

There is a Blood Commentary on this article in this issue.
Key Points
KDM1A/LSD1 is a new therapeutic target for AML; we have identified SNAI1 as a pathological modulator of KDM1A/LSD1 target selection in AML.
Targeting the SNAI1-LSD1 complex or its downstream targets may be a novel and potent therapeutic strategy for the treatment of AML.
Abstract
Modulators of epithelial-to-mesenchymal transition (EMT) have recently emerged as novel players in the field of leukemia biology. The mechanisms by which EMT modulators contribute to leukemia pathogenesis, however, remain to be elucidated. Here we show that overexpression of SNAI1, a key modulator of EMT, is a pathologically relevant event in human acute myeloid leukemia (AML) that contributes to impaired differentiation, enhanced self-renewal, and proliferation of immature myeloid cells. We demonstrate that ectopic expression of Snai1 in hematopoietic cells predisposes mice to AML development. This effect is mediated by interaction with the histone demethylase KDM1A/LSD1. Our data shed new light on the role of SNAI1 in leukemia development and identify a novel mechanism of LSD1 corruption in cancer. This is particularly pertinent given the current interest surrounding the use of LSD1 inhibitors in the treatment of multiple different malignancies, including AML.
Visual Abstract

Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous disease with an average 5-year overall survival (OS) of <40%. Comprehensive genomic profiling of AML patients has resulted in a clearer understanding of the recurrent genetic lesions that underpin the development and pathogenesis of this aggressive leukemia.1,2 The frequent mutation of epigenetic regulators, such as DNMT3a, TET1/2, and IDH1/2, highlights a critical role for deregulated epigenetic mechanisms in AML pathogenesis.3 In contrast to genetic changes, epigenetic modifications are potentially reversible and thus provide unique opportunities for targeted therapy.4 Lysine-specific demethylase 1A (LSD1/KDM1A), hereafter referred to as LSD1, is an H3K4Me1/2 histone demethylase that regulates gene expression through its involvement in various transcriptional complexes such as CoREST and the nucleosome remodelling and deacetylase complex.5,6 LSD1 has emerged as a viable therapeutic target in AML7 because its activity is frequently perturbed in this disease, and studies have demonstrated that LSD1 inhibition and/or downregulation can induce AML cell differentiation in vitro and reduce tumor burden in vivo.8,9 However, the mechanisms by which LSD1 activity is perturbed in AML, and the identity of the key downstream events that contribute to AML pathogenesis, remain unclear.
Epithelial-to-mesenchymal transition (EMT) modulators of the SNAIL (SNAI1/2/3) and ZEB (ZEB1/2) families are key regulators of epithelial tumor biology by facilitating cancer cell invasion and metastasis, acquiring cancer stem cell properties, and activating survival pathways responsible for increased chemotherapy and radiotherapy resistance.10,11 In hematological malignancies, however, the role of these proteins has been largely overlooked because of their perceived lack of relevance in non-EMT contexts. Recently, we and others have begun to show that deregulated expression of EMT modulators represents a previously unrecognized pathogenic event in acute leukemia.12-14 Increased levels of ZEB1 in AML are associated with a more aggressive and invasive phenotype and subsequently poorer OS,13 and ZEB2 has been shown to be a novel regulator of AML differentiation and proliferation12 as well as a driver of early thymic progenitor T-cell acute lymphoblastic leukemia.14 The mechanisms by which EMT modulators contribute to leukemia development and pathogenesis, however, remain to be elucidated.
In this current study, we have discovered a novel oncogenic role for SNAI1 in AML development and show that increased expression of EMT modulators, such as SNAI1, are key contributors to the perturbation of LSD1 activity that is critical for AML pathogenesis.
Materials and methods
Cell culture
RIEP-modified human AML cell lines15 were maintained in RPMI medium supplemented with 10% fetal bovine serum (Sigma), penicillin (100 U/mL-1) and streptomycin (100 μg/mL-1), and 2 mM of l-glutamine (Gibco). HPC-7 cells were kindly provided by Leif Carlsson (Umea University, Umea, Sweden) and grown in Iscove modified Dulbecco medium supplemented with 10% fetal bovine serum (Sigma), penicillin (100 U/mL-1) and streptomycin (100 μg/mL-1), 2 mM of l-glutamine (Gibco), 0.05 mM of 2-mercaptoethanol, and 100 ng/mL of mouse stem cell factor (mSCF; peprotech). Fetal liver cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% (volume/volume) fetal calf serum, mSCF (100 ng/mL), mouse interleukin-6 (mIL-6; 10 ng/mL), mouse Flt3L (5 ng/mL), and mouse thrombopoietin (mTPO; 50 ng/mL).
Retroviral production and cell transduction
The top 3 predicted short hairpin RNAs (shRNAs) for SNAI1 (shSNAI1.774, shSNAI1.1577, and shSNAI1.1633) were cloned into the LMP-miR-E vector,16 and murine wild-type (WT) and mutant Snai1 complementary DNA (cDNA) were subcloned with a 5′FLAG tag into the MSCV-IRES-GFP vector (Addgene). HEK293T packaging cells were transfected with target viral vectors and packaging plasmids using Lipofectamine reagent (Invitrogen), and supernatant was collected after 48 hours and stored in aliquots at −80°C degrees. Retroviral transduction of fetal liver cells and HPC7 cells was performed in 12-well plates coated with 32 μg/mL of retronectin (Takara Bio) using the spin infection protocol. Briefly, retroviral supernatant was spun onto retronectin-coated plates for 2 hours, followed by addition of cells in culture medium supplemented with 4 μg/mL of polybrene (Sigma).
Flow cytometry
Cells were run on an LSR Fortessa (BD Biosciences) flow cytometric machine and data analyzed using FACSDiva or FlowJo software (BD Biosciences). Dead cells were excluded from analysis using either propidium iodide or the fixable viability dye eFluor780 or eFluor450 (eBioscience). Antibodies used for flow cytometry are listed in supplemental Table 6.
Human patient samples
Human patient samples were obtained and used in accordance with the Declaration of Helsinki guidelines, and investigations were performed only after local ethical committee approval and with informed written patient consent. Patient sample details are provided in supplemental Table 1.
Mice and animal procedures
All animal experiments were performed according to the regulations and guidelines in the Australian Code for the care and use of animals for scientific purposes 2013 and approved by the Alfred Medical Research and Education Precinct Animal Ethics Committee. Bone marrow transplantation and Cre induction experiments are described in the supplemental Methods.
Mouse pathology and blood cell analysis
Hematopoietic organs were fixed in 10% buffered formalin and stained with hematoxylin and eosin for histopathological examination. Spleen and bone marrow cytospins (Cytospin 4; Thermo Fisher Scientific) and peripheral blood smears were stained with Wright-Giemsa. Submandibular blood samples were collected into EDTA-coated tubes, and differential counts were performed on a HemaVet 950FS automated blood analysis machine (Drew-Scientific).
Quantitative Real-time polymerase chain reaction
RNA was isolated using the RNeasy midi- or minikit (Qiagen), and RNA concentration was measured on the NanoDrop 1000 Spectrophotometer according to the manufacturer’s instructions (Bio-Rad). cDNA was generated using the SuperScript III reverse transcriptase kit according to the manufacturer’s instructions (Sigma). Relative gene expression of SNAI1 was calculated using the δ-Ct method.17
Methylcellulose culture and replating
Methylcellulose cultures were incubated for 7 days at 37°C and 5% carbon dioxide in duplicate 1.1-mL, 35-mm dishes of Methylcult 3234 (Stem Cell Technologies) supplemented with 100 ng/mL of SCF, 10 ng/mL of IL-3, and 4 IU/mL of erythropoietin. Methylcellulose colonies were dissociated into Dulbecco’s modified Eagle medium/10% fetal calf serum and stained for flow cytometric analysis or cytocentrifuged for cytological analysis (antibodies used are given in supplemental Table 1). For replating assays, cells were washed out of methylcellulose using 6× phosphate-buffered saline (PBS) washes and then replated in Methocult 3234 and cultured for another 7 days.
RNA-seq analysis
At 7 to 10 days posttransduction, RNA was extracted using an RNeasy midikit and quality assessed using a BioAnalyzer machine (Agilent). Library preparation was performed using the Truseq stranded messenger RNA (mRNA) kit (Illumina), and single-end 100-bp reads were generated on an Illumina HiSeq 2500. Details of RNA sequencing (RNA-seq) data analysis are included in the supplemental Methods. RNA-seq data are available from Gene Expression Omnibus (GEO; GSE132724).
ChIP
Chromatin immunoprecipitation (ChIP) assays were performed as previously described18 with 2 × 107 cells per condition using an antibody against H3K4me1 (Abcam #ab8895), H3K4me2 (Millipore #07-030), SNAI1 (Cell Signaling #3879), or LSD1 (Abcam #ab17721). More detailed protocols are given in the supplemental Methods.
ChIP-seq
Library preparation for H3K4me1/2 ChIP samples was performed by BGI (Hong Kong) using a variation of Illumina’s standard protocol. The libraries were sequenced by BGI using a HiSeq2500 analyzer. Library preparation for the Snai1 ChIP samples was performed using the Illumina Truseq ChIP library preparation kit (Illumina), and sequencing was performed on an Illumina NextSeq analyzer. ChIP-seq data are available from GEO (GSE132990).
Western blotting and immunoprecipitation
Western blot analysis was performed using standard protocols with the following antibodies: rabbit polyclonal α-SNAI1 (C15D3; Cell Signaling Technology #3879), HRP-conjugated α-Β-Actin (Sigma #ab49900), and secondary HRP-conjugated rabbit α-mouse (Calbiochem #401353) and goat α-rabbit (Calbiochem #402335) antibodies. Detection was performed using an ECL detection kit (GE Healthcare). Immunoprecipitations were performed using the α-LSD1 antibody (Abcam #ab17721) conjugated to Dynabeads Protein G (Invitrogen #1003D). More detailed protocols are given in the supplemental Methods.
ATAC-seq
A total of 100 000 cells per sample were collected and washed with PBS, pelleted by centrifugation, and washed with lysis buffer (10 mM of tris[hydroxymethyl]aminomethane hydrochloride; pH, 7.4; 10 mM of sodium chloride; 3 mM of magnesium chloride; 0.1% Tween 20) after a 3-minute incubation on ice. The tagmentation reaction (1× Tagment DNA buffer, 0.3× PBS, 0.1% Tween 20, and 2.5 μL of Tn5 [Illumina]) was performed at 37°C for 30 minutes using a thermocycler in a 50-μL volume as previously described.19 Reactions were immediately purified using a MinElute PCR Kit (Qiagen) and underwent 13 cycles of amplification with indexing adapters before being sequenced on the NovaSeq 6000 platform (Illumina). Replicate sample libraries (3×) were generated on successive days. Assay for transposase-accessible chromatin (ATAC)–seq data are available from GEO (GSE147873).
Results
Increased SNAI1 expression plays a key role in human AML pathogenesis
Using the BloodSpot database, we discovered that SNAI1 expression is significantly increased in AML compared with normal hematopoietic stem/progenitor cells irrespective of presence of genetic abnormality/driver mutation (supplemental Figure 1A). To confirm this result, we performed quantitative real-time polymerase chain reaction (qRT-PCR) on 6 AML patient samples (supplemental Table 1) and 3 human CD34+ control hematopoietic stem/progenitor cell preparations and found that SNAI1 mRNA levels were on average ∼12-fold higher in the AML samples (Figure 1A). A recent study by Shousha et al20 also found an increased level of SNAI1 in AML patients compared with healthy controls. In their study, they observed only a 2.6-fold increase in SNAI1 mRNA; however, they analyzed whole-blood samples rather than bone marrow, which may explain their lower fold change result. Furthermore, we could also observe a clear increase in protein levels for SNAI1 in a separate cohort of 4 of 6 primary AML patient samples compared with normal CD34+ cells (Figure 1B) and in 3 of 6 AML cell lines (Figure 1C). Because of dynamic and rapid degradation of SNAI1 protein, primary AML and CD34+ cells had to first be treated with a proteasome inhibitor (MG132) for 24 hours to stabilize the SNAI1 protein before western blot analysis.
Figure 1.
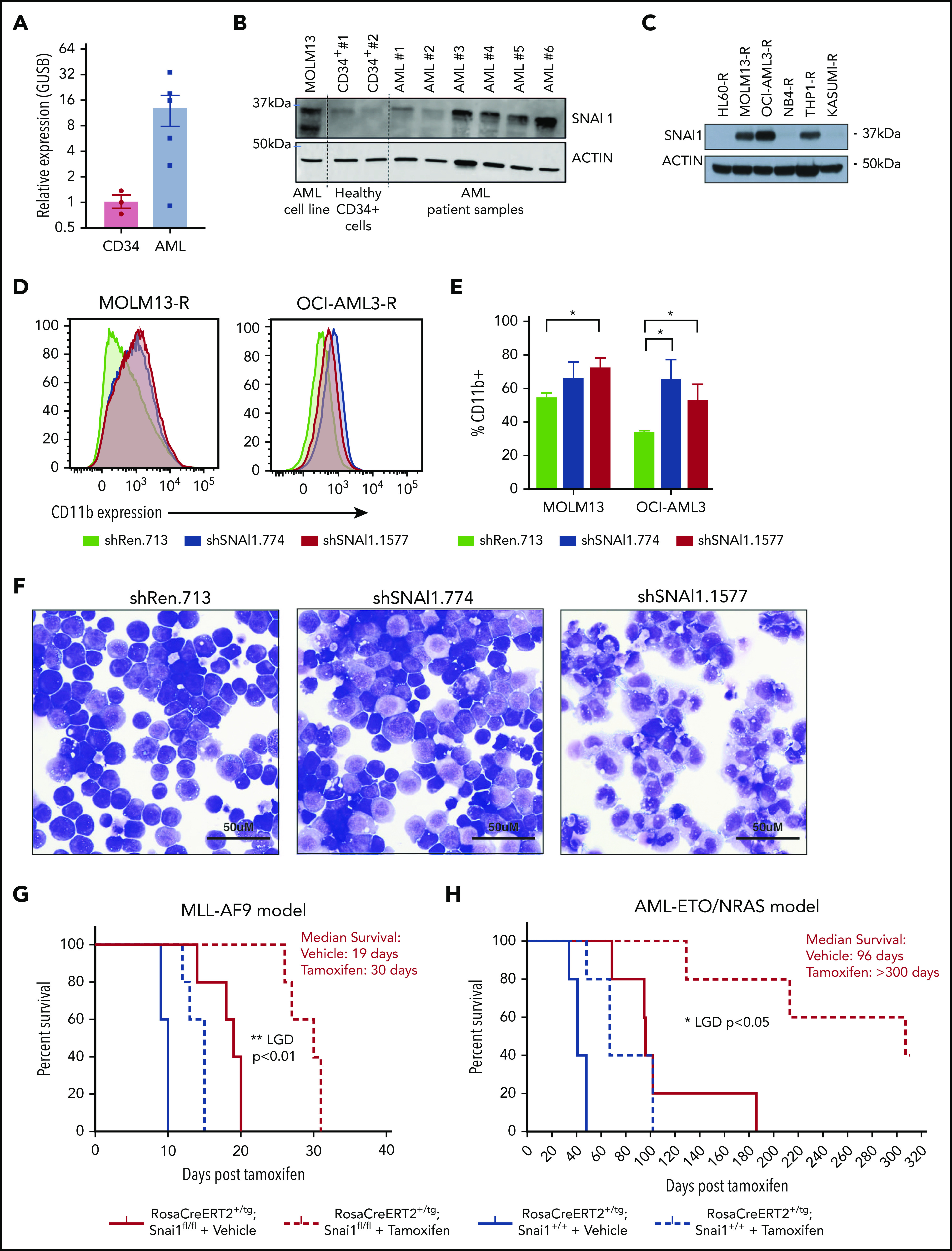
A putative role for SNAI1 in human AML. (A) Quantitative real-time PCR analysis showing SNAI1 mRNA is expressed ∼12-fold higher in human AML patient samples compared with normal hematopoietic stem and progenitor cells (CD34+, n = 3; AML, n = 6; P < .05 Mann-Whitney nonparametric 1-tailed test). SNAI1 expression is normalized to the expression of the housekeeping gene GUSB. (B) SNAI1 protein expression is higher in AML patient samples compared with healthy CD34+ control cells. The MOLM13 cell line is also shown as a comparison. (C) Western blot analysis showing MOLM13-R, OCI-AML3-R, and THP1-R AML cell lines express SNAI1 protein, whereas HL60-R, NB4-R, and Kasumi-R do not. (D) shRNA-mediated SNAI1 knockdown in OCI-AML3-R and MOLM13-R AML cell lines results in upregulation of the myeloid maturation marker CD11b (red and blue lines) compared with the control shRen.713 shRNA (black line). (E) Quantification of the percentage of CD11b+ cells in shRen.713 infected cells (black bars) compared with shSNAI1 infected cells (red bars). Data are represented as mean + standard error of the mean; n = 3 independent replicates. (F) Wright-Giemsa staining analysis of MOLM13-R cells shows evidence of myeloid differentiation, such as increased cytoplasmic/nuclear ratio and presence of cytoplasmic granules, upon SNAI1 knockdown in shSNAI1.774 and shSNAI1.1577 cells compared with control shRen.713 cells. Kaplan-Meier plots showing that Cre (tamoxifen)–mediated loss of Snai1 in MLL-AF9 (G) and AML-ETO/NRAS–driven (H) AML models leads to a significant reduction in the survival of recipient mice as determined by leukemia growth delay (LGD)52 analysis (AML-ETO, P < .05; MLL-AF9, P < .01). Red lines indicate Snai1fl/fl cells, and black lines indicate Snai1+/+ cells. Dotted lines indicate mice treated with tamoxifen vs solid lines indicated mice treated with vehicle. Data are from 5 recipient mice per cohort, each transplanted with 300 000 (MLL-AF9) or 500 000 (AML-ETO/NRAS) AML cells combined from 2 to 3 primary tumors. *P < .05 Student 2-sided unpaired t test, **P < .01 Mantel-Cox test.
Using 2 independent AML patient gene expression data sets (available from the online databases PROGgene21,22 and UCSC Cancer Browser23), we identified a significant correlation between high SNAI1 expression (above the median) in AML patients with normal cytogenetics and reduced OS (supplemental Figure 1B). This survival advantage was only observed in patients with normal-/intermediate-risk cytogenetics, and no correlation with common AML mutations could be observed (data not shown).
To determine how increased SNAI1 expression contributes to AML biology, we performed shRNA-mediated SNAI1 knockdown using existing optimized miR-E-shRNA murine retroviral vectors16 in 6 human AML cell lines that were modified (AML-RIEP) to express the retroviral ecotropic receptor (rtTA3-IRES-EcoR-PGK-Puro).15 Three of these AML-RIEP cell lines (OCI-AML3-R, MOLM13-R, and THP1-R) expressed SNAI1 protein, whereas the other 3 cell lines (HL60-R, NB4-R, and KASUMI-R) were negative for SNAI1 under steady-state conditions (Figure 1C). We tested the top 3 predicted shRNAs for human SNAI1 according to Fellman et al16 in MOLM13-R cells and identified a 60% to 90% knockdown at the RNA level for all 3 shRNAs (supplemental Figure 1C). A concomitant reduction in SNAI1 protein level was seen for shSNAI1.774 and shSNAI1.1577 vs the control shREN.713, an shRNA targeting the Renilla gene (supplemental Figure 1D). Because no effect on SNAI1 protein levels was seen for shSNAI1.1663, we excluded this from future experiments. We successfully transduced 5 of 6 of these AML cell lines (THP1-R was refractory to transduction, suggesting the inserted ecotropic receptor gene had been shut down or lost) with either 1 of the 2 best-performing shRNAs (shSNAI1_774 and shSNAI1_1577) or a control shRNA targeting the Renilla gene (shRen_713). Two weeks posttransduction, shRNA-expressing OCI-AML3-R and MOLM13-R (SNAI1+; Figure 1C) cells showed significantly increased levels of CD11b on their surface (indicative of myeloid maturation; Figure 1D-E), whereas no change in CD11b expression was observed in the SNAI1− (Figure 1C) NB4-R, HL60-R, or Kasumi-R cell lines (supplemental Figure 1E-F). The increase in CD11b expression observed in the MOLM13-R cell line further correlated with morphological changes consistent with partial myeloid differentiation, including an increased cytoplasmic/nuclear ratio and presence of cytoplasmic granules (Figure 1F).
Snai1 expression is required for AML pathogenesis driven by common AML oncogenes
To investigate whether endogenous expression of Snai1 is required for the pathogenesis of common AML oncogenes, we generated mouse AMLs driven by either the MLL-AF9 (t9;11) or AML/ETO (t8;21)/NRAS human oncogenes on a tamoxifen-inducible RosaCreERT2-Snai1fl/fl background.24 Tumor latency was significantly increased for the MLL-AF9 model when Snai1 was deleted (median survival of 30 days for tamoxifen compared with 19 days for vehicle-treated mice; Figure 1G red lines). The delay in development of the AML-ETO/NRAS model was even more profound (96 days for vehicle-treated and >300 days for tamoxifen-treated mice), with 3 of 5 mice remaining alive at the end of the experiment (300 days posttransplantation; Figure 1H red lines). Importantly, the delay in tumor latency after Snai1 deletion was significantly longer in both models than that induced by Cre toxicity alone25 (Figure 1G-H black lines).
Ectopic Snai1 induces myeloproliferation and predisposes mice to leukemia development
To determine the effect of increased Snai1 expression on hematopoietic development, we used our published conditional gain-of-function Snai1 transgenic mice26 (supplemental Figure 2A). Expression of the Snai1-IRES-EGFP transgene from the Rosa26 promoter was induced specifically in the hematopoietic lineage by breeding transgenic mice onto a Vav-iCretg/+ transgenic background.27 Vav-iCre+/−Snai1tg/+ (Snai1 heterozygous) or Vav-iCre+/−Snai1tg/tg (Snai1 homozygous) transgenic mice were born at normal Mendelian ratios (data not shown). The levels of Snai1 mRNA in the bone marrow of transgenic mice were approximately eightfold (Snai1tg/+) to 15-fold (Snai1tg/tg) greater than those observed in WT littermate controls (Figure 2A), comparable to the increased levels of SNAI1 we observed in human AML samples.
Figure 2.
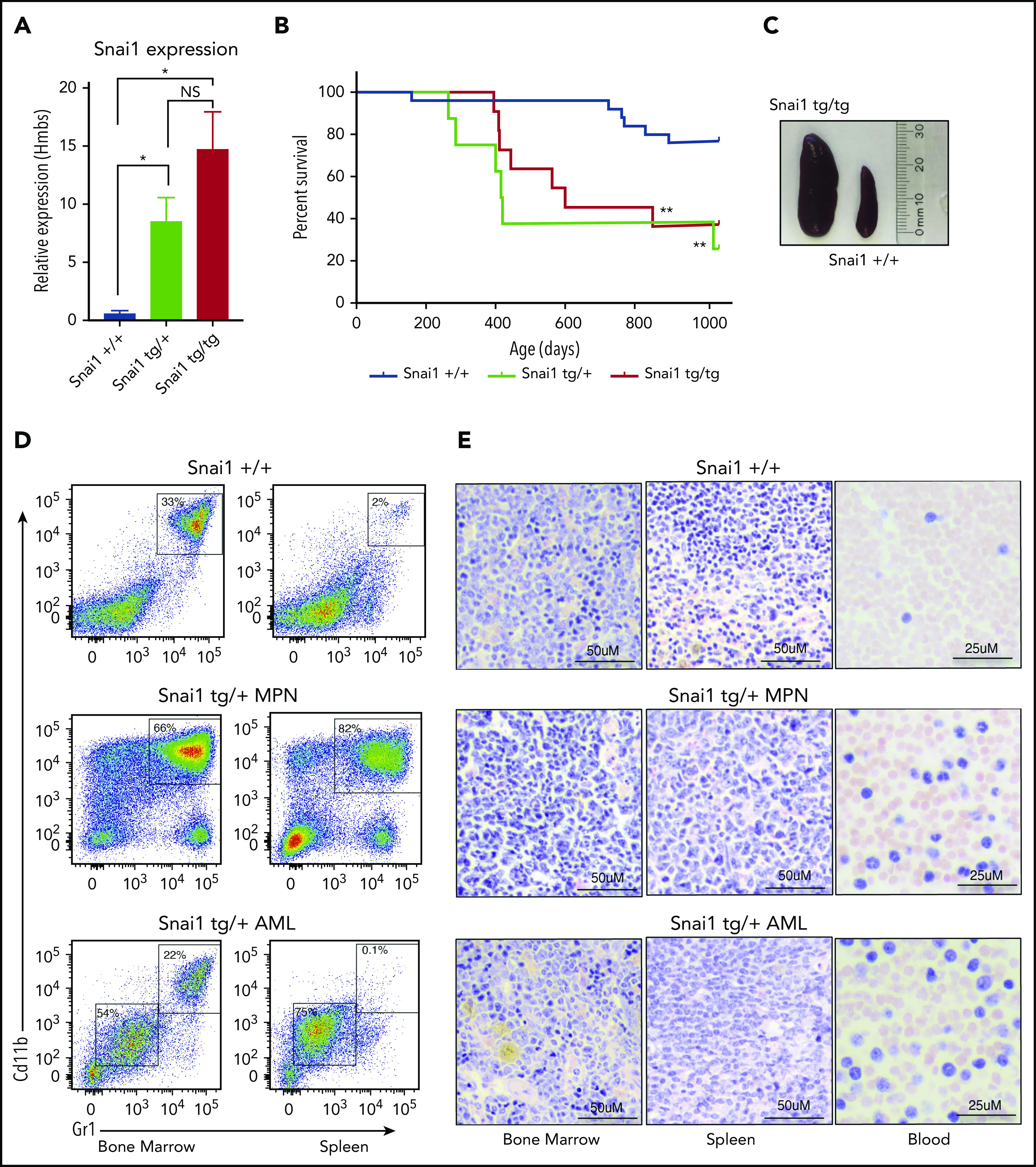
Enforced hematopoietic expression of Snai1 predisposes mice to AML development. (A) Quantitative real-time PCR analysis showing Snai1 mRNA levels were approximately eightfold to 15-fold higher in transgenic mouse bone marrow, as compared with WT controls (Snai1+/+, n = 3; Snai1tg/+, n = 3; Snai1tg/tg, n = 3). Data are presented as mean + standard error of the mean. (B) Kaplan-Meier plot showing Snai1 transgenic mice become moribund from ∼12 months of age, with a median age of 601 days for Snai1tg/tg and 418 days for Snai1tg/+. No significant difference in survival was observed between heterozygous and homozygous transgenic mice. (C) Pictograph showing splenomegaly in an Snai1tg/tg mouse. (D) Representative flow cytometric plots with predominant CD11b+GR1+ cell populations in both the bone marrow and spleen. Flow cytometric profiles of CD11b and GR1 expression on bone marrow and spleen cells are shown for a representative WT mouse in the top panel, a moribund mouse with a myeloproliferative disease in the middle panel, and a mouse with AML in the bottom panel. (E) Hematoxylin and eosin staining of bone marrow, spleen, and peripheral blood histological sections of the same mice as in panel D are shown. A representative WT mouse is depicted in the top panel. A representative mouse with myeloproliferative disease is shown in the middle panel, with evidence of granulocytic cell infiltration in the spleen and blood, increased numbers of granulocytic precursor and mature cells in the bone marrow and spleen, and reduced erythropoiesis. In the bottom panel, a representative mouse with AML is shown as evidenced by myeloid blast cell infiltration into bone marrow, spleen, and peripheral blood, along with loss of normal hematopoiesis in the bone marrow and spleen. *P < .05 1-tailed Student unpaired t test, **P < .01 Mantel-Cox test.
We aged a cohort of Snai1 transgenic mice up to 34 months of age (952 days) and found that from 12 months onward, mice progressively became moribund with evidence of anemia and splenomegaly (Figure 2B-C). In Snai1 transgenic mice analyzed upon terminal disease development (Snai1tg/+, n = 7; Snai1tg/tg, n = 2), flow cytometry of hematopoietic organs revealed an expanded population of myeloid cells with variable levels of CD11b+GR1+ expression (Figure 2D; supplemental Figure 2B) or, in 1 case, cells expressing CD71+Ter119+ (supplemental Figure 2C). Histological analysis revealed the presence of myeloproliferation in all mice, including hypercellularity and disordered architecture of bone marrow and spleen, increased granulopoiesis, expanded erythropoiesis, and an increase in immature myeloid forms (Figure 2E; supplemental Figure 2Di). Three mice further demonstrated clear AML development, with excessive blast cells evident in bone marrow, spleen, and/or peripheral blood (Figure 2E; supplemental Figure 2Dii). Two of the AMLs (1 Snai1tg/+ and 1 Snai1tg/tg) as well as one of the myeloproliferative diseases (Snai1tg/+) could be transplanted into NSG mice, with recipient mice showing development of a disease similar to that of the primary donors (supplemental Figure 2E).
Ectopic Snai1 significantly perturbs myeloid cell development
To gain a better understanding of how Snai1 expression predisposes to leukemia, we analyzed myeloid development in preleukemic Snai1 transgenic mice at 8 months of age. Peripheral blood counts of transgenic animals did not differ from those of WT littermate controls (supplemental Figure 3A), and hematopoietic organ cellularity and architecture were also normal (supplemental Figure 3B-C). Flow cytometric analysis of bone marrow identified a slight but nonsignificant increase in the number of hematopoietic stem cell (HSC)–enriched LSK cells, as well as a significant expansion of the myeloid progenitor (MP) compartment in Snai1tg/tg mice (Figure 3A-B). Within the LSK compartment, Snai1tg/tg mice further displayed a significant increase in the number of short-term HSCs (ST-HSCs) but no difference in multipotent progenitors (MPPs) or long-term HSCs (LT-HSCs) (Figure 3C-D). Within the myeloid progenitor compartment, both the Snai1tg/+ and Snai1tg/tg mice were found to have a significant increase in the number of granulocyte macrophage progenitor cells (GMPs) but no change in the number of common myeloid progenitor cells (CMPs). A slight but significant decrease in the number of megakaryocyte erythroid progenitor cells (MEPs) was observed in the Snai1tg/+ mice (Figure 3E-F).
Figure 3.
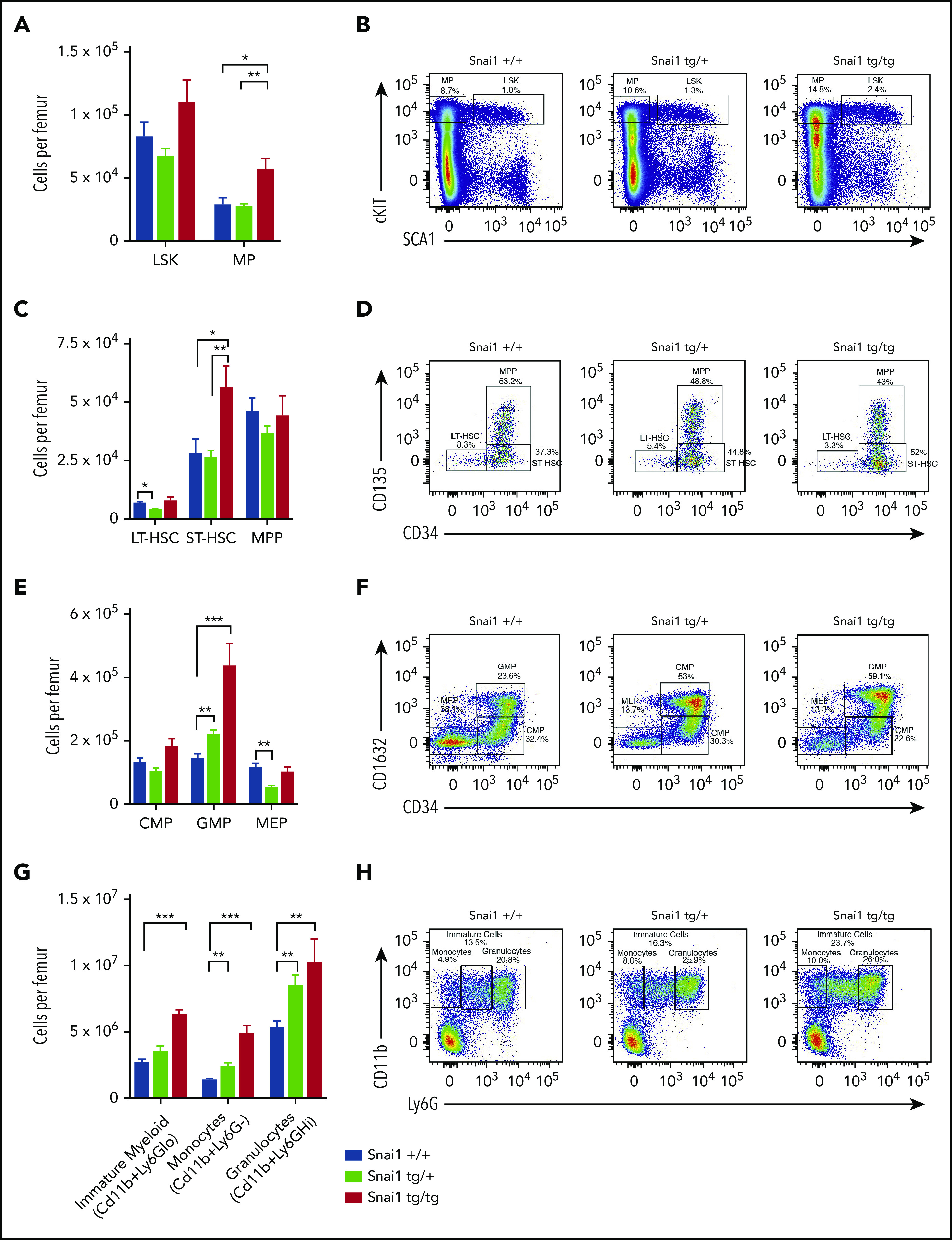
Snai1 overexpression perturbs myeloid lineage development. (A) Immunophenotypic characterization of hematopoietic stem (LSK) and myeloid progenitor (MP) cell populations within the bone marrow of Snai1 transgenic mice. (B) Flow cytometric analysis showing representative dot plots of LSK gated cells. (C) Within the stem cell compartment, there was a significant increase in the number of short-term HSCs (ST-HSCs; LIN−cKIT+SCA1+CD34+CD135−) but no change in the number of long-term HSCs (LT-HSCs; lin−cKit+Sca1+CD34−-CD135−) or multipotent progenitors (MPPs; LIN−cKIT+SCA1+CD34+CD135+). (D) Representative dot plots of LT-HSC, ST-HSC, and MPP gated cells are shown. (E) A significant increase in the number of common myeloid progenitors (CMPs; LIN−cKIT+SCA1−CD34+CD16/32−) GMPs (LIN−cKIT+SCA1−CD34+CD16/32+) was observed; however, the megakaryocyte/erythroid progenitor (MEP; LIN−cKIT+SCA1−CD34−CD16/32−) population was slightly reduced (significant only in the heterozygous mice). (F) Representative FACs plots of myeloid progenitor gating are shown. (G) A significant increase in the number of immature (CD11b+Ly6Glo) and mature myeloid cells (monocytes CD11b+GR1− and granulocytes CD11b+Ly6Ghi) was also evident in the bone marrow of Snai1 transgenic mice. (H) Flow cytometric analysis showing representative dot plots of myeloid gated cells. (A,C,E,G) Data are represented as mean + standard error of the mean for Snai1+/+ (n = 13), Snai1tg/+ (n = 8), and Snai1tg/tg (n = 7) biological replicates.*P < .05, **P < .01, ***P < .001 Student 2-sided unpaired t test.
Further downstream of the GMPs, we identified a significant increase in the number of myeloid lineage cells (CD11b+) in the bone marrow and spleen of Snai1 transgenic mice. Ly6G levels on Cd11b+ cells can distinguish immature myeloid cells and monocytes, which are Ly6Glow or Ly6G−, respectively, from mature granulocytes, which are Ly6Ghigh. All myeloid cell populations were found to be expanded in both Snai1tg/+ and Snai1tg/tg mice, suggesting an overall increase in myeloid cell output (Figure 3G-H).
Snai1-induced perturbed myelopoiesis is a cell-intrinsic phenotype
To further explore this perturbed myeloid differentiation phenotype and compare Snai1 transgenic stem and progenitor cell fitness with that of WT control cells, we performed a competitive bone marrow transplantation. Test bone marrow from 6- to 8-month-old Snai1+/+, Snai1tg/+, or Snai1tg/tg mice (CD45.2+) was transplanted alongside competitor bone marrow from Snai1+/+ C567BL/6 (CD45.1+) mice into lethally irradiated C57BL/6 mice (supplemental Figure 4A). Mice were then analyzed at 12 and 20 weeks posttransplantation.
Particularly evident in the spleen, both the Snai1tg/+ and Snai1tg/tg cells contributed to a significantly higher proportion of myeloid cells at both time points compared with Snai1+/+ cells (supplemental Figure 4B-C). These data demonstrate that the perturbed myelopoiesis in the Snai1 transgenic animals is a cell-intrinsic effect.
In the bone marrow, there was no significant difference in the contribution of Snai1tg/+ or Snai1tg/tg cells to the stem and progenitor cell compartment, with the exception of a mild increase in the contribution of the Snai1tg/+ cells to the GMP lineage at 12 weeks posttransplantation (supplemental Figure 4D-E). These data indicate that the changes in the stem and progenitor populations seen in the transgenic mice may be compensatory because of an altered bone marrow microenvironment.
To confirm that the myeloid differentiation defects were indeed cell intrinsic, we performed methylcellulose colony-forming assays. Whole bone marrow from Snai1tg/+ and Snai1tg/tg mice generated normal numbers of hematopoietic colonies compared with Snai1+/+ mice (data not shown). However, the colony type was significantly skewed toward the granulocyte/macrophage lineage, with a higher number of colony-forming unit granulocyte/macrophage (CFU-GM) and a concomitant reduction in colony-forming unit granulocyte/erythroid/macrophage/megakaryocyte (CFU-GEMM) in both the Snai1tg/+ and Snai1tg/tg cultures (Figure 4A). Furthermore, cytocentrifuge and flow cytometric analysis of cells washed out of methylcellulose revealed an increase in the number of immature myeloid cells (red arrows) within the transgenic colonies and a reduction in the number of mature granulocytes (green arrows) compared with WT colonies (Figure 4B), suggesting terminal myeloid differentiation was impaired. Colony size also seemed larger in the transgenic cultures, with the number of cells per colony being higher in the Snai1 transgenic cultures, although this was only significant for the Snai1tg/tg cultures (Figure 4C).
Figure 4.
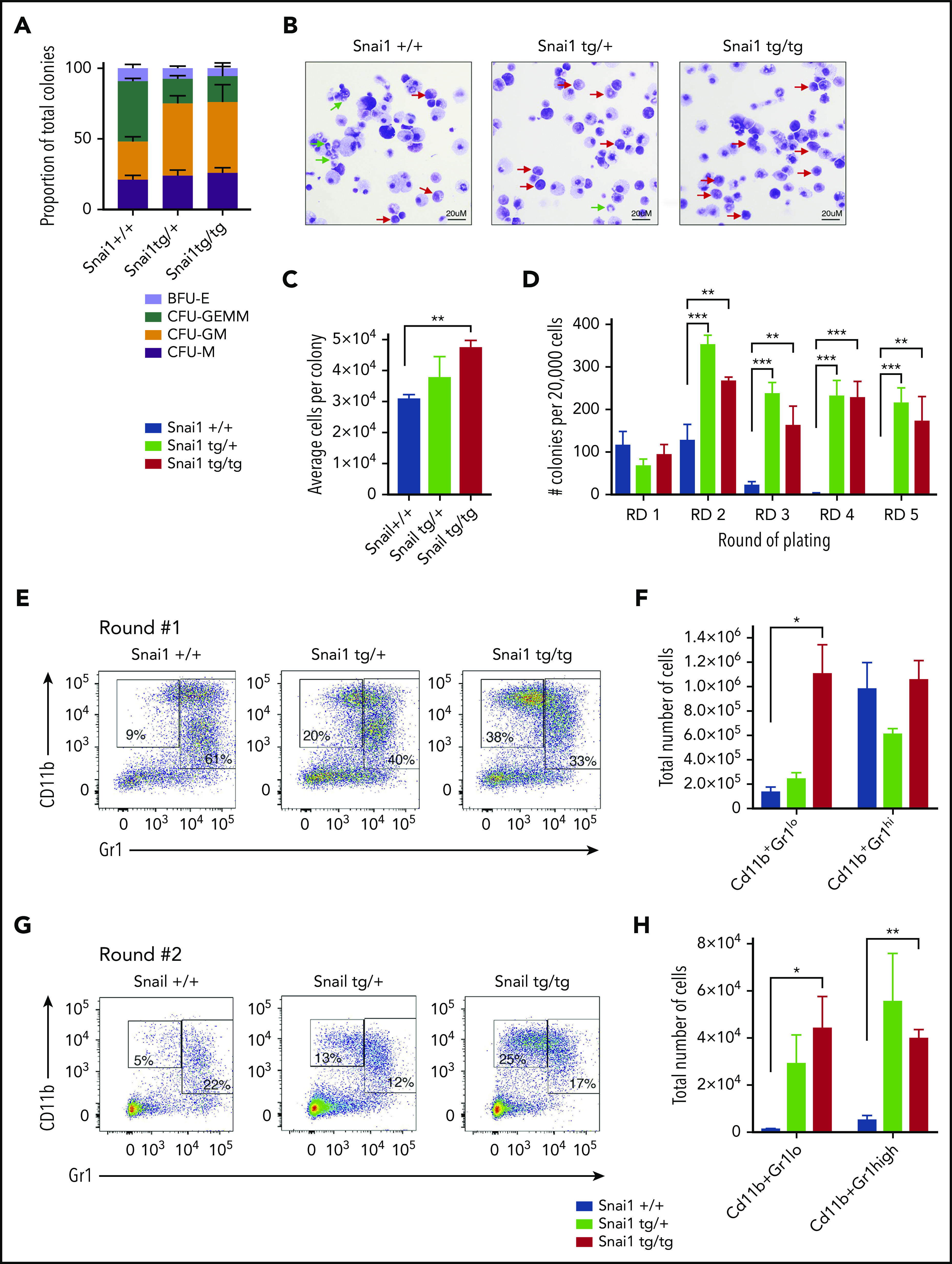
Snai1 overexpression induces self-renewal potential in immature myeloid cells and impairs granulocytic differentiation. (A) Snai1tg/+ and Snailtg/tg bone marrow generate significantly more colony-forming unit (CFU) granulocyte/macrophage (GM) colonies and significantly fewer CFU granulocyte/erythroid/macrophage/megakaryocyte (GEMM) colonies as compared with Snai1+/+ bone marrow. (B) Wright-Giemsa staining analysis of Snai1tg/+ and Snailtg/tg methylcellulose colony cytospins showing almost complete lack of mature granulocytes (green arrows) and an increase in immature myeloid cells (red arrows) as compared with Snai1+/+ colonies. (C) The number of cells per colony (calculated as total number of cells per total number of colonies) was significantly increased in Snailtg/tg cultures compared with Snai1+/+ cultures. Snai1tg/+ cultures also showed a trend toward an increased number of cells per colony; however, this was not significant. (D) Quantification of bone marrow methylcellulose colonies showing that Snai1tg/+ hematopoietic progenitor cells have increased self-renewal capability compared with Snai1+/+ controls, with Snai1tg/+ and Snai1tg/tg cells able to generate colonies in methylcellulose up to 5 rounds of replating. Snai1+/+ cells were only able to replate up to 3 rounds. (E) Flow cytometric analysis of methylcellulose-derived hematopoietic cells showing a higher percentage of immature and mature myeloid cells generated from Snai1tg/+ and Snai1tg/tg progenitor cells at the first round of culture. (F) Quantification of immature (CD11b+GR1lo) and mature (CD11b+GR1hi) myeloid cell populations in Snai1+/+, Snai1tg/+, and Snai1tg/tg cultures showing a significant increase in immature myeloid cells in the Snai1tg/tg cultures at the first round of culture. (G-H) A similar increase in immature myeloid cells was also observed after the second round of replating. (A,C,D,F,H) Data are represented as mean + standard error of the mean; n = 3 biological replicates. *P < .05, **P < .01, ***P < .001 Student 2-tailed unpaired t test.
Ectopic Snai1 increases self-renewal of myeloid progenitors
Given that Snai1 expression has previously been associated with promoting stemness in mammary tumors,28 we sought to determine whether the expanded myeloid progenitor cell compartment of transgenic mice displayed increased self-renewal capacity. We performed serial replating methylcellulose assays on bone marrow from Snai1+/+, Snai1tg/tg, and Snai1tg/+ mice and observed that although colony numbers after the first round of colony formation were similar between WT and transgenic cultures, the subsequent rounds of replating all resulted in an increase in the number of colonies generated by the transgenic cells (Figure 4D). Furthermore, although transgenic cells were capable of replating up to at least 5 rounds in methylcellulose, all self-renewal capacity was exhausted in the WT cells after the third round of replating (Figure 4D). The colonies generated at round 2 were again larger in the transgenic cultures, with a concomitant increase in the total number of cells per culture dish (data not shown). Immunophenotypic analysis of cells washed out of methylcellulose revealed a significant increase in the number of immature (CD11b+, GR1lo) myeloid cells in the transgenic colonies after round 1 of replating (Figure 4E-F), which was also evident after round 2 (Figure 4G-H). Morphological analysis further demonstrated that WT colonies from round 2 consisted predominantly of mast cells (supplemental Figure 4F green arrows), whereas transgenic cultures still retained a large number of immature myeloid cells (supplemental Figure 4F red arrows).
Ectopic SNAI1 perturbs normal myeloid differentiation via its interaction with the histone demethylase LSD1
In epithelial tumor contexts, SNAI1 is known to exert its predominantly gene-repressive functions via interactions with histone modifying complexes, including the CoREST/HDAC complex.29,30 A key component of this complex is the histone demethylase LSD1, which has been shown to directly bind SNAI1 via its SNAG (SNAIL/GFI) protein interaction domain.30 LSD1 is an integral cofactor of the SNAG domain–containing hematopoietic transcription factors GFI1/1B and has also been shown to cooperate with other key hematopoietic transcription factors, including RUNX1, GATA2, SCL/TAL1, and SALL4.31-34 Through these interactions, LSD1 is essential for maintaining normal hematopoietic stem cell function and self-renewal, as well as for driving myeloid differentiation.34-36 Importantly, loss of LSD1 function in mice results in similar myeloid differentiation defects (eg, impaired granulocytic development and expansion of immature myeloid cells and GMPs), as we observed in our Snai1 transgenic mice, suggesting that increased expression of SNAI1 in the hematopoietic system may affect LSD1 function.35,36 To test whether ectopic SNAI1 can physically interact with LSD1 in the hematopoietic context, we performed coimmunoprecipitation studies in the murine hematopoietic progenitor cell line, HPC7, which had been transduced with a retroviral vector encoding FLAG-tagged SNAI1.37 Immunoprecipitation with an anti-LSD1 antibody was able to pull down both endogenous and ectopic SNAI1 (Figure 5A), consistent with previous studies in epithelial cells.30 To determine whether this SNAI1/LSD1 interaction is required for the myeloid defects induced by Snai1, we cloned either WT Snai1 (MIG-Snai1) or a mutant form of Snai1 (MIG-mut5Snai1), which is unable to bind LSD1, into an MSCV-IRES-GFP retroviral vector backbone (Figure 5B). The mutant Snai1 cDNA encodes a protein with a single amino acid change at position 5 (A>F) of the N-terminal SNAG domain that blocks LSD1 binding but does not impair protein stability, localization, or folding30 (Figure 5C; data not shown). Coimmunoprecipitation analysis confirmed the inability of this mutant form of SNAI1 to bind LSD1 (Figure 5A). Next, we generated mouse bone marrow chimeras using C57BL/6 fetal liver cells transduced with MIG-Snai1, MIG-mut5Snai1, or empty vector MIG-EV retrovirus. At 12 weeks posttransplantation, GFP+ cells from MIG-Snai1 bone marrow chimeric mice displayed the same myeloid developmental defects and cell expansion we had previously identified in the Snai1 transgenic mice (Figure 5D-G blue bars). The phenotype, however, was much more profound in the retroviral setting, with an expansion of immature myeloid cells and a reduction in mature granulocytes also readily apparent upon morphological analysis of GFP+ bone marrow cells from MIG-Snai1 mice (Figure 5H red arrows). These data further confirm that the Snai1-induced myeloid defects are due to cell-intrinsic effects of Snai1 expression on myeloid cell development.
Figure 5.
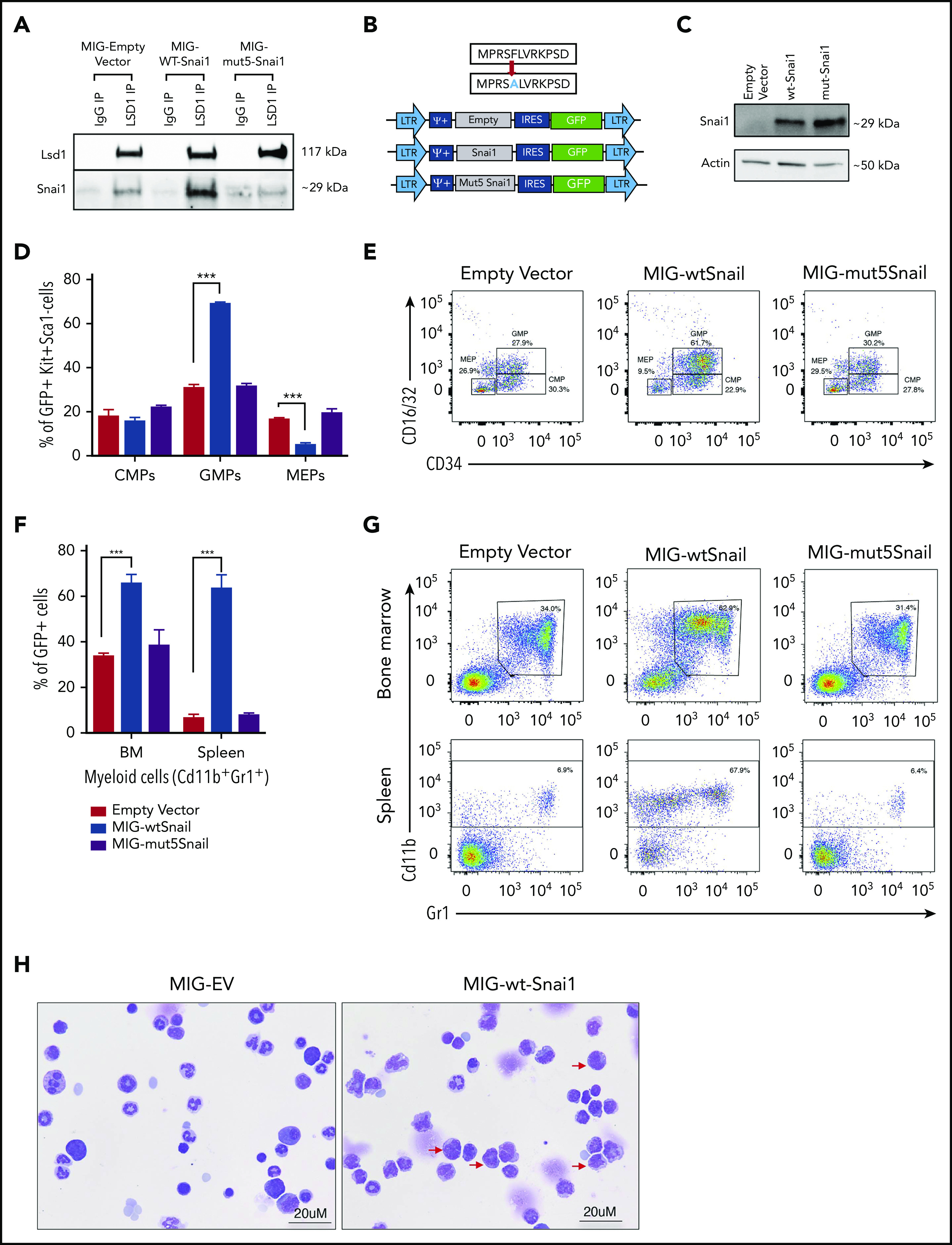
SNAI1 requires interaction with LSD1 to induce myeloid development defects. (A) Western blot analysis showing that LSD1 immunoprecipitation is able to pull down SNAI1 in the mouse hematopoietic progenitor cell line (HPC7). Empty vector control (MIG-EV)–transduced cells and MIG-mut5Snai1–transduced cells show a low level of endogenous SNAI1 pulldown, whereas the MIG-Snai1–transduced cells show a much higher level of SNAI1 pulldown because of the overexpressed WT-SNAI1 protein also being pulled down. The overexpressed mut5SNAI1 protein is not able to be pulled down by LSD1. (B) Overview (upper panel) of the mutant version of Snai1 that was generated with a phenylalanine (F) to alanine (A) amino acid change at position 5 of the SNAI1 protein. WT Snai1 cDNA and the mutant Snai1 cDNA were individually cloned into the MSCV-IRES-GFP retroviral vector. An empty MSCV-IRES-GFP vector was used as a transduction control. (C) MIG-Snai1– and MIG-mut5-Snai1–transduced cells both show high levels of SNAI1 protein, whereas the endogenous SNAI1 protein is unable to be detected in the empty vector–transduced cells. The western blot also demonstrates that the F→A mutation in the mut5-SNAI1 protein does not affect its overall protein stability or antibody recognition. (D) Flow cytometric quantification of the GFP+ bone marrow cell population in MIG-Snai1–recipient mice at 12 weeks posttransplantation, showing a significantly increased proportion of granulocyte/macrophage progenitor cells (GMPs) and a significantly decreased proportion of megakaryocyte/erythroid progenitor cells (MEPs) compared with GFP+ cells in MIG-EV–recipient mice (blue bars compared with black bars). The common myeloid progenitor cell (CMP) population was not different between the 2 mouse cohorts (left panel). No difference was observed in MIG-mut5Snai1 bone marrow compared with MIG-EV control bone marrow (purple bars compared with black bars). (E) Representative myeloid progenitor flow cytometric plots from empty vector, MIG-Snai1, and MIG-mut5Snail mice. (F) A significant increase in the proportion of mature myeloid cells was also observed within the GFP+ cell population in MIG-Snai1–recipient mouse bone marrow and spleen (blue bars compared with black bars). These myeloid abnormalities were completely absent in the MIG-mut5Snai1–recipient mice (purple bars). Data are represented as mean + standard error of the mean (SEM); n = 3 biological replicates. (G) Representative myeloid cell flow cytometric plots from bone marrow and spleens of MIG-EV, MIG-Snai1, and MIG-mut5Snail mice. (H) Wright-Giemsa staining of GFP+ bone marrow cytocentrifuge preparations shows normal myeloid development in MIG-EV–recipient mice, whereas in MIG-Snai1–recipient mice, there is a significant increase in the number of immature myeloid cells (red arrows), and a significant reduction of mature granulocytes. (D-E) Data are presented as mean + SEM; n = 3 mice from each cohort ***P < .001 Student 2-sided unpaired t test.
Notably, the myeloid defects observed in the MIG-Snai1 mice were completely absent in the MIG-mut5Snai1 mice (Figure 5D-G purple bars). Combined, these data verify that the 5F>A mutation in the SNAG domain completely abolishes the ability of SNAI1 to perturb myeloid development both in vivo and in vitro, signifying a requirement for LSD1 binding in SNAI1-induced myeloid defects.
Ectopic SNAI1 induces an altered myeloid differentiation gene expression program through perturbed LSD1 activity
Increased levels of SNAI1 in stem/progenitor cells may: (1) sequester LSD1 away from its normal hematopoietic interacting partners (ie, GFI1/1b, RUNX1, GATA2, SCL/TAL1, and SALL431-34), leading to reduced demethylation of its H3K4me1/2 histone substrates at promoter/enhancer elements of target genes, and/or (2) result in binding of SNAI1/LSD1 complexes at promoters and enhancers containing SNAI1 E-box binding motifs. LSD1 may then demethylate SNAI itself and/or enhance the demethylation of H3K4me1/2 marks specifically at SNAI1 target genes. To investigate these proposed models further, we performed RNA and ChIP-seq as well as ATAC-seq analysis on the HPC7 cell line transduced with either MIG-Snai1, MIG-mut5Snai1, or MIG-EV retroviral vectors.
Using a false discovery rate (FDR) cutoff of P < .05, we identified 474 significantly upregulated genes (the second highest being Snai1 itself) and 361 significantly downregulated genes upon ectopic expression of WT Snai1 (supplemental Figure 5A; supplemental Table 2). In contrast, the mut5-Snai1–expressing cells showed no differentially expressed genes (other than mutant Snai1 itself) compared with MIG-EV-transduced control cells (supplemental Figure 5B).
Differentially expressed genes (FDR < 0.05) in the MIG-Snai1–transduced HPC7 cells cover a broad spectrum of biological processes, including, but not limited to, alterations in cytokine signaling (eg, Il1b, Il1r2, Csf1r, and Csf2rb), transcriptional regulation (eg, AP-1 family members Jun/Fos, Hes1, Cebpb, and Gata1), migration/adhesion/invasion (eg, Mmp8/9/19, Ccl2/Ccr2, and Cxcr4), and emerging tumor suppressor genes (eg, Ssbp2, Zdhhc14, Sik1, and Hook138-41; supplemental Figure 5C [activated genes, red; repressed genes, blue). Unbiased gene set enrichment analysis using the Hallmark Gene Set panel in the Molecular Signatures Database found that the top gene sets enriched in differentially expressed genes (FDR < 0.05) from MIG-Snai1 HPC7 cells included the TNFα-NFκΒ, Il2-STAT5, KRAS, apoptosis, hypoxia, p53 signaling, and expected EMT pathway components (Figure 6A; supplemental Table 3), all of which have been previously implicated in AML development/progression.42-46 This analysis further revealed upregulation of a distinct myeloid differentiation gene expression program in MIG-Snai1 cells (Figure 6B), which was further demonstrated by an increased expression of key myeloid cell surface markers (CD11b, Ly6G, and Ly6C) as detected by flow cytometric analysis (supplemental Figure 6A).
Figure 6.
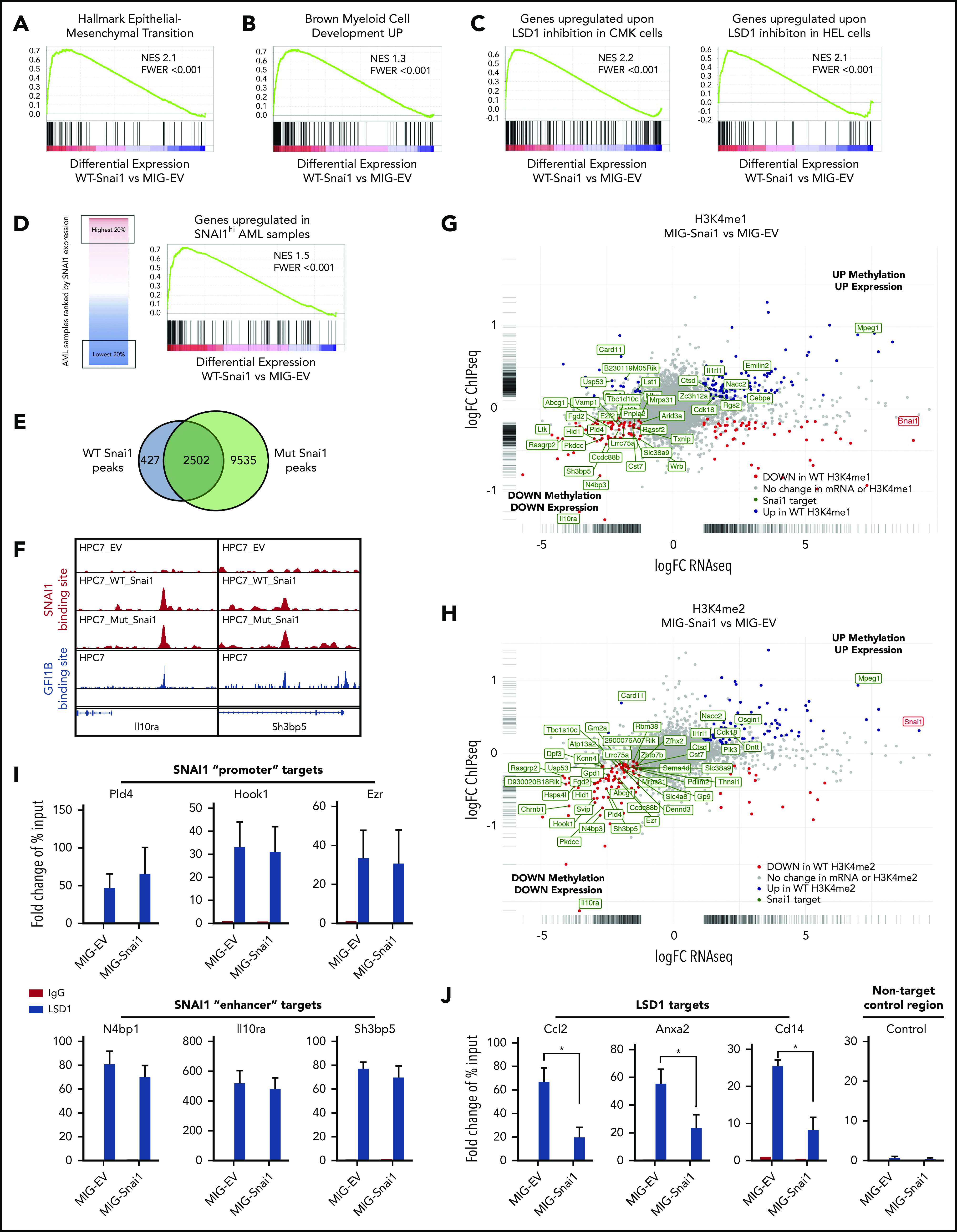
Genomic analysis of HPC7 cells transduced with either MIG-Snai1, MIG-mut5Snai1, or MIG-EV. Gene set enrichment analysis (GSEA) on RNA-seq data from MIG-Snai1 vs MIG-EV identified a significant correlation between genes upregulated in HPC7 cells expressing Snai1 (MIG-Snai1) and genes involved in the EMT (GSEA: hallmark epithelial mesenchymal transition) (A), genes upregulated upon myeloid development (GSEA: brown myeloid cell development up), genes upregulated upon LSD1 inhibition in HEL and CMK leukemia cell lines (GSE68348) (C), and genes upregulated in SNAI1 high AML patient samples (GSE10358) (D). (E) Overlap between WT SNAI1 and mut5-SNAI1 binding sites in HPC7 cells indicates that mut5SNAI1 protein is still capable of binding DNA. (F) Sample IGV tracks showing clear overlap of WT SNAI1 and mut5SNAI1 binding sites in 2 representative gene regulatory elements for Il10ra and Sh3bp5, as well as with published binding sites for GFI1B.49 FC-FC plot showing differentially expressed genes on the x-axis and differential H3K4me1 (G) or H3K4me2 (H) methylation levels on the y-axis. A correlation between differential methylation and differential expression in MIG-Snai1 cells is evident in both plots. Genes in green have an identified SNAI1 binding site in the ChIP-seq data. Red and blue dots indicate genes with significantly reduced or significantly increased respectively gene expression and methylation in MIG-Snai1 cells using a P value cut off <.05. (I) LSD1 ChIP–quantitative PCR (qPCR) results for 6 SNAI1 target genes that have reduced gene expression and reduced H3K4 methylation in MIG-Snai1 HPC7 cells. ChIP-qPCR data show binding of LSD1 directly overlapping SNAI1 binding sites in both the MIG-EV and MIG-Snai1 cells at all sites analyzed. (J) LSD1 ChIP-qPCR results for 3 LSD1 target sites showing significantly reduced LSD1 binding upon SNAI1 expression in MIG-Snai1 HPC7 cells. Control immunoglobulin G (IgG) samples were used as a control for nonspecific ChIP enrichment, and a nontarget control region was used to show specific pulldown at LSD1-bound sites compared with other sites within the genome. Data analyzed using a Mann-Whitney 1-tailed t test. *P < .01.
In agreement with our working model of LSD1 inhibition by SNAI1, we observed a significant correlation between genes upregulated as a result of LSD1 chemical inhibition in 2 human AML cell lines (HEL and CMK) and those upregulated by Snai1 overexpression in HPC7 cells (Figure 6C). Notably, we also identified a significant correlation between those genes upregulated in MIG-Snai1 HPC7 cells and genes upregulated in AML patient samples expressing high levels of endogenous SNAI1 (Figure 6D; patient data taken from GSE10358).
To determine the genome-wide effects of Snai1 overexpression on methylation level of the LSD1 histone substrates H3K4me1 and H3K4me2, we performed ChIP-seq analysis using antibodies for the H3K4me1 and H3K4me2 marks. We limited our analysis to regions surrounding the annotated transcription start site (± 5 kb) encompassing proximal and distal regulatory regions, where predominant peaks were observed (supplemental Figure 6B). This analysis identified 629 and 127 genes with associated differential H3K4me1 or H3K4me2 methylation, respectively (supplemental Table 4). Because these 2 histone marks are commonly associated with transcriptional activation, an increased level of methylation at promoters and/or enhancers would be expected to correspond to an increase in gene expression and vice versa. We indeed saw that 21% (135 of 629) of genes with H3K4me1 changes and 48% (61 of 127) of genes with H3K4me2 changes showed significant differential expression in our RNA-seq analysis (supplemental Table 4). Furthermore, we observed a clear trend for increased levels of methylation to correspond with increased gene expression and decreased levels of methylation to correspond with reduced gene expression, which was particularly evident for the H3K4me2 mark (supplemental Figure 6C).
Because LSD1 is predominantly associated with gene repression (via demethylation of the active H3K4me1/2 marks), we hypothesized that genes with decreased methylation (and decreased gene expression) may be SNAI1 targets that had become demethylated (and therefore repressed) as a result of binding of LSD1 to SNAI1-bound promoter or enhancer sites. To test this, we performed SNAI1 ChIP-seq analysis to identify SNAI1-bound sites within the MIG-Snai1 HPC7 cells. MACS2 analysis identified 2932 peaks in MIG-Snai1 cells, 12 054 peaks in MIG-Mut5-Snai1 cells, and 137 peaks in MIG-EV cells (supplemental Table 5). Importantly, a majority of WT SNAI1 peaks overlapped entirely with the Mut5-SNAI1 peaks (Figure 6E-F), demonstrating that the mutant form of SNAI1 maintains DNA binding ability but is unable to regulate gene transcription. MEME motif discovery analysis47 further revealed that 92% of WT SNAI1 binding sites contained a central canonical E-box sequence (CAGGTG), which is known to be the preferred DNA binding motif of SNAI1 (supplemental Figure 6D).48 In contrast, only 62% of Mut5-SNAI1 binding sites contained this E-box sequence (data not shown), suggesting that reduced specificity of binding may be partly responsible for the increased number of ChIP-seq peaks identified in MIG-Mut5-Snai1 cells. A higher level of expression and stability of the Mut5-SNAI1 protein (data not shown) may also explain the increased peak number. SNAI1 binding sites were predominantly promoter associated (44%) or intergenic (40%), with only 16% of peaks being intragenic (supplemental Figure 6E). WT SNAI1 was bound to 15% of differentially expressed genes (126 of 835), a majority of which were repressed genes (supplemental Figure 6F; 90 of 126 were repressed compared with 36 of 126 being activated). When we overlaid WT SNAI1 binding sites onto our gene expression FC vs histone methylation FC chart (Figure 6G-H), we could clearly see an extensive overlap of WT SNAI1 binding sites in the genes with decreased histone methylation and decreased gene expression. In contrast, we saw very little overlap of binding to activated genes.
ChIP–quantitative PCR for LSD1 in our MIG-Snai1 HPC7 cells further confirmed that LSD1 was indeed also bound at downregulated SNAI1 target sites (Pld4, Hook1, Ezr, N4bp1, Il10ra, and Sh3bp5; Figure 6I), and ATAC-seq analysis demonstrated that WT SNAI-bound gene promoters frequently had reduced chromatin accessibility correlating with reduced transcriptional activity (supplemental Figure 6G). In MIG-EV cells, LSD1 was also found to be bound at these sites, suggesting that it may be recruited in the absence of ectopic SNAI1 by another hematopoietic cofactor, such as GFI1B. Indeed, using previously published data,49 we were able to identify an overlapping binding site for GFI1B in 50% of WT SNAI1-bound sites that had significant changes in H3K4me2 methylation (Figure 6F; supplemental Table 4). These data suggest possible competition between SNAI1 and GFI1B for LSD1 binding at these sites, particularly as they interact with LSD1 via exactly the same SNAG protein domain.
Finally, ChIP–quantitative PCR for LSD1 at 3 upregulated LSD1 targets with increased H3K4me1/2 methylation (Ccl2, Cd14, and Anxa2) confirmed that there was a significant reduction of LSD1 binding at these sites in MIG-Snai1 HPC7 cells (Figure 6J), supporting our model that overexpressed SNAI1 can also sequester LSD1 away from its normal gene targets.
Discussion
Deregulated expression of EMT modulators is emerging as a novel theme in AML biology; however, our understanding of how these key developmental regulators and epithelial tumor oncogenes contribute to malignancy of the hematopoietic system is still lacking. In this current study, we have demonstrated a previously unknown association between ectopic EMT factor expression and altered LSD1 activity during malignant transformation of hematopoietic and myeloid stem/progenitors.
SNAI1 is overexpressed throughout a broad spectrum of primary AML patient samples, irrespective of presence of driver mutation or genetic abnormality, and higher levels of SNAI1 are correlated with decreased OS in this disease (also recently demonstrated for ZEB113). Furthermore, we have shown that increased expression of SNAI1 is functionally relevant for AML biology and contributes to the differentiation block in AML cells in a fashion similar to that demonstrated for ZEB2.12 Using in vivo transgenic and retroviral Snai1 overexpression systems, we discovered that increased expression of Snai1 drives an expanded myelopoiesis, enhances self-renewal and proliferative capacity of immature myeloid cells, and ultimately results in the development of a myeloproliferative-like disease that can transform into AML over a prolonged period of time (Figure 7). We identified several AML-relevant biological pathways that are upregulated by SNAI1, including the TNFα-NFκβ pathway42 and key cytokine/signaling pathways such as those involving RAS and IL-2/Stat5,43-45 as well as decreased expression of emerging (and thus less well studied) AML tumor suppressors such as Ssbp2.38 The altered expression of these key pathways likely cooperates with Snai1 overexpression to drive full-scale transformation of immature myeloid cells that have enhanced self-renewal properties.
Figure 7.
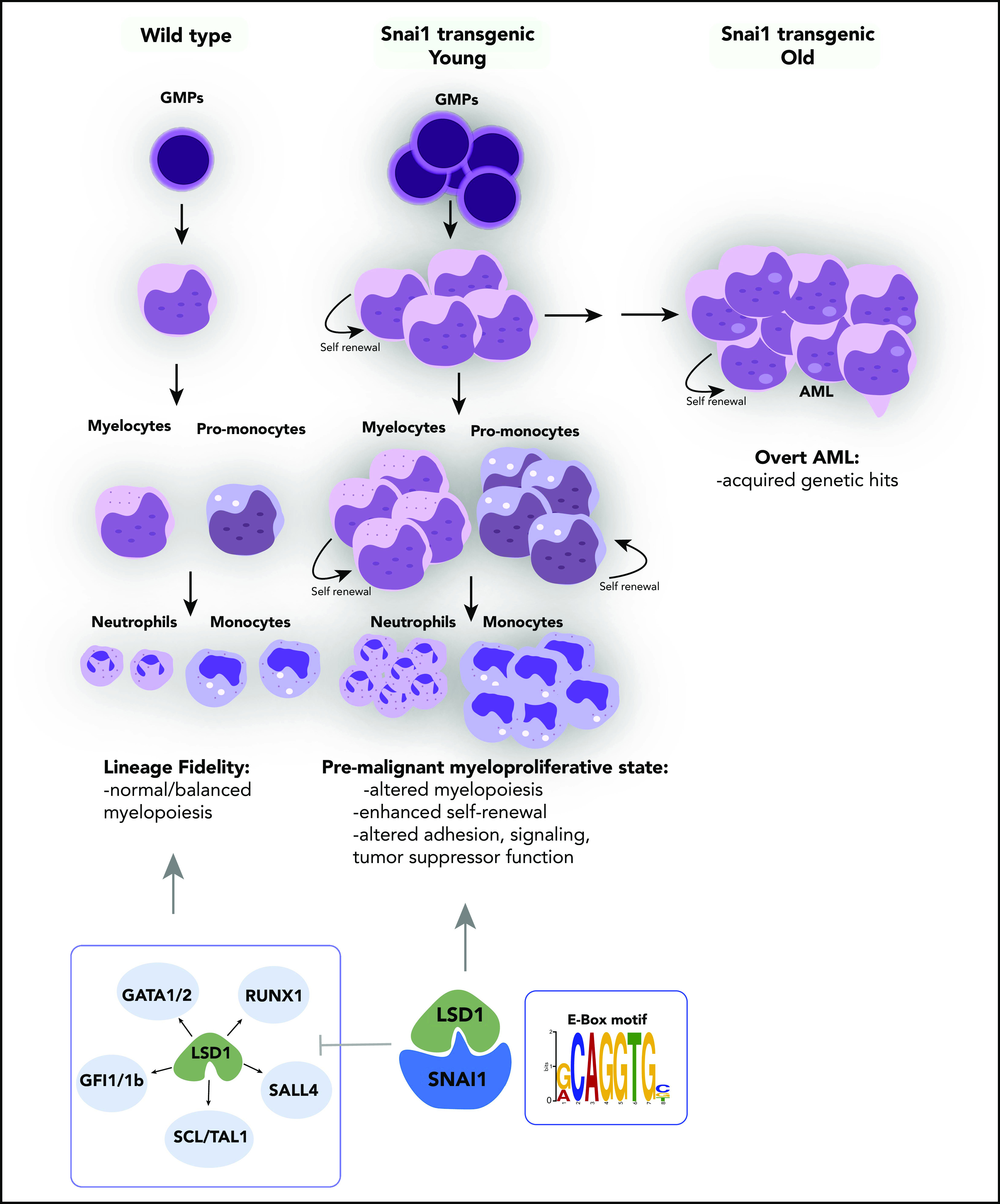
Proposed model of the effect of ectopically expressed SNAI1 on normal hematopoiesis and LSD1 function. During normal hematopoiesis, balanced myeloid development is observed, resulting in normal numbers of mature myeloid cells. Ectopic SNAI1 in hematopoietic cells interacts with LSD1 and subsequently leads to inhibition promotion of myeloid differentiation along the granulocyte and macrophage lineages. This complex drives expanded myeloid cell differentiation and production of excessive numbers of mature myeloid cells, resulting in myeloproliferative phenotypes. SNAI1/LSD1 complex also imbues enhanced self-renewal capacity on immature myeloid cells, allowing these cells to expand and potentially accumulate additional mutations that can result in AML development over an extended period of time. We propose that the interaction between SNAI1 and LSD1 drives myeloid developmental defects through physical interaction at SNAI1 gene targets containing the canonical E-box motif and subsequent modulation of LSD1 function and inhibition of LSD1 binding to its normal hematopoietic transcription factor partners, such as GATA1/2, SALL4, and GFI1/1b, and subsequently compromising the function of these key hematopoietic transcription factors.
Interestingly, we found that SNAI1-induced myeloid developmental changes were completely dependent on the SNAG domain–mediated interactions with LSD1. This interaction led to not only loss of LSD1 demethylase activity at normal hematopoietic gene targets, but also acquisition or modulation of LSD1 demethylase/gene repression activity at other gene targets that were also bound by SNAI1. In both scenarios, competition for LSD1 binding between SNAI1 and other LSD1 cofactors, such as GFI1B, likely also contributes to the phenotype; however, additional studies will be required to test this further.
LSD1 has garnered much interest over the past few years as a putative therapeutic target in multiple cancer types, including solid tumors and AML, because of its frequently altered expression and/or activity in malignant cells.50,51 The mechanism of action of LSD1 in AML, however, seems to be in conflict with its known biological roles during normal hematopoietic development. Although loss of LSD1 in hematopoietic cells results in derepression of stem cell–associated genes, acquisition of enhanced stem cell function, and reduced hematopoietic cell differentiation,35 inhibition/loss of LSD1 in AML cells instead drives myeloid differentiation and switches off expression of oncogenic stem cell genes.9 Presumably, these conflicting roles suggest that LSD1 function can be altered by malignant mechanisms such as the expression of key AML oncogenes. Indeed, Harris et al8 showed that LSD1 function specifically at MLL-AF9–bound promoters was important for sustaining leukemia stem cell activity in a mouse model of AML. Our data now suggest that during malignant hematopoiesis, ectopic expression of SNAI1 (and potentially other EMT modulators) is able to perturb normal LSD1 function, not only by interfering with its ability to interact with its normal hematopoietic partners, such as GFI1B (Figure 7), but also by coopting its histone demethylase activity to repress expression of SNAI1 target genes that play key roles in regulating adhesion, signaling, and tumor suppressor functions.
Although our study has focused primarily on the role of LSD1/SNAI1 interactions, it should be noted that other known interactions between SNAI1 and epigenetic modifiers, such as HDAC1/2 and SIN3A, may also be altered upon aberrant SNAI1 expression. Further investigation into the common/specific target genes and pathways regulated by SNAI1/LSD1 and other SNAI/ZEB-containing repression complexes, as well as their functional characterization, is therefore warranted and will enable a greater understanding of the mechanism by which LSD1 and EMT transcription factors contribute to AML biology. Given the known dose-limiting effects of LSD1 inhibition on normal blood cell development, our study suggests that targeting the SNAI1/LSD1 complex or its downstream targets, rather than LSD1 itself, may be a more viable therapeutic option in AML.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Lief Carlsson (Umea University, Sweden) for providing the HPC7 cells and Andrew Wei (Australian Centre for Blood Diseases, Monash University, Australia) for providing access to primary AML samples.
This work was supported by the Multi-modal Australian Sciences Imaging and Visualisation Environment high-performance computing facility and National eResearch Collaboration Tools and Resources project and funded by project grants G1141081 (J.J.H.); 1102589, 1139787, 11398111, and 1160110 (J.E.P.); program grants 1016647 and 1113577 (B.T.K.), and Senior Research Fellowship (D.J.C) and Independent Research Institutes Infrastructure Support Scheme grant 361646 (B.T.K.) from the Australian National Health and Medical Research Council. T.L. received funding from vzw Kinderkankerfonds and D.B. received funding from Cancer Institute NSW. J.J.H. also received funding from the Canadian Institute of Health Research (project grant 419220). S.G. and P.V.V. received funding from the European Hematology Association (EHA), the Research Foundation Flanders (FWO), the Basic Research Fund of Ghent University and the Belgian Foundation Against Cancer. J.E.P. is also supported by the Anthony Rothe Leukemia Foundation, Translational Cancer Research Network, Cancer Institute NSW, NSW Health Pathology, and South Eastern Area Laboratory Services.
Footnotes
Contact the corresponding author for original data.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.L.C., S.G., and J.J.H. were responsible for study conception and design; C.L.C., J.W., O.K., A.B., T.N., C.D.M., A.R.M., S.S., K.G., W.R.L.M., A.N.Q.V., T.L., B.L., B.J.S., H.M., K.H., Y.H., J.A.I.T., and K.K. acquired the data; C.L.C., K.G., S.H.-z., M.J.D., M.E.R., N.C.W., S.G., and J.J.H. analyzed and interpreted the data; A.P. provided histological review of pathology specimens; C.L.C., S.G., and J.J.H. wrote, reviewed, and revised the manuscript; and P.V.V., E.T., D.J.C., D.B., M.P.M., E.P.O., R.A.D., G.B., J.E.P., J.Z., and B.T.K provided other critical samples, reagents, and/or technical support.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jody J. Haigh, CancerCare Manitoba, ON5029, 675 McDermot Ave, Winnipeg, MB, Canada, R3E 0V9; e-mail: jody.haigh@umanitoba.ca; and Catherine L. Carmichael, Australian Centre for Blood Diseases, Monash University, 99 Commercial Rd, Prahran, Melbourne, VIC, Australia, 3004; e-mail: catherine.carmichael@monash.edu.
REFERENCES
- 1.Ley TJ, Miller C, Ding L, et al. ; Cancer Genome Atlas Research Network . Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia [published correction appears in N Engl J Med. 2013;369(1):98]. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Gerstung M, Bullinger L, et al. . Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenblatt SM, Nimer SD. Chromatin modifiers and the promise of epigenetic therapy in acute leukemia. Leukemia. 2014;28(7):1396-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai CT, So CW. Epigenetic therapies by targeting aberrant histone methylome in AML: molecular mechanisms, current preclinical and clinical development. Oncogene. 2017;36(13):1753-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi Y, Lan F, Matson C, et al. . Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941-953. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Sawada J, Sui G, et al. . Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422(6933):735-738. [DOI] [PubMed] [Google Scholar]
- 7.Magliulo D, Bernardi R, Messina S. Lysine-specific demethylase 1A as a promising target in acute myeloid leukemia. Front Oncol. 2018;8:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris WJ, Huang X, Lynch JT, et al. . The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells [published correction appears in Cancer Cell. 2012;21(6):856]. Cancer Cell. 2012;21(4):473-487. [DOI] [PubMed] [Google Scholar]
- 9.Schenk T, Chen WC, Göllner S, et al. . Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16(6):488-494. [DOI] [PubMed] [Google Scholar]
- 11.Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol. 2019;21(1):102-112. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Mar BG, Zhang H, et al. . The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood. 2017;129(4):497-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stavropoulou V, Kaspar S, Brault L, et al. . MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30(1):43-58. [DOI] [PubMed] [Google Scholar]
- 14.Goossens S, Radaelli E, Blanchet O, et al. . ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat Commun. 2015;6:5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuber J, Shi J, Wang E, et al. . RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fellmann C, Hoffmann T, Sridhar V, et al. . An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5(6):1704-1713. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402-408. [DOI] [PubMed] [Google Scholar]
- 18.Diffner E, Beck D, Gudgin E, et al. . Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia [published correction appears in Blood. 2014;123(18):2901]. Blood. 2013;121(12):2289-2300. [DOI] [PubMed] [Google Scholar]
- 19.Corces MR, Trevino AE, Hamilton EG, et al. . An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shousha WG, Ramadan SS, El-Saiid AS, Abdelmoneim AE, Abbas MA. Expression and clinical significance of SNAI1 and ZEB1 genes in acute myeloid leukemia patients. Mol Biol Rep. 2019;46(4):4625-4630. [DOI] [PubMed] [Google Scholar]
- 21.Goswami CP, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma. 2013;3(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. 2014;14:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J, Sanborn JZ, Benz S, et al. . The UCSC Cancer Genomics Browser. Nat Methods. 2009;6(4):239-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray SA, Oram KF, Gridley T. Multiple functions of Snail family genes during palate development in mice. Development. 2007;134(9):1789-1797. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Choi PS, Casey SC, Felsher DW. Activation of Cre recombinase alone can induce complete tumor regression. PLoS One. 2014;9(9):e107589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyabi O, Naessens M, Haigh K, et al. . Efficient mouse transgenesis using Gateway-compatible ROSA26 locus targeting vectors and F1 hybrid ES cells. Nucleic Acids Res. 2009;37(7):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Boer J, Williams A, Skavdis G, et al. . Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33(2):314-325. [DOI] [PubMed] [Google Scholar]
- 28.Mani SA, Guo W, Liao MJ, et al. . The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29(35):4896-4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y, Wu Y, Li J, et al. . The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010;29(11):1803-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Fu X, Huo B, et al. . GATA2 regulates GATA1 expression through LSD1-mediated histone modification. Am J Transl Res. 2016;8(5):2265-2274. [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Deng C, Hu X, et al. . Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene. 2012;31(48):5007-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Souto J, Liao W, et al. . Histone lysine-specific demethylase 1 (LSD1) protein is involved in Sal-like protein 4 (SALL4)-mediated transcriptional repression in hematopoietic stem cells. J Biol Chem. 2013;288(48):34719-34728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27(4):562-572. [DOI] [PubMed] [Google Scholar]
- 35.Kerenyi MA, Shao Z, Hsu YJ, et al. . Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife. 2013;2:e00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sprüssel A, Schulte JH, Weber S, et al. . Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26(9):2039-2051. [DOI] [PubMed] [Google Scholar]
- 37.Pinto do O P, Wandzioch E, Kolterud A, Carlsson L. Multipotent hematopoietic progenitor cells immortalized by Lhx2 self-renew by a cell nonautonomous mechanism. Exp Hematol. 2001;29(8):1019-1028. [DOI] [PubMed] [Google Scholar]
- 38.Liang H, Samanta S, Nagarajan L. SSBP2, a candidate tumor suppressor gene, induces growth arrest and differentiation of myeloid leukemia cells. Oncogene. 2005;24(16):2625-2634. [DOI] [PubMed] [Google Scholar]
- 39.Sun X, Zhang Q, Chen W, et al. . Hook1 inhibits malignancy and epithelial-mesenchymal transition in hepatocellular carcinoma. Tumour Biol. 2017;39(7):1010428317711098. [DOI] [PubMed] [Google Scholar]
- 40.Yang L, Xie N, Huang J, et al. . SIK1-LNC represses the proliferative, migrative, and invasive abilities of lung cancer cells. OncoTargets Ther. 2018;11:4197-4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeste-Velasco M, Mao X, Grose R, et al. . Identification of ZDHHC14 as a novel human tumour suppressor gene. J Pathol. 2014;232(5):566-577. [DOI] [PubMed] [Google Scholar]
- 42.Kagoya Y, Yoshimi A, Kataoka K, et al. . Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J Clin Invest. 2014;124(2):528-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allan JN, Roboz GJ, Askin G, et al. . CD25 expression and outcomes in older patients with acute myelogenous leukemia treated with plerixafor and decitabine. Leuk Lymphoma. 2018;59(4):821-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wingelhofer B, Maurer B, Heyes EC, et al. . Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32(5):1135-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou JD, Yao DM, Li XX, et al. . KRAS overexpression independent of RAS mutations confers an adverse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget. 2017;8(39):66087-66097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Post SM, Kornblau SM, Quintás-Cardama A. p53 pathway dysfunction in AML: beyond TP53 mutations. Oncotarget. 2017;8(65):108288-108289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bailey TL, Boden M, Buske FA, et al. . MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202-W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiang C, Ayyanathan K. Characterization of the E-box binding affinity to snag-zinc finger proteins. Mol Biol (Mosk). 2012;46(6):907-914. [PubMed] [Google Scholar]
- 49.Wilson NK, Foster SD, Wang X, et al. . Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532-544. [DOI] [PubMed] [Google Scholar]
- 50.Niebel D, Kirfel J, Janzen V, Höller T, Majores M, Gütgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood. 2014;124(1):151-152. [DOI] [PubMed] [Google Scholar]
- 51.Schulte JH, Lim S, Schramm A, et al. . Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69(5):2065-2071. [DOI] [PubMed] [Google Scholar]
- 52.Szymanska B, Wilczynska-Kalak U, Kang MH, et al. . Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS One. 2012;7(3):e33894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.