during the post-processing step of the initial calls. First, we filtered mutations that overlap with low complexity regions. Second, we excluded regions with excessive read depth, as those regions are probably associated with spurious mappings. Third, we required the mutations to have multiple observations of the alternate (nonreference) allele in reads from both DNA strands. Last, we used the P value cutoff of 0.01. SNVs passing these filters were intersected with annotations in the dbSNP database (build 149). Calls matching both the position and allele of known dbSNP entries were removed (fig. S1A).
Identification of SVs
Delly (version 0.7.2) and Lumpy (version 0.2.13) were used to call SVs. We used the default parameter settings of both software with the exception of setting minimum mapping quality threshold to zero as advised by the Complete Genomics data analysis pipeline (https://target-data.nci.nih.gov/Public/Resources/WGS/CGI/READMEs/). The initial SVs called by both software were retained for further filtering. First, we removed SVs in which break points are located in repetitive regions. Second, we removed SVs that are also identified in the baseline genomes that consist of 261 WGS samples from the 1000 Genome Project (www.internationalgenome.org/data). Last, we selected SVs with at least seven supporting reads as the final set of SVs.
RNA-seq data analysis
Raw reads from RNA-seq were mapped to the reference human genome (release GRCh37) using STAR with default parameter setting. Transcripts were assembled using Cufflinks using mapped fragments outputted by STAR. RefSeq (GRCh37) was used for the annotation of known transcripts. Normalized transcript abundance was computed using Cufflinks and expressed as FPKM (fragments per kilobase of transcripts per million mapped reads).
Weighted elastic net model as a general framework for predicting putative causal mutations disrupting promoter regulation
For a given gene promoter, we consider all types of mutations that could potentially disrupt its regulation, including SNVs and small indels, CNVs, inversions, and translocations. These mutations could either disrupt the function of the cis-regulatory sequences per se or disrupt the interactions between the enhancers and the promoters. For the latter category of mutations, SVs could hijack enhancer(s) for a given promoter. We define potential enhancer hijacking as existing enhancer relocated to new region with a nearby promoter (<200 kbp). For each promoter, its potential enhancers are detected on the basis of patient SV data. We developed a regression-based approach to identify specific mutations that are associated with gene expression change. We used elastic net to implement the regression analysis. Elastic net combines the strength of ridge regression and least absolute shrinkage and selection operator (LASSO). It can enforce sparsity, has no limitation on the number of selected variables, and encourages grouping effect in the presence of highly correlated predictors.
For each gene, let us consider a regression model with n observations (i.e., n patients). Suppose that xi = (xij, ⋯xnj)T, j = 1, ⋯, p are the predictors and y = (y1, ⋯yn)T is the gene expression of the target promoters. X = [x1, ⋯, xp] denotes the predictor matrix. We treat noncoding mutations affecting the same enhancer/promoter/EP interaction as the same predictor. The rationale for this is that because they affect the same enhancer/promoter/EP interaction, the impact on gene expression (the independent variable) is likely to be the same. The total number of predictors is the number of mutations summed over all promoters/enhancers regulating the given gene. The regression model can be expressed as y = Xβ + ϵ, where β = (β1, ⋯, βp)Tand the noise term ε ∼ N(0, σ2In). The elastic net model is defined as
where λ1 and λ2 are tuning parameters that balance the goodness-of-fit and complexity of the model. A model fitting procedure produces the estimate of β, , and the corresponding P value for each regression coefficient of the predictor. Putative causal mutations are predicted on the basis of the regression P value of the corresponding predictor. We fit one model for each gene. Multiple testing is corrected on the basis of the total number of predictors in a cancer type, using the Benjamini-Hochberg method.
Because the EP links are computationally predicted using the IM-PET algorithm, we address the uncertainty of the prediction by using a weighted elastic net approach. Specifically, we associate the probability score of an EP prediction (computed using IM-PET) to the coefficient of the predictor in the model to enforce penalty on predictors caused by uncertainty in EP predictions. The weighted elastic net model is as follows
where wi > 0, j = 1, ⋯, p are weight based on the EP prediction score computed using IM-PET.All fitted models for five cancer types are summarized in a table that is available via the following link: https://chopri.box.com/s/eoibo98p93syx9qkkbbm2fdle5kcajeh.
CRISPR/Cas9-mediated translocation
Single-guide RNAs (sgRNAs) targeting the break points were designed using CRISPOR (http://crispor.tefor.net) and cloned into the CRISPR vector pX459 (Addgene plasmid no. 448139). CRISPR/Cas9-mediated translocation in Ba/F3 cells was performed as described in a previous study (47) with some modifications. Plasmids were cotransfected to Ba/F3 cells by electroporation. Dead cells were removed by centrifugation at 300g and room temperature for 5 min 72 hours after electroporation. Cell concentration and viability were measured using Countess II (Life Technologies). Live cells were resuspended in Ba/F3 conditional medium to a concentration of 5 cells/ml. One hundred microliters of cell suspension was transferred to a 96-well plate and cultured for 2 to 3 weeks for selection of single-cell clones. Genomic DNA was isolated using a Quick-DNA 96 kit (ZYMO) and used for screening for clones with translocation by polymerase chain reaction (PCR) (table S4). Clones with translocation were further confirmed by Sanger sequencing.
Generation of cell lines with stably expressed Cas9 endonuclease
The B-ALL cell lines NALM-6 and REH were lentivirally transduced with plasmid expressing Cas9 nuclease (lentiCRISPR v2; Addgene plasmid no. 52961), as described below at a multiplicity of infection (MOI) of 0.5. Cells then underwent 14 days of antibiotic selection with medium containing puromycin (0.25 μg/ml) to generate cell lines that stably express Cas9. Both cell lines were cultured in RPMI medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml). All cell lines were validated by American Type Culture Collection (ATCC) short tandem repeat (STR) profiling and confirmed to be mycoplasma free.
Lentivirus production and transduction
Lentivirus was produced by transfecting human embryonic kidney (HEK) 293FT cells with packaging plasmids pMD2.G and psPAX2 and the individual CRISPR components [Cas9-expressing plasmid or sgRNA-expressing plasmid: MCB306 (Addgene no. 89360) or MCB320 (Addgene no. 89359)]. Lentivirus-laden supernatant was harvested at 48 and 72 hours after transfection, and viral supernatant was filtered through 0.45-μmpolyvinylidene difluoride filter (Millipore) and concentrated using ultracentrifugation at 25,000 rpm for 2 hours at 4°C. Virus was then titered via transduction of target cell line (REH or NALM-6).
Cells were lentivirally transduced by spinfection in the presence of polybrene (8 μg/ml) (Millipore) at 1000g for 2 hours. Wild-type cells were transduced with virus containing plasmid expressing Cas9 at an MOI of 0.5. Cells stably expressing Cas9 were then transduced with plasmid containing sgRNA targeting CHD4 or nontargeting control at an MOI of 0.4. Three days after virus transduction, green fluorescent protein (GFP)–positive (transduced) cells were sorted by fluorescence-activated cell sorting (FACS) and seeded into six-well plates for recovery before the downstream analyses.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH grants GM104369, GM108716, CA226187, CA233285 (to K.T.), and K08CA184418 (to S.K.T.) and a pilot grant from the Children’s Hospital of Philadelphia Research Institute (to K.T. and S.K.T.). Author contributions: B.H. and K.T. designed the overall study, analyzed data, and wrote paper. P.G. performed most of the experiments. C.-H.C. and Y.-Y.D. performed competition growth assays. C.C., Y.-Y.D., G.C., H.K., S.K.T., and S.P.H. helped with experimental design, data interpretation, and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Data and software used in this study are listed in Materials and Methods and table S1. The PANGEA software is deposited at https://github.com/tanlabcode/PANGEA and publicly available. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba3064/DC1
REFERENCES AND NOTES
- 1.Ma X., Liu Y., Liu Y., Alexandrov L. B., Edmonson M. N., Gawad C., Zhou X., Li Y., Rusch M. C., Easton J., Huether R., Gonzalez-Pena V., Wilkinson M. R., Hermida L. C., Davis S., Sioson E., Pounds S., Cao X., Ries R. E., Wang Z., Chen X., Dong L., Diskin S. J., Smith M. A., Guidry Auvil J. M., Meltzer P. S., Lau C. C., Perlman E. J., Maris J. M., Meshinchi S., Hunger S. P., Gerhard D. S., Zhang J., Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gröbner S. N., Worst B. C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V. A., Johann P. D., Balasubramanian G. P., Segura-Wang M., Brabetz S., Bender S., Hutter B., Sturm D., Pfaff E., Hübschmann D., Zipprich G., Heinold M., Eils J., Lawerenz C., Erkek S., Lambo S., Waszak S., Blattmann C., Borkhardt A., Kuhlen M., Eggert A., Fulda S., Gessler M., Wegert J., Kappler R., Baumhoer D., Burdach S., Kirschner-Schwabe R., Kontny U., Kulozik A. E., Lohmann D., Hettmer S., Eckert C., Bielack S., Nathrath M., Niemeyer C., Richter G. H., Schulte J., Siebert R., Westermann F., Molenaar J. J., Vassal G., Witt H.; ICGC Ped Brain-Seq Project; ICGC MMML-Seq Project, Burkhardt B., Kratz C. P., Witt O., van Tilburg C. M., Kramm C. M., Fleischhack G., Dirksen U., Rutkowski S., Frühwald M., von Hoff K., Wolf S., Klingebiel T., Koscielniak E., Landgraf P., Koster J., Resnick A. C., Zhang J., Liu Y., Zhou X., Waanders A. J., Zwijnenburg D. A., Raman P., Brors B., Weber U. D., Northcott P. A., Pajtler K. W., Kool M., Piro R. M., Korbel J. O., Schlesner M., Eils R., Jones D. T. W., Lichter P., Chavez L., Zapatka M., Pfister S. M., The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Lorbeer F. K., Hockemeyer D., TERT promoter mutations and telomeres during tumorigenesis. Curr. Opin. Genet. Dev. 60, 56–62 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Oldridge D. A., Wood A. C., Weichert-Leahey N., Crimmins I., Sussman R., Winter C., McDaniel L. D., Diamond M., Hart L. S., Zhu S., Durbin A. D., Abraham B. J., Anders L., Tian L., Zhang S., Wei J. S., Khan J., Bramlett K., Rahman N., Capasso M., Iolascon A., Gerhard D. S., Guidry Auvil J. M., Young R. A., Hakonarson H., Diskin S. J., Thomas Look A., Maris J. M., Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 528, 418–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansour M. R., Abraham B. J., Anders L., Berezovskaya A., Gutierrez A., Durbin A. D., Etchin J., Lawton L., Sallan S. E., Silverman L. B., Loh M. L., Hunger S. P., Sanda T., Young R. A., Look A. T., Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Cancer Genome Consortium , International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Northcott P. A., Lee C., Zichner T., Stütz A. M., Erkek S., Kawauchi D., Shih D. J. H., Hovestadt V., Zapatka M., Sturm D., Jones D. T. W., Kool M., Remke M., Cavalli F. M. G., Zuyderduyn S., Bader G. D., VandenBerg S., Esparza L. A., Ryzhova M., Wang W., Wittmann A., Stark S., Sieber L., Seker-Cin H., Linke L., Kratochwil F., Jäger N., Buchhalter I., Imbusch C. D., Zipprich G., Raeder B., Schmidt S., Diessl N., Wolf S., Wiemann S., Brors B., Lawerenz C., Eils J., Warnatz H.-J., Risch T., Yaspo M.-L., Weber U. D., Bartholomae C. C., von Kalle C., Turányi E., Hauser P., Sanden E., Darabi A., Siesjö P., Sterba J., Zitterbart K., Sumerauer D., van Sluis P., Versteeg R., Volckmann R., Koster J., Schuhmann M. U., Ebinger M., Grimes H. L., Robinson G. W., Gajjar A., Mynarek M., von Hoff K., Rutkowski S., Pietsch T., Scheurlen W., Felsberg J., Reifenberger G., Kulozik A. E., von Deimling A., Witt O., Eils R., Gilbertson R. J., Korshunov A., Taylor M. D., Lichter P., Korbel J. O., Wechsler-Reya R. J., Pfister S. M., Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan R. J. H., Drier Y., Whitton H., Cotton M. J., Kaur J., Issner R., Gillespie S., Epstein C. B., Nardi V., Sohani A. R., Hochberg E. P., Bernstein B. E., Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov. 5, 1058–1071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tasian S. K., Loh M. L., Understanding the biology of CRLF2-overexpressing acute lymphoblastic leukemia. Crit. Rev. Oncog. 16, 13–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zook J. M., Chapman B., Wang J., Mittelman D., Hofmann O., Hide W., Salit M., Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Perry J. A., Kiezun A., Tonzi P., Van Allen E. M., Carter S. L., Baca S. C., Cowley G. S., Bhatt A. S., Rheinbay E., Pedamallu C. S., Helman E., Taylor-Weiner A., McKenna A., DeLuca D. S., Lawrence M. S., Ambrogio L., Sougnez C., Sivachenko A., Walensky L. D., Wagle N., Mora J., de Torres C., Lavarino C., Dos Santos Aguiar S., Yunes J. A., Brandalise S. R., Mercado-Celis G. E., Melendez-Zajgla J., Cárdenas-Cardós R., Velasco-Hidalgo L., Roberts C. W. M., Garraway L. A., Rodriguez-Galindo C., Gabriel S. B., Lander E. S., Golub T. R., Orkin S. H., Getz G., Janeway K. A., Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. U.S.A. 111, E5564–E5573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts K. G., Li Y., Payne-Turner D., Harvey R. C., Yang Y.-L., Pei D., McCastlain K., Ding L., Lu C., Song G., Ma J., Becksfort J., Rusch M., Chen S.-C., Easton J., Cheng J., Boggs K., Santiago-Morales N., Iacobucci I., Fulton R. S., Wen J., Valentine M., Cheng C., Paugh S. W., Devidas M., Chen I.-M., Reshmi S., Smith A., Hedlund E., Gupta P., Nagahawatte P., Wu G., Chen X., Yergeau D., Vadodaria B., Mulder H., Winick N. J., Larsen E. C., Carroll W. L., Heerema N. A., Carroll A. J., Grayson G., Tasian S. K., Moore A. S., Keller F., Frei-Jones M., Whitlock J. A., Raetz E. A., White D. L., Hughes T. P., Guidry Auvil J. M., Smith M. A., Marcucci G., Bloomfield C. D., Mrózek K., Kohlschmidt J., Stock W., Kornblau S. M., Konopleva M., Paietta E., Pui C.-H., Jeha S., Relling M. V., Evans W. E., Gerhard D. S., Gastier-Foster J. M., Mardis E., Wilson R. K., Loh M. L., Downing J. R., Hunger S. P., Willman C. L., Zhang J., Mullighan C. G., Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 371, 1005–1015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Firpi H. A., Ucar D., Tan K., Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 26, 1579–1586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He B., Chen C., Teng L., Tan K., Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. U.S.A. 111, E2191–E2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan A., Zhang X., dbSUPER: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44, D164–D171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drier Y., Cotton M. J., Williamson K. E., Gillespie S. M., Ryan R. J. H., Kluk M. J., Carey C. D., Rodig S. J., Sholl L. M., Afrogheh A. H., Faquin W. C., Queimado L., Qi J., Wick M. J., El-Naggar A. K., Bradner J. E., Moskaluk C. A., Aster J. C., Knoechel B., Bernstein B. E., An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 48, 265–272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peifer M., Hertwig F., Roels F., Dreidax D., Gartlgruber M., Menon R., Krämer A., Roncaioli J. L., Sand F., Heuckmann J. M., Ikram F., Schmidt R., Ackermann S., Engesser A., Kahlert Y., Vogel W., Altmüller J., Nürnberg P., Thierry-Mieg J., Thierry-Mieg D., Mariappan A., Heynck S., Mariotti E., Henrich K.-O., Gloeckner C., Bosco G., Leuschner I., Schweiger M. R., Savelyeva L., Watkins S. C., Shao C., Bell E., Höfer T., Achter V., Lang U., Theissen J., Volland R., Saadati M., Eggert A., de Wilde B., Berthold F., Peng Z., Zhao C., Shi L., Ortmann M., Büttner R., Perner S., Hero B., Schramm A., Schulte J. H., Herrmann C., O’Sullivan R. J., Westermann F., Thomas R. K., Fischer M., Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian M., Zhang H., Kham S. K.-Y., Liu S., Jiang C., Zhao X., Lu Y., Goodings C., Lin T.-N., Zhang R., Moriyama T., Yin Z., Li Z., Quah T. C., Ariffin H., Tan A. M., Shen S., Bhojwani D., Hu S., Chen S., Zheng H., Pui C.-H., Yeoh A. E.-J., Yang J. J., Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 27, 185–195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander T. B., Gu Z., Iacobucci I., Dickerson K., Choi J. K., Xu B., Payne-Turner D., Yoshihara H., Loh M. L., Horan J., Buldini B., Basso G., Elitzur S., de Haas V., Zwaan C. M., Yeoh A., Reinhardt D., Tomizawa D., Kiyokawa N., Lammens T., de Moerloose B., Catchpoole D., Hori H., Moorman A., Moore A. S., Hrusak O., Meshinchi S., Orgel E., Devidas M., Borowitz M., Wood B., Heerema N. A., Carrol A., Yang Y.-L., Smith M. A., Davidsen T. M., Hermida L. C., Gesuwan P., Marra M. A., Ma Y., Mungall A. J., Moore R. A., Jones S. J. M., Valentine M., Janke L. J., Rubnitz J. E., Pui C.-H., Ding L., Liu Y., Zhang J., Nichols K. E., Downing J. R., Cao X., Shi L., Pounds S., Newman S., Pei D., Guidry Auvil J. M., Gerhard D. S., Hunger S. P., Inaba H., Mullighan C. G., The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562, 373–379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dege C., Hagman J., Mi-2/NuRD chromatin remodeling complexes regulate B and T-lymphocyte development and function. Immunol. Rev. 261, 126–140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sperlazza J., Rahmani M., Beckta J., Aust M., Hawkins E., Wang S. Z., Zu Zhu S., Podder S., Dumur C., Archer K., Grant S., Ginder G. D., Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. Blood 126, 1462–1472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pugh T. J., Morozova O., Attiyeh E. F., Asgharzadeh S., Wei J. S., Auclair D., Carter S. L., Cibulskis K., Hanna M., Kiezun A., Kim J., Lawrence M. S., Lichenstein L., McKenna A., Pedamallu C. S., Ramos A. H., Shefler E., Sivachenko A., Sougnez C., Stewart C., Ally A., Birol I., Chiu R., Corbett R. D., Hirst M., Jackman S. D., Kamoh B., Khodabakshi A. H., Krzywinski M., Lo A., Moore R. A., Mungall K. L., Qian J., Tam A., Thiessen N., Zhao Y., Cole K. A., Diamond M., Diskin S. J., Mosse Y. P., Wood A. C., Ji L., Sposto R., Badgett T., London W. B., Moyer Y., Gastier-Foster J. M., Smith M. A., Auvil J. M. G., Gerhard D. S., Hogarty M. D., Jones S. J. M., Lander E. S., Gabriel S. B., Getz G., Seeger R. C., Khan J., Marra M. A., Meyerson M., Maris J. M., The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuang L., Ma Y., Wang Q., Zhang J., Zhu C., Zhang L., Xu X., Atg3 overexpression enhances bortezomib-induced cell death in SKM-1 cell. PLOS ONE 11, e0158761 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moignard V., Macaulay I. C., Swiers G., Buettner F., Schütte J., Calero-Nieto F. J., Kinston S., Joshi A., Hannah R., Theis F. J., Jacobsen S. E., de Bruijn M. F., Göttgens B., Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat. Cell Biol. 15, 363–372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vassen L., Beauchemin H., Lemsaddek W., Krongold J., Trudel M., Möröy T., Growth factor independence 1b (gfi1b) is important for the maturation of erythroid cells and the regulation of embryonic globin expression. PLOS ONE 9, e96636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thivakaran A., Botezatu L., Hönes J. M., Schütte J., Vassen L., Al-Matary Y. S., Patnana P., Zeller A., Heuser M., Thol F., Gabdoulline R., Olberding N., Frank D., Suslo M., Köster R., Lennartz K., Görgens A., Giebel B., Opalka B., Dührsen U., Khandanpour C., Gfi1b: A key player in the genesis and maintenance of acute myeloid leukemia and myelodysplastic syndrome. Haematologica 103, 614–625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hnisz D., Abraham B. J., Lee T. I., Lau A., Saint-André V., Sigova A. A., Hoke H. A., Young R. A., Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damm F., Thol F., Hollink I., Zimmermann M., Reinhardt K., van den Heuvel-Eibrink M. M., Zwaan C. M., de Haas V., Creutzig U., Klusmann J.-H., Krauter J., Heuser M., Ganser A., Reinhardt D., Thiede C., Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: A study of the AML-BFM and DCOG study groups. Leukemia 25, 1704–1710 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Patel J. L., Schumacher J. A., Frizzell K., Sorrells S., Shen W., Clayton A., Jattani R., Kelley T. W., Coexisting and cooperating mutations in NPM1-mutated acute myeloid leukemia. Leuk. Res. 56, 7–12 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Gröschel S., Sanders M. A., Hoogenboezem R., de Wit E., Bouwman B. A. M., Erpelinck C., van der Velden V. H. J., Havermans M., Avellino R., van Lom K., Rombouts E. J., van Duin M., Döhner K., Beverloo H. B., Bradner J. E., Döhner H., Löwenberg B., Valk P. J. M., Bindels E. M. J., de Laat W., Delwel R., A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Sima J., Gilbert D. M., Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 25, 93–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tasian S. K., Hunger S. P., Genomic characterization of paediatric acute lymphoblastic leukaemia: An opportunity for precision medicine therapeutics. Br. J. Haematol. 176, 867–882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieraccioli M., Nicolai S., Pitolli C., Agostini M., Antonov A., Malewicz M., Knight R. A., Raschellà G., Melino G., ZNF281 inhibits neuronal differentiation and is a prognostic marker for neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 115, 7356–7361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dzieran J., Rodriguez Garcia A., Westermark U. K., Henley A. B., Eyre Sánchez E., Träger C., Johansson H. J., Lehtiö J., Arsenian-Henriksson M., MYCN-amplified neuroblastoma maintains an aggressive and undifferentiated phenotype by deregulation of estrogen and NGF signaling. Proc. Natl. Acad. Sci. U.S.A. 115, E1229–E1238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faber Z. J., Chen X., Gedman A. L., Boggs K., Cheng J., Ma J., Radtke I., Chao J.-R., Walsh M. P., Song G., Andersson A. K., Dang J., Dong L., Liu Y., Huether R., Cai Z., Mulder H., Wu G., Edmonson M., Rusch M., Qu C., Li Y., Vadodaria B., Wang J., Hedlund E., Cao X., Yergeau D., Nakitandwe J., Pounds S. B., Shurtleff S., Fulton R. S., Fulton L. L., Easton J., Parganas E., Pui C.-H., Rubnitz J. E., Ding L., Mardis E. R., Wilson R. K., Gruber T. A., Mullighan C. G., Schlenk R. F., Paschka P., Döhner K., Döhner H., Bullinger L., Zhang J., Klco J. M., Downing J. R., The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 48, 1551–1556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmlund T., Lindberg M. J., Grander D., Wallberg A. E., GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia 27, 578–585 (2013). [DOI] [PubMed] [Google Scholar]
- 37.De Cecco L., Capaia M., Zupo S., Cutrona G., Matis S., Brizzolara A., Orengo A. M., Croce M., Marchesi E., Ferrarini M., Canevari S., Ferrini S., Interleukin 21 controls mRNA and microRNA expression in CD40-activated chronic lymphocytic leukemia cells. PLOS ONE 10, e0134706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izycka-Swieszewska E., Wozniak A., Kot J., Grajkowska W., Balcerska A., Perek D., Dembowska-Baginska B., Klepacka T., Drozynska E., Prognostic significance of HER2 expression in neuroblastic tumors. Modern Pathol. 23, 1261–1268 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Thomas H., Beck K., Adamczyk M., Aeschlimann P., Langley M., Oita R. C., Thiebach L., Hils M., Aeschlimann D., Transglutaminase 6: A protein associated with central nervous system development and motor function. Amino Acids 44, 161–177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Algarni A. S., Hargreaves A. J., Dickenson J. M., Activation of transglutaminase 2 by nerve growth factor in differentiating neuroblastoma cells: A role in cell survival and neurite outgrowth. Eur. J. Pharmacol. 820, 113–129 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Stransky N., Cerami E., Schalm S., Kim J. L., Lengauer C., The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Sun Y., Sun Y., Meng L., Xu X., First case of AML with rare chromosome translocations: A case report of twins. BMC Cancer 18, 458 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu G., Ma Z., Hu W., Wang D., Gong B., Fan C., Jiang S., Li T., Gao J., Yang Y., Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 8, e2700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L., Luquette L. J., Gehlenborg N., Xi R., Haseley P. S., Hsieh C.-H., Zhang C., Ren X., Protopopov A., Chin L., Kucherlapati R., Lee C., Park P. J., Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153, 919–929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janoueix-Lerosey I., Hupé P., Maciorowski Z., La Rosa P., Schleiermacher G., Pierron G., Liva S., Barillot E., Delattre O., Preferential occurrence of chromosome breakpoints within early replicating regions in neuroblastoma. Cell Cycle 4, 1842–1846 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Kim P., Cheng F., Zhao J., Zhao Z., ccmGDB: A database for cancer cell metabolism genes. Nucleic Acids Res. 44, D959–D968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torres R., Martin M. C., Garcia A., Cigudosa J. C., Ramirez J. C., Rodriguez-Perales S., Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 5, 3964 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Weirauch M. T., Yang A., Albu M., Cote A. G., Montenegro-Montero A., Drewe P., Najafabadi H. S., Lambert S. A., Mann I., Cook K., Zheng H., Goity A., van Bakel H., Lozano J.-C., Galli M., Lewsey M. G., Huang E., Mukherjee T., Chen X., Reece-Hoyes J. S., Govindarajan S., Shaulsky G., Walhout A. J. M., Bouget F.-Y., Ratsch G., Larrondo L. F., Ecker J. R., Hughes T. R., Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang W., Bojorquez-Gomez A., Velez D. O., Xu G., Sanchez K. S., Shen J. P., Chen K., Licon K., Melton C., Olson K. M., Yu M. K., Huang J. K., Carter H., Farley E. K., Snyder M., Fraley S. I., Kreisberg J. F., Ideker T., A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat. Genet. 50, 613–620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weddington N., Stuy A., Hiratani I., Ryba T., Yokochi T., Gilbert D. M., ReplicationDomain: A visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics 9, 530 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansen R. S., Thomas S., Sandstrom R., Canfield T. K., Thurman R. E., Weaver M., Dorschner M. O., Gartler S. M., Stamatoyannopoulos J. A., Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U.S.A. 107, 139–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grant C. E., Bailey T. L., Noble W. S., FIMO: Scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leiserson M. D. M., Wu H.-T., Vandin F., Raphael B. J., CoMEt: A statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 16, 160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juven-Gershon T., Cheng S., Kadonaga J. T., Rational design of a super core promoter that enhances gene expression. Nat. Methods 3, 917–922 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doench J. G., Fusi N., Sullender M., Hegde M., Vaimberg E. W., Donovan K. F., Smith I., Tothova Z., Wilen C., Orchard R., Virgin H. W., Listgarten J., Root D. E., Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaba3064/DC1