

Abstract

Ammonium salts are used as phase-transfer catalysts for fluorination with alkali metal fluorides. We now demonstrate that these organic salts, specifically azetidinium triflates, are suitable substrates for enantioselective ring opening with CsF and a chiral bis-urea catalyst. This process, which highlights the ability of hydrogen bonding phase-transfer catalysts to couple two ionic reactants, affords enantioenriched γ-fluoroamines in high yields. Mechanistic studies underline the role of the catalyst for phase-transfer, and computed transition state structures account for the enantioconvergence observed for mixtures of achiral azetidinium diastereomers. The N-substituents in the electrophile influence the reactivity, but the configuration at nitrogen is unimportant for the enantioselectivity.
Asymmetric phase-transfer catalysis (PTC) is one of the most practical methods for enantioselective synthesis.1 For many years, PTC approaches to asymmetric fluorinations have used F2-derived electrophilic reagents and cationic or anionic chiral species for effective phase-transfer.2 Inspired by nature’s fluorinase,3 we reported a complementary hydrogen bonding phase-transfer catalysis (HB-PTC) manifold that employed alkali metal fluorides for asymmetric nucleophilic fluorinations.4 Specifically, a chiral N-alkylated bis-urea served as the hydrogen bond donor (HBD) catalyst to bring KF or CsF in solution. The process involves a chiral urea–fluoride complex that is capable of ion-pairing with in situ-formed meso-episulfonium or -aziridinium ions. The ensuing enantioselective desymmetrization afforded enantioenriched β-fluorosulfides and β-fluoroamines. To date, all enantioselective fluorinations carried out under PTC use nonionic substrates, including β-keto esters, alkenes, β-bromosulfides or β-chloroamines. An unexplored scenario in asymmetric C–F bond construction under PTC is the use of two ionic reactants. We became interested in this challenge as we envisioned that enantioselective desymmetrization of achiral azetidinium salts with fluoride would afford γ-fluoroamines of high value for medicinal chemistry.5 Azetidinium salts6 with non-nucleophilic counteranions are bench-stable solids7a and can be prepared from commercially available7b or readily synthesized7c azetidines. Few methods are available to access enantioenriched γ-fluoroamines,8 and strategies for the enantioselective installation of CH2F are scarce.9
In 2018, Sun and co-workers reported the desymmetrization of azetidinium salts with mercaptobenzothiazoles and a chiral phosphate catalyst (Scheme 1A, left).10 This pioneering study encouraged experimentation applying this anionic PTC approach (CAPT) with TBAF or CsF; none of our attempts yielded γ-fluoroamines (Scheme S6). This result prompted the use of HB-PTC as an alternative manifold. Mechanistically, achiral azetidinium salts could themselves act as phase-transfer agents enabling solubilization of solid alkali metal fluorides as azetidinium fluorides. Indeed, ammonium salts,11a−11c pyridinium salts,11d and imidazolium-based ionic liquids11e have been used as phase-transfer catalysts for non-enantioselective fluorination reactions with KF or CsF (Scheme 1B, left).11f,11g Such a cationic phase-transfer catalysis (CPTC) scenario would transform in situ-formed azetidinium fluoride into racemic γ-fluoroamine. We envisioned that HB-PTC using a chiral bis-urea catalyst could offer a viable approach for the desymmetrization of achiral azetidinium salts with alkali metal fluorides (Scheme 1A,B, right). This scenario is not without challenges because the use of two preformed ionic reactants implies a high concentration of ions in solution, a drastic change compared with transformations featuring transiently formed ion pairs.4a,4b Significant variation in the fluorination kinetics and competitive binding events (e.g. azetidinium counteranion X– vs F–) can be expected.12 Computational studies indicated that a neutral chiral N-methyl-bis(urea)4a binds a CsF unit more strongly than 1,1-dimethylazetidinium ion in 1,2-dichloroethane (1,2-DCE) (ΔGurea = −69 kJ/mol, ΔGazet = −14 kJ/mol; Scheme 1C).13a
Scheme 1. (A) Desymmetrization of Azetidinium Salts; (B) R4N+X– as a Catalyst (CPTC) versus R4N+X– as a Substrate (HB-PTC); (C) Computational Binding Studies (R4N+X– Treated as a Dissociated Species).

Encouraged by these findings,13b we surmised that azetidinium salts (R4N+X–) could undergo enantioselective fluorination with an alkali metal fluoride (M+F–) in the presence of a chiral HBD catalyst (urea*) if orchestrated hydrogen bonding and ion metathesis generate the soluble chiral ion pair [R4N]+[F·urea*]−. This species could undergo C–F bond formation with release of the enantioenriched γ-fluoroamine and the catalyst. Herein we describe the development of this unusual PTC process featuring two ionic reactants and demonstrate that achiral azetidinium salts are amenable to desymmetrization with CsF and a chiral BINAM-derived bis-urea catalyst.
Preliminary investigations unveiled details of the impact of the structural features of azetidinium salts on the reactivity (Table 1). 3-Phenyl N,N-dibenzyl and N-methyl-N-benzylazetidinium triflates afforded traces of product with CsF and catalyst (S)-A (Table 1, entries 1 and 2). When N-methyl-N-benzhydryl substrate 1a was employed, the desired γ-fluoroamine was obtained in poor yield and enantioselectivity (Table 1, entry 3). Notably, 1a reacted with CsF in 1,2-DCE in the absence of catalyst to afford γ-fluoroamine (±)-2a in 8% yield (Table S1). When this reaction was carried out using the N-alkyl-bis(urea) catalyst (S)-B, (S)-C, or (S)-D, 2a was obtained in moderate yield and enantiomeric ratio (e.r.) (Table 1, entries 4–6). Solvent screening showed the superiority of 1,2-DCE (up to 81:19 e.r.; Table 1, entries 7–9). In addition to the benzhydryl group, the second N-substituent also influenced the reactivity and enantioselectivity, with benzyl and ethyl being superior to methyl (up to 96:4 e.r.; Table 1, entries 10–13). After optimization, the reaction of 1aa (1:1.1 d.r.) in 1,2-DCE with CsF (2 equiv) and N-isopropyl-bis(urea) catalyst (S)-D (5 mol%) at room temperature afforded γ-fluoroamine 2aa in 98% yield with 96:4 e.r. (Table 1, entry 13). N-Ethylazetidinium triflate 1ab required a longer reaction time (72 h) and a higher catalyst loading (10 mol%) to afford 2ab (93% yield, 96:4 e.r.; Table 1, entry 11). These findings were encouraging because azetidinium salts can be used as mixtures of diastereomers, and both benzhydryl and benzyl groups are cleavable, releasing a primary amine that is amenable to myriad transformations.
Table 1. Optimization of the Reaction Conditionsa.

| entry | R1 | R2 | cat. | solvent | yieldb | e.r.c |
|---|---|---|---|---|---|---|
| 1 | Bn | Bn | A | CH2Cl2 | traces | – |
| 2 | Me | Bn | A | CH2Cl2 | traces | – |
| 3 | Me | Bzh | A | CH2Cl2 | 14% | 55:45 |
| 4 | Me | Bzh | B | CH2Cl2 | 20% | 55:45 |
| 5 | Me | Bzh | C | CH2Cl2 | 20% | 75:25 |
| 6 | Me | Bzh | D | CH2Cl2 | 45% | 74:26 |
| 7 | Me | Bzh | D | CHCl3 | 56% | 67:33 |
| 8 | Me | Bzh | D | 1,2-DFB | 47% | 79:21 |
| 9 | Me | Bzh | D | 1,2-DCE | 51% | 81:19 |
| 10 | Et | Bzh | D | 1,2-DCE | 40% | 96:4 |
| 11d,e | Et | Bzh | D | 1,2-DCE | 93% | 96:4 |
| 12 | Bn | Bzh | D | 1,2-DCE | >95% | 96:4 |
| 13d,f | Bn | Bzh | D | 1,2-DCE | 98% | 96:4 |
Reaction conditions: 0.05 mmol of 1, 0.25 M, 10 mol% cat., stirring at 900 rpm, 24 h.
Determined by 19F NMR spectroscopy with 4-fluoroanisole as an internal standard.
Enantiomeric ratios were determined by HPLC using a chiral stationary phase.
Yield of isolated product.
72 h, 10 mol% cat.
48 h, 5 mol% cat.
The benefit of the N-benzhydryl group on reactivity prompted further investigation.14 Fluorination reactions performed on differently N,N-disubstituted azetidinium salts under homogeneous conditions (TBAF·3H2O, 1,2-DCE, no catalyst) showed benzhydryl to be superior to all other N-substituents (Scheme S4). The increased reactivity of N-benzhydrylazetidinium salts is therefore unconnected with phase-transfer. Computed transition state (TS) structures for the fluorination of seven azetidinium ions by free fluoride (homogeneous conditions) showed that the increased experimental yields are consistent with smaller computed activation barriers. N-Benzhydrylazetidinium ions have barriers to fluorination that are ∼6 kJ/mol lower than those for the corresponding N-benzyl substrates. This can be traced to increased reactant strain: N-benzhydrylazetidinium ions have more elongated C–N bonds and earlier fluoride delivery TS positions compared with the methyl or benzyl substrates (Figure 1).
Figure 1.
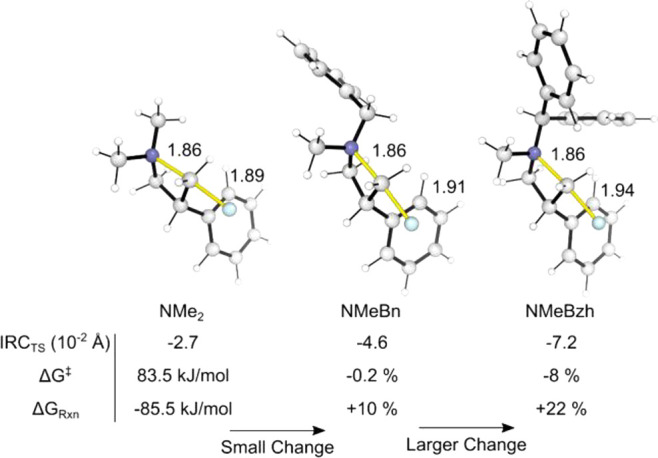
Effect of the N-benzhydryl group on the reactivity (Me, Bn and Bzh series). The intrinsic reaction coordinate position was calculated as the C–N distance minus the C–F distance.15
The scope of γ-fluoroamine synthesis was examined next (Scheme 2). High yields and enantioselectivities were obtained with 3-arylazetidinium triflates. Substrates bearing aromatic groups with electron-withdrawing and electron-donating substituents at the meta or para position were converted in excellent yields and enantioselectivities (2aa–2ha, up to 99% yield, 97.5:2.5 e.r.). Heteroaromatic groups such as thiophene (2pa), pyrazole (2qa), and indole (2ra) were compatible, representing pharmaceutically relevant motifs (up to 99% yield, 94:6 e.r.). Additional highlights are the suitability of N-allylazetidinium salts (2ac, 2ic), the tolerance of the reaction to 3-aryloxy (2ia–2ja, up to 99% yield, 93.5:6.5 e.r.), 3-alkoxy (2ka–2mb), and 3-phthalimido (2sa) groups, and the synthesis of enantioenriched γ-fluoroamines 2oa and 2va–2ua featuring a tetrasubstituted stereogenic carbon. Furthermore, 2ta bearing an ester group stands out as an immediate precursor to enantioenriched fluorinated β-lactams and β-amino acids.16 Tertiary fluoride 2wa was accessed in good yield with moderate enantioselectivity. A single recrystallization of 2sa, 2ta and 2wa afforded these fluorinated amines in high enantioselectivity or as a single enantiomer. Substrates mono- or bis-alkylated at position 3 were less successful (Scheme S7).13a Single-crystal X-ray diffraction analysis of 2ma·HCl and 3ab·HCl (Scheme 3A) enabled the assignment of the absolute configuration ((S)-catalyst affords (S)-product).17
Scheme 2. Reaction Scope.

Reactions were performed on a 0.1 mmol scale, except for 1sa, 1ta, and 1wa (0.5–1 mmol scale). Absolute configurations were assigned by analogy to (S)-2ma for all products except 2oa and 2ua–2wa featuring a tetrasubstituted stereogenic carbon. The e.r. values in parentheses were obtained after a single recrystallization.
20 mol% cat.
Scheme 3. (A) Gram-Scale Reactions and Deprotection. (B) Enantioselective Synthesis of Fluorinated Lorcaserin.

This new catalytic protocol enabled the preparative-scale synthesis of γ-fluoroamines 3aa and 3ab (Scheme 3A). The reaction of 1 g of 1aa was conducted with a lower catalyst loading (3 mol%) and no compromise in e.r. relative to the smaller-scale reaction. Deprotection and a single recrystallization gave the primary γ-fluoroamine 3aa with 99:1 e.r. A similar protocol afforded enantioenriched secondary γ-fluoroamine 3ab with 98.5:1.5 e.r. The synthesis of the fluorinated analogue of lorcaserin,18 a selective serotonin 2C receptor agonist that is FDA-approved for chronic weight management, illustrates the value of the method for accessing valuable pharmaceutical motifs (Scheme 3B).
Further experimentation was undertaken to gain more insight into this process. (i) The reaction of 1aa with 1 equiv of [(S)-D·F]−[nBu4N]+ formed in situ or preformed from nBu4N+F–·3H2O in 1,2-DCE (0.25 M) afforded 2aa in 30% yield with 96:4 e.r. This result confirms the involvement of [(S)-D·F]− for enantiocontrol (Scheme S5) and highlights the detrimental impact of water on the yield (Table S5). (ii) Exchanging OTf− of 1aa with PF6− gave 2aa with identical e.r. (96:4) but in only 31% yield (Table S6). This observation indicates that the counteranion influences the efficacy of phase-transfer and advocates against anion-binding catalysis. This is further supported by NMR studies showing the stronger binding preference of the catalyst for fluoride compared with other anions (F– ≫ TfO– ≈ BF4– > PF6–).13a (iii) The linear relationship between the enantiopurities of the catalyst and product supports the involvement of a single urea catalyst in the enantiodetermining step (Table S7). (iv) When diastereomerically pure N-methyl-substituted cis-1a or trans-1a was subjected to asymmetric fluorination under the standard reaction conditions, fluoroamine (S)-2a was formed with comparable e.r. values (80.5:19.5 and 80:20, respectively; Table S4).
Finally, we turned our attention to the origin of enantioconvergence for this transformation. The TSs for fluorination of 1a mediated by catalyst (S)-D were computed using density functional theory (DFT).19 An ensemble of TSs were optimized for both cis and trans substrates (Figure 2). With the cis substrate (major diastereomer), both TSMajor-cis and TSMinor-cis feature a face-to-face π interaction between the phenyl group at the 3-position of the substrate and the catalyst (Figure 2Ai), orienting the substrate in the catalytic pocket. In contrast, the benzhydryl groups point in different directions. In TSMajor-cis, the azetidinium ion adopts its favored conformation, with an intramolecular CH···π interaction worth approximately 2–5 kJ/mol (Figure 2Aii). In TSMinor-cis, this is compensated by an aromatic edge-to-face interaction of the benzhydryl group with the catalyst BINAM backbone (Figure 2Aiii). When computed with an N-ethyl substituent (1ab), the (S)-catalyst affords the (S)-product with 91:9 e.r., consistent with the experimental enantioselectivity of 96:4 e.r. Comparison of the lowest-energy TS, TSMajor-cis, to the TS for the major product with the trans substrate, TSMajor-trans, shows remarkable similarity, with excellent superposition of the catalyst, fluoride, and substrate. The only exception is the reversal of the configuration at nitrogen, which causes the sterically demanding benzhydryl group to point in a different direction (Figure 2B). The enantioconvergence of the diastereomers originates from the projection of the azetidinium N-substituents away from the catalyst and the resulting indifference to the configuration at nitrogen.20
Figure 2.
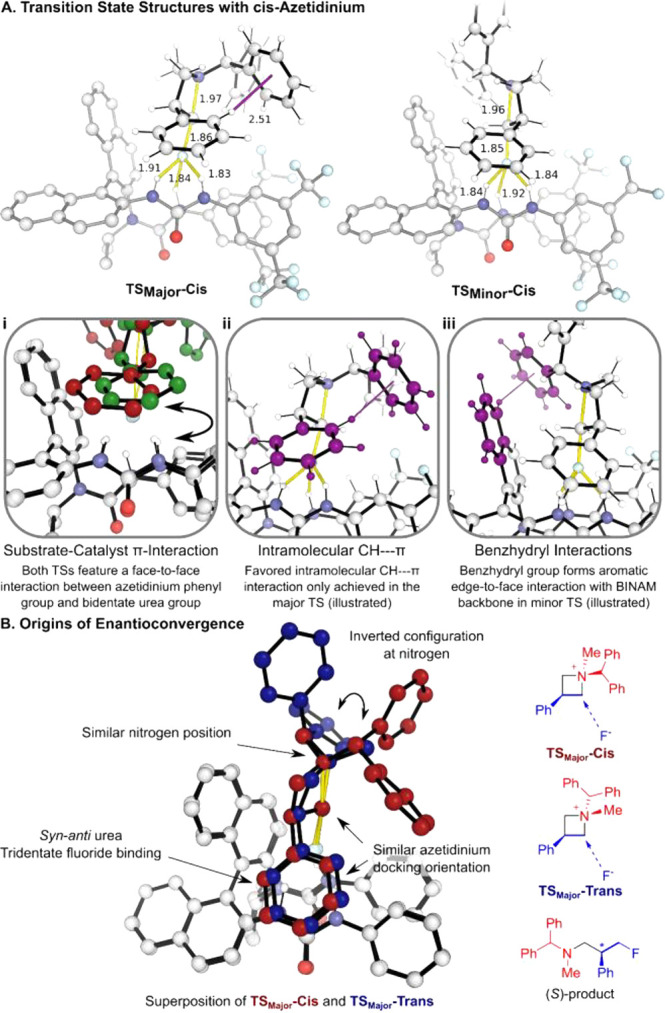
Computed TSs for fluorination of 1a. (A) TSs with cis-1a and key structural features. (B) Origin of the enantioconvergence of cis-1a and trans-1a demonstrated through the structural similarity of the most favorable TSs for the two diastereomers of the substrate.
In conclusion, this study provides new insights into HB-PTC and its application to high-value fluorine-containing molecules. Neutral N-alkyl-bis(urea) catalysts are more effective at fluoride binding than azetidinium ions, a feature enabling efficient enantioselective ring opening with CsF for the synthesis of γ-fluoroamines. As the use of ammonium salts as substrates is uncommon in asymmetric catalysis,21 the principles outlined here may encourage further studies to transform ionic starting materials into enantioenriched products by applying HB-PTC.
Acknowledgments
We thank Dr. J. M. Brown for insightful discussions, the University of Oxford Advanced Research Computing facility (http://dx.doi.org/10.5281/zenodo.22558), and the Extreme Science and Engineering Discovery Environment (allocation TG-CHE180056). This work was supported by the EU H2020 Research and Innovation Programme (Marie Skłodowska-Curie Agreements 721902, 675071, and 789553), the Engineering and Physical Sciences Research Council (EP/R010064, SBM-CDT EP/L015838/1), and the European Research Council (Agreement 832994).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c05131.
Author Contributions
⊥ G.R. and D.M.H.A. contributed equally.
The authors declare no competing financial interest.
Notes
The crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (1973306 and 1973307).
Supplementary Material
References
- a Ooi T.; Maruoka K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem., Int. Ed. 2007, 46, 4222. 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]; b Shirakawa S.; Maruoka K. Recent Developments in Asymmetric Phase-transfer Reactions. Angew. Chem., Int. Ed. 2013, 52, 4312. 10.1002/anie.201206835. [DOI] [PubMed] [Google Scholar]; c Phipps R. J.; Hamilton G. L.; Toste F. D. The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis. Nat. Chem. 2012, 4, 603. 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- For selected examples using cationic salts, see:; a Wang X.; Lan Q.; Shirakawa S.; Maruoka K. Chiral Bifunctional Phase-Transfer Catalysts for Asymmetric Fluorination of β-keto Esters. Chem. Commun. 2010, 46, 321. 10.1039/B920099A. [DOI] [PubMed] [Google Scholar]; b Kim D. Y.; Park E. J. Catalytic Enantioselective Fluorination of β-Keto Esters by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salts. Org. Lett. 2002, 4, 545. 10.1021/ol010281v. [DOI] [PubMed] [Google Scholar]; For anionic salts, see:; c Rauniyar V.; Lackner A. D.; Hamilton G. L.; Toste F. D. Asymmetric Electrophilic Fluorination Using an Anionic Chiral Phase-transfer Catalyst. Science 2011, 334, 1681. 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]; d Yang X.; Phipps R. J.; Toste F. D. Asymmetric Fluorination of α-Branched Cyclohexanones Enabled by a Combination of Chiral Anion Phase-Transfer Catalysis and Enamine Catalysis using Protected Amino Acids. J. Am. Chem. Soc. 2014, 136, 5225. 10.1021/ja500882x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dong C.; Huang F.; Deng H.; Schaffrath C.; Spencer J. B.; O’Hagan D.; Naismith J. H. Crystal Structure and Mechanism of a Bacterial Fluorinating Enzyme. Nature 2004, 427, 561. 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]; b Zhu X.; Robinson D. A.; McEwan A. R.; O’Hagan D.; Naismith J. H. Mechanism of Enzymatic Fluorination in Streptomyces Cattleya. J. Am. Chem. Soc. 2007, 129, 14597. 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]; c O’Hagan D.; Deng H. Enzymatic Fluorination and Biotechnological developments of the fluorinase. Chem. Rev. 2015, 115, 634. 10.1021/cr500209t. [DOI] [PubMed] [Google Scholar]
- a Pupo G.; Ibba F.; Ascough D. M. H.; Vicini A. C.; Ricci P.; Christensen K. E.; Pfeifer L.; Morphy J. R.; Brown J. M.; Paton R. S.; Gouverneur V. Asymmetric Nucleophilic Fluorination under Hydrogen Bonding Phase-transfer Catalysis. Science 2018, 360, 638. 10.1126/science.aar7941. [DOI] [PubMed] [Google Scholar]; b Pupo G.; Vicini A. C.; Ascough D. M. H.; Ibba F.; Christensen K. E.; Thompson A. L.; Brown J. M.; Paton R. S.; Gouverneur V. Hydrogen Bonding Phase-Transfer Catalysis with Potassium Fluoride: Enantioselective Synthesis of β-Fluoroamines. J. Am. Chem. Soc. 2019, 141, 2878. 10.1021/jacs.8b12568. [DOI] [PubMed] [Google Scholar]; For initial studies on hydrogen-bonded alcohol/urea complexes, see:; c Engle K. M.; Pfeifer L.; Pidgeon G. V.; Giuffredi G. T.; Thompson A. L.; Paton R. S.; Brown J. M.; Gouverneur V. Coordination Diversity in Hydrogen- bonded Homoleptic Fluoride–alcohol Complexes Modulates Reactivity. Chem. Sci. 2015, 6, 5293. 10.1039/C5SC01812A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pfeifer L.; Engle K. M.; Pidgeon G. V.; Sparkes H. A.; Thompson A. L.; Brown J. M.; Gouverneur V. Hydrogen-bonded Homoleptic Fluoride–Diarylurea Complexes: Structure, Reactivity, and Coordinating Power. J. Am. Chem. Soc. 2016, 138, 13314. 10.1021/jacs.6b07501. [DOI] [PubMed] [Google Scholar]
- a Rowley M.; Hallett D. J.; Goodacre S.; Moyes C.; Crawforth J.; Sparey T. J.; Patel S.; Marwood R.; Patel S.; Thomas S.; Hitzel L.; O’Connor D.; Szeto N.; Castro J. L.; Hutson P. H.; MacLeod A. M. 3-(4-Fluoropiperidin-3-yl)-2-phenylindoles as High Affinity, Selective, and Orally Bioavailable h5-HT2A Receptor Antagonists. J. Med. Chem. 2001, 44, 1603. 10.1021/jm0004998. [DOI] [PubMed] [Google Scholar]; b Yang Z.-Q.; Barrow J. C.; Shipe W. D.; Schlegel K. -A. S.; Shu Y.; Yang F. V.; Lindsley C. W.; Rittle K. E.; Bock M. G.; Hartman G. D.; Uebele V. N.; Nuss C. E.; Fox S. V.; Kraus R. L.; Doran S. M.; Connolly T. M.; Tang C.; Ballard J. E.; Kuo Y.; Adarayan E. D.; Prueksaritanont T.; Zrada M. M.; Marino M. J.; Graufelds V. K.; DiLella A. G.; Reynolds I. J.; Vargas H. M.; Bunting P. B.; Woltmann R. F.; Magee M. M.; Koblan K. S.; Renger J. J. Discovery of 1,4-Substituted Piperidines as Potent and Selective Inhibitors of T-Type Calcium Channels. J. Med. Chem. 2008, 51, 6471. 10.1021/jm800830n. [DOI] [PubMed] [Google Scholar]; c Shipe W. D.; Barrow J. C.; Yang Z.-Q.; Lindsley C. W.; Yang F. V.; Schlegel K. -A. S.; Shu Y.; Rittle K. E.; Bock M. G.; Hartman G. D.; Tang C.; Ballard J. E.; Kuo Y.; Adarayan E. D.; Prueksaritanont T.; Zrada M. M.; Uebele V. N.; Nuss C. E.; Connolly T. M.; Doran S. M.; Fox S. V.; Kraus R. L.; Marino M. J.; Graufelds V. K.; Vargas H. M.; Bunting P. B.; Hasbun-Manning M.; Evans R. M.; Koblan K. S.; Renger J. J. Design, Synthesis, and Evaluation of a Novel 4-Aminomethyl-4-fluoropiperidine as a T-Type Ca2+ Channel Antagonist. J. Med. Chem. 2008, 51, 3692. 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- For non-asymmetric ring opening of azetidinium ions with different nucleophiles, see:; a Couty F.; David O.; Durrat F.; Evano G.; Lakhdar S.; Marrot J.; Vargas-Sanchez M. Nucleophilic Ring-Opening of Azetidinium Ions: Insights into Regioselectivity. Eur. J. Org. Chem. 2006, 2006, 3479. 10.1002/ejoc.200600200. [DOI] [Google Scholar]; b Couty F.; Durrat F.; Evano G.; Marrot J. Ring expansions of 2-alkenylazetidinium salts—a new route to pyrrolidines and azepanes. Eur. J. Org. Chem. 2006, 2006, 4214. 10.1002/ejoc.200600362. [DOI] [Google Scholar]; c Couty F.; Durrat F.; Evano G. Regioselective Nucleophilic Opening of Azetidinium Ions. Synlett 2005, 2005, 1666. 10.1055/s-2005-871537. [DOI] [Google Scholar]; d De Rycke N.; David O.; Couty F. Assessing the Rates of Ring-Opening of Aziridinium and Azetidinium Ions: A Dramatic Ring Size Effect. Org. Lett. 2011, 13, 1836. 10.1021/ol200348k. [DOI] [PubMed] [Google Scholar]; e Gaertner V. R. Ring-opening Alkylations and Equilibria Involving 1,1-Diethyl-3-substituted-azetidinium Cations. Tetrahedron Lett. 1967, 8, 343. 10.1016/S0040-4039(00)71547-4. [DOI] [Google Scholar]; f O’Brien P.; Phillips D. W.; Towers T. D. An azetidinium ion approach to 3-aryloxy-3-aryl-1-propanamines. Tetrahedron Lett. 2002, 43, 7333. 10.1016/S0040-4039(02)01724-0. [DOI] [Google Scholar]; g Krawiecka B.; Jeziorna A. Stereocontrolled Synthesis of 3-Amino-2-hydroxyalkyl Diphenylphosphine Oxides Mediated by Chiral Azetidinium Salts and Epoxyamines. Tetrahedron Lett. 2005, 46, 4381. 10.1016/j.tetlet.2005.04.075. [DOI] [Google Scholar]; h Concellòn J. M.; Bernad P. L.; Perez-Andrés J. A. Nucleophilic Ring Closure and Opening of Aminoiodohydrins. Tetrahedron Lett. 2000, 41, 1231. 10.1016/S0040-4039(99)02250-9. [DOI] [Google Scholar]; For non-asymmetric nucleophilic ring opening with fluoride, see:; i Wiemer J.; Steinbach J.; Pietzsch J.; Mamat C. Preparation of a Novel Radiotracer Targeting the EphB4 Receptor via Radiofluorination Using Spiro Azetidinium Salts as Precursor. J. Labelled Compd. Radiopharm. 2017, 60, 489. 10.1002/jlcr.3526. [DOI] [PubMed] [Google Scholar]; l Kiesewetter O. D.; Eckelman W. C. Utility of Azetidinium Methanesulfonates for Radiosynthesis of 3-[18F]Fluoropropyl Amines. J. Labelled Compd. Radiopharm. 2004, 47, 953. 10.1002/jlcr.884. [DOI] [Google Scholar]; For a desymmetrization reaction using sulfur nucleophiles, see ref (10a).
- a All of the azetidinium triflate salts employed in this study were bench-stable solids.; b All major chemical suppliers have 30–50 azetidines in their catalogues.; c For a recent example of the synthesis of tetrasubstituted azetidines, see:Fawcett A.; Murtaza A.; Gregson C. H. U.; Aggarwal V. K. Strain-Release-Driven Homologation of Boronic Esters: Application to the Modular Synthesis of Azetidines. J. Am. Chem. Soc. 2019, 141, 4573. 10.1021/jacs.9b01513. [DOI] [PubMed] [Google Scholar]
- a O’Reilly M. C.; Lindsley C. W. A General, Enantioselective Synthesis of β- and γ-Fluoroamines. Tetrahedron Lett. 2013, 54, 3627. 10.1016/j.tetlet.2013.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tanaka J.; Suzuki S.; Tokunaga E.; Haufe G.; Shibata N. Asymmetric Desymmetrization via Metal-Free C–F Bond Activation: Synthesis of 3,5-Diaryl-5-fluoromethyloxazolidin-2-ones with Quaternary Carbon Centers. Angew. Chem., Int. Ed. 2016, 55, 9432. 10.1002/anie.201603210. [DOI] [PubMed] [Google Scholar]; For a recent non-asymmetric report, see:; c Chen Y.-Q.; Singh S.; Wu Y.; Wang Z.; Hao W.; Verma P.; Qiao J. X.; Sunoj R. B.; Yu J.-Q. Pd-catalyzed Gamma-C(sp3)-H Fluorination of Free Amines. J. Am. Chem. Soc. 2020, 142, 9966. 10.1021/jacs.9b13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mizuta S.; Shibata N.; Goto Y.; Furukawa T.; Nakamura S.; Toru T. Cinchona Alkaloid-Catalyzed Enantioselective Monofluoromethylation Reaction Based on Fluorobis(phenylsulfonyl) methane Chemistry Combined with a Mannich-type Reaction. J. Am. Chem. Soc. 2007, 129, 6394. 10.1021/ja071509y. [DOI] [PubMed] [Google Scholar]; b Furukawa T.; Shibata N.; Mizuta S.; Nakamura S.; Toru T.; Shiro M. Catalytic Enantioselective Michael Addition of 1-Fluorobis(phenylsulfonyl)methane to α,β-Unsaturated Ketones Catalyzed by Cinchona Alkaloids. Angew. Chem., Int. Ed. 2008, 47, 8051. 10.1002/anie.200802904. [DOI] [PubMed] [Google Scholar]; c Alba A.-N.; Companyo X.; Moyano A.; Rios R. Formal Highly Enantioselective Organocatalytic Addition of Fluoromethyl Anion to α,β-Unsaturated Aldehydes. Chem. - Eur. J. 2009, 15, 7035. 10.1002/chem.200900991. [DOI] [PubMed] [Google Scholar]; d Ullah F.; Zhao G.-L.; Deiana L.; Zhu M.; Dziedzic P.; Ibrahem I.; Hammar P.; Sun J.; Cordova A. Enantioselective Organocatalytic Conjugate Addition of Fluorocarbon Nucleophiles to α,β-Unsaturated Aldehydes. Chem. - Eur. J. 2009, 15, 10013. 10.1002/chem.200901260. [DOI] [PubMed] [Google Scholar]; e Fukuzumi T.; Shibata N.; Sugiura M.; Yasui H.; Nakamura S.; Toru T. Fluorobis(phenylsulfonyl) methane: A Fluoromethide Equivalent and Palladium-Catalyzed Enantioselective Allylic Monofluoromethylation. Angew. Chem., Int. Ed. 2006, 45, 4973. 10.1002/anie.200600625. [DOI] [PubMed] [Google Scholar]; f Liu W.-B.; Zheng S.-C.; He H.; Zhao X.-M.; Dai L.-X.; You S.-L. Iridium-Catalyzed Regio- and Enantioselective Allylic Alkylation of Fluorobis(phenylsulfonyl)methane. Chem. Commun. 2009, 43, 6604. 10.1039/b914315g. [DOI] [PubMed] [Google Scholar]; For a recent review on the topic, see:; g Yang X.; Wu T.; Phipps R. J.; Toste F. D. Advances in Catalytic Enantioselective Fluorination, Mono, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826. 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Qian D.; Chen M.; Bissember A. C.; Sun J. Counterion-Induced Asymmetric Control in Ring-Opening of Azetidiniums: Facile Access to Chiral Amines. Angew. Chem., Int. Ed. 2018, 57, 3763. 10.1002/anie.201712395. [DOI] [PubMed] [Google Scholar]; For selected desymmetrizations of oxetanes, see:; b Wang Z.; Chen Z.; Sun J. Catalytic Enantioselective Intermolecular Desymmetrization of 3-Substituted Oxetanes. Angew. Chem. 2013, 125, 6817. 10.1002/ange.201300188. [DOI] [PubMed] [Google Scholar]; c Strassfeld A. D.; Wickens Z. K.; Picazo E.; Jacobsen E. N. J. Am. Chem. Soc. 2020, 142, 9175. 10.1021/jacs.0c03991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sasson Y.; Negussie S.; Royz M.; Mushkin N. Tetramethylammonium Chloride as a Selective and Robust Phase Transfer Catalyst in a Solid–Liquid Halex Reaction: The Role of Water. Chem. Commun. 1996, 297. 10.1039/CC9960000297. [DOI] [Google Scholar]; b Macfie G.; Brookes B. A.; Compton R. G. Reactions at Solid-Liquid Interfaces. The Mechanism and Kinetics of the Fluorination of 2,4-Dinitrochlorobenzene Using Solid Potassium Fluoride in Dimethylformamide. J. Phys. Chem. B 2001, 105, 12534. 10.1021/jp012612r. [DOI] [Google Scholar]; c Bram G.; Loupy A.; Pigeon P. Easy and Efficient Heterogeneous Nucleophilic Fluorination without Solvent. Synth. Commun. 1988, 18, 1661. 10.1080/00397918808081327. [DOI] [Google Scholar]; d Brunelle D. J.; Singleton D. A. N-Alkyl-4-(N′,N′-Dialkylamino)pyridinium Salts: Thermally Stable Phase Transfer Catalysts for Nucleophilic Aromatic Displacement. Tetrahedron Lett. 1984, 25, 3383. 10.1016/S0040-4039(01)91026-3. [DOI] [Google Scholar]; e Kim D. W.; Chi D. Y. Polymer-Supported Ionic Liquids: Imidazolium Salts as Catalysts for Nucleophilic Substitution Reactions Including Fluorination. Angew. Chem., Int. Ed. 2004, 43, 483. 10.1002/anie.200352760. [DOI] [PubMed] [Google Scholar]; For selected examples of crown ether and glycols as solid–liquid phase-transfer catalysts for KF, see:; f Liotta C. L.; Harris H. P. Chemistry of Naked Anions. I. Reactions of the 18-Crown-6 Complex of Potassium Fluoride with Organic Substrates in Aprotic Organic Solvents. J. Am. Chem. Soc. 1974, 96, 2250. 10.1021/ja00814a044. [DOI] [Google Scholar]; g Deutsch J.; Niclas H.-J. A Convenient and New Modified Preparation of Nitrosubstituted Aryl Fluorides. Synth. Commun. 1991, 21, 505. 10.1080/00397919108016776. [DOI] [Google Scholar]
- Turnover of the productive fluorination pathway will be influenced by a change in substrate cation concentration (i.e., preformed azetidinium versus in situ-formed episulfonium). Previous work4a showed that the episulfonium ion pair lies at +48 kJ/mol relative to the nonionized precursor, corresponding to an equilibrium constant of 4 × 10–9 at room temperature. High concentrations of the substrate counteranion will also increase competitive binding of the hydrogen bonding catalyst, potentially hindering HB-PTC and/or facilitating alternative anion-binding pathways. See:; a Kozuch S.; Shaik S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101. 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]; b Kozuch S.; Shaik S. Kinetic-Quantum Chemical Model for Catalytic Cycles: The Haber-Bosch Process and the Effect of Reagent Concentration. J. Phys. Chem. A 2008, 112, 6032. 10.1021/jp8004772. [DOI] [PubMed] [Google Scholar]; c Uhe A.; Kozuch S.; Shaik S. Automatic Analysis of Computed Catalytic Cycles. J. Comput. Chem. 2011, 32, 978. 10.1002/jcc.21669. [DOI] [PubMed] [Google Scholar]
- a See the Supporting Information for details.; b Favorable binding thermodynamics is necessary but not sufficient to ensure catalytic activity. Under a Curtin–Hammett scenario, the levels of enantioselectivity and the differential reactivities of azetidinium ions are determined by the relative stabilities of competing TSs.
- N-Benzhydrylazetidinium salts were also featured in the study by Sun and co-workers.10a
- A larger variation in key metrics occurs when Bn is changed to Bzh. ΔG⧧ was measured relative to TBAF + Azet+. ΔGRxn measures the release of ring strain of the ion upon fluorination with TBAF.
- a Kunieda T.; Nagamatsu T.; Higuchi T.; Hirobe M. Highly Efficient Oxazolone-derived Reagents for Beta-Lactam Formation from Beta-amino Acids. Tetrahedron Lett. 1988, 29, 2203. 10.1016/S0040-4039(00)86712-X. [DOI] [Google Scholar]; b Weiner B.; Szymański W.; Janssen D. B.; Minnaard A. J.; Feringa B. L. Chem. Soc. Rev. 2010, 39, 1656. 10.1039/b919599h. [DOI] [PubMed] [Google Scholar]
- Low-temperature single-crystal X-ray diffraction data were collected using a Rigaku Oxford Diffraction SuperNova diffractometer. Raw frame data were reduced using CrysAlisPro, and the structures were solved using Superflip. See:; a Palatinus L.; Chapuis G. SUPERFLIP - A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Crystallogr. 2007, 40, 786. 10.1107/S0021889807029238. [DOI] [Google Scholar]; Successive refinement was performed with CRYSTALS. See:; b Parois P.; Cooper R. I.; Thompson A. L. Crystal Structures of Increasingly Large Molecules: Meeting the Challenges with CRYSTALS Software. Chem. Cent. J. 2015, 9, 30. 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cooper R. I.; Thompson A. L.; Watkin D. J. CRYSTALS Enhancements: Dealing with Hydrogen Atoms in Refinement. J. Appl. Crystallogr. 2010, 43, 1100. 10.1107/S0021889810025598. [DOI] [Google Scholar]; Full refinement details are given in the Supporting Information.
- a Smilovic I. G.; Cluzeau J.; Richter F.; Nerdinger S.; Schreiner E.; Laus G.; Schottenberger H. Synthesis of Enantiopure Antiobesity Drug Lorcaserin. Bioorg. Med. Chem. 2018, 26, 2686. 10.1016/j.bmc.2018.02.038. [DOI] [PubMed] [Google Scholar]; b Zhu Q.; Wang J.; Bian X.; Zhang L.; Wei P.; Xu Y. Novel Synthesis of Antiobesity Drug Lorcaserin Hydrochloride. Org. Process Res. Dev. 2015, 19, 1263. 10.1021/acs.oprd.5b00144. [DOI] [Google Scholar]
- Calculations were performed using Gaussian 16, rev. A.03 for optimization and frequency calculations, with single-point energy corrections performed using ORCA 4.2.0. The CPCM(1,2-DCE)/ωB97X-D3/(ma)-def2-TZVPP//CPCM(1,2-DCE)/M06-2X/def2-SVP(D) level of theory was used for all of the calculations. Full computational details, including precise definitions of the basis sets, are provided in the Supporting Information. Thermochemistry was evaluated at 298.15 K at 1 M standard concentration.
- The full ensemble of computed TSs for the reactions of cis-1a and trans-1a are provided in the Supporting Information.
- For rare examples, see ref (10a) and:West T. H.; Daniels D. S. B.; Slawin A. M. Z.; Smith A. D. An Isothiourea-Catalyzed Asymmetric [2,3]-Rearrangement of Allylic Ammonium Ylides. J. Am. Chem. Soc. 2014, 136, 4476. 10.1021/ja500758n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.