

Abstract
In proteins where conformational changes are functionally important, the number of accessible states and their dynamics are often difficult to establish. Here we describe a novel 19F-NMR spectroscopy approach to probe dynamics of large membrane proteins. We labeled a glutamate transporter homologue with a 19F probe via cysteine chemistry and with a Ni2+ ion via chelation by a di-histidine motif. We used distance-dependent enhancement of the longitudinal relaxation of 19F nuclei by the paramagnetic metal to assign the observed resonances. We identified two outward- and one inward-facing states of the transporter, in which the substrate-binding site is near the extracellular and intracellular solutions, respectively. We then resolved the structure of the unanticipated second outward-facing state by Cryo-EM. Finally, we showed that the rates of the conformational exchange are accessible from measurements of the metal-enhanced longitudinal relaxation of 19F nuclei.
Introduction
The rapid expansion of protein structure determinations now often results in reports of multiple conformations of the same protein, providing snapshots of functional states, but their temporal relationship is usually unknown. NMR spectroscopy is a versatile approach to probe protein dynamics 1, but large assemblies, such as membrane proteins, present challenges because of severe broadening and overlap of resonances. TROSY NMR addresses the issue 2, but requires protein perdeuteration, which is cumbersome and not always possible for membrane proteins 3.
19F-NMR provides high sensitivity, robust chemical shift dispersion, the absence of background signals, and does not require perdeuteration 4,5. However, assigning resonances to structural states is challenging. Typically, the assignment relies on population shifts in response to ligand binding or mutations and requires prior information from other techniques 6. Alternatively, differences in solvent paramagnetic resonance enhancement (PRE) 7 are used, but this approach only works when states feature distinct solvent exposure of the 19F probe. Here, we measured 19F longitudinal relaxation rates (R1) and their distance-dependent enhancement by paramagnetic ions chelated by di-histidine motifs to assign resonances to different states and to estimate the rates of their conformational exchange.
We used a membrane transporter, GltPh, forming homo-trimeric ~300 kDa protein/detergent particles 8 to develop the strategy. GltPh is an aspartate/sodium symporter from Pyrococcus horikoshii, homologous to human glutamate transporters essential for glutamatergic neurotransmission in the brain 9. Human glutamate transporters and GltPh utilize trans-membrane ionic gradients, primarily of sodium (Na+) ions, to drive concentrative uptake of their substrates 10,11. GltPh transports its cargo by an “elevator” mechanism, whereby the substrate-binding transport domain transitions between outward- and inward-facing states (OFS and IFS, respectively) relative to the stationary trimerization scaffold (Fig. 1a). The dynamics of these transitions likely determine the rate of transport 12,13. We labeled a cysteine introduced into the transport domain of GltPh with a 19F trifluoroethanethio (TET) moiety 14 (Extended Data Fig.1a ) and placed a pair of histidines into a transmembrane helix (TM) of the scaffold domain to chelate a nickel (Ni2+) ion at a site where the distance to the 19F probe changes upon state transition (Fig. 1a). By measuring Ni2+-mediated enhancement of 19F longitudinal relaxation rates, we assigned the three resonances observed in the 19F spectrum to one IFS and two distinct OFS conformations. Guided by the NMR experiments, we determined the Cryo-EM structures of the two outward-facing states. Finally, the Ni2+-enhanced R1 relaxation rates could be used to estimate the exchange rate between the OFS and IFS conformations.
Figure 1: Design for 19F and Ni2+ labeling of GltPh for R1 PRE.
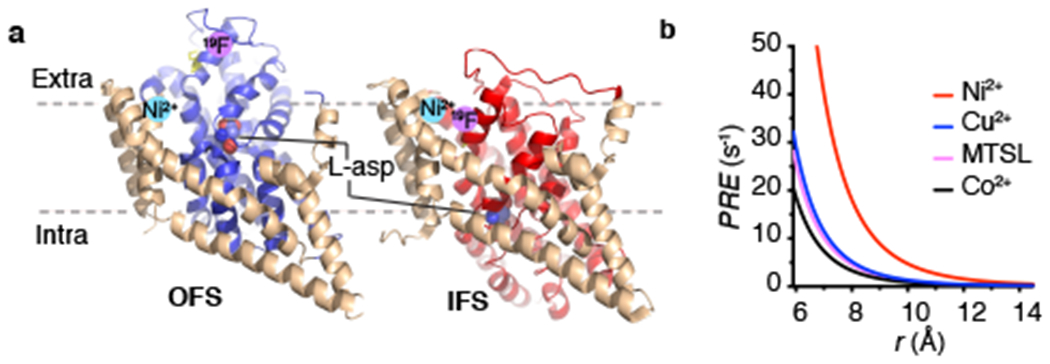
a, Cartoon representations of the structures of L-asp bound GltPh in the OFS (PDB accession code 2NWX) and IFS (accession code 3KBC). The structurally rigid scaffold domain is colored wheat and the dynamic transport domain is colored blue and red in the OFS and IFS, respectively. The substrate L-asp is shown as spheres. Pink and cyan circles represent the expected locations of the 19F label and bound Ni2+ ion, respectively. Only one of the three protomers was shown. b, Dependence of the longitudinal R1 PRE on the distance between 19F and paramagnetic centers based on Equation 1, assuming τc is 213 ns, S2 is 0.1 and τi is 20 ps. Other parameters are given in Supplementary Note. MTSL, (1-Oxyl-2,2,5,5-tetramethylpyrroline-3- methyl)methanethiosulfonate. Lines are colored red (Ni2+), blue (Cu2+), magenta (MTSL), black (Co2+).
Results
Expected Ni2+-mediated 19F R1 relaxation enhancement
PRE results from a dipole-dipole interaction between spins of an unpaired electron and a nucleus, which depends on the inverse sixth power of the distance, r, between them. It affects the rates of both longitudinal (R1 PRE) and transverse relaxation (R2 PRE). The R2 PRE of 1H nuclei is often used to obtain distance information for structure determination, identification of protein-protein interfaces, and detection of invisible transient states 15. However, the transverse relaxation time of 19F nuclei in large membrane proteins is very short, only a few milliseconds 7, rendering the R2 PRE effect insignificant and challenging to quantify 16.
In contrast, R1 PRE is relatively insensitive to the protein size. Its magnitude is described by the Solomon−Bloembergen equations 17,18:
| (1) |
where γ is the nuclear gyromagnetic ratio and JMF is the spectral density function accounting for the local side chain motions (Supplementary Note). Because PRE depends quadratically on the gyromagnetic ratio, it is weak for 13C and 15N nuclei but is significant for 1H and 19F 19. Retrieving the distance-dependent paramagnetic component from the R1 PRE for 1H nuclei is difficult because of the interference from 1H-1H cross relaxation 15. 19F longitudinal relaxation occurs in proteins on a seconds time scale and, because it is dominated by dipole-dipole interactions 20 with negligible heteronuclear 19F-1H cross relaxation 21,22, is effectively mono-exponential 21.
Paramagnetic divalent ions can be chelated by pairs of histidines placed into i and i+4 positions of a helix 23, and the resultant PRE can be measured by taking the difference of the relaxation rates in the presence of an ion, R1, Ni and in its absence, R1,ref:
| (2) |
Based on Equation 1, Ni2+ is particularly advantageous compared to other paramagnetic centers because it leads to a larger R1 PRE effect (Fig. 1b).
19F-NMR spectra of GltPh reveal multiple conformations
We set out to use 19F R1 PRE to detect and distinguish the two key functional states of GltPh, the OFS and the IFS (Fig. 1a). We introduced a single cysteine mutation M385C into the transport domain for labeling with TET and a double histidine mutation Y215H/E219H (dHis) into TM5 of the scaffold domain to generate a coordination site for a Ni2+ ion (Fig. 1a). dHis/M385C GltPh proteins were efficiently labeled with TET group 14,24, and remained active in L-aspartate (L-asp) uptake experiments in proteoliposomes (Extended Data Fig. 1).
1D 19F-NMR spectra of dHis/M385C-TET GltPh (Fig. 2) in the presence of saturating concentrations of Na+ ions and L-asp showed three partially overlapping resonances, S1-S3. Notably, dHis/M385C-TET GltPh in complex with a non-transportable blocker DL-threo-3-methyl aspartic acid (TMA) 25 displayed a similar spectrum. Thus, the 19F-NMR probe revealed three distinct conformations of the transporter that did not undergo rapid exchange on the NMR time scale. Indeed, slow interconversion between the OFS and the IFS on the order of seconds is expected for GltPh based on single-molecule FRET (smFRET) and atomic force microscopy (AFM) studies 12,26,27. Earlier measurements also showed that Na+ and L-asp-bound GltPh samples the OFS and the IFS with comparable probabilities 12,28. Therefore, it is possible that S1, S2, and S3 peaks, which all show similar populations, correspond to either OFS or IFS. To assign the resonances to specific states, we first examined how distinct ligands changed their populations. In the absence of L-asp, GltPh binds Na+ with about 3-fold higher affinity in the OFS than in the IFS, suggesting that Na+ alone should favor the OFS 29. The 19F-NMR spectrum of dHis/M385C-TET GltPh in the presence of 0.6 M Na+ and in the absence of L-asp showed a diminished S1 peak (Fig. 2b), suggesting that the peak corresponded to the IFS. Consistently, the S1 peak also disappeared in the presence of a competitive blocker DL-threo-β-benzyloxyaspartic acid (TBOA) (Fig. 2c), which favors the OFS 30. The S2 and S3 peaks persisted under these conditions, suggesting that both corresponded to the OFS. If so, the relative populations of the IFS and OFS of Na+/L-asp-bound dHis/M385C-TET GltPh calculated from the areas of the 3 peaks, were 30 ± 1.4 % and 70 ± 6 %, respectively, consistent with previous measurements 12,28. To support our peak assignments, we collected 1D 19F spectra for dHis/M385C-TET GltPh bearing additional mutations that favor the IFS, K290A and R276S/M395R (termed RSMR from here on, for brevity) 12,26. Both mutants showed similar peak positions to dHis/M385C-TET GltPh, but dramatically increased intensities of peak S1, consistent with this resonance corresponding to the IFS (Fig. 2e,f and Supplementary Fig. 1).
Figure 2: 19F-NMR spectra of dHis/M385C-TET GltPh and its mutants.
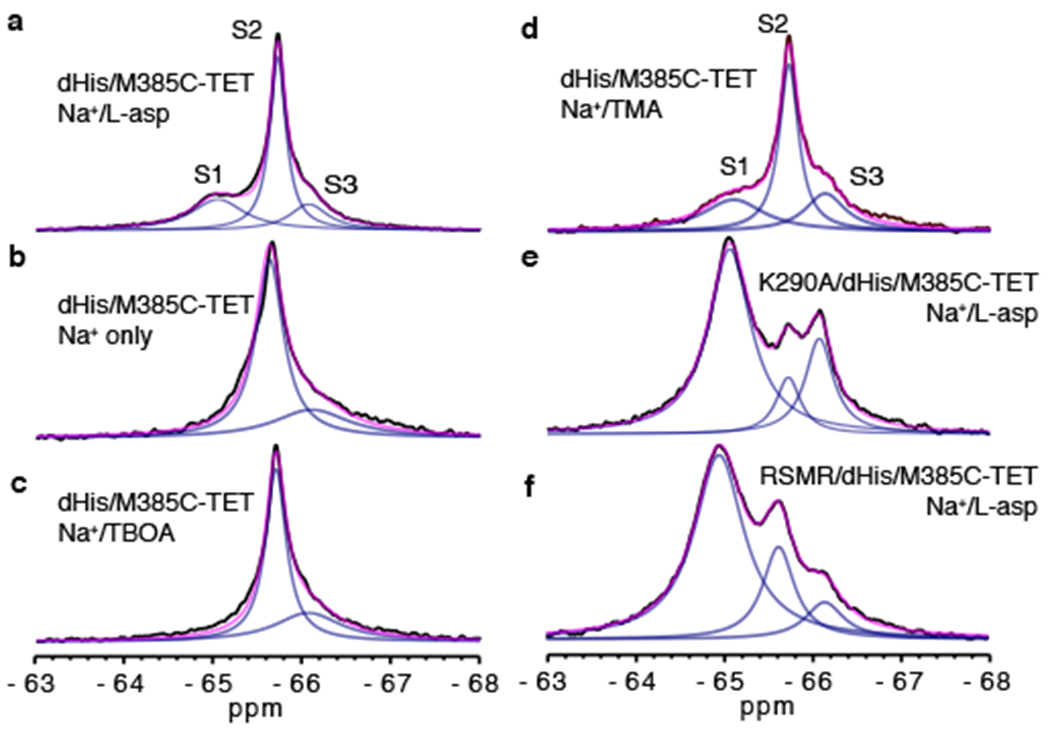
1D spectra of dHis/M385C-TET GltPh (WT) recorded at 293 K in the presence of 200 mM Na+ and 10 μM L-asp (a), 0.6 M Na+ only (b), 200 mM Na+ and 1 mM TBOA (c) or 200 mM Na+ and 110 μM TMA (d). 1D spectra of K290A/dHis/M385C-TET (e) and RSMR/dHis/M385C-TET (f) GltPh mutants in the presence of 200 mM Na+ and 10 μM L-asp. The spectra were deconvoluted into Lorentzian peaks S1, S2 and S3. Raw data are black, fits are magenta and deconvoluted peaks are blue.
R1 PRE differentiates outward- and inward-facing states
To measure Ni2+-mediated PRE, we added 3 molar equivalents of NiSO4 to dHis/M385C-TET GltPh. Unexpectedly, the S1 peak showed increased intensity, corresponding to a population increase to 60 ± 4.5 % (Fig. 2a, 3a). In contrast, the control M385C-TET GltPh lacking the dHis motif did not show this population change (Extended Data Fig. 2). Thus, it appears that Ni2+ binding to the dHis motif in TM5 favors the IFS by ~1.3 kT. The dHis motif in the scaffold is adjacent to the transport domain, and its local environment may differ in the OFS and the IFS, leading to higher affinity for Ni2+ in the latter. Titrating dHis/M385C-TET GltPh with Ni2+ and plotting the intensity of the S1 peak yielded a binding isotherm (Fig. 3a) from which we estimated an affinity for Ni2+ of 34 ± 16 μM, within the range reported for transition metal ions binding to dHis motifs 31.
Figure 3: 19F peak assignment using Ni2+-mediated PRE.
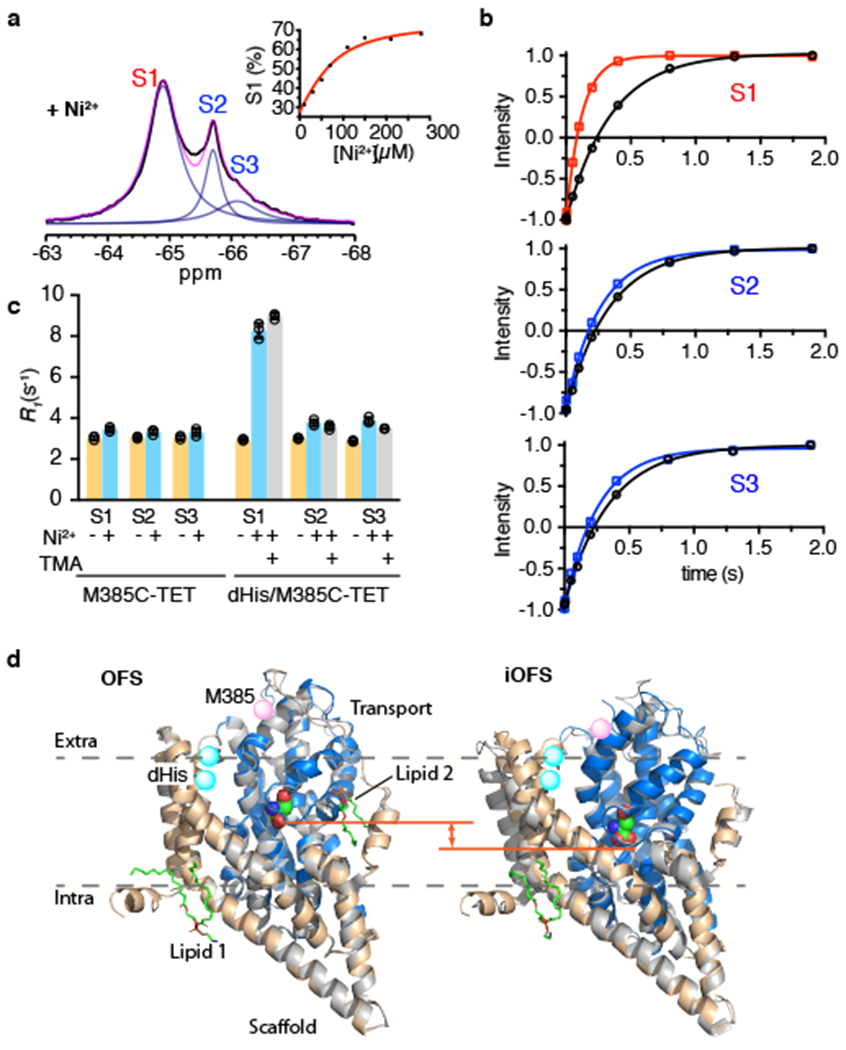
a, 1D 19F spectra of Na+/L-asp-bound dHis/M385C-TET GltPh recorded at 293 K in the presence of 3 molar equivalents of Ni2+ ions. Raw data are black, fits are magenta and deconvoluted peaks are blue. Inset: Ni2+ titration of 69 μM protein. Data are plotted as peak S1 population. Solid red line is the fit to the quadratic binding equation. b, Representative R1 relaxation traces for S1 (top), S2 (middle) and S3 peak (bottom) in the absence (black) and in the presence of Ni2+ ions (color). Solid lines through the data correspond to mono-exponential fits and the results of the fits are shown in Supplementary Table 1. Three independently prepared samples yielded similar results. c, R1 rates of S1, S2 and S3 peaks of M385C-TET and dHis/M385C-TET GltPh constructs in the absence (yellow) and presence of Ni2+ ions and bound to L-asp (cyan) or TMA (gray). Individual data points are shown and the bars indicate means ± s.d. (N = 3). d, Structural comparison between Cryo-EM and crystal structures of GltPh in the OFS (left) and iOFS (right) conformations. Single protomers are viewed in the plain of the membrane and are depicted in cartoon representation. The crystal structures are colored gray, and the Cryo-EM structures are in color with scaffold domain wheat and the transport domain blue. Bound L-asp is shown as spheres and is colored by atom type. Two structurally well-defined lipid molecules are in stick representation, colored by atom type, and labeled Lipid 1 and 2. M385 is rendered as pink spheres. The location of the dHis motif is highlighted as cyan spheres. The orange line indicates the position of the HP2 tips.
Next, we measured R1 relaxation rates for each peak in the absence and presence of Ni2+ ions. We observed a relatively strong PRE of 5.4 ± 0.4 s−1 for peak S1, while peaks S2 and S3 showed weak PRE effects of 0.7 ± 0.3 and 1.0 ± 0.3 s−1, respectively (Fig. 3b and c). When L-asp was replaced by the non-transportable blocker TMA, we observed similar PRE values (Fig. 3c and Supplementary Table 1). In contrast, M385C-TET GltPh without the dHis motif did not show significant R1 PRE in the presence of Ni2+ ions for either of the peaks (Fig. 3c). Using Equation 1 and PRE values measured for dHis/M385C-TET GltPh in the presence of TMA, we estimated Ni2+ to 19F distances to be 9.6 Å for the S1 peak and 14 Å for the S2 and S3 peaks. The PRE-derived distance for the S1 peak is consistent with the distance range between the Ni2+ binding site and the 19F label calculated from molecular dynamics (MD) simulations (see Online Methods) of the IFS structure (mean of 7.7 ± 1.9 Å) (Extended Data Fig. 1e). The PRE-derived distance for the S2 and S3 peaks is significantly larger than for S1 as would be expected for the OFS. It is somewhat smaller than the distance derived from MD simulations of the OFS (mean of 19.4 ± 2.3 Å) (Extended Data Fig. 1e), perhaps because we are beyond the limit of the sensitivity of the measurement (Fig. 1b). Collectively, the data allow unambiguous assignment of the S1 peak to the IFS and the S2 and S3 peaks to the OFS.
Detection of two distinct OFS conformations was surprising but not inconsistent with previous findings. Earlier crystal structures elucidated an OFS conformation but also captured an outward-facing intermediate state (iOFS) of a GltPh mutant, in which the transport domain shifted inward somewhat 32. Intrigued by the NMR results, we used Cryo-EM to visualize conformations sampled by GltPh. We imaged wild type GltPh reconstituted into nanodiscs in the presence of saturating concentrations of L-asp and Na+ ions (Supplementary Table 2). 3D classification and preliminary refinement applying C3 symmetry (Extended Data Fig. 3) was followed by symmetry expansion. The focused 3D classification without alignments revealed two structural classes that were refined and postprocessed to the final resolution of 3.1 and 3.6 Å, respectively. Notably, we observed no protomers in the IFS, consistent with the membrane environment favoring the OFS 26. Class 1 was structurally indistinguishable from the OFS of the transporter 30, while Class 2 closely resembled the iOFS (Fig. 3d) 32. The iOFS was previously interpreted as an on-pathway intermediate state between the OFS and the IFS. Its most interesting feature is the imperfectly packed domain interface, which provides a rationale for the hypothesis that translocation intermediates might form chloride channels 32,33. All glutamate transporters can serve as glutamate-gated chloride channels, but the mechanism of this channel activity is still unknown. Interestingly, in the Cryo-EM structures, we found a well-structured lipid molecule inserted into a crevice between the transport and scaffold domains in the OFS, but in the iOFS, it was either absent or disordered.
To assign these OFS and iOFS states to our two OFS 19F NMR signals, we considered the effects of the K290A mutation, which eliminates a salt bridge between K290 in the transport domain and E192 in the scaffold in the OFS, and should therefore destabilize it. We observed increased intensities of peaks S1 and S3 relative to peak S2 in the spectrum of K290A/dHis/M385C-TET GltPh protein (Fig. 2e). We therefore posit that peak S2 corresponds to the OFS, while peak S3 corresponds to the iOFS. The distance between the 19F probe and the bound Ni2+ ion is decreased by ~3 Å in the iOFS structure compared to the OFS, but remains consistent with our PRE measurements. The observation of the iOFS along with the OFS by 19F NMR in solution strongly supports the iOFS as a mechanistically relevant state.
Exchange rates from enhanced R1 relaxation
We next performed PRE measurements on the K290A and RSMR mutants, which exchange more rapidly between the IFS and OFS than the wild type GltPh 12,26. We observed weaker PREs for peak S1 of K290A and the RSMR than for the wild type dHis/M385C-TET GltPh, and, conversely, stronger PREs for peaks S2 and S3 (Fig. 4, Fig. 5 and Supplementary Table 1). In contrast, when we repeated the PRE measurements for the K290A mutant bound to TMA, which blocks transport, we observed PREs similar to those measured for the wild type transporter (Fig. 4b, 4d and Supplementary Table 1), indicating that the different states are not structurally perturbed by these mutations. Therefore, the altered PRE effects observed in the more dynamic mutants are likely due to chemical exchange between the peaks.
Figure 4: Paramagnetic R1 relaxation and conformational exchange of K290A mutant.
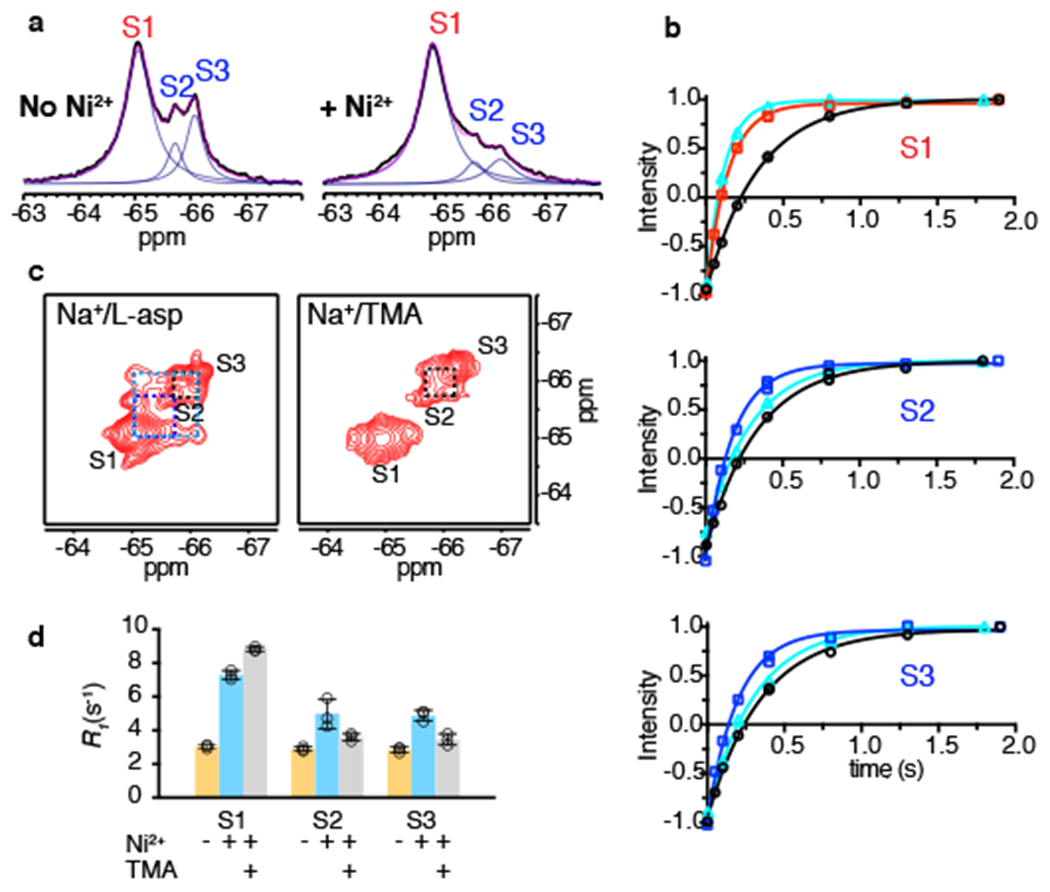
a, 1D 19F spectra of K290A/dHis/M385C-TET GltPh in the absence (left) and presence (right) of Ni2+ ions. Raw data are black, fits are magenta and deconvoluted peaks are blue. b, Representative R1 relaxation traces of S1 (top), S2 (middle) and S3 (bottom) in the absence of Ni2+ ions (black), or in the presence of Ni2+ and bound to L-asp (red for S1, blue for S2 and S3) or TMA (cyan). Solid lines represent mono-exponential fits with fitted R1 values shown in Supplementary Table 1. Three independently prepared samples yielded similar results. c, 19F-19F EXSY spectra of K290A/dHis/M385C-TET GltPh bound to L-asp (left) or TMA (right). Mixing time was set to 0.4 s. Dashed lines indicate cross peaks for S1 and S2 (dark blue), S1 and S3 (light blue) and S2 and S3 (black). d, R1 rates of S1, S2 and S3 peaks of K290A/dHis/M385C-TET in the absence (yellow) and presence of Ni2+ ions and bound to L-asp (cyan) or TMA (gray). Individual data points are shown and the bars indicate means ± s.d. (N = 3).
Figure 5: Paramagnetic R1 relaxation and conformational exchange of RSMR mutant.
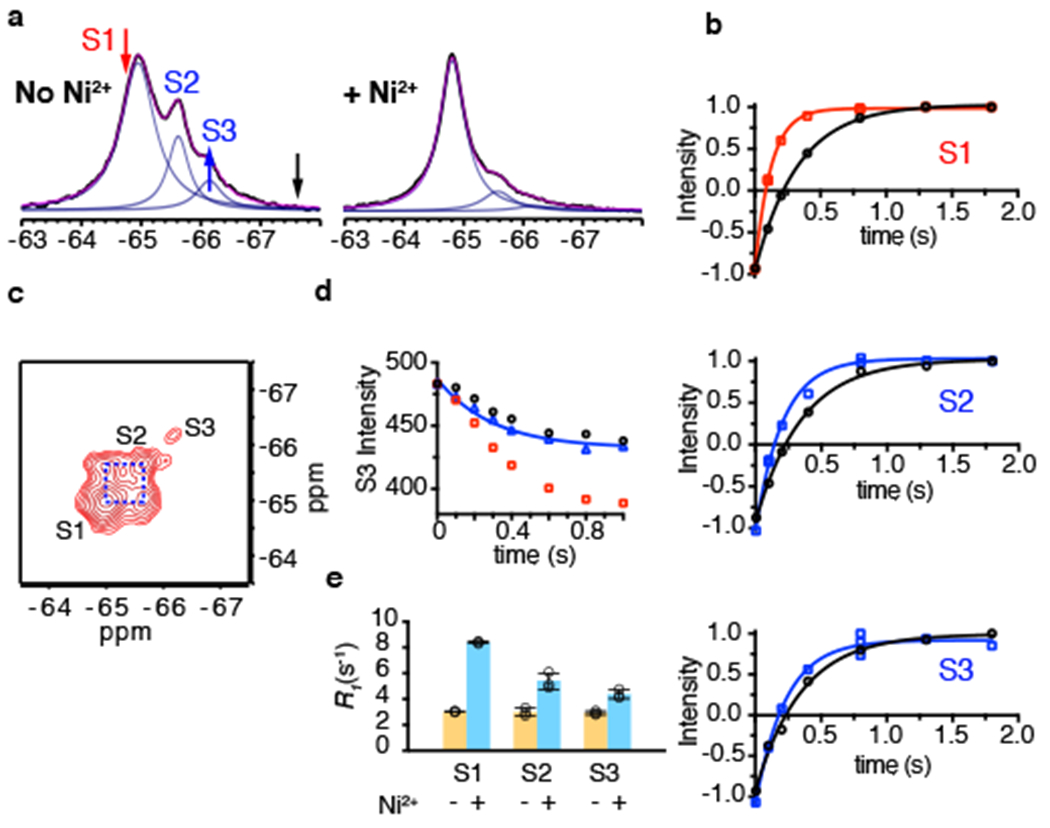
a, 1D 19F NMR spectra of RSMR/dHis/M385C-TET in the absence (left) and presence (right) of Ni2+ ions. Raw data are black, fits are magenta and deconvoluted peaks are blue. The red, black and blue arrows indicate the saturation pulse, control pulse and observed peak, respectively, in the STD experiment in panel d. b, Representative R1 relaxation traces of S1 (top), S2 (middle) and S3 (bottom) peaks in the absence (black) and presence (red for S1 and blue for S2 and S3) of Ni2+ ions. Solid lines represent mono-exponential fits with fitted R1 values shown in Supplementary Table 1. All measurements are in the presence of 200 mM NaCl and 10 μM L-asp. Three independently prepared samples yielded similar results. c, 19F-19F EXSY spectrum of RSMR/dHis/M385C-TET GltPh in the presence of 200 mM NaCl and 10 μM L-asp recorded with mixing time of 0.4 s. d, Decay of the S3 peak upon saturating the S1 peak (red arrow in a) in the STD experiment (red squares). To account for the off-resonance saturation effect, a control experiment (black circles) was performed at an equidistant frequency to S3 peak (black arrow in a). The effective decay curve (blue triangle) is fit to the Equation 5, with results given in Supplementary Table 1. Two independently prepared samples yielded similar results. e, R1 rates of S1, S2 and S3 peaks of RSMR/dHis/M385C-TET in the absence (yellow) and presence of Ni2+ ions with NaCl and L-asp (cyan). Individual data points are shown and the bars indicate means ± s.d. (N = 3).
Considering a two-state model where states A and B have 19F resonances with strong and weak PREs, respectively,
| (3) |
the relaxation processes of the A and B resonances are expected to be bi-exponential because the spins exchange between the two environments (Supplementary Note Equations 11, 12). The time evolution of peak B magnetization depends on the intrinsic relaxation rates of the spin in states A and B (R*1,A and R*1,B, respectively), on the populations of the states (fA and fB), and on the microscopic rate constants (kAB and kBA). We measured R*1,A and R*1,B separately in the presence of the blocker TMA, and integrating peaks in 1D 19F-NMR spectra yielded fA and fB. The relaxation curve of state B can be fitted to the following equation with only two unknown parameters:
| (4) |
Where R1,A and R1,B and , and are defined in the Supplementary Note.
For the dHis/M385C-TET GltPh, the R1 PREs measured for each of the three peaks under paramagnetic conditions were similar in the presence of saturating L-asp or TMA. Therefore, as expected, the L-asp bound dHis/M385C-TET transporter underwent only slow exchange between the OFS and the IFS with negligible contributions to the R1 PREs. Consistently, when we recorded 2D 19F-19F EXSY (EXchange SpectroscopY) spectra 34, we observed only very weak paired cross peaks and only between peaks S1 and S2 at 25 °C (Extended Data Fig. 4). The intensity of EXSY cross-peaks depends on rates of the chemical exchange between the corresponding states, and we estimate the upper limit for the OFS and IFS exchange at ~ 0.3 s−1, consistent with earlier measurements 12,26,27.
In the more dynamic K290A mutant, we considered pairwise equilibria between peaks S1 and S2 and between peaks S1 and S3. S1 (IFS) corresponds to the high-PRE state A, while S2 (OFS) and S3 (iOFS) correspond to the low-PRE state B in Scheme 1. We then fitted R1 relaxation curves for peaks S2 and S3 using Equation 4 and obtained exchange rates, defined as kAB+kBA, of 5.4 ± 2.1 s−1 between S1 and S2 and 3.0 ± 1.2 s−1 between S1 and S3 (Supplementary Table 1). 19F-19F EXSY experiments without Ni2+ ions resolved symmetric off-diagonal cross peaks between all three resonances in K290A mutant (Fig. 4c). Notably, when EXSY spectra were obtained in the presence of the blocker TMA, cross peaks between S1 and S2 peaks and between S1 and S3 peaks were abolished, while the cross peaks between S2 and S3 peaks were still observed. These results are consistent with the assignment of peaks S2 and S3 to the OFS and iOFS, and of peak S1 to the IFS. The average exchange rates obtained from the EXSY experiments, 1.7 s−1 (peaks S1 and S2) and 1.3 s−1 (peaks S1 and S3) were smaller than those from the paramagnetic R1 relaxation measurements. The differences seem to be due mostly to the faster transition rates from the OFS and iOFS (peaks S2 and S3) to the IFS (peak S1) when bound to Ni2+ ions (Supplementary Table 1).
For the RSMR mutant, the exchange rates determined from paramagnetic R1 measurements were 7 ± 2.6 s−1 (peaks S1 and S2) and 2 ± 1.6 s−1 (peaks S1 and S3) (Fig. 5b and Supplementary Table 1). Because of the low population of the S3 peak in the presence of Ni2+ ions, R1 relaxation measurements were of lower precision, but still suggested that the chemical exchange occurred. EXSY spectra also showed large cross peaks between peaks S1 and S2 with an exchange rate of ~5 s−1 (Fig. 5c and Supplementary Table 1). Peak S3 was poorly populated and we could not detect cross peaks between S3 and S1. Instead, we estimated the conformational exchange rate between peaks S1 and S3 using saturation transfer difference (STD) 35,36 at ~0.6 s−1 (Fig. 5d).
When chemical exchange is significantly faster than the intrinsic relaxation of the high-PRE state R1,A*, the measured R1 rate becomes the same for states A and B, and is uninformative about the exchange rate (Fig. 6a and Supplementary Note Equation 25). Therefore, the upper limit of the measurable chemical exchange rate depends on the intrinsic R1,A* rate of the high-PRE state and, to a lesser extent, on the populations of the states. In our experiments on dHis/M385C-TET GltPh in complex with Ni2+ ions, the R1,A* rate is ~9 s−1, and faster chemical exchange would not be measurable (Fig. 6a). However, R1,A* relaxation rates increase steeply with decreasing distance. For example, if the distance in the high-PRE state were 6 Å, R1* would approach 100 s−1 based on Equation 1, allowing measurements of correspondingly faster exchange rates (Fig. 6a). To test this, we moved the labeling site from M385C to A381C, where the distance between 19F nucleus and the chelated Ni2+ ion in the IFS is expected to be ~6 Å based on the crystal structure 37. 19F spectra of dHis/A381C-TET GltPh exhibited three peaks similar to those observed for dHis/M385C-TET GltPh but with greater overlap (Fig. 6b), and an additional peak further downfield, which we termed S0. In the presence of 3 molar equivalents of Ni2+ ions, the populations of peaks S0 and S1 increased relative to peaks S2 and S3, consistent with our results for dHis/M385C-TET GltPh and suggesting that S0 represents an IFS variant. PRE measurements in the presence of TMA revealed dramatically faster R1,A* rates of ~125 and 110 s−1 for peaks S0 and S1 (Fig. 6c and Supplementary Table 1), which correspond to distances between Ni2+ ion and 19F nucleus of 5.8 and 5.9 Å, respectively, similar to the expected distances for the IFS. Thus, strategic positioning of the dHis motif relative to the 19F probe can be used to adjust the intrinsic high-PRE R1,A* relaxation rate to tune the range of the measurable conformational exchange rates.
Figure 6: Intrinsic relaxation rate R1,A* determines the range of accessible exchange rates.
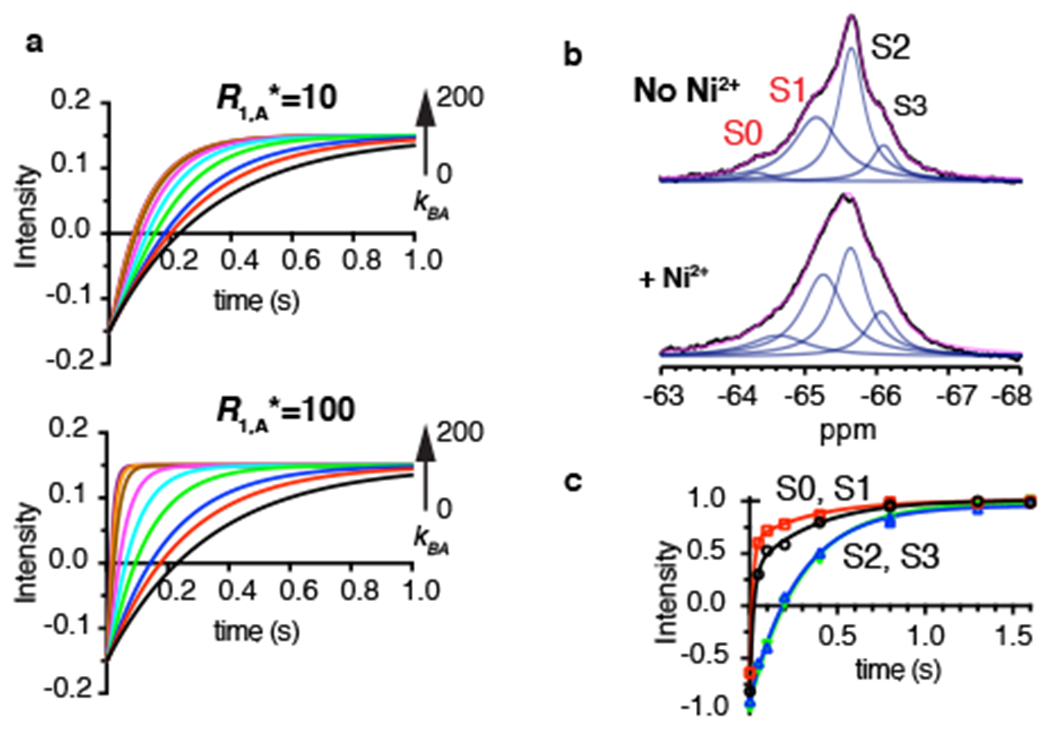
a, Simulated R1 relaxation curves of spin B (R1,B* = 3.0 s−1) in exchange with A with R1,A* = 10 s−1 (top) or 100 s−1 (bottom). The fraction of spins in state B was set to 0.15 and the rate of transition from state B to state A, kBA, varied from 0 to 200 s−1 (black 0, red 1, blue 2, green 5, cyan 10, magenta 20, brown 50, orange 100, purple 200). b, 1D 19F NMR spectra of dHis/A381C-TET GltPh in the absence (top) and presence (bottom) of 3 molar equivalents of Ni2+ ions. Raw data are black, fits are magenta and deconvoluted peaks are blue. c, Paramagnetic R1 relaxation curve of the S0 (black circles), S1 (red squares), S2 (blues triangles) and S3 (green reverse triangles) peaks of dHis/A381C-TET GltPh in the presence of Ni2+ ions. All measurements were performed in the presence of 200 mM NaCl and 10 μM L-asp. Solid lines represent bi-exponential fits for the S0 and S1 peaks and mono-exponential fits for the S2 and S3 peaks. The fitted parameters for S0 are: kfast = 124.7 ± 32.0 s−1, kslow = 2.2 ± 1.7 s−1; S1: kfast = 97.5 ± 5.4 s−1, kslow = 2.8 ± 0.5 s−1; S2: k = 5.4 ± 0.8 s−1; S3: k = 4.5 ± 0.5 s−1. The bi-exponential nature of R1 relaxations of peaks S0 and S1 may reflect the presence of small but significant chemical exchange with the low-PRE peaks. Two independently prepared samples yielded similar results.
Discussion
1D 19F NMR of dHis/M385C-TET GltPh bound to Na+ ions and L-asp showed three resonances, S1, S2 and S3, which we interpret to correspond to three distinct conformations of the transporter that do not rapidly exchange with each other on the 19F NMR time scale. Measurements of Ni2+-mediated PREs allowed us to unambiguously assign peak S1 as an IFS and peaks S2 and S3 as two OFS conformations. The effects of mutations and ligands on the observed populations are consistent with these assignments. Our spectra alone do not provide structural details of the observed states, but they can guide high-resolution structural studies employing other techniques. Thus, prompted by our NMR results, we examined GltPh structure by Cryo-EM and observed the presence of two outward-facing states: the OFS and iOFS, to which we assign peaks S2 and S3, respectively. While the populations of the states might not be accurately estimated from Cryo-EM imaging, it is notable that particles corresponding to the OFS were more abundant than particles corresponding to iOFS. Consistently, higher populations of the peak S2 (OFS) compared to the peak S3 (iOFS) are observed in 19F NMR spectra. 19F NMR can also identify mutations or ligands that favor specific states to facilitate their characterization via other approaches. For example, while both TBOA and TMA are nontransportable blockers 25,30, our experiments show that TMA does not have a conformational preference, while TBOA shifts the distributions strongly toward OFS conformations. Similarly, Na+ ions also shift the wild type dHis/M385C-TET and all mutants examined toward the outward-facing states (Supplementary Fig. 1).
Ni2+-mediated PRE has the potential to serve as a “molecular ruler”, yielding distances between the metal ion and the 19F nucleus. Other methods, such as smFRET and double electron-electron resonance (DEER), can also measure distances between fluorophore or spin labels under favorable circumstances. Unlike these methods, which are most useful in the 20 to 80 Å distance range, R1 PRE-based measurements are most applicable to shorter distances, under 20 Å, making them complementary to other approaches. In many membrane proteins, the shorter distance range is advantageous because the proteins are relatively small, making it difficult to identify suitable labeling sites separated by the requisite longer distances. Distances estimated from our R1 PRE measurements were in reasonable agreement with the values extracted from molecular dynamic simulations of the corresponding states based on existing crystal structures. Nevertheless, there are several factors that may introduce errors into the distance estimates. First, the distances are affected by the rotamers sampled by the TET probe. Similar issues are present in DEER and FRET measurements, where frozen rotamers of spin probes or preferred orientations of fluorophores can result in significant changes to the measured distances. In NMR measurements, rotamers are sampled on fast time scales compared to the experimental measurement and an averaged position of the probe is obtained. Distances calculated from Equation 1 are also affected by the uncertainty in the order parameter, S2, and the internal motion time, τi, of the TET probe, which stems in part from the fact that these have not been independently determined for the TET side chain, but rather approximated using values typical for methionine.
Comparing the magnitudes of the Ni2+-mediated PREs in the wild type dHis/M385C-TET GltPh to those in more dynamic mutants, we observed that in the latter, the PREs for peaks S2 and S3 were larger in the presence of the substrate L-asp, but not when the transporters were bound to the blocker TMA. These increased PREs for the OFS resonances of the dynamic mutants are attributed to the chemical exchange with the IFS, which shows a stronger PRE. From the paramagnetic R1 measurements, we obtained the pairwise chemical exchange rates between the resonances with high and low PREs, which agree well with those estimated from EXSY and STD. The advantages of using paramagnetic R1 measurements to follow conformational exchange over methods relying on 2D measurements are their rapidity and greater tolerance to spectral overlap. Consequently, PRE-based detection of conformational dynamics provides a viable method to screen for modulators of membrane transporters, channels and receptors. Furthermore, as R1 does not increase continually with molecular weight, this method is applicable to larger systems for which R2 becomes very large, which makes commonly used approaches such as CPMG measurements 38 quite challenging. Notably, the method is also complementary to single-molecule spectroscopic approaches, such as smFRET, which distinguishes states with significantly different distances between the labeling sites. Because 19F NMR is sensitive to changes in the local electrochemical environments, it can distinguish states such as the iOFS and the OFS that do not show large structural movements.
Our analysis shows that both OFS and iOFS conformations of the K290A and RSMR mutants of GltPh exchange with the IFS. However, it is not possible to unambiguously establish the order of the events. Thus, the OFS conformation may either transition first into the iOFS and from there into the IFS, or alternately transition directly into the IFS with rare visits of the iOFS state. Interestingly, the exchange rates between the S1 (IFS) and S2 (OFS) peaks are higher than between the S1 and S3 (iOFS) peaks for both mutants. This may suggest that iOFS is not an obligatory intermediate between the OFS and the IFS states. Notably, data interpretation for GltPh might be further complicated by the kinetic complexity revealed by smFRET recordings for this transporter 26, which may be masked by ensemble averaging in 19F NMR experiments. In simpler cases of two-state equilibria between the outward- and inward-facing states, our method could provide a complete thermodynamic and kinetic description.
In summary, our method provides an avenue to assign resonance peaks observed in 19F NMR spectra to specific known structures. As in our case, the method can also reveal additional, as yet uncharacterized states in the functional conformational ensemble and provide information regarding their dynamics. It should be broadly applicable to the study of membrane proteins undergoing conformational transitions on sub-second to second time scales.
Online Methods
Protein expression, labeling and purification
The fully functional cysteine-free GltPh C321A with seven additional histidines, which significantly improve expression yield 39, is referred to the wild type throughout the paper for brevity. M385C, Y215H/E219H (dHis) and other mutations were introduced using QuikChange kit (Qiagen). The protein constructs with carboxyl-terminal thrombin cleavage site and (His)8-tag were purified as described previously 39. Briefly, the plasmids were transformed into E. coli DH10-B cells (Invitrogen). Cells were grown in LB media supplemented with 0.2 mg/l of Ampicillin (Goldbio) at 37 °C until OD600 of 1.0. Protein expression was induced by adding 0.2 % arabinose (Goldbio) for 16 hr at 24 °C. The cells were harvested by centrifugation and re-suspended in 20 mM Hepes, pH7.4, 200 mM NaCl, 1 mM L-asp, 1 mM EDTA. The suspended cells were broken by Emulsiflex C3 high pressure homogenizer (Avestin Inc.) in the presence of 0.5 mg/ml lysozyme (Goldbio) and 1 mM Phenylmethanesulfonyl fluoride (PMSF, MP Biomedicals). After centrifugation for 15 min at 5000 g at 4 °C to remove the debris, membranes were pelleted by centrifugation at 125000 g for 60 min. The membranes were homogenized in 20 mM Hepes, pH 7.4, 200 mM NaCl, 1 mM L-asp, 10 mM EDTA, and 10 % sucrose. The suspension was centrifuged at 125000 g for 60 min. The crude membranes were collected and homogenized in Buffer A, containing 20 mM Hepes, pH7.4, 200 mM NaCl, 1 mM L-asp, at 8 ml per gram of membranes. For fluorine labeling 14,24, membranes were solubilized in 40 mM n-dodecyl-β-D-maltopyranoside (DDM, Anatrace, Inc.) for 2 hours at 4 °C in the presence of 2 mM 2,2’-dithiodipyridine (DTDP, Sigma Aldrich), which serves to activate the cysteine on GltPh by forming thiopyridine adduct. The mixture was centrifuged for 60 min at 125000 g, the supernatant was diluted 4 times with Buffer A and incubated with Ni-NTA resin (Qiagen) for 1 hour at 4 °C. The resin was washed with 5 volumes of Buffer A with 1 mM DDM to remove unreacted DTDP. The resin slurry was supplemented with 2 mM trifluoroethanethiol (Sigma Aldrich) and incubated with mixing at 4 °C overnight to form the TET-labeled GltPh. After washing the resin with 10 volumes of Buffer A containing 1 mM DDM and 25mM imidazole, the labeled protein was eluted in the same buffer containing 250 mM imidazole. The protein samples were concentrated to ca. 10 mg/ml using concentrators with 100 kDa MW cutoff (Amicon). Protein concentration was determined by UV absorbance at 280 nm using extinction coefficient of 57400 M−1 cm−1 and MW of 44.7 kDa. The (His)8-tag was cleaved by thrombin (Sigma) using 20 U per 1 mg GltPh in the presence of 5 mM CaCl2 at room temperature overnight. The reaction was stopped by addition of 10 mM EDTA and 1 mM PMSF. The protein was further purified by size exclusion chromatography (SEC) on Superdex 200 Increase 10/300 GL column (GE Healthcare Life Sciences). GltPh bound to L-Asp condition was prepared using SEC in buffer containing 20 mM Hepes, pH7.4, 100 mM NaCl, 10 μM L-Asp, 1 mM DDM. To prepare substrate-free GltPh, the SEC buffer with 20 mM Hepes, pH 7.4, 50 mM KCl, 1 mM DDM was used. NaCl and ligands in appropriate concentrations were then added into the eluted protein solution. The protein was then concentrated and either used immediately or snap-frozen in liquid nitrogen and then stored at −80 °C. To evaluate the efficiency of TET labeling, labeled and unlabeled protein samples were incubated with 10 molar equivalents of fluorescein-5-malaimide in the presence of 2% SDS for 4h at room temperature. The reaction mixtures were than analyzed by SDS-PAGE followed by fluorescence imaging.
Transport activity assay.
Labelled M385C and dHis M385C GltPh proteins were reconstituted into liposomes and 3H L-asp uptake was measured as previously described 30. Briefly, liposomes were prepared from a 3:1 (w/w) mixture of E. coli total lipid extract and egg yolk phosphotidylcholine (Avanti Polar Lipids) in 20 mM Tris/HEPES, pH7.4, and containing 200 mM KCl and 100 mM CholineCl. Liposomes were destabilized by addition of Triton X-100 at detergent to lipid ratio of 0.5:1 (w/w). GltPh proteins were added at final protein to lipid ratio of 1:2000 (w/w) and incubated for 30 min at room temperature. Detergents were removed by repeated incubations with Bio-Beads™ SM-2 resin (Bio-Rad). The proteoliposomes were subjected to three freeze-thaw cycles and extruded through 400 nm filters before the uptake assay. Uptake reaction was started by diluting the proteoliposomes 100-fold into reaction buffer containing 20 mM HEPES/Tris pH 7.4, 200 mM KCl, 100 mM NaCl, the indicated amounts of L-asp (L-asp concentrations above 1 μM were supplemented with 5 μM cold L-asp, maximally diluting 3H-L-asp 10-fold) and 0.5 μM valinomycin. Uptake was performed for 1 min at 35 °C.
19F NMR spectroscopy
19F-NMR experiments were performed using a Bruker Avance IIIHD 500 MHz spectrometer equipped with a TCI 1H-19F/13C/15N triple resonance cryogenic probe (Bruker Instruments). 50 μM TFA and 10 % D2O were included in the sample and used as chemical shift reference (−75.4 ppm) and lock reagent, respectively. 1D 19F-NMR spectra were recorded with 4096 points and spectral width (SW) of 40 ppm. Acquisition time was 109 ms under these settings. Carrier frequency was set to −70 ppm. The number of scans was set between 512 and 4096 depending on the sample conditions. The recycle delay was 0.6 s for 1D 19F spectra except when otherwise indicated. 90° pulse length was calibrated for each sample and was typically 11.4 μs. All the spectra were recorded without 1H decoupling. 1D spectra were processed using MestRaNova 12.0.0 software (Mestrelab Research) employing a 20 Hz exponential windows function, zero filling to 16 k points. The spectra were baseline-corrected, the peaks fitted to Lorentzian peak shapes and assessed based on fit residuals.
19F longitudinal relaxation rates were measured by the inversion recovery method. The recycle delay was 1.8 s, which is ~5 times T1. Eight different delays between 0.05 ms and 1.8 s were randomly sampled, with one point repeated 3 times. The peak intensities were obtained from spectral deconvolution. The I = I0 (1 − 2exp(−R1t)). The R1 PREs were obtained by subtracting the relaxation rate measured in the absence of Ni2+ ion from the rate measured in the presence of the ion. The errors were calculated by error propagation. All experiments were repeated at least twice.
19F-19F EXSY experiments were recorded with 4096 points and spectral width of 40 ppm in the direct dimension and with 24 complex points and SW of 5 ppm in the indirect dimension. Recycle delay was 1.2 s. Typically, 512 to 1024 scans per increment were accumulated depending on sample concentrations. The spectra were processed using Topspin 3.5 software (Brucker Instruments). The indirect dimension was zero-filled to 128 points and a Gaussian window function was applied with line broadening factor (LB) of −20 Hz and gaussian multiplication factor (GM) of 0.02. In the direct dimension, the GM window function was applied with LB of −20 Hz and GB of 0.01. The processed spectra were analyzed using Sparky software (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco) and the peak intensities were obtained by integration with Lorentzian fit. A spectrum with mixing time was 0.4 s, unless otherwise stated, and a spectrum with 0 s mixing time were recorded. The peak volumes were fitted using EXSYcalc software (Mestrelab Research).
19F Saturation transfer difference (STD) spectra were recorded using a train of 50 ms 180° Gaussian shaped pulses with total duration of 0, 100, 200, 300, 400, 600 and 1000 ms. For each duration, on and off resonance irradiation pulses (as shown in Figure 5a) were alternately interleaved to generate saturation and control datasets 35,36. The corrected intensities of the observed peak A upon saturation duration on peak B were fitted to the following equation to determine kAB:
| (5) |
where R1 is the longitudinal relaxation rate of the observed peak. The backward rate is obtained using the equilibrium populations of A and B, i.e., kBA = kAB*fA/fB. The fitting was performed in Prism 6 (GraphPad Software Inc.)
Molecular dynamics simulations
The molecular models of the outward-facing GltPh bound to 3 Na+ ions and L-asp was constructed from PDB id: 2NWX, and of the inward-facing GltPh bound to 3 Na+ ions and L-asp from PDB id: 3KBC. Missing loops and residues were added with the Modeler software package 40. The system was immersed in a POPC membrane in a neutralizing solution of NaCl at 0.15 M using the CHARMM-GUI membrane builder web interface 41, yielding a simulation box with dimensions: 165 × 175 × 86 Å3 and about 250,000 atoms for each system. CHARMM36 parameters 42 were used for protein and lipids in all simulations. A standard equilibration protocol developed for various membrane systems 43 was applied as follows: the solvent and membrane were equilibrated with harmonic restraint applied to protein and heavy atoms (except those modeled). These restraints were gradually decreased from 20 kcal/mol/Å2 to 0.1 kcal/mol/Å2 over the course of 5 ns simulation, followed by an additional 13 ns with restraints on the Cα atoms and bound sodium atoms, and an additional 6 ns without any restraints. The last frames of these equilibration phases were used to add the NMR probe and metal ions as follow: M385 was mutated to cysteine modified with NMR probe TET, and residues 215 and 219 were mutated to histidine. The force field parameters for the TET probe, with net zero charge, was adapted from CHARMM generalized force field parameters for difuorobenzylphosphonate and the regular methionine amino acid. To mimic the Ni2+ site between H215 and H219, harmonic restraints (10 kcal/mol/Å2) were used to restrain Zn2+ (with available CHARMM parameters) near these His residues. The systems were then minimized and the final production runs, about 100 ns for each system, were performed using NAMD software version 2.13 44 at 310 K in NPT ensemble with 2 fs time steps. Particle mesh Ewald 45 was used for long range electrostatic interactions. Short range electrostatic interactions were calculated using Lennard-Jones potential with truncation distance cutoff at 11 Å with smoothing starting at 8 Å. Pressure was controlled with Langevin barostat at 1 bar with oscillation period of 200 fs and damping time of 100 fs. The X, Y, Z cell dimensions were allowed to fluctuate independently. Temperature was controlled with Langevin thermostat with damping coefficient of 0.1 1/ps. The visualization and quantitative analyses of all simulations are done with VMD 46 and MDTraj software 47.
Cryo-EM structure determination
GltPh reconstitution into nanodiscs:
Membrane scaffold protein MSP1E3 was expressed and purified from E. coli and GltPh was reconstituted into lipid nanodiscs as previously described, with modifications 48. Briefly, E. coli polar lipid extract and egg phosphatidylcholine in chloroform (Avanti) were mixed at 3:1 (w:w) ratio and dried on rotary evaporator and under vacuum overnight. The dried lipid film was resuspended in buffer containing 200 mM NaCl, 1 mM L-asp, 80 mM DDM, 20 mM Hepes/Tris, pH 7.4 by 10 freeze/thaw cycles resulting in 20 mM lipid stock. Purified GltPh protein in DDM was mixed with MSP1E3 and lipid stock at 1:1:50 molar ratio at the final lipid concentration of 5 mM and incubated at 21 °C for 30 min. Biobeads SM2 (Bio-Rad) were added to one third of the reaction volume and the mixture was incubated at 21 °C for 2 hr on a rotator. Biobeads were replaced and incubated at 4 °C overnight. The reconstitution mixture was cleared by centrifugation at 100,000 g and GltPh-containing nanodiscs were purified using a Superose 6 Increase 10/300 GL column (GE Lifesciences) pre-equilibrated with buffer containing 200 mM NaCl, 1 mM L-asp, 20 mM Hepes/Tris, pH7.4.
Cryo-EM data collection:
To prepare cryo-grids, 3.5 μL of GltPh-containing nanodiscs (6 mg/mL) supplemented with 1.5 mM fluorinated Fos-Choline-8 (Anatrace) was applied to a glow-discharged UltrAuFoil R1.2/1.3 300-mesh gold grid (Quantifoil) and incubated for 20 s under 100 % humidity at 15 °C. Following incubation, grids were blotted for 2 s and plunge frozen in liquid ethane using a Vitrobot Mark IV (FEI). Cryo-EM imaging data were acquired on a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 kV with a K3 Summit direct electron detector (Gatan). Automated data collection was carried out in super-resolution counting mode using SerialEM software 49 with a magnification of 81,000 x, which corresponds to a calibrated pixel size of 1.06 Å on the specimen and 0.53 Å for super-resolution images. Each movie had a total dose of 50.1615 e−/Å2 distributed over 30 frames (1.672 e−/ Å2/frame) with an exposure time of 8.1 s (269 ms/frame) and a defocus range of −1.5 to −2.5 μm.
Image processing:
The frame stacks were motion corrected using MotionCorr2 50 and contrast transfer function (CTF) estimation was performed using CTFFIND4 51. All processing steps were done using RELION 3.0 unless otherwise indicated 52. Dogpicker 53 as part of the Appion processing package 54 was used for reference-free particle picking. Picked particles were then extracted and subjected to 2D classification to generate 2D class-averages which were used as templates for automated particle picking in Relion. The particles were extracted using a box size of 256 Å with 2x binning and subjected to 2 rounds of 2D classification ignoring CTFs until the first peak. 926,308 particles selected from 2D classification were further classified into 6 classes without applying symmetry using an initial model from a density map previously obtained for a GltPh-containing nanodisc (unpublished) filtered to 40 Å. 342,356 particles from the best class showing a trimeric transporter arrangement were re-extracted, unbinned, and subjected to 3D refinement applying C3 symmetry. After conversion, the refinement was continued with a mask excluding the nanodisc, resulting in a 3.35 Å resolution map. The resolution of the refined map was assessed using Relion postprocessing and gold standard FSC value 0.143 using a mask that excluded the nanodisc. The 3D refinement with C3 symmetry produced a map of GltPh in the OFS with bound substrate closely resembling the PDB model 2NWX. To probe for conformational heterogeneity, we employed the symmetry expansion implemented in Relion. 1,027,068 protein subunits were rotated to the same position and subjected to a focused 3D classification without alignment with T=40 into 10 classes. The local mask was generated using Chain A of PDB model 2NWX and included only densities from one subunit on the refence map. Nine classes were either low resolution or pictured GltPh in the OFS conformation. One class, which contained 11.7% of symmetry expanded particles, showed a different conformation, closely resembling the iOFS conformation. The best OFS class (94,731 particles) and the iOFS class (120282 particles) were separately subjected to a final focused 3D refinement with C1 using a mask to exclude the nanodisc. The local angular searches in this refinement were conducted only around the expanded set of orientations to prevent contributions from neighbor subunits in the same particle. The resulting maps were postprocessed in Relion using the same mask as in 3D classification after symmetry expansion. The final resolution at gold standard FSC value 0.143 was estimated as 3.1 Å for the OFS map and 3.6 Å for iOFS map. Local resolution variations were estimated using ResMap 55.
Model building and refinement:
For atomic model building, one subunit of 2NWX GltPh crystal structure was docked into the density map of the OFS using UCSF Chimera 56. For iOFS, one subunit of 3V8G crystal structure was docked into the density. For each model after the first round of real-space refinement using Phenix 57, miss-aligned regions were manually adjusted and missing side chains and residues were manually added in COOT 58. Phosphatidylethanolamine is used as a model lipid to be placed into densities which resemble lipid molecules with its acyl chains or ethanolamine heads truncated to fit the visible densities. Models were iteratively refined applying secondary structure restraints and validated using Molprobity 59. For further cross validation and to check for overfitting, all atoms of each model were randomly displaced by 0.3 Å and each resulting model was refined against the first half-map obtained from processing. FSC between the refined models and the half-maps used during the refinement were calculated and compared to the FSC between the refined models and the other half-maps. In addition, the FSC between the refined model and sum of both half-maps was calculated. The resulting FSC curves were similar showing no evidence of overfitting.
Data availability
Atomic coordinates for the Cryo-EM structures of the OFS and iOFS states have been deposited in the Protein Data Bank under accession codes 6UWF and 6UWL, respectively, and the corresponding Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-20922 and EMD-20923, respectively. The other data that support the findings of this study are available from the corresponding authors upon reasonable request.
Extended Data
Extended Data Fig. 1. Protein purity, and 3H L-asp uptake and simulated 19F-Ni2+ distance distribution.
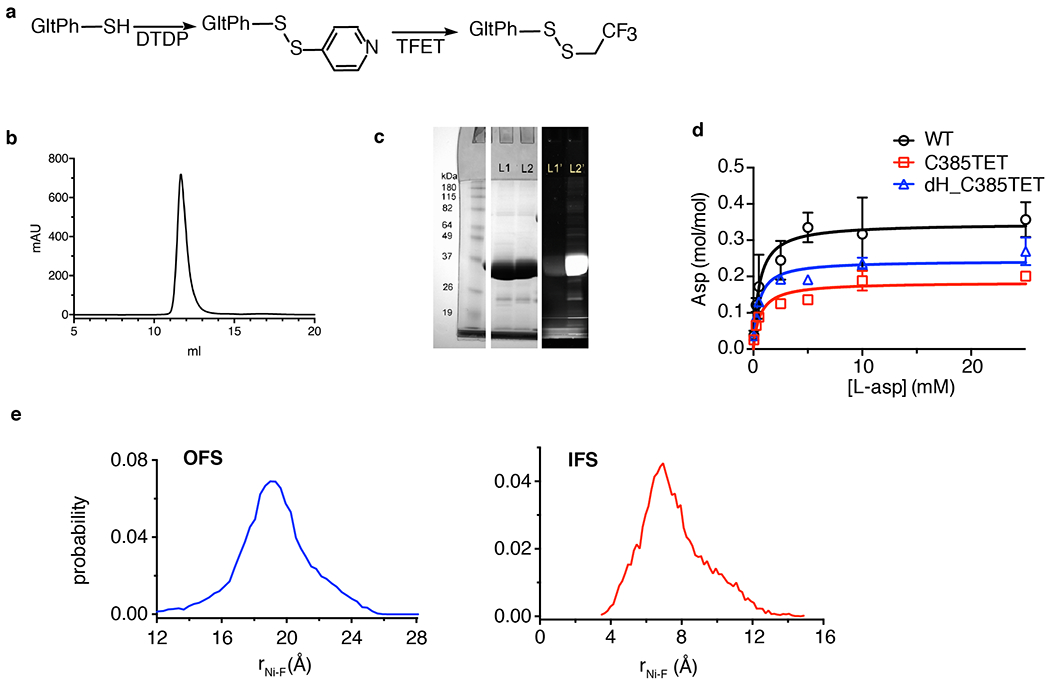
a, Scheme for site-specifically introducing 19F label into M385C GltPh mutant. b, Representative size exclusion chromatography elution profile of M385C-TET GltPh. More than 3 independent samples were repeated with similar results. c, SDS-PAGE gel imaged by Coommasie blue staining (middle) and fluorescence (right) and of M385C GltPh labeled with fluorescein-5-malaimide before (lane 1) and after (lane 2) labeling with TFET. Protein samples were incubated with 10-fold excess of fluorescein-5-maleimide for 4h prior to analysis. Two independent samples were prepared and yielded similar results. d. Michaelis-Menten kinetics of 3H L-Asp uptake for wide type (WT) GltPh (black circle), M385C-TET (red square), and dHis/M385C-TET GltPh (blue triangle). Data shown are means ± s.d. (N = 3 biological replicates). e, Distance probability distributions between 19F and Ni2+ calculated from 100 ns of the molecular dynamics simulation trajectories. To mimic experimental conditions M385 was mutated to NMR probe TET, residues 215 and 219 were mutated to histidine, and Zn2+ ion was constrained between these histidines (see Online Methods for details). The distance distributions were calculated for all three protomers and shown curves are averaged values from three protomers in the OFS (left) and the IFS (right). DTDP: 2,2’-dithiodipyridine; TFET: trifluoroethanethiol.
Extended Data Fig. 2. Specific Ni2+ binding to dHis/M385C-TET GltPh mutant.
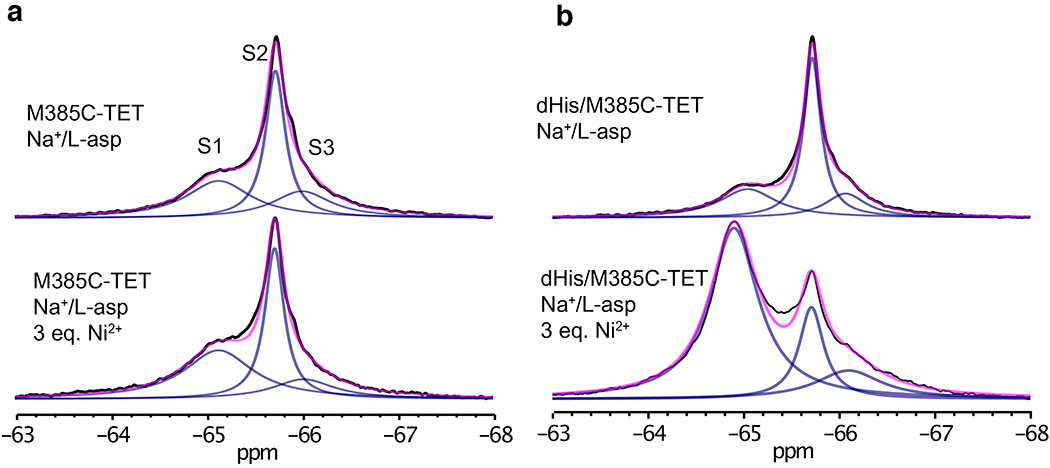
1D 19F-NMR spectra of M385C-TET GltPh (a) and dHis/M385C-TET GltPh (b) without (up) and with (bottom) 3 molar equivalents of Ni2+ ions. Spectra were recorded at 293K in the presence of NaCL and L-asp. Raw data are black, fits are magenta and deconvoluted peaks are blue. Note: the dHis/M385C-TET spectra are the same as the ones shown in Fig. 2a and Fig. 3a in the main text.
Extended Data Fig. 3. Cryo-EM data processing.
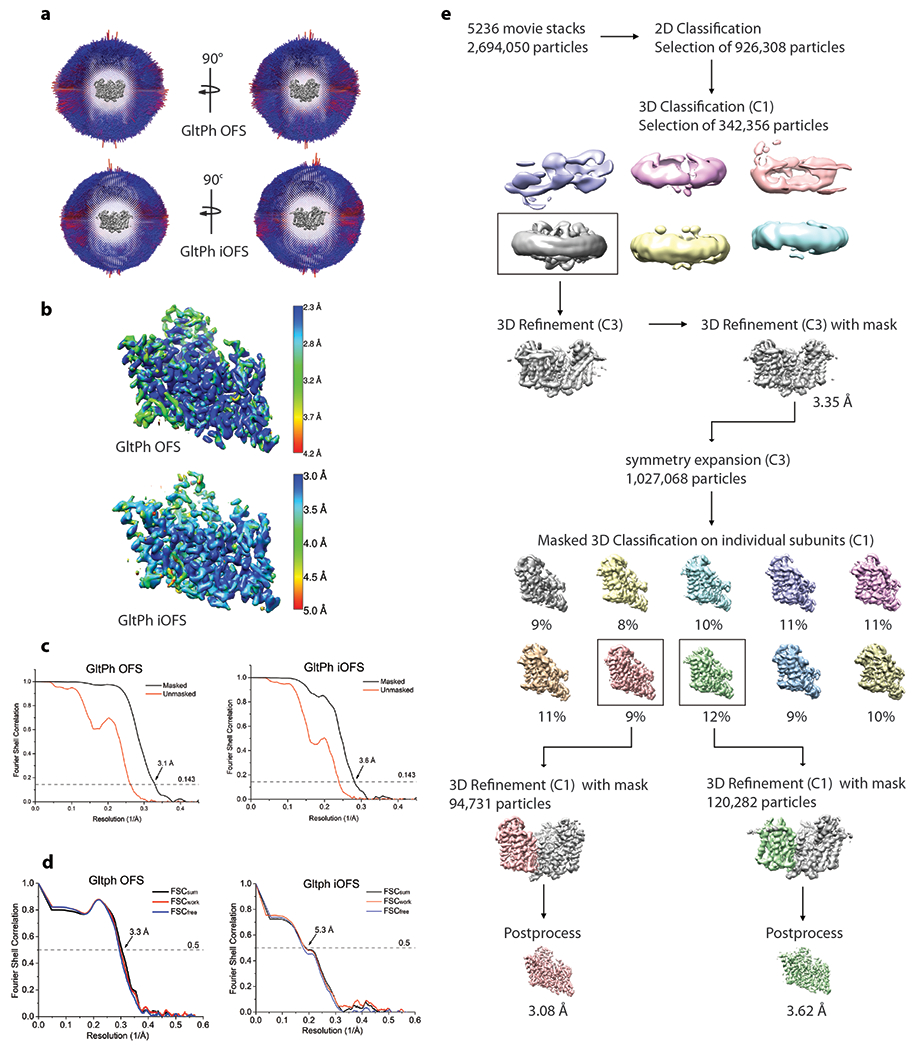
a, Angular distribution of particles contributing to the final reconstitution. Number of views at each angular orientation is represented by length and color of cylinders where red indicates more views. b, Final maps after Relion post-processing colored according to local resolution estimation using ResMap. c, Fourier shell correlation (FSC) curves indicating the resolution at the 0.143 threshold of final masked (black) and unmasked (orange) maps of GltPh OFS (left) and iOFS (right). d, FSC curves from cross validation of refined GltPh OFS (left) and iOFS (right) models compared to the masked half-map 1 (Orange traces: FSCwork, used during validation refinement), masked half map 2 (Blue traces: FSCfree, not used during validation refinement), and the masked summed map (Black traces: FSCsum). e, Data processing flow chart for GltPh reconstituted into nanodisc in the presence of NaCl and L-asp.
Extended Data Fig. 4. 2D 19F EXSY spectrum of dHis/M385C-TET GltPh.
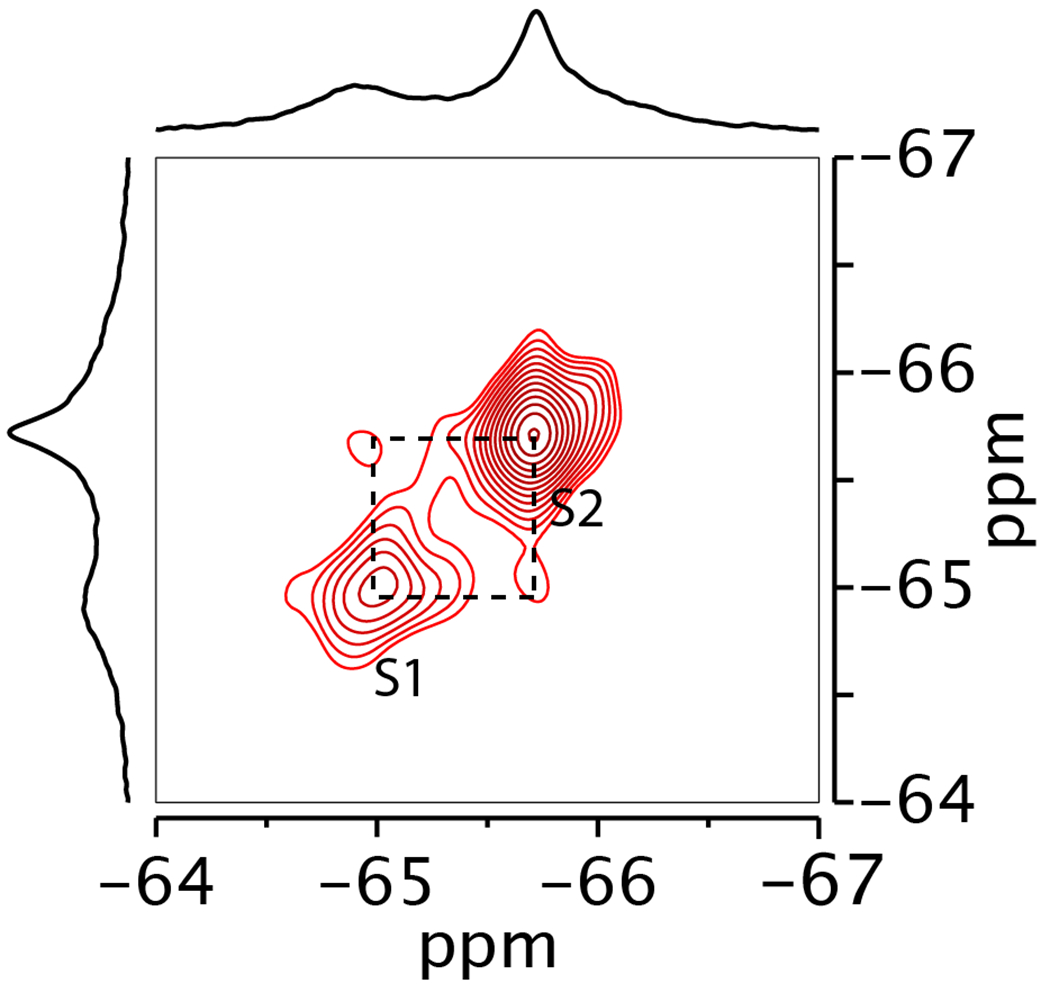
Spectrum was recorded with mixing time of 0.4 s in the presence of 200 mM Na+ and 10 μM L-asp at 298K.
Supplementary Material
Acknowledgements
The authors thank Dr. X. Yao for helpful discussions, Dr. M. Goger and Dr. S. Bhattacharya for help with setting up NMR, and Dr. M. A. Cuendet for help with setting up initial MD simulations. We thank Dr. C. Xu and Dr. K. Song at the UMass cryo-EM facility for help with electron microscopy data collection. We also thank W. Eng for assistance with protein expression. This work was supported by NIH grants R37NS085318 and R01NS064357 (OB), R37AG019391 (DE) and S10OD016320 (CB). OB, DE and CB are members of the New York Structural Biology Center (NYSBC) which is supported in part by NIH Grant P41 GM118302 (CoMD/NMR: Center on Macromolecular Dynamics by NMR Spectroscopy), ORIP/NIH facility improvement grant CO6RR015495 and NIH grant S10OD018509. The coordinates of the structures and the density maps have been submitted to Protein Data Bank under the accession codes of 6UWF and 6UWL.
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Palmer AG Enzyme Dynamics from NMR Spectroscopy. Acc. Chem. Res. 48, 457–465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pervushin K, Riek R, Wider G & Wuthrich K Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Y & Kalodimos CG NMR Studies of Large Proteins. J. Mol. Biol. 429, 2667–2676 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Danielson MA & Falke JJ Use of 19F NMR to probe protein structure and conformational changes. Annu. Rev. Biophys. Biomol. Struct. 25, 163–195 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Susac L, Eddy MT, Didenko T, Stevens RC & Wuthrich K A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 115, 12733–12738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manglik A et al. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 161, 1101–1111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Pietrantonio C, Pandey A, Gould J, Hasabnis A & Prosser RS Methods Enzymol. 615, 103–130 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Yernool D, Boudker O, Folta-Stogniew E & Gouaux E Trimeric Subunit Stoichiometry of the Glutamate Transporters from Bacillus caldotenax and Bacillus stearothermophilus. Biochemistry 42, 12981–12988 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Danbolt NC Glutamate uptake. Prog. Neurobiol. 65, 1–105 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Zerangue N & Kavanaugh MP Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Levy LM, Warr O & Attwell D Stoichiometry of the Glial Glutamate Transporter GLT-1 Expressed Inducibly in a Chinese Hamster Ovary Cell Line Selected for Low Endogenous Na-Dependent Glutamate Uptake. J. Neurosci. 18, 96209628 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akyuz N, Altman RB, Blanchard SC & Boudker O Transport dynamics in a glutamate transporter homologue. Nature 502, 114-+118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erkens GB, Hänelt I, Goudsmits JMH, Slotboom DJ & van Oijen AM Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature 502, 119–123 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Liu JJ, Horst R, Katritch V, Stevens RC & Wuthrich K Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clore GM & Iwahara J Theory, Practice, and Applications of Paramagnetic Relaxation Enhancement for the Characterization of Transient Low-Population States of Biological Macromolecules and Their Complexes. Chem. Rev. 109, 4108–4139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bondarenko V et al. 19F Paramagnetic Relaxation-Based NMR for Quaternary Structural Restraints of Ion Channels. ACS Chem. Biol. 14, 2160–2165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solomon I Relaxation Processes in a System of Two Spins. Phys. Rev. 99, 559–565 (1955). [Google Scholar]
- 18.Bloembergen N Proton Relaxation Times in Paramagnetic Solutions. J Chem. Phys. 27, 572–573 (1957). [Google Scholar]
- 19.Matei E & Gronenborn AM 19F Paramagnetic Relaxation Enhancement: A Valuable Tool for Distance Measurements in Proteins. Angew. Chem. Int. Ed. 55, 150–154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hull WE & Sykes BD Fluorotyrosine alkaline phosphatase: Internal mobility of individual tyrosines and the role of chemical shift anisotropy as a 19F nuclear spin relaxation mechanism in proteins. J. Mol. Biol. 98, 121–153 (1975). [DOI] [PubMed] [Google Scholar]
- 21.Hull WE & Sykes BD Dipolar nuclear spin relaxation of 19F in multispin systems. Application to 19F labeled proteins. J. Chem. Phys. 63, 867–880 (1975). [Google Scholar]
- 22.Gerig JT Fluorine-proton Overhauser effects in fluorine-labeled macromolecular systems. J. Am. Chem. Soc. 99, 1721–1725 (1977). [Google Scholar]
- 23.Todd RJ, Van Dam ME, Casimiro D, Haymore BL & Arnold FH Cu(II)-Binding properties of a cytochrome c with a synthetic metal-binding site: His-X3-His in an α-helix. Proteins 10, 156–161 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Didenko T, Liu JJ, Horst R, Stevens RC & Wuthrich K Fluorine-19 NMR of integral membrane proteins illustrated with studies of GPCRs. Curr. Opin. Struct. Biol. 23, 740–747, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McIlwain BC, Vandenberg RJ & Ryan RM Characterization of the Inward- and Outward-Facing Substrate Binding Sites of the Prokaryotic Aspartate Transporter, GltPh. Biochemistry 55, 6801–6810 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Akyuz N et al. Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature 518, 68–73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruan Y, Miyagi A, Wang X, Chami M, Boudker O, and Scheuring S Direct visualization of glutamate transporter elevator mechanism by high-speed AFM. Proc. Natl. Acad. Sci. USA 114, 1584–1588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Georgieva ER, Borbat PP, Ginter C, Freed JH & Boudker O Conformational ensemble of the sodium-coupled aspartate transporter. Nat. Struct. Mol. Biol. 20, 215–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reyes N, Oh S & Boudker O Binding thermodynamics of a glutamate transporter homolog. Nat. Struct. Mol. Biol. 20, 634–640 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boudker O, Ryan RM, Yernool D, Shimamoto K & Gouaux E Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Suh S-S, Haymore BL & Arnold FH Characterization of His-X3-His sites in α-helices of synthetic metal-binding bovine somatotropin. Protein Eng. Des. Sel. 4, 301–305 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Verdon G & Boudker O Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 19, 355–357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machtens JP et al. Mechanisms of Anion Conduction by Coupled Glutamate Transporters. Cell 160, 542–553 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M & Skelton NJ in Protein NMR Spectroscopy (Second Edition), 333–404 (Academic Press, 2007). [Google Scholar]
- 35.Mayer M & Meyer B Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 38, 1784–1788 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Spoerner M, Hozsa C, Poetzl JA, Reiss K, Ganser P, Geyer M, and Kalbitzer HR Conformational states of human rat sarcoma (Ras) protein complexed with its natural ligand GTP and their role for effector interaction and GTP hydrolysis. J. Biol. Chem. 285, 39768–39778, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes N, Ginter C & Boudker O Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye L, Van Eps N, Zimmer M, Ernst OP & Prosser RS Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Yernool D, Boudker O, Jin Y & Gouaux E Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Sali A & Blundell TL Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 41.Jo S, Lim JB, Klauda JB & Im W CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 97, 50–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klauda JB et al. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi L, Quick M, Zhao Y, Weinstein H & Javitch JA The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 30, 667–677 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips JC et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 46.Humphrey W, Dalke A & Schulten K VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 47.McGibbon Robert T. et al. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 109, 1528–1532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie TK et al. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, eLife.42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voss NR, Yoshioka CK, Radermacher M, Potter CS & Carragher B DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Afonine PV et al. phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 43, 669–676 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates for the Cryo-EM structures of the OFS and iOFS states have been deposited in the Protein Data Bank under accession codes 6UWF and 6UWL, respectively, and the corresponding Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes EMD-20922 and EMD-20923, respectively. The other data that support the findings of this study are available from the corresponding authors upon reasonable request.