

Abstract
SARS-related coronaviruses poses continual threat to humanity by rapidly mutating and emerging as severe pandemic outbreaks, including the current nCoV-19 pandemic. Hence a rapid drug repositioning and lead identification strategy are required to mitigate these outbreaks. We report a pharmacophore and molecular dynamics-based approach for drug repositioning and lead identification against dual targets (3CLp and PLp) of SARS-CoV-2. The pharmacophore model of 3CLp inhibitors was apolar with two aromatic and two H-bond acceptors, whereas that of PLp was relatively polar, bearing one aromatic and three H-bond acceptors. Pharmacophore-based virtual screening yielded six existing FDA-approved drugs and twelve natural products with both the pharmacophoric features. Among them are nelfinavir, tipranavir and licochalcone-D, which has shown better binding characteristics with both the proteases compared to lopinavir. The molecular dynamics revealed that the connecting loop (residues 176–199) of 3CLp is highly flexible, and hence, inhibitors should avoid high-affinity interactions with it. Lopinavir, due to its high affinity with the loop region, exhibited unstable binding. Further, the van der Waals size of the 3CLp inhibitors positively correlated with their binding affinity with 3CLp. However, the van der Waals size of a ligand should not cross a threshold of 572Å3, beyond which the ligands are likely to make high-affinity interaction with the loop and suffer unstable binding as observed in the case of lopinavir. Similarly, the total polar surface area of the ligands were found to be negatively correlated with their binding affinity with PLp.
Keywords: Pharmacophore, SARS-CoV-2 inhibitors, 3CL-protease, PL-protease, molecular dynamics, protease inhibitors, anti-HIV drugs, natural products
Graphical Abstract

Communicated by Ramaswamy H. Sarma
Introduction
An epidemic of severe acquired respiratory syndrome (SARS) occurred in the year 2003 with 8000+ infected cases across 26 countries, since then smaller incidents of infections continued to occur (Mackenzie et al., 2004). The recently emerged novel coronavirus (nCoV-19), a mutated and more contagious sister of SARS has caused a global pandemic concern, with 8.7 million infected cases and 4.7 lakh deaths across 216 countries within six months from dawn of its spread (WHO. Coronavirus). The enhanced infection rate of SARS-CoV-2 compared to other SARS-related viruses has been attributed to its unique furin cleavage site at the interface of S1/S2 subunit in the spike protein (Walls et al., 2020). The development of anti-SARS therapeutics remains a challenging task due to various reasons including rapid mutation characteristic of viruses, contagious handling hazard and rapid infection rate. SARS-related coronaviruses including SARS-CoV-2 belongs to the family of RNA viruses under the subclass of β-coronaviridae, which encodes their replication machinery thereby increasing their survival fitness. Its genome consists of 14 open reading frames (ORF), that encodes a single polyprotein which is processed to produce different structural (membrane and spike glycoprotein) and functional (replicase/transcriptase, proteases) viral proteins (Woo et al., 2010).
The 3CL-protease (a C30 endopeptidase) and PL-protease (papain-like protease) belong to the class of cysteine proteases that processes the C-terminal of replicase apo-polyprotein to release the functional replicase. Unlike other corona viruses, β-coronaviruses including nCoV-19, encodes only one papain-like protease (PLp) instead of two, and one 3CL-protease (3CLp) (Fehr & Perlman, 2015; Woo et al., 2010). The prime functional role of these proteases makes them an attractive target for anti-viral drug design. However, the uncertainty due to rapid mutation still poses a problem. Hence we strategize on identifying inhibitors that possess dual inhibition capability of both the PLp and 3CLp. This dual inhibition strategy can improve the anti-viral efficacy and also avert the problem of resistance due to mutation, wherein the molecule could still exert its action, even if one of the targets get mutated. Pharmacophore based drug design has shown to be a better approach to develop ligands bearing active interactive features of different targets than other strategies (Arooj et al., 2013; Jana & Singh, 2019; Mitra et al., 2019; Thangapandian et al., 2011; Xu et al., 2015). Pharmacophore can be defined as a common denominator of molecular interactive features including electronic and steric, that is shared by a set of ligands that are active against a particular target (Liu et al., 2020).
In this study, we developed two pharmacophore models (Ph-1 and Ph-2) obtained from reported specific inhibitors of SARS-CoV 3CLp and PLp, respectively. Further, both the pharmacophore models were individually used as constraints to screen against FDA-approved drug and natural product databases. The overlapping hits were further shortlisted based on drug-likeness and toxicity filters. The resulting shortlisted candidates were docked into both the proteases and the final selected candidates with higher docking scores were subjected to molecular dynamics simulation to understand their binding potential and interaction with the target, as well as their stability. The study aims to identify existing drugs that can be potentially repositioned against SARS-CoV-2 as well as suggest natural compounds as a prospective leads for dual inhibition of these two proteases (Figure 1). Furthermore, molecular dynamics study was performed to understand the structural dynamics of these proteases, which can help in designing stable binding inhibitors.
Figure 1.

Graphical abstract of workflow followed in the study.
Materials and methods
Data set and pharmacophore modelling
Two pharmacophore models were developed using the previously reported inhibitors for both these proteins (Arooj et al., 2013; Mitra et al., 2019). In the case of 3CLp (PDB: 6LU7) the respective inhibitors (supplementary material Table 1) were docked into the active site using AutoDockVina to obtain their bioactive conformation, which is termed as set-1 ligands. The homology model of SARS-CoV-2 PLp (SMTL ID: 6w9c.1) was obtained from SWISS MODEL repository (Bienert et al., 2017). The reported PLp inhibitors (supplementary material Table 2) were docked into the active site of the modeled SARS-CoV-2 PLp and the binding pose with the highest energy was considered as bio-active conformation, which is termed as set-2 ligands.
Table 1.
Final shortlisted drugs from ZINC drug database, as potential repositioning candidates.
| ZINC I.D. | Name | Structure | Binding energy (kcal/mol) |
|
|---|---|---|---|---|
| 3CLp | PLp | |||
| ZINC32350 | Cinoxacin | 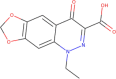 |
–6.6 | –7.3 |
| ZINC6627681 | Trimethoprim | 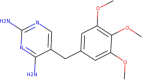 |
–6.7 | –6.8 |
| ZINC3833846 | Nelfinavir | 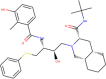 |
–8.2 | –7.8 |
| ZINC76945632 | Novobiocin | 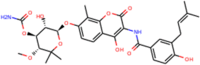 |
–8.4 | –7.9 |
| ZINC537891 | Ofloxacin |  |
–7.4 | –6.9 |
| ZINC100016058 | Tipranavir |  |
–8.0 | –7.9 |
| Control | Lopinavir | – | –7.4 | –7.2 |
Table 2.
Final shortlisted natural products from ZINC natural product database, as potential lead compounds.
| ZINC I.D. / Name | Structure | Binding energy (kcal/mol) |
|
|---|---|---|---|
| 3CL-PRO | PL-PRO | ||
| ZINC000079188744 |  |
–8.2 | –7.9 |
| ZINC000070670121 |  |
–8.0 | –8.6 |
| ZINC000070669963 | 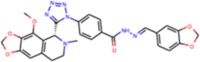 |
–8.1 | –8.6 |
| ZINC000001105784 | |
–8.6 | –8.2 |
| ZINC000004098633 Pieceid | 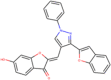 |
–7.8 | –8.0 |
| ZINC000004262788 | 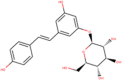 |
−8.5 | –8.4 |
| ZINC000006483512 Wedelolactone | 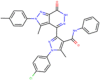 |
–8.5 | –7.8 |
| ZINC000008771878 | 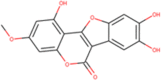 |
–8.0 | –7.6 |
| ZINC000011867619 | 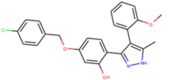 |
–8.8 | –7.9 |
| ZINC000013378583 Boeravinone F | 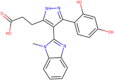 |
–8.8 | –8.0 |
| ZINC000014762510 Lichochalcone D | 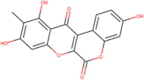 |
–8.9 | –8.7 |
| ZINC000068583835 | 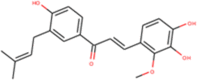 |
–8.8 | –8.5 |
Both the ligand sets were submitted to PharmaGist, an open-source webserver that uses a deterministic algorithm to identify the overlapping potential pharmacophoric points between the set of ligands (Schneidman-Duhovny et al., 2008). The pharmacophore with highest alignment score was selected for further study. The molecular similarity between the obtained pharmacophoric features and their ligands in the respective datasets was calculated using molecular overlay module of the Discovery Studio (San Diego; Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017). This software calculates the similarity between the pharmacophores in a fingerprint-based approach and reports the overlap as similarity, in the scale of 0 to 1.
Pharmacophore based virtual screening and molecular docking
The best pharmacophore models (Ph-1 and Ph-2) obtained from PharmaGist were used as constraints to screen against ZINC database using ZincPharmer (Koes & Camacho, 2012). The databases of natural products and approved drugs were screened separately using both the pharmacophore models. A combined hits approach was used where the hits that overlapped in both the pharmacophore screens were considered as potential hits that can act against both the targets (Xu et al., 2015). The potential hit lists were further filtered based on ADME, drug-likeness and toxicity criteria using Swiss-ADME (Daina et al. 2017) to yield the shortlisted hits.
The shortlisted hits were docked into the active sites of SARS-CoV-2 3CLp (PDB: 6LU7) and PLp. The receptors fordocking were prepared using UCSF-Chimera Dock Prep module (Pettersen et al., 2004). The multiple ligands docking was performed using PyRx for virtual screening (Dallakyan & Olson, 2015). A cubic grid box of 30 Å centered at (–14.0102, 12.7531, 67.4105) for 3CL-protease and 25 Å for PLp, was considered to encompass the binding site. The compounds that have binding energy lesser than or equal to that of reference is considered as final shortlisted potential candidates. Here lopinavir was used as the reference.
Molecular simulation and analysis
In order to further understand the role of pharmacophores in stabilizing the ligand-protein interactions, all atom molecular dynamics simulations was performed using GROMACS2018 with AMBER03 forcefield (Abraham et al., 2015; Duan et al., 2003). The pose with best binding energy was selected and the receptor was extracted from the docked complex and processed through H++ server [ver.3.2] using default parameters (Anandakrishnan et al., 2012). The ligand topologies was generated using ACPYPE (Sousa da Silva & Vranken, 2012). In case of 3CLp, the respective complex topologies were created by combining protein and ligand topologies. Complexes as well as the protein were placed in individual cubic periodic boxes, with a minimum distance of 2 nm from the box edges, filled with water (TIP3P) and neutralized and saturated with a salt concentration of 0.15 M NaCl. Energy minimization, equilibration (NVT and NPT for 500 ps each) and production run (50 ns) were carried out with the parameters and conditions as described elsewhere (Lemkul, 2019; Naresh et al., 2015). In the case of PLp, the modelled protein was first subjected to a production run of 20 ns and a snapshot of the protein at 20 ns was extracted and used to dock the ligands of interest. The complexes thus formed were subjected to a production run of 30 ns, as mentioned above. The RMSD, RMSF, number of hydrogen bonds were obtained from tools provided in the GROMACS suite. g_mmpbsa module was used to carry out the MM-PBSA analysis on 50 snapshots extracted from the trajectory post convergence (Open Source Drug Discovery Consortium, 2014).
Results and discussion
There are currently no reported inhibitors available against 3CLp and PLp of SARS-CoV-2 since it is a newly emerged pathogen. However, there exists a handful of reported inhibitors against SARS-CoV. A preliminary two-dimensional and three-dimensional (3D) structural comparison of SARS-CoV-2 3CLp (PDB: 6LU7) and SARS-CoV-1 3CLp (PDB: 5N19), indicated that they share 96% similarity, particularly the active site structure was found to be conserved. The homology model of SARS-CoV-2 PL-protease (SMTL ID: 6w9c.1) was obtained from SWISS model database which was found to share 89% similarity with SARS-CoV-1 PLp. This modelled PLp was subjected to equilibration and MD simulations pre-run for 20 ns to optimize the active site side-chain rotamers. The pharmacophore models were developed from SARS-CoV 3CLp and PLp experimental inhibitors reported in the literature (Báez-Santos et al., 2014; Blanchard et al., 2004; Chen et al., 2005; Cho et al., 2013; Kim et al., 2014; Kuo et al., 2009; Liu et al., 2005; Park et al., 2012a; Park et al., 2012b; Park et al., 2016; Park et al., 2017; Pillaiyar et al., 2016; Ramajayam et al., 2010a; Ramajayam et al., 2010b; Song et al., 2014; Yuan et al., 2017).
Pharmacophore modeling of SARS-CoV-2 proteases
The 3CLp is a C30-endopeptidase that belongs to the class of cysteine proteases where HIS-41 and CYS-145 were found to be the catalytic dyad involved in proteolytic action (Huang et al., 2004). The pharmacophore model, Ph-1 (Figure 2(a)) obtained from a set of reported 15 different 3CLp inhibitors (supporting material Table 1) suggests, two aromatic centroids (AR) and two hydrogen bond acceptors (ACC). The catalytic mechanism studies have found that 3CLp bears a non-canonical substrate specificity, requiring a PHE at the second position upstream of the cleavage point as well as at the third downstream position (Muramatsu et al., 2016). The two aromatic features obtained in the pharmacophore model represent them. The molecular similarity of the reported inhibitors with respect to the developed pharmacophore model Ph-1, showed good correlation (R = 0.90) with the binding energies of the same (Figure 2(b)).
Figure 2.

(a) Pharmacophore modeling of the reported 3CLp inhibitors from their bio-active conformation (i) alignment of reported inhibitors displaying their overlapping pharmacophoric points, (ii) the derived Pharmacophore model-1(Ph-1); (b) The plot indicates the correlation between the binding energies of the reported inhibitors and their pharmocophoric similarity with respect to Ph-1 (R = 0.90); (c) (i) pharmacophore modelling of the reported PLp inhibitors from their bio-active conformation (i) alignment of reported inhibitors displaying their overlapping pharmacophoric points, (ii) the derived pharmacophore model-2 (Ph-2); (d) the plot indicates the correlation between the binding energies of the reported inhibitors and their pharmocophoric similarity with respect to Ph-2 (R = 0.92).
Similarly the 3D pharmacophore model, Ph-2 (Figure 2(c)) developed from 17 different PLp inhibitors (supporting material Table 2) is composed of one aromatic centroid (AR) and three hydrogen bond acceptors (ACC). The PLp is a part of the nsp3 polyprotein complex which consists of papain-like protease domain that recognizes LXGG motif and cleaves it releasing various other non-structural proteins including nsp1, nsp2 and nsp3 from viral polyprotein complex (Báez-Santos et al., 2015). The main catalytic diad in action was found to be CYS and HIS, while an ASP was thought to be involved in an analogous role as in the serine proteases (Ménard et al., 1990). The molecular similarity of the reported PLp-inhibitors with respect to the developed pharmacophore model Ph-2, also showed a good correlation (R = 0.92) with the binding energies of the same (Figure 2(d)).
It is observed in both the cases, that the pharmacophoric similarity of the inhibitors with respect to their obtained pharmacophore models, exhibited good correlation to their binding energies. This could be due to the fact that, the inhibitors were docked into their respective targets, and the bio-active conformation obtained was used for pharmacophore elucidation. The pharmacophore obtained for 3CLp is found to be more apolar (bearing two aromatic and two acceptor features), when compared to the pharmacophore of PLp (which bears one aromatic and three acceptors). The difference in their polarity could be due to the nature of amino acids present in the active site and entrance of these proteins. The active site of 3CLp is present near the inter-domain cleft consisting of both aromatic (PHE, TYR) and polar (CYS, HIS) amino acids, while that of PLp is solvent exposed predominantly consisting of polar amino acids including CYS, HIS, SER and THR.
Pharmacophore based virtual screening
Pharmacophore based virtual screening of ZINC drug database yielded six existing drug candidates that overlapped in both the pharmacophore screens (Table 1) (Figure 3(a)). These drugs included cinoxacin, nelfinavir, tipranavir, ofloxacin, trimethoprim and novobiocin. Further, docking of these candidates against SARS-CoV-2 3CLp and PLp indicated that nelfinavir, tipranavir, novobiocin and ofloxacin, possessed better binding potential when compared to lopinavir (binding energy = –7.3 kcal/mol). Here, lopinavir a known anti-viral protease inhibitor, is used as the reference, since there are no known drugs against SARS-CoV proteases, as well as this drug is being considered as a part of the current treatment strategy (Cao et al., 2020).
Figure 3.

Results of pharmacophore-based virtual screening of (a) (i) ZINC drug database, (ii) top two candidates based on docking score and drug-likeness; (b) (i) natural product databases, (ii) top two candidates based on docking score and drug-likeness. Ph-1 = pharmacophore model based on 3CLp inhibitors, Ph-2 = pharmacophore model based on PLp. The circles indicate the total number of hits occurred individually in each pharmacophore screen, while the intersection indicates the overlap candidates.
In order to mitigate the rapid spread and decrease of the disease burden, existing drugs including anti-retroviral protease inhibitors are being tested as potential candidates for re-positioning against SARS-CoV-2, amongst which ritonavir/lopinavir (Kaletra®) combination has been approved for clinical management. However, recent clinical and in vitro studies have shown that lopinavir is less effective against SARS-CoV-2 (Cao et al., 2020; Sheahan et al., 2020). Our study suggests that nelfinavir and tipranavir (existing anti-HIV protease inhibitors) which bears the desired pharmacophoric features could be considered as potential candidates for inhibiting SARS-CoV-2 proteases. A recent high-throughput screening of anti-retroviral protease inhibitors have also identified that tipranavir and nelfinavir to be potent inhibitors against SARS-CoV-2 when compared to lopinavir. In addition, nelfinavir has also shown potent anti-viral activity against SARS-CoV-1 (Yamamoto et al., 2020, 2004). Novobiocin and ofloxacin belongs to the class of broad-spectrum anti-bacterial agents. Studies have shown that ofloxacin also exhibits potent anti-viral activity against influenza and vaccinia virus (Ikeda et al., 1987). Similarly, novobiocin also has shown potent anti-viral activity in vitro and in vivo against zika and vaccinia viruses (Sekiguchi & Shuman, 1997; Yuan et al., 2017). This suggests that apart from exhibiting anti-bacterial actions these drugs may also exhibit anti-viral activity.
The pathogenesis of SARS-CoV-2 manifests in various ways, where more than 50% of the deaths and complications are caused due to lung damage and bacterial co-infection leading to pneumonia (Provisional Death Counts for Coronavirus Disease (COVID-19), 2020; Vincent & Taccone, 2020). Hence the use of broad-spectrum anti-bacterial agents such as novobiocin and ofloxacin might improve the treatment outcome by preventing the lower respiratory tract infections including bacterial pneumonia as well as interfere with SARS-CoV-2 proteins. A similar approach where the combination of azithromycin and hydroxychloroquine have been shown to lead to a better recovery rate when compared to hydroxychloroquine alone(Gautret et al., 2020). Thus, nelfinavir, tipranavir, ofloxacin and novobiocin could be considered as potential re-positioning candidates for SARS-CoV-2 treatment.
Similarly, the pharmacophore based virtual screening of ZINC Natural product database yielded 157 overlapped hits (Figure 3(b)) which bears both the pharmacophoric features. Further, they were also screened for toxicity and drug-likeness score which led to 28 candidates that satisfied these conditions. The final 12 potential natural product candidates were shortlisted based on the docking scores (Table 2). The notable ones include wedelolactone (present in extracts of Eclipta alba—also known as bhringraj), piecied (analog of resveratrol present in grapes), boeravinone-F (present in extracts of Boerhaavia diffusa Linn. also known as punarnava), and licochalcone-D (present in extracts of Glycyrrhiza glabra, also known as licorice). These natural products have been reported to have broad-spectrum anti-viral activities against phyto-viruses, enteroviruses and hepatitis virus (Manvar et al., 2012; Wang et al., 2015). The extracts and phytoconstituents of Glycyrrhiza glabra, have shown to have potent broad-spectrum anti-viral activity against various viruses, including SARS-related coronaviruses including SARS-CoV (Cinatl et al., 2003; Fiore et al., 2008; Hoever et al., 2005; Wang et al., 2015). The coumarin and stilbene scaffolds present in boeravinone and pieceid, respectively indicate that these candidates might show activity against SARS-CoV-2 (Hassan et al., 2016; Wahedi et al., 2020). It is heartening to note that these natural products identified in the study have also shown in vitro anti-viral activity against several viruses and hence can be considered as potential lead candidates for designing SARS-CoV-2 inhibitors.
Molecular dynamics and analysis
The top two candidates with best docking binding energy were selected from the final shortlisted candidates, namely nelfinavir and tipranavir from FDA-approved drugs and licochalcone-D and wedelolactone from natural product database for molecular dynamics simulations studies with both the proteases. Lopinavir was used as reference and the receptors alone (3CLp and PLp) were also subjected to simulations.
The RMSD of 3CLp complexed with lopinavir showed a significant deviation between 6–13 ns, beyond which the complex continued to have only partially stable RMSD (Figure 4(a)). While other complexes including nelfinavir, tipranavir, licochalcone-D and wedelolactone reached convergence within 4 ns and continued to remain stable. Ligand interaction plots and H-bond analysis (Figure 4(d)) throughout the trajectory indicated that all the ligands were making a minimum of two hydrogen bonds and two aromatic π-π interactions as suggested by the pharmacophore model Ph-1. The residues, HIE 41, MET 49 and MET 165 contribute to significant favorable interactions with all the ligands, while ASP 187 and GLU 166 are making unfavorable interactions. This abnormal spike in RMSD and the relative instability of lopinavir is consistent with previous reports (Wang, 2020), in the subsequent section we explain the cause for the same, to aid in designing effective 3CLp inhibitors.
Figure 4.

Molecular dynamic simulations of selected final candidates against 3CLp (a) RMSD plot; (b) RMSD plot of ligand backbone (vs.) protein backbone; (c) average energy per residue plot indicating the energies of interacting residue of lopinavir (LOP), tipranavir (TIP), nelfinavir (NEL), licochalcone (LIC) and, wedelolactone (WED); (d) average of H-bonds throughout the trajectory of 3CLp complexes; (e) binding energy of 3CLp complexes obtained from MM-PBSA post-convergence.
Principle component analysis indicated that the cause for the observed fluctuations in lopinavir global RMSD is due to the twisting and stretching movement of the flexible connecting loop (residue 176–199). Lopinavir was making a highly favorable interaction (–8.19 kJ—attractive force) with the connecting loop residue GLN 189 (which is located in the middle of the loop) (Figure 5(a); (ii)). This interaction could cause a restriction in the movement of the loop which could be one of the reasons for the observed fluctuations in the global RMSD. This was also confirmed by comparing the RMSD plot between the backbone of the main protein and the ligand (lopinavir) with including and excluding the connecting loop (Figure 5(a); (iii)).
Figure 5.

(a) 3CLp depicting the connecting loop regions (red, residues 176–199), and main regions (green, residues 1–176); (b) energy per residue interaction plot of lopinavir obtained from MM-PBSA. Arrow indicating the higher favorable interaction of GLN189; (c) individual RMSD plot of lopinavir showing the RMSD of the main region (black), connecting loop region (yellow) and ligand backbone (green); (d) energy per residue interaction plot of tipranavir obtained from MM-PBSA. Arrow indicating the unfavorable interaction of GLN189.
MM-PBSA binding energy analysis (Figure 4(c)) showed that lopinavir (–34.57 ± 3.18 kcal/mol) and tipranavir (–33.77 ± 4.03 kcal/mol) have higher binding energy when compared to nelfinavir and licochalcone-D which have an average binding energy of –18.07 ± 2.97 kcal/mol. For all the ligands the major energy contribution comes from van der Waals interaction (supplementary material S1). Though lopinavir had good binding energy, the relative instability caused due to the high-affinity interaction with the connecting loop might render it ineffective. Licochalcone-D which has less favorable interaction (0 to –1 kJ) with the loop, shows only a slight deviation in the RMSD. Here, the contribution of GLN189 is relatively low when compared to other strong interactions from residues within the pocket, thereby retaining the molecule inside the pocket. In the case of tipranavir the connecting loop residues, ASP187 and ARG188 are making unfavorable contact (+3.21 kJ—repulsive force) which pushes the drug into the pocket (Figure 5(b)).These observations suggest that while designing 3CLp inhibitors, care must be taken so that the inhibitor avoids making high-affinity interaction with the connecting loop residues (176 to 199). Similar conclusions were also found in the case of HIV-protease flaps (Meagher & Carlson, 2004). The observed instability of lopinavir could be one plausible reason for its observed weak activity (Cao et al., 2020; Sheahan et al., 2020; Yamamoto et al., 2020).
The RMSD of all the PLp-complexes reached convergence between 2 and 4 ns (Figure 6(a)). The ligand RMSD (Figure 6(c)) of nelfinavir, tipranavir and licochalcone-D were stable; however, the RMSD of lopinavir and wedelolactone were relatively unstable, particularly lopinavir was showing a significant spike (>1.0 Å) at 25 ns. MM-PBSA (Figure 6(b)) indicates that lopinavir (–19.2 kcal/mol) and nelfinavir (–16.9 kcal/mol) have high binding energy, while tipranavir and licochalcone were having a binding energy of –13.5 kcal/mol. Energy decompositi on analysis (supplementary material in S1) indicated that van der Waals energy played a significant contribution. Further it was also observed that polar solvation energy (+47.25 kJ—unfavorable) of lopinavir was very high when compared to other energy contributions including van der Waals energy (–41.68 kJ) indicating that binding of lopinavir is significantly influenced by solvent, which might be one of the reasons for the observed instability in its RMSD. The energy per residue plot (Figure 6(d)) indicated that PRO247, PRO248 and TYR264 contributes to favorable interactions whereas ASP164, ASP302, and GLU167 show unfavorable interactions. Nelfinavir and tipranavir were found to make high energy favorable interactions with more residues when compared to other candidates (Figure 6(e,f)).
Figure 6.

Molecular dynamic simulations of selected final candidates against PLp (a) global RMSD plot; (b) Binding energy of PLp complexes obtained from MM-PBSA; (c) RMSD plot of ligand backbone (vs.) protein backbone of PLp complexes; (d) average energy per residue plot indicating the energies of interacting residue of lopinavir (LOP), tipranavir (TIP), nelfinavir (NEL), licochalcone (LIC) and, wedelolactone (WED); Energy per residue 3D interaction plot of obtained from MM-PBSA, (e) lopinavir, (f) nelfinavir.
Effect of van der Waal size and polar surface area in binding stability
The van der Waals size of the reported 3CLp inhibitors was found to correlate well (R = 0.90) with their binding affinity such that ligands with higher van der Waals size exhibited better binding affinity towards 3CLp. But if the van der Waals size of a ligand exceeds the threshold value of 572 A3, which is the active site pocket volume of 3CLp, then it is highly likely that it may make high-affinity interaction with the connecting loop (supporting material Figure). This was observed in the case of lopinavir (607.2 A3), whereas tipranavir (522.1 A3), nelfinavir (522.4 A3) and licochalcone-D (327.8 A3) with sizes lower than this threshold value were found to bind stably.
In the case of PLp the binding affinity of the reported PLp inhibitors were found to negatively correlate with their total polar surface area (tPSA) (R = –0.81) which could be due to the fact that molecules with a high polar surface area might suffer higher solvation energy (unfavourable) thereby decreasing the binding affinity with the protein. This is also observed in the case of lopinavir (supporting information Figure S1) which has relatively higher polar surface area as well as high solvation energy (47.2 kJ/mol) when compared to other candidates (26.8 kJ/mol). These observations suggest that van der Waals size and total polar surface area of the ligands might play a significant role in their binding with 3CLp and PLp, respectively. This difference in variables could be attributed to the active site nature of these two enzymes, where the active site of 3CLp is located at the interdomain cleft whereas that of PLp is solvent-exposed. However, there could be exceptions as in the case of wedelolactone.
Conclusion
The negative binding energies of the final shortlisted candidates with 3CLp were as follows: lopinavir (–34.57 ± 3.42 kcal/mol) > tipranavir (–33.77 ± 4.02 kcal/mol) > nelfinavir (–20.54 ± 1.73 kcal/mol) > licochalcone-D (–18.90 ± 1.97 kcal/mol) > wedelolactone (–11.24 ± 2.08 kcal/mol), whereas those with PLp were: lopinavir (–19.23 ± 0.40 kcal/mol > nelfinavir (–16.48 ± 0.55 kcal/mol) > tipranavir (–13.76 ± 0.60 kcal/mol) > licochalcone-D (–13.26 ± 0.37 kcal/mol) > wedelolactone (–8.82 ± 0.40 kcal/mol). It should be noted that lopinavir, despite having lowest negative binding energy, was found to have unstable binding with both the proteases, which might be one of the reasons for its low activity observed towards SARS-CoV-2 (Choy et al., 2020). Hence, nelfinavir and tipranavir could be considered as potential repositioning candidates alternate to lopinavir. This also correlates with recent in vitro high-throughput screening studies which indicated that, nelfinavir and tipranavir can inhibit SARS-CoV-2 with good activity (Yamamoto et al., 2020). Also, the binding energy of licochalcone-D and wedelolactone indicates that these natural products can also have inhibitory activity against SARS-CoV-2 proteases. In addition, the anti-bacterial agents like ofloxacin, cinoxacin, novobiocin and trimethoprim can also be considered as adjuvant in combinatorial therapy against SARS-CoV-2. Similarly, the 12 natural products (Table 2) identified including licochalcone-D and wedelolactone could be considered as potential lead candidates for designing inhibitors against SARS-CoV-2. The molecular dynamic studies have shown that the active site of 3CLp is dynamic, due to the flexible connecting loop (residue 177–200) which acts like a lid, similar to HIV protease dynamics (Torbeev et al., 2009). The 3CLp inhibitors should avoid making high affinity interaction with this loop in order to bind stably with the protein. The ligands having higher van der Waals size might have better binding affinity towards 3CLp. However, the van der Waals size should not be greater than 572 A3, since crossing this threshold might likely increase the chance of high-affinity interaction with the flexible loop. Since the active site of PLp is highly solvent exposed, the ligands having higher total polar surface area might suffer greater solvation (polar solvation energy) effect thereby decreasing their binding stability. Hence it is suggested that, the ligands having lesser total polar surface area might have better binding affinity with PLp.
Supplementary Material
Acknowledgements
The authors thank the computational facility at the COSMOS lab of CRIF, Sri Sathya Sai Institute of Higher Learning, for providing computational facilities to carry out the work.
Glossary
Abbreviations
- SARS
severe acquired respiratory syndrome
- SARS-CoV-2
severe acquired respiratory syndrome-coronavirus-2
- nCoV-19
novel coronavirus-2019
- 3CLp
3-chymotrypsin-like protease
- PLp
papain-like protease
- Ph-1
pharmacophore model based on 3CLp inhibitors
- Ph-2
pharmacophore model based on PLp inhibitors
- LOP
lopinavir
- TIP
tipranavir
- NEL
nelfinavir
- WED
wedelolactone
- LIC
licochalcone-D
- tPSA
total polar surface area
Disclosure statement
None to declare.
References
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Anandakrishnan, R., Aguilar, B., & Onufriev, A. V. (2012). H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Research, 40(Web Server issue), W537–W541. 10.1093/nar/gks375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arooj, M., Sakkiah, S., Cao, G. Ping., & Lee, K. W. (2013). An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLos One, 8(4), e60470. 10.1371/journal.pone.0060470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos, Y. M., Barraza, S. J., Wilson, M. W., Agius, M. P., Mielech, A. M., Davis, N. M., Baker, S. C., Larsen, S. D., & Mesecar, A. D. (2014). X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-like Proteases. Journal of Medicinal Chemistry, 57(6), 2393–2412. https://doi.org/ 10.1021/jm401712t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos, Y. M., St. John, S. E., & Mesecar, A. D. (2015). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert, S., Waterhouse, A., de Beer, T. A. P., Tauriello, G., Studer, G., Bordoli, L., & Schwede, T. (2017). The Swiss-model repository-new features and functionality. Nucleic Acids Research, 45(D1), D313–D319. 10.1093/nar/gkw1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard, J. E., Elowe, N. H., Huitema, C., Fortin, P. D., Cechetto, J. D., Eltis, L. D., & Brown, E. D. (2004). High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chemistry & Biology, 11(10), 1445–1453. 10.1016/j.chembiol.2004.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, B., Wang, Y., Wen, D., Liu, W.Wang, Jingli, Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., … Wang, C. (2020). A trial of lopinavir–ritonavir in adults hospitalized with severe covid-19. New England Journal of Medicine, 382(19), 1787–1799.. 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L., Chen, S., Gui, C., Shen, J., Shen, X., & Jiang, H. (2006). Discovering severe acute respiratory syndrome coronavirus 3CL protease inhibitors: virtual screening, surface plasmon resonance, and fluorescence resonance energy transfer assays. Journal of Biomolecular Screening, 11(8), 915–921. 10.1177/1087057106293295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L.-R., Wang, Y.-C., Lin, Y. W., Chou, S.-Y., Chen, S.-F., Liu, L. T., Wu, Y.-T., Kuo, C.-J., Chen, T. S.-S., & Juang, S.-H. (2005). Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorganic & Medicinal Chemistry Letters, 15(12), 3058–3062. 10.1016/j.bmcl.2005.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, J. K., Curtis-Long, M. J., Lee, K. H., Kim, D. W., Ryu, H. W., Yuk, H. J., & Park, K. H. (2013). Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorganic & Medicinal Chemistry, 21(11), 3051–3057. 10.1016/j.bmc.2013.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy, K.-T., Wong, Y.-L., Kaewpreedee, P., Sia, S. F., Chen, D., Hui, K. P. Y., Chu, D. K. W., Chan, M. C. W., Cheung, P.-H., Huang, X., Peiris, M., & Yen, H.-L. (2020). Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Research, Antiviral Res, 178, 104786. https://doi.org/ 10.1016/j.antiviral.2020.104786 32251767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl, J., Morgenstern, B., Bauer, G., Chandra, P., Rabenau, H., & Doerr, H. W. (2003). Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. The Lancet, 361(9374), 2045–2046. 10.1016/S0140-6736(03)13615-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. 10.1038/srep42717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan, S., & Olson, A. J. (2015). Small-molecule library screening by docking with PyRx . Methods in Molecular Biology, 1263, 243–250. 10.1007/978-1-4939-2269-7_19 [DOI] [PubMed] [Google Scholar]
- Duan, Y., Wu, C., Chowdhury, S., Lee, M. C., Xiong, G., Zhang, W., Yang, R., Cieplak, P., Luo, R., Lee, T., Caldwell, J., Wang, J., & Kollman, P. (2003). A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of Computational Chemistry, 24(16), 1999–2012. 10.1002/jcc.10349 [DOI] [PubMed] [Google Scholar]
- Fehr, A. R., & Perlman, S. (2015). Coronaviruses: An overview of their replication and pathogenesis. Methods in Molecular Biology (Clifton, N.J.), 1282, 1–23. 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore, C., Eisenhut, M., Krausse, R., Ragazzi, E., Pellati, D., Armanini, D., & Bielenberg, J. (2008). Antiviral effects of Glycyrrhiza species. Phytotherapy Research, 22(2), 141–148. 10.1002/ptr.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautret, P., Lagier, J.-C., Parola, P., Hoang, V. T., Meddeb, L., Mailhe, M., Doudier, B., Courjon, J., Giordanengo, V., Vieira, V. E., Dupont, H. T., Honoré, S., Colson, P., Chabrière, E., La Scola, B., Rolain, J.-M., Brouqui, P., & Raoult, D. (2020). Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. International Journal of Antimicrobial Agents, 105949, 105949. 10.1016/j.ijantimicag.2020.105949 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hassan, M. Z., Osman, H., Ali, M. A., & Ahsan, M. J. (2016). Therapeutic potential of coumarins as antiviral agents. European Journal of Medicinal Chemistry, 123, 236–255. 10.1016/j.ejmech.2016.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoever, G., Baltina, L., Michaelis, M., Kondratenko, R., Baltina, L., Tolstikov, G. A., Doerr, H. W., & Cinatl, J. (2005). Antiviral activity of glycyrrhizic acid derivatives against SARS-coronavirus . Journal of Medicinal Chemistry, 48(4), 1256–1259. 10.1021/jm0493008 [DOI] [PubMed] [Google Scholar]
- Huang, C., Wei, P., Fan, K., Liu, Y., & Lai, L. (2004). 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry, 43(15), 4568–4574. 10.1021/bi036022q [DOI] [PubMed] [Google Scholar]
- Ikeda, S., Yazawa, M., & Nishimura, C. (1987). Anti-viral activity and inhibition of topoisomerase by ofloxacin, a new quinolone derivative. Antiviral Research, 8(3), 103–113. 10.1016/0166-3542(87)90064-7 [DOI] [PubMed] [Google Scholar]
- Jana, S., & Singh, S. K. (2019). Identification of selective MMP-9 inhibitors through multiple e-pharmacophore, ligand-based pharmacophore, molecular docking, and density functional theory approaches. Journal of Biomolecular Structure & Dynamics, 37(4), 944–965. 10.1080/07391102.2018.1444510 [DOI] [PubMed] [Google Scholar]
- Kim, D. W., Seo, K. H., Curtis-Long, M. J., Oh, K. Y., Oh, J.-W., Cho, J. K., Lee, K. H., & Park, K. H. (2014). Phenolic phytochemical displaying SARS-CoV papain-like protease inhibition from the seeds of Psoralea corylifolia. Journal of Enzyme Inhibition and Medicinal Chemistry, 29(1), 59–63. 10.3109/14756366.2012.753591 [DOI] [PubMed] [Google Scholar]
- Koes, D. R., & Camacho, C. J. (2012). ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Research, 40(Web Server issue), W409–W414. 10.1093/nar/gks378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., & Lynn, A., Open Source Drug Discovery Consortium. (2014). g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Kuo, C.-J., Liu, H.-G., Lo, Y.-K., Seong, C.-M., Lee, K.-I., Jung, Y.-S., & Liang, P.-H. (2009). Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Letters, 583(3), 549–555. 10.1016/j.febslet.2008.12.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul, J. (2019). From proteins to perturbed hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1.0]. Living Journal of Computational Molecular Science, 1(1), 5068. 10.33011/livecoms.1.1.5068 [DOI] [Google Scholar]
- Liu, J., Zhu, Y., He, Y., Zhu, H., Gao, Y., Li, Z., Zhu, J., Sun, X., Fang, F., Wen, H., & Li, W. (2020). Combined pharmacophore modeling, 3D-QSAR and docking studies to identify novel HDAC inhibitors using drug repurposing. Journal of Biomolecular Structure & Dynamics, 38(2), 533–547. 10.1080/07391102.2019.1590241 [DOI] [PubMed] [Google Scholar]
- Liu, Y.-C., Huang, V., Chao, T.-C., Hsiao, C.-D., Lin, A., Chang, M.-F., & Chow, L.-P. (2005). Screening of drugs by FRET analysis identifies inhibitors of SARS-CoV 3CL protease. Biochemical and Biophysical Research Communications, 333(1), 194–199. 10.1016/j.bbrc.2005.05.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie, J. S., Drury, P., Ellis, A., Grein, T., Leitmeyer, K. C., Mardel, S., Merianos, A., Olowokure, B., Roth, C., Slattery, R., Thomson, G., Werker, D., & Ryan, M. (2004). The Who response to SARS and preparations for the future. In Learning from SARS: Preparing for the next disease outbreak. Workshop summary. National Academies Press. [PubMed] [Google Scholar]
- Manvar, D., Mishra, M., Kumar, S., & Pandey, V. N. (2012). Identification and evaluation of anti hepatitis C virus phytochemicals from Eclipta alba. Journal of Ethnopharmacology, 144(3), 545–554. 10.1016/j.jep.2012.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher, K. L., & Carlson, H. A. (2004). Incorporating protein flexibility in structure-based drug discovery: Using HIV-1 protease as a test case. Journal of the American Chemical Society, 126(41), 13276–13281. 10.1021/ja0469378 [DOI] [PubMed] [Google Scholar]
- Ménard, R., Khouri, H. E., Plouffe, C., Dupras, R., Ripoll, D., Vernet, T., Tessier, D. C., Lalberté, F., Thomas, D. Y., & Storer, A. C. (1990). A protein engineering study of the role of aspartate 158 in the catalytic mechanism of papain. Biochemistry, 29(28), 6706–6713. 10.1021/bi00480a021 [DOI] [PubMed] [Google Scholar]
- Mitra, K., Chadha, A., & Doble, M. (2019). Pharmacophore based approach to screen and evaluate novel Mycobacterium cell division inhibitors targeting FtsZ - A modelling and experimental study. European Journal of Pharmaceutical Sciences: Official Journal of the European Federation for Pharmaceutical Sciences, 135, 103–112. 10.1016/j.ejps.2019.04.023 [DOI] [PubMed] [Google Scholar]
- Muramatsu, T., Takemoto, C., Kim, Y.-T., Wang, H., Nishii, W., Terada, T., Shirouzu, M., & Yokoyama, S. (2016). SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proceedings of the National Academy of Sciences of the United States of America, 113(46), 12997–13002. 10.1073/pnas.1601327113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naresh, K. N., Sreekumar, A., & Rajan, S. S. (2015). Structural insights into the interaction between molluscan hemocyanins and phenolic substrates: An in silico study using docking and molecular dynamics. Journal of Molecular Graphics & Modelling, 61, 272–280. 10.1016/j.jmgm.2015.07.006 [DOI] [PubMed] [Google Scholar]
- Park, J.-Y., Jeong, H. J., Kim, J. H., Kim, Y. M., Park, S.-J., Kim, D., Park, K. H., Lee, W. S., & Ryu, Y. B. (2012. a). Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biological & Pharmaceutical Bulletin, 35(11), 2036–2042. 10.1248/bpb.b12-00623 [DOI] [PubMed] [Google Scholar]
- Park, J.-Y., Kim, J. H., Kim, Y. M., Jeong, H. J., Kim, D. W., Park, K. H., Kwon, H.-J., Park, S.-J., Lee, W. S., & Ryu, Y. B. (2012. b). Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorganic & Medicinal Chemistry, 20(19), 5928–5935. 10.1016/j.bmc.2012.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J.-Y., Ko, J.-A., Kim, D. W., Kim, Y. M., Kwon, H.-J., Jeong, H. J., Kim, C. Y., Park, K. H., Lee, W. S., & Ryu, Y. B. (2016). Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. Journal of Enzyme Inhibition and Medicinal Chemistry, 31(1), 23–30. 10.3109/14756366.2014.1003215 [DOI] [PubMed] [Google Scholar]
- Park, J.-Y., Yuk, H. J., Ryu, H. W., Lim, S. H., Kim, K. S., Park, K. H., Ryu, Y. B., & Lee, W. S. (2017). Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. Journal of Enzyme Inhibition and Medicinal Chemistry, 32(1), 504–512. 10.1080/14756366.2016.1265519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., & Jung, S.-H. (2016). An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. Journal of Medicinal Chemistry, 59(14), 6595–6628. 10.1021/acs.jmedchem.5b01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provisional Death Counts for Coronavirus Disease (COVID-19) [WWW Document]. (2020). Retrieved December 4, 2020, from https://www.cdc.gov/nchs/nvss/vsrr/covid19/index.htm.
- Ramajayam, R., Tan, K.-P., Liu, H.-G., & Liang, P.-H. (2010. a). Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorganic & Medicinal Chemistry, 18(22), 7849–7854. 10.1016/j.bmc.2010.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramajayam, R., Tan, K.-P., Liu, H.-G., & Liang, P.-H. (2010. b). Synthesis, docking studies, and evaluation of pyrimidines as inhibitors of SARS-CoV 3CL protease. Bioorganic & Medicinal Chemistry Letters, 20(12), 3569–3572. 10.1016/j.bmcl.2010.04.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Dror, O., Inbar, Y., Nussinov, R., & Wolfson, H. J. (2008). PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Research, 36(Web Server issue), W223–W228. 10.1093/nar/gkn187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi, J., & Shuman, S. (1997). Novobiocin inhibits vaccinia virus replication by blocking virus assembly. Virology, 235(1), 129–137. 10.1006/viro.1997.8684 [DOI] [PubMed] [Google Scholar]
- Sheahan, T. P., Sims, A. C., Leist, S. R., Schäfer, A., Won, J., Brown, A. J., Montgomery, S. A., Hogg, A., Babusis, D., Clarke, M. O., Spahn, J. E., Bauer, L., Sellers, S., Porter, D., Feng, J. Y., Cihlar, T., Jordan, R., Denison, M. R., & Baric, R. S. (2020). Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nature Communications, 11, 1–14. 10.1038/s41467-019-13940-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. H., Kim, D. W., Curtis-Long, M. J., Yuk, H. J., Wang, Y., Zhuang, N., Lee, K. H., Jeon, K. S., & Park, K. H. (2014). Papain-like protease (PLpro) inhibitory effects of cinnamic amides from Tribulus terrestris fruits. Biological & Pharmaceutical Bulletin, 37(6), 1021–1028. 10.1248/bpb.b14-00026 [DOI] [PubMed] [Google Scholar]
- Sousa da Silva, A. W., & Vranken, W. F. (2012). ACPYPE - AnteChamber PYthon Parser interfacE. BMC Research Notes, 5, 367. 10.1186/1756-0500-5-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangapandian, S., John, S., Sakkiah, S., & Lee, K. W. (2011). Molecular docking and pharmacophore filtering in the discovery of dual-inhibitors for human leukotriene A4 hydrolase and leukotriene C4 synthase. Journal of Chemical Information and Modeling, 51(1), 33–44. 10.1021/ci1002813 [DOI] [PubMed] [Google Scholar]
- Torbeev, V. Y., Raghuraman, H., Mandal, K., Senapati, S., Perozo, E., & Kent, S. B. H. (2009). Dynamics of “flap” structures in three HIV-1 protease/inhibitor complexes probed by total chemical synthesis and pulse-EPR spectroscopy. Journal of the American Chemical Society, 131(3), 884–885. 10.1021/ja806526z [DOI] [PubMed] [Google Scholar]
- Vincent, J.-L., & Taccone, F. S. (2020). Understanding pathways to death in patients with COVID-19. The Lancet. Respiratory Medicine, 8(5), 430–432. 10.1016/S2213-2600(20)30165-X32272081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahedi, H. M., Ahmad, S., & Abbasi, S. W. (2020). Stilbene-based natural compounds as promising drug candidates against COVID-19. Journal of Biomolecular Structure & Dynamics, 1–10. 10.1080/07391102.2020.176274332345140 [DOI] [PubMed] [Google Scholar]
- Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., Mcguire, A. T., & Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell, 181(2), 281–292.e6. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. (2020). Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. Journal of Chemical Information and Modeling, 60(6), 3277–3286. 10.1021/acs.jcim.0c00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Yang, R., Yuan, B., Liu, Y., & Liu, C. (2015). The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharmaceutica Sinica B, 5(4), 310–315. 10.1016/j.apsb.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Coronavirus [WWW Document]. (n.d.). Retrieved April 8, 2020, from https://www.who.int/emergencies/diseases/novel-coronavirus-2019
- Woo, P. C. Y., Huang, Y., Lau, S. K. P., & Yuen, K.-Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses, 2(8), 1804–1820. 10.3390/v2081803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y., Liu, X., Wang, Y., Zhou, N., Peng, J., Gong, L., Ren, J., Luo, C., Luo, X., Jiang, H., Chen, K., & Zheng, M. (2015). Combinatorial pharmacophore modeling of multidrug and toxin extrusion transporter 1 inhibitors: A theoretical perspective for understanding multiple inhibitory mechanisms. Scientific Reports, 5, 13684–13613. 10.1038/srep13684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, N., Matsuyama, S., Hoshino, T., & Yamamoto, N. (2020). Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. BioRxiv. 10.1101/2020.04.06.026476 [DOI] [Google Scholar]
- Yamamoto, N., Yang, R., Yoshinaka, Y., Amari, S., Nakano, T., Cinatl, J., Rabenau, H., Doerr, H. W., Hunsmann, G., Otaka, A., Tamamura, H., Fujii, N., & Yamamoto, N. (2004). HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochemical and Biophysical Research Communications, 318(3), 719–725. 10.1016/j.bbrc.2004.04.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S., Chan, J. F.-W., den-Haan, H., Chik, K. K.-H., Zhang, A. J., Chan, C. C.-S., Poon, V. K.-M., Yip, C. C.-Y., Mak, W. W.-N., Zhu, Z., Zou, Z., Tee, K.-M., Cai, J.-P., Chan, K.-H., de la Peña, J., Pérez-Sánchez, H., Cerón-Carrasco, J. P., & Yuen, K.-Y. (2017). Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antiviral Research, 145, 33–43. 10.1016/j.antiviral.2017.07.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.