

Abstract
Diabetic heart disease (DHD) can be classified as a primary consequence from several pathophysiological manifestation of diabetes mellitus (DM) on cardiac tissues or secondarily in extracardiac tissues and is encountered as either primary or secondary complications of DM. Endothelitis is inflammation of the vascular endothelium and is likely to be seen in the majority of patients who start to manifest an end organ complication of DM in this case DHD. Diabetes is a leading cause for many cardiovascular syndromes and diseases including congestive heart failure (CHF) however much remains unknown about the transition from diagnosed DM to clinical state and the contribution of the various mechanical and counterregulatory systems in the manifested complaint. Diastolic heart failure or heart failure with preserved ejection fraction (DHF/HFpEF), accounts for half of all CHF presentations, has DM as a major contributor, however, there remain large gaps in clinical and pathophysiological understanding. This review aims to explore the microscopic aspects in diabetic endothelitis and provide a clinical link to with context to HFpEF.
Keywords: Cardiovascular disease, Diabetic heart disease, Diabetes mellitus, Diastolic heart failure, Endothelitis, Heart failure with preserved ejection fraction, Inflammation
Core tip: We discuss the concept of diabetes mellitus and inflammation in the endothelium of blood vessels or “diabetic endothelitis”. The vascular endothelium permeates every organ in the body. Macro and microvascular inflammation in coronary and related arterial beds contributes to diabetic heart diseases such as congestive heart failure. Heart failure with preserved ejection fraction is an important and poorly understood condition. In this review we provide a basic science perspective and a clinical link to this problem.
INTRODUCTION
Basic sciences are the doorway for forensic analysis of clinical syndromes. Since the early community heart studies such as Framingham Heart Studies two clear delineations of congestive heart failure were observed, systolic heart failure where all cases had diastolic impairment and isolated heart failure where systolic function appeared preserved from all available diagnostic tools hence diastolic heart failure or Heart Failure with preserved Ejection Fraction (DHF/ HFpEF)[1]. The former is well studied however the latter still struggles from a fixed definition, aetiology, pathophysiology and diagnostic taxonomy and a single proven prognostic therapy[2]. The bench will play a critical role in understanding this syndrome. Diabetes Mellitus (DM) and its common end organ complication of ”endothelitis” are seen in all diabetic heart disease (DHD) and is a valid area to focus to identify a bedside link for HFpEF.
The vascular endothelium, that forms the lining of all blood and lymphatic vessels, is uniquely vulnerable to the effects of chronic or intermittent hyperglycaemia[3]. Being largely dependent on glycolytic metabolism for generating adenosine triphosphate rather than oxidative phosphorylation, the uptake of glucose into endothelial cells is not downregulated as ambient glucose levels rise. This means that increasing glycolytic flux increasingly generates toxic intermediates including reactive dicarbonyls and reactive oxygen species (ROS). This glucotoxicity, along with the additional impacts of lipotoxicity, endoplasmic reticulum (ER) stress, inflammasome activation, oxidative and shear stress in diabetes induce pathophysiological changes in the vascular endothelium that are best characterised as “endothel-it is”. These changes include increased adhesion and extravasation of leucocytes, production of chemokines/cytokines, exudation of plasma, altered vasomotor tone and haemostasis, endothelial senescence and apoptosis, endothelial to mesenchymal transition (EndoMT) and neo-angiogenesis that contribute not only to accelerated atherosclerosis but also the development of progression of heart failure in diabetes[3-6] (Figure 1). Moreover, although originally considered a consequence of hypertrophy and overload, HFpEF is increasingly viewed as a “microvascular” disorder driven by endothelitis[7]. In this paper, we explore the key inflammatory changes in the vascular endothelium and their potential role in DHD. We also provide a short hypothetical perspective of a contextual bedside translational strategy to advance a clinical focus for diabetic endothelitis (“diabetic endothelitis” is a term the authors use to describe endothelial injury associated with the chronic inflammatory milieu of diabetes. The exacts proponents of the injury and its manifestations are the subject of this paper and ongoing works. Conventional vascular inflammation often associated with connective tissue diseases are well described. The endothelium itself is a component of the vasculature, is less describe in that sense. When endothelial function is altered the term “endothelial dysfunction” is used.).
Figure 1.

Endothelitis is associated with a range of dysfunctional changes that contribute the development and progression of cardiovascular disease. EndoMT: Endothelial-mesenchymal transition; EPC: Endothelial progenitor cell.
CELLULAR BASIS OF DIABETIC INFLAMMATION IN THE ENDOTHELIUM AND DYSFUNCTION
Endothelitis, leukocyte recruitment and infiltration
Activation of the vascular endothelium plays a key initiating role in the leucocyte adhesion and the subsequent development of the nascent atherosclerotic plaque. Intrinsically, monolayer of endothelial cells forms a critical interface between circulating immune cells and the tissues of the body. The luminal expression of chemokines, like macrophage chemoattractant protein (MCP-1), attracts leucocytes, which then roll, arrest and bind to an activated endothelium expressing adhesion molecules, including selectins, intercellular adhesion molecule (ICAM)-1, and vascular cell adhesion molecule (VCAM)-1, Junctional Adhesion Molecules (JAMs), P-selectin and E-selectin, and subsequently migrate into the sub-epithelium and beyond. Subepithelial macrophages become engorged with cholesterol creating lipid-laden macrophages known as foam cells, creating fatty streaks and eventually the necrotic core of atheromatous plaques after their cellular death. In diabetes, the rate at which circulating monocytes enter the atherosclerotic lesion is increased, while plaque regression is reduced[8]. The increased transit of activated inflammatory cells into the vessel wall in diabetes, not only leads to more atherosclerosis, but as a result, atherosclerosis is more complex, multivessel with more unstable plaque in diabetes. Diabetic heart failure in diabetes is also characterised by a leukocytic infiltration, including activated monocytes, T-cells and dendritic cells[9,10]. As with atherosclerosis, these cells largely originate in the bloodstream and therefore must bind to and cross an activated endothelium expressing adhesion molecules to reach the heart in a targeted way.
Endothelitis and endothelial dysfunction
Endothelial dysfunction is one end-result of the phenotypic changes associated with endothelitis. Although there are very many dysfunctional microvascular changes associated with endothelitis, the best characterised is an impairment of endothelium - dependent nitric oxide (NO)- mediated vasodilatation. In healthy vessels, increased shear stress triggers flow-mediated vasodilatation due to increased synthesis and release of NO, the principal regulator of vascular tone, that acts on underlying smooth muscle to relax blood vessels. In patients with diabetes, vaso-relaxation is significantly impaired or even paradoxically reversed. This is partly due to impaired formation of NO due to uncoupling of endothelial nitric oxide synthase and a decrease in tetrahydrobiopterin and L-arginine, the co-factor and the substrate, respectively, for NO synthesis. At the same time, the bioavailability of NO is reduced due to quenching by ROS and reactive dicarbonyls. In addition, elevated levels of asymmetric dimethylarginine, function as an endogenous competitive inhibitor of NO activity.
Endothelial dysfunction can be measured using invasive tests by selective infusion of acetylcholine into the epicardial coronary arteries. However, it is more commonly estimated using non-invasive testing, including flow-mediated dilation (FMD), low-flow-mediated constriction of the brachial artery measured by ultrasound, and peripheral arterial tonometry (EndoPAT) using the finger pulse wave amplitude in response to reactive hyperaemia. More recently, changes in endothelial-mediated blood flow can be identified using monitored by positron emission tomography. Each of these non-invasive are correlated with the results of invasive testing as well as future cardiac outcomes. However, each phenomenon is only partly NO determined. Moreover, none have a clear place in guiding treatment or prognosis in the clinic.
Compromised endothelium - dependent vasodilatation is thought to be an important contributor to increased myocardial ischemia in diabetes. In particular, myocardial ischemia in individuals with relatively normal epicardial coronary arteries (also known as microvascular angina) appears to be partly driven by impaired endothelium-mediated vasomotion in the cardiac microvasculature. For example, 10-year data from the Women's Ischemia Syndrome Evaluation (WISE) study, found that half of individuals undergoing angiography for investigation of chest pain, but found not to have obstructive coronary disease, had endothelial dysfunction[11], and those that did had worse clinical outcomes. This is not simply an epiphenomenon, as abnormal vasomotion may also be observed before the development of atherosclerotic lesions or overt cardiovascular disease. For example, in individuals at moderate CV risk but without macrovascular disease, endothelial dysfunction is associated with an increased incidence of CV events[12].
The role of microvascular ischemia/hypoxia in the pathogenesis of diabetic heart failure remains controversial. While there is a clear association between endothelial dysfunction and diastolic dysfunction in heart disease, this may simply be because of common risk factors (e.g., diabetes) rather than a linked pathogenesis. Certainly, reduced oxygen delivery impairs myocyte relaxation, especially in the setting of increased oxygen demands associated with increased intramyocardial tension. Chronic ischemia may lead to subclinical micro-scars replacing small foci of dead cardiomyocytes, that cumulatively result in myocardial stiffening. Endothelium-mediated control of the venous vascular tone is also important for the regulation of cardiac filling pressures.
CONSEQUENCES OF ENDOTHELITIS
Endothelitis and increased vascular permeability
The best-known pathophysiological change associated with microvascular dysfunction in diabetes is the accompanying increase in vascular permeability. The healthy endothelium provides an effective semipermeable barrier that prevents the exudation of plasma contents despite high intravascular pressures. In hyperglycaemia, increased permeability and loss of endothelial barrier function, results in extravasation of circulating elements (e.g., large molecular weight proteins, lipoprotein particles, clotting factors) into the interstitial space and tissue oedema. This is simply observed as hard exudates in the retina or with increased albuminuria, both of which are strongly associated with and increased risk of cardiovascular events and heart failure. This is because impaired barrier functions in one site predict endothelial dysfunction at other sites (the so called “Steno hypothesis”).
Increased vascular permeability in diabetes is likely the functional end result of multiple pathophysiological changes in the endothelium including thinning and changes in the composition of the surface glycocalyx, rearrangements of cell-to-cell junctions (paracellular trafficking) and altered vesicular trafficking (transcytosis) associated with endothelitis. One of the most important regulators of barrier function is considered to be Vascular Endothelial Growth Factor (VEGF). The induction of VEGF in diabetes directly increases vascular permeability[13]. Outside of diabetes (e.g., in sepsis), loss of barrier function may also be partly VEGF-dependent.
Endothelitis, thrombo-resistance and fibrinolysis
A healthy endothelium creating an anticoagulant surface for blood flow that inhibits the formation of intraluminal clots (known as thromboresistance). By contrast, an inflamed endothelium is thrombogenic in a number of different ways. For example, endothelial dysfunction is associated with a reduction in expression of the membrane bound anticoagulant glycoprotein, thrombomodulin. At the same time, the release of soluble thrombomodulin is increased, leading to increased circulating levels in patients with diabetes, especially those with vascular complications. Indeed, soluble thrombomodulin levels in diabetes closely correlate with other markers of endothelitis, including circulating cytokines, oxidative stress markers, vascular permeability (e.g., albuminuria, retinal exudates) and impaired FMD. At the same time, an inflamed endothelium also liberates thrombogenic molecules including plasma factor VIII, von Willebrand factor, fibronectin, inhibitor of plasminogen activator type 1, and thrombospondin. Platelet aggregation is also enhanced in diabetes. Although a healthy endothelium produces anti-aggregants, including such as NO and PGI2, to attenuate this process as a negative feedback mechanism. However, both are reduced in the setting of endothelitis.
Endothelitis and vascular rarefaction
Compounding the obvious tissue hypoxia associated with diabetes, there is often a reduction in microvascular density (known as rarefaction or capillary dropout) that is triggered by endothelitis[14]. This phenomenon has been best described in the diabetic retina, where rarefaction and pericyte dropout is thought to be a key driver of tissue hypoxia and subsequent problematic neo-angiogenesis. However, in the diabetic heart as well, endothelitis also leads to capillary rarefaction, associated with decreased contractility and increased left ventricular wall stiffness[8]. Autopsy studies in individuals specifically with heart failure also demonstrate reduced microvascular density in the heart alongside more severe fibrosis when compared to healthy controls[15,16]. Equally in hypertension, the density of microvascular networks in the heart is reduced[17]. In each case rarefaction is thought to be partly driven by inflammatory changes that lead to an imbalance between inadequate angiogenesis/regeneration and augmented vascular destruction/regression due to apoptosis and/or senescence.
Endothelial senescence is an irreversible phenotypic transition that leads to cell-cycle arrest. It can be initiated by inflammation (e.g., activation of NF-κB) as well as result in a pro-inflammatory pro-atherosclerotic, and prothrombotic state characterised by increased cytokine production and expression of adhesion molecules (i.e., endothelitis) which in turn leads to even more senescence[18]. Exposure to high glucose levels also triggers endothelial senescence[13]. This is probably mediated on a number of levels including the induction of cellular/DNA damage by reactive intermediates (e.g., ROS, dicarbonyls), telomere shortening, mitochondrial damage, replicative cell turnover, and epigenetic reprogramming including the persistent activation of activation of NF-κB[19].
Endothelial senescence is thought to contribute to age-related vascular dysfunction and myocardial hypoxia partly by impairing the repair capacity of the endothelial lining, leading ultimately to microvascular rarefaction. Inflammatory changes associated with senescence also potentially contribute to atherosclerosis, thrombophilia and plaque instability. In addition, recent studies also suggest endothelial senescence may be involved in heart failure. For example, in an accelerated model of ageing triggered by augmented endothelial senescence, both diastolic and endothelial dysfunction are also increased[20]. Moreover, knocking-out endothelial p53 prevents endothelial senescence as well as myocardial fibrosis induced following pressure overload in mice[21].
Senescence ultimately ends with programmed cell death (apoptosis) of endothelial cells. Whether this is directly triggered by senescence or indirectly due to exposure to inflammation and oxidative damage in senescent cells is unclear. Of course, apoptosis can also be triggered independent of senescence, particularly associated with endothelitis triggered by glucotoxicity, shear stress, angiotensin II, cholesterol and oxidative stress. Increased endothelial cell apoptosis is observed in the endothelium covering unstable atherosclerotic plaques and in atherosclerosis-prone regions of the vasculature[16]. Apoptosis not only leads to disruption of the endothelial barrier with leak of plasma proteins and exposure of a thrombogenic subendothelial matrix. Apoptotic endothelial cells are themselves pro-coagulant and pro-adhesive for leukocytes and platelets. Moreover, products released from apoptotic cells including DNA, ATP, microRNA both free and within micro-vesicles derived from cell fragmentation communicate endothelial dysfunction to other cells[22]. Quantitative measurement of these microparticles may be a useful biomarker for endothelial damage, as well as their response to treatment.
ENDOTHELIUM, MYOCARDIUM AND VASCULATURE
Endothelitis and EndoMT
The central players in cardiac fibrogenesis are myofibroblasts (Figure 2). These can be derived from the activation of local fibroblasts and the recruitment of circulating precursors. In addition, inflamed endothelial cells are able to transform to become myofibroblasts via a process known as endothelial-mesenchymal transition (EndoMT)[23]. Indeed, to 27%-35 % of the total pool of cardiac fibroblasts during cardiac fibrosis may be derived from endothelial cells[24]. This TGF-β1-dependent transformation and their subsequent detachment and migration into the cardiac interstitium leads to the generation of increased amounts of extracellular matrix proteins, contributing to diastolic stiffening. In addition, products released from active myofibroblasts have paracrine actions on cardiomyocyte to induce apoptosis[25]. Augmented EndoMT has been documented in the diabetic kidney and retina, where they contribute to increased tissue fibrosis[26,27]. Little is known about the role of endoMT in the diabetic heart although the same factors that promote endoMT at other sites (e.g., TGF β-Smad3) are also increased in the diabetic myocardium as the content of cardiac myofibroblasts is also increased.
Figure 2.
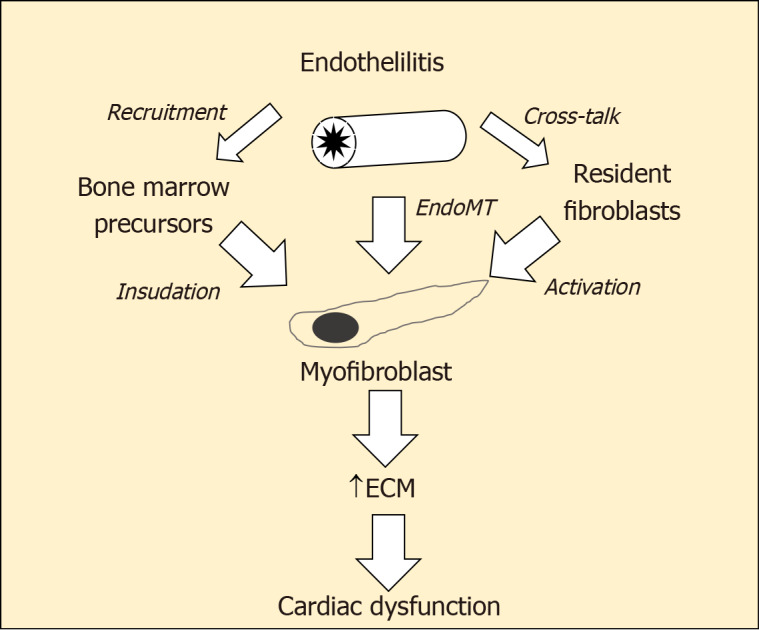
Myofibroblasts the central players in cardiac fibrogenesis. ECM: Extracellular matrix; EndoMT: Endothelial-mesenchymal transition; EPC: Endothelial progenitor cell.
Endothelitis and endothelial progenitor cells
Circulating endothelial progenitor cells (EPCs) normally participate in the process of new blood vessel formation and vascular repair. In the setting of cardiac ischaemia, the mobilisation of EPCs is thought to contribute to optimal remodelling. Elevated number and the function of EPCs is generally correlated with better endothelial function. Indeed, EPCs have been suggested as useful additional marker of endothelial function. In diabetes, functionality of EPCs is weakened, including impaired migration, mobilization, adhesion and homing, reduced ability to proliferate, differentiate and survive, leading to reduced reparative capacity. In addition, EPCs numbers are paradoxically normal or sometime s reduced. In particular, EPC numbers also fall in patients with diabetic kidney disease, especially with increasing albuminuria or reducing renal function.
Cross talk between endothelium and cardiomyocytes
The endothelium not only regulates and redistributes regional blood flow. Endothelial cells also have significant paracrine effects to regulate regional function. For example, it is well established that vascular smooth muscle function is modified through the altered production of vasoactive substances by endothelial cells (e.g., NO). In addition, in the heart, diffusible factors released from the coronary and endocardial endothelium also modulate underlying cardiomyocyte contractility and remodelling. These factors include NO, prostaglandins, endothelins, interleukins, and other “angiocrine” small-molecules[28]. For example, NO released from the endothelium stimulates an earlier onset of relaxation favouring diastolic filling[29]. In co-culture experiments, endothelial cells modulate the contraction and relaxation of cardiomyocytes, and this is substantially altered by the induction of endothelial inflammation (e.g., pre-exposure to TNF-α or interleukin-1β). Consequently, the impact of impaired NO generation associated with endothelial dysfunction extends beyond simply changes in local blood flow to real-time cardiac dynamics.
The paracrine signals emerging from endothelitis also alters the perivascular environment to activate pathways that ultimately converge on converge on myocardial fibrosis. A quiescent endothelium releases factors that suppress fibrogenesis including bone morphogenetic protein 9 (BMP9), while an activated endothelium releases pro-fibrotic cytokines including TGF-β and IL-33, MCP-1, endothelin-1 (ET-1). In addition, paracrine and autocrine signalling of the renin-angiotensin-aldosterone system (RAAS), and its primary mediator Ang II, are known to have a direct influence on the progression of the atherosclerotic process[30], reactive ventricular hypertrophy, and myocardial fibrosis. Diabetes is classically associated with activation of the cardiac RAAS, at least partly through the development of endothelitis. Moreover, blockade of the RAAS in the setting of diabetes, using ACE inhibitors angiotensin receptor blockers or mineralocorticoid receptor blockade, has clear effects on vascular remodelling and myocardial fibrosis, over and above their effects on blood pressure.
The beating heart has only a limited capacity for energy substrate synthesis or storage, relying instead on blood flow and transit across the endothelium for its energy needs. Changes in metabolic status (e.g., feeding or fasting state) and therefore optimal substrate utilisation are communicated both to and from the vascular endothelium. Beyond regulating blood flow and the trans-endothelial supply of metabolic substrates, paracrine signals from the endothelium also have a major impact on cardiac metabolism, including NO, insulinotropic factors, growth factors and enzymes[4]. In addition, endothelial lipase (EL) activity appears to play an important role in liberating free fatty acids from high density lipoproteins to be used in cardiac metabolism, as genetic depletion of endothelial lipase results in heart failure due to reduced fatty acid uptake in the heart[31]. In cardiac hypertrophy, EL activity is increased, possibly to facilitate increased fat supply to the myocardium. Serum EL concentrations in human plasma are associated with circulating inflammatory markers, while plasma levels are increased in diabetes, heart failure and atherosclerosis. This increase in circulating EL may partly reflect the loss from endothelial sites associated with endothelitis that modulates cardiac metabolism and contributes in the long term to dysfunction.
CONNECTING THE DOTS FROM THE LAB TO THE CLINIC PATIENT A FOCUS ON HFPEF
HFpEF is an evolving syndrome where observations have been predominately clinical and some pathophysiological connect. In the transition from Diabetes to HFpEF, knowing the canvas is large and will advance, we highlight 5 areas relevant to our regional context that the authors feel are worth exploring. Creating a clinical link for DM, DHD and finally HFpEF will be difficult, highlights the complexity of the syndrome, but is critical as more elements of the basic science map are revealed. In a recent review we discussed the importance of the primary step of confirming the diagnosis CHF, with solid proof, i.e. elevation of left ventricular end diastolic pressure (LVEDP). A rise in LVEDP in the absence of systolic impairment, is then the result of a complex interplay of diastolic phase that is unable to contribute (is impaired/ or failing) to overall cardiac output directly or through adequate counterregulatory compensation or the lack of it (dysfunction). A direction to approach this interplay has been published and we refer readers to Figure 1 in reference 2. From this complex canvas we extract several clinical observations and areas we feel relevant for further study (Figure 3). We break this down in an inflammatory context:
Figure 3.

Theoretical bench to bedside flow chart for some commonly encountered clinical scenarios. 1Cytoskeleton (e.g., Rho kinase activity), contractile apparatus (e.g., Titin phosphorylation), mitochondrial energetics, AGE. 2Ventricular atrial coupling; Microvasculature; Pericardial restraint; Chronotropic reserve. ADHF: Acute decompensated heart failure; AGE: Advanced glycation end products; ANP: Atrial natriuretic peptide; BNP: Brain natriuretic peptide; IL-1α: Interleukin 1 alpha; CRI: Chronic renal impairment/insufficiency; IS: Indoxyl sulphate; PBUT: Protein bound uremic toxins; PCS: P-cresyl sulphate; RAAS: Renin angiotensin aldosterone syndrome; SNSA: Sympathetic nervous system activity.
Inflammation: Being selective and finding where to focus in this vast area is important. For example, chronic sterile inflammation has key pathways triggered by dying cells such as IL-1α[32], suggests opportunities exist for identifying other key pathways.
Clinical scenario: (1) Acute Decompensated Heart Failure (ADHF) – is the drawing board to for clinical studies. Often patients present with CHF and are discharged after a short stay. Trials have predominately focused on relieving congestion. The future landscape should be greater forensic interrogation of molecular and clinical observations; (2) Risk factors – pregnancy and future risk of HFpEF, hypertension and metabolic syndromes suggest a haemodynamic connect. Aging is associated diastolic impairment on echocardiography although the clinical manifestation varies suggesting a more complex interaction[33,34]; (3) Obesity (flux) – is difficult to determine if it’s a risk factor, association, driver of cardiac decompensation (ADHF) or a combination. However rapid changes in weight in either direction can provide exponential clinical changes in symptoms; and (4) Secondary endorgan – Chronic renal impairment (CRI) and retention of protein bound uremic toxins (PBUT), which have robust pro-inflammatory properties but have been relatively immune to treatments[35].
Tissue/systems: Four arms the mechanical, cytoskeletal (structure), conduction and heart, the interconnectivity (hemodynamical and molecular) shape the clinical picture of HFpEF.
Pathophysiological changes: When tissue and systems are exposed to disease or altered state a series of changes occur that is initially transient then becomes permanent. This remodelling leads to heart failure but also feedback loops. In time an equilibrium sets in that changes baseline levels e.g., biomarkers like natriuretic peptides.
Questions: Based on points 1-4 here we need to devise how we examine these areas. Four pressing areas are needed: Firstly – a basic science program including animal models, omics and biobanks to that have close relationship with clinician and scientists; Secondly are diagnostic biomarkers, by understanding disease baseline and changes when under stress to help with bedside stress testing diagnosis and monitoring treatment response[30]; Finally as there are so more causative confounders and counterregulatory pathways identifying key pathways will help focus clinical questions.
In summary while the above list is superficial however the idea of creating flow loops is vital as HFpEF remains a syndrome at its infancy. Much is not known and observations and idea are still welcome additions to this science[36-38].
CONCLUSION
There is an undeniable link between systemic inflammation and primary metabolic comorbidities including diabetes, obesity, obstructive sleep apnoea and secondary associations including aging, hypertension, lifestyle. These are all associated with endothelitis and the resulting vascular dysfunction is a “common soil” for atherogenesis and cardiac failure[39,40]. The cellular mechanism for inflammation in the subendothelial space that leads to atherogenesis and the inflammatory changes in the failing heart begins with (1) Dysfunctional endothelial changes that facilitate the recruitment of inflammatory cells; (2) Furthermore, increased barrier permeability, reduced thromboresistance, and altered paracrine signalling associated with endothelitis contributes to and compounds adverse vascular and cardiac remodelling in diabetes; and (3) In so far as, endothelitis is a driving force of progressive cardiac dysfunction in diabetes, then targeting microvascular dysfunction should provide benefits for patients with diabetes. The bench to bedside link can be seen when optimal glucose and blood pressure control have microvascular as well as macrovascular benefits, albeit largely when instituted early in the course of disease and continued for long periods of time (> 10 years). In addition, blockade of the RAAS in patients with a high cardiovascular risk or CHF is unequivocally associated with improved clinical outcomes, independent of blood pressure reduction, and over short trial intervals. The MICRO-HOPE study, treatment with ramipril was associated with a reduction in CHF hospitalisation and major acute coronary events. With increasing understanding of “diabetic endothelitis“ and its contribution to HFpEF specifically, in the future a more direct targeting of endothelial dysfunction with therapies including anti-inflammatory therapies, oral nitrite/nitrate, guanylyl cyclase activators and phosphodiesterase inhibitors may prove useful. An improved understanding of these sciences will also allow opportunities to advance clinical and pathophysiological understanding; and an improved ability to clinically correlate haemodynamic observations to monitor progress perhaps with novel diagnostic biomarkers.
Footnotes
Conflict-of-interest statement: All co-authors have won independent and governmental research funding. None pose a conflict of interest for this review.
Manuscript source: Invited manuscript
Peer-review started: March 25, 2020
First decision: June 5, 2020
Article in press: July 18, 2020
Specialty type: Cardiac and cardiovascular systems
Country/Territory of origin: Australia
Peer-review report’s scientific quality classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Bolshakova GB, Hamad ARA S-Editor: Gong ZM L-Editor: A P-Editor: Li JH
Contributor Information
Merlin C Thomas, Department of Diabetes, Monash University, Melbourne 3004, Victoria, Australia. merlin.thomas@monash.edu.
Pupalan Iyngkaran, Werribee Mercy Sub School, School of Medicine Sydney, University of Notre Dame, Northcote 3070, Victoria, Australia.
References
- 1.Iyngkaran P, Liew D, Neil C, Driscoll A, Marwick TH, Hare DL. Moving From Heart Failure Guidelines to Clinical Practice: Gaps Contributing to Readmissions in Patients With Multiple Comorbidities and Older Age. Clin Med Insights Cardiol. 2018;12:1179546818809358. doi: 10.1177/1179546818809358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iyngkaran P, Thomas MC, Neil C, Jelinek M, Cooper M, Horowitz JD, Hare DL, Kaye DM. The Heart Failure with Preserved Ejection Fraction Conundrum-Redefining the Problem and Finding Common Ground? Curr Heart Fail Rep. 2020;17:34–42. doi: 10.1007/s11897-020-00454-2. [DOI] [PubMed] [Google Scholar]
- 3.Kim P, Chu N, Davis J, Kim DH. Mechanoregulation of Myofibroblast Fate and Cardiac Fibrosis. Adv Biosyst. 2018;2 doi: 10.1002/adbi.201700172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pi X, Xie L, Patterson C. Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ Res. 2018;123:477–494. doi: 10.1161/CIRCRESAHA.118.313237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. 2019;40:1–8. doi: 10.1038/s41401-018-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stone WL, Basit H, Burns B. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020. [Google Scholar]
- 7.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 8.Flynn MC, Pernes G, Lee MKS, Nagareddy PR, Murphy AJ. Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Front Pharmacol. 2019;10:666. doi: 10.3389/fphar.2019.00666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2016;90:84–93. doi: 10.1016/j.yjmcc.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urbina P, Singla DK. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2014;307:H762–H772. doi: 10.1152/ajpheart.00367.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasdai D, Holmes DR, Jr, Higano ST, Burnett JC, Jr, Lerman A. Prevalence of coronary blood flow reserve abnormalities among patients with nonobstructive coronary artery disease and chest pain. Mayo Clin Proc. 1998;73:1133–1140. doi: 10.4065/73.12.1133. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J Am Heart Assoc. 2015;4 doi: 10.1161/JAHA.115.002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res. 2010;87:262–271. doi: 10.1093/cvr/cvq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinkel R, Howe A, Renner S, Ng J, Lee S, Klett K, Kaczmarek V, Moretti A, Laugwitz KL, Skroblin P, Mayr M, Milting H, Dendorfer A, Reichart B, Wolf E, Kupatt C. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J Am Coll Cardiol. 2017;69:131–143. doi: 10.1016/j.jacc.2016.10.058. [DOI] [PubMed] [Google Scholar]
- 15.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, Rasmusson BY, Whitehead KJ, Salama ME, Selzman CH, Stehlik J, Clayson SE, Bristow MR, Renlund DG, Li DY. Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol. 2010;56:382–391. doi: 10.1016/j.jacc.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feihl F, Liaudet L, Waeber B, Levy BI. Hypertension: a disease of the microcirculation? Hypertension. 2006;48:1012–1017. doi: 10.1161/01.HYP.0000249510.20326.72. [DOI] [PubMed] [Google Scholar]
- 18.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985) 2009;106:326–332. doi: 10.1152/japplphysiol.91353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers SC, Zhang X, Azhar G, Luo S, Wei JY. Exposure to high or low glucose levels accelerates the appearance of markers of endothelial cell senescence and induces dysregulation of nitric oxide synthase. J Gerontol A Biol Sci Med Sci. 2013;68:1469–1481. doi: 10.1093/gerona/glt033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, Lemmens K, Van Craenenbroeck EM. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail. 2017;10 doi: 10.1161/CIRCHEARTFAILURE.116.003806. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida Y, Shimizu I, Katsuumi G, Jiao S, Suda M, Hayashi Y, Minamino T. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J Mol Cell Cardiol. 2015;85:183–198. doi: 10.1016/j.yjmcc.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. 2019;76:1093–1106. doi: 10.1007/s00018-018-2983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Nkennor B, Mastikhina O, Soon K, Nunes SS. Endothelium-mediated contributions to fibrosis. Semin Cell Dev Biol. 2020;101:78–86. doi: 10.1016/j.semcdb.2019.10.015. [DOI] [PubMed] [Google Scholar]
- 24.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 25.Sniegon I, Prieß M, Heger J, Schulz R, Euler G. Endothelial Mesenchymal Transition in Hypoxic Microvascular Endothelial Cells and Paracrine Induction of Cardiomyocyte Apoptosis Are Mediated via TGFβ1/SMAD Signaling. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18112290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abu El-Asrar AM, De Hertogh G, van den Eynde K, Alam K, Van Raemdonck K, Opdenakker G, Van Damme J, Geboes K, Struyf S. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT) Exp Eye Res. 2015;132:179–189. doi: 10.1016/j.exer.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Bertram JF. Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology (Carlton) 2010;15:507–512. doi: 10.1111/j.1440-1797.2010.01319.x. [DOI] [PubMed] [Google Scholar]
- 28.Segers VFM, Brutsaert DL, De Keulenaer GW. Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front Physiol. 2018;9:382. doi: 10.3389/fphys.2018.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 30.Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, Toffoli B, Nguyen-Huu TP, Head GA, Fu Y, Chin-Dusting J, Cooper ME, Tikellis C. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res. 2010;107:888–897. doi: 10.1161/CIRCRESAHA.110.219279. [DOI] [PubMed] [Google Scholar]
- 31.Wan A, Rodrigues B. Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc Res. 2016;111:172–183. doi: 10.1093/cvr/cvw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 33.Nanayakkara S, Marwick TH, Kaye DM. The ageing heart: the systemic and coronary circulation. Heart. 2018;104:370–376. doi: 10.1136/heartjnl-2017-312114. [DOI] [PubMed] [Google Scholar]
- 34.Nanayakkara S, Haykowsky M, Mariani J, Van Empel V, Maeder MT, Vizi D, Kaye DM. Hemodynamic Profile of Patients With Heart Failure and Preserved Ejection Fraction Vary by Age. J Am Heart Assoc. 2017;6 doi: 10.1161/JAHA.116.005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savira F, Magaye R, Hua Y, Liew D, Kaye D, Marwick T, Wang BH. Molecular mechanisms of protein-bound uremic toxin-mediated cardiac, renal and vascular effects: underpinning intracellular targets for cardiorenal syndrome therapy. Toxicol Lett. 2019;308:34–49. doi: 10.1016/j.toxlet.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol. 2016;17:906–913. doi: 10.1038/ni.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Soudi A, Kaaij MH, Tas SW. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun Rev. 2017;16:951–962. doi: 10.1016/j.autrev.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis. 2016;248:97–109. doi: 10.1016/j.atherosclerosis.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graziani F, Varone F, Crea F, Richeldi L. Treating heart failure with preserved ejection fraction: learning from pulmonary fibrosis. Eur J Heart Fail. 2018;20:1385–1391. doi: 10.1002/ejhf.1286. [DOI] [PubMed] [Google Scholar]
- 40.Colombo PC, Onat D, Sabbah HN. Acute heart failure as "acute endothelitis"--Interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail. 2008;10:170–175. doi: 10.1016/j.ejheart.2007.12.007. [DOI] [PubMed] [Google Scholar]