

Synthetic attC sites can be embedded into virtually any sequence and constitute a structure-specific DNA recombination system.
Abstract
Recombination systems are widely used as bioengineering tools, but their sites have to be highly similar to a consensus sequence or to each other. To develop a recombination system free of these constraints, we turned toward attC sites from the bacterial integron system: single-stranded DNA hairpins specifically recombined by the integrase. Here, we present an algorithm that generates synthetic attC sites with conserved structural features and minimal sequence-level constraints. We demonstrate that all generated sites are functional, their recombination efficiency can reach 60%, and they can be embedded into protein coding sequences. To improve recombination of less efficient sites, we applied large-scale mutagenesis and library enrichment coupled to next-generation sequencing and machine learning. Our results validated the efficiency of this approach and allowed us to refine synthetic attC design principles. They can be embedded into virtually any sequence and constitute a unique example of a structure-specific DNA recombination system.
INTRODUCTION
DNA recombination is one of the basic tools used in genetic engineering. However, site-specific recombination systems require the recombination sites to have a consensus sequence that cannot be easily modified or must even be kept constant (1), while homologous recombination systems are based on high sequence similarity between target sites of large size (2). These requirements limit the possibilities of inserting recombination sites into DNA regions that already carry a function, such as protein coding sequences or promoters. The development of a site-specific yet non–sequence-specific recombination system would allow embedding recombination sites into virtually any DNA sequence.
We decided that such a recombination system could be developed on the basis of the bacterial integron platform. Integrons play a major role in the dissemination of antibiotic resistance genes among clinically relevant Gram-negative pathogens (3). The functionality of integrons relies on the activity of the integron integrase (hereafter integrase), a site-specific tyrosine recombinase capable of excising genes from the integron in the form of circular cassettes and reinserting these cassettes into the platform (Fig. 1A). Cassette excision involves the recombination of two adjacent attC sites, whereas their insertion mainly occurs through recombination of an attC site within the cassette with an attI site located in the integron platform (Fig. 1A). While attI sites resemble canonical tyrosine recombinase sites and are recombined in the form of double-stranded DNA (4), attC sites are atypical: The bottom strand of their DNA folds into a hairpin-like structure, which is then recombined by the integrase as a folded single strand (Fig. 1B) (5, 6). Tyrosine recombinases usually show high sequence specificity toward their substrates (7). The integrase, however, is able to efficiently recombine attC sites with highly variable sequences: For instance, class 1 integrase IntI1 recombines equally well attCaadA7 and attCereA2, which have only 58% sequence identity (8) and can site-specifically recombine sedentary chromosomal integron attC sites that considerably differ both in size [from 53 to 152 nucleotide (nt)] and in sequence (8, 9). The specificity of the reaction is ensured by the conserved structure of folded attC sites (Fig. 1B) (6, 8, 9). Moreover, their recombination can reach frequencies on the order of 10−1 (8), making them perfect candidates for the development of a highly efficient structure-specific recombination system.
Fig. 1. Integron attC recombination sites.
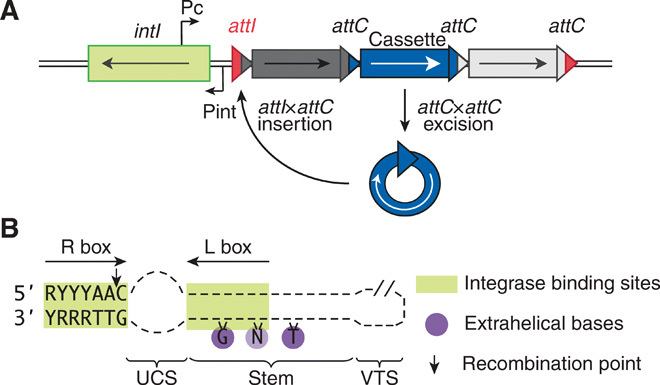
(A) Schematic of the integron system. Pint, integrase promoter; intI, integrase gene; Pc, cassette promoter; attI, cassette insertion site; attC, cassette attachment site. (B) Schematic of a folded bottom strand of attC recombination site.
attC sites appear to have very few sequence-level constraints (Fig. 1B). Their recombination occurs between the A and the C within the consensus sequence 5′-RYYYAAC-3′ of the integrase binding site named R box (10). There are two unpaired bases within the hairpin stem, called the extrahelical bases (EHBs) and which are typically G and T (9). The rest of the sequence is not conserved: Even the sequence of the second integrase binding site (L box) is degenerate. Instead, loss-of-function approaches have shown that integrase binding and recombination depends on structural features of attC sites: the EHBs, the unpaired central spacer (UCS), and the variable terminal structure (VTS) (Fig. 1B) (8, 9, 11).
Here, we designed a structure-specific recombination system based on attC sites. We started with an assumption that if all important attC features have been characterized, then any DNA sequence having these features could be recognized and recombined by the integrase. We designed an algorithm that generates synthetic attC sites with conserved structural features and minimal constraints on the sequence level and demonstrated that all 14 generated sites are functional, with recombination efficiencies reaching up to 60%. To show the potential of synthetic attC sites as bioengineering tools, we embedded them into three peptide linkers and four regions of β-galactosidase and tested their functionality. However, the variability of synthetic attC site recombination frequencies suggested that more factors contribute to attC site recombination than previously thought. Through a combination of experimental and computational approaches, we uncovered additional features that could not be identified through conventional methods and refined synthetic attC site design principles.
RESULTS
Algorithm generating synthetic attC sites
In our previous works, we demonstrated the determinant role of the R box and structural features in attC site recombination (6, 8, 9, 12, 13). To test whether any DNA sequence having these properties could be recognized and recombined by IntI1, we designed an algorithm that generates synthetic attC sites with arbitrary sequences constrained to have these features. Since we did not know a priori the extent to which each of the identified constraints was important, we developed two versions of the algorithm.
The first version generated synthetic attC sites with constraints based on empirical results (Fig. 2A and table S1). This corresponds to the minimal constraints that we deemed necessary for a functional attC site based on our previous studies (6, 8, 9, 12, 13). The second version of the algorithm used more relaxed constraints based on bioinformatic analysis of 263 natural attC sites from class 1 mobile integrons in the INTEGRALL database (14) and reflected the variability observed among them (Fig. 2B).
Fig. 2. Synthetic attC site recombination.
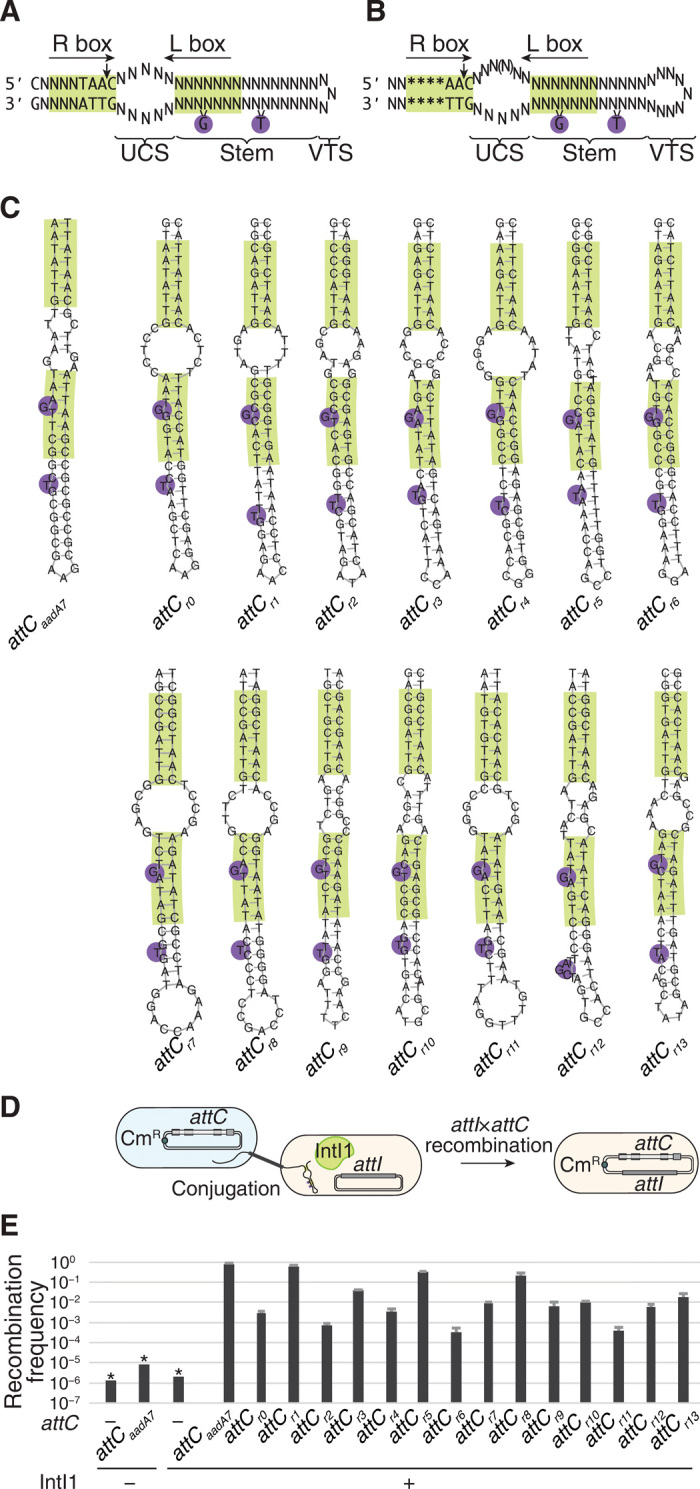
(A and B) Schematic of the bottom strands of attC sites with constraints used by the two versions of the algorithm generating synthetic sites: with constraints based on empirical results (A) and with constraints based on bioinformatic analysis of wild-type attC sites (B). N, arbitrary base. *Base generated according to the probability distribution of each base in the sequence of the R box in wild-type attC sites (table S1). (C) Predicted structures of the paradigmatic attCaadA7 site and synthetic attC sites, with seven sites generated by each version of the algorithm. All structural predictions were performed using ViennaRNA 2.1.8 package. (D) Schematic of the suicide conjugation assay to measure attC site recombination frequency. A plasmid carrying an attC site is conjugated into a strain that does not have the machinery for its replication. However, the plasmid and the chloramphenicol resistance marker (CmR) that it carries can be maintained in the recipient strain through attI1×attC recombination. The recombination frequency can be measured as the ratio of chloramphenicol-resistant cells to all recipient cells. (E) Recombination frequencies of an empty vector in the presence or absence of IntI1 integrase (negative controls), attCaadA7 (positive control), and synthetic attC sites. Values represent means of three independent experiments; error bars represent mean absolute error. Asterisks (*) indicate that the recombination frequency was below detection level, indicated by the bar height.
Recombination of synthetic attC sites
Both versions of the algorithm were used to generate several synthetic attC sites: seven with each version (Fig. 2C). We then synthesized the corresponding DNA sequences, cloned each of them into the pSW23T vector, and transformed into the Escherichia coli ß2163 strain that expresses the π protein to maintain this vector (Supplementary Materials and table S2) (15). These strains were then tested using the previously developed suicide recombination protocol that measures the frequency of attI1×attC recombination (Fig. 2D and Supplementary Materials) (6). We used the wild-type attCaadA7 site (16) as a positive control in this assay.
Our results showed that all synthetic attC sites generated by the algorithm were functional (Fig. 2E). This confirmed our hypothesis that the constraints used in our algorithm were sufficient to generate attC sites that could be recognized and recombined by the IntI1 integrase. Moreover, the difference in recombination frequencies of attC sites generated by the two versions of the algorithm was not statistically significant (t test, P > 0.4), suggesting that the imposition of strict constraints was not necessary.
Synthetic attC sites embedded into protein linkers
Having freed synthetic attC sites from most sequence-related constraints, we decided to test whether the sequence can now be used to encode proteins. To reconcile both protein coding and attC structure-related constraints within the same DNA fragment, we decided to use in silico directed evolution. Starting with a large set of synthetic attC sites, we evolved them to encode a protein sequence with desired properties, here, encoding peptides with sequences similar to those of unstructured linkers from the database (17). We then selected three synthetic sites attCL1–3 that obtained the best scores (Fig. 3, A to C, and Supplementary Materials). Like other synthetic attC sites, they were all functional in recombination, with attCL1 and attCL3 sites recombining with more than 10−1 frequency (Fig. 3D). To test whether the resulting sequences were also functional as peptide linkers, we used them to fuse two domains of Bordetella pertussis adenylate cyclase, used as a bacterial two-hybrid system (18, 19). The cyclic adenosine monophosphate (AMP) production by the enzyme requires a close interaction between both domains and can be measured by an enzyme-linked immunosorbent assay (ELISA)–based assay (Supplementary Materials). The linker-like sequence of the protease processing site p5 from the HIV was used as a positive control (19), and the same sequence with a frameshift was used as a negative control. All three peptide-encoding synthetic attC sites were able to act as peptide linkers, reconstituting a fully functional enzyme (Fig. 3E).
Fig. 3. Synthetic attC sites embedded into protein coding regions.
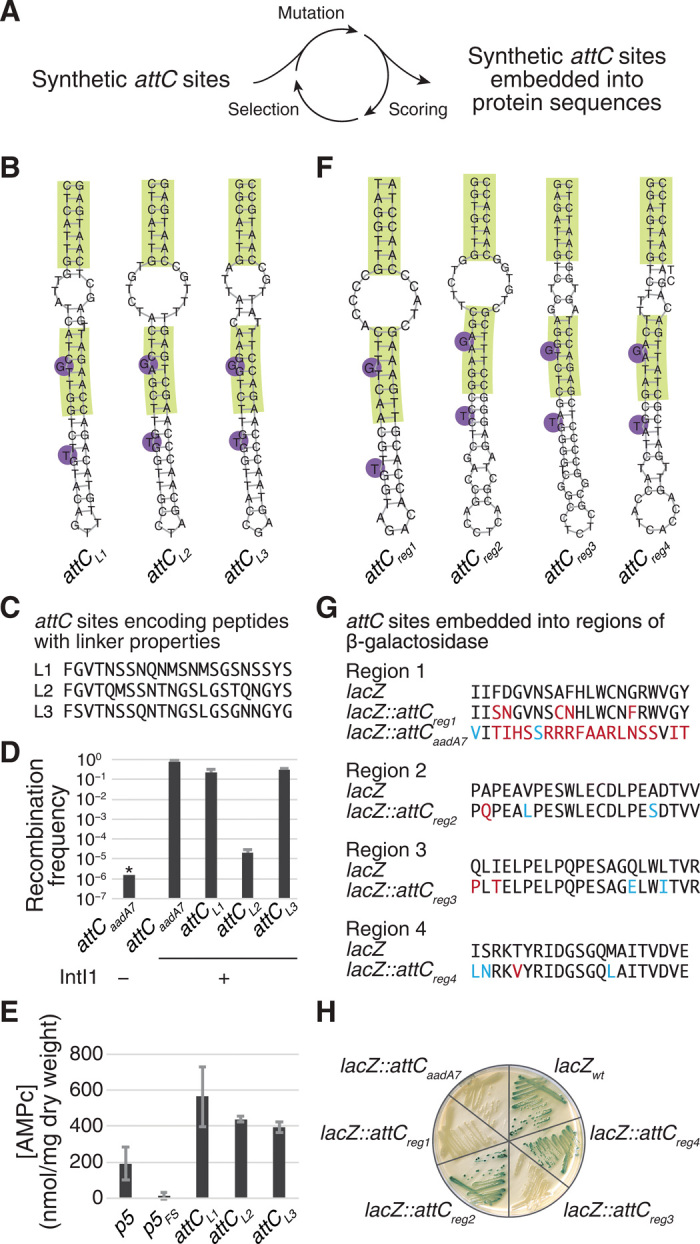
(A) In silico directed evolution approach that was used to generate synthetic attC sites with peptide linker properties (B-E) and to embed synthetic attC sites into lacZ (F-H). (B) Structures of the three synthetic attC sites encoding peptide linkers L1, L2, and L3, predicted using ViennaRNA 2.1.8. (C) Protein sequences of encoded peptide linkers. (D) Recombination frequencies of attCaadA7 (positive control) and synthetic attC sites encoding peptide linkers. Values represent means of three independent experiments; error bars represent mean absolute error. Asterisk (*) indicates that the recombination frequency was below detection level, indicated by the bar height. (E) Results of a bacterial two-hybrid assay with the two domains of Bordetella pertussis adenylate cyclase fused either with a natural linker (p5, positive control), a natural linker with a frameshift mutation (p5FS, negative control), or synthetic attC sites encoding peptide linkers. Values represent means of three independent experiments; error bars represent mean absolute error. (F) Predicted structures of the synthetic attC sites embedded into four regions of the lacZ gene encoding β-galactosidase, predicted using ViennaRNA 2.1.8. (G) Protein sequences of the four β-galactosidase target regions and sequences after attC site embedding. Blue, mutations that preserve the amino acid physicochemical properties; red, other nonsilent mutations. (H) Strains with the four synthetic attC sites embedded into lacZ and streaked on an LB agarose plate with X-gal and isopropyl-β-D-thiogalactopyranoside. Blue color indicates a functional β-galactosidase, as in lacZwt. White color indicates that an embedded attC site perturbed the function of the β-galactosidase, as in lacZ::attCaadA7.
In a similar manner, synthetic attC sites can be embedded into virtually any desired protein sequence with minimal changes to its amino acid sequence. The redundancy of the genetic code allows enough flexibility that modifications to the DNA sequence that introduce attC site properties on the structural level do not necessarily cause changes to the encoded protein. Allowing a slightly variable VTS size between 3 and 21 nt [for reference, VTS varies between 3 and 100 nt in natural class 1 attC sites (20)] allows additional flexibility when finding a sequence that accommodates both constraints. As an example, we generated four synthetic attC sites embedded into different locations of the lacZ gene that encodes β-galactosidase. In each case, the best-scoring attC sites introduced only three to five amino acid changes, some of which preserve the main physicochemical properties (Fig. 3, F to H, and Supplementary Materials). In comparison, in-frame substitution of lacZ region 1 by the natural attCaadA7 site leads to 19 amino acid changes over 21 positions (Fig. 3G). However, even small changes in the amino acid sequence of an enzyme can affect its functionality. By streaking E. coli strains expressing lacZ variants, we observed that two of four synthetic attC sites embedded into lacZ preserved the β-galactosidase’s ability to produce a blue pigment upon the addition of X-galactosidase (X-gal) (Fig. 3H), confirming that they can be successfully embedded into proteins.
For synthetic attC sites to become a truly useful tool, their recombination frequencies have to reach at least the order of 10−4, frequency comparable to the commonly used Lambda Red system (21), and preferably even higher. Regardless of the algorithm version used, we observed a high variability in recombination frequencies among synthetic attC sites: Similarly to wild-type attC sites (8), their values varied over several orders of magnitude (Figs. 2E and 3D and fig. S1). This result underlined our incomplete knowledge of the constraints required for generating efficiently recombining synthetic attC sites. We therefore decided to undertake a large-scale mutagenesis study of a synthetic attC site to deduce additional relationships between attC site structure and functionality, which would allow us to improve the efficiency of synthetic sites.
Large-scale mutagenesis of a synthetic attC site
We selected attCr0 for our mutational analysis: a synthetic attC site with a recombination frequency in the lower range. We decided to construct a library consisting of all sequences with a single mutation within the constant region of the attCr0 site, as well as sequences containing all possible combinations of two mutations (Fig. 4A, blue region). This region covered the entire sequence that could be recovered, since upon recombination, the 5′ extremity of an attC site (upstream of the recombination point) is exchanged with a partner site. Using a custom-designed degenerate oligonucleotide attCr0_library (table S2), we cloned a library of 162 single and 12,879 pairwise mutants within the constant region of attCr0 into the pSW23T vectors and transformed it into the ß2163 strain.
Fig. 4. Enrichment of attCr0 mutant library in sites with higher recombination frequencies.
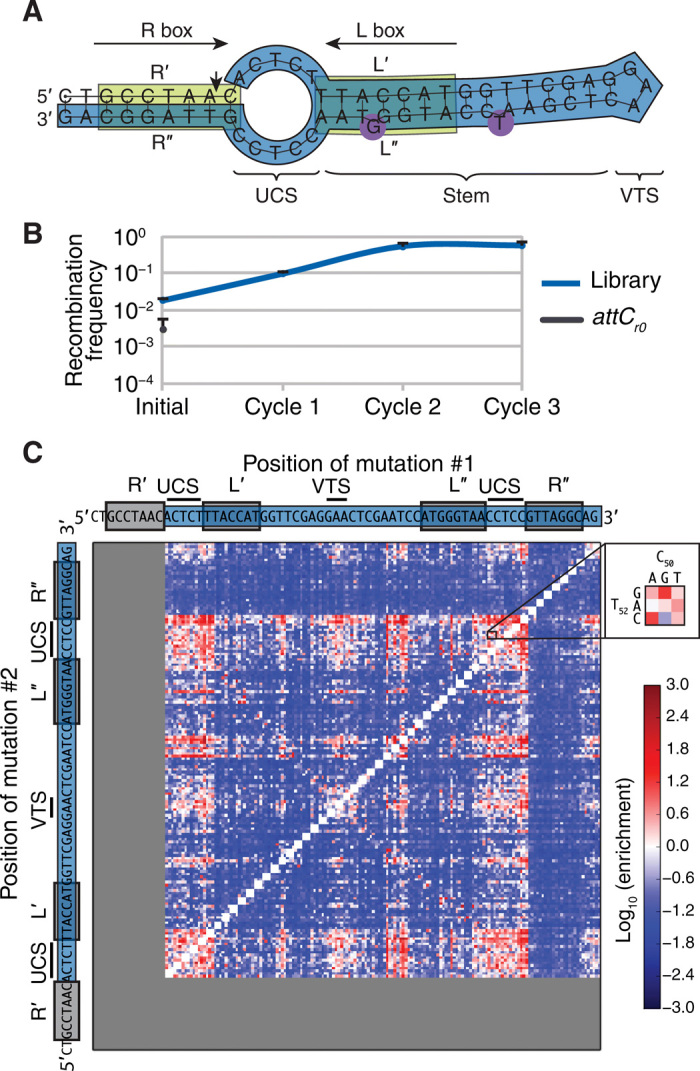
(A) Schematic of the folded bottom strand of attCr0 used for library construction. The region submitted to mutational analysis is shown in blue. (B) Recombination frequencies of the library throughout cycles of recombination (blue). The recombination frequency of attCr0 is used for comparison (black). Values represent means of three independent experiments; error bars represent mean absolute error. (C) Heat map of enrichment values for all pairwise attCr0 mutants. At the intersection of nucleotides depicted along each axis, a series of points represent enrichment values corresponding to all pairwise mutants of these nucleotides. Inset: Example for all pairwise mutants of C50 and T52.
To assess the recombination efficiency of each mutant within the library, we performed an assay based on the suicide conjugation protocol (6, 22), where all the variants were present in a pool (Supplementary Materials and fig. S2). In this assay, we expected the more recombinogenic sites to be enriched in the final pool compared to less efficient attC sites. To validate our experimental design, we performed several cycles of this competition assay and measured the overall recombination frequency of the library throughout these cycles (Fig. 4B). The recombination frequency of the library increased over two orders of magnitude, reaching a plateau at 6 × 10−1 after the second enrichment cycle. This indicated that the library first got enriched and then saturated in highly reactive attCr0 mutants, according to the design of our competition assay.
We then computed the enrichment of each site by comparing its relative abundance in the library before and after the assay, as measured by next-generation sequencing (NGS) (fig. S3). To justify the use of the enrichment value as a proxy for recombination frequency, we tested the recombination of six double mutants using the suicidal conjugation assay and confirmed that the enrichment values were indeed highly correlated with the recombination frequencies (Pearson r = 0.92, P < 0.01; fig. S4).
The enrichment values varied over six orders of magnitude between the most enriched and the most depleted mutants (Fig. 4C). The heat map of double mutant enrichment values showed some regions corresponding to mutants with highly increased recombination frequencies (bright red) and others corresponding to those where recombination was impaired (deep blue), but the pattern did not provide a straightforward answer as to which attC site mutations were responsible for better recombination and why (Fig. 4C).
Machine learning predictions of attC site recombination
To analyze the results corresponding to 162 single and 12,879 double attCr0 mutants, we decided to use a machine learning (ML) approach. Our objective was twofold: to test whether a regression algorithm (23) could predict the recombination frequencies of attC sites based on their sequence and structure and, if so, to deduce the properties on which these predictions are based.
A regression algorithm is a supervised ML technique for estimating an unknown continuous function based on a finite number of data points. Here, a data point is an attCr0 mutant, the input is the set of sequence- and structure-related features, and the output is the enrichment value. Once the model is learnt from the training dataset, it can be tested on the unseen data (test dataset) to predict the output, and its performance can be evaluated.
To use regression algorithms, we first needed to define a number of features to describe each attCr0 mutant. Since the integrase recognizes the structure of the bottom strand of attC sites (6), we based our features on the predicted folding of the bottom strand of each mutant using the RNAfold program from the ViennaRNA 2.1.8 package (Supplementary Materials) (24). Our list included several global features describing attC site folding, such as Gibbs free energy (∆G) of the structures, the probability to fold a functional structure (pfold), i.e., a structure with correctly folded integrase binding sites (25, 26), and others. It also included a set of characteristics for each particular base, such as the nucleotide present at that position, its pairing probability, and positional entropy. The positional entropy of a base reflects how unstable it is: Low positional entropy means that the base is stabilized in only one major state (either paired with a particular base or unpaired), whereas high positional entropy means that the base can be found in various states within the thermodynamic ensemble of possible structures.
As a second step, we prepared the data for ML by discarding features with zero variance, normalizing feature and enrichment values, and equilibrating our dataset to contain an equal number of enriched and depleted mutants (Supplementary Materials). Last, we used four different regression algorithms: decision tree regression (27), ridge regression (28), support vector regression (29), and random forest regression (30), available in the Python scikit-learn library (31). To evaluate their performance, we ran every algorithm 50 times, each time splitting the data points into training and test datasets and performing fivefold cross-validation. Among the four algorithms, random forest regression showed the best performance, attaining a Pearson r = 0.81 (Fig. 5, A and B, and table S3, A and B).
Fig. 5. Analysis of ML results.
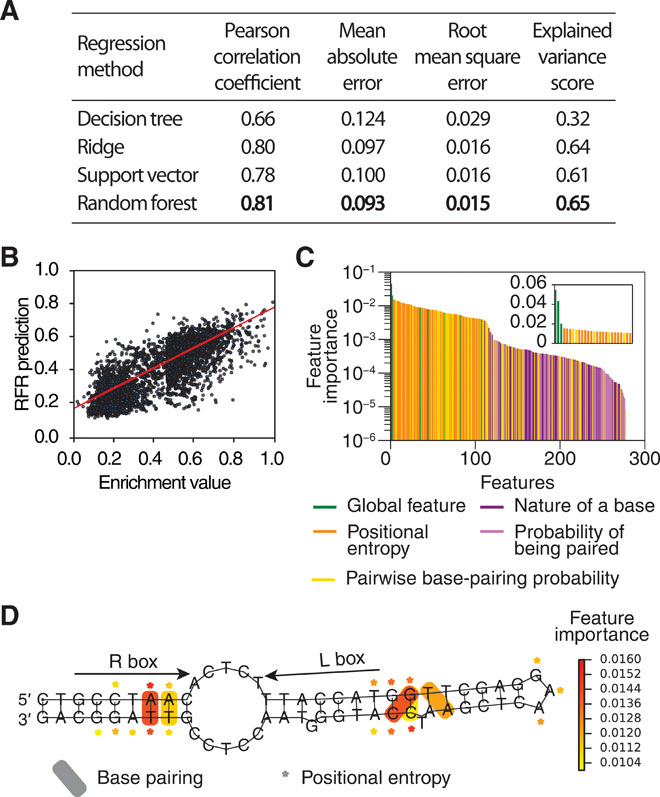
(A) Performance of four different ML algorithms in regression, measured as described in Supplementary Materials. (B) Correlation between the measured and the predicted enrichment values given by the random forest regression (RFR) algorithm for attCr0 site mutants from the library test dataset (Pearson r = 0.81, P < 0.01). (C) Features used by RFR, ranked according to their importance measure. Inset: Most important features (score > 0.01). (D) Mapping of the most important nonglobal features (score > 0.01) onto the predicted structure of attCr0.
It is known that feature selection, a common procedure in ML, can provide better interpretability, avoid overfitting, and simplify the algorithm through dimensionality reduction. Random forest regression performs its own feature selection, but other three algorithms could potentially benefit from a preliminary feature selection step. We tested two feature selection methods and one dimensionality reduction method [k best features (32), manual feature selection strategy, and principal component analysis (33); Supplementary Materials] on the three algorithms. However, they did not achieve results comparable to those obtained by random forest regression (table S3B). This confirmed that the recombination frequency of attC sites is a multifactorial function that depends on a wide array of properties.
On the example of this library of synthetic attC site mutants, we have demonstrated that it is possible to predict with high accuracy the recombination frequency of attC sites (through the proxy for the enrichment value) based on their structure.
Biological interpretation of ML results
Since the ML algorithm performed well in cross-validation tests on the dataset of attCr0 mutants, we concluded that it did not overfit the data but instead “learned” the relationship between the features of attC sites and their recombination frequencies. We tried to deduce these relationships by analyzing features that were most relevant for the prediction. None of the feature selection methods succeeded in identifying a set of features sufficient to construct a good predictive model, suggesting that most of the features were meaningful. However, random forest regression algorithm allows ranking features according to their relevance to the model, using the feature importance measure that assigns higher scores to the most important features (table S3C).
Three global features had the highest importance scores: the ∆G of the thermodynamic ensemble of folded molecules, the ∆G of the minimal free energy structure (see Supplementary Materials), and the number of hydrogen bonds (Fig. 5C and inset, in green). However, these features alone did not account for the high correlation score achieved by the algorithm.
They were closely followed by features corresponding to positional entropies and base-pairing probabilities (orange and yellow) and a long tail of other features (purple) with gradually decreasing importance (Fig. 5C). We observed that features ranked as most important ones (importance score > 0.01; inset of Fig. 5C) were clustered in two regions of the folded attC site structure: one in the R box and another just outside of the L box, around the second EHB (Fig. 5D). The importance of the R box was previously shown experimentally (12, 13). However, the location of the second region was unexpected: It lay outside integrase binding sites, and none of the previous studies attributed any particular role to this part of the hairpin stem.
To understand how its structure influenced recombination, we analyzed the correlation of the most important features within this region with the enrichment values. First, we focused on positional entropies of bases within the apical stem. All positional entropies identified as important features for ML correlated negatively with the enrichment values, suggesting that a stable stem is more favorable for recombination (Fig. 5D, Supplementary Materials, and fig. S5). In addition, the stem should include the two EHBs located at the 6–base pair (bp) distance that is important for wild-type site recombination by IntI1 (34).
Second, we looked at base-pairing features. We observed that recombination was improved in attCr0 mutants where mutations caused changes in the structure that shifted both EHBs by one position toward the apex of the stem (Supplementary Materials and fig. S5). On the basis of these observations, we hypothesized that two design strategies could increase attC site recombination. Hypothesis 1: The positional entropies of bases in the apical stem should be reduced to stabilize the hairpin structure, maintaining a 6-bp distance between the EHBs. Hypothesis 2: In addition, the positions of both EHBs should be shifted by one position toward the apex of the hairpin relative to the initial design, respectively, 9 and 16 nucleotides away from the R box.
Generalization of synthetic attC site design principles
We decided to test these hypotheses on three synthetic attC sites that showed the lowest recombination frequencies (below 10−3): attCr2, attCr6, and attCr11 (Fig. 2E). For each site, we constructed two mutants: one only stabilizing the apical stem and the other also shifting the EHBs by one position (Fig. 6, A to C).
Fig. 6. Confirmation of ML-derived hypotheses for three synthetic attC sites.
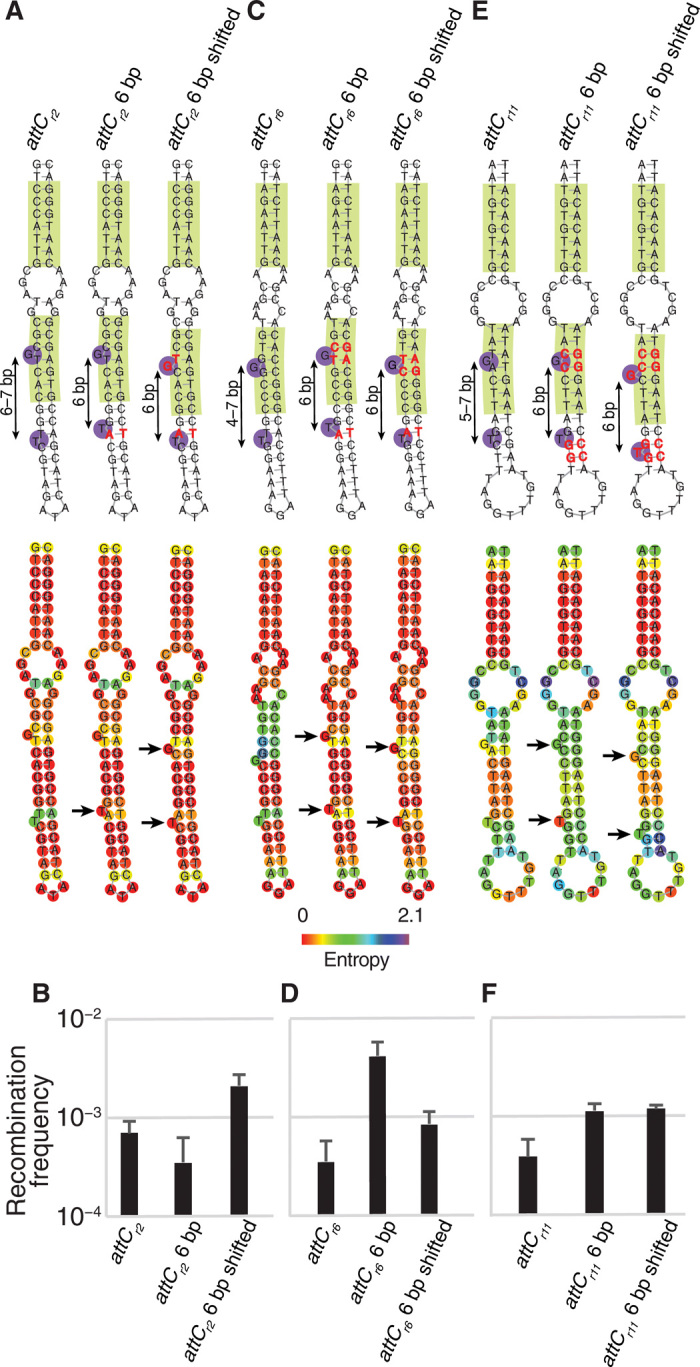
Structural predictions, positional entropies of bases, and recombination frequencies of initial and mutated sites attCr2 (A and B), attCr6 (C and D), and attCr11 (E and F). Mutations are shown in red. Arrows indicate EHBs that were stabilized in low entropy state through mutations. All structural predictions were performed using ViennaRNA 2.1.8 package. Recombination values represent means of three independent experiments; error bars represent mean absolute error.
In attCr2, the apical stem was already relatively stable, and its further stabilization did not increase the recombination frequency but instead slightly lowered it (Fig. 6, A and B). However, when both EHBs were shifted toward the apex, the recombination frequency increased threefold (Fig. 6, A and B), which was not the case for single EHB shifts (fig. S6, A and B). Thus, best results were achieved through combination of the two design strategies.
In attCr6, the initial distance between the two EHBs varied between 4 and 7 bp, and the entropies of surrounding regions were relatively high (Fig. 6C). The stabilization of the stem increased the recombination frequency 12-fold, while concomitant shift of the two EHBs only led to a twofold increase (Fig. 6D). For attCr6, the first design strategy was very successful, while the second did not bring any additional benefit.
In attCr11, the positional entropy of the entire apical stem was quite high (Fig. 6E). The stabilization of the stem in any of the two conformations with a 6 bp distance between the EHBs increased the recombination threefold (Fig. 6F). Again, the 6-bp distance between the EHBs was crucial since mutants that did not maintain it did not perform as well (fig. S6, C and D). As for attCr6, the first design strategy was successful for attCr11, while the additional strategy did not further improve the results.
Among the two hypotheses based on the analysis of attCr0 site mutants, the first was corroborated on three other synthetic sites and consistently led to higher recombination frequencies, while the second was only confirmed for one other attC site. This validates our approach in using feature importance to construct biologically relevant hypotheses and shows the potential of using it to interpret ML results in a broader context.
DISCUSSION
Synthetic attC sites as a bioengineering tool
In this work, we designed an algorithm that can generate any number of synthetic attC sites with very few constraints in their sequences and showed that they can be embedded into protein coding sequences in a way that preserves the protein’s function. The development of such a site-specific yet non–sequence-specific recombination system allows the encryption of recombination sites into any chosen DNA sequence.
The recombination assay results have shown that all generated sites are functional, although their recombination frequencies vary over several orders of magnitude. Despite the initial variability, we could substantially improve the recombination of the least efficient sites up to frequencies above 10−3 through a small (four to eight) number of mutations (Fig. 6). This validated our bottom-up approach as complementary to the previous mutational studies, as it allowed us to identify complex features that were impossible to uncover through direct loss-of-function approaches.
Our large-scale mutational analysis suggests that a more efficient version of a synthetic attC site can lie only one or two mutations away and that multiple solutions to this optimization problem are available. Moreover, the use of consecutive enrichment cycles (Fig. 4B) allows us to easily select these highly efficient mutants without the need for costly NGS or time-consuming data analysis. This holds the potential of further improving the recombination frequency of any synthetic attC site above 10−1 for the use in a highly efficient structure-specific recombination system.
As shown here, synthetic attC sites can be embedded within a DNA sequence that already carries a function (e.g., encodes a protein) at the cost of only three to five amino acid changes, which often preserve the physicochemical properties (Fig. 3G). While this number of mutations is very low, in some cases, they can be sufficient to disrupt an enzyme’s functionality (Fig. 3H), especially if they are introduced at or near its catalytic site. Because our algorithm allows introducing an attC into virtually any protein sequence, regions that are less sensitive to mutations, such as peptide linkers, would be better candidates for embedding attC sites. In addition, when the algorithm is run several times, it typically converges to slightly different solutions, meaning that several options for embedding an attC site into a protein sequence exist. Depending on the particular application, additional considerations can be taken into account when choosing the best design, such as avoiding key residues if they are known.
The use of synthetic attC sites would be of particular interest for generating large-scale libraries of multidomain or multimodular proteins, such as polyketide synthases (PKSs) or nonribosomal peptide synthetases (NRPSs). PKSs and NRPSs are multimodular enzymatic assembly lines that produce a vast diversity of natural products. Combinatorial engineering of these enzymes through module exchange is a highly appealing strategy to access new chemical compounds (35, 36). The possibility of embedding synthetic attC sites into PKS and NRPS genes opens possibilities for combinatorial shuffling of their modules: Once synthetic attC sites are embedded into homologous regions of several modules, integrase-mediated recombination would allow us to excise, insert, and shuffle their modules in vivo. We have previously shown that the integron’s gene shuffling activity can be efficiently used to improve biosynthetic pathways in vivo (37). By embedding attC sites into protein sequences, this engineering approach could be extended to multimodular biosynthetic gene clusters.
This tool could be used in various Gram-negative bacteria, where mobile class 1 integrons are widespread. Integron recombination relies on the integrase for the initial strand exchange that leads to an atypical Holliday junction (aHJ) structure, which is then resolved through replication (38). Since the exact proteins involved in replicative resolution are not known, it is difficult to predict whether it would be applicable in a broader host range. For instance, in eukaryotic cells the aHJ intermediate might be recognized as sign of damage and repaired before replication can resolve it (39).
attC site recombination is a multifactorial function
Our previous studies have already suggested that attC site recombination frequency depends on numerous properties related to their sequence, structure, and stability (6, 8, 9, 11, 12, 25, 26, 40, 41). Most of these factors apply to both attI×attC and attC×attC recombination reactions. In addition, recombination frequency between two attC sites depends on folding properties of both sites, the distance between them, and their orientation relative to the replication fork (26). To evaluate synthetic attC sites in a context that does not depend on the partner attC site, in this work, we used attI×attC recombination assays. Our results confirmed that even in this reaction, recombination frequency is a multifactorial function: Most features contributed to the predictive power of the random forest regression (Fig. 5C), and the use of fewer features led to poorer results (table S3, A and B).
attC site properties that we identified here as beneficial for recombination constitute an addition to those previously identified. To understand the extent to which these new findings are important, they must be put into context of previous discoveries. Since the suicide recombination assay is a reference method for assessing attC site recombination (6), we were able to compare the importance of different structure- or sequence-related properties on the order-of-magnitude scale, among several studies.
On one hand, we have observed large changes in recombination frequency (one to three orders of magnitude) upon modification of essential attC site elements: the 5′-AAC-3′ triplet of the R box (12), the stem (6, 42), the EHBs (8, 9), and the overall recombinogenic folding (25). On the other hand, we have identified features of attC sites that are highly conserved and play an important role in recombination, and yet, their disruption does not produce more than twofold differences in recombination: the high GC content in the apical part of the stem (40), the nucleotide skew within the unpaired regions (8), and the overall propensity to form a straight hairpin (41). The present study identified attC site mutations that are responsible for substantial (up to 12-fold) changes in recombination frequency, which places the uncovered properties among the most important ones.
These properties have not been previously identified through classical loss-of-function approaches (6, 8, 9, 11–13, 22, 25, 34). A direct analysis of EHB positional entropies in synthetic attC sites would not have revealed their role on recombination either since their values are not correlated with recombination (fig. S7). This lack of correlation is probably due to other properties having more dominant effects on recombination, which underscores the difficulty of analyzing the contribution of each individual factor in a multifactorial function and the advantage of the methodology described here.
Overall, the large-scale mutational analysis coupled to NGS and ML approaches allowed us to uncover several important properties of attC sites. Using these results, we attained recombination frequencies above 10−3 for each of the 14 designed attC site or its mutant, proving that synthetic attC sites with very few constraints in their sequences can indeed be efficiently recombined by the IntI1 integrase.
ML analysis of large-scale mutational datasets
ML is recognized as one of the most efficient methods for analyzing complex problems (43) and is rapidly gaining importance in bioinformatics (44). Since attC site recombination is a multifactorial problem, the use of ML was a compelling strategy to construct a predictive model of this biological function.
Here, ML allowed us to achieve both of our objectives: We constructed a predictive model of attC site recombination frequency and were able to draw biological conclusions from this analysis. However, this approach also has its limitations: The interpretation of ML performance is known to be nonstraightforward, and its generalization is not always possible. To overcome this limitation, we performed feature importance analysis to make hypotheses that were then experimentally tested. Contrary to the purely hypothesis-driven research largely used in genetics in the previous decades, the data-driven approach is particularly well suited to complex multifactorial problems. We propose that the method described here could be used to study other complex genetic systems such as recombination sites or functional DNA and RNA structures. While attC sites are exceptional in terms of their structure-based recognition, the general pipeline consisting of mutational analysis followed by selection, NGS, ML, and feature importance evaluation could also be applied to more conventional genetic elements recognized on the basis of their sequence. In these cases, an exhaustive mutational analysis of a smaller region would be more suitable than a pairwise mutational screening described here. Overall, it is a relatively low-cost, versatile, and not labor-intensive approach that can yield a vast amount of data and allow its in-depth analysis.
MATERIALS AND METHODS
Bacterial strains and media
Detailed information on bacterial strains, media, and antibiotic concentrations used in this study can be found in the Supplementary Materials.
Plasmids
Synthetic attC sites and their mutants were constructed by annealing two overlapping phosphorylated oligonucleotides (table S2), fully complementary except for the overhangs corresponding to either Eco RI and Bam HI restriction sites for testing recombination or Dpn I and Kpn I restriction sites for testing linker properties or by polymerase chain reaction (PCR) amplification of the vector plasmid for constructing sites embedded into lacZ. In the first case, the sites were then ligated into p9276, a derivative of the pSW23T vector (15) in the direction allowing the delivery of their bottom strands, previously digested with Eco RI/Bam HI. The resulting plasmids were then transformed into the ß2163 strain (table S2B) (15). In the second case, the sites were then ligated into p9983, pKAC::p5 vector (18) previously digested with Dpn I/Kpn I. The resulting plasmids were then transformed into the adenylate cyclase deficient strain DHM1 (table S2B) (45). In the third case, the sites were assembled into p1370, a derivative of the pSU vector (42). It expresses lacZ from a pLac promoter and was amplified by PCR with corresponding primers. The resulting plasmids were then transformed into b-galactosidase deficient strain MG1656 (46) for lacZ expression tests and into DH5α strain for the recombination assay (table S2B).
Library construction and competition assay
A custom oligonucleotide attCr0_library was PCR-amplified with primers Gibson1 and Gibson2. The pSW23T vector p4383 was PCR-amplified with primers Gibson3 and Gibson4. The two products were then purified, joint together through Gibson assembly (47), and transformed into the ß2163 strain (15). All oligonucleotide sequences are provided in table S2. A detailed protocol of the competition assay is provided in Supplementary Materials and fig. S2. To measure the enrichment value of each mutant, we performed NGS of the library before and after the recombination assay using the Ion Proton technology.
ML algorithms and performance measures
All algorithms are available in the sklearn library (31). For the ridge regression algorithm (from the “linear_model” package), the alpha parameter that controls the regularization strength was set to 1.0, while other parameters were kept at default values. The support vector regression algorithm (“SVR” from the “SVM” package) was trained using regression mode with radial basis function kernel, with other parameters kept at default values. For the decision tree regressor algorithm (from the “tree” package), all parameters were kept at default values. For the random forest regressor algorithm (from the “ensemble” package), the parameter max_depth (the maximum depth of the tree) was set to 30, and the parameter max_features (the maximum number of features to be considered when looking for the best split) was set to 10. All other parameters were kept at default values, and a total of 1000 trees were constructed. The exact formulas for the evaluation of regression model performance are available in Supplementary Materials.
Random forest regression feature importance measures
To compute how much each feature contributed to the dataset separation, we computed its importance score in each decision tree and averaged it across all trees in the forest using the “feature\_importances\_” attribute of the sklearn library and across the 50 runs of the algorithm. The importance score of each feature is a numeric value in the interval [0, 1], and the sum of all scores is equal to 1.
Supplementary Material
Acknowledgments
We would like to thank M. Weigt for fruitful discussions and D. Ladant for providing vectors for the ELISA-based linker assay, as well as C. Bouchier and S. Kennedy from the Biomics Pole of Institut Pasteur for performing the sequencing and for general help. Funding: This work was supported by Institut Pasteur, Centre National de la Recherche Scientifique, French Government’s Investissement d’Avenir program Laboratoire d’Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR-10-LABX-62- IBEID), the French National Research Agency (ANR-12-BLAN-DynamINT), Paris Descartes University, and Ecole Doctorale Frontieres du Vivant, Fondation pour la Recherche Medicale (FDT20150532465). Author contributions: D.M., A.N., J.B., M.S.G., J.C., and D.B. designed the research. A.N., M.S.G., C.L., J.C., and L.S. designed and performed the in vivo experiments. A.N. and J.B. designed and performed the machine learning experiments. A.N. and J.B. wrote the draft of the manuscript. All authors read, amended the manuscript, and approved its final version. Competing interests: D.M., A.N., D.B., and M.S.G. are authors of the patent WO/2018/019991-EP3497217 that describes the algorithm that generates synthetic attC sites and its applications. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The algorithm for generating synthetic attC sites as well as data used for ML and the implementations of the algorithms can be found at www.lcqb.upmc.fr/attCsynth. The algorithm that generates synthetic attC sites and embeds them into protein sequences has been submitted to the Agence pour la Protection des Programmes under IDDN FR.001.210001.000.S.P.2017.000.31235. This algorithm and the patent WO/2018/019991-EP3497217 that describes its principles and uses are subject to licensing for any commercial use. The algorithm, bacterial strains, and plasmids can be provided by Institut Pasteur pending scientific review and a completed material transfer agreement. Requests for the material should be submitted to mazel@pasteur.fr or mta@pasteur.fr.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaay2922/DC1
REFERENCES AND NOTES
- 1.Olorunniji F. J., Rosser S. J., Stark W. M., Site-specific recombinases: Molecular machines for the genetic revolution. Biochem. J. 473, 673–684 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Sharan S. K., Thomason L. C., Kuznetsov S. G., Court D. L., Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escudero J. A., Loot C., Nivina A., Mazel D., The integron: Adaptation on demand. Microbiol. Spectr. 3, MDNA3–0019–2014 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Escudero J. A., Loot C., Parissi V., Nivina A., Bouchier C., Mazel D., Unmasking the ancestral activity of integron integrases reveals a smooth evolutionary transition during functional innovation. Nat. Commun. 6, 10937 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francia M. V., Zabala J. C., de la Cruz F., García Lobo J. M., The IntI1 integron integrase preferentially binds single-stranded DNA of the attC site. J. Bacteriol. 181, 6844–6849 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouvier M., Demarre G., Mazel D., Integron cassette insertion: A recombination process involving a folded single strand substrate. EMBO J. 24, 4356–4367 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grindley N. D. F., Whiteson K. L., Rice P. A., Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75, 567–605 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Nivina A., Escudero J. A., Vit C., Mazel D., Loot C., Efficiency of integron cassette insertion in correct orientation is ensured by the interplay of the three unpaired features of attC recombination sites. Nucleic Acids Res. 44, 7792–7803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouvier M., Ducos-Galand M., Loot C., Bikard D., Mazel D., Structural features of single-stranded integron cassette attC sites and their role in strand selection. PLOS Genet. 5, e1000632 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall R. M., Brookes D. E., Stokes H. W., Site-specific insertion of genes into integrons: Role of the 59-base element and determination of the recombination cross-over point. Mol. Microbiol. 5, 1941–1959 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Johansson C., Kamali-Moghaddam M., Sundström L., Integron integrase binds to bulged hairpin DNA. Nucleic Acids Res. 32, 4033–4043 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frumerie C., Ducos-Galand M., Gopaul D. N., Mazel D., The relaxed requirements of the integron cleavage site allow predictable changes in integron target specificity. Nucleic Acids Res. 38, 559–569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald D., Demarre G., Bouvier M., Mazel D., Gopaul D. N., Structural basis for broad DNA-specificity in integron recombination. Nature 440, 1157–1162 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Moura A., Soares M., Pereira C., Leitão N., Henriques I., Correia A., INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25, 1096–1098 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Demarre G., Guerout A.-M., Matsumoto-Mashimo C., Rowe-Magnus D. A., Marliere P., Mazel D., A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPα) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol. 156, 245–255 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Mazel D., Dychinco B., Webb V. A., Davies J., Antibiotic resistance in the ECOR collection: Integrons and identification of a novel aad gene. Antimicrob. Agents Chemother. 44, 1568–1574 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George R. A., Heringa J., An analysis of protein domain linkers: Their classification and role in protein folding. Protein Eng. 15, 871–879 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Karimova G., Pidoux J., Ullmann A., Ladant D., A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 5752–5756 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dautin N., Karimova G., Ullmann A., Ladant D., Sensitive genetic screen for protease activity based on a cyclic AMP signaling cascade in Escherichia coli. J. Bacteriol. 182, 7060–7066 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cury J., Jové T., Touchon M., Néron B., Rocha E. P., Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 44, 4539–4550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu D., Ellis H. M., Lee E.-C., Jenkins N. A., Copeland N. G., Court D. L., An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 5978–5983 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demarre G., Frumerie C., Gopaul D. N., Mazel D., Identification of key structural determinants of the IntI1 integron integrase that influence attC × attI1 recombination efficiency. Nucleic Acids Res. 35, 6475–6489 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishop C. M., Pattern recognition. Mach. Learn. 128, 1–58 (2006). [Google Scholar]
- 24.Lorenz R., Bernhart S. H., Siederdissen C. H. z., Tafer H., Flamm C., Stadler P. F., Hofacker I. L., ViennaRNA package 2.0. Algorithms Mol. Biol. 6, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loot C., Bikard D., Rachlin A., Mazel D., Cellular pathways controlling integron cassette site folding. EMBO J. 29, 2623–2634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loot C., Nivina A., Cury J., Escudero J. A., Ducos-Galand M., Bikard D., Rocha E. P., Mazel D., Differences in integron cassette excision dynamics shape a trade-off between evolvability and genetic capacitance. MBio 8, e02296-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R. J. Lewis, An introduction to classification and regression tree (CART) analysis, in Annual Meeting of the Society for Academic Emergency Medicine, (San Francisco, CA, 2000), pp. 1–14. [Google Scholar]
- 28.Hoerl A. E., Kennard R. W., Ridge regression: Biased estimation for nonorthogonal problems. Technometrics 12, 55–67 (1970). [Google Scholar]
- 29.V. Vapnik, Statistical Learning Theory (Wiley, 1998). [Google Scholar]
- 30.Breiman L., Random forests. Mach. Learn. 45, 5–32 (2001). [Google Scholar]
- 31.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay É., Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 32.Saeys Y., Inza I., Larrañaga P., A review of feature selection techniques in bioinformatics. Bioinformatics 23, 2507–2517 (2007). [DOI] [PubMed] [Google Scholar]
- 33.I. Jolliffe, Principal Component Analysis (Wiley Online Library, 2002). [Google Scholar]
- 34.Larouche A., Roy P. H., Effect of attC structure on cassette excision by integron integrases. Mob. DNA 2, 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menzella H. G., Reid R., Carney J. R., Chandran S. S., Reisinger S. J., Patel K. G., Hopwood D. A., Santi D. V., Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 23, 1171–1176 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Bozhüyük K. A. J., Fleischhacker F., Linck A., Wesche F., Tietze A., Niesert C. P., Bode H. B., De novo design and engineering of non-ribosomal peptide synthetases. Nat. Chem. 10, 275–281 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Bikard D., Julié-Galau S., Cambray G., Mazel D., The synthetic integron: An in vivo genetic shuffling device. Nucleic Acids Res. 38, e153 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loot C., Ducos-Galand M., Escudero J. A., Bouvier M., Mazel D., Replicative resolution of integron cassette insertion. Nucleic Acids Res. 40, 8361–8370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matos J., West S. C., Holliday junction resolution: Regulation in space and time. DNA Repair 19, 176–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grieb M. S., Nivina A., Cheeseman B. L., Hartmann A., Mazel D., Schlierf M., Dynamic stepwise opening of integron attC DNA hairpins by SSB prevents toxicity and ensures functionality. Nucleic Acids Res. 45, 10555–10563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukhortava A., Pöge M., Grieb M. S., Nivina A., Loot C., Mazel D., Schlierf M., Structural heterogeneity of attC integron recombination sites revealed by optical tweezers. Nucleic Acids Res. 47, 1861–1870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biskri L., Bouvier M., Guerout A.-M., Boisnard S., Mazel D., Comparative study of class 1 integron and Vibrio cholerae superintegron integrase activities. J. Bacteriol. 187, 1740–1750 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.J. G. Carbonell, R. S. Michalski, T. M. Mitchell, in Machine Learning: An Artificial Intelligence Approach, R. S. Michalski, J. G. Carbonell, T. M. Mitchell, Eds. (Springer, 1983), pp. 3–23. [Google Scholar]
- 44.P. Baldi, S. Brunak, Bioinformatics: The Machine Learning Approach (MIT Press, ed. 2, 2001). [Google Scholar]
- 45.G. Karimova, D. Ladant, A. Ullmann, L. Selig, P. Legrain, Bacterial two-hybrid system for protein-protein interaction screening, new strains for use therein, and their applications, U.S. Patent 0045237 (2002).
- 46.Espeli O., Moulin L., Boccard F., Transcription attenuation associated with bacterial repetitive extragenic BIME elements. J. Mol. Biol. 314, 375–386 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. III, Smith H. O., Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Plackett R. L., Karl Pearson and the chi-squared test. Int. Stat. Rev. 51, 59–72 (1983). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/30/eaay2922/DC1